

Abstract
Opioids are typically used for the treatment of pain related to disease or surgery. In the body, they enter the bloodstream and interact with a variety of immune and neurological cells that express the μ-, δ-, and κ-opioid receptors. One blood-borne cell-like body that is not well understood in the context of opioid interactions is the platelet. The platelet is a highly sensitive anucleate cell-like fragment responsible for maintaining hemostasis through shape change and the secretion of chemical messengers. This research characterizes platelet opioid receptors, how specific receptor agonists impact platelet exocytosis, and the role of the κ-and μ-receptors in platelet function. Platelets were found to express all three opioid receptors, but upon stimulation with their respective agonist no activation was detected. Furthermore, exposure to the opioid agonists did not impact traditional platelet secretion stimulated by thrombin, a natural platelet activator. In addition, data collected from knockout mice suggest that the opioid agonists may be interacting nonspecifically with platelets. Dark-field images revealed differences in activated platelet shape between the κ- and μ-knockout platelets and the control platelets. Finally, κ-knockout platelets showed variations in their ability to adhere and aggregate compared to control platelets. Overall, these data show that platelet function is not likely to be heavily affected by blood-borne opioids.
Keywords: opioids, platelets, degranulation, opioid receptors
Introduction
Opioids, a class of drug molecules including heroin, opium, codeine, morphine, oxycodone, and fentanyl, are used for the treatment of moderate to severe pain caused primarily by cancer, surgery, and arthritic diseases.1,2 In 2019, around 153 million opioid prescriptions were written in the United States.3 In addition, the abuse of opioids, including prescription and illicit opioids, has resulted in over 450 000 deaths between 1991 and 2018.4 Upon introduction into the body, opioids bind to at least one of the three G protein-coupled receptors (mu (μ), kappa (κ), and/or delta (δ)) on various cell types.5−8 In addition, opioids are able to cross-react between the receptors, activating different signaling pathways. One example includes morphine, which binds to the μ- and δ-opioid receptors. However, the affinity of morphine, a common pain reliever, is 50× greater for the μ-receptor than the δ-receptor.9,10 In contrast, agonists for the κ-receptor have been noted to play a more direct role in the immune system. Several immune cells are known to express opioid receptors, and many, including natural killer cells, mast cells, and neutrophils, have shown a cell response when exposed to opioids over an extended period of time.5,7,11−13
One immune cell-like body that has been largely overlooked in opioid research is the platelet. Platelets play a crucial role in hemostasis but are also intimately involved in serious health risks including stroke and myocardial infarction upon unwanted clotting or excessive bleeding. Upon activation, platelets secrete chemical messengers from three distinct granule types, δ-granules, α-granules, and lysosomes (Figure 1). δ-Granules contain small molecules including serotonin, adenosine diphosphate (ADP), adenosine triphosphate (ATP), Ca2+, and histamine, all of which play a role in vascular constriction, inflammation, and activation. α-Granules contain platelet factors (PFs) including PF4, clotting proteins, and adhesion molecules. Lysosomes, which are rare in platelets, contain hydrolases including β-hexosaminidase (β-Hex).14 Due to the heavy use of intravenously injected opioids during surgery, with 40–60% of the opioid content entering the bloodstream, platelets have ample opportunity to interact directly with these drugs.7
Figure 1.

Diagram of general platelet degranulation. (A) Resting platelets contain granules, represented by the beige spheres that have different chemical messengers inside of them, as represented by the blue triangles, yellow stars, and green squares. (B) Platelets are stimulated by an agonist such as thrombin. (C) Granule membranes fuse with the platelet plasma membrane, releasing the chemical messengers to the platelet exterior. Image created with BioRender.
Past platelet–opioid interaction research has been focused on studying the effects of various opioid anesthetic agents on perioperative bleeding.15−17 More recent research has focused on opioid and opioid addiction therapies’ effect on platelet aggregation, platelet cell count, and mean platelet volume.18−20 To the best of our knowledge, only two papers have studied platelets directly by observing the binding attachment of naloxone, an opioid antagonist, on human platelets.21,22 The purpose herein is to explore the fundamentals of how the opioid receptors and receptor agonists impact platelet function by establishing which G protein-coupled opioid receptors are present on platelets, how stimulation of each individual receptor impacts granule secretion, and which roles the receptors play in normal platelet function. For stimulation, to prevent cross-reaction when studying receptors, the agonists [d-Ala2, NMe-Phe4, Gly-ol5]enkephalin (DAMGO), [d-Pen2,5]enkephalin, [d-Pen2,d-Pen5]enkephalin (DPDPE), or U-50488 were used to stimulate the μ-, δ-, and κ-receptors, respectively. Platelet cholesterol concentration and secreted serotonin levels were measured to track granular secretion trends. To determine the role of individual receptors, κ- (Jackson Laboratory B6.129S2-Oprk1tm1kff/j) and μ- (Jackson Laboratory B6.129S2-OPRM1TM1KFF/J) opioid receptor knockout (KO) mice were studied as well; due to limited availability, δ-KO mouse studies could not be included in this work.
Results and Discussion
Literature precedent on the effects of opioids and their receptors on platelets have focused on the aftereffects of anesthetic agents on clotting. While this is important to understand for surgical use, it is also important to understand the fundamentals of how the agonists are interacting with the platelets by leveraging analytical and biological chemistry techniques. This knowledge of the role each opioid receptor has on platelet function can then be exploited to predict how certain drugs will interact with platelets and influence platelet function.
Verification of Opioid Receptors on Platelets
Platelets are sensitive to a vast array of molecules that cause activation, including thrombin, collagen, and ADP. These compounds induce different coagulation pathways, causing varied amounts of aggregation and exocytosis from the α-, δ-, and lysosome granules. Activation also induces upregulation of different fibrinogen binding sites and cytoskeletal rearrangement for better adhesion/interaction.23 In addition to natural stimulants, many drugs affect normal platelet function, varying from direct platelet activation to prevention of exocytosis.24 To fully understand the effects of opioids on platelets, it is important to first determine which receptors the platelets express. Previous literature has demonstrated that platelets are impacted by the antagonist naloxone, which primarily binds to the μ-receptor, but also has low affinity for the δ- and κ-receptors.22
Western blots performed as part of this study using μ-, δ-, and κ-antibodies demonstrated the presence of all three receptors on control mouse platelets. In addition, Western blots on the μ- and κ-KO mice demonstrated that the κ- KO mice had a knocked-out κ receptor and still contained both the μ- and δ-receptor (data shown in Figure SI 1). However, the same experiments on the μ-KO mouse demonstrated that all three receptors were still present. A different antibody and a new μ-KO mouse were purchased for confirmation with the same outcome. In addition, tail clippings from the original mice were sent to TransnetYX (Cordova, TN) to confirm the μ-gene was knocked out. While the gene knockout was confirmed, the results indicated all three receptors were still present in the Western blot (Figure SI 1). This suggests either that both μ-antibodies are binding nonspecifically or that there may be a μ-like receptor expressed by the platelet.
Platelet Secretion in Response to Opioid Agonists
The role of each receptor in platelet granule secretion was analyzed using receptor-specific agonists. For initial experiments, control platelets were incubated for 2 h with the opioid agonists DAMGO, DPDPE, or U-50488 that target the μ-, δ-, and κ-receptors, respectively. The 2 h incubation time was chosen for two reasons. The first reason is that the agonist can stay in the bloodstream for several hours to a day. We wanted to capture this longer length of time but limited the time in order to not damage the platelets as they sat out at room temperature. The second reason is that a typical surgery can last several hours, and we wanted to capture any bleeding problems that may be seen during surgery or directly post surgery. The supernatant was analyzed for serotonin from δ-granule secretion after the platelets were spun down (Figure 2A). The platelets were resuspended and then stimulated with thrombin, a natural platelet stimulant, for 20 min. The supernatant was collected and analyzed (Figure 2B). Secretion from δ-granules was not apparent in platelets incubated with the agonists alone, nor did these agonists influence natural platelet stimulation by thrombin. A calibration curve was constructed using serotonin standards (Figure SI 2), from which the serotonin concentration of each sample was extrapolated.
Figure 2.

Effects of opioid agonists on wild type platelet secretion and response to the natural stimulant thrombin. Control platelets were incubated with Tyrode’s buffer (negative control), DAMGO (μ opioid receptor agonist), DPDPE (δ opioid receptor agonist), or U-50488 (κ opioid receptor agonist) for 2 h. The platelets were pelleted, and the supernatant serotonin concentration was analyzed. Each condition had five biological replicates. The platelet pellet was resuspended in (A) Tyrode’s buffer (F(3,21)=0.8590, p ≥ 0.01) or (B) stimulated with thrombin for 20 min, and the serotonin release was measured. (F(4,20)=167, p ≤ 0.01) ++++p ≤ 0.01 vs Tyrode’s buffer condition.
These data suggest that opiates do not cause the platelets to secrete unnecessarily, and they do not impact the ability of the platelets to secrete δ-body granules under normal stimulating conditions. These data are comparable to previously published data examining postoperative bleeding and clotting, which did not show significant differences when comparing platelets that had and had not come into contact with opioids.15−17 However, there was a nonstatistically significant decrease in serotonin secretion in the agonist-activated platelet supernatant compared to our negative control, Tyrode’s buffer (Figure 2A). These data suggest that the opioids may help stabilize unstimulated platelets, but upon thrombin stimulation, the total amount of serotonin secreted is not impacted. The facts that the differences are not statistically different and that the measured serotonin concentrations are above the limit of detection but not the limit of quantification suggest that further measurements are necessary to better understand this phenomenon. To further investigate this trend, the first part of the experiment was run again with a 30 min thrombin or opioid agonist stimulation (Figure 3A). A shortened incubation time was used to better understand the immediate impacts of opioids on platelet secretion and to limit platelets from uptaking secreted components. To help normalize the various secretion tests performed, which might have slight platelet concentration variations, the total protein content was measured. Again, the nonstatistically significant decrease in serotonin secretion from δ-granules was detected. Some of the supernatant from the platelet suspension was also used to analyze the amount of platelet factor 4 secreted from α-granules (Figure 3B) and β-Hex from lysosomes (Figure 3C). For both α-granules and lysosomes, there was no significant difference in content secretion with opioid stimulation compared to the negative control (p > 0.05). This indicates that platelets interacting with opioids are not always more stable, as appeared possible from subtle differences among δ-granule measurements, but also may have little to no secretion unless a stimulant like thrombin is present. The data in Figure 3 clearly demonstrate that stimulation with thrombin leads to a significant increase in δ-granule and lysosome secretion compared to the negative control and agonist conditions.
Figure 3.

Wild type mouse platelet granule secretion, with respect to total protein content, upon opioid agonist stimulation: (A) δ-granule secreted serotonin (F(4,15) = 13.36, p ≤ 0.0001), (B) α-granule-secreted PF4 (F(4,14) = 1.258, p = 0.3324), and (C) lysosome-secreted β-Hex from C57 control mouse platelets after a 30 min stimulation (F(4,15) = 131.2, p ≤ 0.0001). All data is relative to the BCA-based protein content to normalize for variable platelet count. **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 vs thrombin condition. n = 4 for all conditions except Tyrode’s buffer, which had n = 10.
The roles of the individual receptors and specific agonist–receptor interactions were analyzed using κ- and μ-KO mouse platelets (unfortunately, we did not have access to δ-KO mice for comparison when the experiments were being performed, though they are commercially available) to see if KO platelets would react differently from control platelets. δ-Granule secretion response was monitored in lysed (Figure 4A), resting (Figure 4B), and thrombin-stimulated (Figure 4C) platelets relative to the total amount of serotonin they contained (to ensure that the KO platelets were functioning normally). Comparing the percent of total serotonin secreted gives general insight into whether the biophysical characteristics of the granule secretion process may be affected by knocking out the opioid receptors. The control C57 mouse platelets were found to contain the greatest amount of serotonin, which was significantly higher than that for both κ- (p ≤ 0.0001) and μ- (p ≤ 0.001) KO mouse platelets. μ-KO mouse platelets had slightly more serotonin than the κ-KO mouse platelets (p ≤ 0.05). While there is literature precedent suggesting that serotonin uptake and transport can be impacted in the presence of some opioids, to the best of our knowledge, there have not been similar studies of platelet function with opioid receptor KO mice.25 Even with this difference in total serotonin content, all three platelet populations reacted similarly in Tyrode’s buffer and with stimulation by thrombin, releasing similar percentages of their total serotonin content. Without doing single cell analysis, we cannot confirm that granule secretion, trafficking, and kinetics are the same, but the similar response on a bulk cell level supports the fact that knocking out the receptors did not impact the platelets’ ability to secrete granular content. Cholesterol concentrations in platelets were also measured because cholesterol has been shown to play a role in granular secretion.26−28 In this work, there was no significant difference in the amount of platelet cholesterol in the various strains of mice (Figure SI 3).
Figure 4.

Knockout mouse serotonin concentration and secretion in response to thrombin. Initial experiments were performed to determine if the knockout platelets responded differently than control platelets in control environments. The total concentration of serotonin was measured after (A) lysing the platelets with 0.5 μM HClO4, (B) incubating them in a nonstimulant Tyrode’s buffer, (C) or stimulating them with the natural stimulant thrombin. The knockout mice showed significant changes in their total concentration of serotonin (A, F(2,7) = 55.14, p ≤ 0.0001). However, the percentage of serotonin released in a resting stage (B, F(2,9) = 1.342, p = 0.3089) and upon stimulation (C, F(2,9) = 0.6464, p = 0.5466) was statistically indistinguishable for the knockout platelets compared to the control C57 mouse platelets. *p ≤ 0.05, ***p ≤ 0.001, and p ≤ 0.0001 vs indicated position. n = 4 for all conditions except for PF4 Tyrode’s condition with n = 3.
When the κ- and μ-KO mice platelets (Figure 5) were exposed to the same treatment as the C57 mice platelets (Figure 3), similar trends were seen and, in some cases, enhanced. For κ-KO platelets, the agonists had a nonsignificant average decrease of δ-granule secretion compared to Tyrode’s buffer (Figure 5A). The μ-KO platelet response to agonists had a significantly (p ≤ 0.001) decreased level of δ-granule secretion compared to Tyrode’s buffer (Figure 5D). For the μ-KO platelets, the μ-agonist DAMGO had a statistically significant impact on δ-granule secretion when compared to stimulation by U-50488. DAMGO caused over twice the amount of serotonin to be secreted compared to U-50488 stimulation (p ≤ 0.05). This increased change in secretion of serotonin upon DAMGO exposure compared to the other agonists is still seen (not significantly) in the control and κ-KO platelets, but to a lesser extent, suggesting that some μ-like receptors are still located on the platelet surface (which was one hypothesis generated from the Western blot data) or that the agonist is interacting nonspecifically with platelets.
Figure 5.

KO platelet granule secretion upon opioid agonist stimulation: (A, D) δ-granule-secreted serotonin, (B, E) α-granule-secreted PF4, and (C, F) lysosome-secreted β-Hex release from κ-knockout (A–C) and μ-knockout (D–F) mice platelets after a 30 min thrombin stimulation. One-way ANOVA was performed on each data set. (A) F(4,15) = 12, p = 0.0001; (B) F(4,14) = 4.277, p = 0.0181, (C) F(4,13) = 9.206, p = 0.0009, (D) F(4,13) = 109.1, p ≤ 0.0001, (E) F(4,15) = 426.8, p ≤ 0.0001), (F) F(4,15) = 525, p ≤ 0.0001. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 vs thrombin condition. +++p ≤ 0.001 and ++++p ≤ 0.0001 vs Tyrode’s buffer condition. #p ≤ 0.05 and ##p ≤ 0.01 vs indicated conditions. n = 4 for all conditions except PF4 Tyrode’s buffer with n = 3.
For α-granule secretion (Figure 5B, E), the differences in C57 PF4 secretion in the presence of U-50488 were also measured in the κ-KO and μ-KO platelets, with an average increased difference of 0.03 ng PF4/mg protein in both samples compared to Tyrode’s buffer. The κ-KO platelet average difference was 0.02 and 0.05 ng PF4/mg protein in secretion from DAMGO and DPDPE, respectively, compared to Tyrode’s buffer. Lysosome secretion was not statistically different between C57 (Figure 3C) and μ-KO platelets (Figure 5F). However, in lysosome secretions from the κ-KO platelets, there was a significant (p ≤ 0.05) increase (∼0.03 ng PF4/mg protein) for all three agonists compared to Tyrode’s buffer (Figure 5C). These data suggest that there might be a role for the κ-receptor in hindering lysosome secretion when exposed to opioids, whereas the μ-receptor could play a potential role in δ-granule secretion when exposed to opioids. To confirm this association, a more focused study is needed to determine if/how the receptors interact with the granule populations of interest. Finally, even though Western blot analysis revealed that the κ-receptor was knocked out as expected, the U50488 κ-agonist did not induce a difference in response compared to the other agonists and showed similar secretion trends compared to the C57 and μ-KO platelets. This suggests that the κ-agonist is interacting with the same receptor as the other agonists or a receptor that has a similar pathway for granule secretion. While several sources suggest that U-50488 is highly selective for the κ-receptor, several commercial sources for U-50488 as well as several published papers suggest that U-50488 could block either Na+ or Ca2+ channels at high concentrations.29,30 Nonselective impacts on Ca2+ channels could account for the degranulation trends measured here and will be the subject of future research.
The total amounts of each receptor located on the KO and control platelets were not analyzed. Therefore, it is not known if knocking out one receptor may have changed the ability of the other receptors to respond to their agonist.
Platelet Adhesion, Aggregation, and Activation Variations in Knockout Mouse Platelets
Another important component in hemostasis is the platelets’ ability to adhere to both the endothelial cell wall and each other to form clots and prevent bleeding. Using a microfluidic channel coated with endothelial cells to mimic vasculature (Figure 6A), platelets were flowed through the straight channel device (Figure 6B). Data were obtained after no stimulation (Tyrode’s buffer) (Figure 6C) or after activation with ADP, an agonist which helps with platelet shape change/adhesion (Figure 6D). The number of adherent KO platelets were counted and compared to the number of C57 platelets that adhered. In nonactivated platelets, only κ-KO mouse platelets adhered significantly less than control platelets (p ≤ 0.05). Even after activation with ADP, κ-KO platelets still had decreased adhesion compared to both the μ-KO and C57 control platelets (p ≤ 0.05 and p ≤ 0.01, respectively).
Figure 6.

Measurement of platelet adhesion in a microfluidic channel coated with endothelial cells. (A) Straight channel platelet adhesion polydimethylsiloxane-based microfluidic device coated with endothelial cells. (B) Platelets were flowed across the device, and adherent platelets were counted using bright field microscopy. The scale bar for the inset micrograph represents a length of 64 μm. Statistical analysis of adherent platelets was performed either when platelets were (C) not activated or (D) activated with 5 μM ADP, a natural platelet stimulant, before being flowed through the device. *p ≤ 0.05, **p ≤ 0.01 vs indicated position. n = 4 for all conditions except for PF4 Tyrode’s buffer condition with n = 3.
To further investigate possible reasons for the κ-KO platelets’ decreased ability to adhere, both the rate of aggregation (Figure 7) and activated platelet images (Figure 8) were acquired. Both aggregation behavior and morphological image analysis showed differences between the KO platelets and the control platelets. For aggregation experiments, the change in light transmission through 2 × 108 suspended platelets per milliliter was measured after thrombin stimulation.
Figure 7.
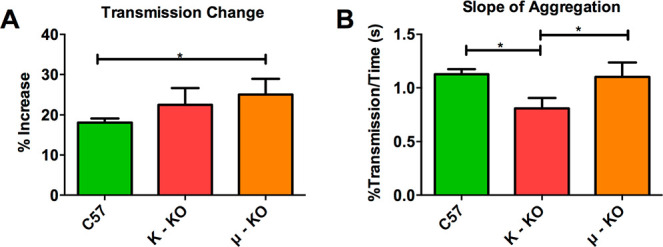
Platelet aggregation measurements upon stimulation with 4.8 U/mL thrombin. The change in (A) light transmission (F(2,9) = 4.668, p = 0.04707) and (B) the rate of change (F(2,8) =10.64, p = 0.0056) were measured. *p ≤ 0.05 vs indicated position. Each condition had five biological replicates.
Figure 8.

Representative activated platelet dark-field scattering image for (A) C57 control mice, (B) κ-KO mice, and (C) μ-KO mice. Microscope image exposure time for the knockout platelets was increased compared to the control mouse platelets due to lower scattering efficiency.
Compared to control platelets, μ-KO platelets had a larger change in total transmission (Figure 7A), which correlates to more platelets aggregating and falling out of solution. The μ-KO platelet suspension had an increased transmission of 25.1% compared to 22.5% and 18.1% for κ-KO and control platelets, respectively. This difference was statistically significant for comparison of C57 and μ-KO platelets (p ≤ 0.05). Surprisingly, the overall change in aggregation behavior did not impact the rate of aggregation, with both μ-KO and C57 platelets having a 1.1% change in transmission each second. On the other hand, the κ-KO platelets took significantly longer to aggregate than both C57 (p ≤ 0.01) and μ-KO platelets (p ≤ 0.05) with a change in transmission of 0.82%/s. This slower aggregation could be one possible reason that fewer κ-KO platelets adhered to the microfluidic device in the time frame and distance measured.
Another factor that has the potential to impact adhesion behavior is activation shape. Images of control (27 images, 2 biological replicates), κ-KO (30 images, 2 biological replicates), and μ-KO (25 images, 2 biological replicates) platelets with a minimum of 20 platelets per image were visualized using dark-field microscopy to characterize platelet shape without need for a fluorescent label (Figure 8). Additional representative images are available in the Supporting Information (Figure SI 4). Visual comparison was used to characterize the varying activation shapes of the C57, κ-KO, and μ-KO platelets. Qualitatively, neither the κ- nor μ-KO platelets were able to scatter light as efficiently as the C57 platelets, although no statistical analysis was done to compare their light scattering efficiency. Thus, to obtain an image of these activated KO platelets, exposure time was increased compared to the control platelets. The activation shapes of the different types of platelets were visually distinct. C57 platelets (Figure 8A) often showed two to three filopodia spread across the platelet while still maintaining a smaller circular center. The κ-KO platelets displayed a more flattened look with a larger lamellipodia structure and small filopodia surrounding the entire platelet, giving the platelets a spiky appearance (Figure 8B). Finally, μ-KO platelets were intermediate in appearance between the κ-KO and C57 platelets. They contained distinct filopodia like the C57 platelets but also contained a greater number of these features than C57 platelets and had a more spread-out appearance (Figure 8C). The platelets’ ability to flatten out and form filopodia and lamellipodia affects how they adhere. When a platelet’s shape flattens out, it enables platelets to adhere more closely to the endothelial cell surface. The filopodia bind fibrin strands, and the long extensions help intertwine the platelets while forming a clot, whereas the lamellipodia bind to the wounded surface to close vasculature leaks.28,31 An experiment performed by Stenberg et al.31 studied Wistar Furth rats whose platelets had hereditary macrothrombocytopenia. These platelets have previously demonstrated decreased adhesion and result in prolonged bleeding times. To further explore these phenomena, Stenberg et al. studied the shape change and formation of filopodia and lamellipoida and compared this shape change to normal rat platelets. They found that the Wistar Furth rats had few and short filopodia but these were still able to eventually reach the fully spread platelet stage. They also hypothesized that the limited filopodia formed more fragile clots which increased bleeding time.28 Like the Wistar Furth rat platelets, the κ-KO platelets also demonstrated these smaller filopodia, suggesting that the decrease in adhesion and increase in aggregation time are likely due to the shape of the activated platelet.
Overall, due to their widespread use and public health implications, the mechanisms by which opioids interact with platelets and other blood cells warrant further study. For example, a recent study demonstrated that the μ-receptor agonist fentanyl may interfere with antiplatelet drugs.32 The work documented herein explored opioid receptor agonist effects on murine platelet function in genetic knockouts of μ- and κ-receptors. Following this precedent study, further extensions of this work will delve deeper into opioid receptor expression as well as explore opioid receptor antagonists such as naloxone and naltrexone, the latter of which has been demonstrated to suppress platelet aggregation.33
Conclusion
In conclusion, platelets express opioid receptors; however, wild type platelets incubated with opioid receptor agonists do not degranulate in direct response to the opioid agonists, and the agonists do not affect platelet δ-granule secretion upon subsequent thrombin stimulation. When the receptors are knocked out, several unique features are seen, suggesting subtle roles for the opioid receptors. κ-KO mouse platelets display decreased lysosome secretion upon opioid agonist exposure compared to the wild type control. In addition, the κ-KO platelets are not able to adhere as well, take longer to aggregate, and have a more spread out appearance with less distinct filopodia than control platelets. The μ-KO mouse platelets have inhibited δ-granule secretion to a greater extent upon opioid agonist stimulation than either the κ-KO or wild type platelets. There is also a difference in the final platelet shape, which does not affect adhesion properties but has slightly enhanced aggregation. These data suggest that normal healthy platelet function will not likely be adversely affected when exposed to drugs containing opioids, including morphine and ketamine, during surgery. However, one caveat is that these experiments do not mimic long-term platelet exposure to opioids nor do they account for how platelet behavior may change downstream of other cell types that are impacted by opioids.
Methods
Reagents
All chemicals for the buffers and HPLC mobile phase were purchased from Sigma-Aldrich. Western blot stock solutions were purchased from Bio-Rad laboratories unless otherwise indicated. All chemicals are detailed below and were used without further purification.
Platelet Isolation
Control C57BL/6J, μ-KO B6.129S2-Oprm1tm1Kff, and κ-KO B6.129S2-OPRK1TM1KFF/j mice were purchased from The Jackson Laboratory. Blood was collected via cardiac puncture following University of Minnesota IACUC protocol #1403-31383A. Briefly, mice were euthanized via CO2 asphyxiation, and then a syringe filled with 200 μL of acid citrate dextrose (ACD) was used to draw blood via cardiac puncture. The blood was diluted with Tyrode’s buffer (NaCl, 137 mM; KCl, 2.6 mM; MgCl2, 1.0 mM; d-glucose, 5.6 mM; N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) 5.0 mM; and 12.1 mM NaHCO3, with pH adjusted to 7.3) and centrifuged at 130g for 10 min with reduced braking to prevent platelet activation. The top platelet-rich plasma layer was collected, and additional ACD and Tyrode’s buffer were added. Platelets were pelleted at 524g for 10 min with reduced braking. The pellet was resuspended in fresh Tyrode’s buffer, and platelets were counted using a hemocytometer. All platelets were diluted to the lowest platelet density for each experiment, which typically ranged between 1 × 107 and 1 × 108 platelets/mL.
Western Blot
Platelets were repelleted at 524g and resuspended in 200 μL of Tyrode’s buffer to increase concentration. For opioid receptor control, brain cells were extracted from C57BL/6J after CO2 euthanasia; the brain collected was broken into small chunks by vigorous pipetting, in Tyrode’s buffer, and frozen at −80 °C. Both platelets recently collected from mouse donors and samples frozen at −80 °C were tested for Western blotting experiments. After thawing, both platelets and brain cells were roughly pipetted and sonicated for 10 min. A 5% by volume solution of 2-mercaptoethanol in Laemmli buffer was added to the cell solution in a 1:1 ratio. The sample was vortexed for 30 s, put into a heating block, and boiled for 5 min.
Next, 12.5% Criterion Tris-HCl Gel purchased from Bio-Rad Laboratories was inserted into a gel electrophoresis housing containing 1× Tris/Glycine/SDS (TGS). Samples and a precision plus protein standard dual color ladder from Bio-Rad were pipetted into their respective wells. The power was set at 110 V, and the gel was run until the bottom of the ladder was near the end of the gel (around 90 min). A blotting sandwich was set up, keeping the components wet with cold Towbin buffer (1× Tris/Glycine (TG) in 20% methanol). The sandwich was placed in the transfer apparatus and inserted into the transfer cell filled with cold Towbin buffer and a stir bar. The power supply was set at 500 mA or ∼100 V and ran for 120 min in a cold room on a stir plate.
The nitrocellulose membrane was incubated with 5% (w/v) powder skim milk in 1× Tris buffered saline with 0.1% Tween-20 (TBS/T) buffer for 60 min on a shaker at 300 rpm. The membrane was washed 3× for 5 min in TBS/T buffer at 300 rpm and finally placed into bags containing one of the antibodies in 4% powdered skim milk in TBS/T buffer. The μ, δ, and κ antibodies (Millipore Sigma anti-μ opioid receptor AB1580-1 rabbit polyclonal, Thermo Fisher Scientific PA5-26138 OPRM1 antibody, Millipore Ab1560 rabbit anti-δ opioid receptor polyclonal antibody, and Abcam anti-κ opioid receptor antibody ab10566) were diluted 1:1000. The bags were placed in a cold room overnight with gentle rotation. The membrane was removed and washed 3× with TBS/T buffer on a shaker at 300 rpm and finally incubated with Rockland Immunochemical’s secondary anti-rabbit HRP-conjugated antibody (1:5000 dilution) in 4% powdered skim milk for 60 min. After incubation, the membranes were washed 3× with TBS/T buffer in a rotator at 300 rpm for 5 min.
While washing the membrane, a mixture of 1:1 luminol solution and stable peroxide solution from the Super Signal West Femto Maximum Sensitivity Substrate kit (Pierce) was mixed. The membranes were laid onto plastic wrap, and the substrate was pipetted dropwise over the proteins/antibodies. After 5 min, the membrane was dabbed to remove excess reagent and wrapped with the plastic wrap. X-ray film was placed over the membrane and developed using a SRX101A medical film processor (Konica Minolta Medical and Graphic Inc.)
Platelet Stimulation Procedure for Serotonin Detection
A volume of 125 μL of platelet suspension was put into 1.7 mL Eppendorf tubes. Platelets were then incubated with 125 μL of opioid agonist in Tyrode’s buffer at a final concentration of either 10 nM DAMGO, 3 μM DPDPE, 100 nM U-50488, or Tyrode’s buffer. Platelets were pelleted at 1200g, and the supernatant was collected for serotonin detection caused by the stimulant. Platelets were resuspended in 125 μL of Tyrode’s buffer and incubated for 15 min before 125 μL of 1 U/mL thrombin was added for a final concentration of 0.5 U/mL thrombin. After 20 min of thrombin stimulation, platelets were spun down at 1200g, and the supernatant was collected again to determine the total amount of serotonin in platelets.
Serotonin Detection Using HPLC
200 μL of the supernatant was filtered using a Millipore 96 well Multi-Screen HTS filter plate (Billerica, Ma) with a 0.45 μm pore size. The supernatant was filtered through at 3000g for 5 min, and 180 μL of it was combined with 20 μL of 5 μM dopamine internal standard in 0.5 M perchloric acid. The serotonin was detected using a previously developed HPLC method using electrochemical detection.23,34 Briefly, a Waters 2465 electrochemical detector with a glassy carbon electrode was attached to a Agilent 1200 HPLC with a 5 μm 4.6 × 150 mm C18 column (Eclipse XDB-18). The samples were auto injected into the mobile phase (11.6 mg/L of sodium octyl sulfate, 170 μL/L dibutylamine, 55.8 mg/L Na2EDTA, 10% methanol, 203 mg/L anhydrous sodium acetate, 0.1 M citric acid, and 120 mg/L sodium chloride) flowing at 2 mL/min. The dopamine (an internal standard) and serotonin spikes were detected using a potential of 700 mV vs Ag/AgCl electrodes and a current range of 50 nA. Concurrently, a calibration curve was run with serotonin concentrations varying from 0 to 1 μM serotonin and 0.5 μM dopamine internal standard in 0.5 μM perchloric acid.
PF4 ELISA Assay
Alpha granule content secretion was detected as directed using an enzyme-linked mouse immunoassay kit for PF4 (R&D Systems). Briefly, supernatants were diluted with Calibrator Diluent RD5-26 (1×). Assay Diluent RD1-40 and the sample were added to each well in the provided plate. The plate was incubated for 2 h, and the wells were washed. Mouse PF4 Conjugate was added to each well, incubated, and washed. Finally, a substrate solution was put in the wells for incubation, and then a quenching solution was added. The optical density was determined with a BioTek Synergy 2 96-well plate reader.
Β-Hexosaminidase Assay
An absorbance assay for β-Hex was prepared as previously described.35 Briefly, a solution of 1 mM p-nitrophenyl acetyl-d-glucosamine in 0.1 M citrate buffer was added to 40 μL of mouse platelet supernatant, as collected according to the platelet isolation section and incubated for an hour. Ice cold 0.1 M carbonate buffer was added to stop the reaction. The absorbance was read at 405 nm with background subtraction at 630 nm.
BCA Protein Assay
Platelet protein content was measured using the Pierce BCA Protein Assay kit (Thermo Scientific) following included directions for the microplate procedure. Briefly, after collecting the supernatant used in experiments from Figures 2 and 4, the pellet was homogenized in 25 μL of Tyrode’s buffer. A volume of 200 μL of working reagent was added to each well, and the plate was incubated for 30 min at 37 °C. The plate was read at 562 nm on a BioTek Synergy 2 96-well plate reader. Due to small pellets, when the supernatant was removed, a small portion was left inside each vial in order to not disturb the pellet. Therefore, upon the addition of Tyrode’s buffer, there may have been slight variations in the total pellet dilution.
Platelet Adhesion
Platelet adhesion studies were performed on a straight channel microfluidic device as previously described.36,37 Briefly, channels were coated with a confluent monolayer of hy926 human endothelial cells (ATCC). Platelets were labeled with CMFDA (5-chloromethylfluorescein diacetate) dye (2 μM, 20 min) for easier visualization and then stimulated with 5 μM ADP or left unstimulated in the Tyrode’s buffer. ADP was utilized because it can initiate adhesion without secretion, therefore decoupling secretion and adhesion in this assay.35 The cells were flowed through the device at 30 μL/h for 20 min. The device was washed with Tyrode’s buffer, and the number of adherent cells was counted using a Nikon Microscope with a QuantEM Photometrics CCD camera. Metamorph ver. 7.7.5 was used as the imaging and analysis software.
Platelet Aggregation
A volume of 500 μL of 2 × 108 platelets was placed in a small glass tube with a stir bar. The tube was placed into a Chrono-Log Whole Blood Lumi aggregometer interfaced with Aggro/Link software. A tube with Tyrode’s buffer was placed into the aggregometer for baseline comparisons. After the absorbance stabilized at 100%, 120 μL of 25 U/mL thrombin was added for a final concentration of 4.8 U/mL thrombin. The percent of absorbance over time was measured until the absorbance no longer changed. The data was then converted to percent of transmittance. To calculate the change in transmission, the percent of transmittance was averaged before stimulation and then the average percent transmittance after the decrease was subtracted. For rate of aggregation calculations, the time was considered from when the thrombin was injected until the % transmittance reached the average final value.
Platelet Dark Field Imaging
A volume of 4 μL of platelet suspension was deposited on a glass slide, and a coverslip was gently placed on top before being sealed with clear nail polish. Platelet samples were allowed to settle and activate before imaging. Images were captured on an Olympus microscope from CytoViva with an UplanFLM 100× oil immersion objective and Dage XL camera. The image exposure time was adjusted manually in the Exponent 7 software and had to be increased for both the μ- and κ-KO platelet images based on lower overall scattering signals.
Data Analysis
GraphPad Prism 6 was used to analyze all statistical data. All significance was determined using one-way ANOVA. Figure error bars show standard deviation. Figure 3 had 4 biological replicates for all conditions except Tyrode’s buffer, which had 10 biological replicates. Figures 4–6 and 8 had 4 biological replicates except for the PF4 Tyrode’s conditions, which only had 3 biological replicates due to limited space. Figure 7 had 5 replicates for each condition. For dark-field imaging, 2 slides were prepared for each type of platelet and ∼10–15 images were recorded for each slide.
Acknowledgments
The authors would like to acknowledge funding from the NIH biotechnology training grant (# 5T32GM008347-23) for S.M.G., the NIH New Innovator Award (DP2-OD004258) for B.M.M. and D.K., the UMN Doctoral Dissertation Fellowship for A.F.M., the UMN Lando program for D.H.F., the UMN MRSEC (DMR-2011401) for J.H., and Minnesota Nano Center for access to microfluidic fabrication equipment.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmeasuresciau.1c00012.
Western blots, calibration curve for HPLC data, additional dark-field platelet images (PDF)
Author Present Address
† Boston Scientific, 4100 Hamline Ave N., St. Paul, MN 55112
Author Present Address
‡ Boston Scientific, 3 Scimed Place, Maple Grove, MN 55311
The authors declare no competing financial interest.
Supplementary Material
References
- Kristian Stromgaard P. K.-L.Textbook of Drug Design and Discovery; CRC Press, 2016. [Google Scholar]
- Solomon D. H.; Avorn J.; Wang P. S.; Vaillant G.; Cabral D.; Mogun H.; Stürmer T. Prescription Opioid Use among Older Adults with Arthritis or Low Back Pain. Arthritis Rheum. 2006, 55 (1), 35–41. 10.1002/art.21697. [DOI] [PubMed] [Google Scholar]
- U.S. Opioid Dispensing Rate Maps. Centers for Disease Control and Prevention, https://www.cdc.gov/drugoverdose/maps/rxrate-maps.html (accessed 2021-03-07).
- Understanding the Epidemic. Centers for Disease Control and Prevention, https://www.cdc.gov/drugoverdose/epidemic/index.html (accessed 2021-03-07).
- Smith H. S. The Metabolism of Opioid Agents and the Clinical Impact of Their Active Metabolites. Clin. J. Pain 2011, 27 (9), 824–838. 10.1097/AJP.0b013e31821d8ac1. [DOI] [PubMed] [Google Scholar]
- Farr S. J.; Otulana B. A. Pulmonary Delivery of Opioids as Pain Therapeutics. Adv. Drug Delivery Rev. 2006, 1076–1088. 10.1016/j.addr.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Pathan H.; Williams J. Basic Opioid Pharmacology: An Update. Br. J. Pain 2012, 6 (1), 11–16. 10.1177/2049463712438493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachia E.; Ahmedzai S. H. Transdermal Opioids for Cancer Pain. Curr. Opin. Support. Palliat. Care 2011, 5 (1), 15–19. 10.1097/SPC.0b013e3283437a39. [DOI] [PubMed] [Google Scholar]
- Dhawan B.; Cesselin F.; Raghubir R.; Reisine T.; Bradley P.; Portoghese P.; Hamon M. International Union of Pharmacology. XII. Classification of Opioid Receptors. Pharmacol. Rev. 1996, 48 (4), 567–592. [PubMed] [Google Scholar]
- Kieffer B. L. Opioids: First Lessons from Knockout Mice. Trends Pharmacol. Sci. 1999, 19–26. 10.1016/S0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- Manning B. M.; Hebbel R. P.; Gupta K.; Haynes C. L. Carbon-Fiber Microelectrode Amperometry Reveals Sickle-Cell-Induced Inflammation and Chronic Morphine Effects on Single Mast Cells. ACS Chem. Biol. 2012, 7 (3), 543–551. 10.1021/cb200347q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo R.; de Leon-Casasola O.; Benyamin R. Opioid Therapy and Immunosuppression. Am. J. Ther. 2004, 11 (5), 354–365. 10.1097/01.mjt.0000132250.95650.85. [DOI] [PubMed] [Google Scholar]
- Bidlack J. M. Detection and Function of Opioid Receptors on Cells from the Immune System. Clin. Diagn. Lab. Immunol. 2000, 719–723. 10.1128/CDLI.7.5.719-723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson A.Platelets, 2nd ed.; Michelson A., Ed.; Elsevier, 2007. [Google Scholar]
- Gotta A. W.; Gould P.; Sullivan C. A.; Goldiner P. L. The Effect of Enflurane and Fentanyl Anaesthesia on Human Platelet Aggregation in Vivo. Can. Anaesth. Soc. J. 1980, 27 (4), 319–322. 10.1007/BF03007451. [DOI] [PubMed] [Google Scholar]
- Kozek-Langenecker S. A. The Effects of Drugs Used in Anaesthesia on Platelet Membrane Receptors and on Platelet Function. Curr. Drug Targets 2005, 3 (3), 247–258. 10.2174/1389450023347759. [DOI] [PubMed] [Google Scholar]
- Gibbs N. M.The Effect of Anaesthetic Agents on Platelet Function. Anaesthesia and Intensive Care; SAGE PublicationsSage UK: London, 1991; pp 495–520. 10.1177/0310057x9101900402 [DOI] [PubMed] [Google Scholar]
- Haghpanah T.; Afarinesh M.; Divsalar K. A Review on Hematological Factors in Opioid-Dependent People (Opium and Heroin) after the Withdrawal Period. Addict. Health 2010, 2 (1–2), 9–16. [PMC free article] [PubMed] [Google Scholar]
- Shankar H.; Murugappan S.; Kim S.; Jin J.; Ding Z.; Wickman K.; Kunapuli S. P. Role of G Protein-Gated Inwardly Rectifying Potassium Channels in P2Y 12 Receptor-Mediated Platelet Functional Responses. Blood 2004, 104 (5), 1335–1343. 10.1182/blood-2004-01-0069. [DOI] [PubMed] [Google Scholar]
- Sheu J.-R.; Lee Y.-M.; Lee L.-W.; Luk H.-N.; Yen M.-H. Inhibitory Mechanisms of Naloxone on Human Platelets. Clin. Exp. Pharmacol. Physiol. 1998, 25 (7–8), 585–591. 10.1111/j.1440-1681.1998.tb02256.x. [DOI] [PubMed] [Google Scholar]
- Williams J. P.; Thompson J. P.; McDonald J.; Barnes T. A.; Cote T.; Rowbotham D. J.; Lambert D. G. Human Peripheral Blood Mononuclear Cells Express Nociceptin/Orphanin FQ, but Not μ, δ, or κ Opioid Receptors. Anesth. Analg. 2007, 105 (4), 998–1005. 10.1213/01.ane.0000278865.11991.9d. [DOI] [PubMed] [Google Scholar]
- Mehrishi J. N.; Mills I. H. Opiate Receptors on Lymphocytes and Platelets in Man. Clin. Immunol. Immunopathol. 1983, 27 (2), 240–249. 10.1016/0090-1229(83)90074-0. [DOI] [PubMed] [Google Scholar]
- Zucker M. B.; Nachmias V. T. Platelet Activation. Arteriosclerosis 1985, 5, 2–18. 10.1161/01.ATV.5.1.2. [DOI] [PubMed] [Google Scholar]
- Scharf R. E. Drugs That Affect Platelet Function. Semin. Thromb. Hemostasis 2012, 38 (8), 865–883. 10.1055/s-0032-1328881. [DOI] [PubMed] [Google Scholar]
- Barann M.; Stamer U. M.; Lyutenska M.; Stüber F.; Bönisch H.; Urban B. Effects of Opioids on Human Serotonin Transporters. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015, 388 (1), 43–49. 10.1007/s00210-014-1056-3. [DOI] [PubMed] [Google Scholar]
- Ge S.; White J. G.; Haynes C. L. Critical Role of Membrane Cholesterol in Exocytosis Revealed by Single Platelet Study. ACS Chem. Biol. 2010, 5 (9), 819–828. 10.1021/cb100130b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruba S. M.; Koseoglu S.; Meyer A. F.; Meyer B. M.; Maurer-Jones M. A.; Haynes C. L. Platelet Membrane Variations and Their Effects on δ-Granule Secretion Kinetics and Aggregation Spreading among Different Species. Biochim. Biophys. Acta, Biomembr. 2015, 1848 (7), 1609–1618. 10.1016/j.bbamem.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkenstaedt-Quinn S. A.; Gruba S. M.; Haynes C. L. Variations in Fusion Pore Formation in Cholesterol-Treated Platelets. Biophys. J. 2016, 110 (4), 922–929. 10.1016/j.bpj.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis J. R.; Whately E.; Brust A.; Inserra M. C.; Asvadi N. H.; Lewis R. J.; Alewood P. F.; Cabot P. J.; Vetter I. Activation of κ Opioid Receptors in Cutaneous Nerve Endings by Conorphin-1, a Novel Subtype-Selective Conopeptide, Does Not Mediate Peripheral Analgesia. ACS Chem. Neurosci. 2015, 6 (10), 1751–1758. 10.1021/acschemneuro.5b00113. [DOI] [PubMed] [Google Scholar]
- B H.; V R.-V. The κ-Opioid Receptor Agonist U-50488 Blocks Ca2+ Channels in a Voltage- and G Protein-Independent Manner in Sensory Neurons. Reg. Anesth. Pain Med. 2013, 38 (1), 21–27. 10.1097/AAP.0B013E318274A8A1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg P. E.; Barrie R. J.; Pestina T. I.; Steward S. A.; Arnold J. T.; Murti A. K.; Hutson N. K.; Jackson C. W. Prolonged Bleeding Time With Defective Platelet Filopodia Formation in the Wistar Furth Rat. Blood 1998, 91 (5), 1599–1608. 10.1182/blood.V91.5.1599. [DOI] [PubMed] [Google Scholar]
- Kuczyńska K.; Boncler M. Emerging Role of Fentanyl in Antiplatelet Therapy. J. Cardiovasc. Pharmacol. 2020, 76 (3), 267–275. 10.1097/FJC.0000000000000880. [DOI] [PubMed] [Google Scholar]
- García-Sevilla J. A.; Ulibarri I.; Giralt M. T.; Areso P.; Oliveros R. G.; Gutiérrez M. Chronic Naltrexone Suppresses Platelet Aggregation Induced by Adrenaline and 5-Hydroxytryptamine in Former Heroin Addicts. J. Neural Transm. 1988 732 1988, 73 (2), 157–160. 10.1007/BF01243386. [DOI] [PubMed] [Google Scholar]
- Brass L. F.; Tomaiuolo M.; Stalker T. J. Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response. Hematol./Oncol. Clin.North Am. 2013, 27, 381–409. 10.1016/j.hoc.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseoglu S.; Meyer A. F.; Kim D.; Meyer B. M.; Wang Y.; Dalluge J. J.; Haynes C. L. Analytical Characterization of the Role of Phospholipids in Platelet Adhesion and Secretion. Anal. Chem. 2015, 87 (1), 413–421. 10.1021/ac502293p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruba S. M.; Meyer A. F.; Manning B. M.; Wang Y.; Thompson J. W.; Dalluge J. J.; Haynes C. L. Time- and Concentration-Dependent Effects of Exogenous Serotonin and Inflammatory Cytokines on Mast Cell Function. ACS Chem. Biol. 2014, 9 (2), 503–509. 10.1021/cb400787s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lin Y. S.; Haynes C. L. On-Chip Evaluation of Shear Stress Effect on Cytotoxicity of Mesoporous Silica Nanoparticles. Anal. Chem. 2011, 83 (22), 8377–8382. 10.1021/ac202115a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.