

Abstract
The olfactory system’s ability to detect and discriminate between the vast array of chemicals present in the environment is critical for an animal’s survival. In mammals, the first step of this odor processing is executed by olfactory sensory neurons, which project their axons to a stereotyped location within the olfactory bulb (OB) to form glomeruli. The stereotyped positioning of glomeruli within the OB suggests an importance for this organization in odor perception. However, because the location of only a limited subset of glomeruli has been determined it has been challenging to determine the relationship between glomerular location and odor discrimination. Using a combination of single-cell RNA sequencing (scRNAseq), spatial transcriptomics, and machine learning, we have generated a map of the majority of glomerular positions within the mouse OB. These observations significantly extend earlier studies and suggest an overall organizational principle within the OB that may be used by the brain to assist in odor decoding.
Introduction
Animals detect and interpret the vast array of chemical signals present in their external surroundings to find food and mates and to avoid predators and infection. To ensure survival, animals must be able to detect and discriminate between millions of distinct chemicals that may differ from one another by a single atom. In mammals, this exquisite precision is accomplished primarily through the actions of the olfactory system, which deploys an impressive complement of odorant receptors (ORs) expressed by olfactory sensory neurons (OSNs), which detect volatile chemicals and relay this information to the higher brain regions responsible for creating odor percepts and generating behavioral responses1.
In the mouse, each OSN expresses only one of the approximately 1000 OR genes encoded by the murine genome, and all OSNs that express the same OR project their axons to one of two insular anatomic structures located within the OB known as glomeruli2–5. The location of a particular glomerulus is stereotyped and is highly similar across different animals of the same species; the majority of prior studies have found that the location of a given glomerulus across different animals is within ~100 μm, although at least one study has found that this distance may be as great as 400 μm 4,6–12. Odor binding to an OR triggers a biochemical signal transduction cascade culminating in the release of neurotransmitter within the glomerulus13. Each OR possesses a unique chemoreceptive field, and a given chemical compound will therefore induce a unique pattern of glomerular activation across presentations of the same odor at the same concentration11,14–17. The result of this organization is that each odor at a given concentration maps reliably across animals to the same unique pattern of spatial activation within the brain, (although there is evidence that experience can alter these patterns18), and that to decipher which odor it has smelled and how to respond appropriately15,19,20, an animal, in theory, needs only to interpret this pattern of glomerular activation.
Despite its discovery several decades ago and the theoretical advantages to odor discrimination afforded by a stereotyped glomerular map, it has largely remained unclear whether this stereotypy is utilized by the animal to form accurate odor percepts. Recently however, several observations have begun to suggest that the theoretical advantages for odor perception afforded by a stereotyped glomerular map are indeed actualized by the olfactory system. One key finding that supports this idea is the stereotyped location of glomeruli4,6. This observation extends beyond the mouse; it holds true for all mammalian and fish species that have been examined and has been proposed to be a universal feature of vertebrate olfactory systems21,22. Moreover, invertebrate olfactory systems, which evolved independently from those of vertebrates, also appear to possess largely spatially invariant glomerular maps23–26. The parallel evolution of precise glomerular positioning in insects and vertebrates suggests that the evolutionary forces that created these stereotyped glomerular maps were likely driven by some functional and/or computational gain.
A second observation that the glomerular map itself encodes information arises from the finding that the physical properties of the OR expressed by an OSN dictate, at least in part, where within the OB the OSNs coalesce their axons to form a glomerulus27,28. Changing the amino acid sequence of an OR alters the location of the corresponding glomerulus within the OB29. As the sequence of the OR determines the odors it detects, these observations suggest that the Cartesian characteristics of the map may reflect odor detection properties of the olfactory system. Consistent with this idea, previous work suggests that at least in some instances, glomeruli that detect odorants with similar chemical structures may be located in close proximity to one another19,30,31. Moreover, it has been postulated that odorants that elicit similar odor-driven behaviors may be detected by neighboring glomeruli. For instance, glomeruli that respond to predator-derived odorants, which induce fear responses, appear to cluster in the dorsal regions of the OB32. Moving these glomeruli impairs behavioral responses to these odorants suggesting that the location of glomeruli may be important for facilitating the decoding of odor identity33,34.
Nonetheless, our understanding of how the brain accomplishes this computation and transforms patterns of glomerular activation into accurate internal representations of the animal’s external surroundings remains poorly understood. A major obstacle to addressing these questions is that the precise locations of only a handful of glomeruli are known. This lack of knowledge has made it challenging to decipher the molecular mechanisms that control the generation of the glomerular map. While a relatively small number of genes that influence the patterning of glomeruli within the mouse OB have been identified, the molecular mechanisms underlying the formation of the glomerular map remain largely unknown35–38.
Here, using a combination of scRNAseq, spatial transcriptomics, and machine learning we have mapped the locations of 654 glomeruli in the mouse OB. We find that each type of OSN expresses a unique transcriptional program—beyond simply the OR it expresses—that distinguishes it from all other types of OSN. This unique transcriptional program is highlighted by a subset of axon guidance genes and together is sufficient to predict where within the OB, the OSN projects its axon to form a glomerulus. This work significantly advances our understanding of how the olfactory system is organized to facilitate odor decoding.
Results
Assessment of OSN transcriptional heterogeneity
All OSNs expressing the same OR project their axons to a glomerulus whose position is largely spatially invariant. Moreover, each type of OSN is uniquely defined at a molecular level by the OR it expresses. Together, these properties suggest the intriguing possibility that the axon of each OSN may be guided to the correct location through a hard-wired transcriptional program that is unique to each type of OSN and that the OSN’s transcriptional program is tied to the properties of the OR that the OSN expresses. To explore whether OSNs that display a specific OR express a distinct transcriptional program from OSNs that display a different OR, we used fluorescence activated cell sorting (FACS) and Olfr1507-IRES-GFP, Olfr160-IRES-tauCherry, and Olfr73-IRES-GFP mice to isolate pure populations of OSNs that express Olfr1507, Olfr160, or Olfr73, respectively (Extended Data Fig.1a)29,39,40. Bulk RNA sequencing was performed on RNA extracted from three independent biological replicates of sorted OSNs from each of the three mouse lines (Fig.1a). Each sample expressed high levels of genes that define mature OSNs, including, Omp and Cnga2, and low levels of genes like Gap43, which are found in immature OSNs, indicating that we had efficiently isolated mature OSNs (Extended Data Fig.1b)41–43. Moreover, each OSN population selectively expressed its corresponding OR, indicating that OSNs bearing distinct ORs were captured (Extended Data Fig.1c).
Figure 1: RNA sequencing of FACS isolated OSNs reveals transcriptional heterogeneity between different types of OSNs.
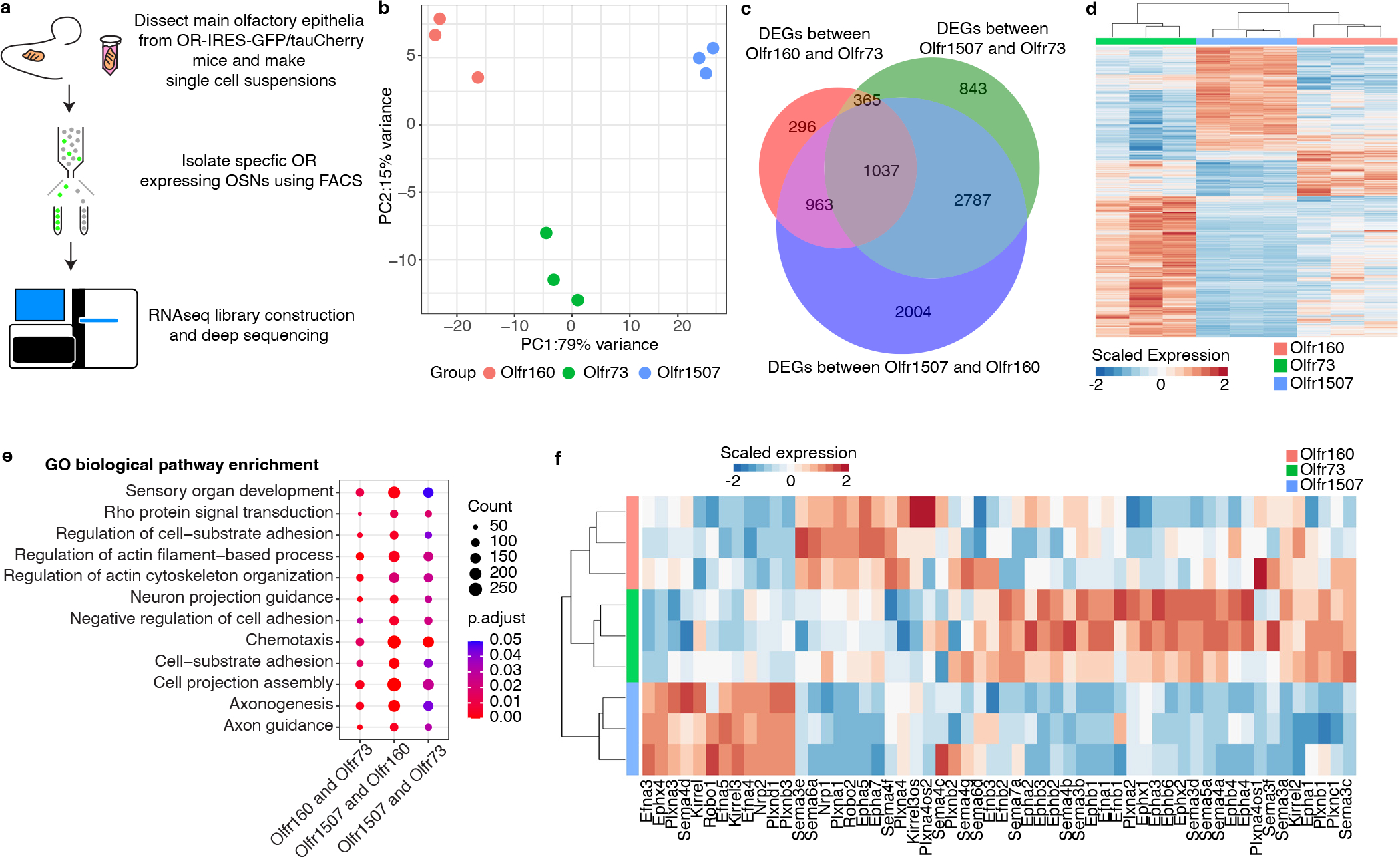
a, Schematic representation of the workflow used to identify RNA transcripts expressed by OSNs displaying different ORs. b, PCA applied to genes whose expression varied between OSNs expressing either the Olfr1507, Olfr160, or Olfr73 ORs. Each circle on the plot represents an independent biological replicate. c, Venn diagram representation of genes whose expression varies between populations of OSNs expressing the Olfr1507, Olfr160, or Olfr73 ORs. is indicated on the Venn diagram. d, Heat map of all of the differentially expressed genes between three independent biological replicates of OSNs expressing Olfr1507, Olfr160, or Olfr73. e, GO analysis performed upon the genes whose expression differed between OSNs expressing Olfr1507, Olfr160, and Olfr73. Genes involved in axonal pathfinding processes are amongst the most differentially expressed genes between these populations of OSN. One-sided Fisher’s exact test with Benjamini-Hochberg correction was used to account for multiple comparisons. f, Heat map of the most differentially expressed axon guidance genes (curated from differentially expressed genes) between three independent biological replicates of OSNs expressing the Olfr1507, Olfr160, or Olfr73. This analysis reveals that biological replicates expressing the same OR have similar patterns of genes that regulate axon guidance processes. The differentially expressed genes described in this figure were identified using a two-sided Wald test, Benjamini-Hochberg correction was used to account for multiple comparisons. Genes with Padj < 0.05 were considered to be differentially expressed (Supplementary table 1).
To determine whether these different populations of OSNs express distinct transcriptional programs, a principal components analysis (PCA) was applied to genes whose expression varied across the samples. Biological replicates expressing the same OR clustered together and were separated from samples expressing other ORs (Fig.1b), and several thousand genes were consistently differentially expressed by these three different types of OSNs (Fig.1c,d and Supplementary Table 1). Gene ontology (GO) analysis of the differentially expressed genes revealed that genes involved in axonal projection and axon guidance were enriched amongst the differentially expressed genes (Fig.1e). This observation was confirmed through an examination of individual gene implicated in axonal pathfinding (Fig.1f).
Together, these results suggest that different types of OSNs may express distinct patterns of genes. To systematically investigate transcriptional heterogeneity between OSNs, we performed scRNAseq on cells isolated from the olfactory epithelia of three adult female and three adult male mice (Extended Data Fig.2a). After filtering out low-quality cells, we identified a total of 45,317 cells that clustered into 16 different cell types, including 20,375 OSNs, which clearly expressed a single OR (Extended Data Fig.2b–d and Fig.3a,b). As previously reported, some OR genes were detected more frequently than others, likely owing to their proximity to cis-acting transcriptional regulatory sequences44–46 (Extended Data Fig.3c,d). Expression data from these OSNs were projected into two-dimensional space using Uniform Manifold Approximation and Projection (UMAP) algorithms, which cluster cells with similar transcriptional programs47. This clustering efficiently segregated both dorsal and ventral OB-projecting OSNs and anterior and posterior OB-projecting OSNs indicating that this approach accurately captures differences in gene expression (Extended Data Fig.4a). An examination of OSNs expressing ten random ORs revealed that cells expressing the same OR cluster together on the UMAP plot separately from cells expressing a different OR (Fig.2a), suggesting that each type of OSN might possess a unique transcriptome.
Figure 3: Spatial transcriptomic based reconstruction of the mouse glomerular map.
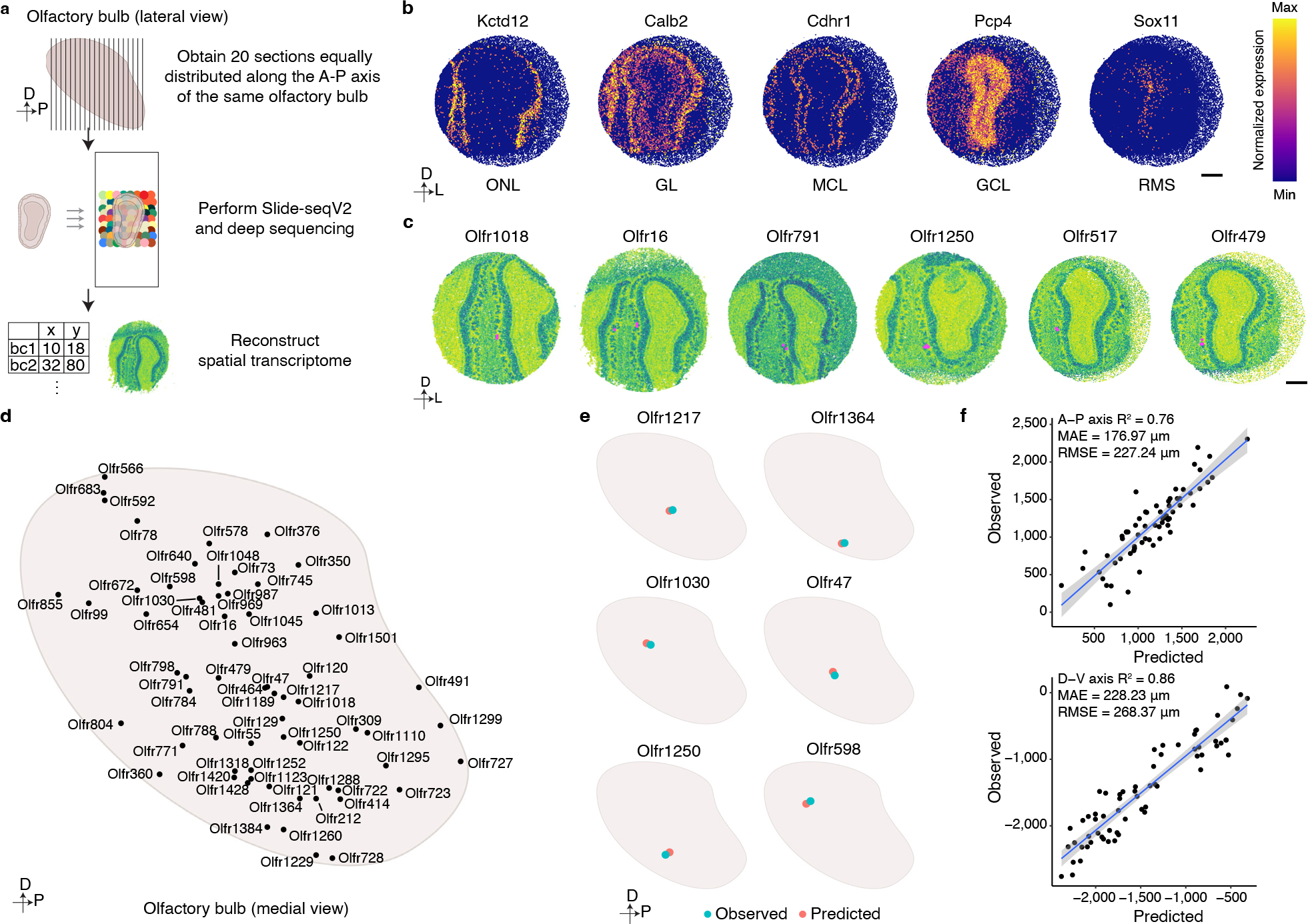
a, Schematic representation of the workflow used to generate a spatial transcriptomics-based reconstruction of gene expression within the mouse OB. Twenty sections evenly spaced along the anterior-posterior axis of the OB were subjected to Slide-seqV2 technology, and a custom analysis pipeline was then used to reconstruct a spatial transcriptomic map of the OB. b, Representative heatmaps of the expression of genes that define particular cellular layers within the OB: Kctd12-Olfactory Nerve Layer (ONL); Calb2-Glomerular Layer (GL); Cdhr1-Mitral Cell Layer (MCL); Pcp4-Granule Cell Layer (GCL); Sox11-Rostral Migratory Stream (RMS). Scale bar: 500 μm. c, Representative images of six glomeruli detected by our Slide-SeqV2 experiments. Instances in which proximal beads reveal expression of the same OR (magenta) were used to identify glomerular positions (see methods). Scale bar: 500 μm. d, Schematic representation of the positions of all the glomeruli identified through this spatial transcriptomics approach. e, A ridge regression model generated using the spatial transcriptomics-identified glomerular positions and the scRNAseq-determined OSN transcriptomes accurately predicts glomerular positions. Six different examples of the observed (blue) and predicted (red) glomeruli are depicted. f, Quantification of the accuracy of the ridge regression model for predicting glomerular position. The scatter plot depicts the positions of all 65 observed and predicted glomerular positions on both the anterior-posterior (upper panel) and dorsal-ventral (bottom panel) axes. Data were fitted with a regression line (blue) +/− SEM (light blue).
Figure 2: scRNAseq reveals that OSNs that display different ORs express unique transcriptional programs.
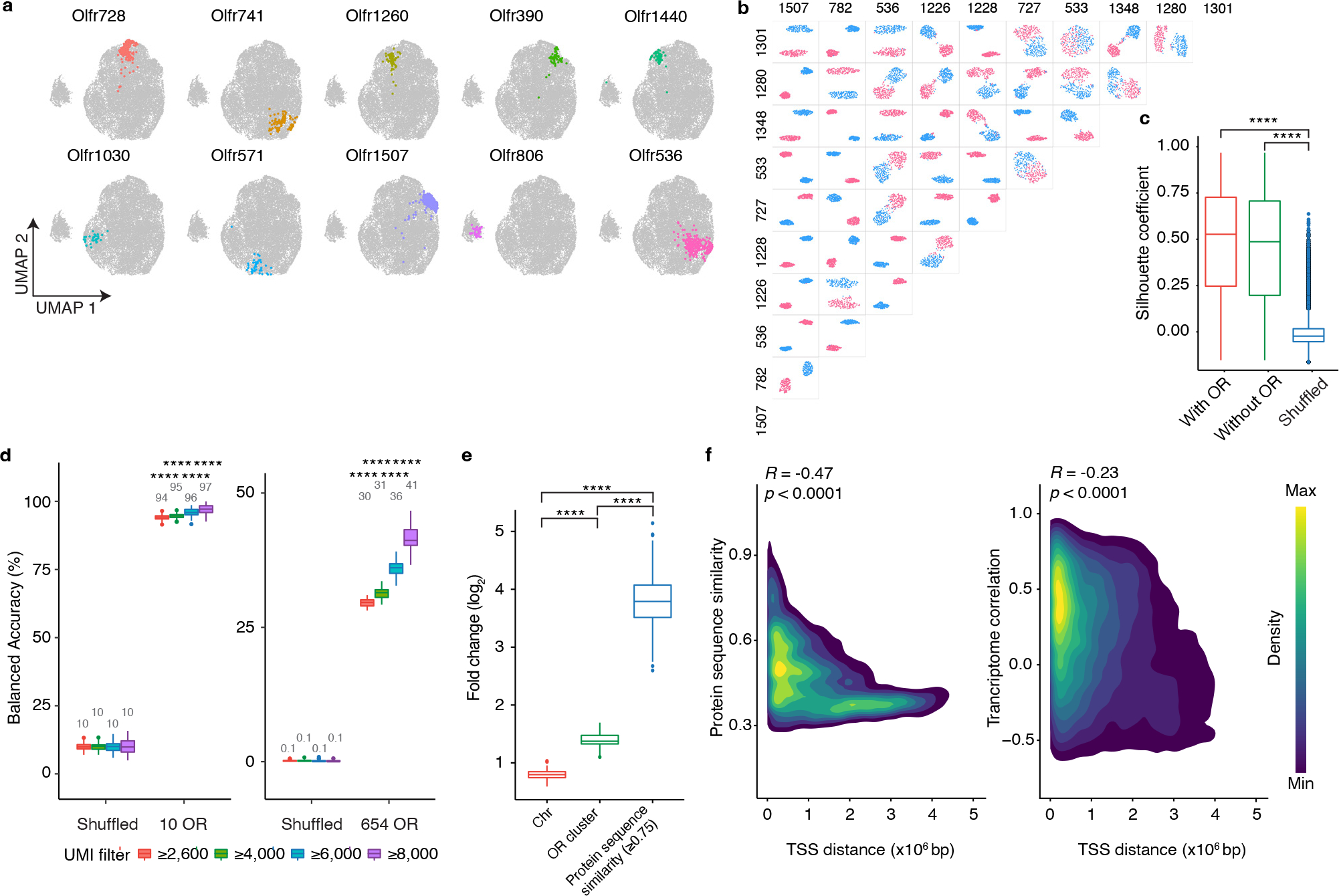
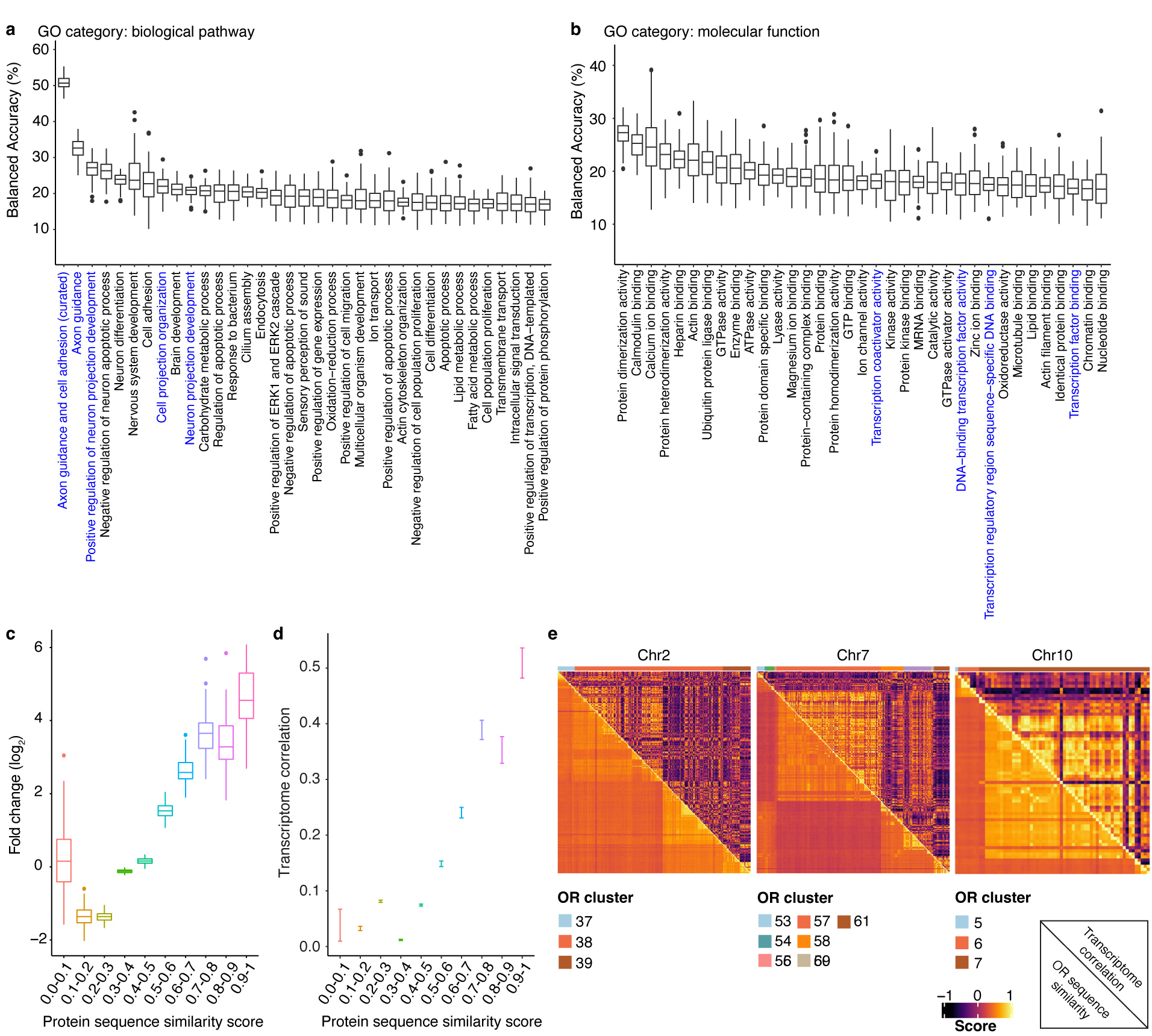
a, OSNs expressing each of ten random ORs are pseudo-colored on a UMAP plot. b, UMAP plots regenerated in a pairwise manner using each of the types of OSNs expressing the ten most frequently occurring ORs; OSNs expressing one OR are colored red and OSNs expressing the other OR are colored blue. c, To quantify the differences observed in location within UMAP space for different types of OSN, the Silhouette coefficients were calculated in a pairwise manner for all 654 types of OSN for which at least seven cells were sequenced using the complete gene expression matrix (with OR), the gene expression matrix with OR genes excluded (without OR), or the OR genes excluded expression matrix in which OR identity was randomly shuffled (Shuffled). N = 213,533 pairs, **** p < 0.0001 by two-sided Wilcoxon rank sum test, Benjamini-Hochberg correction was performed to account for multiple comparisons. d, SVM classifier analyses for OSNs expressing the 10 most frequently occurring OR types (left panel) and all 654 types of OSN for which at least seven cells were sequenced (right panel). Filtering OSNs that are inputted into the classifier with a unique molecular identifier (UMI) threshold increases the accuracy of the model. n = 100 independent tests, **** p < 0.0001 by two-sided Wilcoxon rank sum test comparing the actual data to the corresponding shuffled groups. e, The incorrect prediction results from d were collected and the likelihood for failed trials to be on the same chromosome, contained within the same genomic cluster, or to possess a protein similarity score > 0.75 with the observed OR was calculated. Data plotted as fold-change relative to chance. N = 100 independent tests, **** p < 0.0001 by two-sided Wilcoxon rank sum test, Benjamini-Hochberg correction was performed to account for multiple comparisons. f, Density map plot of the Pearson correlation between the genomic distance between the transcriptional start sites (TSS) of different ORs and their protein similarity score (left panel) or the transcriptome correlation of the OSNs that express those ORs (right panel). Only the genomic clusters containing at least 10 ORs were included in this analysis (N = 792 ORs on 19 clusters). Boxplots in this figure represent Q1 (quartile 1)-1.5*IQR (Interquartile range), Q1, median, Q3 (quartile 3), and Q3+1.5*IQR, data beyond the whisker were plotted as individual dots. Details of the statistical tests described in this figure are provided in Supplementary data 1.
To investigate this possibility in more detail, we selected the ten most frequently occurring types of OSNs in our data set and generated UMAP plots in a pairwise manner for all 45 possible combinations. In each instance, OSNs expressing one OR clearly clustered separately from OSNs expressing another OR (Fig.2b). To exclude the possibility that these differences in transcriptional profiles could be accounted for simply by the expression of the OR genes themselves, OR genes were excluded from the analysis and OSN clustering was reanalyzed. Even in the absence of OR gene information, OSNs that express the same OR clustered together and were separated from OSNs expressing different ORs (Extended Data Fig.4b–d). We next extended this analysis to include all 654 types of OSNs for which at least 7 cells were sequenced and calculated the separation between each type of OSN in a pairwise manner. Even with OR gene expression information withheld, each type of OSN clustered separately from all other types (Fig.2c) suggesting that each type of OSN expresses a unique transcriptional program that distinguishes it from all other OSNs. Interestingly, an analysis of the genes that are differentially expressed between distinct types of OSN, reveals that the vast majority of these differentially expressed genes are detected in nearly all types of OSN; there are very few genes whose expression is unique to a limited subset of OSN types (Extended Data Fig.4e). This observation suggests that rather than a small number of genes defining each type of OSN, there is instead a programmatic alteration in the level of expression of a larger set of genes.
If this conclusion is correct, it should be possible to infer the OR that a given OSN displays solely by examining the expression levels of the non-OR genes it expresses. To test this corollary, we selected the ten most frequently occurring types of OSNs in our data set, determined the genes whose expression was most variable between these cells (with OR gene information removed), and then computed the top 10 principal components explaining this variation. This information was then inputted into a support vector machine (SVM) classifier that was trained on 75% of the data. When the remaining 25% of OSNs were tested, the model correctly predicted the OR that each cell expressed with ~95% accuracy (Fig.2d). When this analysis was extended to include all types of OSNs for which at least 7 cells had been sequenced, the classifier still correctly predicted which OR was expressed with approaching 50% accuracy (Fig.2d).
The observation that OSNs that express a given OR express distinct programs of gene expression raises the question of when during the development of the OSN this defined pattern of transcription emerges. To address this subject, we analyzed single cell transcriptomes of immature OSNs by taking all immature OSNs for which a specific OR was expressed dominantly compared to other ORs, and asking whether the SVM classifier could predict which OR was expressed from the other genes it expressed. In contrast to what we observed with mature OSNs, the classifier was unable to correctly predict the expressed OR (0.85% accuracy for immature OSNs). This result is consistent with prior observations indicating that OR choice in mammals is stochastic, and that following the expression of a particular OR, the transcription of specific axon guidance genes, including kirrels, ephrins, plexins, and neuropilins within the OSN can be altered through differences in intrinsic activity exhibited by the OR28,35,36,38,48. Together, these results suggest that each type of OSN possesses a unique transcriptome that emerges only after OR choice has been made.
To determine which genes are most distinct between types of OSNs, we asked how well the SVM classifier could predict which OR is expressed using different subsets of genes. To this end, we selected all GO terms that contain at least 150 genes and asked how well the SVM model identified the expressed OR using only genes found within that GO category. This analysis revealed that genes associated with axon guidance processes most strongly influence the prediction of which OR is expressed (Extended Data Fig.5a and Supplementary Table 2). To a lesser extent, genes involved in transcriptional regulatory processes also contributed to the classifier’s success (Extended Data Fig.5b). These observations are consistent with recent work in the fly, which found that each type of OSN possesses a unique transcriptome, highlighted by the differential expression of transcription factors and axon guidance genes49.
Interestingly, in instances in which the classifier incorrectly predicted the OR expressed by an OSN, there was a strong bias toward it selecting an OR that was found within close proximity within the genome to the correct OR (Fig.2e). Previous reports have found that OR genes in the same genomic cluster share epigenetic regulatory features as well as more similar protein coding sequences50–52. Therefore, the transcriptional similarity between OSNs expressing ORs from the same genomic cluster could reflect either an influence of genomic location or protein sequence similarity (Fig.2f). To investigate these possibilities, we asked whether protein sequence similarity between ORs correlated with the transcriptomes of their respective OSNs. This analysis revealed that the more similar the sequences of the displayed ORs, the more similar the transcriptomes expressed by their OSNs (Extended Data Fig.5c,d). In fact, protein sequence similarity appears to be a stronger predictor of transcriptome correlation than genomic location (Fig.2e). Consistent with this hypothesis, an examination of the transcriptional correlation between OSNs expressing ORs located within the same genomic cluster reveals that even within a given genomic cluster, OSNs expressing ORs with more similar protein sequences have more similar transcriptional programs (Extended Data Fig.5e).
Spatial transcriptomic analysis of glomerular locations in the OB
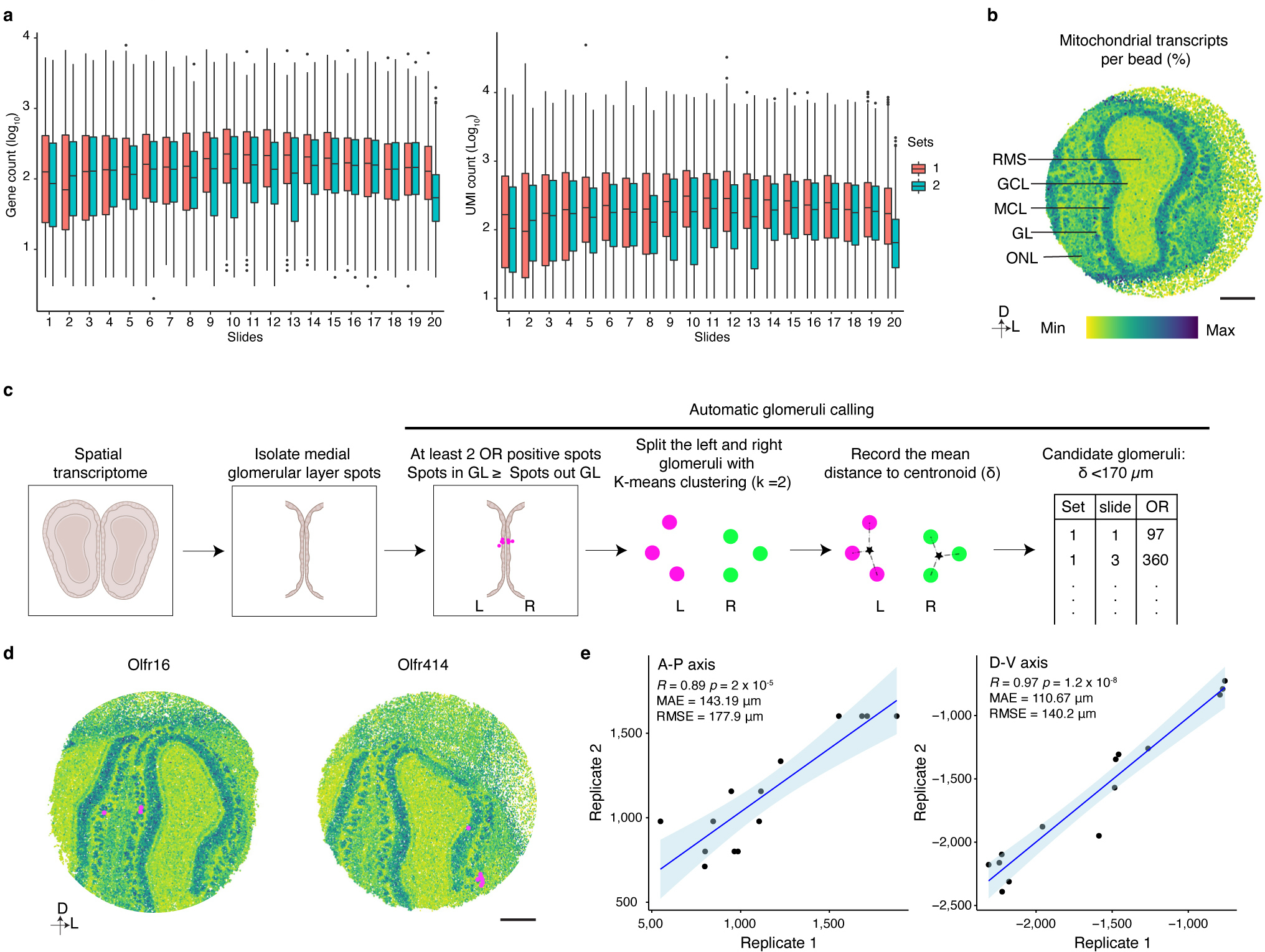
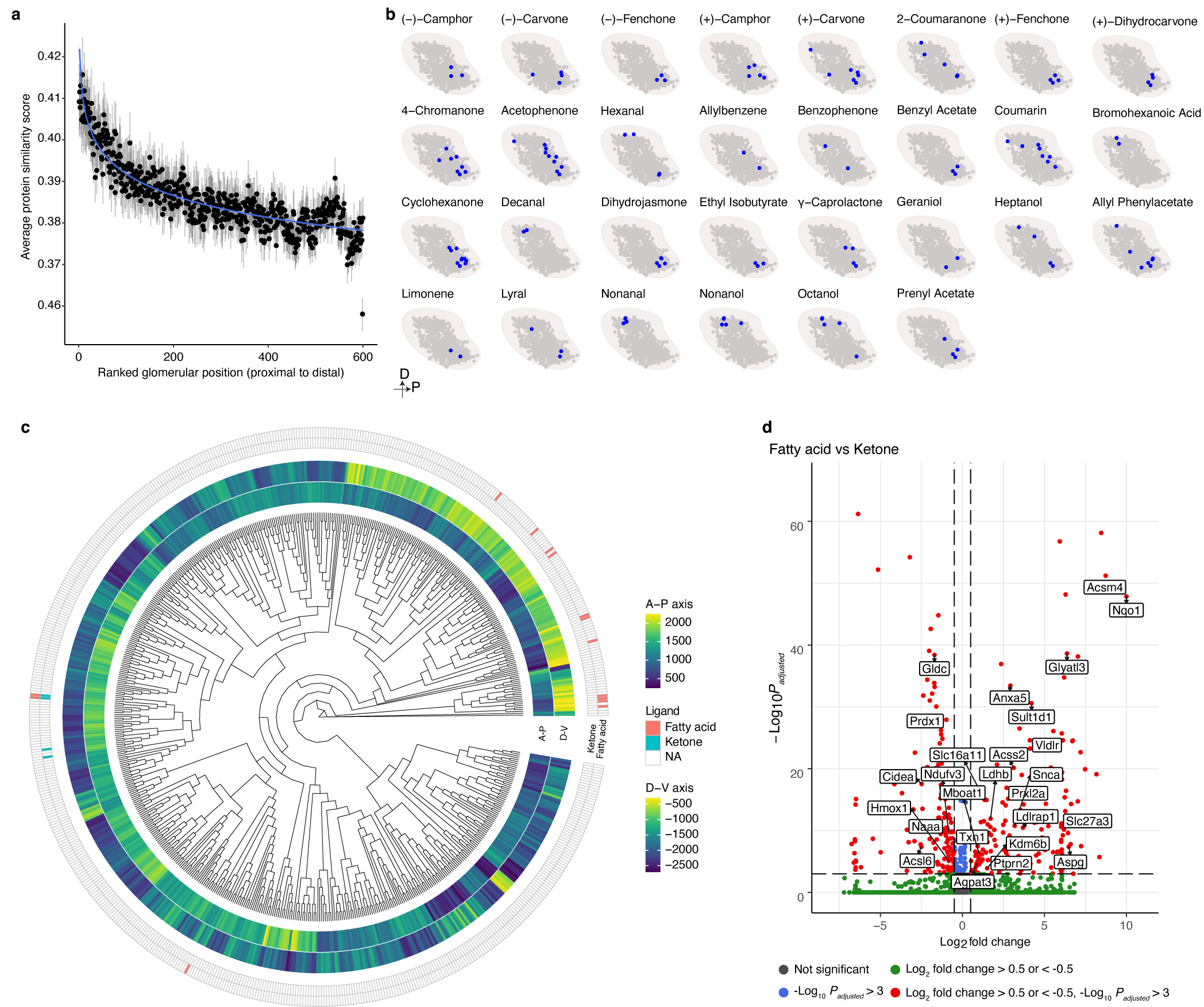
Together, these results reveal that each type of OSN expresses a unique, stereotyped transcriptional program, and that the genes expressed by different types of OSN are most closely associated with axon guidance processes. These observations are consistent with the hypothesis that the transcriptional program expressed by an OSN can be used to determine where an OSN will project its axon to form a glomerulus. However, only a handful of glomerular positions has been mapped experimentally, making it challenging to examine the correlation between OSN transcriptomes and their glomerular locations. To overcome this obstacle, we took advantage of spatial transcriptomics and the fact that OR mRNAs are found in OSN axonal termini to map the location of additional glomeruli within the mouse OB6. Using Slide-seqV2, spatial transcriptomic information was obtained from 40 sections of the murine OB that were evenly distributed along the anterior-posterior axis from independent mice (Fig.3a)53. An analysis of the resultant sequencing data revealed that the different cellular layers of the OB could be readily distinguished using known markers of these structures, indicating that this approach accurately captured the spatial location of mRNA transcripts (Fig.3b and Extended Data Fig.6a,b). To determine the location of glomeruli, we identified multiple, independent spots in close proximity to each other within the glomerular layer of the OB in which the same OR gene was detected (Fig.3c and Extended Data Fig.6c). By this approach we ascertained the location of 65 glomeruli that were evenly distributed in both the anterior-posterior and dorsal-ventral axes (Fig.3d). There was not a strong bias in detecting glomeruli formed by OSNs that expressed more frequently occurring ORs (Extended Data Fig.3c). In the two cases (Olfr16 and Olfr414), where the location of these glomeruli had been mapped using alternative methods, there was a high concordance in their mapped Cartesian coordinates (Extended Data Fig.6d)7,54. In addition, in the 14 instances in which the same glomerulus was detected in independent biological replicates, there was high agreement in its location across the data sets. The average distance between observed locations across biological replicates for these 14 glomeruli was 143 microns in the A-P axis and 111 microns in the D-V axis, which represents similar variance in glomerular positioning to what we determined using fluorescent OR reporter mouse lines and to what has been previously reported (Extended Data Fig.6e and 7a,b)7. Together, these observations suggest that Slide-seqV2 accurately captured the anatomic locations of glomeruli whose positions were previously unknown and provided us a substrate upon which we could test our hypothesis that the unique transcriptional identity of an OSN could be used to predict its glomerular location.
To test this hypothesis, we first trained a linear model to predict the glomerular location of a given type of OSNs from its transcriptional profile (excluding the OR gene). We selected all but one of the 65 glomeruli detected by the Slide-seqV2 spatial transcriptomics and used a cross-validation scheme and ridge regression to determine the best set of hyper-parameters from the transcriptomes of the OSNs that explained glomerular position. The position of the held-out glomerulus was then predicted using the model. This procedure was iterated such that the locations of all 65 glomeruli were predicted. Glomerular location predicted by the model and its actual observed anatomic position were in high concordance in each instance (average difference in distance of ~200 microns in the A-P and D-V axes). Given that a glomerulus has a diameter of ~100 microns and that the average differences in glomerular position across animals is at least 1 glomerular distance, these observations indicate that the model is able to accurately infer glomerular location based on the transcriptional profile of its associated OSNs (Fig.3e,f)7. The accuracy of the model compares favorably with previous studies that have predicted glomerular position using odor receptive fields9,55.
Recreation of the mouse glomerular map
To extend our model beyond the 65 glomeruli identified by spatial transcriptomics profiling, we applied it to all 654 types of OSN for which we had obtained scRNAseq data from at least 7 cells to create a map consisting of 654 glomeruli (Fig.4a). There are four independent lines of evidence suggesting that this glomerular map generated by the ridge regression model accurately recapitulates the actual orientation of glomeruli within the OB. Firstly, light sheet based microscopic imaging of Olfr1507-IRES-GFP, Olfr160-IRES-tauCherry, and Olfr73-IRES-GFP mouse lines revealed that in all three instances the actual location of these glomeruli closely matches the location predicted by our model (Fig.4a). Secondly, glomeruli formed by OSNs expressing class I ORs localized nearly exclusively to the anterior and dorsal regions of the OB. By contrast, the glomeruli formed by OSNs expressing class II ORs were found in more posterior and ventral regions of the OB (Fig.4b). This segregation of Class I and II ORs matches previously reported data56. Thirdly, a number of genes have been previously reported to exist in a gradient across the OB in a manner that correlates with their glomerular positions. In our model, Acsm4, Nrp1, Nrp2, and Plxna1 display gradients of expression that matches what has been reported previously (Fig.4c) 36,57.
Figure 4: Organizing principles revealed through the reconstruction of a map of mouse glomerular positions.
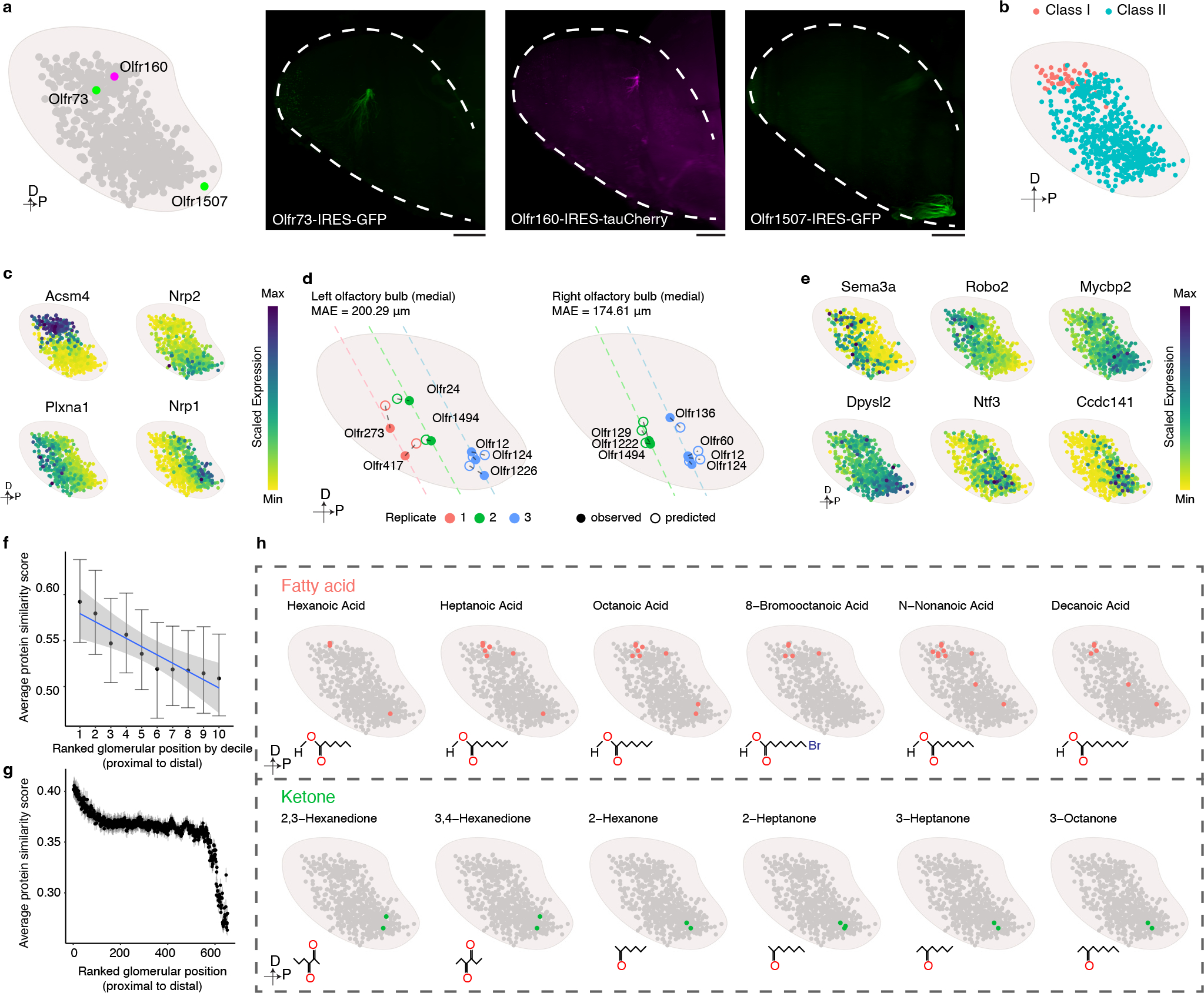
a, Using mice harboring Olfr1507-IRES-GFP, Olfr160-IRES-tauCherry, or Olfr73-IRES-GFP alleles and light sheet microscopy we empirically determined that the locations of these three glomeruli (right panels) are consistent with our prediction (left panel). Similar results were observed from each mouse line with at least 3 biological repeats. Scale bar: 500 μm. b, The distribution of Class I (Red)- and Class II (Blue) glomeruli on the predicted glomerular map. c, Scaled expression of Acsm4, Nrp2, Plxna1, and Nrp1. d, Location of the indicated glomeruli determined by MERFISH (●) relative to their position predicted by the model (○). Dashed line indicates the slide position. e, Scaled expression of axon guidance genes obtained from the scRNAseq data for each of the OSNs that form glomeruli in our model. f, Glomeruli formed by OSNs expressing ORs from the same genomic cluster (only the clusters with at least 10 corresponding glomeruli detected were included, N = 19) were iteratively rank ordered according to their distance from each other and then the average protein sequence similarity was calculated for each ranked glomerular position. To enable comparison across genomic clusters that contain different numbers of ORs, glomerular position was binned by decile and calculated for each genomic cluster separately. The data are presented as the mean +/− bootstrapped 95% CI. Data were fitted with a linear model (blue), and the +/− 95% CI (gray). g, Glomeruli (N = 654) were iteratively rank ordered according to their distance from each other, and the average protein sequence similarity was calculated for each ranked glomerular position. The data are presented as the mean +/− bootstrapped 95% CI. h, Published databases of the chemoreceptive fields of mouse ORs were used to create a schematic representation of the glomeruli that respond to the indicated fatty acid (green) and ketone (red) odorants.
Lastly, we employed multi-plexed error-robust fluorescent in situ hybridization (MERFISH) technology to independently assess the locations of glomeruli58. By this approach, we were able to reliably determine the positions of 11 glomeruli from three independent sections (Extended Data Fig.7c). The empirical position of these glomeruli matches the location predicted by our model (Fig.4d). Together, these results suggest that the glomerular map generated by the ridge regression model accurately reflects what exists within the mouse OB.
These analyses reveal the most comprehensive map of the mouse OB to date and allow for an examination of the logic underlying glomerular positioning. To this end, we first determined the genes, which most strongly contribute to the model’s ability to predict glomerular location. A GO analysis of these genes revealed that there was the strongest enrichment for genes involved in axon guidance and axon development (Extended Data Fig.8a). Interestingly, the genes that we identified by this approach include both known regulators of OSN axon guidance such as Sema3a, Robo2, Dlx5, Efna5, and Mycbp2, as well as genes that have not previously been implicated in OSN axon guidance, including Dpysl2, Ntf3, Nexn, Etv1, and Ccdc141 (Fig.4e and Extended Data Fig.8b), which are expressed in a gradient manner across the OB in either the A-P or D-V axes 37,59,60.
We next asked beyond this axon guidance gene expression program whether there were additional features of the OSNs (and the ORs that they express), which correlate with their position in the OB. A visual examination revealed that glomeruli formed by OSNs expressing ORs found within the same genomic cluster often group together within the OB (Extended Data Fig.9). Even for OSNs expressing ORs found within the same genomic cluster, those expressing ORs with more similar protein sequences are closer to each other than those expressing ORs with more divergent sequences (Fig.4f). Interestingly, a closer inspection of the entire glomerular map reveals that the property of OSNs expressing ORs with higher sequence similarity forming glomeruli that are closer in Cartesian space is limited to the local environment of the glomerulus. Once it extends beyond the nearest approximately few dozen glomeruli the correlation between glomerular distance and OR protein sequence similarity declines exponentially (Fig.4g and Extended Data Fig.10a). These observations suggest that a local spatial environment of glomeruli is created that are formed by OSNs with similar protein sequences, and thus by extension presumably more similar ligand specificities.
Although it is difficult to comprehensively test this prediction as the ligands for only a relatively limited subset of odorants have been tested, we nonetheless queried a database of OR-ligand relationships that have been previously experimentally validated and asked whether local clusters of glomeruli may exist that detect odors with similar structures. This does indeed appear to be the case, as glomeruli that respond to ketones are clustered in the ventral, posterior bulb; by contrast, glomeruli that detect fatty acids tend to localize to the dorsal, anterior OB (Fig.4h and Extended Data Fig.10b,c). These findings are consistent with prior functional imaging experiments in the mouse and rat OBs, which have revealed a correlation between the proximity of two glomeruli and their likelihood of responding to the same class of odorant19,21,30,31,61. It is important to note that this correlation in glomerular responses has been reported to fall apart as the distance between the glomeruli widens beyond ~1 mm, which is consistent with our observation that OR protein sequence similarity decays exponentially after a few dozen glomerular distances9. Together, these observations suggest that although it remains controversial whether chemotopy exists in the bulb, that nonetheless, the location of a glomerulus within the OB may convey information about the set of odorants that it is able to detect.
Discussion
The work presented above represents a powerful example of how high-resolution genomic technologies can be used to reconstruct or even predict complex tissue organization. We have for the first time created a detailed map of the positions of the majority of glomeruli within the OB, increasing our knowledge of mouse glomerular positioning by nearly two orders of magnitude. These results reveal that glomeruli formed by OSNs expressing more similar ORs are closer to one another. These observations extend earlier pioneering studies performed on a much smaller scale and suggest an overall organizational principle within the OB51,52. Further, we show that the transcriptional profiles of OSNs expressing similar ORs are more similar to one another. These results may provide a mechanistic basis for the observation that glomeruli that respond to similar odorants are close to one another in physical space and, more broadly, suggest that the organization of glomeruli within the OB may be leveraged to assist the brain in computing odor identity.
This observation suggests that understanding the mechanisms by which the glomerular map is generated is critical for elucidating how the olfactory system has evolved to optimize its capacity for odor perception. Our finding that each OSN possesses a unique transcriptional identity provides an important clue as to how that map may be generated. As the OR expressed by an OSN correlates with the transcriptional program of that cell there is a clear path upon which evolution can act to couple OR identity (and thus ligand specificity) and glomerular positioning. Consistent with this idea, we have found that the glomeruli located in dorsal, anterior OB respond preferentially to fatty acid compounds. Intriguingly, the OSNs that form these glomeruli preferentially express a number of genes that participate in fatty acid metabolism (Extended Data Fig.10d), suggesting a link between the transcriptional programs of these OSNs and their functional properties.
Although significantly more experimentation will be required to determine the role of these genes in olfactory processes and to determine if this observation extends to other types of OSNs, it is nonetheless intriguing to speculate that there may be a generalizable, close evolutionary link between the properties of the OR, the transcriptomes of the OSNs in which they are expressed, and the odor sensing properties of the associated glomeruli. The observation that new OR genes arise during evolution through tandem duplication and thus cluster with other ORs within the genome that share similar protein sequence, suggests a mechanism by which an animal can modulate its OR repertoire while maintaining its ability to decode the identity of the odorants it is sensing62. As tandemly duplicated OR genes integrate into the genome, they are likely to be regulated by the same set of enhancer elements, which our work suggests correlates strongly with their glomerular positioning. Our work, in combination with numerous previous studies, suggest that the OR protein sequences themselves play a role in regulating the expression of axonal guidance genes and thus glomerular location27–29. As OR protein sequence also dictates the array of chemicals that an OR can bind, together these properties suggest a mechanism through which it might be possible to obtain a form of chemotopic organization within the glomeruli map, whereby, at least locally, nearby glomeruli detect similar types of odorants. However, it is important to note that this feature is likely restricted to a local environment and that the extent to which chemotopy exists within the bulb is highly controversial9,11,17,19,21,55,63. The staggering size of chemical space and the large number of ORs in the mammalian genome has made it technically challenging to comprehensively catalog OR-ligand relationships. In addition, chemicals have many different features in addition to basic functional groups, and it is still unclear which of these are most critical to odor perception. Despite these challenges, we believe the uncovering of the glomerular map will facilitate resolving some of the controversies surrounding chemotopical organization in the OB.
Together, we believe the observations described here shed significant light on long standing questions within the field of olfaction. This work provides a detailed map of the mammalian OB and provides a substrate upon which questions about how this organization influences odor perception and the mechanisms by which this map is created and maintained. In the future, work describing the molecular basis by which this map is generated promises to offer significant insight into how the olfactory system is able to execute the remarkable task of detecting and discriminating millions of compounds.
Methods
Mice
Olfr1507-IRES-GFP mice were a gift from Gilad Barnea (Brown University), Olfr73-IRES-GFP mice were a gift from Bob Datta (Harvard University). Olfr160-IRES-tauCherry (stock no. 029637) and C57BL/6J (stock no. 000664) mice were obtained from Jackson Laboratories. 4 to 6 month mice, both genders, were used in all experiments in this study. All animal care and use procedures were followed in accordance with federal guidelines (12 hr. light/12 hr. dark cycle, 20–23°C, 30–70% humidity) and approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee (Protocol A-2645-18).
Generating olfactory epithelial single cell suspensions
Mice were euthanized according to our IACUC protocol, and then main olfactory epithelia were dissected and quickly immersed in ice-cold HBSS (Worthington). Olfactory epithelia were separated from the bone under a dissection microscope and then were minced into small pieces using forceps. Each epithelium was then washed once with HBSS and resuspended in 2 ml of EBSS containing papain (20 U/mL, Worthington) and DNase I (2000 U/mL, Worthington). After 40 minutes of incubation with gentle agitation at 37°C, the suspension was washed with 5 ml of PBS + 0.02% BSA twice and passed through a 30 μm cell strainer (Miltenyi Biotec).
Bulk RNAseq
For each biological repeat, single cell suspensions of the main olfactory epithelium were generated with multiple OR-IRES-GFP/tauCherry mice (mixed genders) according to the protocol described above. The cell suspension was stained with 3 μM of DAPI and subjected to FACS to isolate the GFP/tauCherry high and DAPI low population of cells. 5000 cells were collected for each sample and RNA was extracted with a RNeasy micro kit (QIAGEN). A Single Cell/Low Input RNA Library Prep Kit (NEB) was used to construct sequencing libraries following the manufacturer’s guidelines with one alteration, which was to increase the insert length to ~300 base pair. Libraries were sequenced using paired end 150 cycle reads on an Illumina Hiseq4000 (Novogene). The sequencing reads were processed using the DolphinNext RNAseq pipeline1 (https://dolphinnext.umassmed.edu/index.php?np=1&id=732). The default settings were employed, except STAR v2.6.1 and RSEM v1.3.1 were used for alignment and quantification, respectively. Trancriptome build: gencode M252,3. The count matrix was loaded to R (4.0.0 or later), and DEseq2 (1.30.1) was used to normalize the matrix and perform differential gene expression analysis4. The clusterProfiler (3.18.1) and org.Mm.eg.db (3.12.0) annotation databases were used for GO term (http://geneontology.org/) analysis5–7.
Single-cell RNA sequencing
Olfactory epithelial single cell suspensions were prepared using 3 male and 3 female mice according to the protocol described above. Single-cell sequencing libraries were prepared using the Chromium Single Cell 3’ Reagent Kit V3 (10X Genomics) according to the manufacturer’s guidelines. Libraries were sequenced using 150 cycles of paired end reads on Illumina Hiseq4000 and Novaseq6000 instruments (Novogene). The sequencing reads were processed using the DolphinNext Single Cell-10X Genomics pipeline (https://dolphinnext.umassmed.edu/index.php?np=1&id=420, default settings except STAR v2.6.1 was used for alignment, transcriptome build: gencode M251,2). The OR_deconvolution script (https://github.com/elisadonnard/ORdeconvolution) was used to correct mis-mapped OR reads.
Seurat (v4.0.3) run in R was used for data analysis8. Cells with fewer than 1500 unique molecular identifiers (UMIs), more than 50000 UMIs, fewer than 500 genes detected, more than 1% hemoglobin transcripts, or more than 10% mitochondrial transcripts were removed. Python (3.7.6 or higher) package Scrublet (0.2.2) was used to calculate a doublet score, and cells with high doublet scores and/or expressing multiple markers of different cell types were removed9. The filtered count matrix was then normalized with log-transformation and the top 2000 variable genes were selected to perform PCA. Harmony was used to correct batch effects in PCA space10. The number of PCs used for generating UMAP plots was determined by the “Elbowplot” function. Wilcoxon rank sum test-based differential gene expression analysis (“FindAllMarkers” function) was used to identify cell types.
To confidently isolate the population of OSNs that expressed a single OR, the mature OSN cluster was isolated, cells with fewer than 2600 UMIs, more than 15000 UMIs, or expressing any other types of olfactory receptor (Taar, Ms4a, V1r, V2r, Fpr, Tas1r, Tas2r) were removed. Finally, only the cells expressing a single OR (UMI>1) were used for all future OSN analyses. The OR identity of the OSNs was thus determined by the OR transcripts detected by this single cell sequencing analysis and only OSNs whose OR identity was clear from this approach were carried forward for future analyses.
The OR identity of immature OSNs was determined using the dominantly detected OR from scRNAseq (the OR with the highest normalized expression with a value > 2).
Silhouette coefficient calculation
The single cell transcriptomes for each pairwise combination from all 654 types of OSNs for which we had sequenced at least 7 cells were isolated and then normalized using log-transformation. The top 2000 variable genes (OR genes were removed prior to gene selection) were selected to perform PCA. Harmony was used to correct batch effects. The distances (cosine) between cells in PCA space were calculated and used to calculate the silhouette coefficient of the clustering between OR pairs.
OR identity prediction
The normalized single cell transcriptomes of target OSN groups (either those expressing the top ten most frequently occurring ORs or all 654 types of OSNs for which we had sequenced at least seven cells) were in silico isolated then subsetted by the most differentially expressed genes (1121 for the top ten OR groups, 4521 for the 654 OR groups, (OR genes were removed prior to gene selection)). The gene expression matrix was then split such that for each OSN type 75% was used as a training dataset and 25% as a testing dataset. To balance the contribution of each type of OSN in the PCA, each type of OSN from the training data was grouped to the same number of cells and then the expression was averaged. The averaged expression matrix was then scaled and used to perform the PCA. The training data and test data were then projected to the PCA space created by averaging the training cells. A matrix consisting of the specific number of PCs (top 8 PCs for the top 10 OR groups; top 18 PCs for the 654 OR group) and the OR identity of each cell was used to build the prediction model. Random under sampling was applied to the training data to balance the weighting of each group of OSN during model building. A support vector classifier was implemented with the “SVC” class (C = 0.01, kernel = “linear”, class_weight = “balanced”, decision_function_shape = “ovo”) from Scikit-Learn (0.24.2)11. At least 100 iterations of random split and model building were performed for each sample group. The prediction results and cell barcodes were recorded for analyzing the correlations between the observed and predicted OR.
Protein similarity score
Fasta files containing mouse protein sequences were downloaded from the Ensembl database (GRCm38.p6, release 102). OR protein sequences were extracted and pairwise protein similarity scores were calculated using the “parSeqSim” function in the protr package (1.6–2)12.
Transcriptome correlation
The harmony-corrected first 50 PCs of all 654 groups of OSNs were averaged by OR group. The averaged PCs matrix was then used to calculate the Pearson correlation coefficient in a pairwise manner for all 654 OR groups.
OR cluster assignment
The assignment of each OR to a genomic cluster was performed according to a previous study13.
Slide-seqV2
Male mice were euthanized according to our IACUC protocol. The anterior part of the brain containing the OB was dissected and placed in a plastic mold filled with OCT (Sakura Finetek from VWR), The molds were then frozen with dry ice pre-chilled 2-methylbutane (Fisher Scientific). The samples were incubated in −80°C overnight prior to cryosectioning. 20 OB sections were obtained with equal spacing (130 μm) along the anterior-posterior axis from each animal. In total, two sets of 20 slides were obtained from different animals. All of the Slide-seqV2 procedures were performed and the sequence libraries was generated as previously described36. Libraries were sequenced using 100 cycles of paired end reads on Illumina Novaseq6000, targeting 200 million reads per sample. The sequence reads were processed with slideseq-tools (0.2.1) to generate the expression matrix and beads coordinates (https://github.com/MacoskoLab/slideseq-tools). The expression matrix and the beads coordinates were loaded in R and analyzed with Seurat package (v4.0.3). The raw count matrices were log-transformed, and the expression of OB layer marker genes and the mitochondrial transcript percentage were plotted along with spatial coordinates to identify the layers.
Glomerular map reconstruction
For each Slide-seqV2 data set, all the slides were aligned according to the shape of OB, the transcriptomes of beads localized in the medial glomeruli layer were isolated and subjected to automatic glomeruli calling: (1) For a given OR candidate, the OR transcripts need be detected by at least 2 beads within the glomeruli layer, and the number of OR positive beads in the glomeruli layer needs to be higher than all other layers. (2) The OR positive beads were split into left and right OB by applying k-means clustering (k=2). (3) The average distance between each bead and their centroid was calculated. (4) The candidates with an average distance to centroid smaller than 170 μm were then considered to represent real glomerular signal and were carried forward for subsequent analysis (Extended Data Fig.6c). For each glomerular-localized OR signal, the average position of the spots was projected to the dorsal-ventral axis to obtain the y-coordinate and the slide position was used to represent the x-coordinate. The medial site of the detected glomeruli was used for further analysis, since the glomerular positions are mirror-symmetric. The coordinates of Slide-seqV2 replicate 1 were transformed to the coordinates of Slide-seqV2 replicate 2 using the Kabsch algorithm. The top 50 PCs of the OSN transcriptome were z-score normalized using the “StandardScaler” class from Scikit-Learn (0.24.2)11. Synthetic training data points (to ensure an equal number of training instances for each OR) were generated using the Synthetic Minority Oversampling Technique (“SMOTE” class from the “imbalanced-learn” library (0.8.0)). Ridge regression was implemented with the “Ridge” class from Scikit-Learn11 to learn the correlation between the position of the candidate glomeruli detected by the Slide-seqV2 and its corresponding OSN transcriptome. The remaining OSN transcriptomes were then applied to the model to generate the “complete” glomerular map.
MERFISH
MERFISH was performed as previously described, according to the manufacturer’s instructions (Vizgen)14. Briefly, mice were euthanized and brains were immediately placed in cold OCT. Embedded brains were then frozen at −20°C and transferred to −80°C for storage. Briefly, a 10 μm slice of the tissue was sectioned and placed upon a functionalized coverslip covered with fluorescent beads. Once adhered to the coverslip the tissue was fixed (4% PFA in 1x PBS, 15 minutes, room temperature) followed by 3 washes with 1x PBS. After aspiration, 70% ethanol was added to permeabilize the tissue for 24 hours. After a wash with Formamide Wash Buffer (30% formamide in 2x saline sodium citrate (SSC)), the sample was incubated with a custom MERFISH probe library (Vizgen) and left to hybridize for 48 hours. The sample was then washed and incubated at 47°C with Formamide Wash Buffer twice, and then the tissue was embedded in a polyacrylamide gel followed by incubation with tissue clearing solution (2xSSC, 2% SDS, 0.5% v/v Triton X-100, and 1:100 proteinase K) overnight at 37°C. Then, the tissue was washed and hybridized for 15 min with the first hybridization buffer, which contained the readout probes associated with the first round of imaging. After washing, the coverslip was assembled into the imaging chamber and placed into the microscope for imaging. MERFISH imaging was performed on an automated Vizgen Alpha Instrument using imaging buffers, hybridization buffers, and parameter files provided by Vizgen. Briefly, the sample was loaded into a flow chamber connected to the Vizgen Alpha Instrument. First, a low-resolution mosaic image was acquired (DAPI channel) with a low magnification objective (10x). Then the microscope was switched to a high magnification objective (60x) and seven 1.5μm z-stack images of each FOV position were generated in 750nm, 650nm, and 560nm channels. A single image of the fiducial beads on the surface of the coverslip was acquired and used as a spatial reference. After each round of imaging, the readout probes were extinguished, and the sample was hybridized with another set of readout probes. This process was repeated until combinatorial FISH was completed. Raw data were decoded using the MERLIN pipeline (v0.1.6, provided by Vizgen) using the codebook for the library used.
Ligand response distribution on predicted glomeruli map
Ligand-OR pair information was compiled from the Olfactory Receptor DataBase (ORDB, https://senselab.med.yale.edu/ordb/)15. All ligand-OR pairs were then filtered to include only those ORs present in the reconstructed glomerular map. Ligands recognized by only one OR in our model were removed.
Light-sheet fluorescent microscope imaging
Male OR-IRES-GFP/tauCherry mice were euthanized according to our IACUC protocol. Mice were perfused with 20ml ice-cold 4% PFA PBS solution. The OB and the anterior part of the brain were dissected and incubated in 4% PFA in PBS at 4°C overnight. The iDISCO protocol was used to clear the tissue (https://idisco.info/idisco-protocol/)16. Anti-GFP (Aves, GFP-1020, 1:2000) and anti-mCherry/RFP (Rockland, 600-401-379, 1:200) and Alexa Fluor® 488 AffiniPure Alpaca Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch, 611-545-215, 1:300) Alexa Fluor® 488 AffiniPure Goat Anti-Chicken IgY (IgG) (H+L) (Jackson ImmunoResearch, 103-545-155, 1:300) were used to stain and boost the fluorescent signal. The cleared and stained tissue was imaged using a light-sheet microscope (Ultramicroscope II; LaVision Biotec). InspectorPro software (LaVision BioTec) and Imaris x64 software (version 8.0.1, Bitplane) were used to acquire and analyze the images, respectively. The 3D reconstructed OB was aligned based on its shape and orientation, and the glomerular fluorescent signal was used to render the 3D surfaces.
Statistical information
For bulk RNAseq experiments, a two-tailed Wald test with Benjamini-Hochberg correction on multiple comparisons was used to identify differentially expressed genes between genotypes. The genes with an adjusted p value lower than 0.01 were considered to be significantly differentially expressed genes. These genes were than used to perform GO enrichment analysis with one-sided Fisher’s exact test, Benjamini-Hochberg correction was performed to account for multiple comparisons. GO terms with an adjusted p value greater than 0.05 were not considered.
For scRNAseq analyses, the Wilcoxon rank sum test was used to determine differentially expressed genes among different cell types; genes with a Benjamini-Hochberg correction adjusted p-value lower than 0.05 were considered to be significantly differentially expressed and were used for downstream analysis. For the silhouette coefficient analysis, OR identity prediction, and OR feature enrichment fold change, Wilcoxon rank sum test with Benjamini-Hochberg correction was used to compare the mean between two groups of samples. A Pearson correlation was used to compare the correlation among OR protein similarity, transcriptome similarity, and the distance between transcription start site.
For calculating the gene contribution to the ridge model in the glomerular reconstruction analysis, the PCs with a significant coefficient (p <0.05) in the ridge regression model were extracted. The weighted loading of each gene in each PC was calculated by multiplying the gene loading (absolute value) with the coefficient (absolute value). For each gene, the sum of weighted loading of each gene across all significant PCs are ranked. The gene contribution of the prediction power on both axes are calculated. For each axis, the top 350 ranked genes were used to perform the GO analysis with one-sided Fisher’s exact test and Benjamini-Hochberg correction. The GO terms with an adjusted p value greater than 0.05 were discarded. The top ten GO terms with the highest gene ratios were selected for visualization.
No statistical tests were used to predetermine sample sizes, but our sample sizes were similar to those reported in previous publications17–20. Samples were assigned by genotype, gender and batch. Data were analyzed in an automated manner whenever possible in the absence of human supervision to ensure blinding. In randomly subsetted data analyses, data points selected were chosed by algorithm without human supervision. Besides the low quality cells filtered out from the single-cell RNAseq data, no samples were discarded in this study. Before performing statistical analysis, Shapiro-Wilk normality test and Q-Q plot were used to determine the normality of the data and a Levene’s test was performed to determine the variance homogeneity of the data. All the statistical details (effect size, test statistics and exact p-value) of each test performed in this study are provided in Supplementary data 1.
Data availability
All RNA sequencing data described in this manuscript are deposited at GEO accession GSE169021. The positions of the reconstructed glomerular map can be found at:
https://github.com/Greerlab/OSN_2021_paper.
The processed MERFISH data can be found at:
https://zenodo.org/record/5745947#.YaeQ-PHMIqs
Fig.1 and Extended Data Fig.1 are associated with GSE169010; Fig.2–4, Extended Data Fig.2–5,8,10 are associated with GSE169011; Fig.3 and Extended Data Fig.6 are associated with GSE169012.
Code availability
All the scripts used for this study can be found at:
Extended Data
Extended Data Figure 1: FACS isolation of specific OR-expressing OSNs using OR-IRES-GFP/mCherry knockin mice.
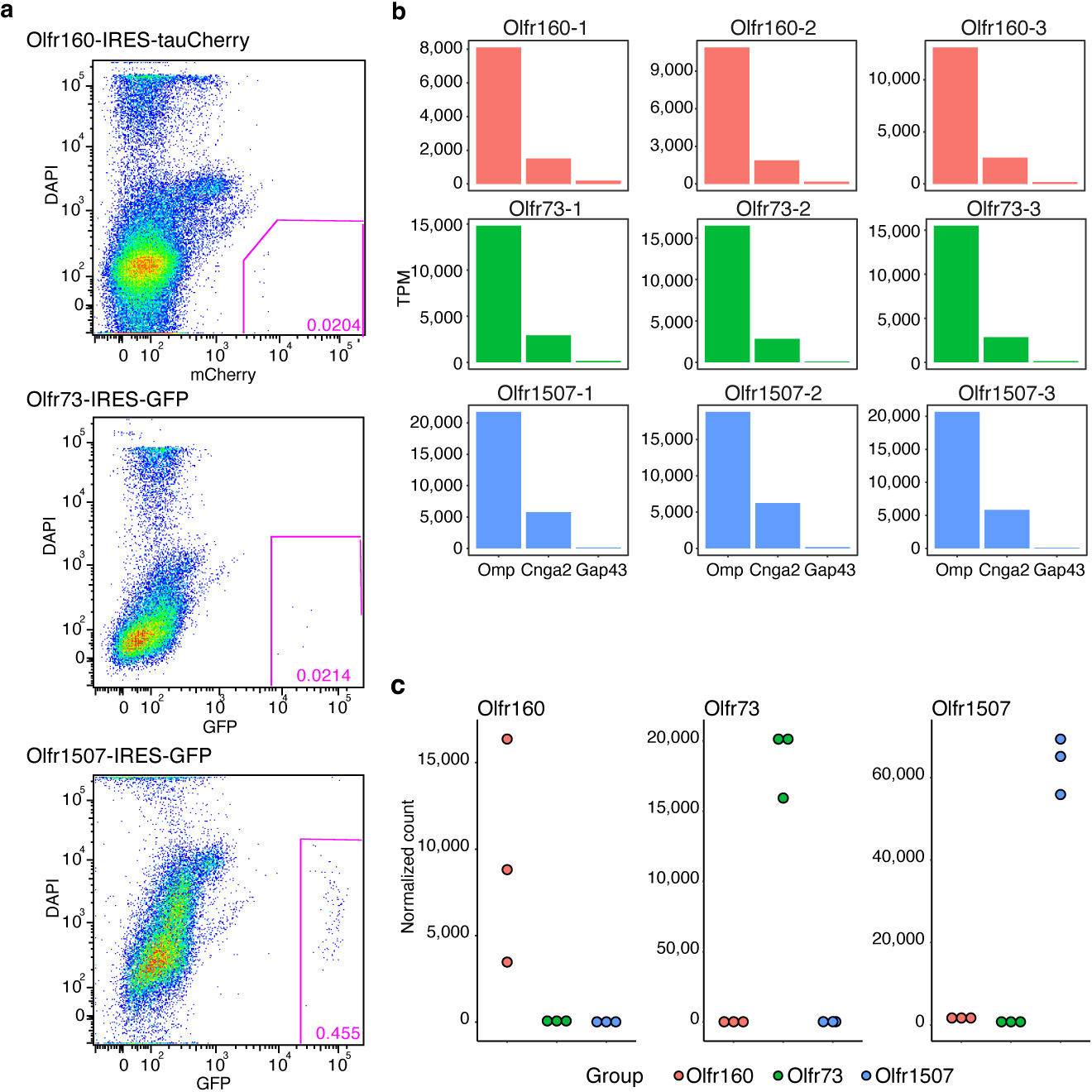
a, Scatter plots of FACS isolated, dissociated olfactory epithelial cells from mice harboring the Olfr160-IRES-tauCherry (top), the Olfr73-IRES-GFP (middle), or the Olfr1507-IRES-GFP (bottom) alleles. The gate used to isolate ~100% pure populations of fluorescent OSNs is indicated. b, Mature OSN markers (e.g., Omp and Cnga2) but not immature OSN markers (e.g., Gap43) are detected in FACS isolated samples. c, Each OSN population selectively expresses its corresponding OR.
Extended Data Figure 2: scRNAseq reveals diverse cell types in the main olfactory epithelium.
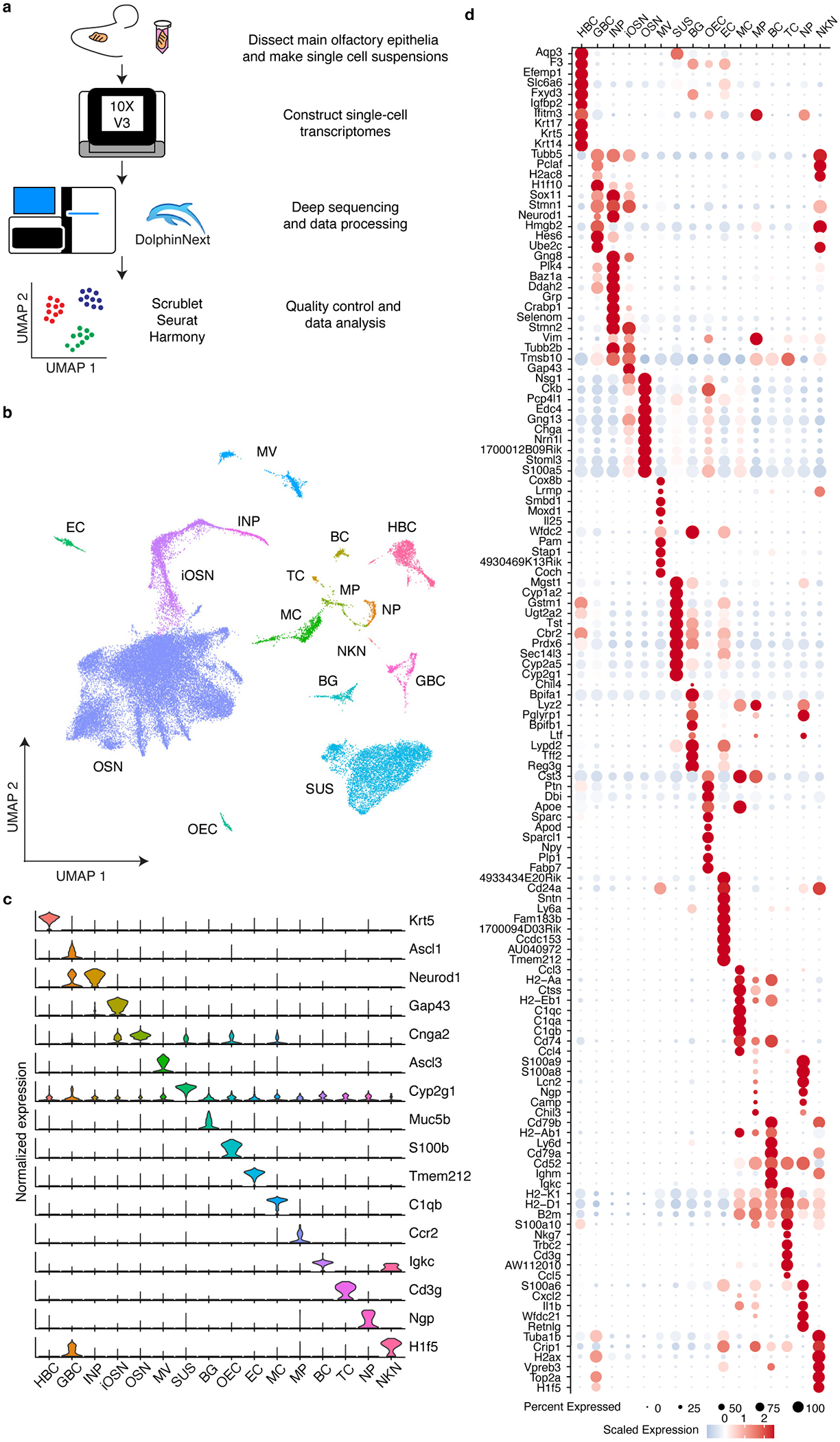
a, Schematic representation of the workflow used to perform scRNAseq on cellular populations isolated from the mouse main olfactory epithelium. b, The distribution of different cell types on the UMAP projection plots. HBC: horizontal basal cell; GBC: Globose basal cell; INP: Immediate neuronal precursor; iOSN: immature olfactory sensory neuron; OSN: Olfactory sensory neuron; MV: Microvillar cell; SUS: Sustentacular cell; BG: Bowman’s gland; OEC: Olfactory ensheathing cell; EC: Ependymal cell; MC: myeloid cell; MP: Macrophage; BC: B cell; TC: T cell; NP: Neutrophil; NKN: Unknown. c, Violin plots of the genes used to assign cell types. d, Dot plot of a subset of the differentially expressed genes between cell types. For each gene depicted, the size of the circle corresponds to the percentage of sequenced cells in which transcripts that map to that gene were detected, and the color reflects the average expression level of the gene within those cells.
Extended Data Figure 3: Quality control analysis of OSN scRNAseq.
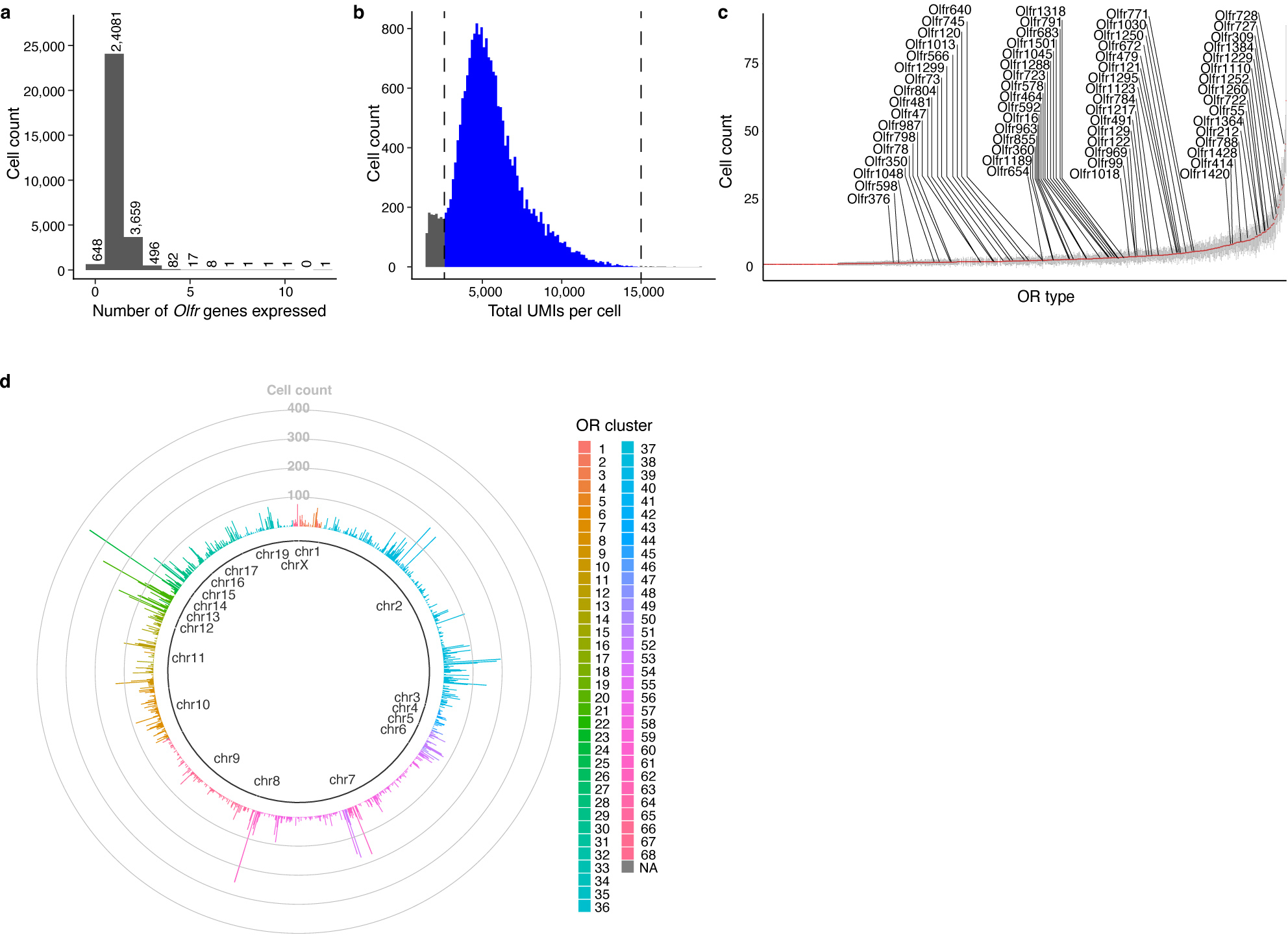
a, Histogram of the number of distinct OR expressed by each of the mature OSNs captured in scRNAseq experiments. b, Histogram of the number of UMIs detected by each of the single OR expressing OSNs captured in scRNAseq experiments, the cells with 15000 > UMI > 2600 were used for the future analyses (blue area). c, Plot of the number of cells sequenced that express each distinct OR, the OR transcript detected by Slide-seqV2 are labeled. Data are plotted as mean +/− SD from six independent experiments. d, Schematic of each cluster of OR genes and their chromosomal location and how many cells were detected that express each of these ORs.
Extended Data Figure 4: UMAP projections reveal distinct transcriptomes expressed by different types of OSN.
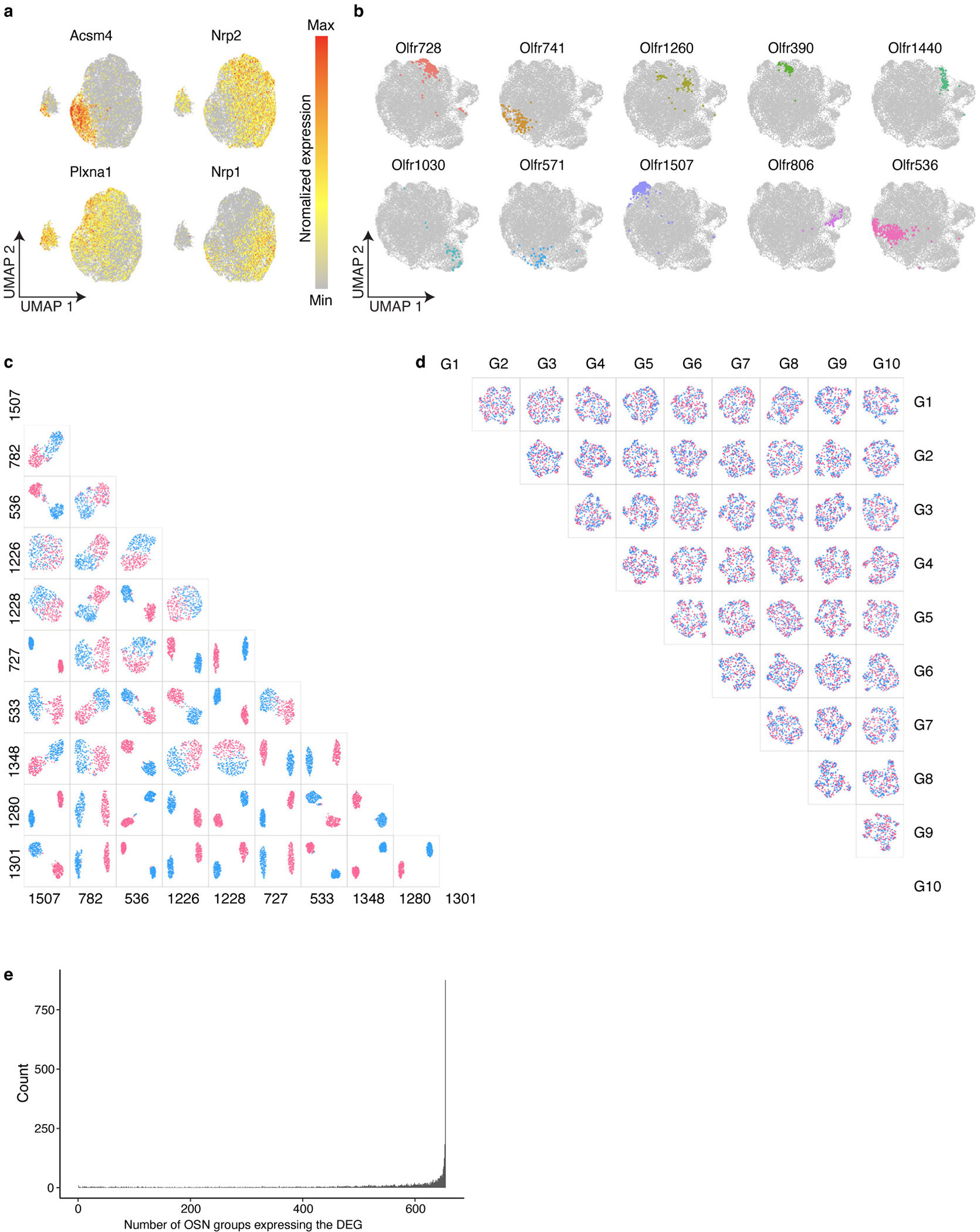
a, Normalized expression of the Acsm4, Nrp2, Plxna1, and Nrp1 genes on UMAP projection plots of OSN scRNAseq data. b, UMAP plots of OSNs were regenerated with all OR gene expression information removed. OSNs expressing each of ten random ORs selected in Fig.2a are pseudo colored in the UMAP plot, revealing that cells displaying the same OR cluster together even in the absence of OR gene expression information. c, UMAP plots were regenerated in a pairwise manner using each of the groups of OSNs expressing the ten most frequently occurring ORs with OR gene expression information removed prior to clustering. In each pairwise analysis all of the OSNs expressing one OR are colored red, and all of the OSNs expressing the other OR are colored blue to make it easier to visualize their separation in UMAP space. d, “Shuffled” control for the pairwise OSN UMAP segregation analysis. The analysis was performed as in c, except OR identity was randomly assigned prior to analysis. e, Analysis of all of the differentially expressed genes identified in a pairwise manner for each of the 654 types of OSN. For each differentially expressed gene, we determined for each of the 654 types of OSN in how many of those types of OSN is that gene detected in at least 10% of the sequenced cells.
Extended Data Figure 5: Pairwise analysis reveals OSN features that correlate with transcriptomic similarity.
a, Analysis of the ability to predict OR identity using different GO biological pathway term gene sets. The boxplots depict the bootstrapped data from 100 iterations of the prediction for each GO term, The top 30 terms are ordered from the highest mean accuracy to the lowest. In addition to the GO terms found in the axon guidance biological category, we hand curated a list of axon guidance genes and adhesion molecules (Supplementary Table 2), removing transcription factors and odorant receptor genes themselves and included this refined set of axon guidance/adhesion molecule genes in the analysis. b, Analysis of the ability to predict OR identity using different GO molecular pathway term gene sets. The boxplots depict the bootstrapped data from 100 iterations of the prediction for each GO term, The top 30 terms are ordered from the highest mean accuracy to the lowest. c, Incorrectly predicted ORs are likely to possess similar protein sequence to the correct OR. The OR protein similarity scores of incorrectly predicted OR pairs from Fig.2d were calculated and binned (N = 100 independent tests). The boxplot depicts the likelihood of a given OR protein similarity score distribution as the fold-change relative to random OR pairs. d, The transcriptomic correlation between a pair of OSNs positively correlates with the protein sequence similarity of the ORs they express. Transcriptomic correlation and OR protein sequence similarity of all the pairwise combination of 654 types of OSN were calculated and binned. The plot depicts the mean +/− SEM for each protein similarity score bin. e, Heat map of each set of OSNs expressing a specific OR reveals high transcriptional correlation (upper triangle) and OR protein sequence similarity (lower triangle) between OSNs expressing ORs located within the same genomic cluster. OSNs are ordered by genomic location and the color bar indicates the genomic cluster. Boxplots in this figure represent Q1–1.5*IQR, Q1, median, Q3, and Q3+1.5*IQR, data beyond the whisker were plotted as individual dots.
Extended Data Figure 6: Slide-seqV2 enables high quality spatial transcriptomic analysis of the mouse OB.
a, Plot of the number of genes (left panel) or UMIs (right panel) detected per bead on each slide of the Slide-seqV2 experiments (N ranges from 30041 to 56016, the exact number of beads on each individual slide is provided in Supplementary data 1), Boxplots indicate Q1–1.5*IQR, Q1, median, Q3, and Q3+1.5 *IQR; data beyond the whisker were plotted as individual dots. b, Mitochondrial transcript levels as a marker to define layer organization in the OB. Scale bar: 500 μm. c, Schematic of automated calling of glomerular positions in Slide-seqV2 experiments. d, Slide-seqV2 identified glomerular positions match glomerular positions that have been empirically determined. Scale bar: 500 μm. e, The glomerular positions of 14 ORs detected in two replicates of 20 Slide-seqV2 experiments exhibit high spatial correlation. 13 of the 14 glomerular positions identified in two, independent biological replicates were used as the anchors to align the two OBs. The held-out glomerulus was then transformed based on the anchors and the position was recorded. This process was iterated for all 14 glomeruli. The correlation between the position of the held-out glomeruli on the A-P and D-V axes across both replicates is shown. Data were fitted with a regression line (blue), +/− 95% CI (light blue).
Extended Data Figure 7: Validation of the predicted glomerular map.
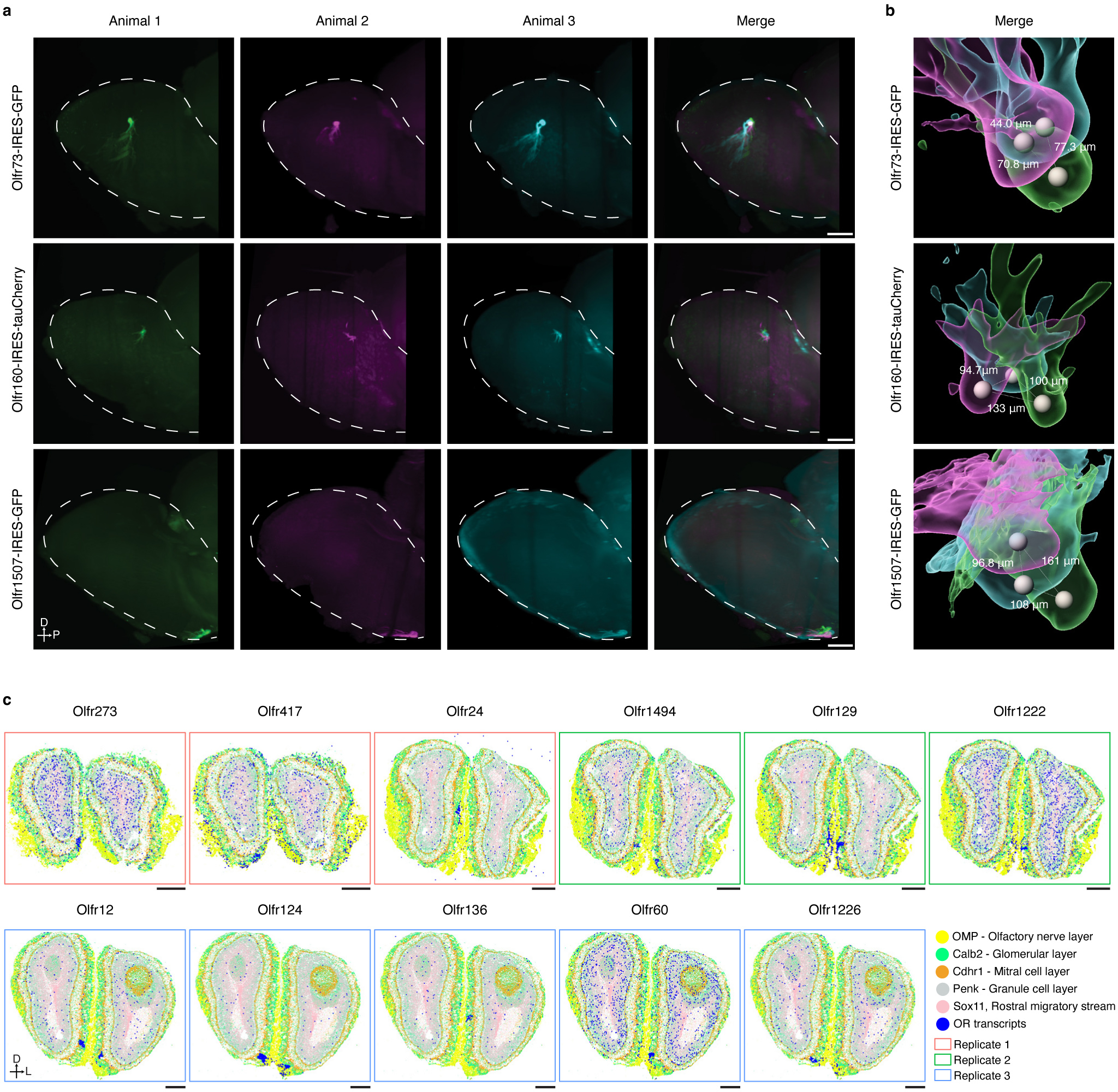
a, Light sheet microscopic images of intrinsic fluorescent signals from the OBs of three independent mice harboring the Olfr73-IRES-GFP, Olfr160-IRES-tauCherry, or the Olfr1507-IRES-GFP alleles. Similar results were observed from each mouse line with at least 3 biological repeats per line. Scale bar: 500 μm. b, Reconstruction of the signals detected in a. The distance between the centroid positions of the same glomerulus detected in independent biological replicates is depicted. c, Example signal captured by MERFISH for each of the ORs depicted. Each spot represents an individual mRNA molecule that was detected by MERFISH in these experiments. From the three sections analyzed, OR signal was reliably detected in a total of 11 glomeruli, which is approximately the number expected given that ~300 OR probes were used in these experiments and the fact that sectioning the entire olfactory bulb at this thickness would result in ~300 sections. Scale bar: 500 μm.
Extended Data Figure 8: An axon guidance gene expression program strongly correlates with glomerular positioning.
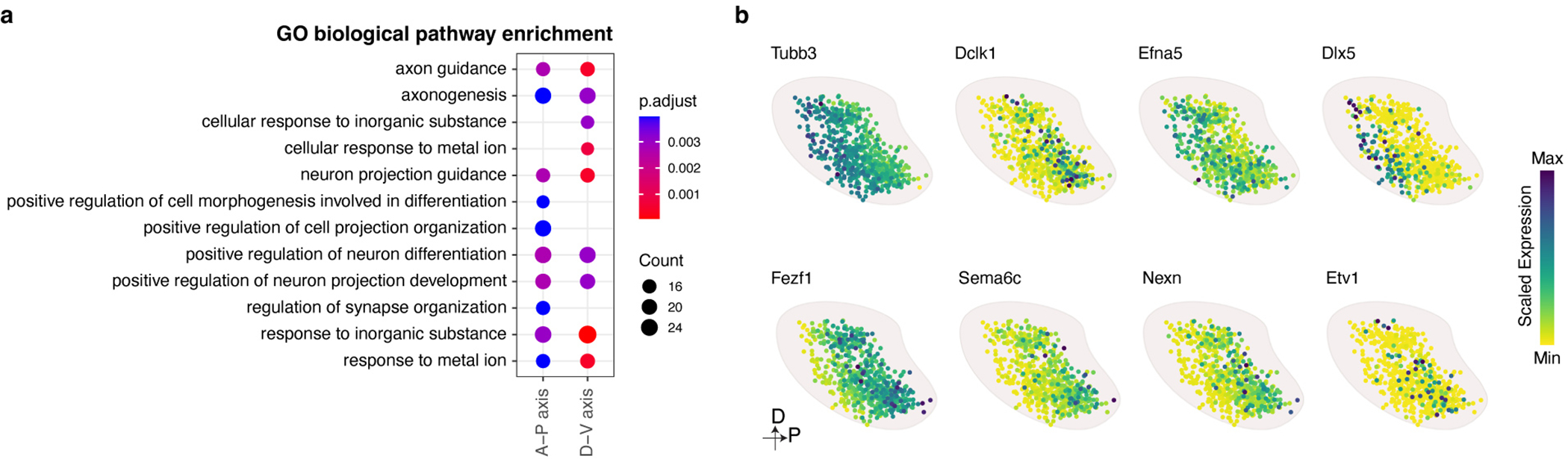
a, GO analysis of the genes that most contribute to the ability of the model to predict glomerular position. The top five GO terms in both the dorsal-ventral and anterior-posterior axes are shown. b, Scaled expression of axon guidance genes obtained from the scRNAseq data for each of the OSNs that form glomeruli in our model.
Extended Data Figure 9:
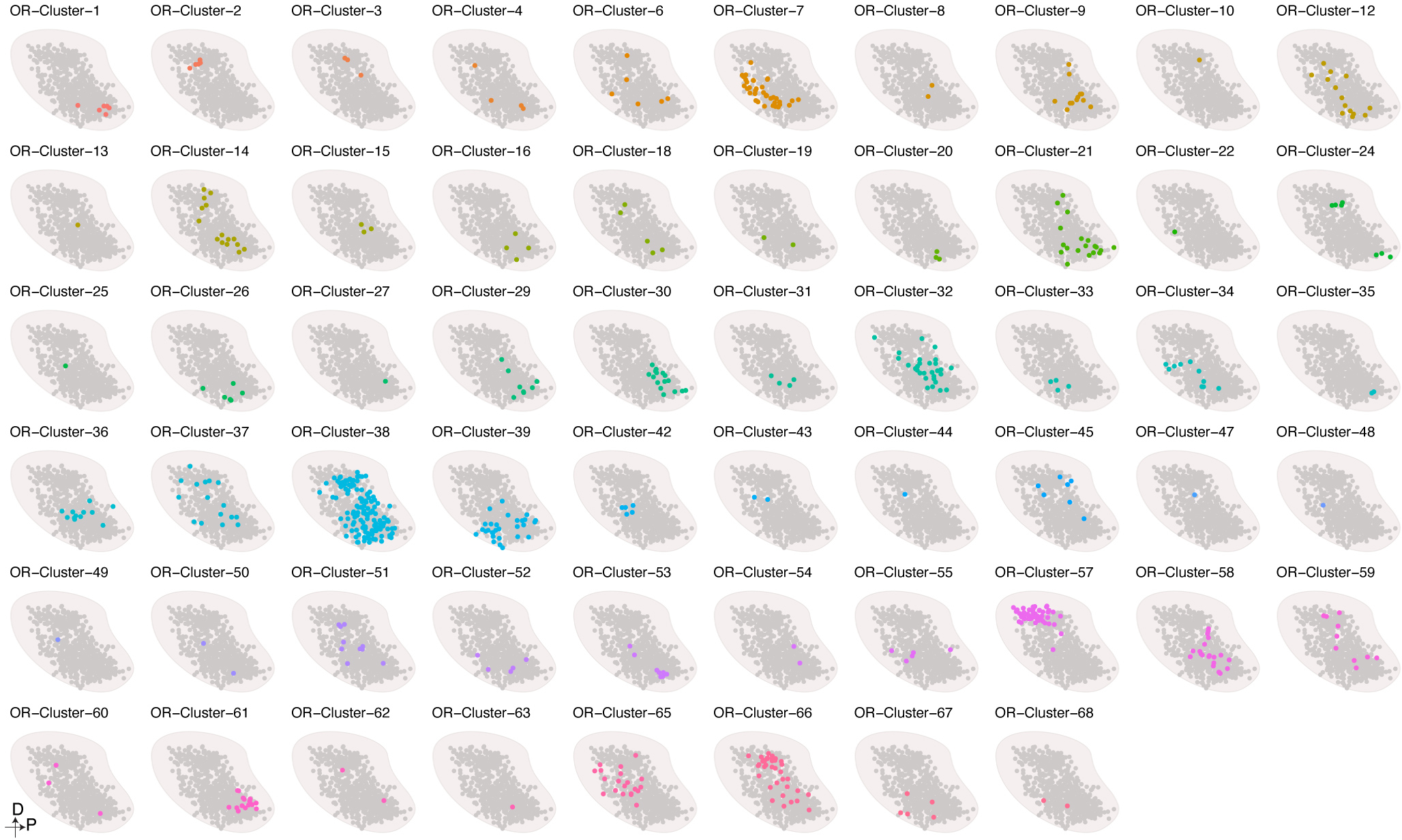
OSNs expressing ORs from the same genomic cluster form glomeruli in nearby locations within the OB.
Extended Data Figure 10: Correlation between glomerular location and odor detecting properties.
a, Only glomeruli formed by OSNs expressing class II ORs were used for this analysis (N = 601). The data are presented as the mean with +/− bootstrapped 95% CI. Data were fitted with an exponential model (blue). OR proteins share significant similarity with one another to facilitate common signaling pathways. The majority of the differences plotted here occur in the ligand binding regions of the ORs. b, The distribution of the glomeruli responding to the indicated ligands on the predicted glomerular map. The data presented in Fig.4h are excluded here. c, Dendrogram of the OSN transcriptome. The coordinate of the predict glomerular positions on the A-P and D-V axis and the chemical responses to fatty acid odorants and ketones in Figure 4h are indicated d, Volcano plot of the differentially expressed genes between OSNs that respond to the fatty acids and ketones indicated in Fig.4h, the labeled genes are genes involved in lipid metabolism, two-sided likelihood-ratio test, Bonferroni correction for multiple comparisons.
Supplementary Material
The code needed to reproduce this work and detailed results of all of the statistical analyses related to Fig.2c–f and Fig.6a
The differentially expressed genes between OSNs expressing Olfr73, Olfr160, or Olfr1507.
The list of axonal guidance genes used in the OSN transcriptome identity prediction analysis.
Acknowledgement
We thank Roger Davis, Michael Lee, Alexandra Byrne, Jay Bikoff, Judy Lieberman, Tatsuya Tsukahara, David Brann, Bob Datta and members of the Greer Lab for helpful comments on the manuscript. We thank the Bio Imaging Group and Felipe Henriques for microscopy assistance and the Flow Cytometry Core for aid in running the FACS experiments. We thank Shannon Becker for assistance in performing MERFISH. We would like to thank Bob Datta and Gilad Barnea for providing mouse lines. P.L.G. is supported by fellowships from the Smith Family Foundation, the Searle Scholars Program, the Rita Allen Foundation, the Whitehall Foundation, and by grants DP2 OD027719-01 and NIH 5 KL2 TR001455-04 from the National Institutes of Health. Z.W. and G.A. are partially supported by NIH grant HG009446. D.M.B is a Biogen Fellow of the Life Sciences Research Foundation and an Interdisciplinary Scholar of the Wu Tsai Neurosciences Institute at Stanford University. Production of Slide-seq data was supported by NIH grant RF1MH124598 to E.Z.M. and F.C. D.P.S. and C.E.B are supported by the Massachusetts Life Sciences Center (MLSC), and D.P.S. is supported by NIH/NIMH R01 MMH113743, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests
The authors report no competing interests.
Supplementary Information
Reference
- 1.Axel R The molecular logic of smell. Sci Am 273, 154–159, doi: 10.1038/scientificamerican1095-154 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Buck L & Axel R A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65, 175–187, doi: 10.1016/0092-8674(91)90418-x (1991). [DOI] [PubMed] [Google Scholar]
- 3.Chess A, Simon I, Cedar H & Axel R Allelic inactivation regulates olfactory receptor gene expression. Cell 78, 823–834, doi: 10.1016/s0092-8674(94)90562-2 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Ressler KJ, Sullivan SL & Buck LB Information coding in the olfactory system: evidence for a stereotyped and highly organized epitope map in the olfactory bulb. Cell 79, 1245–1255, doi: 10.1016/0092-8674(94)90015-9 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Serizawa S et al. Mutually exclusive expression of odorant receptor transgenes. Nat Neurosci 3, 687–693, doi: 10.1038/76641 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Vassar R et al. Topographic organization of sensory projections to the olfactory bulb. Cell 79, 981–991, doi: 10.1016/0092-8674(94)90029-9 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Zapiec B & Mombaerts P Multiplex assessment of the positions of odorant receptor-specific glomeruli in the mouse olfactory bulb by serial two-photon tomography. Proc Natl Acad Sci U S A 112, E5873–5882, doi: 10.1073/pnas.1512135112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mombaerts P et al. Visualizing an olfactory sensory map. Cell 87, 675–686, doi: 10.1016/s0092-8674(00)81387-2 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Soucy ER, Albeanu DF, Fantana AL, Murthy VN & Meister M Precision and diversity in an odor map on the olfactory bulb. Nat Neurosci 12, 210–220, doi: 10.1038/nn.2262 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Schaefer ML, Finger TE & Restrepo D Variability of position of the P2 glomerulus within a map of the mouse olfactory bulb. J Comp Neurol 436, 351–362 (2001). [PubMed] [Google Scholar]
- 11.Wachowiak M & Cohen LB Representation of odorants by receptor neuron input to the mouse olfactory bulb. Neuron 32, 723–735, doi: 10.1016/s0896-6273(01)00506-2 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Oka Y et al. Odorant receptor map in the mouse olfactory bulb: in vivo sensitivity and specificity of receptor-defined glomeruli. Neuron 52, 857–869, doi: 10.1016/j.neuron.2006.10.019 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Berkowicz DA, Trombley PQ & Shepherd GM Evidence for glutamate as the olfactory receptor cell neurotransmitter. J Neurophysiol 71, 2557–2561, doi: 10.1152/jn.1994.71.6.2557 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Malnic B, Hirono J, Sato T & Buck LB Combinatorial receptor codes for odors. Cell 96, 713–723, doi: 10.1016/s0092-8674(00)80581-4 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Belluscio L & Katz LC Symmetry, stereotypy, and topography of odorant representations in mouse olfactory bulbs. J Neurosci 21, 2113–2122 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wachowiak M, Denk W & Friedrich RW Functional organization of sensory input to the olfactory bulb glomerulus analyzed by two-photon calcium imaging. Proc Natl Acad Sci U S A 101, 9097–9102, doi: 10.1073/pnas.0400438101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma L et al. Distributed representation of chemical features and tunotopic organization of glomeruli in the mouse olfactory bulb. Proc Natl Acad Sci U S A 109, 5481–5486, doi: 10.1073/pnas.1117491109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kass MD, Rosenthal MC, Pottackal J & McGann JP Fear learning enhances neural responses to threat-predictive sensory stimuli. Science 342, 1389–1392, doi: 10.1126/science.1244916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchida N, Takahashi YK, Tanifuji M & Mori K Odor maps in the mammalian olfactory bulb: domain organization and odorant structural features. Nat Neurosci 3, 1035–1043, doi: 10.1038/79857 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Rubin BD & Katz LC Optical imaging of odorant representations in the mammalian olfactory bulb. Neuron 23, 499–511, doi: 10.1016/s0896-6273(00)80803-x (1999). [DOI] [PubMed] [Google Scholar]
- 21.Takahashi YK, Kurosaki M, Hirono S & Mori K Topographic representation of odorant molecular features in the rat olfactory bulb. J Neurophysiol 92, 2413–2427, doi: 10.1152/jn.00236.2004 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Fuss SH & Korsching SI Odorant feature detection: activity mapping of structure response relationships in the zebrafish olfactory bulb. J Neurosci 21, 8396–8407 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galizia CG & Menzel R Odour perception in honeybees: coding information in glomerular patterns. Curr Opin Neurobiol 10, 504–510, doi: 10.1016/s0959-4388(00)00109-4 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Lei H, Christensen TA & Hildebrand JG Spatial and temporal organization of ensemble representations for different odor classes in the moth antennal lobe. J Neurosci 24, 11108–11119, doi: 10.1523/JNEUROSCI.3677-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Couto A, Alenius M & Dickson BJ Molecular, anatomical, and functional organization of the Drosophila olfactory system. Curr Biol 15, 1535–1547, doi: 10.1016/j.cub.2005.07.034 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Riabinina O et al. Organization of olfactory centres in the malaria mosquito Anopheles gambiae. Nat Commun 7, 13010, doi: 10.1038/ncomms13010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feinstein P, Bozza T, Rodriguez I, Vassalli A & Mombaerts P Axon guidance of mouse olfactory sensory neurons by odorant receptors and the beta2 adrenergic receptor. Cell 117, 833–846, doi: 10.1016/j.cell.2004.05.013 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Nakashima A et al. Agonist-independent GPCR activity regulates anterior-posterior targeting of olfactory sensory neurons. Cell 154, 1314–1325, doi: 10.1016/j.cell.2013.08.033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feinstein P & Mombaerts P A contextual model for axonal sorting into glomeruli in the mouse olfactory system. Cell 117, 817–831, doi: 10.1016/j.cell.2004.05.011 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Meister M & Bonhoeffer T Tuning and topography in an odor map on the rat olfactory bulb. J Neurosci 21, 1351–1360 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson BA & Leon M Chemotopic odorant coding in a mammalian olfactory system. J Comp Neurol 503, 1–34, doi: 10.1002/cne.21396 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayakawa K et al. Innate versus learned odour processing in the mouse olfactory bulb. Nature 450, 503–508, doi: 10.1038/nature06281 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Prince JE, Brignall AC, Cutforth T, Shen K & Cloutier JF Kirrel3 is required for the coalescence of vomeronasal sensory neuron axons into glomeruli and for male-male aggression. Development 140, 2398–2408, doi: 10.1242/dev.087262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho JH, Prince JE, Cutforth T & Cloutier JF The pattern of glomerular map formation defines responsiveness to aversive odorants in mice. J Neurosci 31, 7920–7926, doi: 10.1523/JNEUROSCI.2460-10.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serizawa S et al. A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cell 127, 1057–1069, doi: 10.1016/j.cell.2006.10.031 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Imai T, Suzuki M & Sakano H Odorant receptor-derived cAMP signals direct axonal targeting. Science 314, 657–661, doi: 10.1126/science.1131794 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Cho JH, Lepine M, Andrews W, Parnavelas J & Cloutier JF Requirement for Slit-1 and Robo-2 in zonal segregation of olfactory sensory neuron axons in the main olfactory bulb. J Neurosci 27, 9094–9104, doi: 10.1523/JNEUROSCI.2217-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeuchi H et al. Sequential arrival and graded secretion of Sema3F by olfactory neuron axons specify map topography at the bulb. Cell 141, 1056–1067, doi: 10.1016/j.cell.2010.04.041 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Sosulski DL, Bloom ML, Cutforth T, Axel R & Datta SR Distinct representations of olfactory information in different cortical centres. Nature 472, 213–216, doi: 10.1038/nature09868 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shykind BM et al. Gene switching and the stability of odorant receptor gene choice. Cell 117, 801–815, doi: 10.1016/j.cell.2004.05.015 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Graziadei GA, Stanley RS & Graziadei PP The olfactory marker protein in the olfactory system of the mouse during development. Neuroscience 5, 1239–1252, doi: 10.1016/0306-4522(80)90197-9 (1980). [DOI] [PubMed] [Google Scholar]
- 42.Verhaagen J, Oestreicher AB, Gispen WH & Margolis FL The expression of the growth associated protein B50/GAP43 in the olfactory system of neonatal and adult rats. J Neurosci 9, 683–691 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sautter A, Zong X, Hofmann F & Biel M An isoform of the rod photoreceptor cyclic nucleotide-gated channel beta subunit expressed in olfactory neurons. Proc Natl Acad Sci U S A 95, 4696–4701, doi: 10.1073/pnas.95.8.4696 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuss SH, Omura M & Mombaerts P Local and cis effects of the H element on expression of odorant receptor genes in mouse. Cell 130, 373–384, doi: 10.1016/j.cell.2007.06.023 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Lyons DB et al. An epigenetic trap stabilizes singular olfactory receptor expression. Cell 154, 325–336, doi: 10.1016/j.cell.2013.06.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan M, Vaes E & Mombaerts P Regulation of the probability of mouse odorant receptor gene choice. Cell 147, 907–921, doi: 10.1016/j.cell.2011.09.049 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Leland McInnes JH, Melville James. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv (2018). [Google Scholar]
- 48.Cutforth T et al. Axonal ephrin-As and odorant receptors: coordinate determination of the olfactory sensory map. Cell 114, 311–322, doi: 10.1016/s0092-8674(03)00568-3 (2003). [DOI] [PubMed] [Google Scholar]
- 49.McLaughlin CN et al. Single-cell transcriptomes of developing and adult olfactory receptor neurons in Drosophila. Elife 10, doi: 10.7554/eLife.63856 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godfrey PA, Malnic B & Buck LB The mouse olfactory receptor gene family. Proc Natl Acad Sci U S A 101, 2156–2161, doi: 10.1073/pnas.0308051100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strotmann J et al. Small subfamily of olfactory receptor genes: structural features, expression pattern and genomic organization. Gene 236, 281–291, doi: 10.1016/s0378-1119(99)00275-9 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Tsuboi A et al. Olfactory neurons expressing closely linked and homologous odorant receptor genes tend to project their axons to neighboring glomeruli on the olfactory bulb. J Neurosci 19, 8409–8418 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stickels RR et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol, doi: 10.1038/s41587-020-0739-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi H, Yoshihara S, Nishizumi H & Tsuboi A Neuropilin-2 is required for the proper targeting of ventral glomeruli in the mouse olfactory bulb. Mol Cell Neurosci 44, 233–245, doi: 10.1016/j.mcn.2010.03.010 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Chae H et al. Mosaic representations of odors in the input and output layers of the mouse olfactory bulb. Nat Neurosci 22, 1306–1317, doi: 10.1038/s41593-019-0442-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bozza T et al. Mapping of class I and class II odorant receptors to glomerular domains by two distinct types of olfactory sensory neurons in the mouse. Neuron 61, 220–233, doi: 10.1016/j.neuron.2008.11.010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imai T et al. Pre-target axon sorting establishes the neural map topography. Science 325, 585–590, doi: 10.1126/science.1173596 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090, doi: 10.1126/science.aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwarting GA et al. Semaphorin 3A is required for guidance of olfactory axons in mice. J Neurosci 20, 7691–7697 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.James G, Key B & Beverdam A The E3 ubiquitin ligase Mycbp2 genetically interacts with Robo2 to modulate axon guidance in the mouse olfactory system. Brain Struct Funct 219, 861–874, doi: 10.1007/s00429-013-0540-8 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Igarashi KM & Mori K Spatial representation of hydrocarbon odorants in the ventrolateral zones of the rat olfactory bulb. J Neurophysiol 93, 1007–1019, doi: 10.1152/jn.00873.2004 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Nozawa M, Kawahara Y & Nei M Genomic drift and copy number variation of sensory receptor genes in humans. Proc Natl Acad Sci U S A 104, 20421–20426, doi: 10.1073/pnas.0709956104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yablonka A, Sobel N & Haddad R Odorant similarity in the mouse olfactory bulb. Proc Natl Acad Sci U S A 109, E2916–2917; E2918–2919, doi: 10.1073/pnas.1211623109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Yukselen O, Turkyilmaz O, Ozturk AR, Garber M & Kucukural A DolphinNext: a distributed data processing platform for high throughput genomics. BMC Genomics 21, 310, doi: 10.1186/s12864-020-6714-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323, doi: 10.1186/1471-2105-12-323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287, doi: 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlson M org.Mm.eg.db: Genome wide annotation for Mouse. (2019).
- 7.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, doi: 10.1038/75556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolock SL, Lopez R & Klein AM Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291 e289, doi: 10.1016/j.cels.2018.11.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296, doi: 10.1038/s41592-019-0619-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedregosa F, G. V., Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, and Duchesnay E Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830 (2011). [Google Scholar]
- 12.Xiao N, Cao DS, Zhu MF & Xu QS protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 31, 1857–1859, doi: 10.1093/bioinformatics/btv042 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Monahan K et al. Cooperative interactions enable singular olfactory receptor expression in mouse olfactory neurons. Elife 6, doi: 10.7554/eLife.28620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090, doi: 10.1126/science.aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crasto C, Marenco L, Miller P & Shepherd G Olfactory Receptor Database: a metadata-driven automated population from sources of gene and protein sequences. Nucleic Acids Res 30, 354–360, doi: 10.1093/nar/30.1.354 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renier N et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910, doi: 10.1016/j.cell.2014.10.010 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Saraiva LR et al. Hierarchical deconstruction of mouse olfactory sensory neurons: from whole mucosa to single-cell RNA-seq. Sci Rep 5, 18178, doi: 10.1038/srep18178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivatsan SR et al. Embryo-scale, single-cell spatial transcriptomics. Science 373, 111–117, doi: 10.1126/science.abb9536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stickels RR et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol, doi: 10.1038/s41587-020-0739-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hara T et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779–792 e711, doi: 10.1016/j.ccell.2021.05.002 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The code needed to reproduce this work and detailed results of all of the statistical analyses related to Fig.2c–f and Fig.6a
The differentially expressed genes between OSNs expressing Olfr73, Olfr160, or Olfr1507.
The list of axonal guidance genes used in the OSN transcriptome identity prediction analysis.
Data Availability Statement
All RNA sequencing data described in this manuscript are deposited at GEO accession GSE169021. The positions of the reconstructed glomerular map can be found at:
https://github.com/Greerlab/OSN_2021_paper.
The processed MERFISH data can be found at:
https://zenodo.org/record/5745947#.YaeQ-PHMIqs
Fig.1 and Extended Data Fig.1 are associated with GSE169010; Fig.2–4, Extended Data Fig.2–5,8,10 are associated with GSE169011; Fig.3 and Extended Data Fig.6 are associated with GSE169012.