

Abstract
Local delivery of anticancer agents has the potential to maximize treatment efficacy and minimize the acute and long-term systemic toxicities. Here we used unsupervised systematic evolution of ligands by exponential enrichment (SELEX) to identify four RNA aptamers that specifically recognized mouse and human myeloid cells infiltrating tumors but not their peripheral or circulating counterparts in multiple mouse models and from patients with head and neck squamous cell carcinoma (HNSCC). The use of these aptamers conjugated to doxorubicin enhanced the accumulation and bystander release of the chemotherapeutic drug in both primary and metastatic tumor sites in breast and fibrosarcoma mouse models. In the 4T1 mammary carcinoma model, these doxorubicin-conjugated aptamers outperformed Doxil, the first clinically approved highly optimized nanoparticle for targeted chemotherapy, by promoting tumor regression with no detected toxicity measured as weight loss and by blood chemistry after just 3 administrations. These RNA aptamers recognized tumor infiltrating myeloid cells in a variety of mouse tumors in vivo and from human HNSCC ex vivo. This suggests their use for the detection of myeloid-derived suppressor cells (MDSCs) in human and for a targeted delivery of chemotherapy to the tumor microenvironment in multiple malignancies.
One sentence summary:
Aptamers specific for mouse and human tumor-infiltrating myeloid cells allow delivery of anticancer drugs to the tumor microenvironment in multiple mouse models.
Introduction
Chemotherapy using different cytotoxic agents is the standard of care treatment for most human malignancies. However, these treatments are associated with moderate to serious toxicities that decrease overall quality-of-life for patients with cancer and are thus often dose-limiting and responsible for modest therapeutic efficacy (1, 2). Drug delivery nanoplatforms aimed at concentrating the chemotherapeutic agent(s) at the tumor site(s) are gaining interest as modalities to decrease systemic toxicity and increase efficacy (2–4). However, most nanoplatforms rely either on physical properties of tumor vasculature, such as the enhanced permeability and retention (EPR) effect (5), or on the presence of markers characterizing the neoplastic cells. The EPR effect allows the preferential targeting of primary tumors and established metastases but is poorly effective against unvascularized micro-metastases (6). Targeting an appropriate tumor-associated antigen allows drug delivery to disseminated neoplastic cells but may result in tumor editing (7, 8) rather than eradication due to tumor heterogenicity and the genetic instability of neoplastic cells (9). An alternative and burgeoning strategy is to deliver therapeutic agents to the genetically stable tumor stromal cells that characterize primary tumor, metastases, and pre-metastatic niches (10). Myeloid cells in the tumor microenvironment are such a target because they account for a large proportion of the tumor mass, promote cancer cell survival and metastases (11–16), provide immune protection, and are recruited early during tumor progression in the primary tumor and in the pre-metastatic niches (17). Tumor-infiltrating myeloid cells (TIMCs, comprising myeloid-derived suppressor cells [MDSCs], tumor-associated macrophages [TAMs], neutrophils, and monocytes) express an activated, pro-tumoral phenotype that differentiates them from their systemic counterpart (18–22). As markers expressed on TIMC but not on their circulating counterparts remain to be fully defined, we used an unsupervised approach to generate RNA aptamers against known and unknown TIMC-specific epitopes (23).
RNA aptamers are particularly suitable for drug delivery as they penetrate deeply into tissues, are non-immunogenic, and are easy to manufacture and engineer for improved pharmacokinetic and stability. Furthermore, these reagents can be selected to be rapidly internalized in the target cells (24), thus allowing specific drug absorption in the desired cells. Last, the combinatorial use of aptamers that each bind a different epitope in the target cell should increase the overall specificity and maximize drug delivery in the chosen tissue.
Here, we identified and characterized four RNA aptamers that specifically recognize TIMCs in different preclinical models and in humans.
Results
Identification of aptamers specific for tumor-infiltrating myeloid cells
To simplify the identification and selection of TIMC-specific aptamers, we performed an unsupervised, high-throughput sequencing Cell-SELEX (25) using the MDSC-derived cell line MSC2 (26), followed by an empirical selection of monoclonal aptamer binding TIMC but not the splenic counterpart. MSC2 cells can be considered a suitable TIMC surrogate for initial aptamer screening as these cells are available without requiring any artifact-causing manipulation of tumor-infiltrating cells, and they acquire the suppressive activity only after treatment with IL4 (26). Furthermore, MSC2 cells treated with IL4 were transcriptionally similar to TIMC, both in terms of genome-wide expression and over-expressed genes, whereas untreated MSC2 had gene expression profiles similar to splenic CD11b+ cells (fig. S1).
Aptamers were partitioned through 11 cycles of Cell-SELEX using untreated MSC2 cells as negative selector and IL4-treated MSC2 cells as positive selector or TIMC surrogate. The resulting polyclonal aptamer library displayed increasing specificity with selection cycles (Fig.1A) only for IL4-treated MSC2 cells, illustrating the overall efficacy of the SELEX enrichment (Fig.1A–B). Moreover, when tested on single-cell suspensions obtained from either the tumor or spleen of CT26 tumor-bearing mice, the aptamer library discriminated TIMC by preferentially recognizing MDSCs and macrophages from the tumor but not from the spleen of the same tumor-bearing animals or tumor-free mice (Fig.1C).
Figure 1: Selection of polyclonal aptamers specific for tumor infiltrating myeloid cells.
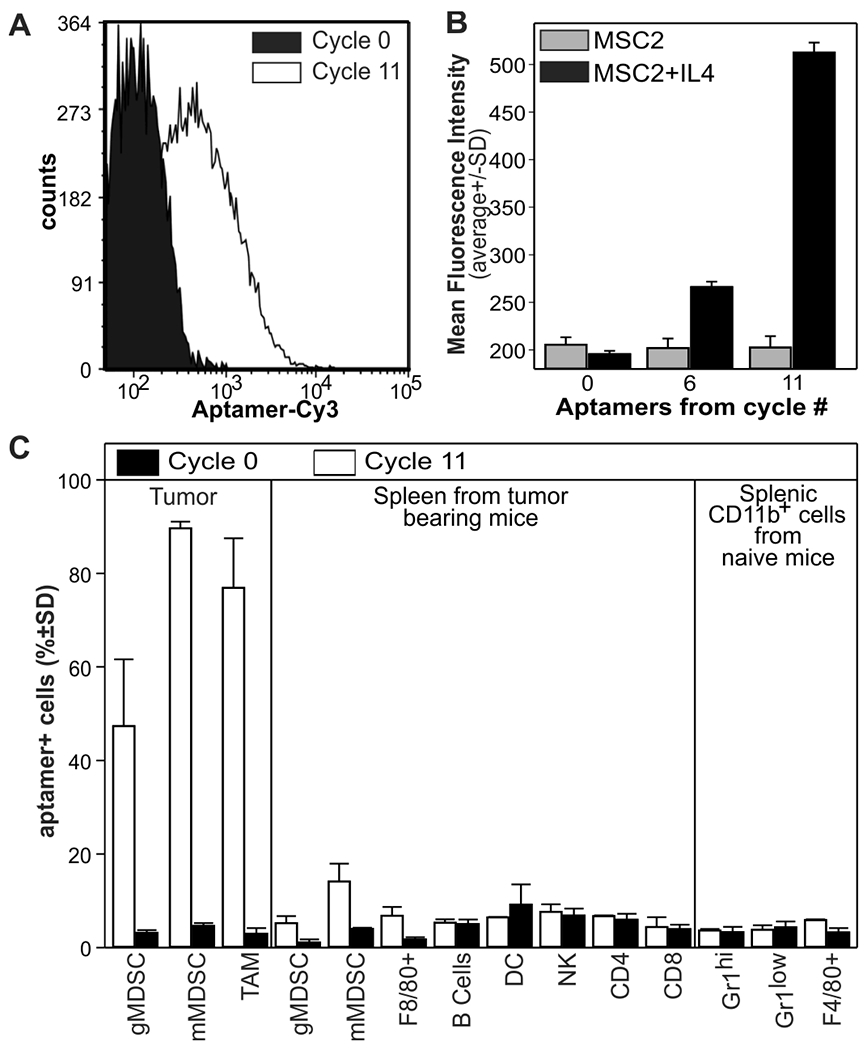
High-throughput Cell-SELEX was performed on MSC2 cells treated with IL4 or left untreated as a proxy of tumor-infiltrating and splenic myeloid cells, respectively. A) FACS analysis of IL4-treated MSC2 stained with the Cy3-labelled RNA aptamers from cycle 0, as control, or cycle 11.B) FACS analysis (N=3) of MSC2 or IL4-treated MSC2 cells with Cy3-labelled polyclonal aptamers from the indicated library. C) Single-cell suspensions from the spleen or tumors of mice bearing CT26 colon carcinomas (0.5 cm in diameter) were stained with a polyclonal aptamer library and counterstained with antibodies against CD11b, Gr1, F4/80, CD11c, CD19, CD49b, CD4, and CD8. Data derived from N=2 experiments with (n=3-5 mice).
Identification of 4 monoclonal aptamers specific for tumor-infiltrating myeloid cells
We identified monoclonal aptamers that can be easily produced and manipulated using a bioinformatics pipeline. Briefly, we high throughput-sequenced polyclonal aptamer libraries from SELEX cycles 1, 6, 10, and 11 corresponding to the first and last cycle and increment in selection stringency and analyzed the resulting data with APTANI, a computational tool to identify target-specific aptamers from SELEX sequencing and secondary structure information (27). Analysis of cycle 11 library resulted in 154 monoclonal aptamers that were enriched in the library by more than 0.01% and that contained 158 secondary RNA motifs with a motif frequency higher than 0.05% of the total number of motifs. From this set of aptamers, we focused on 15 sequences that had an enrichment of at least 0.1% and contained either a large (>=3) number of different secondary RNA motifs or at least 1 motif with a frequency higher than 0.1% (table S1). Frequency analysis showed that most of these aptamers emerged starting from cycle 6, when stringency was gradually increased (fig. S2).
Using flow cytometry, we tested these 15 aptamers against single cell suspensions from tumor and spleen of mice challenged with the 4T1 tumor, with the polyclonal cycle 0 random aptamer library as negative control (Fig. 2A). Although 12 out of 15 aptamers recognized macrophages, granulocytic MDSCs (gMDSC), or monocytic MDSCs (mMDSCs) from the tumor with statistically significant accuracy (as compared to the random aptamer library; p<0.001; Fig. 1D), only 6 (aptamers 3, 6, 11, 12, 14, 15) were specific for tumor-infiltrating myeloid cells and did not recognize any subset of splenic MDSCs nor CD11b−cells (p>0.05). Cluster and secondary structure analysis indicated that aptamers 15 and 11 were similar to aptamers 14 and 12, respectively (fig. S3), thus only aptamer 3, 6, 11, and 14 (Fig. 2B) were chosen for further experiments.
Figure 2: Identification of monoclonal aptamers specific for tumor-infiltrating myeloid cells.
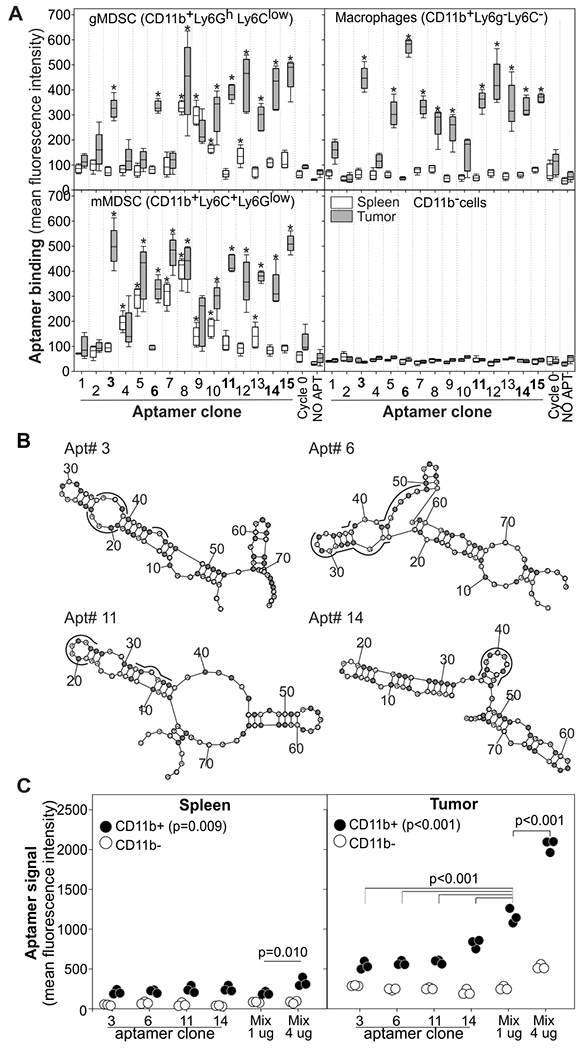
A) Single-cell suspensions from spleens and tumors of mice pooled from 3 mice bearing 4T1 tumors (0.5 cm in diameter) were labelled with antibodies against CD11b, Ly6C, Ly6G and the 15 Cy3-labelled monoclonal aptamers identified by APTANI. Data derived from n=5 biological replicates and N=2 independent experiments. Aptamers 3, 6, 11, and 14 were chosen for further analysis. * = P<0.001 by one-way ANOVA and post-hoc Holm-Sidak comparisons versus control aptamer. B) Secondary structures of aptamers 3, 6, 11, and 14. Binding motifs identified by APTANI are underlined in black (see table S1). Fluorinated nucleotides are highlighted in gray. C) Specificity of an equimolar mixture of the aptamers for tumor-infiltrating myeloid cells. Pooled single cell suspensions of tumor or spleen from 4T1-bearing mice were labeled with aptamers 3, 6, 11, 14, or equimolar mixtures of each aptamer and analyzed by FACS. Data derived from N=3 independent experiments with pooled spleen and tumors from 5 mice. Data were analyzed by one-way ANOVA and post-hoc Holm-Sidak comparisons.
The selected aptamers recognize tumor infiltrating myeloid cells from multiple mouse tumors
We next evaluated the binding properties of these monoclonal RNA aptamers in other tumor models (fig. S4). We stained single cell suspensions from tumor, bone marrow, spleen, and lungs of BALB/c mice bearing the 4T1 mammary carcinoma, the CT26 colon carcinoma, or the RENCA renal carcinoma with aptamer 3, 6, 11, or 14. We performed a similar analysis on single cell suspensions from C57Bl/6 mice bearing B16LU8 melanoma, MCA203 fibrosarcoma, or E0771 mammary carcinoma. The selected aptamers recognized TIMC from all the tumors in both mice strains, whereas no or low staining was observed in myeloid cells from either the spleen or bone marrow, or in T or B cells (fig. S4). Binding was also observed on the myeloid cells from the lung of B16Lu8- and E0771-bearing mice. Considering that lungs are the primary metastatic sites in these models (28, 29)) and the role of myeloid cells in the generation of the premetastatic niche (30), this finding may indicate either a targeting of the premetastatic lesions or passive lung entrapping of the aptamers in these metastatic models. In summary, in all analyzed tumor samples, aptamer 3, 6, 11 and 14 recognized TIMCs regardless of the mouse strain or tumor type. The selected aptamers recognized different tumor-infiltrating myeloid subsets in different cancer types.
We further evaluated specificity by immunofluorescence microscopy using 4T1 tumors and tissues arrays from naïve mice (fig. S5). Although all the selected aptamers recognized cells in the tumors (fig. S5A), most of the tissues were negative for the aptamer binding with a few exceptions: aptamer 3 recognized hepatocytes at low intensity, aptamer 6 showed binding to epithelial cells in the uterus and low binding to pancreatic acinar tissue, and aptamer 14 recognizes intestinal villi and showed a nuclear staining of cerebellum, pancreatic acinar tissue, stomach and spleen (fig. S5B).
We next evaluated whether an equimolar mixture of aptamers 3, 6, 11, and 14 was more efficient in recognizing TIMCs as compared to each individual aptamer. FACS analysis on cells from the spleen and from the tumor of 4T1 bearing mice revealed a clear additive effect when using a mixture of the four aptamers (Fig. 2C).
The selected aptamers recognize human myeloid cells in tumors but not blood of patients with head and neck squamous cell carcinoma.
Having seen that the selected aptamers discriminated between splenic and tumor-infiltrating myeloid cells in mice, we tested whether they cross-reacted with human myeloid cells and whether they could discriminate human TIMCs from their circulating counterparts. Briefly, single-cell suspensions from the tumor or the blood of patients with recurrent HNSCC were stained with antibodies against CD33, HLADR, CD11b, CD14, CD15, and IL4Ra, counterstained with PE conjugated monoclonal aptamers and vital dye, and analyzed by flow cytometry (Fig.3A and fig. S6). Although no or low staining was detected in PBMCs and non-myeloid cells, at least two aptamers recognized myeloid cells infiltrating the tumor from all the analyzed patients to different degrees. In particular, at the tumor site the selected aptamers recognized the majority of mMDSC and CD14−CD15−macrophages/iMDSC and a subset of gMDSC (Fig.3). Within each subset, the aptamers recognized mostly the IL4Ra+ cells (fig. S6) that we have previously shown to be the suppressive myeloid cells and that correlate with tumor recurrence in HNSCC (31).
Figure 3: Monoclonal RNA aptamers preferentially recognize human tumor-infiltrating myeloid cells over circulating myeloid cells from patients with recurrent HNSCC.
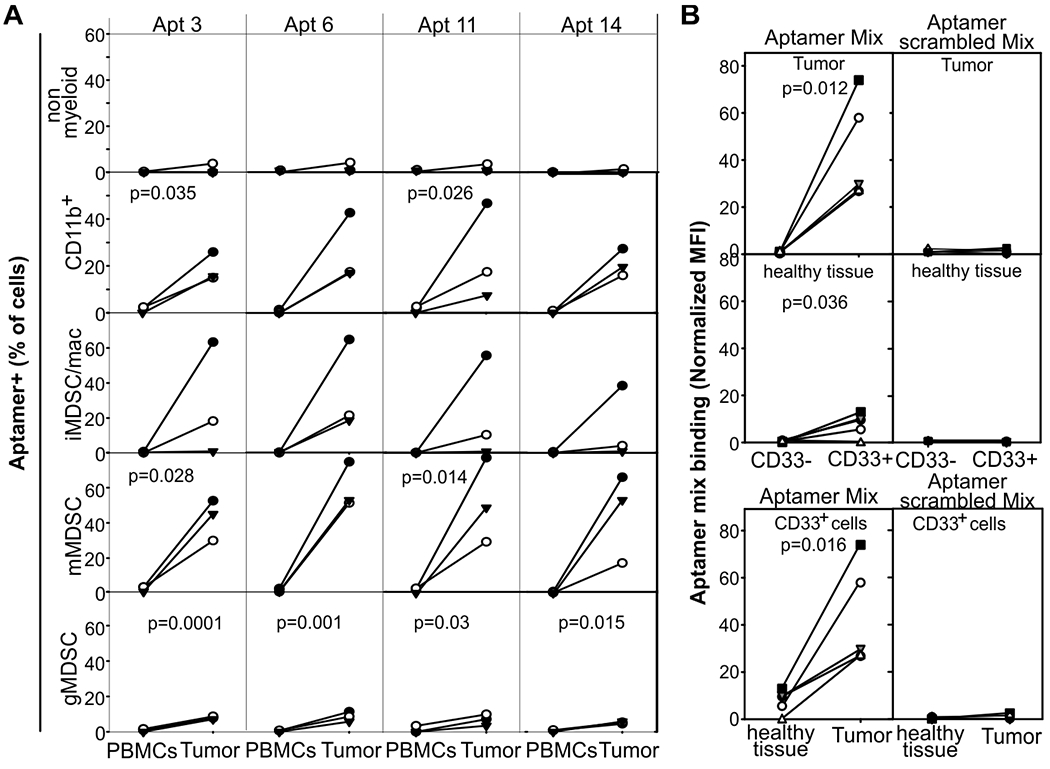
A) Single-cell suspensions from blood and the tumor of patients (n=3) with recurrent HNSCC were stained with AF647-labelled aptamers, anti-CD33, anti-CD14, anti-CD124 antibodies, and vital dye, and analyzed by flow cytometry. B) Image cytometry: Paraffin-embedded tumor specimens from patients (n=5) with HNSCC undergoing salvage surgery were stained with an equimolar mixture of Cy3-aptamer (TIMC specific or scrambled control), anti-CD33 antibody, and DAPI, and analyzed by cell profiler and FCS-express V6 after gating on the “tumor area” or “healthy tissue area” and either on CD33+ or CD33− cells. Aptamer MFI was normalized on the MFI of the all the cells in the corresponding region of interest. Approximately 106 and 105 cells were analyzed in the “tumor area” or “healthy tissue area”, respectively. Significant paired t-tests are reported.
We obtained similar results by image cytometry (Fig. 3B and fig S7). Briefly, paraffin-embedded tissue from patients with recurrent HNSCC were stained with an equimolar mixture of the 4 Cy3-labelled aptamers or with a mixture of scrambled aptamer as negative control. Specimens were counter-stained with DAPI and anti-CD33 antibody, images were acquired by a high-resolution scanner, and data fed into cellprofiler and FCS-express for final analysis. Aptamers showed higher binding to CD33+ cells infiltrating the tumor compared to CD33− cells in the tumor and CD33+ cells present in the surrounding healthy tissue (Fig. 3B and fig. S7). To further evaluate aptamer specificity across human tissues, we stained sections of lung and kidney (fig. S8) and tissue microarrays (fig. S9, brain, heart, skeletal muscle, liver, pancreas and skin) from healthy donors. No aptamer binding was observed in any of the tissues evaluated. In summary, the selected aptamers cross-reacted with human myeloid cells and preferentially recognized myeloid cells infiltrating HNSCC.
Annexin A4 and vimentin are the putative ligands for aptamers 3 and 11
Aptamer-based immunoprecipitation and mass spectrometry identified annexin A4 (ANXA4 isoform X1 P97495) and vimentin (VIM P20152) as putative targets for aptamer 3 and 11, respectively (Fig. 4A). No targets have been identified thus far for aptamers 6 and 14. We first evaluated affinity by flow cytometry using IL4-treated MSC2 and epoxybeads conjugated with relevant or irrelevant recombinant proteins. We also used scrambled aptamers as additional negative controls (Fig. 4A–C and fig. S10A,B). These analyses confirmed the specificity of the aptamer for the IL4-treated MSC2 and indicated a KD in the high nanomolar range of aptamer 3 and 11 for ANXA4 and VIM, respectively. Because the presence of a fluorochrome can alter the interaction between molecules, we evaluated binding by surface plasmon resonance (SPR) using aptamers as ligands and recombinant proteins as analytes. This label-free analysis confirmed the good affinity of aptamers 3 and 11 for ANXA4 and VIM, respectively (Fig.4A and fig. S10C). We tested the specificity of aptamer 3 for ANXA4 and aptamer 11 for vimentin by competitive experiments using the relevant recombinant proteins (Fig. 4D). The addition of the recombinant proteins reduced aptamer binding to IL4-treated MSC2 in a dose-dependent manner. Last, we further tested specificity with validated shRNAs (32) (Fig. 4E). Whereas vimentin silencing reduced aptamer 11 binding (Fig. 4E), ANXA4 silencing resulted in the death of IL4-treated MSC2 cells (fig. S10D).
Figure 4: Characteristics of aptamers that preferentially recognize tumor-infiltrating myeloid cells.
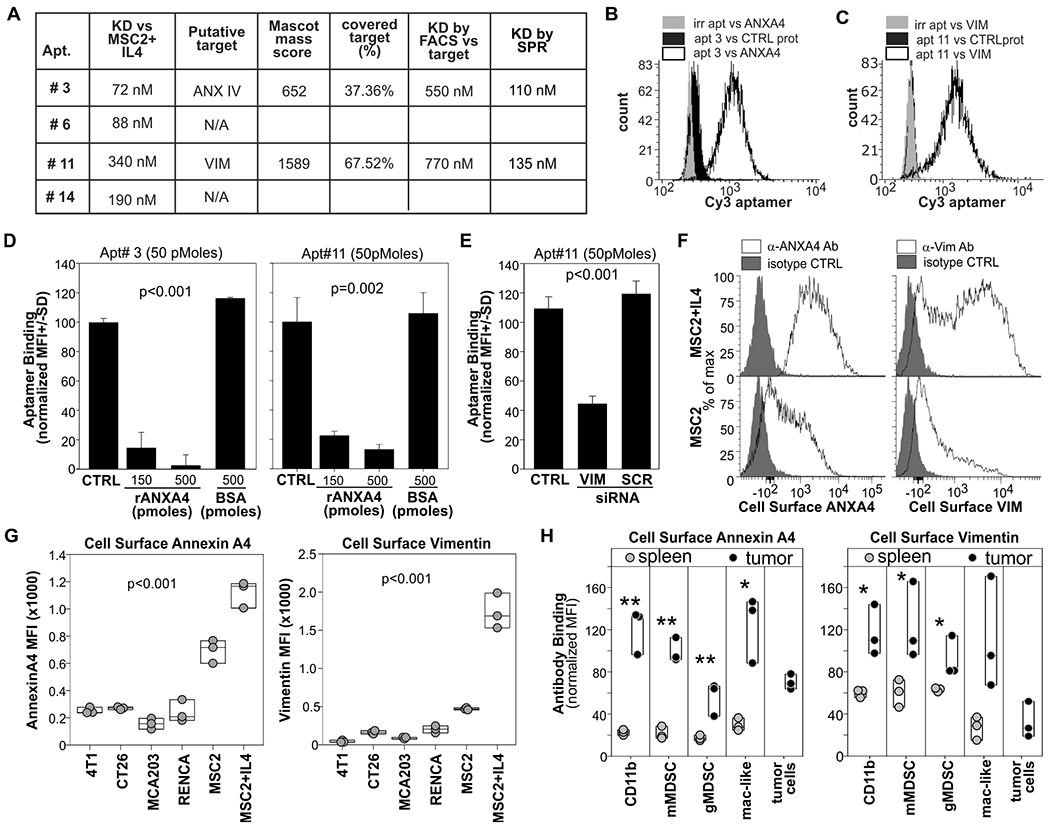
A) Affinity of the aptamers for IL4-treated MSC2 cells was determined by flow cytometry. Putative targets (ANXA4 and VIM) were identified via aptamer-based immunoprecipitation, SDS page, and mass spec analysis. Dissociation constants (KDs) against ligands were determined by FACS against recombinant protein conjugated to epoxy beads and by and surface plasmon resonance (SPR) analysis using biotinylated aptamer as ligand and recombinant proteins as analytes. B) Epoxy beads conjugated with ANXA4 or VCAM as control were stained with Cy3-labelled aptamer 3 or Cycle 0 aptamer library and analyzed by FACS. C) Epoxy beads conjugated with rVIM or irrelevant protein were stained with Cy3-labelled aptamer 11 or cycle 0 library and analyzed by FACS. D) ANXA4 and vimentin competitive assay. 5x105 IL4-treated MSC2 were stained with aptamer 3 or aptamer 11 in the presence or in the absence of recombinant ANXA4 or vimentin, respectively. Mean fluorescence intensity (MFI) is reported. E) MCS2 cells were transfected via 4PD nanoparticles with shRNA against vimentin or a scrambled shRNA. 4 days later cells were stained with Cy3-labelled aptamer 11 and DAPI. F) MSC2 or IL4 treated MSC2 were stained withanti-VIM or anti-ANXA4 antibodies or isotype control G) The indicated cell lines were surface-stained with anti-VIM or anti ANXA4 antibodies or isotype control. H) Single-cell suspensions of tumor or spleen of MCA203 tumor-bearing mice with antibodies against CD11b, Ly6g, Ly6c and either vimentin or ANXA4. P= One-way ANOVA p value. *= p<0.05, **= p<0.001. Data derived from at least N=2 independent experiments.
We then evaluated cell surface expression of ANXA4 and Vimentin with specific antibodies by flow cytometry on MSC2, IL4-treated MSC2 cells, and different mouse tumor cell lines (Fig. 4 F, G). As expected for the putative targets of TIMC specific aptamers, both proteins were significantly (p<0.001) upregulated on the cell surface of MSC2 after IL4 treatment (Fig. 4 F, G) and we found low surface binding in the tested tumor cell lines. Similar results were obtained from the analysis of splenic and tumor infiltrating myeloid cells (Fig. 4H), indicating higher expression of both vimentin and ANXA4 in TIMCs compared to their splenic counterpart.
Aptamers specifically recognize tumor-associated myeloid cells in vivo
To evaluate whether the aptamers can accumulate at the tumor site in vivo, mice orthotopically implanted with the 4T1-luciferase mammary carcinoma or tumor-free mice were treated intravenously with a mixture of aptamers 3, 6, 11, and 14 conjugated with Streptavidin-Alexa Fluor 750 or Alexa Fluor 647 (Fig. 5A). These fluorochromes allow detection of aptamer biodistribution by in vivo imaging system (IVIS) or flow cytometry, respectively. IVIS analysis revealed that the selected aptamers rapidly accumulated at the tumor site and in the liver (Fig. 5 B,C) and, subsequently, in the bladder. A signal from the TIMC-specific aptamers was detectable in the tumor 8 days after injection, whereas a fluorescence signal in other tissues disappeared more rapidly indicating a preferential retention of the aptamers for the tumor microenvironment. Flow cytometry analysis (Fig. 5 D–F) confirmed the IVIS results and indicated that aptamers in the tumor microenvironment preferentially bind to CD11b+ myeloid cells (Fig. 5D), particularly to mMDSC and TAM. Time course analysis revealed that a population of aptamer-positive gMDSCs appeared at later time points in the tumor and spleen of treated mice, (Fig. 5E–F), supporting the notion that mMDSCs can differentiate into gMDSCs (33, 34). To further evaluate the specificity of the aptamer for TIMC, we treated 4T1-bearing mice with an anti-Gr1 antibody (35) to deplete myeloid cells before IVIS and flow cytometry evaluation (Fig. 5G). As expected, treatment with the anti-Gr1 antibody significantly (p<0.0001) reduced tumor infiltrating CD11b+ myeloid cells (Fig. 5H). This reduction in TIMC correlated with a reduction in aptamer signal as evaluated by IVIS (Fig. 5I). In summary, systemically administered TIMC-specific aptamers rapidly accumulate at the tumor site and recognize TIMC in vivo.
Figure 5: Aptamers preferentially target tumor stroma in vivo.
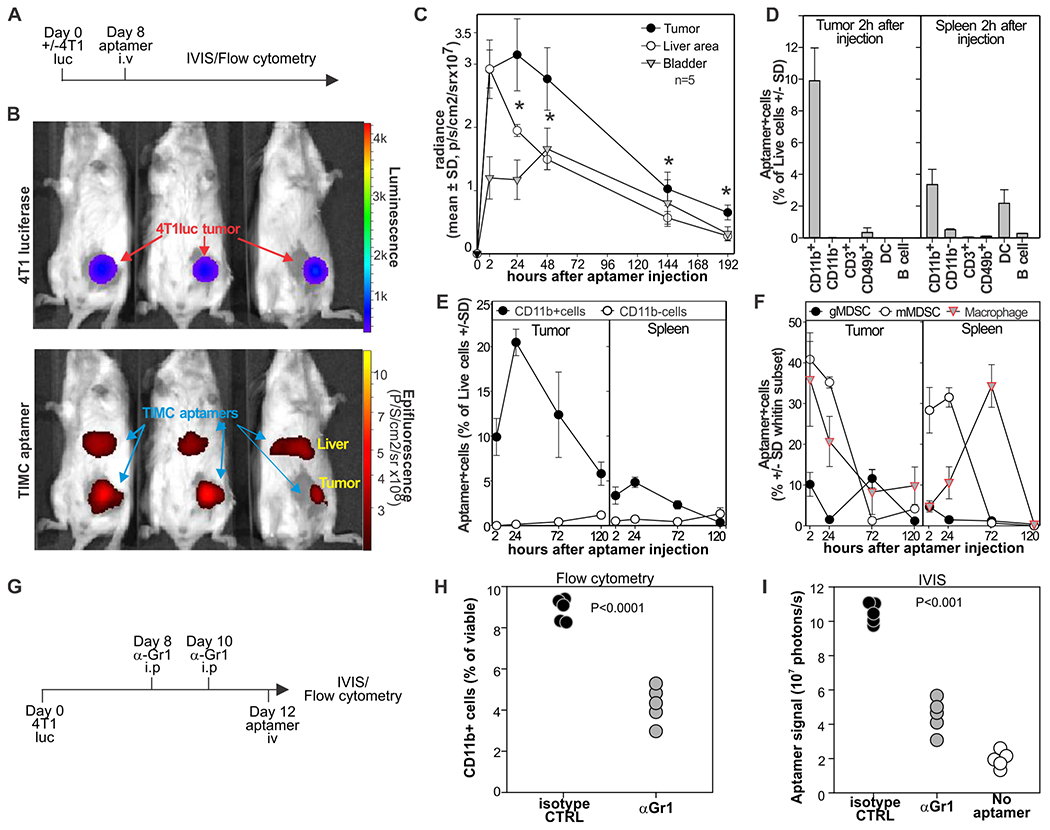
A) Aptamers target tumors in vivo. Mice bearing 4T1-luciferase mammary carcinomas were injected intravenously with an equimolar mixture of biotinylated aptamers 3, 6, 11, and 14 conjugated with AF750 streptavidin. B) Bio-distribution was evaluated by IVIS 2h later. C) Time course analysis or aptamer bio-distribution detected by IVIS. D) Mice (n=5) bearing 4T1 mammary carcinomas were injected i.v. with aptamers 3, 6, 11, and 14 loaded on AF-647 streptavidin. After two hours, indicated organs were harvested and counterstained with antibodies against CD11b, CD19, CD49b, CD11c, and CD3. DC, dendritic cells. E) Mice (n=5) were treated as in (D), and aptamer distribution was evaluated by flow cytometry on CD11b+ and CD11b− cells at different time points. F) Mice (n=5) were treated as in (D), and aptamer distribution was evaluated by flow cytometry on gMDSC (CD11b+Ly6g+cells), mMDSC (CD11b+Ly6c+cells), or macrophages (CD11b+F4/80+Ly6g−Ly6c− cells)s. G-I) 4T1-bearing mice were treated i.p with anti-GR1 antibody or rat IgG antibody 8 and 10 days after challenge (G). Two days later, mice were injected i.v. with AF750-conjugated aptamers imaged by IVIS (I), euthanized and tumors analyzed by FACS for the presence of CD11b+cells (H). * = one-way ANOVA p<0.001. Data derived from N=2–3 experiments each with n=3 mice.
TIMC-specific aptamers maximize the delivery of doxorubicin at tumor sites
As TIMC-specific aptamers can target the tumor microenvironment, we evaluated their drug delivery ability using doxorubicin (DOX) as a chemotherapeutic agent. Briefly, aptamers were extended at the 3’ terminus with a GC rich tail facilitating DOX intercalation (36, 37) and optimal loading conditions were evaluated by fluorescence spectroscopy (Fig. 6A,B). Because aptamer formulation can affect their pharmacokinetics and biodistribution, 4T1-bearing mice were i.v. treated with DOX conjugated to either TIMC-specific aptamers or scrambled aptamers. We then evaluated aptamer biodistribution by qRT-PCR at different timepoints (Fig. 6C), which confirmed our results from IVIS and flow cytometry. However, we observed no aptamer accumulation in the liver (Fig. 6C), suggesting the involvement of streptavidin or fluorochrome in the hepatic entrapment observed in previous experiments.
Figure 6: TIMC-specific aptamers increase doxorubicin concentration at the tumor site.
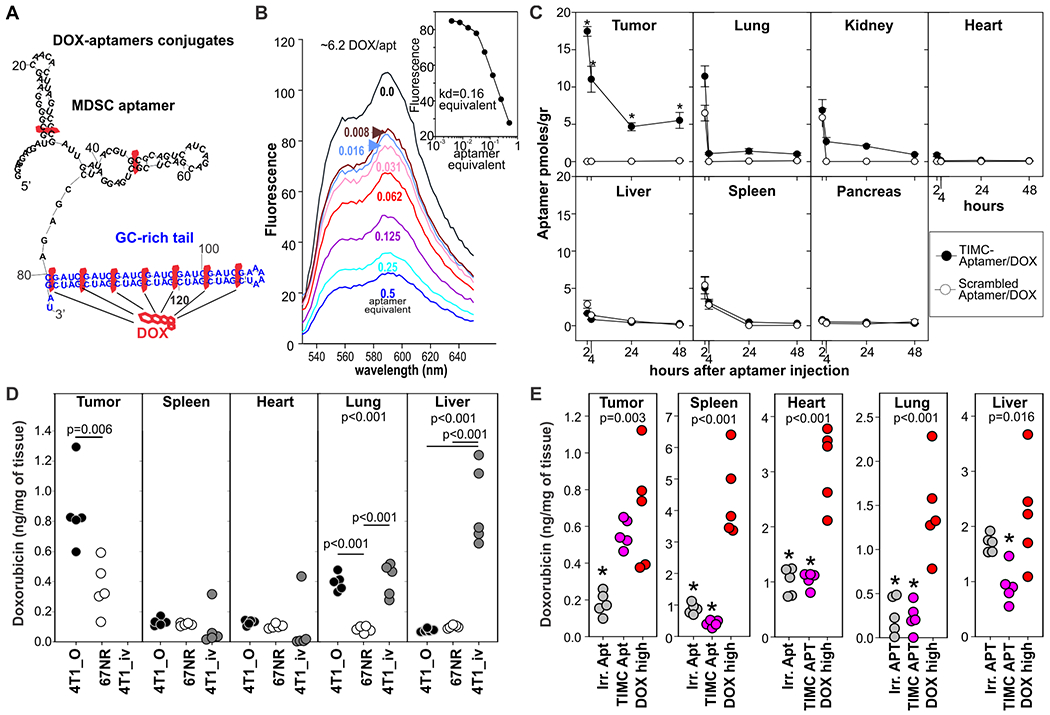
A) TIMC-specific aptamers were conjugated to DOX by extending the 3’ end with a GC rich tail. B) Fluorescence spectra of DOX solution (1.5 μM) with increasing molar ratios of the TIMC aptamer equimolar mixture (top to bottom: 0, 0.008, 0.016, 0.032, 0.062, 0.125, 0.25, and 0.5 equivalent). Inset: A Hill plot for the aptamer titration (Kd=0.16 equivalent; 6.2 dox molecules/aptamer). C) BALB/c mice (n=5) bearing the orthotopic 4T1 tumors were injected i.v. with DOX (0.35mg/kg) conjugated with TIMC-specific or scrambled aptamers. At the indicated time point mice were euthanized and aptamer biodistribution in different organs was evaluated by qRT-PCR. D) BALB/c mice (n=5) were challenged with orthotopic 4T1 mammary carcinomas (4T1_o; primary tumor: breast, metastases: lung) or intravenously (4T1_iv; metastasis in the lung and in the liver). An additional group of mice was challenged orthotopically with the non-metastatic, 4T1-derived cell line 67NR (only primary tumor). 10 days later, mice were treated i.v. with DOX (0.35 mg/kg) conjugated with TIMC-specific aptamers. DOX bio-distribution was evaluated 24 h later. E) Bio-distribution of free DOX or DOX given via TIMC-specific or scrambled aptamers. Mice (n=5) bearing 4T1 mammary carcinomas (0.5 cm in diameter) in the 3rd mammary gland were given i.v. free doxorubicin (DOX high 3.5 mg/kg), DOX-conjugated TIMC-aptamer (TIMC apt; 0.35mg/kg) or scrambled aptamer (Irr. Apt)-conjugated doxorubicin. 24h later doxorubicin was quantified in tissues by spectrometry after acid alcohol extraction. Data derived from N=1–2 experiments each with n=5 mice *= p<0.05 in multiple pairwise comparison vs dox high group (Holm-Sidak method).
To evaluate whether DOX-aptamer conjugates could target primary and metastatic lesions, an equimolar mixture of the four aptamers was loaded with DOX and injected intravenously into mice previously challenged with: 4T1 mammary carcinoma injected into the mammary gland (primary tumor in breast, metastasis in lung); 4T1 administered intravenously, resulting in tumors in the liver and lungs; or the non-metastatic 4T1-variant 67NR injected into the mammary gland, resulting in localized tumors. 24 hours after injection, we evaluated the bio-distribution of DOX-aptamer conjugates by spectrophotometer analysis (Fig. 6D). The aptamers delivered DOX to both the primary mammary tumor and to metastatic sites. In mice with non-metastatic 67NR tumors, DOX was found only in cancerous breast tissue. In mice with metastatic 4T1 injected intravenously, DOX was found in the liver and lungs, whereas in mice with 4T1 injected into the mammary gland, DOX accumulated at the primary tumor site and in the lung, a primary metastatic site in this model. Additional experiments indicated that the DOX concentration at the tumor site of mice treated intravenously with 0.35 mg/kg (a “low dose”) of DOX-aptamer complexes was similar to those of mice treated with 3.5 mg/kg (a “high dose”) of free DOX, whereas in all other evaluated compartments the overall DOX concentration was significantly (p<0.001) lower in the mice treated with DOX-aptamer complexes (Fig. 6E). These results indicate that DOX conjugation does not affect the aptamer specificity and that TIMC-specific aptamers can deliver chemotherapeutic agents to primary and metastatic sites.
TIMC-specific aptamers increase doxorubicin’s therapeutic index
We next compared the therapeutic efficacies of DOX delivered by TIMC-specific aptamers, by PEGylated liposomes (Doxil, the first clinically available nanoparticle that became the gold standard for DOX treatment (38, 39)), and as unconjugated molecules. Briefly, 4T1-bearing BALB/c mice were treated with unconjugated DOX at high or low doses; with low doses of DOX conjugated to the TIMC-specific aptamer; or with Doxil (low dose - 0.35 mg/kg). Treatment was repeated 2 and 6 days later. As additional controls, mice were treated with unconjugated aptamers, irrelevant aptamers conjugated with DOX, or left untreated. No significant (P>0.05) anti-tumor effect was observed in mice treated with either high doses of free DOX or Doxil (Fig. 7A). Conversely, low doses of DOX delivered via TIMC-specific aptamers significantly (P<0.001) delayed tumor progression and resulted in 40% of treated mice having no clinically detectable tumor 60 days after treatment (Fig. 7A). We then evaluated the therapeutic efficacy of DOX conjugated aptamers in the C57Bl/6-derived MCA203 fibrosarcoma model (Fig. 7B) whose tumors are characterized by a reduced infiltration of myeloid cell infiltrate compared to the 4T1 carcinoma (5.6±1.7% vs 22.9±4.5% of viable cells, respectively). Whereas no antitumor effect was observed with high doses of free DOX or scrambled aptamer/DOX complexes, significant (p<0.001) tumor growth reduction and increased survival was observed in the mice treated with low doses of DOX conjugated to TIMC specific aptamers (Fig. 7B).
Figure 7: TIMC-specific aptamers increase the doxorubicin therapeutic index.
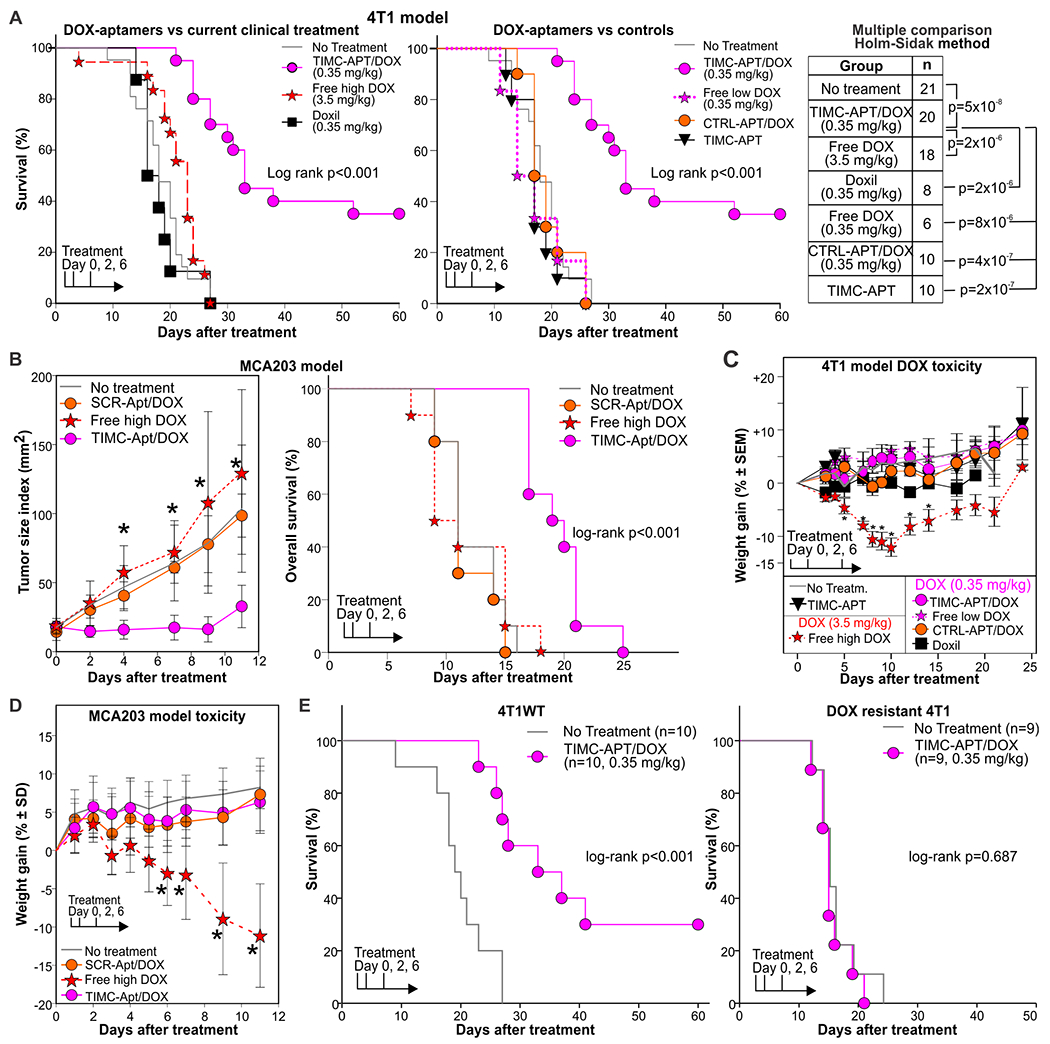
A) Mice bearing 4T1 tumors (~0.5 cm in diameter) in the 3rd mammary gland were treated i.v. with free doxorubicin (3.5 mg/kg or 0.35 mg/kg), Doxil (0.35 mg/kg), or doxorubicin conjugated to a TIMC-specific aptamer mix (0.35 mg/kg). Additional controls included DOX-conjugated scrambled aptamers, unconjugated TIMC-specific aptamers, and untreated mice. Treatment was repeated 2 and 6 days after initial injection. Mice were euthanized when the tumor index reached 1.2 cm2. Log-rank and post-hoc multiple comparison analysis (Holm-Sidak method) is reported. Individual tumor growth curves are shown in fig. S6. B) C57Bl/6 mice were challenged with MCA203 fibrosarcoma. When tumors reached ~0.5cm in diameter, mice were treated with vehicle, high doses of free DOX (3.5 mg/kg), or low doses of DOX (0.35 mg/kg) conjugated either to TIMC-specific or scrambled aptamer. C, D) Weight loss as measure of toxicity is reported for mice bearing the 4T1 (C) and MCA203 (D) tumors. E) BALB/c mice were challenged with the 4T1wt breast cancer or with a DOX-resistant variant of 4T1. When tumors reached 0.5 cm in diameter, mice were treated with TIMC-specific aptamers or left untreated. Survival was monitored. Data are cumulative of 2-3 independent experiments.
Furthermore, although high doses of free DOX resulted in high toxicity as determined by significant (p<0.01) body weight loss and 15% treatment-related mortality, no toxicity was observed with DOX-loaded aptamers (Fig. 7 C, D). Increased concentrations of amylase and aspartate transaminase and a reduction of white and red blood cells suggested pancreatic, hepatic, and bone marrow toxicities in the free DOX-treated group, whereas no differences from untreated controls were observed in the mice treated with DOX loaded aptamers (fig. S12). However, cytological signs of toxicity were not observed in H&E stained sections of internal organs such as heart, liver, kidney, and pancreas harvested 2 days after the last treatment in any of the experimental groups.
DOX can depleted (40) MDSCs or impair their function (41) and depletion or inactivation of MDSCs can delay tumor progression (19)., Thus, we asked whether the observed anti-tumor efficacy was due to a cytotoxic activity of DOX-conjugated aptamer on TIMC or rather to DOX uptake by TIMC and its subsequent bystander release and cytotoxicity to neoplastic cells. To this aim, we evaluated the efficacy of treatment using a DOX-resistant 4T1 cell line (DOXR-4T1) and the DOX-sensitive parental cell line. DOX-loaded aptamer treatment confirmed its anti-tumor efficacy on the DOX-sensitive parental cell line, whereas no therapeutic effect was observed in mice challenged with DOX-resistant 4T1 tumors (Fig. 7C). Taken together, these data support the notion that TIMC-specific aptamers can increase the DOX therapeutic index by releasing this chemotherapeutic agent in the tumor micro-environment.
Discussion
Systemic chemotherapy remains one of the most important treatments for cancer therapy, but it comes with systemic side effects, such as cardiotoxicity and profound neutropenia that impose limits on the use of this therapeutic option (42). Additionally, chemotherapy induces long-term morbidity that decreases the quality of life of cancer survivors (43). Targeted delivery of chemotherapeutics using antigens present on neoplastic cells was proposed as a magic bullet approach (44); however, considering the heterogeneity and genomic instability of malignant cells, these approaches will likely result in tumor editing rather than tumor eradication (45).
Here, we present a strategy by which chemotherapeutic agents can be concentrated in the tumor and metastatic sites by physically targeting the pro-tumoral activated phenotype of tumor- infiltrating myeloid cells. Myeloid cells are the most abundant innate immune cells present in the stroma of several types of mouse and human cancer (46–48). Myeloid cell presence in human tumors correlates with increased vascular density, higher metastatic spread, and a worse clinical outcome, and their presence is necessary for tumor progression (49, 50). At the tumor site, myeloid cells acquire unique antigenic profiles and functional characteristics that differentiate them from their systemically circulating counterparts, and fulfill immune suppressive, tolerogenic, and pro-tumoral roles (20, 51, 52). Although many approaches have been and are being tested to inhibit the mechanisms that myeloid cells employ to promote tumor progression or their interaction with the neoplastic cells (53–55), to our knowledge no reagent able to discriminate between tumor infiltrating and circulating myeloid cells is yet available. Furthermore, although aptamers and peptides able to bind both tumor-associated and circulating MDSCs in mice have been isolated (56–58) and employed for MDSC depletion (57, 58) or to improve Doxil delivery (56), these reagents did not preferentially recognize TIMCs. Indeed, when used for Doxil delivery an important accumulation of DOX in the heart, spleen, and liver was noted (56).
We isolated four RNA aptamers that were specific for the tumor-infiltrating myeloid cells but not for their circulating counterparts, regardless of the used tumor model or mouse strain. The combinatorial use of these four aptamers shows an additive specificity for TIMC and offers an efficient delivery of the chemotherapeutic agent doxorubicin in our proof-of-principle experiments, to both primary and metastatic tumor sites and with minimal accumulation in other tissues. In therapeutic settings our approach was superior to free doxorubicin or Doxil (38, 39) (today’s gold standard for doxorubicin targeted delivery) and resulted in tumor regression in approximately 40% of treated mice with no observed systemic toxicity.
We identified ANXA4 and vimentin as putative ligands for aptamer 3 and aptamer 11, respectively. In line with the anti-apoptotic role reported for ANXA4 (59), attempts at silencing this gene resulted in the death of IL4-treated MSC2. ANXA4 is a calcium-dependent, phospholipid-binding protein that promotes membrane fusion and exocytosis (60) and that is overexpressed in activated MDSCs, TIMC, and activated M2 macrophages (51, 61). Although localized in the cytoplasm in resting macrophages, ANXA4 translocates to the membrane during activation (62, 63). Vimentin is present in the tumor stroma of different cancers, is poorly expressed in cultured tumor cells (64–66), and has been involved in epithelial-mesenchymal transition and metastasis (67). Vimentin plays a key role in monocyte differentiation and in the production of reactive oxygen species and, under pro-inflammatory stimuli, translocates to the membrane of activated macrophages (64–66, 68, 69). We observed an increase of both ANXA4 and vimentin on the surface of MSC2 after activation with IL4 and the upregulation of both protein at the tumor site. Both proteins are expressed in human malignancies and their expression correlates with a worse prognosis in renal, breast and ovarian cancer according to the TCGA data and the Protein Atlas. Thus, both identified targets support the specificity of the aptamers for TIMC activated phenotypes.
The isolated TIMC specific aptamers cross-reacted with and preferentially recognized human MDSC in the tumors but not blood of patients with HNSCC, suggesting their possible use for MDSCs detection or as tumor targeting agents in human malignancies. The specificity of the aptamer for tumor stroma was further supported by absence of aptamer signal in different human tissues evaluated by immunofluorescence microscopy.
There are several limitations to this study. First, although we showed that the aptamers can recognize TIMC from patients with HNSCC, we have not yet tested these reagents in other human malignancies. Second, although we identified the putative targets for aptamers 3 and 11, we were unable to identify the cognate ligands for aptamers 6 and 14. This may limit future preclinical development and approval of aptamers 6 and 14 for clinical use. Last, we focused on the delivery of doxorubicin, but other aptamer formulations might be needed to allow the delivery of multiple drugs for possible targeted combinatorial therapy.
In any case, our data indicate that it is possible to potentiate the therapeutic index of chemotherapeutic agents by targeting the surface protein profile of tumor-infiltrating myeloid cells, allowing the bystander release of the drug within the tumor microenvironment. The combinatorial use of several different aptamers as a means of chemotherapeutic agent delivery, each targeting different TIMC specific epitopes, increases treatment specificity, improves efficacy, and reduces toxicity of chemotherapeutic agents as compared with the systemic delivery of these agents in current, clinically available treatment modalities.
Materials and Methods
Study design
The goal of this study was to identify RNA aptamers able to discriminate myeloid cells infiltrating the tumor from those in the periphery. Aptamers were selected by unsupervised Cell-SELEX, bioinformatics analysis, and empirical testing by flow cytometry and immunofluorescence analysis. Each experiment has been performed at least twice by two independent experimenters and, if possible, the same phenomenon was evaluated using two independent techniques to eliminate assay-specific artifacts. Flow cytometry was performed on a daily-calibrated flow cytometer, using titrated and validated antibodies, vital dye, automatic compensation using single-cell color, and full-minus-one (FMO) or control aptamers (cycle 0, irrelevant or scrambled aptamer) as negative controls. For in vivo experiments, mice were randomized before treatment and tumor measurement was taken by an experimenter blinded to treatment. Unless otherwise specified, mice were euthanized when tumors reached ~1.2 cm diameter, or if they lost more than 20% of initial body weight or showed clinical signs of treatment related toxicity (lethargy, ruffled coat). Data are cumulative and derived from 2 to 3 replicate experiments each with 3-5 mice per group. Group size was determined by power analysis using the effect size from pilot experiments. Outliers were always included. In one experiment (Fig. 7E), two mice were removed and euthanized for reasons unrelated to the study (wounds from fights).
Mice
All animal experiments were approved by the Division of Veterinary Resources and the Institutional Animal Care & Use Committee of the University of Miami. 8-10 week-old BALB/c and C57Bl/6J mice were purchased from Jackson Laboratories and maintained in pathogen-free animal facilities at the University of Miami on a chlorophyll-free diet. Mice were allowed to acclimate for at least one week before experiments, ear-tagged, and randomized after tumor inoculation. Balb/c mice were injected orthotopically with 3x105 4T1 cells, 3x105 4T1 cells, or 4T1Thy1.1 Luciferase cells, or injected subcutaneously (s.c.) with 5x105 CT26 or 3x105 RENCA cells in 100 μl of PBS. C57Bl/6 mice were injected in the mammary gland with 2x105 E0771 cells, s.c. with 5x105 MCA203 cells, or 5x105 CT26 cells. In some experiments (Fig.5G–H), myeloid cells were depleted by injecting 100μg anti-Gr1 (clone RB6-8C5) i.p. 8 and 10 days after challenge. Tumor growth and mice weights are reported. Mice were evaluated at least 3 times a week and humanely euthanized when tumor size index reached 150 mm2 or if they lost >20% of initial weight due to chemotherapy or tumor growth, in compliance with the IACUC policy and animal protocol. Data are expressed as tumor size index, defined as the product of the main diameter with the perpendicular one.
Aptamer-doxorubicin treatment and toxicology studies
Aptamer-DOX was prepared by constructing a DNA template with the appropriate aptamer followed by a GC-rich primer sequence downstream (GC rich Sul3’, table S2). RNA was transcribed from this template and purified as described above. DOX (Sigma) was mixed with the RNA sequence as needed. Doxil (ALZA corporation) was purchased through the University of Miami pharmacy.
BALB/c or C57Bl/6J mice were injected in the mammary gland with the 4T1 luciferase thy1.1 tumor cell line, or s.c. with the MCA203 fibrosarcoma cell line, respectively. Mice were treated intravenously with DOX or DOX-aptamer complexes starting when tumors reached 5 mm of diameter and repeated 2 and 6 days later. In some experiments, mice were euthanized 2 days after the last treatment, and blood and internal organs were harvested for toxicology studies. Blood was processed by the University of Miami pathology core for complete chemistry and cell subset count using standard procedures. Tissues were snap frozen, stained with H&E, and analyzed by an experienced pathologist blinded to treatment group.
IVIS analysis
Isofluorane-anesthetized tumor-bearing mice were analyzed by the In Vivo Imaging System (Xenogen IVIS Spectrum - Perkin Elmer) 2h after i.v. injection of 5’ biotinylated aptamers conjugated with Alexa Fluor-750 (AF750) streptavidin (13.6 pmol/g) with an imaging stage heated at 37°C. 15’ before imaging mice were injected intraperitoneal with D-luciferine (150 μg/g). AF750 fluorescence was read at 800 nm after excitation at 748 nm. The raw signal was subject to spectral unmixing to remove background fluorescence signal and AF750 fluorescence was quantified with Living Image v4.3 software (Perkin Elmer).
Detection of aptamer in tissues by qRT-PCR
Internal organs were rinsed in PBS, weighed, and 50-60 mg of each tissue were mechanically dissociated using an electric micro-mortar (VWR) in 2 volumes of Trizol (Thermofisher). 8 additional volumes of Trizol were then added to each mixture and RNA was extracted as per manufacturer instructions. 100-300 ng of RNA were amplified in qRT-PCR by using the SUL3’short and SUL5’ oligonucleotides (table S2) using the iTAq Universal sybr Green one-step kit (Biorad) with the following cycling conditions: 1) 50°C for 50’ (Retrotranscription), 2) 95°C for 5’, 3) 30x(95°C for 5”, 53°C for 15”, and 72°C for 60”). Aptamer concentration was determined by interpolation with a calibration curve (range 3x10−12 to 3x10−20 moles) generated under the same conditions using the same aptamer mixture. Data were normalized on the original RNA yield and organ weight. Data were derived from 2 experiments with at least 3 biological replicates. The limit of detection of our qRT-PCR is estimated as approximately 0.03 fentomoles of aptamer in 300 ng of total RNA (~3,000-26,000 aptamer molecules/μg of tissue).
Statistical analysis
All values depicted represent mean ± standard deviation of biological replicates unless otherwise indicated in the figure legend. Statistical calculations were performed by a person blinded to the treatment group using Sigmaplot 12.5 (Systat software). One-way ANOVA with Holm Sidak test for multiple pairwise comparison or two-tailed unpaired Student’s t-test were applied as indicated in the figure legends after normality was evaluated by the Shapiro-Wilk test. ANOVA on ranks followed by Tukey test for multiple comparisons was used for those data that failed the normality test. Variances were similar between groups in each experiment unless otherwise stated. In vivo experiments included cohorts of the size indicated in each figure legend (at least 6 mice per group). In vitro analyses and in vivo experiments were repeated two to five times to ensure reproducible conclusions; the exact number of repetitions is stated in each figure legend. Log-rank test was used for survival analysis followed by all pairwise multiple comparison procedures (Holm-Sidak method). Data from multiple experiments were averaged unless otherwise indicated in the figure legends. No experimental data points were excluded from analysis. Sample size was chosen by power analysis using effect size determined by pilot experiments or prior experience of the authors.
Supplementary Material
Fig. S1: MSC2 cells treated with IL4 are transcriptionally similar to TIMC.
Fig. S2: Frequency variation of the 15 selected aptamers during HT-Cell SELEX.
Fig. S3: Cluster analysis of the 15 aptamers selected with APTANI.
Fig. S4: Aptamer 3, 6, 11, and 14 recognize TIMC among different tumors and different genetic backgrounds.
Fig. S5: Aptamer 3, 6, 11 and 14 show low or no binding to other mouse tissues.
Fig. S6: Aptamers 3, 6, 11, and 14 recognize TIMC in patients with HNSCC. Flow cytometry.
Fig. S7: Aptamers 3, 6, 11, and 14 recognize TIMC in patients with HNSCC. Image cytometry.
Fig. S8: TIMC-specific aptamers do not bind lung or kidney from healthy donors.
Fig. S9: TIMC-specific aptamers do not bind to human tissue microarrays.
Fig.S10: Affinity of aptamers 3, 6, 11, and 14 for IL4-treated MSC2 or their putative ligand.
Fig. S11: TIMC-specific aptamers are superior to Doxil against the 4T1 breast cancer.
Fig. S12: TIMC-aptamer approach reduces doxorubicin toxicity.
Table S1: Sequence, frequency, and motifs of 15 aptamers selected by APTANI.
Table S2: List of oligonucleotides used.
Table S3: List of antibodies used.
Data file S1: Data from primary figures.
Acknowledgments:
We thank Svetlana Speransky for help with immunofluorescence on human tissues, Carmen Gomez for help distinguishing human tumors from surrounding healthy tissue, and Oliver Umland for help with flow cytometry analysis.
Funding:
This work is supported by the DOD-BRCA idea award W81XWH-11-1-0478 (to P.S.), the UM-Sylvester Cancer Center, the AIRC Special Program Molecular Clinical Oncology “5 per mille”, and the Italian Epigenomics Flagship Project (Epigen; to S.B).
Footnotes
Competing interests: A.D., S.Z., J.C, D.V.S. V.B., S.B and P.S. are named as inventors in non-provisional US patent application # 62/815,142 entitled “RNA aptamer and uses thereof” and filed by the University of Miami and regarding the use of RNA aptamer to target myeloid cells. PS is a consultant for Nanovax, LLC.
Data and materials availability:
All data associated with this study can be found in the paper or supplementary materials. Microarray data are available at GSE110774. HT-sequencing data are available at GSE113378.
References and notes
- 1.Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, Moreira PI, Doxorubicin: the good, the bad and the ugly effect. Current medicinal chemistry 16, 3267–3285 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Kydd J, Jadia R, Velpurisiva P, Gad A, Paliwal S, Rai P, Targeting Strategies for the Combination Treatment of Cancer Using Drug Delivery Systems. Pharmaceutics 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinnear C, Moore TL, Rodriguez-Lorenzo L, Rothen-Rutishauser B, Petri-Fink A, Form Follows Function: Nanoparticle Shape and Its Implications for Nanomedicine. Chemical reviews 117, 11476–11521 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Bahrami B, Hojjat-Farsangi M, Mohammadi H, Anvari E, Ghalamfarsa G, Yousefi M, Jadidi-Niaragh F, Nanoparticles and targeted drug delivery in cancer therapy. Immunology letters 190, 64–83 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Tiet P, Berlin JM, Exploiting homing abilities of cell carriers: Targeted delivery of nanoparticles for cancer therapy. Biochemical pharmacology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC, Challenges and key considerations of the enhanced permeability and retention (EPR) effect for nanomedicine drug delivery in oncology. Cancer research 73, 2412–2417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Scholler J, Lacey SF, Melenhorst JJ, Nazimuddin F, Perazzelli J, Christian DA, Hunter CA, Porter DL, June CH, Grupp SA, Gill S, Treatment of leukemia antigen-loss relapses occurring after CD19-targeted immunotherapies by combination of anti-CD123 and anti-CD19 chimeric antigen receptor T cells. Journal for Immunotherapy of Cancer 3, O5–O5 (2015). [Google Scholar]
- 8.Kelderman S, Schumacher TNM, Haanen JBAG, Acquired and intrinsic resistance in cancer immunotherapy. Molecular Oncology 8, 1132–1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Attarwala H, Role of antibodies in cancer targeting. Journal of Natural Science, Biology, and Medicine 1, 53–56 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szot C, Saha S, Zhang XM, Zhu Z, Hilton MB, Morris K, Seaman S, Dunleavey JM, Hsu K-S, Yu G-J, Morris H, Swing DA, Haines DC, Wang Y, Hwang J, Feng Y, Welsch D, DeCrescenzo G, Chaudhary A, Zudaire E, Dimitrov DS, St. Croix B, Tumor stroma–targeted antibody-drug conjugate triggers localized anticancer drug release. The Journal of Clinical Investigation 128, 2927–2943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condamine T, Ramachandran I, Youn JI, Gabrilovich DI, Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med 66, 97–110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marvel D, Gabrilovich DI, Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 125, 3356–3364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arina A, Corrales L, Bronte V, Enhancing T cell therapy by overcoming the immunosuppressive tumor microenvironment. Semin Immunol, (2016). [DOI] [PubMed] [Google Scholar]
- 14.Ugel S, De Sanctis F, Mandruzzato S, Bronte V, Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 125, 3365–3376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zilio S, Serafini P, Neutrophils and Granulocytic MDSC: The Janus God of Cancer Immunotherapy. Vaccines 4, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serafini P, Myeloid derived suppressor cells in physiological and pathological conditions: the good, the bad, and the ugly. Immunol Res 57, 172–184 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Kitamura T, Qian B-Z, Pollard JW, Immune cell promotion of metastasis. 15, 73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Achyut BR, Arbab AS, Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. OncoTargets and therapy 9, 1047–1055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ugel S, De Sanctis F, Mandruzzato S, Bronte V, Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. The Journal of Clinical Investigation 125, 3365–3376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kusmartsev S, Gabrilovich DI, STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol 174, 4880–4891 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Kumar V, Patel S, Tcyganov E, Gabrilovich DI, The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends in immunology 37, 208–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, Ochoa AC, Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. OncoImmunology 6, e1344804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catuogno S, Esposito CL, Aptamer Cell-Based Selection: Overview and Advances. Biomedicines 5, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Rossi J, Aptamers as targeted therapeutics: current potential and challenges. Nature reviews. Drug discovery 16, 181–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamada M, In silico approaches to RNA aptamer design. Biochimie, (2017). [DOI] [PubMed] [Google Scholar]
- 26.Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, Young HA, Zanovello P, Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol 165, 6723–6730 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Caroli J, Taccioli C, De La Fuente A, Serafini P, Bicciato S, APTANI: a computational tool to select aptamers through sequence-structure motif analysis of HT-SELEX data. Bioinformatics 32, 161–164 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ewens A, Mihich E, Ehrke MJ, Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res 25, 3905–3915 (2005). [PubMed] [Google Scholar]
- 29.Bronte V, Cingarlini S, Apolloni E, Serafini P, Marigo I, De Santo C, Macino B, Marin O, Zanovello P, Effective genetic vaccination with a widely shared endogenous retroviral tumor antigen requires CD40 stimulation during tumor rejection phase. J Immunol 171, 6396–6405 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Doak GR, Schwertfeger KL, Wood DK, Distant Relations: Macrophage Functions in the Metastatic Niche. Trends in cancer 4, 445–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weed DT, Vella JL, Reis IM, De la Fuente AC, Gomez C, Sargi Z, Nazarian R, Califano J, Borrello I, Serafini P, Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res 21, 39–48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desclaux M, Teigell M, Amar L, Vogel R, Gimenez YRM, Privat A, Mallet J, A novel and efficient gene transfer strategy reduces glial reactivity and improves neuronal survival and axonal growth in vitro. PLoS One 4, e6227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zilio S, Vella JL, De la Fuente AC, Daftarian PM, Weed DT, Kaifer A, Marigo I, Leone K, Bronte V, Serafini P, 4PD Functionalized Dendrimers: A Flexible Tool for In Vivo Gene Silencing of Tumor-Educated Myeloid Cells. J Immunol 198, 4166–4177 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, Sarnaik A, Horna P, Sotomayor E, Gabrilovich DI, Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nature immunology 14, 211–220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P, Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 96, 3838–3846 (2000). [PMC free article] [PubMed] [Google Scholar]
- 36.Stuart CH, Horita DA, Thomas MJ, Salsbury FR, Lively MO, Gmeiner WH, Site-Specific DNA–Doxorubicin Conjugates Display Enhanced Cytotoxicity to Breast Cancer Cells. Bioconjugate Chemistry 25, 406–413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagalkot V, Farokhzad OC, Langer R, Jon S, An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed Engl 45, 8149–8152 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D, Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Experimental Hematology & Oncology 1, 10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bulbake U, Doppalapudi S, Kommineni N, Khan W, Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 9, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belyaev NN, Abdolla N, Perfilyeva YV, Ostapchuk YO, Krasnoshtanov VK, Kali A, Tleulieva R, Daunorubicin conjugated with alpha-fetoprotein selectively eliminates myeloid-derived suppressor cells (MDSCs) and inhibits experimental tumor growth. Cancer immunology, immunotherapy : CII 67, 101–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N, Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res 74, 104–118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ansari L, Shiehzadeh F, Taherzadeh Z, Nikoofal-Sahlabadi S, Momtazi-Borojeni AA, Sahebkar A, Eslami S, The most prevalent side effects of pegylated liposomal doxorubicin monotherapy in women with metastatic breast cancer: a systematic review of clinical trials. Cancer gene therapy 24, 189–193 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Gegechkori N, Haines L, Lin JJ, Long-Term and Latent Side Effects of Specific Cancer Types. The Medical clinics of North America 101, 1053–1073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toporkiewicz M, Meissner J, Matusewicz L, Czogalla A, Sikorski AF, Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: principles, hopes, and challenges. International journal of nanomedicine 10, 1399–1414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkatesan S, Swanton C, Taylor BS, Costello JF, Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harbor perspectives in medicine 7, a026617 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sica A, Bronte V, Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 117, 1155–1166 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erez N, Coussens LM, Leukocytes as paracrine regulators of metastasis and determinants of organ-specific colonization. Int J Cancer 128, 2536–2544 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pollard JW, Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4, 71–78 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD, Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med 362, 875–885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Patel L, Pienta KJ, CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev 21, 41–48 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V, Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 116, 2777–2790 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kusmartsev S, Nagaraj S, Gabrilovich DI, Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol 175, 4583–4592 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V, Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Frontiers in immunology 9, 398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamada S, Masamune A, Shimosegawa T, Novel therapeutic strategies targeting tumor-stromal interactions in pancreatic cancer. Frontiers in physiology 4, 331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veglia F, Perego M, Gabrilovich D, Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu H, Mai J, Shen J, Wolfram J, Li Z, Zhang G, Xu R, Li Y, Mu C, Zu Y, Li X, Lokesh GL, Thiviyanathan V, Volk DE, Gorenstein DG, Ferrari M, Hu Z, Shen H, A Novel DNA Aptamer for Dual Targeting of Polymorphonuclear Myeloid-derived Suppressor Cells and Tumor Cells. Theranostics 8, 31–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, Qian J, Hailemichael Y, Nurieva R, Dwyer KC, Roth J, Yi Q, Overwijk WW, Kwak LW, Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nature medicine 20, 676–681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roth F, De La Fuente AC, Vella JL, Zoso A, Inverardi L, Serafini P, Aptamer-Mediated Blockade of IL4Ralpha Triggers Apoptosis of MDSCs and Limits Tumor Progression. Cancer Res 72, 1373–1383 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Yao H-S, Sun C, Li X-X, Wang Y, Jin K-Z, Zhang X-P, Hu Z-Q, Annexin A4-nuclear factor-κB feedback circuit regulates cell malignant behavior and tumor growth in gallbladder cancer. Scientific reports 6, 31056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boye TL, Maeda K, Pezeshkian W, Sønder SL, Haeger SC, Gerke V, Simonsen AC, Nylandsted J, Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nature Communications 8, 1623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, Ten Hacken NH, Cobos Jimenez V, Kootstra NA, Hamann J, Greaves DR, Locati M, Mantovani A, Gordon S, Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–69 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Diakonova M, Gerke V, Ernst J, Liautard JP, van der Vusse G, Griffiths G, Localization of five annexins in J774 macrophages and on isolated phagosomes. Journal of Cell Science 110, 1199–1213 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, Ten Hacken NH, Cobos Jiménez V, Kootstra NA, Hamann J, Greaves DR, Locati M, Mantovani A, Gordon S, Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–e69 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Benes P, Maceckova V, Zdrahal Z, Konecna H, Zahradnickova E, Muzik J, Smarda J, Role of vimentin in regulation of monocyte/macrophage differentiation. Differentiation 74, 265–276 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM, Vimentin is secreted by activated macrophages. Nat Cell Biol 5, 59–63 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Lou Y, Preobrazhenska O, auf dem Keller U, Sutcliffe M, Barclay L, McDonald PC, Roskelley C, Overall CM, Dedhar S, Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev Dyn 237, 2755–2768 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Pena EQ, Taube JH, Chang JT, Miura N, Porter W, Sphyris N, Andreeff M, Mani SA, GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 34, 2958–2967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bilalic S, Michlmayr A, Gruber V, Buchberger E, Burghuber C, Bohmig GA, Oehler R, Lymphocyte activation induces cell surface expression of an immunogenic vimentin isoform. Transplant immunology 27, 101–106 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S, Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(−) novel stem-like hepatocellular carcinoma cells expressing EMT markers. International journal of cancer 137, 491–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pulaski BA, Ostrand-Rosenberg S, Mouse 4T1 breast tumor model. Current protocols in immunology Chapter 20, Unit 20.22 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I, Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 203, 2691–2702 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nanni P, de Giovanni C, Lollini PL, Nicoletti G, Prodi G, TS/A: a new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clinical & experimental metastasis 1, 373–380 (1983). [DOI] [PubMed] [Google Scholar]
- 73.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM, Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res 40, 2142–2146 (1980). [PubMed] [Google Scholar]
- 74.Murphy GP, Hrushesky WJ, A murine renal cell carcinoma. Journal of the National Cancer Institute 50, 1013–1025 (1973). [DOI] [PubMed] [Google Scholar]
- 75.Barth RJ, Bock SN, Mulé JJ, Rosenberg SA, Unique murine tumor-associated antigens identified by tumor infiltrating lymphocytes. The Journal of Immunology 144, 1531–1537 (1990). [PubMed] [Google Scholar]
- 76.Aslakson CJ, Miller FR, Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52, 1399–1405 (1992). [PubMed] [Google Scholar]
- 77.Layzer JM, Sullenger BA, Simultaneous generation of aptamers to multiple gamma-carboxyglutamic acid proteins from a focused aptamer library using DeSELEX and convergent selection. Oligonucleotides 17, 1–11 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Ahmed M, Monsky WE, Girnun G, Lukyanov A, D’Ippolito G, Kruskal JB, Stuart KE, Torchilin VP, Goldberg SN, Radiofrequency thermal ablation sharply increases intratumoral liposomal doxorubicin accumulation and tumor coagulation. Cancer Res 63, 6327–6333 (2003). [PubMed] [Google Scholar]
- 79.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: MSC2 cells treated with IL4 are transcriptionally similar to TIMC.
Fig. S2: Frequency variation of the 15 selected aptamers during HT-Cell SELEX.
Fig. S3: Cluster analysis of the 15 aptamers selected with APTANI.
Fig. S4: Aptamer 3, 6, 11, and 14 recognize TIMC among different tumors and different genetic backgrounds.
Fig. S5: Aptamer 3, 6, 11 and 14 show low or no binding to other mouse tissues.
Fig. S6: Aptamers 3, 6, 11, and 14 recognize TIMC in patients with HNSCC. Flow cytometry.
Fig. S7: Aptamers 3, 6, 11, and 14 recognize TIMC in patients with HNSCC. Image cytometry.
Fig. S8: TIMC-specific aptamers do not bind lung or kidney from healthy donors.
Fig. S9: TIMC-specific aptamers do not bind to human tissue microarrays.
Fig.S10: Affinity of aptamers 3, 6, 11, and 14 for IL4-treated MSC2 or their putative ligand.
Fig. S11: TIMC-specific aptamers are superior to Doxil against the 4T1 breast cancer.
Fig. S12: TIMC-aptamer approach reduces doxorubicin toxicity.
Table S1: Sequence, frequency, and motifs of 15 aptamers selected by APTANI.
Table S2: List of oligonucleotides used.
Table S3: List of antibodies used.
Data file S1: Data from primary figures.
Data Availability Statement
All data associated with this study can be found in the paper or supplementary materials. Microarray data are available at GSE110774. HT-sequencing data are available at GSE113378.