

Abstract
Specific biological properties of those circulating cancer cells that are the origin of brain metastases (BM) are not well understood. Here, single circulating breast cancer (BC) cells were fate-tracked during all steps of the brain metastatic cascade in mice after intracardial injection over weeks. A novel in vivo two-photon microscopy methodology was developed that allowed to determine the specific cellular and molecular features of BC cells that homed in the brain, extravasated, and successfully established a brain macrometastasis. Those BM-initiating breast cancer cells (BMICs) were mainly originating from a slow-cycling subpopulation that included only 16–20% of all circulating cancer cells. BMICs showed enrichment of various markers of cellular stemness. As a proof-of-principle for the principal usefulness of this approach, expression profiling of BMICs vs. non-BMICs was performed, which revealed up-regulation of NDRG1 in the slow-cycling BMIC subpopulation in one BM model. Here, BM development was completely suppressed when NDRG1 expression was downregulated. In accordance, in primary human BC, NDRG1 expression was heterogeneous, and high NDRG1 expression was associated with shorter metastasis-free survival. In conclusion, our data identifies temporary slow-cycling BC cells as the dominant source of brain and other metastases and demonstrates that this can lead to better understanding of BMIC-relevant pathways, including potential new approaches to prevent BM in patients.
Keywords: Brain metastases, breast cancer, circulating tumor cells, in vivo two-photon microscopy, NDRG1, slow-cycling, cancer stem cells
Introduction
Up to 40% of patients with metastatic breast cancer (BC) develop brain metastases (BM), and intracranial progression is the cause of death in approximately half of them [1]. Given the limited therapeutic possibilities in established breast cancer BM, prevention of BM outgrowth by interfering with the early steps of the brain metastatic cascade is a novel and promising approach, with the potential of high effectivity if the mandatory steps of the brain metastatic cascade are targeted [2–4]. In this context, the brain metastasis-initiating cancer cell (BMIC) would be a promising target for BM prevention in patients, but its specific biological characteristics remain obscure.
A high level of cellular heterogeneity is found within solid tumors, and the existence of a distinct subpopulation of cancer cells with disease-initiating and -perpetuating properties that also shows a particular high resistance to therapies is now confirmed by multiple studies [5]. Thus, the basic rationale for the study reported here was the hypothesis that specific cellular properties of metastasis-initiating cancer cells might be more easily revealed than that of tumor-initiating cells in primary tumors: during the challenging, multistep nature of organ colonization, metastasizing cancer cells might just lack the time to adapt to the foreign soil before they die [2]. In this study, by establishing a novel in vivo two-photon microscopy methodology (MPLSM) to unequivocally identify BMICs from breast cancer cell lines and characterize their role in BM formation, we demonstrate that those BMICs can be readily identified in the circulating cancer cell population. BMICs are rare, slow-cycling, and harbor a specific molecular profile. Finally, specific targeting of BMICs can effectively prevent BM formation.
Materials and Methods
Cell lines
The human breast cancer cell lines JIMT1 (estrogen receptor negative, HER2 overexpressing, trastuzumab resistant) and MDA-MB-231 (triple negative) were transfected with lentiviral vectors as described previously to order to generate a stably enhanced green fluorescent protein (EGFP) or tdTomato expressing cell line for in vivo imaging [6]. Human breast cancer sublines JIMT1 br3 (referred to in the manuscript as JIMT1br) and MDA-MB-231br were kept under standard media conditions (DMEM, 10% FBS, P/S). Both cell lines were regularly checked for Mycoplasma infections by PCR according to guideline of the German Cancer Research Center (DKFZ), and for cellular identity using multiplex human cell line authentication test (last time, 09.06. 2017).
PKH26 membrane staining
PKH26 as well as PKH67 cell membrane staining was performed according to manufactures protocol (Sigma Aldrich, Merck KGaA Darmstadt, Germany). The homogeneity of the staining was controlled under the microscope. PHK26 membrane was reduced with every cell division resulting in a heterogeneous population of slow- (cells with remaining PKH26 membrane staining) and fast- (cells without any remaining PKH26 membrane staining) cycling cells.
Definition of slow- versus fast-cycling cells in vitro
Using PKH26 membrane staining as described above a heterogeneous population of slow- and fast-cycling cells could be identified. Using ImageJ (National Institutes of Health, Bethesda, MD) image analyzing software any PHK26 red fluorescent signal that was greater than background (after positive verification by image inspection) was defined as PKH26 positive in vitro as well as in vivo. A continuous decline in PKH26 membrane dye staining intensity was observed during in vitro growth. To be able to further characterize the slow-cycling vs. fast-cycling cancer cell subpopulation in subsequent experiments, we pragmatically chose day 4 as the cut-off time point, since here (a) the slow-cycling cells during in vitro growth were technically still detectable, and in a sufficient number for further analysis, but (b) the fast-cycling cancer cells during in vitro growth have already clearly separated.
Four days after staining 16–20% of cells still showed PKH26 membrane staining and were defined as ‘slow-cycling cells’ as defined by FACS as well as MPLSM. Cells without any detectable remaining PKH26 membrane staining after four days were defined as ‘fast-cycling cells’ (Fig. 1D. Established proliferation marker carboxyfluorescein diacetate, succinimidyl ester (CSFE), as well as transduction with a TET off regulated H2B-GFP reporter for nuclear GFP labelling, which can selectively suppressed by application of doxycycline, were used as previously described to verify the slow cycling properties of PKH26 retaining cells [7]. For experiments analyzing the population of slow- and fast-cycling cell separately cell were sorted by flow cytometry (FACS) using the cell sorter BD FACSAria™ III (BD Biosciences, Heidelberg, Germany) according to their PKH26 signal (present for slow-cycling cells; absent for fast-cycling cells). Slow-cycling cells were defined as the 16% with the highest PKH26 fluorescence intensity of the whole population, and fast-cycling cells as the 16% with the lowest intensity. A second green membrane staining (PKH67; Sigma Aldrich, Merck KGaA Darmstadt, Germany) was used to determine the cell division rate and show that the two populations retain their slow and fast cycling properties.
Figure 1: Combined in vitro / in vivo model to study the role of slow- and fast-cycling cancer cells in brain metastasis formation.

(A) Experimental strategy and procedures. Different cancer cell subpopulations are labeled in vitro (fast- vs. slow-cycling cells by PKH26, and live stemness reporters), and their characteristics is further analyzed in vitro (overlap with other labels indicating a certain population or a certain relative gene expression, cell growth), and in vivo (likelihood to master all steps of the brain metastatic cascade; overall brain metastatic efficacy). Results from different brain metastatic capacities of slow- vs. fast-cycling cancer cells are then used for identification of key molecular differences of these cell populations, finally used for knock-down studies of candidate gene(s) that can be tested regarding their influence on the brain metastatic cascade. MPLSM: multiphoton laser-scanning microscopy.(B) Representative images of a slow-cycling MDA-MB-231 breast cancer cell (remaining PKH26 staining; arrow), followed by repetitive in vivo MPLSM through all steps of the brain metastatic cascade: intravascular arrest (Days 1,3 – single cells), extravasation and colonization of the perivascular niche (Day 6 – up to 5 cells), micro- (Days 14, 21 – up to 50 cells) and macrometastasis (Day 28 - > 50 cells) formation. Green, cytoplasm of GFP-positive cancer cell(s); blue, brain microvessels labeled by TRITC angiogram; red, PKH26 staining labeling slow-cycling cancer cells. scale bars: 30μm. (C) Representative images of a fast-cycling MDA-MB-231 cell (absence of PKH26 staining) through the early steps of the brain metastatic cascade (Day 1 – single cells), until its death on day 6. scale bars: 30μm. (D) Percentage of all slow-cycling and fast-cycling MDA-MB-231 breast cancer cells, in vitro at the day of intracardial injection, and in vivo one day after. At day 1, all cancer cells were still in the state of intravascular arrest. Included cells day 0 MDA-MB-231 n=784; included cells on day 1 MDA-MB-231 n=138 (p<0.001; Chi Square test). (E) Slow-cycling JIMT1 breast cancer cell mastering all steps of the brain metastatic cascade; intravascular arrest (day 1 & day 3), extravasation and colonization of the perivascular niche (day 6), micrometastasis (day 9), macrometastasis (day 14 & day 21 & day 28). Green, cancer cell(s); blue, brain microvessels. scale bars: 30μm. (F) Fast-cycling JIMT1 breast cancer cell mastering intravascular arrest (day 1) and extravasation (day 6), but disappears afterwards until day 14. (G) Percentage of all slow-cycling and fast-cycling JIMT1 breast cancer cells, in vitro at the day of intracardial injection, and in vivo one day after. At day 1, all cancer cells were still in the state of intravascular arrest. Included cells day 0: Jimt1 n=1254; included cells on day 1: Jimt1 n=238 (p<0.001; Chi Square test). B-G: data obtained by in vivo MPLSM; scale bars: 30μm. 3 replicates per experiment.
In vivo Multiphoton Laser Scanning Microscopy (MPLSM)
For in vivo multiphoton laser-scanning microscopy (MPLSM), a ZEISS LSM 7MP equipped with a Coherent Chameleon Ultra II laser was used as described previously [8]. To obtain angiograms, 100 µl of 5 mg/ml tetramethylrhodamine-isothiocyanate‒Dextran (TRITC dextrane; average MW: 500,000, Sigma-Aldrich) or fluorescein isothiocyanate–Dextran (FITC dextran; MW: 500,000, Sigma-Aldrich) was injected into the tail vein shortly before each imaging depending on the florescent signal of the used cells (GFP or tdTomato). The signals of the used fluorophores were differentiated as appropriate by different excitation wavelengths (750 nm: PKH26; 850 nm: GFP, TRITC-dextran; 950 nm: tdTomato; FITC-dextran). PKH26 dye retention was evaluated using the 750 nm wavelength. Remaining membrane could be reliable identified by an overlapping signal in the 750 nm (PKH26 signal) wavelength and the 850 nm wavelength channel visualizing the GFP signal of the tumor cell. Thereby, also small remaining membrane dye could be assigned to a particular cell due to the visual overlap in the two independent channels. A BP500–550 filter was used for the green fluorophores, and a BP575–610 filter was used for the red fluorophores. Standard gains were set between 700 and 750, and standard z-interval was 3 μm. Laser power was tuned as low as possible ranging from 1% on the very surface to 100% in the deepest regions of interest. Absence of phototoxicity was controlled by careful re-examination of pre-imaged regions vs. those regions without pre-imaging over weeks. For in vivo imaging, mice were narcotized with isoflurane (1% to 2% in 100% O2), guaranteeing a pain free intervention for the animal. Mice were fixed using a custom-build fixation system with a titan ring, and body temperature was measured by rectal thermometer and kept constant by a heating- pad system.
In vivo quantification of the brain metastatic cascade
MPLSM through the chronic cranial window allowed investigation of all single steps of the brain metastatic cascade, including intravascular arrest, extravasation, colonization of the perivascular niche, and establishment of micro- and macrometastasis. Brain-colonizing cancer cells did not show intravascular proliferation after heart injection, suggesting that pivotal properties of injected cells stay rather stable until after extravasation [2]. Micrometastasis were defined as a tumor cell cluster of more than 4 cells and a diameter smaller than 143 µm, which is the mouse correlate of a human micrometastasis just detectable with MRI (of 2 mm diameter); these are present between day 10 and 14 after intracardial injection [2]. A macrometastasis was defined as a tumor cell cluster with more than 50 cells, or a diameter larger than 143 µm, presenting from day 10 onwards, reflecting to a human macrometastasis defined as visible in MRI imaging (around 1 mm) [8]. Number of cells in a micro- or macrometastasis was estimated by surface. Number of cells per micro- or macrometastasis size was available from a previous work, in which we could correlate the metastasis size with the cell count due to double fluorescent cells (RFP cytoplasm, GFP nucleus) [8]. Imaging of the same areas was performed on days 1, 3, 6, 9, 14, 21 and 28 after heart injection to follow the single steps tough each step. Superficial angiograms (providing a stable superficial “road map” as no change is observed over time) and stereotactic coordinates allowed the re-locating of the areas of interest over the entire imaging period, and ensured that the same cells identified on day 1 after injection were followed and evaluated for their successful brain metastasis formation. An average of 6 to 8 regions were imaged per mouse.
Quantification of slow- and fast-cycling cells in vivo
In vivo MPLSM through a chronic cranial window after heart injection of PKH26 membrane staining cells (JIMT1 or MDA-MB-231; 4 days after PKH26 membrane staining; n=4 mice per cell line) allowed to analyze differences with respect to cellular behaviour during the brain metastastic cascade between slow- and fast-cycling cells. Remaining PKH26 membrane staining was detected by MPLSM (wavelength: 750 nm) in intravascular arrested cells (day 1 after injection), and as outlined above, cells with remaining staining were defined as slow-cycling, while cells without any remaining PKH26 signal were classified as fast-cycling. Therefore, all cells were defined on day 1 either as slow- or as fast-cycling, and could be followed over all steps of the brain metastatic cascade, including their efficacy to perform every single step of the brain metastatic cascade.
Stemness reporter systems
The JIMT1 cell line was transduced with the retroviral vector pQCXIN-ZsGreen-cODC to study S26 proteasome activity as a marker of stemmness (kind gift of Frank Pajonk, David Geffen School of Medicine at UCLA, Los Angeles, California) as previously published [9]. Stable transfectants were selected with G418 (Invitrogen). Efficacy control of the lentiviral transduction was possible, as the vector included a RFP signal as well as the dynamic GFP signal in dependence of the S26 proteasome activity. > 90% of cells presented with RFP signal after transduction. To secure a homogenous cell population, FACS sorting was additionally performed. Dynamic S26 proteasome activity could be studied as expression of the fusion protein ZsGreen-cODC results in as fluorescent fusion protein that accumulates in cells in the absence of S26 proteasome activity, while cells without fluorescence protein accumulation indicate normal proteasome activity [9].
In vivo quantification of S26 proteasome low cells
MPLSM trough the chronic cranial window was performed on days 1,3,6,9,14,21 and 28 to quantify the success of the single steps of the brain metastatic cascade and cells according to their S26 proteasome activity. Low S26 proteasome activity could be detected by expression of ZsGreen-cODC (850 nm wavelength) in vivo, allowing investigation of a change of activity over time, and also analysis of heterogeneous activity within a cell convolute.
Illumina gene expression profiling
To address gene expression differences between slow- and fast-cycling cells, cells were separated 4 days after PKH26 membrane staining and FACS sorting as described above. RNA was isolated using a RNeasy Kit (Qiagen). RNA samples (pprox. 3 ng per sample) were used for gene expression profiling assay by Illumina BeadArray (Illumina Human HT 12 v4 R2). Negative control probes were used for background correction and both negative and positive controls were used for normalization [10]. LIMMA analysis was used to analyzed gene expression differences between slow- and fast-cycling cells [11]. The p-values were adjusted by Benjamin & Hochberg correction to avoid the problem of multiple testing [12]. Genes that showed a fold change between the two groups of more than 1.5 considered regulated biologically relevant and proceeded for pathway analysis. IPA (Qiagen, Hilden, Germany) was then performed with the resulting dataset using standard parameters. For gene set enrichment analysis (GSEA), the GSEA software tool downloaded from the homepage of the Broad Institute (http://software.broadinstitute.org/gsea) was used. Fifty hallmark gene sets as well as the C2 and C5 gene sets were used for exploratory testing of pathway enrichments in the full dataset of microarray genes. The focus was set to gene sets involving cell cycle and proliferation. The number of gene set permutations was set to 1,000. The datasets generated during the current study are available from the corresponding author on reasonable request.
Generation of NDRG1 knock-down cell lines
Knock-down of NDRG1 in JIMT1 and MDA-MB-231 cells was performed as described previously using two short-hairpin RNA (shRNA) sequences targeting different site [13]. Two shRNA constructs targeting the 3´ -untranslated region (UTR) of NDRG1 were used (shRNA NDRG1 43: 5’- TGCATTATTGGCATGGGAACTTCAAGAGAGTTCCCATGCCAATAATGCTTTTTTC-3’; shRHNA NDRG1 47: 5’- TATGCAGAGTAACGTGGAAGTTCAAGACTTCCACGTTACTCTGCATTTTTTTC-3’). The shRNA NDRG1 43 was used for NDRG1 knock down in Jimt1 cells which were investigated using MPLSM. Both shRNA hairpins were used for NDRG1 knock down in Jimt1 cells, and subsequent analysis of brain metastasis frequency by MRI. The shRNA NDRG1 43 was used for MDA-MB-231 NDRG1 knock down, and subsequent analysis of brain metastasis frequency in histological slides. Non-targeting shRNA (MISSION SHC002) was used as a control in JIMT1 and MDA-MB-231 cells. Efficacy of the knock down was evaluated by western blot analysis (see below).
qPCR analysis
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) to quatify NDRG1 gene expression was done as described previously [13]. NDRG1 expression results were normalized to glyceraldehyde-3- phosphate dehydrogenase (GAPDH). Further, OCT4 and SOX2 gene expression was analyzed in the sorted population of slow- and fast cycling cells by qRT-PCR. Primer sequences are listed in table 1.
Table 1:
Primer Sequences
| NDRG1 forward | TCAAGATGGCGGACTGTG |
| NDRG1 reverse | GAAGGCCTCAGCGAGCTT |
| GAPDH forward | CTCTCTGCTCCTCCTGTTCGAC |
| GAPDH reverse | TGAGCGATGTGGCTCGGCT |
| OCT4 forward | ATGTGGGGCTCACCCTGGGG |
| OCT4 reverse | CTTCTGCAGCAAGGGCCGCA |
| SOX2 forward | GCCGAGTGGAAACTTTTGTCG |
| SOX2 reverse | GGCAGCGTGTACTTATCCTTCT |
NDRG1 western blot
Protein expression of NDRG1 was analyzed by western blot in slow- versus fast-cycling cells as well as to verify the knock-down as previously descried (NDRG1 antibody, goat, dilution 1:2500, Abcam, Cambridge, UK) [13].
In vivo brain metastatic behavior of NDRG1 knock-down cells
MPLSM trough the chronic cranial window was again used on days 1,3,6,9,14,21 and 28 was again used to quantify the success of each step of the brain metastatic cascade of JIMT1 GFP NDRG1 knock-down cells (n= 4 animals) and JIMT1 GFP control cells (n= 4 animals).
Immunohistochemical analysis in human primary breast cancer and brain metastasis tissue
Formalin fixed and paraffin embed tissue of human breast cancer brain metastases and primary breast cancer was retrieved from the Biobank of the Medical University of Vienna and the National Center for Tumor disease Heidelberg. 4um slides were cut with a standard microtome for further processing. In brief, immunohistochemical staining was performed with an automated Bentana BenchMark XT (Ventana Medical Systems,Tuscon, AZ, USA) immunostainer. The anti-NDRG1 clone HPA006881 (Sigma Aldrich; dilution 1:70) was used. Quantification was performed as previously published scoring the membranous expression of NDRG1 on tumor cells [14]. In brief, only full, strong and complete membranous staining was scored and groups were defined as NDRG1 low (absent or only single cells) or NDRG1 high (at least 10% of tumor cells) for correlation of NDRG1 expression and clinical data. The cytoplasmic staining, presenting around necrotic areas was not included in the analysis. For HIF1a and ki67 analysis, slides underwent heat-induced epitope retrieval in pH6.0 citrate buffer (HIF1a: 92 minutes, Ki67: 20 min). Afterward sections are incubated with antibody (HIF1a: polyclonal rabbit purified Anti-Human HIF 1 alpha/ 610959 BD Transduction LaboratoriesTM; Ki67: monoclonal mouse Ki67 Clone MIB-1/ M7240 Dako). Estrogen receptor (ER) status was assessed by immunohistochemistry using the CONFIRM SP1 clone (Ventana, Tucson, AZ); hormone receptor expression was estimated as the percentage of positively stained tumour cells. All patients had a minimum follow up period of ten years. Clinical data (metastatic disease versus non metastatic disease; brain metastases versus no brain metastases) was retrieved by chart review.
Publicly available data sets and co-expression analysis
Gene expression microarray and RNA-seq data of 7 publicly available data sets were obtained from GEO and TCGA databases, respectively. The following data sets were used: GSE65216, GSE41119, GSE20685, GSE58644, GSE16201, GSE54002 and TCGA_BRCA. Moreover, protein levels determined by mass spectrometry were retrieved from TCGA_BRCA data set via cbioportal.org. Expression values of NDRG1 and ESR1 were extracted from these data sets and co-expression analysis were calculated using Spearmańs rank correlation.
Image processing
MPLSM imaging data was acquired by the Zeiss ZEN Software (Zeiss, Germany) and used to analyze the success of the single cells in performing the single steps of the brain metastatic cascade. Slow- and fast-cycling cells were defined using Image J (National Institutes of Health, Bethesda, MD) as described above. To generate the examples pictures displayed in the figures, images were transferred to Imaris (Bitplane, Switzerland) for processing and to generate the displayed single planes and 3D images. If necessary, changes in brightness, contrast, or color balance were made to whole images.
Statistical analysis
Chi square test, Fisher’s Exact test, Wilcoxon signed rank test, t-test and Kruskal-Wallis test were used as appropriate to assess group differences. To test the difference in macrometastasis frequency between slow and fast cycling cells Chi square test was used. Metastasis free survival was defined from diagnosis of primary breast cancer to first diagnosis of distant metastases. For correlation of estrogen receptor and NDRG1 expression in publicly available data the Spearman correlation coefficient was used. A correlation coefficient of (−)0.8 to (−)1 was interpreted as a very strong association, of (−)0.6 – 0.8 as strong, of (−)0.4 to (−)0.6 as moderate, of (−)0.2 to (−)0.4 as a low and (−)0.2 to 0 as no association. Group differences concerning the metastasis free survival were analyzed using the Kaplan Meier product limit method and the log-rank test. Statistical significance was stated for P values < 0.05. All experiments were performed with 3 replicates unless stated otherwise. Animal groups were conducted with 4 animals per group. Box-plots show mean at center line and under and lower maximum. Error bars show standard deviation. Statistical analysis was performed with statistical package for the social sciences (SPSS) 20.0 software (SPSS Inc., Chicago, IL, USA).
Results
A combined in vitro / in vivo model for fate-tracking of slow-cycling cancer cells
To study how distinct cancer cell subpopulations differ in their ability to master all steps of brain colonization, we developed an in vivo multiphoton laser-scanning microscopy (MPLSM) that was initially established in our laboratory [2]. This approach was chosen to follow individual BMICs from two BC cell lines over all steps of the brain metastatic cascade, from first intravascular arrest, extravasation, colonization of the perivascular niche, and establishment of a clinically relevant macrometastasis weeks later. To clarify whether a pre-existing BMIC subpopulation does exist within circulating vs. slow-cycling cancer cells, we used a membrane dye staining method that allows to identify slow-cycling at the moment of vascular arrest (dye retaining) vs. fast-cycling cancer cells, first in vitro, and then, by following their fate after heart injection, in vivo (Fig. 1A; Supplementary Fig. 1A–C).
First, we verified that the PKH26 membrane dye staining method did not influence the growth kinetics of cancer cells, and did faithfully detect the slow-cycling BC cell subpopulation in vitro (Supplementary Fig. 2A–E) [7]. Not unexpectedly, the in vitro slow-cycling BC cells were a dynamic subpopulation: after 21 days of in vitro growth, both fast- and slow-cycling subpopulations had transformed back into the original population again with respect to their cycling behaviour (Supplementary Fig. 2F,G).
Slow-cycling BMICs frequently arrest in the brain
After intracardial injection, cells with slow-cycling vs. fast-cycling behaviour at the moment of vascular arrest could be reliably distinguished from each other after arrest in brain microvessels by in vivo MPLSM (Fig. 1B–G; Supplementary Fig 1D). Quantification of their relative amount revealed that in vitro, just before implantation, the majority (80 and 84% of MDA-MB-231 and JIMT1 cells, respectively) of cancer cells were fast-cycling, and only a minority (16–20%) was slow-cycling, as detected by MPLSM. This proportion was just reversed in vivo: here, slow-cycling cancer cells where the dominating cell population inside brain capillaries 24 hours after intracardial injection (Fig. 1D,G; Fig. 2A; p<0.001 for JIMT1 and p<0.001 for MDA-MB-231, Wilcoxon signed rank test). In synopsis of these numbers, in vitro slow-cycling breast cancer cells are about 4-fold more likely to successfully master a permanent arrest in brain microvessels, which we have identified before as the first mandatory step of the brain metastatic cascade [2]. The cell diameter did not differ between slow- and fast-cycling cells nor did cells with larger cell diameter develop more frequently to macrometastases, which argues against a pure mechanical reason for this difference (Supplementary Fig. 3A). Importantly, a relevant difference in the cell division properties of slow- vs. fast-cycling cancer cells was evident over several days, still detectable 8 days after sorting in vitro (Supplementary Fig. 2F). This is the time needed in vivo for a particular BMIC to successfully master the first steps of brain metastasis: extravasation and perivascular niche colonization. This data speaks for preservation of slow-cycling cellular properties during the time where failure or success of brain colonization is decided [2].
Figure 2: Slow-cycling breast cancer cells are enriched during the brain metastatic cascade.

(A) Quantification of the steps of the brain metastatic cascade in slow-cycling (PKH26 positive) vs. fast-cycling (PKH26 negative) JIMT1 (n=238 tumor cells in n=4 mice) and MDA-MB-231 (n=138 tumor cells in n=4 mice) human breast cancer cells. While slow-cycling cells are only a minority of cancer cells on the injection day, this population is particularly able to master all steps of the brain metastatic cascade successfully, greatly outnumbering its fast-cycling counterparts (p<0.05; Chi Square test). Percentages are given relative to the total number of intravascular arrested cells in the slow-cycling and the fast-cycling group on day 1 after intracardial injection; extravasation: day 3–6; perivascular single cells: day 3–9; micrometastasis: day 9–14; macrometastasis: day 9–29. (B,C) Quantification of the relative tumor cell number in the JIMT1 (B) and MDA-MB-231 (C) cell lines over 28 days in vivo, depending on their cycling properties. (4 mice per group). Significant (p<0.05) differences in the successful establishment of perivascular cells, micro- and macrometastases between slow- and fast-cycling cells could be detected from day 6 on. (D) Whole-mouse imaging without the brain compartment revealed that slow-cycling JIMT1 breast cancer cells give rise to a significantly higher extracranial metastatic burden, compared to fast-cycling or unsorted control cells (n=4 mice per group; p<0.05; t test). Tumor cells were FACS-sorted after PKH26 staining, and injected intracardially. A-C: data obtained by in vivo MPLSM; scale bars: 30μm. 3 replicates per experiment.
Slow-cycling BMICs succeed during all steps of the brain metastatic cascade
To study the impact of slow- vs. fast cycling cellular states on the entire process of brain colonization, repetitive in vivo MPLSM of the same deep brain regions was performed to track the subsequent fate of arrested slow- vs. fast-cycling breast cancer cells (Fig. 1B–G; Fig. 2A,B). A substantial number of slow-cycling BC cells colonized the perivascular niche and mastered to further proliferate to a brain macrometastasis (Fig. 1 B,E; Fig. 2A–C), while almost all fast-cycling cancer cells died in the weeks after extravasation (Fig. 1C,F; Fig. 2A–C). Quantification of the individual fate of 108 brain-arrested fast-cycling cells vs. 268 slow-cycling cells at the moment of vascular arrest from two breast cancer cell lines over all steps of the brain-metastatic cascade confirmed that slow-cycling cancer cells were the only ones that gave rise to brain metastases in the JIMT1 line. Moreover, they were also significantly more likely to grow to a macrometastasis in the MDA-MB-231 line (Fig. 2A–C). Exponential macrometastatic growth was observed after day 21, without any further regression events detected, indicating that the established macrometastasis resembles an irreversible state of tumor growth in the brain.
Further in-depth analysis of the brain metastatic cascade confirmed that slow-cycling cancer cells were more efficient in performing every single, mandatory step of the brain metastatic cascade, namely extravasation, perivascular niche colonization and survival, proliferation to a micrometastasis, and establishment of a brain macrometastasis [2]: slow-cycling cells (as determined at the moment of vascular arrest) mastered every step more successfully (Fig. 2A), and also more rapidly (Fig. 2B, Fig. 2C), compared to fast-cycling cells. It was not the PKHhigh cells which contain the non-cycling population were those who successfully mastered all steps of the brain metastatic cascade, but rather those cancer cells with low/moderate PKH signal (PKHlow ) indicating true slow-cycling behaviour (Supplementary Fig. 3B–D).
Slow-cycling cancer cells are also more likely to form extracranial metastases
In line with the findings for brain colonization, slow-cycling at the moment of vascular arrest cancer cells also formed significant more extracranial, systemic metastatic foci compared to fast-cycling cells over time, as detected by whole body IVIS imaging after intracardial injection of JIMT1 cells (Fig. 2D and Supplementary Fig. 4). This data demonstrates that slow-cycling cancer cells are not only particularly efficient to form brain metastases, but are also more likely to give rise to extracranial metastases.
Stemness markers are enriched in the slow-cycling subpopulation
To further address the biological foundation of the increased ability of slow cycling cells to form brain metastases, we investigated whether slow- or fast cycling cells have higher expression of cellular marker associated with stemness. Two markers of stemness properties, OCT4 and SOX2, were investigated using real time q-PCR, where higher expression in slow cycling compared to fast cycling cells was detected (Fig. 3A). To follow the hypothesis of higher stemness properties in slow- compared to fast cycling cells we applied various lentiviral reporter systems for cellular stemness to study overlap of those markers with the slow- and fast-cycling cancer cell subpopulations. These included reporters for activation of the Oct4/Sox2 complex [15], the Notch pathway [16], the WNT pathway [17], and for reduced 26S proteasome activity [9] (Fig. 3B, C). As expected, a fairly small proportion of cancer cells was reporter positive: expression of these stemness markers was observed in <1% of the entire cancer cell population only (Fig. 3C). Nevertheless, the slow-cycling subpopulation was strikingly enriched for stemness markers, with Oct4/Sox2, NOTCH, and WNT positivity exclusively found in this cancer cell population where the BMICs originate from (Fig. 3C).
Figure 3: Certain cancer cell stemness markers are enriched in BMICs.

(A) Stemness markers in slow- vs. fast-cycling JIMT1 breast cancer cells. qPCR analysis of the stemness markers SOX2 (p=0.02) and OCT4 (p=0.06). (B) Gene expression of OCT4 (p=0.001) and SOX2 (p>0.05) in OCT4/SOX2 reporter positive cells determined by FACS sorting. (C) Slow-cycling cells reveal a marked enrichment of stemness markers compared to fast-cycling cells, as tested by overlap of PKH26 membrane staining and fluorescence signal of stemness reporter systems by FACS analysis (p<0.05). Note that marker-positive cells remain a small cancer cell subpopulation, even in the slow-cycling cells. (D) Quantification of the brain metastatic cascade of JIMT1 cells transduced with pQCXIN-ZsGreen-cODC for in vivo tracking of the tumor cell subpopulation with reduced S26 proteasome activity (n=146 tumor cells, n=4 mice) scale bars: 30μm. (E) One single tumor cell presents with low proteasome activity on day 1 after injection, but is not visible any more after extravasation. (F) One single tumor cell with low proteasome activity occurs in a macrometastasis over time (arrow). D,E,F: in vivo MPLSM; 3 replicates per experiment
Cell cycle analysis revealed that slow-cycling cancer cells are more likely in the G2/M phase, compared to fast-cycling cells (Supplementary Fig. 5A,B). This characteristics has been associated with cancer cell stemness before [18]: slow-cycling cells accumulate in the G2/M phase, allowing the cancer cell to take more time for DNA repair, which results in lower mutation and apoptosis rates [19], and improved cell survival [18].
Low 26S proteasome activity is not defining BMICs
Next, we wanted to provide a proof-of-principle that the dynamic gain and loss of a specific molecular marker, indicative of a particular cellular state, can be followed throughout the entire brain metastatic cascade on a microscopic level in vivo. The reporter system for low 26S proteasome activity [20] which was moderately associated with slow-cycling properties (Fig. 3C) generated sufficient fluorescence signal strength that allowed its use in intravital microscopy. In vivo, of 146 brain-arrested cancer cells tracked, only a single JIMT1 breast cancer cell presented with low proteasome 26S activity on day 1 after injection (Fig. 3D, E). This single cell successfully performed extravasation on day 3, but disappeared afterwards (Fig. 3E). Another single cancer cell with reduced 26S proteasome activity was observed within a larger metastasis that formed from cells without prior reporter positivity, indicating that low 26S proteasome activity is a dynamic feature which can change during BM outgrowth (Fig. 3F). Quantification of the entire brain metastatic cascade confirmed those findings (Fig. 3D). This data suggests that proteasome reporter positivity, detectable in only 1% of all slow-cycling breast cancer cells, is not related to a particular capability of BMICs to successfully establish a brain metastasis.
BMICs are characterized by a distinct gene expression profile
Next, we sought to gain deeper insights into the molecular characteristics of the slow-cycling cancer cell subpopulation that contains the BMICs. Therefore, a comparative gene expression microarray analysis of slow- vs. fast-cycling cells was performed in vitro for the JIMT1 cell line. Ingenuity pathway analysis revealed “cellular growth and proliferation” as the most frequently affected cellular function, with 60 of 88 genes differentially regulated more than 1.5-fold between slow and fast cycling cells, providing a basic methodological validation.
Intriguingly, NDRG1 was among those genes significantly up-regulated in slow-cycling vs. fast-cycling breast cancer cells, with the 17th highest relative overexpression in slow-cycling cells (Fig. 4A and Supplementary Table 1). We chose to further characterize NDRG1 because its overexpression in primary tumors has been shown to be associated with poor prognosis in breast cancer patients, while playing a rather complex role in other cancer entities [14]. Furthermore, it had been associated with cellular stemness states before, albeit with oppositional findings in different tumor entities [21,22]. Increased protein expression of NDRG1 in JIMT1 slow-cycling breast cancer cells was confirmed by FACS (Fig. 4B), qPCR (Fig. 4C) and Western Blot (Fig. 4D). Furthermore, NDRG1 expression was observed in other human and mouse breast cancer cell lines, with considerable high expression in brain seeking ones (Supplementary Fig 6B). All included cell lines were hormone receptor negative. In contrast, in MDA-MB 231 cells which had very low principle NDRG1 expression when compared to JIMT1, no evident association between slow-cycling cellular features (as determined by dye retention) and NDRG1 expression could be detected in vitro (Supplementary Fig 6A,B).
Figure 4: NDRG1 is upregulated in slow-cycling JIMT1 breast cancer cells.

(A) Significant (p<0.05) up-regulation of NDRG1 expression in slow-cycling JIMT1 cells compared to fast-cycling ones. NDRG1 is the 17th highest differentially expressed gene in slow-cycling Jimt1 cancer cells. For a complete gene list, see Supplementary Table 1. (B) Expression of NDRG1 in Jimt1 slow-cycling cells compared to Jimt1 fast-cycling cells in FACS analysis (p<0.01). (C) Relative NDRG1 gene expression in Jimt1 slow- versus fast cycling cells, determined by qPCR (p<0.05). (D) Higher NDRG1 protein expression in Jimt1 slow-cycling compared to fast-cycling cells analyzed with Western Blot. (E) Knock-down of NDRG1 in JIMT1 cells. 3 replicates per experiment
NDRG1-high BMICs depend on NDRG1 proficiency for brain colonization
Next, stable NDRG1 shRNA knock down cell lines were established and validated (Fig. 4E, Supplementary Fig. 7A,B). After orthotopic injection into the mammary fat pat, JIMT1 shNDRG1 tumors were even larger than shRNA control tumors (mean 193 versus 529 mm3 after 3 weeks; p=0.01; Supplementary Fig. 7C,D), demonstrating the uncompromised ability of the shNDRG1 BC cells to grow in vivo. In contrast to this finding from the primary site, shNDRG1 tumor cells showed a reduced ability to achieve a successful intravascular arrest in brain capillaries on day 1 (p=0.047; t test; Fig. 5A). Furthermore, quantification of the brain metastatic cascade of 197 shNDRG1 and 129 shRNA control cancer cells revealed that all steps were significantly compromised in the shNDRG1 JIMT1 breast cancer cells: extravasation (p=0.005; Chi Square test), micro- (p=0.012; Chi Square test) and macrometastasis (p=0.002; Chi Square test) formation (Fig. 5B, C). The overall extravasation rate of NDRG1 knock down cells was lower as the one of control cells (p=0.005; Chi Square test). This difference in metastatic efficiency throughout the entire brain metastatic cascade is reminiscent of that found for the slow-cycling vs. fast-cycling cancer cells (Fig. 2A). Closer examination of the intravital peculiarities that occurred in the shNDRG1 group revealed that one specific deficit was the successful colonization of - and proliferation in - the perivascular niche. As shown exemplary for the shNDRG1 JIMT1 cancer cell that managed to survive longest of all (Fig. 5B), proliferation beyond a three- to four cell state in the perivascular niche was not possible unti day 14, followed by morphological signs of cellular stress (day 21), finally culminating in tumor cell disappearance at day 28; only the typical capillary loop induced by the tumor cell persists at this time point (Fig. 5B). To validate these observations, a second shRNA short hairpin was used to generate NDRG1 knock down cells (Fig. 4E) an tested in vivo. In line with the findings of the in vivo microscopy study, MRT analysis on day 28 revealed that the number of BM was lower in both JIMT1 shNDRG1 cell lines in comparison to the control cell line (p=0.037; Kruskal Wallis test; Fig. 5D). Finally, a knockdown of NDRG1 expression was performed in MDA-MB-231 cells which did not show relevant NDRG1 protein expression or association of cycling behavior with NDRG1 expression, as detailed above (Supplementary Fig. 6A,B). In line, no difference in the median number of brain metastases in the shNDRG1 and the shRNA control group could be observed (p=0.773; Mann Whitney U test; Fig. Supplementary Fig. 6C).
Figure 5: Impact of NDRG1 proficiency on brain metastases formation in vivo.

(A) Impaired ability of JIMT1 NDRG1 knock-down cells to manage the first step of the brain metastatic cascade, which is intravascular arrest (p=0.047; t test); different symbols mark different animals. (B) Intravital imaging of the one JIMT1 shNDRG1 tumor cell that was most successful in mastering the first steps of the brain metastatic cascade, but still dies after day 21. Note the failure of proper perivascular niche colonization, indicated by a roundish tumor cell shape without clear orientation along the blood microvessel in the brain (compare successful brain metastasis of JIMT1, Fig. 2A) scale bars: 30μm. (C) Quantification of shNDRG1 and shRNA control breast cancer cells over the brain metastatic cascade. (n= 326 cells in n=8 animals/n=4 per group; p<0.05; Chi Square test). (D) Number of BM as measured by MRT analysis is lower in both Jimt1 NDRG1 knock-down lines compared to shRNA control cells (n=12 animals/n=4 per group; p=0.037; Kruskal Wallis test). A, B, C: in vivo MPLSM; 3 replicates per experiment
Taken together, the data imply that high NDRG1 expression can be a characteristic of the subpopulation of slow-cycling breast cancer BMICs, and only here NDRG1 deficiency leads to a reduced ability to master crucial steps of the brain metastatic cascade.
NDRG1 expression is associated with metastasis formation in breast cancer patients
Finally, NDRG1 expression was measured in human primary breast cancer tissue, to clarify its prognostic impact and predictive role for brain and extracranial metastasis formation. Heterogeneous expression of NDRG1 protein was observed in primary breast cancer (n=74; Fig. 6A). A similar expression pattern was observed in breast cancer brain metastases specimens (n=61; Fig. 6B). No statistically significant difference in NDRG1 expression was observed between primary and BM specimens (p=0.334). Furthermore, no correlation of membranous NDRG1 expression with ki67 (Spearman correlation coefficient 0.076; p=0.569), the hypoxia marker HIF1 alpha (Spearman correlation coefficient −0.026; p=0.842), nor estrogen receptor expression (p=0.103; Supplementary figure 7 F) could be detected in the primary breast cancer specimens. In addition, publicly available gene expression sets (TCGA_BRCA (RNA-seq); TCGA_BRCA (microarray); TCGA_BRCA (mass spec); GSE65216; GSE41119; GSE20685; GSE58644; GSE16201; GSE54002) were explored for the correlation of estrogen receptor expression and NDRG1 expression. Likewise, only an absent to moderate negative correlation was observed (Spearman correlation coefficient: − 0.422 to −0.308)…”
Figure 6: NDRG1 expression in human breast cancer samples correlates with metastasis formation.
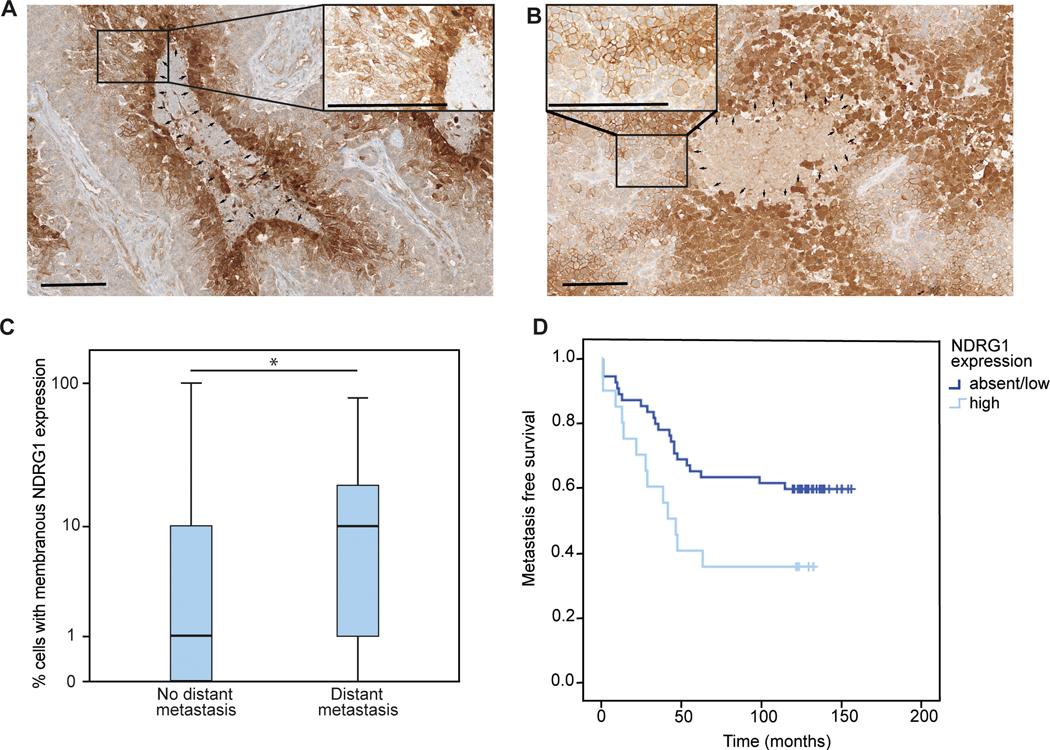
(A,B) Representative immunohistochemical image of NDRG1 expression in a primary breast cancer specimens (A) and a brain metastasis (B), with cyctoplasmatic NDRG1 expression around necrotic areas and unrelated membranous expression on tumor cells (median 7.5%, range 0–100%, n=74). Arrows; magnification x100 and x400; scale bar 200 μm. (C) Median NDRG1 expression in primary breast tumors of patients experiencing distant metastasis (n=35) compared to patients without distant metastases (n=39) during a follow up period of 10 years (p<0.05; Mann Whitney U test). (D) Metastasis-free survival in patients with high NDRG1 expression (n=20) in the primary breast cancer specimen, compared to patients with absent or low NDRG1 expression (n= 54; p=0.035; log rank test)
Importantly, in the group of patients without distant metastasis within 10 years of follow-up, median NDRG1 expression in primary breast cancer tissue was significantly lower (median 1%; range 0–10%), compared to the group of patients experiencing distant metastasis during their course of disease (median 10%; range 0–80%; n=35; p=0.043; luminal A 26.8%; Luminal B: 22.9%; HER2 positive 8.6%; triple negative 40.0%; Fig. 6C). In line, median metastasis free survival was 41 months in the NDRG1 high group (n=20), and not reached yet in the NDRG1 absent/low group (n=54; p=0.035; log rank test; Fig. 6D). Of note, primary tumors from patients experiencing brain metastases (n=20) showed similar high membranous NDRG1 expression levels like those developing extracranial metastases (n=15; Supplementary Fig. 7 E), and patients with visceral metastases did not have higher NDRG1 expression compared to patients presenting only with bone metastases (p=0.111). Taken together, NDRG1 expression was highly heterogeneous between tumor cells and patients, and was associated with impaired prognosis due to more frequent and earlier metastatic spread.
Discussion
Conducting a comprehensive study of the brain metastases-initiating tumor cell subpopulation (BMICs) has met several methodical obstacles in the past, as only a small subfraction of circulating tumor cells will eventually give rise to clinically relevant macrometastases [23]. Intravital microscopy methodologies have significantly contributed to our current understanding of the dynamic behavior and plasticity of cancer cell subpopulations including potential tumor-initiating cells, and also cellular interactions in stem cell niches [24]. To optimally study the cellular and molecular features of this brain metastases-initiating tumor cell subpopulation in breast cancer, we first developed a methodology of combined in vitro and dynamic in vivo characterization of the same cancer cells. Thereby, we were able to study the single steps of brain metastasis development including intravascular arrest, extravasation, survival in the perivascular niche and outgrowth to a macrometastasis as well as the impact of specific biological changes in the single steps of the process. We found that temporary slow-cycling breast cancer cells are the major source of brain metastases. Using this novel experimental set up, NDRG1 protein expression was identified as a molecular characteristic of slow-cycling BMICs in one cell line, and selective knock-down resulted in the inability to form BM in vivo in this model. Higher median NDRG1 expression of primary breast cancers was associated with higher subsequent brain and visceral metastasis formation. All in all, our study provides a proof-of-principle that (brain) metastasis-initiating cells can be reliably identified by a combined in vitro characterization and in vivo microscopy approach, which allows to investigate BMICs in real time, and also to interrogate new molecular candidates in the future.
A higher metastatic potential of slow-cycling cells has been postulated previously [25,26], and brain micrometastases have been demonstrated to be slower-cycling, too [27]. Furthermore, a higher brain tropism of cancer cells cultured under stem-like conditions has been shown [28]. Our study provides the first direct evidence that BMICs have slow-cycling properties, and that this is due to an increased metastatic efficiency over the entire multistep process of brain colonization. An overlap of slow-cycling cancer cells and stemness properties was evident in this and previous studies [29–31]. This overlap is plausible, since stem cells and stem-like tumor cells are considered to be slow-cycling, and it can provide a possible explanation for the successful metastasis-initiating potential of slow-cycling cells[32]. Vice versa, stemness pathways like NOTCH and WNT are known to regulate slow-cycling cellular properties, and in line were also accumulated in the slow-cycling population in the current study [33]. Together this data speaks for (1) slow-cycling breast cancer cells being the BMICs; (2) enrichment of classical stemness markers in BMICs; and thus (3) the existence of a specific cellular state that is required for a cancer cell to become the “seed” for BM formation. Next to better identification of patients at high risk for brain metastatic disease, this information can be used to further determine other biological characteristics of BMICs.
To demonstrate that this approach is indeed possible and feasible, we compared the broad gene expression patterns of slow- vs. fast-cycling breast cancer cells, which revealed significant expression differences of 72 genes. We chose to focus on NDRG1, since high NDRG1 protein expression (as assessed by membranous staining pattern in immunohistochemistry) is a previously identified marker for prognostic assessment of breast cancer patients. NDRG1 expression is even included in the Mammostrat assay, which subdivides patients with early stage breast cancer in low, moderate and high risk as the basis for adjuvant treatment decisions. Therefore, NDRG1 has been clinically established as a marker of poor prognosis in primary breast cancer (22, 33), implying a relevant role for the disease course in humans that generally depends on the extent of future metastasis formation. All this was not the case for the other 71 genes identified. NDRG1 is an interesting gene in cancer, with controversial findings. Initially, various tumor suppressive properties of NDRG1 expression were identified as overexpression was observed to reduce invasion and metastasis of breast, colon, prostate and pancreatic cancer by inhibiting epithelial-mesenchymal transition, cell migration and angiogenesis [34]. Further, NDRG1 was shown to impact a large number of downstream pathways impacting cell proliferation, tumor vascularization, differentiation, and invasion via hypoxia-associated signaling [35]. However, increasing evidence suggests, that NDRG1 could also have pro- metastatic function as in reduced number of bone metastases were observed in a breast cancer model after know-down of NDRG1 [36]. Further, NDRG1 was shown to contribute to breast cancer aggressiveness by regulating the fate of lipids, resulting in elevated rates of metastasis and patient mortality [37]. All in all, the role of NDRG1 as a tumor suppressor (or enhancer) might indeed be tissue-, entity-, and stage- specific [38,39]. A gradual increase in NDRG1 expression from primary tumor to metastases was observed in colorectal cancer, underscoring that NDRG1 signaling as well as downstream mTOR and phosphatidylinositol 3 kinase (PI3K) signaling are involved in the metastatic process [40]. Indeed, the importance of the PI3K/mTOR signaling in brain metastasis growth is supported by growth inhibition after application of a selective PI3K/mTOR inhibitor [8]. In the particular context of breast cancer, NDRG1 expression was associated with markers of poor prognosis such as lack of estrogen and progesterone receptor expression, as well as decreased disease free and overall survival in all breast cancer subtypes [14]. In line, NDRG1 protein expression in primary breast tumors was associated with distant metastasis formation in the cohort analyzed in the current study. Interestingly, no increased NDRG1 expression was observed in human breast cancer BM compared to the primary tumor. This speaks for the plausible assumption that established brain metastases contain again only a small subset of cancer cells with stem-like properties, which has actually been demonstrated before [41]. Thus it suggests a dynamic nature of NDRG1 expression, as a similar phenomenon has been demonstrated for other molecular markers like Lgr5 in colorectal cancer [42], indicating that different genetic drivers maintain primary tumor maintenance, metastasis initiation and metastasis outgrowth. Indeed, knock-down of NDRG1 resulted in the complete inability of breast cancer cells to initiate BM in one of the two used models, while the growth of orthotopical injected tumors in the mammary fat pat was even accelerated, supporting a complex and sometimes even Janus-faced nature of NDRG1. All in all, while the findings of our study support a potential role for NDRG1 for (brain) metastasis formation in subgroups of breast cancer cases, it also underlines the existence of considerable heterogeneity, and provides no definitive solution to this conundrum. In our study, the association of slow cycling cellular behavior in one breast cancer cell line and NDRG1 expression is rather used to provide a proof-of-principle that the newly introduced methodology here has the potency to detect (brain) metastasis-relevant genes for further in-depth analysis.
A limitation of the present study is that dividing cells could not be visualized in real time throughout the experiment. An alternative approach to inject equal numbers of slow- and fast cycling cells would require prior FACS sorting, and this would mean increased stress of the cells and other experimental artefacts. Brain metastasis development is already a rare event in a very healthy, unstressed cell population; only a fraction of < 1% of injected cells shows intravascular arrest and only 1–5% of arrested cancer cells manage to successfully master all steps to a growing macrometastasis. Therefore, considering these limitations, we consider the current set up as the closest model to simulate the fate of a brain metastasis initiating cells entering the blood circulation on the way to form successful brain metastases. A further limitation of our study is the focus on breast cancer. While the importance of slow-cycling cells for tumor initiation and resistance was described for various cancer types, the role of NDRG1 as a prometastatic factor might be rather breast cancer-specific, as NDRG1 appears to play highly heterogeneous roles in other tumor entities as discussed above. Therefore, the association of slow-cycling breast cancer cells and NDRG1 expression in the specific context of BM initiation might be specific for some breast cancers, while it is very likely that other gene expression patterns are relevant for BMICs in other cancer entities [43]. Further, in-depth analysis of the upstream and downstream regulators of NDRG1 would be of general interest. However, the main goal of this study was rather to provide a proof-of-principle that this methodical set up that was newly developed for this study, using repetitive in vivo imaging to identify (brain) metastasis initiating cells and some of their principle biological characteristics, can indeed generate meaningful information about specific characteristics of the cancer cells responsible for metastasis formation. It will also be interesting to investigate in the future whether BMICs show differential interactions with host cells during the brain metastatic process, e.g. CD 8+ T cells [44]. Further, in the present manuscript we concentrated on the single steps of the brain metastatic cascade, but our data implies that slow cycling cellular behavior and NDRG1 expression can also be relevant for the development of extracranial metastases in breast cancer. More research is needed to definitely address this point.
In conclusion, the current study provides a proof-of-principle that metastasis-initiating cancer cells can be identified and characterized in greater detail when the specific advantages of long-term intravital microscopy are combined with in vitro characterizations. Here we demonstrate that slow-cycling breast cancer cells that express stemness markers are the tiny subpopulation of cancer cells that will eventually give rise to brain metastases after mastering the entire brain-metastatic cascade. The introduction of a novel methodology to identify and characterize those BMICs should facilitate a better understanding of their biological nature. Finally, this can help to develop novel concepts how to therapeutically target BMICs more specifically, which would be required for effective BM prevention.
Supplementary Material
Implications.
Cancer cells responsible for successful brain metastasis outgrowth, are slow-cycling and harbor stemness features. The molecular characteristics of these metastasis-initiating cells can be studied using intravital microscopy technology.
Acknowledgments
We thank Julia Meßmer, Dorothee Wilhelm, Ursula Rajky, and Bogdana Kovalchuk for excellent technical assistance. Further, we thank Axel Brenner for critical discussion of the Illumina sequence data and Gerwin Heller for assistance with publicly available gene expression data analysis. Frank Pajonk (David Geffen School of Medicine at UCLA, Los Angeles, California) kindly provided the retroviral vector pQCXIN-ZsGreen-cODC to study S26 proteasome activity.
Funding:
The present study was funded by the German Research fund (Deutsche Forschungsgemeinschaft; DFG grant number WI 1930/5-1 addressed to Frank Winkler), and from the German Cancer Aid (prevent_BM, to Frank Winkler). Anna Sophie Berghoff received a Schrödinger scholarship (J3779-B28) for a PostDoc research term by the Austrian Research Fund (FWF).
Competing interests:
A.S.B. has research support from Daiichi Sankyo (≤ 10000€), Roche (> 10000€) and honoraria for lectures, consultation or advisory board participation from Roche Bristol-Meyers Squibb, Merck, Daiichi Sankyo (all < 5000€) as well as travel support from Roche, Amgen and AbbVie. M.P. has received honoraria for lectures, consultation or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Merck Sharp & Dome. All other authors report no financial conflicts in the context of the present manuscript.
Data and materials availability:
The provided supplementary table provides raw data of the conducted experiments. Any further raw data can be requested from the corresponding author.
References
- [1].Berghoff AS, Schur S, Füreder LM, Gatterbauer B, Dieckmann K, Widhalm G, et al. Descriptive statistical analysis of a real life cohort of 2419 patients with brain metastases of solid cancers. ESMO Open 2016;1:e000024. doi: 10.1136/esmoopen-2015-000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WEF, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16:116–22. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- [3].Preusser M, Winkler F, Collette L, Haller S, Marreaud S, Soffietti R, et al. Trial design on prophylaxis and treatment of brain metastases: lessons learned from the EORTC Brain Metastases Strategic Meeting 2012. Eur J Cancer 2012;48:3439–47. doi: 10.1016/j.ejca.2012.07.002. [DOI] [PubMed] [Google Scholar]
- [4].Valiente M, Ahluwalia MS, Boire A, Brastianos PK, Goldberg SB, Lee EQ, et al. The Evolving Landscape of Brain Metastasis. Trends in Cancer 2018;4:176–96. doi: 10.1016/j.trecan.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Merlano MC, Granetto C, Fea E, Ricci V, Garrone O. Heterogeneity of colon cancer: from bench to bedside. ESMO Open 2017;2:e000218. doi: 10.1136/esmoopen-2017-000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lyle LT, Lockman PR, Adkins CE, Mohammad AS, Sechrest E, Hua E, et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood-Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin Cancer Res 2016;22:5287–99. doi: 10.1158/1078-0432.CCR-15-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Falkowska-Hansen B, Kollar J, Grüner BM, Schanz M, Boukamp P, Siveke J, et al. An inducible Tet-Off-H2B-GFP lentiviral reporter vector for detection and in vivo isolation of label-retaining cells. Exp Cell Res 2010;316:1885–95. doi: 10.1016/j.yexcr.2010.02.015. [DOI] [PubMed] [Google Scholar]
- [8].Osswald M, Blaes J, Liao Y, Solecki G, G�mmel M, Berghoff AS, et al. Impact of blood-brain barrier integrity on tumor growth and therapy response in brain metastases. Clin Cancer Res 2016;22:6078–87. doi: 10.1158/1078-0432.CCR-16-1327. [DOI] [PubMed] [Google Scholar]
- [9].Vlashi E, Lagadec C, Chan M, Frohnen P, McDonald AJ, Pajonk F. Targeted elimination of breast cancer cells with low proteasome activity is sufficient for tumor regression. Breast Cancer Res Treat 2013;141:197–203. doi: 10.1007/s10549-013-2688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shi W, Oshlack A, Smyth GK. Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips. Nucleic Acids Res 2010;38:e204. doi: 10.1093/nar/gkq871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Smyth GK. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat Appl Genet Mol Biol 2004;3:1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- [12].Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003;19:368–75. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- [13].Weiler M, Blaes J, Pusch S, Sahm F, Czabanka M, Luger S, et al. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc Natl Acad Sci U S A 2014;111:409–14. doi: 10.1073/pnas.1314469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nagai MA, Gerhard R, Fregnani JHTG, Nonogaki S, Rierger RB, Netto MM, et al. Prognostic value of NDRG1 and SPARC protein expression in breast cancer patients. Breast Cancer Res Treat 2011;126:1–14. doi: 10.1007/s10549-010-0867-2. [DOI] [PubMed] [Google Scholar]
- [15].Zviran A, Mor N, Rais Y, Gingold H, Peles S, Chomsky E, et al. Deterministic Somatic Cell Reprogramming Involves Continuous Transcriptional Changes Governed by Myc and Epigenetic-Driven Modules. Cell Stem Cell 2019;24:328–341.e9. doi: 10.1016/j.stem.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017;31:355–67. doi: 10.1016/j.ccell.2017.01.007. [DOI] [PubMed] [Google Scholar]
- [17].Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553:96–100. doi: 10.1038/nature25167. [DOI] [PubMed] [Google Scholar]
- [18].Chappell J, Dalton S. Altered cell cycle regulation helps stem-like carcinoma cells resist apoptosis. BMC Biol 2010;8:63. doi: 10.1186/1741-7007-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Harper LJ, Costea DE, Gammon L, Fazil B, Biddle A, Mackenzie IC. Normal and malignant epithelial cells with stem-like properties have an extended G2 cell cycle phase that is associated with apoptotic resistance. BMC Cancer 2010;10:166. doi: 10.1186/1471-2407-10-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vlashi E, Kim K, Lagadec C, Donna L Della, McDonald JT, Eghbali M, et al. In Vivo Imaging, Tracking, and Targeting of Cancer Stem Cells. JNCI J Natl Cancer Inst 2009;101:350–9. doi: 10.1093/jnci/djn509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Y, Zhou Y, Tao F, Chai S, Xu X, Yang Y, et al. N-myc downstream regulated gene 1(NDRG1) promotes the stem-like properties of lung cancer cells through stabilized c-Myc. Cancer Lett 2017;401:53–62. doi: 10.1016/j.canlet.2017.04.031. [DOI] [PubMed] [Google Scholar]
- [22].Wangpu X, Yang X, Zhao J, Lu J, Guan S, Lu J, et al. The metastasis suppressor, NDRG1, inhibits "stemness" of colorectal cancer via down-regulation of nuclear β-catenin and CD44. Oncotarget 2015;6:33893–911. doi: 10.18632/oncotarget.5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee JS, Magbanua MJM, Park JW. Circulating tumor cells in breast cancer: applications in personalized medicine. Breast Cancer Res Treat 2016;160:411–24. doi: 10.1007/s10549-016-4014-6. [DOI] [PubMed] [Google Scholar]
- [24].Suijkerbuijk SJE, van Rheenen J. From good to bad: Intravital imaging of the hijack of physiological processes by cancer cells. Dev Biol 2017;428:328–37. doi: 10.1016/j.ydbio.2017.04.015. [DOI] [PubMed] [Google Scholar]
- [25].Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS- O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- [26].Boral D, Vishnoi M, Liu HN, Yin W, Sprouse ML, Scamardo A, et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun 2017;8:196. doi: 10.1038/s41467-017-00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Singh M, Venugopal C, Tokar T, McFarlane N, Subapanditha MK, Qazi M, et al. Therapeutic Targeting of the Premetastatic Stage in Human Lung-to-Brain Metastasis. Cancer Res 2018;78:5124–34. doi: 10.1158/0008-5472.CAN-18-1022. [DOI] [PubMed] [Google Scholar]
- [28].Ren D, Zhu X, Kong R, Zhao Z, Sheng J, Wang J, et al. Targeting Brain-Adaptive Cancer Stem Cells Prohibits Brain Metastatic Colonization of Triple-Negative Breast Cancer. Cancer Res 2018;78:2052–64. doi: 10.1158/0008-5472.CAN-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010;141:583–94. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zeng L, Zhao Y, Ouyang T, Zhao T, Zhang S, Chen J, et al. Label-retaining assay enriches tumor-initiating cells in glioblastoma spheres cultivated in serum-free medium. Oncol Lett 2016;12:815–24. doi: 10.3892/ol.2016.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis 2009;26:611–23. doi: 10.1007/s10585-009-9260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moore N, Lyle S. Quiescent, Slow-Cycling Stem Cell Populations in Cancer: A Review of the Evidence and Discussion of Significance. J Oncol 2011;2011:1–11. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Srinivasan T, Walters J, Bu P, Than EB, Tung K-L, Chen K-Y, et al. NOTCH Signaling Regulates Asymmetric Cell Fate of Fast- and Slow-Cycling Colon Cancer-Initiating Cells. Cancer Res 2016;76:3411–21. doi: 10.1158/0008-5472.CAN-15-3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kovacevic Z, Fu D, Richardson DR. The iron-regulated metastasis suppressor, Ndrg-1: identification of novel molecular targets. Biochim Biophys Acta 2008;1783:1981–92. doi: 10.1016/j.bbamcr.2008.05.016. [DOI] [PubMed] [Google Scholar]
- [35].Said HM, Safari R, Al-Kafaji G, Ernestus R-I, Löhr M, Katzer A, et al. Time- and oxygen-dependent expression and regulation of NDRG1 in human brain cancer cells. Oncol Rep 2017;37:3625–34. doi: 10.3892/or.2017.5620. [DOI] [PubMed] [Google Scholar]
- [36].Shoda T, Hiraoka K, Hamada T, Hukushima N, Hosoi H, Nagata K, et al. Cap43/NDRG1 expression in breast cancer cells promotes osteolytic bone metastasis by inducing MMPs production via NF-kB activation. Cancer Res 2008;68. [Google Scholar]
- [37].Sevinsky CJ, Khan F, Kokabee L, Darehshouri A, Maddipati KR, Conklin DS. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res 2018;20:55. doi: 10.1186/s13058-018-0980-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Melotte V, Qu X, Ongenaert M, van Criekinge W, de Bruïne AP, Baldwin HS, et al. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 2010;24:4153–66. doi: 10.1096/fj.09-151464. [DOI] [PubMed] [Google Scholar]
- [39].Park KC, Paluncic J, Kovacevic Z, Richardson DR. Pharmacological targeting and the diverse functions of the metastasis suppressor, NDRG1, in cancer. Free Radic Biol Med 2019. doi: 10.1016/j.freeradbiomed.2019.05.020. [DOI] [PubMed] [Google Scholar]
- [40].Wang Z, Wang F, Wang W-Q, Gao Q, Wei W-L, Yang Y, et al. Correlation of N-myc downstream-regulated gene 1 overexpression with progressive growth of colorectal neoplasm. World J Gastroenterol 2004;10:550–4. doi: 10.3748/wjg.v10.i4.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nolte SM, Venugopal C, McFarlane N, Morozova O, Hallett RM, O’Farrell E, et al. A cancer stem cell model for studying brain metastases from primary lung cancer. J Natl Cancer Inst 2013;105:551–62. doi: 10.1093/jnci/djt022. [DOI] [PubMed] [Google Scholar]
- [42].de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017;543:676–80. doi: 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- [43].Jilaveanu LB, Parisi F, Barr ML, Zito CR, Cruz-Munoz W, Kerbel RS, et al. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin Cancer Res 2015;21:2138–47. doi: 10.1158/1078-0432.CCR-14-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, et al. Anti-PD-1/anti-CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci U S A 2018;115:E1540–9. doi: 10.1073/pnas.1714089115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The provided supplementary table provides raw data of the conducted experiments. Any further raw data can be requested from the corresponding author.