

Abstract

Polyelectrolyte complex micelles (PCMs) are a unique class of self-assembled nanoparticles that form with a core of associated polycations and polyanions, microphase-separated from neutral, hydrophilic coronas in aqueous solution. The hydrated nature and structural and chemical versatility make PCMs an attractive system for delivery and for fundamental polymer physics research. By leveraging block copolymer design with controlled self-assembly, fundamental structure–property relationships can be established to tune the size, morphology, and stability of PCMs precisely in pursuit of tailored nanocarriers, ultimately offering storage, protection, transport, and delivery of active ingredients. This perspective highlights recent advances in predictive PCM design, focusing on (i) structure–property relationships to target specific nanoscale dimensions and shapes and (ii) characterization of PCM dynamics primarily using time-resolved scattering techniques. We present several vignettes from these two emerging areas of PCM research and discuss key opportunities for PCM design to advance precision medicine.
1. Introduction
Controlled self-assembly and compartmentalization on the 1–1000 nm length scale in solution have been longstanding goals in nanotechnology, a field that is beginning to address emerging challenges in energy management,1 green catalysis,2 surfactant compatibilizers,3 and human health.4 Polymeric micelles, which undergo microphase separation, have provided a rich array of hierarchical nanoaggregates that have been widely recognized as leading candidates to address these issues. These nanoparticles allow cargo to be packaged into discrete domains that can withstand inhospitable environments and transport molecules across otherwise impermeable barriers. Micelle assembly is commonly driven either by amphiphilic polymer association in selective solvents or by charged polymer interaction in aqueous solution. Significant advances have been made in our fundamental understanding of amphiphilic materials, through foundational works in simulation and modeling,5−7 scaling theories,8−10 self-consistent mean field theory,11−13 and experiments.14−16 In general, by exploiting the synthetic versatility of block copolymers to tune precisely the energetic components of the (i) chain stretching in the core, (ii) excluded volume of the corona, and (iii) interfacial energy of the micelle in solvent, the micellar size, shape, aggregation number, and chain exchange dynamics can be programmed with high specificity and fidelity for intended applications.
Beyond hydrophobic effects in polymers, other driving forces in noncovalent association have emerged to tailor self-assembly further and expand the selection of sophisticated nanostructures. Complex coacervation has emerged as a promising avenue toward self-assembled materials, garnering interest across interdisciplinary fields including the polymer physics, interface and colloid science, and biology communities.17 Oppositely charged polyelectrolytes predominately assemble due to the entropy gain from counterion release,18 resulting in phase-separated polyelectrolyte complex assemblies that exhibit an array of fundamentally unique static and dynamic properties. Polyelectrolyte complex materials can be engineered into polyelectrolyte complex micelles (PCMs) or nanoparticles with a complex core interior and a hydrophilic corona exterior. As shown in Figure 1, PCMs typically employ the coassembly of oppositely charged polymers where at least one polymer in the system has block architecture. In comparison to amphiphilic block copolymer micelles, PCMs are far less quantitatively understood at a molecular level,19−21 as several underlying features complicate the thermodynamic framework of PCMs. For example, the ionic core consists of two distinct polyelectrolytes that, under stoichiometrically equivalent charge matched conditions, form intrinsic ion pairs that act as physical cross-links between polycation and polyanion repeat units. These pairings can be disrupted via the addition of salt or by heating, making PCMs highly responsive to changes in the local environment. The low interfacial tension and water solubility of polyelectrolyte chains in complex coacervates means water is present throughout both the core and corona, further complicating efforts to understand the fundamental physics of these nanoparticles, owing to their intrinsically multicomponent nature.22
Figure 1.

Building blocks and microphase separation process of polyelectrolyte complex micelles (PCMs). For nomenclature, A represents a neutral, hydrophilic block, while B/C represents oppositely charged polyelectrolyte blocks. Typical PCMs consist of an AB diblock polycation and either an AC diblock polyanion or a C homopolyanion.
In this perspective, we discuss a collection of recent research articles that shed new light on design strategies for dilute solutions of PCMs using integrated measurement, analysis, and prediction from experimental and computational tools. Special attention is given to the development of (i) scaling relationships governing size, shape, and morphological transitions of PCMs, and (ii) micellization dynamics in PCM formation/growth, chain exchange, and disassembly pathways. We also provide direct examples of extending polyelectrolyte structure–property principles to impart favorable physiochemical attributes for delivery applications and discuss future directions. Unless specifically mentioned, the studies reviewed here use fully ionized strong polyelectrolytes at stoichiometric charge ratios. Recent reviews on PCM use in gene therapy23−25 and molecular interactions in polyelectrolyte complexation17,20,26,27 can provide further insight into the broad state of this field. These selected works provide blueprints for advancing our fundamental understanding of this important class of self-assembled materials.
2. PCM Structure–Property Relationships
Controlling the size and structure of PCMs is vital to their success as therapeutic delivery vehicles. Nanoparticles that are smaller than ∼10 nm may be removed from the bloodstream by the kidneys, whereas nanoparticles above 200 nm are prone to nonspecific accumulation in the spleen and liver.28 In addition to avoiding renal clearance, nanoparticle design can have profound impacts on nanoparticle biodistribution and cellular uptake. Recently, Ridolfo et al. explored morphology effects in biological settings by comparing the cellular uptake of amphiphilic spherical, worm-like, vesicular, and tubular nanoparticles.29 They found that higher aspect ratio particles such as worms and tubes performed better than spheres and vesicles because higher aspect ratio nanoparticles diffused faster relative to low aspect ratio nanoparticles. These observations should apply to amphiphilic micelles and PCMs equally well, as these observations do not rely on the assembly mechanism. For these reasons, precise control of PCM size and morphology is a key component to developing efficient PCM encapsulants. This section covers structure–property relationships of PCMs with a summary in Table 1.
Table 1. Molecular Architecture Controls Polyelectrolyte Complex Micelle (PCM) Propertiesa.
| property | parameter | notes | references |
|---|---|---|---|
| core radius | neutral block length (NA) | inverse correlation | (36, 59) |
| core radius | charged block length (NB) | direct correlation | (36, 37, 56, 59, 77−79) |
| core radius | homopolymer length (NC) | independentb | (30, 37, 42, 56, 59) |
| corona thickness | neutral block length (NA) | direct correlation | (36, 59−61) |
| aggregation number | neutral block length (NA) | inverse correlation | (30, 59−61) |
| aggregation number | charged block length (NB) | direct correlation | (36, 59, 64) |
| aggregation number | strength of charge | direct correlation | (50) |
| polydispersity | strength of charge | direct correlation | (37, 50) |
| morphology | N/C > 1 | spheroids | (30−32) |
| morphology | N/C < 1 | aggregates, vesicles,c or lamellaec | (30, 33−35, 40, 41) |
| morphology | polyelectrolyte architecture | vesicles, cylinders, and more | (38, 39) |
| morphology | DNA hybridization | ssDNA = spheres | (37, 56) |
| dsDNA = worm-like cylinders | |||
| stability | polymer length | direct correlation | (66) |
| stability | charge density | direct correlation | (67, 68) |
| stability | strength of charge | direct correlation | (50) |
| stability | cross-linking | direct correlation | (73−76) |
The following physical trends are from both experimental and theoretical publications on PCMs.
Under lengths of ∼5000.
Specific cases.
Morphology of PCMs
The length of each polymer block (A, B, or C in Figure 1) can dictate PCM morphology. Roughly, if the length of the neutral block is larger than that of the charged block, i.e., the neutral/charged length ratio (N/C) > 1, self-assembly results in spheroidal micelles for both (AB + AC) and (AB + C) systems.30−32 When N/C < 1 other morphologies can be formed. For most (AB + C) systems, these assemblies are aggregates and complexes similar to bulk assemblies, as the small fraction of neutral polymer does not force microphase separation.30 However, for (AB + AC) systems, interesting morphologies can be formed in the N/C < 1 regime. For example, the Kataoka group observed polyelectrolyte complex vesicles33−35 when N/C ∼ 0.5 compared to spheres for the same system with N/C ∼ 2. Cylindrical and planar assemblies have also been observed when N/C < 1 with a very low poly(ethylene oxide) (PEO, also referred to as poly(ethylene glycol) or PEG) weight fraction.35 PCM theory predicts that for N/C ≪ 1 morphology scales with degree of ionization (f).36 As f increases, morphology changes to lamellae, cylinders, crew-cut spherical micelles (corona thickness ≪ core size), and finally star-like spherical micelles (corona thickness ≫ core size). At very low f, where the free energy gain of complexation is on the order of thermal energy, micellization does not occur and a solution of unimers occurs. Likewise, when charged blocks are very short, experimental results show minimal complexation.30,37 Other factors that can also influence micelle morphology are nonlinear polymer architectures,38,39 nonstoichiometric charge ratios,40,41 salt concentration,31,42 chirality,43 or stimuli-responsive polymers.44,45 The idea of block length ratio evokes analogies to classic packing parameter arguments in hydrophobically driven systems,46,47 suggesting that commonalities exist despite the drastically different driving forces of self-assembly.
Morphological trends based on N/C are clear from the studies discussed above, but experimentally studying systems exactly at the transition (N/C = 1) is quite difficult, due to imprecise polymer synthesis. Recent simulations from the Sing group48 look at a (AB + C) system with exactly matched block lengths in the block copolymer and find that this length ratio is not the sole driving parameter between macro- and microphase separation. For a N/C = 1 system, they predict that at shorter polymer lengths macrophase separation occurs but as length increases past a critical point, microphase separation (micelles) is expected in low salt conditions. This is hard to replicate experimentally, but a reactive polymer system such as poly(allyl glycidyl ether) (PAGE)49,50 is a strong candidate to do so, as reactive polymers are powerful tools for achieving architecturally identical neutral, cationic, or anionic polymers.
While the macromolecules considered here are commonly synthetic polymers, biomolecules drive the design and motivation for hydrophilic PCMs and can add additional layers of complexity, for example, polypeptide chirality controlling the phase of bulk polyelectrolyte complexes.43 There is great interest in PCMs incorporating nucleic acids (often termed “polyplexes”) for therapeutic delivery of cargo like plasmid DNA or small interfering RNA (siRNA).51 Nucleic acids are a densely negatively charged biopolymer, with a phosphate on the backbone between each nucleotide, so they can easily replace a charged block in the general systems scheme described above. Single-stranded nucleic acids behave much like flexible hydrophilic polymers, but double-stranded nucleic acids are substantially more rigid (∼50x longer persistence length) and have a much higher charge density, due to the presence of the complementary strand and formation of a double helix.52,53 The conformational differences between single- and double-stranded nucleic acids drive a morphological shift within PCMs. DNA hybridization in a bulk system of DNA + poly-l-lysine (pLys) forces a phase change between liquid-like coacervates for single-stranded DNA (ssDNA) and solid precipitates for double-stranded DNA (dsDNA),54 driven by the changes in charge density54 and rigidity.55 When single-stranded DNA37,56 or RNA57 is complexed with block copolymers, spheroidal micelles are formed with various charged polymers. However, the double-stranded variant can disrupt micellization as seen with RNA57 or force a shape change to worm-like cylinders37,56,58 with DNA. When N/C ≫ 1, dsDNA micelles are worm-like cylinders formed with DNA lengths ranging from 10 base-pairs (bp) to 1000s of bp.37,56,58 When N/C is ∼1, however, globular micelles can still be formed with dsDNA, as the PEO corona is not crowded enough to force long cylinder formation.58 The distinction between single-stranded and double-stranded nucleic acids is extremely important for therapeutics delivery, as hybridization can drive therapeutic function.
Structural Properties of Spheroidal PCMs
The length of each polymer block in a spheroidal micelle can influence its structural properties including size, aggregation number, and stability. In (AB + C) systems, the PCM core radius (Rcore) is directly proportional to the length of the charged block in the block copolymer (NB),37,56,59 while largely independent of the length of the homopolymer (NC), at least below a large critical length around N ∼ 5000.30,42,59 The size of the neutral block (NA), which forms the corona, has shown to have a minor effect on the size of the core, but noticeably drives the thickness of the corona (H) and therefore the hydrodynamic radius (Rh), or overall size of the micelle, which is a crucial parameter for controlling biodistribution.60,61 Conversely, the aggregation number (P), or number of chains in a given micelle, is shown to decrease as the neutral block size increases for a (AB + C) system.30,59−61 To quantify these physical trends, our group developed experimental scaling laws for Rcore, Rh, H, and P using PCMs containing PEO-block-pLys (PEO-b-pLys) paired with ssDNA or pGlu.59 These scaling laws are shown as black lines in Figure 2 overlaid with accumulated published (AB + C) PCM data representing a variety of synthetic and biological polymers. The data were normalized using the scaling laws for two polymer lengths and plotted against the third length variable. This normalization collapsed all the data to a single trend and is compared to the corresponding scaling law in this figure. NC was found to have no noticeable effect on any physical parameter, which is convenient for creating versatile delivery systems where the C component is often a therapeutic drug or biomolecule. Polyelectrolyte length is generally reported as degree of polymerization, as it is considered here for physical scaling, but contour length or physical size are likely a slightly more accurate factor.
Figure 2.

Aggregated data from published (AB + C) polyelectrolyte complex micelle (PCM) experimental studies using strong polyelectrolytes at stoichiometric charge ratios, overlaid with experimental scaling laws shown as black lines. The data were normalized using scaling laws for two block lengths and plotted against the third block length, collapsing to show scaling for the block length of interest. The available literature provides aggregated data for core size (A–C), hydrodynamic size (D–E), and aggregation number (F). The data represents PCMs from six publications30,37,42,56,59,60 using numerous synthetic and biological polymers and the scaling laws are experimental,59 consistent with theoretical predictions36 for PCMs between the star-like and crew-cut regimes. Adapted from Marras et al.59 Copyright 2021 American Chemical Society.
Theoretical work on PCMs is more precise and predicts similar physical property scaling relationships for two geometrical extremes of PCM structure.36,62,63 For PCMs with fully ionized chains in a “good” aqueous solvent, predictions for Rcore, P, and H at the star-like limit, where (Rcore ≪ H), are shown in eqs 1–3.
| 1 |
| 2 |
| 3 |
Scaling theory for the same conditions, but at the crew-cut limit where (Rcore ≫ H), are shown in eqs 4–6.
| 4 |
| 5 |
| 6 |
The PCMs in Figure 2 consist of fully ionized chains in good solvents and are between the star-like and crew-cut regimes, which is the case for the majority of experimental work. Considering the intermediate regime of these PCMs, the experimental scaling laws59 shown in Figure 2 are consistent with predictions for the two structural limits. Further theoretical predications show dependence on solvent quality, salt concentration, and degree of ionization,36,62−65 but are out of the scope of this structural review. Understanding how PCM structural properties are controlled by polymer structure can accelerate the design process for tailored carriers.
Stability of PCMs
One aspect of PCM stability is measured by its resistance to nanoparticle degradation in the face of increasing ionic strength in solution. Adding excess counterions from salt competes with ion pairing between polymers to disrupt complexation. In bulk systems (B + C), increasing the length of either charged polymer increases the stability,54 which can be easily tested with optical microscopy. This is more difficult to study for nanoparticles that are smaller than the diffraction-limited resolution of optical microscopes, but light scattering and small-angle scattering techniques have been used to show a similar effect for PCMs.42,66 Likewise, increasing charge density increases complex stability67 and can drive micellization in PEC systems with charged biomolecules.68
PCMs are often discussed as two charged, flexible chains coming together in an entropically favorable process; however, the molecular details of each charged group also play a role in the structure and stability of complex formation.69 The Choi group used a functionalizable PEO-b-PAGE for a direct comparison of PCMs comprising charged ammonium (pKa = 11), guanidinium (pKa = 14), carboxylate (pKa = 4), and sulfonate (pKa = 1) groups using thiol–ene click chemistry to attach each of the desired side groups onto otherwise identical polymers. The neutron scattering results revealed an increase in core radius and aggregation number as ion pairing interactions become stronger.50 Our group compared two cationic charged monomers in comparable polymer structures: lysine (primary amine, pKa = 10) and vinylbenzyltrimethylammonium (VBTMA, consisting of a permanently charged ammonium) for complexing DNA at various lengths.37 Despite the permanent charge and additional hydrophobicity imparted by the aromatic moiety, which have been previously shown to strengthen certain PEC systems,70,71 PVBTMA complexed less strongly with DNA, attributed to the steric hindrance in ion pairing.72 Cross-linking cationic polymers using glutaraldehyde,73 disulfide bonds,74,75 or other means76 improves stability after micelle formation and can be reversible. These examples demonstrate that both synthetic polymers and polypeptides are suitable for forming robust PCMs and that the molecular details must be considered in the design process, as they play a vital role in complexation properties and ultimately, functionality.
3. Dynamics of Micellization and Chain Exchange
For nanocarrier applications, understanding the driving forces of micellization, molecular exchange, and evolution is critical for controlling the exposure of the cargo.20,80,81 A more complete understanding of the PCM equilibration process can enable greater control of the physical self-assembly process, nanocarrier stability over time, and encapsulation/release kinetics. In this section, we focus discussions on several recent developments in PCM dynamics using primarily scattering methods. Small-angle scattering is a powerful tool for gathering multiple orders of magnitude of size information simultaneously for an entire solution and in a precise time-resolved manner. Detailed protocols of small-angle X-ray scattering (SAXS) have been recently outlined by our group to assist in the experimental planning and analysis of the SAXS data.82,83Figure 3A shows the model systems used for these studies. Polyelectrolytes include PEO-b-PVBTMA, sodium poly(acrylate) (PAA), poly(ethylene oxide)-block-poly(sodium 4-styrenesulfonate) (PEO-b-PSS), and PSS. We have previously provided experimental details of the controlled synthesis of these polyelectrolytes in water,84,85 so that precise lengths of neutral and charged blocks can be prepared with low dispersity in the molar mass distribution. Depending on the block lengths and pairing of the PEO-b-PVBTMA polycation with PAA, PEO-b-PSS, or PSS polyanion, the assemblies that form can resemble spherical, core/shell PCMs, or polydisperse colloidal aggregates. Both classes of nanostructures will be discussed in detail below, along with open-ended questions that these results raise for the physical chemistry community.
Figure 3.
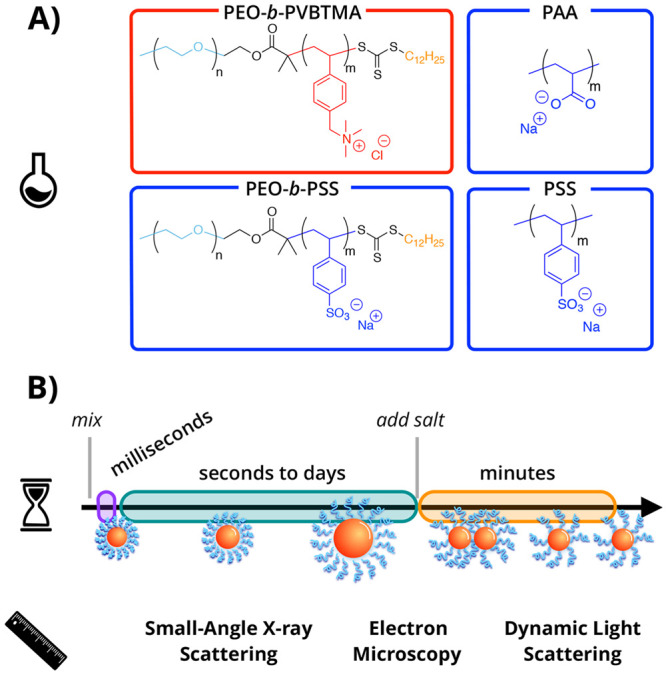
Dynamics of polyelectrolyte complex micelles (PCMs). (A) Chemical structures of poly(ethylene oxide)-block-poly(vinyl benzyl trimethylammonium chloride) (PEO-b-PVBTMA, boxed in red), sodium poly(acrylate) (PAA, boxed in blue), poly(ethylene oxide)-block-poly(sodium 4-styrenesulfonate) (PEO-b-PSS, boxed in blue), and poly(sodium 4-styrenesulfonate) (PSS, boxed in blue). (B) Illustration of the relevant time and length scales investigated in PCM formation (purple), chain exchange (green), and disassembly (orange), ranging from milliseconds to minutes using small-angle X-ray scattering, cryogenic electron microscopy, and dynamic light scattering.
PCM Formation Kinetics
Following established stopped-flow protocols from amphiphilic block copolymer literature, the ultrafast formation of PCMs can be monitored in situ using time-resolved small-angle X-ray scattering (TR-SAXS) with millisecond temporal resolution (Figure 3B). Here, solutions of oppositely charged polymers are loaded into separate syringes, pumped into a turbulent mixer, and dispensed into a capillary cell without further flow for scattering measurements. This technique has provided new physical insights into ionic nanomaterial behavior, including complex coacervate coalescence of poly(allylamine hydrochloride) and PAA as a function of added NaCl salt.86 However, it was only very recently that the initial complexation of block polyelectrolytes has been reported. The chemical and electrostatic nature of the polyelectrolyte pairing appears to greatly influence the kinetic pathway of micellization, demonstrating the importance of mindful polymer selection in constructing PCM nanocarriers. Two independent cases that illustrate completely different pathways are shown in Figure 4.
Figure 4.

Time-resolved small-angle X-ray scattering (TR-SAXS) reveals distinct formation pathways of polyelectrolyte complex micelles (PCMs). (A) For PEO-b-PVBTMA/PAA systems, within 100 ms well-defined spherical micelles incrementally grow into larger micellar entities, as denoted by the black arrow. Adapted from Wu et al.87 Copyright 2020 American Chemical Society. (B) For PEO-b-PVBTMA/PSS systems, within 3 ms aggregates break apart into smaller micellar entities, as denoted by the black arrow. Adapted from Amann et al.89 Copyright 2019 American Chemical Society.
In the first case, Wu and co-workers investigated the spatiotemporal formation kinetics of PEO-b-PVBTMA with PAA.87 Using a stopped-flow apparatus with high-throughput data collection capabilities at the Stanford Synchrotron Radiation Lighthouse (SSRL, SLAC National Accelerator Laboratory),88 they directly observed the assembly kinetics and SAXS profiles of PEO-b-PVBTMA/PAA PCMs via TR-SAXS from 100 ms to 5 s, which exhibited spherical particles (∼q0 power law dependence of intensity for q < 0.01 Å–1) that grew in size over time (Figure 4A). The structural evolution of PCMs was evaluated by determining the apparent Guinier radius of gyration (Rg), which showed incremental micelle growth from Rg ∼ 10 to Rg ∼ 12 nm over 5 s by gradual insertion of either unimer chains or ion-paired clusters. For the second case, Amann and co-workers examined PCMs comprising PEO-b-PVBTMA and PSS.89 Using a SFM-400 stopped-flow apparatus at the European Synchrotron Radiation Facility (ESRF), the researchers reported the TR-SAXS formation of metastable aggregates for internal charge neutralization at 3 ms, preceding rearrangement and pinch-off into small micellar particles over the course of 30 s (Figure 4B). The equilibration data was described by these relaxation processes as a function of the degree of polymerization (N) of PVBTMA from N = 6 to N = 19, where rearrangement of unimer chains or ion-paired clusters becomes increasingly unfavorable as block length increased.
In attempts to account for the differences in kinetic pathways, we have shown that the homopolymers PVBTMA + PAA form liquid-like coacervates, whereas PVBTMA + PSS form solid-like complexes.85 This observation leads us to speculate that the formation kinetics may be strongly dependent on the chemical nature of the polymer constituents, though further work needs to be done to test this hypothesis of whether the complex cores resemble the nature of macroscopic complexes. In addition, block length of the block polyelectrolytes may also affect the rate at which PCMs either grow incrementally or break apart from larger colloidal structures upon complexation. In a previous report, Wu et al. showed that PEO-b-PVBTMA and PEO-b-PSS at N ≈ 50 for the charged blocks form nonequilibrium complexes, far from well-defined spheres by fitting the SAXS data.90 Investigating this system that matches the neutral and charged blocks lengths to PEO-b-PVBTMA/PAA as a direct comparison to the results of Amann et al. is the subject of ongoing work. Altogether, these examples illustrate only two of many possible formation pathways that lead to charge-driven micellization. Expanding TR-SAXS to study more PCM systems and varying parameters like polyelectrolyte selection, block lengths, and molecular architecture can help move PCM design toward more efficacious and predictive encapsulation of cargo.
PCM Chain Exchange
Understanding chain exchange in dilute micelle solutions is crucial toward their development as efficient delivery vehicles, as the rate and method by which sequestered therapeutic molecules are exposed to the surrounding environment will control their efficacy. In general, chain exchange between equilibrium polymeric micelles proceeds via two primary mechanisms: chain expulsion/insertion and micelle fusion/fission.91−93 While chain exchange processes have been probed with amphiphilic polymer assemblies, to the extent of our knowledge very few experimental studies have examined chain exchange for PCMs. Fluorescent imaging is one accessible approach to potentially examine the underlying exchange mechanism of PCMs. Nolles and co-workers have utilized fluorescently labeled proteins in PCMs to probe formation kinetics and exchange dynamics.94 Synchrotron scattering is another potential strategy. We have previously shown how interparticle effects emerge in the form of a structure factor in SAXS profiles for concentrated micelle solutions and thereby maintain PCM stability over time,95 but the molecular details of unimer chain exchange or fusion/fission cannot be unveiled with these experiments, as they lack molecular contrast. Unfortunately, scattering methods with appropriate contrast such as SANS require long time scales and deuterated/hydrogenated systems, limiting their applicability and accessibility. However, advances in molecular dynamic simulations have offered new insights to potential mechanisms of exchange in which electrostatics interplay with other competing noncovalent interactions.
Bos et al. performed coarse-grain dynamics simulations on a model diblock-homopolymer PCM system and outlined physical characteristics influencing the mode of chain exchange and PCM stability.96 In these Langevin dynamics simulations, nonelectrostatic interactions (i.e., hydrogen bonding or hydrophobicity) tended to disfavor the chain expulsion mechanism, as these additional interactions generate an enthalpic cost which counteracts the entropic gain that drives small neutral complex cluster expulsion from PCMs (Figure 5A). Interestingly, these effects depended on the nonelectrostatic interactions (represented by the Lennard-Jones potential εLJ) being intermolecular and changed when the interactions were modified to be solely intramolecular. In the case where one polyelectrolyte displayed significant nonelectrostatic interactions with itself but not the oppositely charged polyelectrolyte, chain expulsion and fusion/fission events increased, suggesting that the PCMs became less stable. Meanwhile, macromolecular design parameters such as block length have divergent effects on the two mechanisms as εLJ was increased, shown in Figure 5B. Chain expulsion was sensitive to the relative length of the charged blocks in the system, displaying a distinct increase for matching lengths relative to unmatched chain lengths. Fusion/fission, on the other hand, appeared insensitive to the ratio of the block lengths, but instead depended on the total length of the homopolymer. Taken together, simulation and experiment provide a strong case for the careful consideration of nonelectrostatic interactions between polyelectrolytes and polyelectrolyte length.
Figure 5.

Chain exchange of polyelectrolyte complex micelles (PCMs) upon formation as a function of electrostatic interactions, nonelectrostatic interactions, and polyelectrolyte length using Langevin dynamics simulations. (A) Histograms of the PCM size distribution varying nonelectrostatic attraction strength between polyelectrolytes at εLJ = 0.05kBT (blue), εLJ = 0.15kBT (red), and εLJ = 0.25kBT (gray); insets show snapshots of the simulated PCMs with Nnegative = Npositive = 20 and Nnetural = 50. (B) Comparison of the number of chain expulsion/insertion and micelle fission/fusion events for PCMs as a function of polyelectrolyte length ratio (Nnegative/Npositive) at increasing nonelectrostatic attraction strengths. Adapted from Bos et al.96 Copyright 2019 American Chemical Society.
Bos, Timmerman, and Sprakel more recently demonstrated the exchange dynamics of PCMs using Förster resonance energy transfer (FRET).80 In this work, poly(ethylene oxide)-b-poly(trimethylammonioethyl methacrylate chloride) (PEO-b-PTMAEMA) and fluorescently labeled poly(3-sulfopropyl methacrylate) (PSPMA) were used as the model system. An analytical model relating the FRET efficiency between fluorophores and the exchange rates of polyelectrolyte chains was developed, in which the broad distribution of detected exchange rate was correlated to changes in salt concentration, polymer length, and micelle concentration. To the extent of our knowledge, this is the first experimental demonstration that PCM equilibration occurs predominately through expulsion/insertion pathways instead of fusion/fission.
For PCMs containing nucleic acids, an important practical consequence of dynamic chain exchange between micellar assemblies is the potential displacement of nucleic acids over time upon immersion in different biological settings. Because of the complicated delivery pathways involved with overcoming various biological barriers, molecular engineering approaches to boost stability in nanocarriers are nontrivial. However, simplified fundamental experiments have the potential to reveal structure–property relationships for PCM stability, answering questions that bring us closer to this goal. For example, can foreign polyelectrolytes with a strong tendency of association into PCM hosts result in mixed micelles? If so, what features are consequential for this feature of macromolecular exchange? To the extent of our knowledge, there are only a handful of published works that have examined such questions. Dautzenberg et al. conducted polyanion exchange reactions involving PCMs containing model oligophosphates and competing higher MW polymers (PSS and DNA) at physiological salt conditions.97 Another study by Harada and Kataoka exploited polymer architecture to show how diblock polyelectrolytes displace homopolyelectrolytes in PCMs formed with an oppositely charged diblock polyelectrolyte, suggesting increased association in (AB + AC) PCM systems compared to (AB + C) systems.98 These examples provide insight on how PCM chain exchange can arise from molecular recognition based on polyelectrolyte compatibility and dynamics, an area in which advances in noninvasive characterization techniques such as small-angle neutron scattering,99 fluorescence microscopy,100 and liquid-phase transmission electron microscopy101 are well-equipped to provide key leaps forward in our understanding of polyelectrolyte complex micellization and chain exchange.
PCM Disassembly During Sudden Environmental Changes
Depending on the intended application, PCMs may encounter gradients in ionic strength, pH, or temperature that may disrupt their structure. It is important to understand what implications dynamic environments may have on PCM stability or disassembly. One straightforward way to evaluate the dissociation of PCMs is through stopped-flow light scattering (Figure 3B), where PCMs can be monitored in situ while a sudden environmental step change, such as a temperature or salt, is introduced.102−104 In this setup, the time-dependent evolution of the scattering intensity is directly proportional to the mean aggregation number (P(t)). Furthermore, the salt-induced temporal dissociation can be fitted by a phenomenological Avrami-type compressed exponential function of the form in eq 7, containing the relaxation time (τ) and the exponential (β) related to nucleation/growth:
| 7 |
Wu et al. examined the disassembly kinetics of PEO-b-PVBTMA/PAA PCMs with stopped-flow light scattering at different temperature and salt conditions.104Table 2 shows the fitting results at (i) 20, 37, and 57 °C, with a constant salt jump of 500 mM NaCl, and at (ii) 300, 400, 500, and 600 mM NaCl, with a constant temperature of 20 °C. As temperature was increased, the dissociation process accelerated as τ decreased from 61.5 to 39.2 min, with β set to 2 assuming second-order kinetics associated with a fission/fusion mechanism of micelle fragmentation and separation. Analogously, by varying the salt-jump concentration and allowing both τ and β to change, higher salt resulted in faster relaxation kinetics, though disassembly was not immediate after the salt-jump. In addition, the β changed from approximately 1 to 2, showing that neither single-chain expulsion/insertion nor the fission–fusion mechanism were the full explanation for PCM disassembly kinetics. Overall, this technique provides a way for researchers to quantitatively assess the robustness of PCMs under changing solution environments.
Table 2. Relaxation Rate and Exponential Fits of Micelle Dissociation at Increasing Temperature and Salt Concentrations (Adapted from Wu et al.104 Copyright 2020 American Chemical Society).
| temp. (°C) | [salt] (mM) | τ (min) | β |
|---|---|---|---|
| 20 | 500 | 61.5 | 2.00 |
| 37 | 500 | 52.2 | 2.00 |
| 57 | 500 | 39.2 | 2.00 |
| 20 | 300 | 51.9 | 0.82 |
| 20 | 400 | 35.2 | 1.43 |
| 20 | 500 | 27.4 | 2.03 |
| 20 | 600 | 10.9 | 1.94 |
4. Applications and Outlook
Fundamental studies on physical scaling and kinetics continue to pique scientific interest and deliver a molecular-level understanding of PCMs with an ultimate goal, from our perspective, of creating tunable polymeric nanoparticles for biomolecule delivery. When model polymers are substituted for therapeutic biomolecules and PCMs are put into complex environments new problems arise and the research adapts. This field has a strong reciprocal connection between fundamental research and application-based science which is apparent in the numerous advances propelling the potential of using PCMs for biomolecule delivery.
Biomolecule Delivery using PCMs
Due to their controllable nanoscale size and morphology, distinctive capability of partitioning hydrophilic cargo, and dynamic responsivity to environmental changes and stimuli, PCMs are well-suited as delivery vehicles for nanoscopic cargo. Nonviral delivery of therapeutics is a critical challenge for nanomedicine and has been evolving for decades.105−108 PCMs are unique among therapeutic nanoparticles in that they are assembled from only hydrophilic materials and thus, are highly hydrated. Compared to hydrophobically driven assemblies, PCMs do not suffer from certain burdensome limitations on biodistribution such as accumulation in the liver.109 They also have a unique ability to sequester hydrophilic cargo, although this is limited to charged cargo or cargo that can be modified to contain charges. Recent studies using PCMs in vivo have laid the foundation for further use in nanomedicine by delivering therapeutic nucleic acids, proteins, and more. Discussed below are select highlights of promising engineering strategies that have sequestered and delivered various therapeutic biomacromolecules.
Perhaps the most straightforward cargos for PCMs are nucleic acids. DNA and RNA, in their single-stranded form, behave much like the linear charged polymers used to sequester them, although molecular details can have a profound effect,37,57,67 as discussed earlier. PCMs have shown promise in delivering antisense oligonucleotides,110,111 microRNA inhibitors,112,113 small interfering RNA,66,67,114,115 messenger RNA,75,116,117 and plasmid DNA75,79,118 by using cationic polyelectrolytes to sequester the inherently anionic nucleic acids in the core of the micelle, protecting the cargo from harsh environments and enzymatic threats. These studies include cellular delivery and animal models, driving PCM research toward real world applications in gene therapy and immunization.
Proteins are incorporated in polyelectrolyte complexes, largely for applications in bulk materials,119 but also in PCMs for delivery.68 Proteins have both anionic and cationic amino acids on their surface, making sequestration in PCMs less straightforward compared to nucleic acids. Strongly charged proteins can form polyelectrolyte complexes rather simply, while proteins that are closer to net-neutral at physiological pH must be converted to a stronger charge. Strategies for charge conversion include adding more charged amino acids to native proteins,120 working at a pH beyond the protein’s isoelectric point or by modifying the actual charged groups on the protein. One method uses citraconic acid to convert primary amines to carboxylic groups on the protein surface, making the protein’s net charge more negative to enable complexation. These groups can be converted back to their original cationic state in acidic conditions like the late endosomal environment. The Kataoka group has demonstrated this strategy by changing the charge of antibodies in order to assemble PCMs with diblock polyelectrolytes including PEO-b-pLys.121−123 Under acidic conditions, natural antibody charge is restored, disrupting micellization and releasing the antibodies while restoring their biological activity. This strategy was successfully implemented for delivery into cancer cells122,123 and to the brain121 using glucose on the PCM surface to cross the blood–brain barrier with glucose transporter proteins. Strengthening association through nonionic methods can also improve protein–polyelectrolyte complex stability, such as adding hydrophobic spacers to polyelectrolytes.124 Complex macromolecules with multiple components like the ribonucleoprotein complex used in CRISPR-Cas9 genome editing can also be sequestered and delivered in PCMs,75,125 expanding their versatility.
Major Challenges and Opportunities
We have covered advancements in tailored PCMs with a focus physical and dynamic characteristics and recent developments toward therapeutics. This success to date, however, is just a fraction of the enormous potential PCMs have in advancing nanomedicine through enhanced biodistribution, targeting, and controlled release for applications including gene therapy, gene manipulation, and protein- or peptide-based drug delivery. The future of PCMs for nanomedicine will build on these efforts and those in other areas of medical research and could have an important role in the accelerating need for nanotherapeutics. The COVID-19 pandemic has accentuated the growing importance of versatile biomolecule carriers. In addition to vaccines, nanoparticle delivery has promising applications in therapies using gene silencing, monoclonal antibody therapy, and small molecule immunotherapy. PCM research has demonstrated the capability to accomplish these tasks using completely hydrophilic components for improved distribution, tailored particle size, shape, stability, release, and custom surface modifications, but many hurdles remain.
PCMs have been shown to be effective in cellular delivery, but the complete pathways are largely unknown and require further investigation. Advancing cell targeting will vastly improve the effectiveness of therapeutic PCMs. Attaching folate to the corona can mediate delivery to cells that overexpress the folate receptor,126,127 like breast cancer. Sequence-defined peptides have been demonstrated for targeting to inflamed vascular endothelial cells.112 Likewise, RGD peptides can promote cell adhesion,128,129 showing biodistribution and targeting cancer cells that upregulate integrins.130 PCM behavior and stability within the weakly acidic environment of an endosome should also be further characterized. RNA, in particular, is sensitive to acidic conditions and must continue to be protected during this stage. Ultimately, the cargo must escape the endosome and be released from the PCM into the cytoplasm, posing a major concern for PCMs. However, preliminary work, using endosomolytic peptides131 and membrane disruption through deprotecting cations,132,133 shows promising results for programming endosomal escape and nucleic acid release.
Selection of physical and chemical polymer attributes is the central tool among the vast PCM design space we have discussed here. These efforts are crucial when designing a delivery system that needs to protect and release cargo in complex environments. Many studies have shown that increasing charge density or polyelectrolyte length will improve salt resistance, indicating a stronger complex. The McCormick group has shown that longer polyelectrolytes increase PCM effectiveness when using siRNA for gene knockdown and silencing applications but with a time delay due to increased binding constants.66 Additionally, lowering polyelectrolyte binding strength by reducing charge density improves siRNA release but makes PCMs more susceptible to enzymatic degradation, ultimately decreasing cell transfection efficiency.67 Furthermore, it is well documented that increasing polycation charge density or molecular weight increases cytotoxicity.134−136 Achieving an acceptable balance between release kinetics, transfection efficiency, and managing cytotoxic effects stresses the importance of polymer selection when designing therapeutic micelles.
We have reviewed the ways polymer selection in core-forming blocks affects PCM size, stability, and efficacy, but polymer choice for neutral blocks is not frequently studied, likely because the current standards work well. Neutral hydrophilic polymers that form nanoparticle coronas are most commonly PEO, which is easily soluble in aqueous solutions, commercially accessible, and shows little concern in vivo, however, better options are seldom explored. Recently zwitterionic polymers have been incorporated into PCMs as net-neutral blocks because of the excellent antiprotein resistance, hydrated lubrication properties, and high biocompatibility.85,137 Protein-resistant corona materials give a route to increased nanoparticle stealth as protein adsorption to the corona is a significant mechanism contributing to nanoparticle expulsion from the bloodstream.138 This field can benefit from a greater understanding of zwitterionic corona behavior and its effect on biomolecule delivery.
The platform for delivering nucleic acids, a variety of proteins, and some small molecule drugs139 using PCMs is already established and unique strategies for selective delivery are continuously being unveiled. Understanding the factors that influence PCM physical properties, stability, and disassembly will be crucial when designing for delivery. Tailored PCMs that protect, deliver, and release therapeutic cargo with control over transport and targeting can enhance precision medicine, driven by advances in structural design.
Acknowledgments
We acknowledge support by the U.S. Department of Commerce, National Institute of Standards and Technology (NIST), as part of the Center for Hierarchical Materials Design (CHiMaD) Award 70NANB19H005. J.M.T. acknowledges support from the NIST-CHiMaD Postdoctoral Fellowship. The authors also thank Dr. Jeffrey Vieregg for helpful feedback on the manuscript.
Biographies

Alexander E. Marras is a postdoctoral researcher at the Pritzker School of Molecular Engineering at The University of Chicago, where he has studied polyelectrolyte complexation with biomacromolecules in the Tirrell group since joining in October 2017. He serves as cochair for the Biomolecular Self-Assembly session at the American Institute of Chemical Engineers meeting, served as Guest Editor of a special issue of Applied Sciences, and served on the User Executive Committee for the Molecular Foundry at Lawrence Berkeley National Laboratory. Marras received his Ph.D. from The Ohio State University as a Presidential Fellow working with Carlos Castro in the Department of Mechanical and Aerospace Engineering. His graduate research in structural DNA nanotechnology focused on the development of DNA-based mechanical mechanisms and methods for their rapid actuation.

Jeffrey M. Ting received his B.S. in Chemical Engineering at the University of Texas and his Ph.D. in Chemical Engineering from the University of Minnesota in 2016, working with Frank S. Bates and Theresa M. Reineke on synthesizing tunable polymers for oral drug delivery. Afterwards, Jeff worked as a NIST-CHiMaD Postdoctoral Fellow in Matthew Tirrell’s group at the University of Chicago Pritzker School of Molecular Engineering, as part of the Center for Hierarchical Materials Design (CHiMaD), supported by the Materials Genome Initiative at the National Institute of Standards and Technology (NIST). He is currently a Senior Polymer Scientist at 3M, as part of the Materials Informatics Group in the Corporate Research Materials Laboratory.

Kaden C. Stevens received his B.S. in polymer science with honors from the University of Southern Mississippi in 2018, working with Prof. Charles McCormick III on stimuli-responsive amphiphilic polymers. He is currently a Ph.D. candidate in the Tirrell Lab at the University of Chicago studying the effect of polyelectrolyte architecture on polyelectrolyte complexes and polyelectrolyte complex micelles.

Matthew V. Tirrell is the Robert A. Millikan Distinguished Service Professor and Dean of the Pritzker School of Molecular Engineering. His research has been in the fields of polymer interfaces, dynamics, fluid phase behavior, and nanomedicine. He is particularly known for work on polymer brushes, surface force measurement, peptide amphiphiles, and polyelectrolyte complex phase behavior. Professor Tirrell simultaneously served as Deputy Laboratory Director for Science (September 2015 to April 2018) at Argonne National Laboratory. Immediately prior to joining the University of Chicago, he was the Arnold and Barbara Silverman Professor and Chair of Bioengineering at the University of California, Berkeley, with additional appointments in chemical engineering and materials science & engineering, as well as a Faculty Scientist appointment at the Lawrence Berkeley National Laboratory. Professor Tirrell completed 10 years as Dean of Engineering at the University of California, Santa Barbara on June 30, 2009. From 1977 to 1999, he was on the faculty of Chemical Engineering and Materials Science at the University of Minnesota, where he served as department head from 1994 to 1999. Tirrell received a B.S. in Chemical Engineering at Northwestern University in 1973 and a Ph.D. in 1977 in Polymer Science from the University of Massachusetts. He has coauthored about 400 papers and one book, and has supervised about 100 Ph.D. students and 50 postdoctoral researchers. Professor Tirrell is a member of the National Academy of Engineering, the National Academy of Sciences, the American Academy of Arts & Sciences, and the Indian National Academy of Engineering and is a Fellow of the American Institute of Medical and Biological Engineers, the AAAS, and the American Physical Society.
Author Present Address
† 3M Company, 3M Center, Saint Paul, MN 55144, United States
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Orilall M. C.; Wiesner U. Block copolymer based composition and morphology control in nanostructured hybrid materials for energy conversion and storage: solar cells, batteries, and fuel cells. Chem. Soc. Rev. 2011, 40 (2), 520–35. 10.1039/C0CS00034E. [DOI] [PubMed] [Google Scholar]
- Dwars T.; Paetzold E.; Oehme G. Reactions in micellar systems. Angew. Chem., Int. Ed. 2005, 44 (44), 7174–99. 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- Mortensen K. PEO-related block copolymer surfactants. Colloids Surf., A 2001, 183–185, 277–292. 10.1016/S0927-7757(01)00546-5. [DOI] [Google Scholar]
- Kataoka K.; Harada A.; Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Delivery Rev. 2001, 47, 113–131. 10.1016/S0169-409X(00)00124-1. [DOI] [PubMed] [Google Scholar]
- McCullagh M.; Prytkova T.; Tonzani S.; Winter N. D.; Schatz G. C. Modeling Self-Assembly Processes Driven by Nonbonded Interactions in Soft Materials. J. Phys. Chem. B 2008, 112, 10388–10398. 10.1021/jp803192u. [DOI] [PubMed] [Google Scholar]
- Haliloğlu T.; Bahar I.; Erman B.; Mattice W. L. Mechanisms of the Exchange of Diblock Copolymers between Micelles at Dynamic Equilibrium. Macromolecules 1996, 29, 4764–4771. 10.1021/ma951301+. [DOI] [Google Scholar]
- Wang Y.; Mattice W. L.; Napper D. H. Simulation of the Formation of Micelles by Diblock Copolymers under Weak Segregation. Langmuir 1993, 9, 66–70. 10.1021/la00025a017. [DOI] [Google Scholar]
- De Gennes P. G. Scaling theory of polymer adsorption. J. Phys. (Paris) 1976, 37 (12), 1445–1452. 10.1051/jphys:0197600370120144500. [DOI] [Google Scholar]
- De Gennes P. G. Conformation of Polymers Attached to an Interface. Macromolecules 1980, 13, 1069–1075. 10.1021/ma60077a009. [DOI] [Google Scholar]
- Halperin A. Polymeric Micelles: A Star Model. Macromolecules 1987, 20, 2943–2946. 10.1021/ma00177a051. [DOI] [Google Scholar]
- Noolandi J.; Hong K. M. Theory of Block Copolymer Micelles in Solution. Macromolecules 1983, 16, 1443–1448. 10.1021/ma00243a007. [DOI] [Google Scholar]
- Leibler L.; Orland H.; Wheeler J. C. Theory of critical micelle concentration for solutions of block copolymers. J. Chem. Phys. 1983, 79, 3550. 10.1063/1.446209. [DOI] [Google Scholar]
- Pépin M. P.; Whitmore M. D. Monte Carlo and Mean Field Study of Diblock Copolymer Micelles. Macromolecules 2000, 33, 8644–8653. 10.1021/ma992135z. [DOI] [Google Scholar]
- Cantu L.; Corti M.; Salina P. Direct measurement of the formation time of mixed micelles. J. Phys. Chem. 1991, 95, 5981–5983. 10.1021/j100168a048. [DOI] [Google Scholar]
- Won Y.-Y.; Davis H. T.; Bates F. S. Molecular Exchange in PEO-PB Micelles in Water. Macromolecules 2003, 36, 953–955. 10.1021/ma021439+. [DOI] [Google Scholar]
- Lund R.; Willner L.; Richter D.; Dormidontova E. E. Equilibrium Chain Exchange Kinetics of Diblock Copolymer Micelles: Tuning and Logarithmic Relaxation. Macromolecules 2006, 39, 4566–4575. 10.1021/ma060328y. [DOI] [Google Scholar]
- Sing C. E.; Perry S. L. Recent progress in the science of complex coacervation. Soft Matter 2020, 16 (12), 2885–2914. 10.1039/D0SM00001A. [DOI] [PubMed] [Google Scholar]
- Fu J.; Schlenoff J. B. Driving Forces for Oppositely Charged Polyion Association in Aqueous Solutions: Enthalpic, Entropic, but Not Electrostatic. J. Am. Chem. Soc. 2016, 138 (3), 980–90. 10.1021/jacs.5b11878. [DOI] [PubMed] [Google Scholar]
- Voets I. K.; de Keizer A.; Cohen Stuart M. A. Complex coacervate core micelles. Adv. Colloid Interface Sci. 2009, 147–148, 300–18. 10.1016/j.cis.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Sproncken C. C. M.; Magana J. R.; Voets I. K. 100th Anniversary of Macromolecular Science Viewpoint: Attractive Soft Matter: Association Kinetics, Dynamics, and Pathway Complexity in Electrostatically Coassembled Micelles. ACS Macro Lett. 2021, 10 (2), 167–179. 10.1021/acsmacrolett.0c00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magana J. R.; Sproncken C. C. M.; Voets I. K. On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers 2020, 12 (9), 1953. 10.3390/polym12091953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggleman R. A.; Kumar R.; Fredrickson G. H. Investigation of the interfacial tension of complex coacervates using field-theoretic simulations. J. Chem. Phys. 2012, 136 (2), 024903. 10.1063/1.3674305. [DOI] [PubMed] [Google Scholar]
- Lachelt U.; Wagner E. Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond). Chem. Rev. 2015, 115 (19), 11043–78. 10.1021/cr5006793. [DOI] [PubMed] [Google Scholar]
- Wei H.; Pahang J. A.; Pun S. H. Optimization of brush-like cationic copolymers for nonviral gene delivery. Biomacromolecules 2013, 14 (1), 275–84. 10.1021/bm301747r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bruggen C.; Hexum J. K.; Tan Z.; Dalal R. J.; Reineke T. M. Nonviral Gene Delivery with Cationic Glycopolymers. Acc. Chem. Res. 2019, 52 (5), 1347–1358. 10.1021/acs.accounts.8b00665. [DOI] [PubMed] [Google Scholar]
- Dignon G. L.; Best R. B.; Mittal J. Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu. Rev. Phys. Chem. 2020, 71, 53–75. 10.1146/annurev-physchem-071819-113553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S.; Leon L. Structural dynamics, phase behavior, and applications of polyelectrolyte complex micelles. Curr. Opin. Colloid Interface Sci. 2021, 53, 101424. 10.1016/j.cocis.2021.101424. [DOI] [Google Scholar]
- Cabral H.; Miyata K.; Osada K.; Kataoka K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118 (14), 6844–6892. 10.1021/acs.chemrev.8b00199. [DOI] [PubMed] [Google Scholar]
- Ridolfo R.; Tavakoli S.; Junnuthula V.; Williams D. S.; Urtti A.; van Hest J. C. Exploring the impact of morphology on the properties of biodegradable nanoparticles and their diffusion in complex biological medium. Biomacromolecules 2021, 22 (1), 126–133. 10.1021/acs.biomac.0c00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burgh S.; de Keizer A.; Cohen Stuart M. A. Complex coacervation core micelles. Colloidal stability and aggregation mechanism. Langmuir 2004, 20 (4), 1073–84. 10.1021/la035012n. [DOI] [PubMed] [Google Scholar]
- Aloi A.; Guibert C.; Olijve L. L. C.; Voets I. K. Morphological evolution of complex coacervate core micelles revealed by iPAINT microscopy. Polymer 2016, 107, 450–455. 10.1016/j.polymer.2016.08.002. [DOI] [Google Scholar]
- Harada A.; Kataoka K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block-Copolymers with Poly(Ethylene Glycol) Segments. Macromolecules 1995, 28 (15), 5294–5299. 10.1021/ma00119a019. [DOI] [Google Scholar]
- Anraku Y.; Kishimura A.; Oba M.; Yamasaki Y.; Kataoka K. Spontaneous formation of nanosized unilamellar polyion complex vesicles with tunable size and properties. J. Am. Chem. Soc. 2010, 132 (5), 1631–6. 10.1021/ja908350e. [DOI] [PubMed] [Google Scholar]
- Koide A.; Kishimura A.; Osada K.; Jang W. D.; Yamasaki Y.; Kataoka K. Semipermeable polymer vesicle (PICsome) self-assembled in aqueous medium from a pair of oppositely charged block copolymers: physiologically stable micro-/nanocontainers of water-soluble macromolecules. J. Am. Chem. Soc. 2006, 128 (18), 5988–9. 10.1021/ja057993r. [DOI] [PubMed] [Google Scholar]
- Wibowo A.; Osada K.; Matsuda H.; Anraku Y.; Hirose H.; Kishimura A.; Kataoka K. Morphology Control in Water of Polyion Complex Nanoarchitectures of Double-Hydrophilic Charged Block Copolymers through Composition Tuning and Thermal Treatment. Macromolecules 2014, 47 (9), 3086–3092. 10.1021/ma500314d. [DOI] [Google Scholar]
- Rumyantsev A. M.; Zhulina E. B.; Borisov O. V. Scaling Theory of Complex Coacervate Core Micelles. ACS Macro Lett. 2018, 7 (7), 811–816. 10.1021/acsmacrolett.8b00316. [DOI] [PubMed] [Google Scholar]
- Marras A. E.; Vieregg J. R.; Ting J. M.; Rubien J. D.; Tirrell M. V. Polyelectrolyte Complexation of Oligonucleotides by Charged Hydrophobic-Neutral Hydrophilic Block Copolymers. Polymers 2019, 11 (1), 83. 10.3390/polym11010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinschulte A. A.; Gelissen A. P. H.; Jung A.; Brugnoni M.; Caumanns T.; Lotze G.; Mayer J.; Pergushov D. V.; Plamper F. A. Facile Screening of Various Micellar Morphologies by Blending Miktoarm Stars and Diblock Copolymers. ACS Macro Lett. 2017, 6 (7), 711–715. 10.1021/acsmacrolett.7b00328. [DOI] [PubMed] [Google Scholar]
- Plamper F. A.; Gelissen A. P.; Timper J.; Wolf A.; Zezin A. B.; Richtering W.; Tenhu H.; Simon U.; Mayer J.; Borisov O. V.; et al. Spontaneous Assembly of Miktoarm Stars into Vesicular Interpolyelectrolyte Complexes. Macromol. Rapid Commun. 2013, 34 (10), 855–860. 10.1002/marc.201300053. [DOI] [PubMed] [Google Scholar]
- Takahashi R.; Sato T.; Terao K.; Yusa S. Intermolecular Interactions and Self-Assembly in Aqueous Solution of a Mixture of Anionic-Neutral and Cationic-Neutral Block Copolymers. Macromolecules 2015, 48 (19), 7222–7229. 10.1021/acs.macromol.5b01368. [DOI] [Google Scholar]
- Takahashi R.; Sato T.; Terao K.; Yusa S. Reversible Vesicle-Spherical Micelle Transition in a Polyion Complex Micellar System Induced by Changing the Mixing Ratio of Copolymer Components. Macromolecules 2016, 49 (8), 3091–3099. 10.1021/acs.macromol.6b00308. [DOI] [Google Scholar]
- van der Kooij H. M.; Spruijt E.; Voets I. K.; Fokkink R.; Cohen Stuart M. A.; van der Gucht J. On the stability and morphology of complex coacervate core micelles: from spherical to wormlike micelles. Langmuir 2012, 28 (40), 14180–91. 10.1021/la303211b. [DOI] [PubMed] [Google Scholar]
- Perry S. L.; Leon L.; Hoffmann K. Q.; Kade M. J.; Priftis D.; Black K. A.; Wong D.; Klein R. A.; Pierce C. F. 3rd; Margossian K. O.; et al. Chirality-selected phase behaviour in ionic polypeptide complexes. Nat. Commun. 2015, 6, 6052. 10.1038/ncomms7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S.; Leon L. Structural transitions and encapsulation selectivity of thermoresponsive polyelectrolyte complex micelles. J. Mater. Chem. B 2019, 7 (41), 6438–6448. 10.1039/C9TB01194C. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Hernandez J.; Lecommandoux S. Reversible inside-out micellization of pH-responsive and water-soluble vesicles based on polypeptide diblock copolymers. J. Am. Chem. Soc. 2005, 127 (7), 2026–7. 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]
- Holder S. J.; Sommerdijk N. A. J. M. New micellar morphologies from amphiphilic block copolymers: disks, toroids and bicontinuous micelles. Polym. Chem. 2011, 2 (5), 1018–1028. 10.1039/C0PY00379D. [DOI] [Google Scholar]
- Srivastava S.; Tirrell M. V. Polyelectrolyte Complexation. Advances in Chemical Physics 2016, 161, 499–544. 10.1002/9781119290971.ch7. [DOI] [Google Scholar]
- Sing C. E. Micro- to macro-phase separation transition in sequence-defined coacervates. J. Chem. Phys. 2020, 152 (2), 024902. 10.1063/1.5140756. [DOI] [PubMed] [Google Scholar]
- Hunt J. N.; Feldman K. E.; Lynd N. A.; Deek J.; Campos L. M.; Spruell J. M.; Hernandez B. M.; Kramer E. J.; Hawker C. J. Tunable, high modulus hydrogels driven by ionic coacervation. Adv. Mater. 2011, 23 (20), 2327–2331. 10.1002/adma.201004230. [DOI] [PubMed] [Google Scholar]
- Heo T. Y.; Kim I.; Chen L.; Lee E.; Lee S.; Choi S. H. Effect of Ionic Group on the Complex Coacervate Core Micelle Structure. Polymers 2019, 11 (3), 455. 10.3390/polym11030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke P. A.; Pun S. H.; Reineke T. M. Advancing polymeric delivery systems amidst a nucleic acid therapy renaissance. ACS Macro Lett. 2013, 2 (10), 928–934. 10.1021/mz400418j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays J. B.; Magar M. E.; Zimm B. H. Persistence Length of DNA. Biopolymers 1969, 8 (4), 531–536. 10.1002/bip.1969.360080410. [DOI] [Google Scholar]
- Tinland B.; Pluen A.; Sturm J.; Weill G. Persistence length of single-stranded DNA. Macromolecules 1997, 30 (19), 5763–5765. 10.1021/ma970381+. [DOI] [Google Scholar]
- Vieregg J. R.; Lueckheide M.; Marciel A. B.; Leon L.; Bologna A. J.; Rivera J. R.; Tirrell M. V. Oligonucleotide-Peptide Complexes: Phase Control by Hybridization. J. Am. Chem. Soc. 2018, 140 (5), 1632–1638. 10.1021/jacs.7b03567. [DOI] [PubMed] [Google Scholar]
- Shakya A.; King J. T. DNA Local-Flexibility-Dependent Assembly of Phase-Separated Liquid Droplets. Biophys. J. 2018, 115 (10), 1840–1847. 10.1016/j.bpj.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueckheide M.; Vieregg J. R.; Bologna A. J.; Leon L.; Tirrell M. V. Structure-Property Relationships of Oligonucleotide Polyelectrolyte Complex Micelles. Nano Lett. 2018, 18 (11), 7111–7117. 10.1021/acs.nanolett.8b03132. [DOI] [PubMed] [Google Scholar]
- Hayashi K.; Chaya H.; Fukushima S.; Watanabe S.; Takemoto H.; Osada K.; Nishiyama N.; Miyata K.; Kataoka K. Influence of RNA Strand Rigidity on Polyion Complex Formation with Block Catiomers. Macromol. Rapid Commun. 2016, 37 (6), 486–93. 10.1002/marc.201500661. [DOI] [PubMed] [Google Scholar]
- Takeda K. M.; Osada K.; Tockary T. A.; Dirisala A.; Chen Q.; Kataoka K. Poly(ethylene glycol) Crowding as Critical Factor To Determine pDNA Packaging Scheme into Polyplex Micelles for Enhanced Gene Expression. Biomacromolecules 2017, 18 (1), 36–43. 10.1021/acs.biomac.6b01247. [DOI] [PubMed] [Google Scholar]
- Marras A. E.; Campagna T. R.; Vieregg J. R.; Tirrell M. V.. Physical Property Scaling Relationships for Polyelectrolyte Complex Micelles. Macromolecules 2021; 10.1021/acs.macromol.1c00743 [DOI]
- Voets I. K.; de Vries R.; Fokkink R.; Sprakel J.; May R. P.; de Keizer A.; Cohen Stuart M. A. Towards a structural characterization of charge-driven polymer micelles. Eur. Phys. J. E: Soft Matter Biol. Phys. 2009, 30 (4), 351–359. 10.1140/epje/i2009-10533-4. [DOI] [PubMed] [Google Scholar]
- Voets I. K.; van der Burgh S.; Farago B.; Fokkink R.; Kovacevic D.; Hellweg T.; de Keizer A.; Cohen Stuart M. A. Electrostatically driven coassembly of a diblock copolymer and an oppositely charged homopolymer in aqueous solutions. Macromolecules 2007, 40 (23), 8476–8482. 10.1021/ma071356z. [DOI] [Google Scholar]
- Zhulina E. B.; Adam M.; LaRue I.; Sheiko S. S.; Rubinstein M. Diblock copolymer micelles in a dilute solution. Macromolecules 2005, 38 (12), 5330–5351. 10.1021/ma048102n. [DOI] [Google Scholar]
- Zhulina E. B.; Borisov O. V. Theory of Block Polymer Micelles: Recent Advances and Current Challenges. Macromolecules 2012, 45 (11), 4429–4440. 10.1021/ma300195n. [DOI] [Google Scholar]
- Kramarenko E. Y.; Khokhlov A. R.; Reineker P. Stoichiometric polyelectrolyte complexes of ionic block copolymers and oppositely charged polyions. J. Chem. Phys. 2006, 125 (19), 194902. 10.1063/1.2387173. [DOI] [PubMed] [Google Scholar]
- Borisov O.; Zhulina E. Effect of salt on self-assembly in charged block copolymer micelles. Macromolecules 2002, 35 (11), 4472–4480. 10.1021/ma010934n. [DOI] [Google Scholar]
- Holley A. C.; Parsons K. H.; Wan W. M.; Lyons D. F.; Bishop G. R.; Correia J. J.; Huang F. Q.; McCormick C. L. Block ionomer complexes consisting of siRNA and aRAFT-synthesized hydrophilic-block-cationic copolymers: the influence of cationic block length on gene suppression. Polym. Chem. 2014, 5 (24), 6967–6976. 10.1039/C4PY00940A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons K. H.; Holley A. C.; Munn G. A.; Flynt A. S.; McCormick C. L. Block ionomer complexes consisting of siRNA and aRAFT-synthesized hydrophilic-block-cationic copolymers II: the influence of cationic block charge density on gene suppression. Polym. Chem. 2016, 7 (39), 6044–6054. 10.1039/C6PY01048B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn J. M.; Kapelner R. A.; Obermeyer A. C. Macro- and Microphase Separated Protein-Polyelectrolyte Complexes: Design Parameters and Current Progress. Polymers 2019, 11 (4), 578. 10.3390/polym11040578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. C.; Fares H. M.; Schlenoff J. B. Ion-Pairing Strength in Polyelectrolyte Complexes. Macromolecules 2017, 50 (3), 1066–1074. 10.1021/acs.macromol.6b02445. [DOI] [Google Scholar]
- Sadman K.; Wang Q. F.; Chen Y. Y.; Keshavarz B.; Jiang Z.; Shull K. R. Influence of Hydrophobicity on Polyelectrolyte Complexation. Macromolecules 2017, 50 (23), 9417–9426. 10.1021/acs.macromol.7b02031. [DOI] [Google Scholar]
- Fernandez-Villamarin M.; Sousa-Herves A.; Porto S.; Guldris N.; Martinez-Costas J.; Riguera R.; Fernandez-Megia E. A dendrimer-hydrophobic interaction synergy improves the stability of polyion complex micelles. Polym. Chem. 2017, 8 (16), 2528–2537. 10.1039/C7PY00304H. [DOI] [Google Scholar]
- Izumrudov V. A.; Zhiryakova M. V.; Kudaibergenov S. E. Controllable stability of DNA-containing polyelectrolyte complexes in water-salt solutions. Biopolymers 1999, 52 (2), 94–108. . [DOI] [PubMed] [Google Scholar]
- Kim B. S.; Chuanoi S.; Suma T.; Anraku Y.; Hayashi K.; Naito M.; Kim H. J.; Kwon I. C.; Miyata K.; Kishimura A.; et al. Self-assembly of siRNA/PEG-b-catiomer at integer molar ratio into 100 nm-sized vesicular polyion complexes (siRNAsomes) for RNAi and codelivery of cargo macromolecules. J. Am. Chem. Soc. 2019, 141, 3699. 10.1021/jacs.8b13641. [DOI] [PubMed] [Google Scholar]
- Miyata K.; Kakizawa Y.; Nishiyama N.; Harada A.; Yamasaki Y.; Koyama H.; Kataoka K. Block catiomer polyplexes with regulated densities of charge and disulfide cross-linking directed to enhance gene expression. J. Am. Chem. Soc. 2004, 126 (8), 2355–61. 10.1021/ja0379666. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ma B.; Abdeen A. A.; Chen G.; Xie R.; Saha K.; Gong S. Versatile Redox-Responsive Polyplexes for the Delivery of Plasmid DNA, Messenger RNA, and CRISPR-Cas9 Genome-Editing Machinery. ACS Appl. Mater. Interfaces 2018, 10 (38), 31915–31927. 10.1021/acsami.8b09642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronich T. K.; Keifer P. A.; Shlyakhtenko L. S.; Kabanov A. V. Polymer micelle with cross-linked ionic core. J. Am. Chem. Soc. 2005, 127 (23), 8236–7. 10.1021/ja043042m. [DOI] [PubMed] [Google Scholar]
- Dey D.; Kumar S.; Banerjee R.; Maiti S.; Dhara D. Polyplex formation between PEGylated linear cationic block copolymers and DNA: equilibrium and kinetic studies. J. Phys. Chem. B 2014, 118 (25), 7012–25. 10.1021/jp501234p. [DOI] [PubMed] [Google Scholar]
- Dhande Y. K.; Wagh B. S.; Hall B. C.; Sprouse D.; Hackett P. B.; Reineke T. M. N-Acetylgalactosamine Block-co-Polycations Form Stable Polyplexes with Plasmids and Promote Liver-Targeted Delivery. Biomacromolecules 2016, 17 (3), 830–840. 10.1021/acs.biomac.5b01555. [DOI] [PubMed] [Google Scholar]
- Sprouse D.; Reineke T. M. Investigating the Effects of Block versus Statistical Glycopolycations Containing Primary and Tertiary Amines for Plasmid DNA Delivery. Biomacromolecules 2014, 15 (7), 2616–2628. 10.1021/bm5004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos I.; Timmerman M.; Sprakel J. FRET-Based Determination of the Exchange Dynamics of Complex Coacervate Core Micelles. Macromolecules 2021, 54 (1), 398–411. 10.1021/acs.macromol.0c02387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R.; Narayanan T.; Yusa S.-i.; Sato T. Kinetics of Morphological Transition between Cylindrical and Spherical Micelles in a Mixture of Anionic–Neutral and Cationic–Neutral Block Copolymers Studied by Time-Resolved SAXS and USAXS. Macromolecules 2018, 51 (10), 3654–3662. 10.1021/acs.macromol.8b00101. [DOI] [Google Scholar]
- Marras A. E.; Vieregg J. R.; Tirrell M. V. Assembly and Characterization of Polyelectrolyte Complex Micelles. J. Visualized Exp. 2020, (157), e60894. 10.3791/60894. [DOI] [PubMed] [Google Scholar]
- Marciel A. B.; Srivastava S.; Ting J. M.; Tirrell M. V. SAXS methods for investigating macromolecular and self-assembled polyelectrolyte complexes. Methods Enzymol. 2021, 646, 223. 10.1016/bs.mie.2020.09.013. [DOI] [PubMed] [Google Scholar]
- Ting J. M.; Wu H.; Herzog-Arbeitman A.; Srivastava S.; Tirrell M. V. Synthesis and Assembly of Designer Styrenic Diblock Polyelectrolytes. ACS Macro Lett. 2018, 7 (6), 726–733. 10.1021/acsmacrolett.8b00346. [DOI] [PubMed] [Google Scholar]
- Ting J. M.; Marras A. E.; Mitchell J. D.; Campagna T. R.; Tirrell M. V. Comparing Zwitterionic and PEG Exteriors of Polyelectrolyte Complex Micelles. Molecules 2020, 25 (11), 2553. 10.3390/molecules25112553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R.; Narayanan T.; Sato T. Growth Kinetics of Polyelectrolyte Complexes Formed from Oppositely-Charged Homopolymers Studied by Time-Resolved Ultra-Small-Angle X-ray Scattering. J. Phys. Chem. Lett. 2017, 8 (4), 737–741. 10.1021/acs.jpclett.6b02957. [DOI] [PubMed] [Google Scholar]
- Wu H.; Ting J. M.; Yu B.; Jackson N. E.; Meng S.; de Pablo J. J.; Tirrell M. V. Spatiotemporal Formation and Growth Kinetics of Polyelectrolyte Complex Micelles with Millisecond Resolution. ACS Macro Lett. 2020, 9 (11), 1674–1680. 10.1021/acsmacrolett.0c00543. [DOI] [PubMed] [Google Scholar]
- Smolsky I. L.; Liu P.; Niebuhr M.; Ito K.; Weiss T. M.; Tsuruta H. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 2007, 40 (s1), s453–s458. 10.1107/S0021889807009624. [DOI] [Google Scholar]
- Amann M.; Diget J. S.; Lyngsø J.; Pedersen J. S.; Narayanan T.; Lund R. Kinetic Pathways for Polyelectrolyte Coacervate Micelle Formation Revealed by Time-Resolved Synchrotron SAXS. Macromolecules 2019, 52 (21), 8227–8237. 10.1021/acs.macromol.9b01072. [DOI] [Google Scholar]
- Wu H.; Ting J. M.; Werba O.; Meng S.; Tirrell M. V. Non-equilibrium phenomena and kinetic pathways in self-assembled polyelectrolyte complexes. J. Chem. Phys. 2018, 149 (16), 163330. 10.1063/1.5039621. [DOI] [PubMed] [Google Scholar]
- Lund R.; Willner L.; Monkenbusch M.; Panine P.; Narayanan T.; Colmenero J.; Richter D. Structural observation and kinetic pathway in the formation of polymeric micelles. Phys. Rev. Lett. 2009, 102 (18), 188301. 10.1103/PhysRevLett.102.188301. [DOI] [PubMed] [Google Scholar]
- Dormidontova E. E. Micellization Kinetics in Block Copolymer Solutions: Scaling Model. Macromolecules 1999, 32, 7630–7644. 10.1021/ma9809029. [DOI] [Google Scholar]
- Aniansson E. A. G.; Wall S. N. Kinetics of step-wise micelle association. J. Phys. Chem. 1974, 78, 1024–1030. 10.1021/j100603a016. [DOI] [Google Scholar]
- Nolles A.; Hooiveld E.; Westphal A. H.; van Berkel W. J. H.; Kleijn J. M.; Borst J. W. FRET Reveals the Formation and Exchange Dynamics of Protein-Containing Complex Coacervate Core Micelles. Langmuir 2018, 34 (40), 12083–12092. 10.1021/acs.langmuir.8b01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Ting J. M.; Weiss T. M.; Tirrell M. V. Interparticle Interactions in Dilute Solutions of Polyelectrolyte Complex Micelles. ACS Macro Lett. 2019, 8 (7), 819–825. 10.1021/acsmacrolett.9b00226. [DOI] [PubMed] [Google Scholar]
- Bos I.; Sprakel J. Langevin Dynamics Simulations of the Exchange of Complex Coacervate Core Micelles: The Role of Nonelectrostatic Attraction and Polyelectrolyte Length. Macromolecules 2019, 52 (22), 8923–8931. 10.1021/acs.macromol.9b01442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg H.; Konak C.; Reschel T.; Zintchenko A.; Ulbrich K. Cationic graft copolymers as carriers for delivery of antisense-oligonucleotides. Macromol. Biosci. 2003, 3 (8), 425–435. 10.1002/mabi.200350014. [DOI] [Google Scholar]
- Harada A.; Kataoka K. Selection between block- and homo-polyelectrolytes through polyion complex formation in aqueous medium. Soft Matter 2008, 4 (1), 162–167. 10.1039/B713853A. [DOI] [PubMed] [Google Scholar]
- Carl N.; Prévost S.; Schweins R.; Huber K. Contrast variation of micelles composed of Ca2+ and block copolymers of two negatively charged polyelectrolytes. Colloid Polym. Sci. 2020, 298 (7), 663–679. 10.1007/s00396-019-04596-1. [DOI] [Google Scholar]
- Qiang Z.; Wang M. 100th Anniversary of Macromolecular Science Viewpoint: Enabling Advances in Fluorescence Microscopy Techniques. ACS Macro Lett. 2020, 9 (9), 1342–1356. 10.1021/acsmacrolett.0c00506. [DOI] [PubMed] [Google Scholar]
- Parent L. R.; Gnanasekaran K.; Korpanty J.; Gianneschi N. C. 100th Anniversary of Macromolecular Science Viewpoint: Polymeric Materials by In Situ Liquid-Phase Transmission Electron Microscopy. ACS Macro Lett. 2021, 10 (1), 14–38. 10.1021/acsmacrolett.0c00595. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen S.; Zhu Z.; Liu S. Stopped-flow kinetic studies of the formation and disintegration of polyion complex micelles in aqueous solution. Phys. Chem. Chem. Phys. 2014, 16 (1), 117–27. 10.1039/C3CP53608D. [DOI] [PubMed] [Google Scholar]
- Meli L.; Santiago J. M.; Lodge T. P. Path-Dependent Morphology and Relaxation Kinetics of Highly Amphiphilic Diblock Copolymer Micelles in Ionic Liquids. Macromolecules 2010, 43 (4), 2018–2027. 10.1021/ma902366c. [DOI] [Google Scholar]
- Wu H.; Ting J. M.; Tirrell M. V. Mechanism of Dissociation Kinetics in Polyelectrolyte Complex Micelles. Macromolecules 2020, 53 (1), 102–111. 10.1021/acs.macromol.9b01814. [DOI] [Google Scholar]
- Park K. Controlled drug delivery systems: past forward and future back. J. Controlled Release 2014, 190, 3–8. 10.1016/j.jconrel.2014.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux J. C. Editorial: Drug Delivery: Too Much Complexity, Not Enough Reproducibility?. Angew. Chem., Int. Ed. 2017, 56 (48), 15170–15171. 10.1002/anie.201709002. [DOI] [PubMed] [Google Scholar]
- Lee Y. W.; Luther D. C.; Kretzmann J. A.; Burden A.; Jeon T.; Zhai S.; Rotello V. M. Protein Delivery into the Cell Cytosol using Non-Viral Nanocarriers. Theranostics 2019, 9 (11), 3280–3292. 10.7150/thno.34412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acar H.; Srivastava S.; Chung E. J.; Schnorenberg M. R.; Barrett J. C.; LaBelle J. L.; Tirrell M. Self-assembling peptide-based building blocks in medical applications. Adv. Drug Delivery Rev. 2017, 110–111, 65–79. 10.1016/j.addr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzer C.; Dirin M.; Winkler A. M.; Baumann V.; Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J. Controlled Release 2015, 203, 1–15. 10.1016/j.jconrel.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Min H. S.; Kim H. J.; Naito M.; Ogura S.; Toh K.; Hayashi K.; Kim B. S.; Fukushima S.; Anraku Y.; Miyata K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem., Int. Ed. 2020, 59 (21), 8173–8180. 10.1002/anie.201914751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi M.; Nagatsugi F.; Sasaki S.; Nagasaki Y.; Kataoka K. Smart polyion complex micelles for targeted intracellular delivery of PEGylated antisense oligonucleotides containing acid-labile linkages. ChemBioChem 2005, 6 (4), 718–25. 10.1002/cbic.200400334. [DOI] [PubMed] [Google Scholar]
- Kuo C. H.; Leon L.; Chung E. J.; Huang R. T.; Sontag T. J.; Reardon C. A.; Getz G. S.; Tirrell M.; Fang Y. Inhibition of atherosclerosis-promoting microRNAs via targeted polyelectrolyte complex micelles. J. Mater. Chem. B 2014, 2 (46), 8142–8153. 10.1039/C4TB00977K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Xiao X.; Zhang B.; Li J.; Zhang Y. A self-assembled peptide nucleic acid-microRNA nanocomplex for dual modulation of cancer-related microRNAs. Chem. Commun. (Cambridge, U. K.) 2019, 55 (14), 2106–2109. 10.1039/C9CC00002J. [DOI] [PubMed] [Google Scholar]
- Konate K.; Dussot M.; Aldrian G.; Vaissiere A.; Viguier V.; Neira I. F.; Couillaud F.; Vives E.; Boisguerin P.; Deshayes S. Peptide-based nanoparticles to rapidly and efficiently ″Wrap’n Roll″ siRNA into cells. Bioconjugate Chem. 2019, 30, 592–603. 10.1021/acs.bioconjchem.8b00776. [DOI] [PubMed] [Google Scholar]
- Christie R. J.; Matsumoto Y.; Miyata K.; Nomoto T.; Fukushima S.; Osada K.; Halnaut J.; Pittella F.; Kim H. J.; Nishiyama N.; et al. Targeted polymeric micelles for siRNA treatment of experimental cancer by intravenous injection. ACS Nano 2012, 6 (6), 5174–89. 10.1021/nn300942b. [DOI] [PubMed] [Google Scholar]
- Koji K.; Yoshinaga N.; Mochida Y.; Hong T.; Miyazaki T.; Kataoka K.; Osada K.; Cabral H.; Uchida S. Bundling of mRNA strands inside polyion complexes improves mRNA delivery efficiency in vitro and in vivo. Biomaterials 2020, 261, 120332. 10.1016/j.biomaterials.2020.120332. [DOI] [PubMed] [Google Scholar]
- Uchida S.; Kinoh H.; Ishii T.; Matsui A.; Tockary T. A.; Takeda K. M.; Uchida H.; Osada K.; Itaka K.; Kataoka K. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 2016, 82, 221–8. 10.1016/j.biomaterials.2015.12.031. [DOI] [PubMed] [Google Scholar]
- Fukushima S.; Miyata K.; Nishiyama N.; Kanayama N.; Yamasaki Y.; Kataoka K. PEGylated polyplex micelles from triblock catiomers with spatially ordered layering of condensed pDNA and buffering units for enhanced intracellular gene delivery. J. Am. Chem. Soc. 2005, 127 (9), 2810–1. 10.1021/ja0440506. [DOI] [PubMed] [Google Scholar]
- Gao S.; Holkar A.; Srivastava S. Protein-Polyelectrolyte Complexes and Micellar Assemblies. Polymers 2019, 11 (7), 1097. 10.3390/polym11071097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapelner R. A.; Obermeyer A. C. Ionic polypeptide tags for protein phase separation. Chemical Science 2019, 10 (9), 2700–2707. 10.1039/C8SC04253E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J. B.; Gonzalez-Carter D.; Tockary T. A.; Nakamura N.; Xue Y. E.; Nakakido M.; Akiba H.; Dirisala A.; Liu X. Y.; et al. Dual-Sensitive Nanomicelles Enhancing Systemic Delivery of Therapeutically Active Antibodies Specifically into the Brain. ACS Nano 2020, 14 (6), 6729–6742. 10.1021/acsnano.9b09991. [DOI] [PubMed] [Google Scholar]
- Kim A.; Miura Y.; Ishii T.; Mutaf O. F.; Nishiyama N.; Cabral H.; Kataoka K. Intracellular Delivery of Charge-Converted Monoclonal Antibodies by Combinatorial Design of Block/Homo Polyion Complex Micelles. Biomacromolecules 2016, 17 (2), 446–53. 10.1021/acs.biomac.5b01335. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Ishii T.; Kim H. J.; Nishiyama N.; Hayakawa Y.; Itaka K.; Kataoka K. Efficient delivery of bioactive antibodies into the cytoplasm of living cells by charge-conversional polyion complex micelles. Angew. Chem., Int. Ed. 2010, 49 (14), 2552–5. 10.1002/anie.200905264. [DOI] [PubMed] [Google Scholar]
- Li K.; Chen F.; Wang Y.; Stenzel M. H.; Chapman R. Polyion Complex Micelles for Protein Delivery Benefit from Flexible Hydrophobic Spacers in the Binding Group. Macromol. Rapid Commun. 2020, 41 (18), 2000208. 10.1002/marc.202000208. [DOI] [PubMed] [Google Scholar]
- Tan Z.; Jiang Y. R.; Ganewatta M. S.; Kumar R.; Keith A.; Twaroski K.; Pengo T.; Tolar J.; Lodge T. P.; Reineke T. M. Block Polymer Micelles Enable CRISPR/Cas9 Ribonucleoprotein Delivery: Physicochemical Properties Affect Packaging Mechanisms and Gene Editing Efficiency. Macromolecules 2019, 52 (21), 8197–8206. 10.1021/acs.macromol.9b01645. [DOI] [Google Scholar]
- Sudimack J.; Lee R. J. Targeted drug delivery via the folate receptor. Adv. Drug Delivery Rev. 2000, 41 (2), 147–62. 10.1016/S0169-409X(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Low P. S.; Henne W. A.; Doorneweerd D. D. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 2008, 41 (1), 120–9. 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Ge Z.; Chen Q.; Osada K.; Liu X.; Tockary T. A.; Uchida S.; Dirisala A.; Ishii T.; Nomoto T.; Toh K.; et al. Targeted gene delivery by polyplex micelles with crowded PEG palisade and cRGD moiety for systemic treatment of pancreatic tumors. Biomaterials 2014, 35 (10), 3416–26. 10.1016/j.biomaterials.2013.12.086. [DOI] [PubMed] [Google Scholar]
- Wang F.; Li Y.; Shen Y.; Wang A.; Wang S.; Xie T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14 (7), 13447–62. 10.3390/ijms140713447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkouhi A. K.; Scholte M.; Storm G.; Haisma H. J. Endosomal escape pathways for delivery of biologicals. J. Controlled Release 2011, 151 (3), 220–8. 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Pittella F.; Zhang M.; Lee Y.; Kim H. J.; Tockary T.; Osada K.; Ishii T.; Miyata K.; Nishiyama N.; Kataoka K. Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials 2011, 32 (11), 3106–14. 10.1016/j.biomaterials.2010.12.057. [DOI] [PubMed] [Google Scholar]
- Takae S.; Miyata K.; Oba M.; Ishii T.; Nishiyama N.; Itaka K.; Yamasaki Y.; Koyama H.; Kataoka K. PEG-detachable polyplex micelles based on disulfide-linked block catiomers as bioresponsive nonviral gene vectors. J. Am. Chem. Soc. 2008, 130 (18), 6001–9. 10.1021/ja800336v. [DOI] [PubMed] [Google Scholar]
- Monnery B. D.; Wright M.; Cavill R.; Hoogenboom R.; Shaunak S.; Steinke J. H. G.; Thanou M. Cytotoxicity of polycations: Relationship of molecular weight and the hydrolytic theory of the mechanism of toxicity. Int. J. Pharm. 2017, 521 (1–2), 249–258. 10.1016/j.ijpharm.2017.02.048. [DOI] [PubMed] [Google Scholar]
- Breunig M.; Lungwitz U.; Liebl R.; Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (36), 14454–14459. 10.1073/pnas.0703882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D.; Li Y.; Ahlemeyer B.; Krieglstein J.; Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24 (7), 1121–31. 10.1016/S0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- Ishihara K. Revolutionary advances in 2-methacryloyloxyethyl phosphorylcholine polymers as biomaterials. J. Biomed. Mater. Res., Part A 2019, 107 (5), 933–943. 10.1002/jbm.a.36635. [DOI] [PubMed] [Google Scholar]
- Blanco E.; Shen H.; Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33 (9), 941. 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. A.; Ni C. H.; Shi G.; Wang J.; Zhang M.; Li W. The polyion complex nano-prodrug of doxorubicin (DOX) with poly(lactic acid-co-malic acid)-block-polyethylene glycol: preparation and drug controlled release. Med. Chem. Res. 2015, 24 (3), 1189–1195. 10.1007/s00044-014-1206-7. [DOI] [Google Scholar]