

Abstract
Aims
The study investigates the role and mechanisms of clinically translatable exercise heart rate (HR) envelope effects, without dyssynchrony, on myocardial ischaemia tolerance compared to standard preconditioning methods. Since the magnitude and duration of exercise HR acceleration are tightly correlated with beneficial cardiac outcomes, it is hypothesized that a paced exercise-similar HR envelope, delivered in a maximally physiologic way that avoids the toxic effects of chamber dyssynchrony, may be more than simply a readout, but rather also a significant trigger of myocardial conditioning and stress resistance.
Methods and results
For 8 days over 2 weeks, sedated mice were atrial-paced once daily via an oesophageal electrode to deliver an exercise-similar HR pattern with preserved atrioventricular and interventricular synchrony. Effects on cardiac calcium handling, protein expression/modification, and tolerance to ischaemia–reperfusion (IR) injury were assessed and compared to those in sham-paced mice and to the effects of exercise and ischaemic preconditioning (IPC). The paced cohort displayed improved myocardial IR injury tolerance vs. sham controls with an effect size similar to that afforded by treadmill exercise or IPC. Hearts from paced mice displayed changes in Ca2+ handling, coupled with changes in phosphorylation of calcium/calmodulin protein kinase II, phospholamban and ryanodine receptor channel, and transcriptional remodelling associated with a cardioprotective paradigm.
Conclusions
The HR pattern of exercise, delivered by atrial pacing that preserves intracardiac synchrony, induces cardiac conditioning and enhances ischaemic stress resistance. This identifies the HR pattern as a signal for conditioning and suggests the potential to repurpose atrial pacing for cardioprotection.
Keywords: Preconditioning, Ischaemia, Pacing, Cardioprotection, Exercise, Heart rate
Graphical abstract
Graphical Abstract.

Introduction
Coronary artery disease results in ischaemic myocardial injury with dysfunction and loss of cardiomyocytes that can cause decreased functional capacity, heart failure, arrhythmias, and death. Significant advances in prevention and mitigation have been made, but the incidence of heart failure and related mortality remains very high. Thus, identification of new, widely applicable, and effective strategies continues to be a priority.
What’s new?
Atrial pacing, with conserved physiologic atrioventricular and interventricular activation, is used to deliver the heart rate (HR) envelope of exercise to sedated mice without inducing the chamber dyssynchrony that is known to have cardiotoxic effects.
When delivered no more than once daily in eight sessions over 2 weeks, the atrial-paced HR envelope results in better myocardial resistance to ischaemic injury than sham pacing.
The magnitude of the cardioprotection is similar to that produced by treadmill exercise or a traditional ischaemic preconditioning protocol.
Atrial pacing mimicking exercise-induced HR acceleration results in changes of myocardial calcium handling and molecular signalling coupled with cardiac conditioning.
The pacing approach to myocardial conditioning has significant potential for clinical translation as its implementation requires only updating software on already existing pacemaker devices, thereby offering a clinically practical approach in contrast to previously identified preconditioning methods.
Myocardial ischaemic preconditioning (IPC) refers to interventions that protect the heart from injury, specifically repeated short episodes of ischaemia resulting in cellular adaptations that protect the myocardium against injury from a subsequent prolonged ischaemic insult with reperfusion (IR).1 While IPC shows great potential in animal models, transition into clinical practice has been difficult, in part due to the difficulty of anticipating spontaneous major ischaemic events and the impracticality of delivering preconditioning protocols involving intermittent coronary occlusion. This has prompted the search for alternative strategies for cardioprotection with better translational potential. One such approach is to create preconditioning ischaemia via increased myocardial demand, rather than restricted supply, by delivering rapid ventricular pacing, a technique that can increase myocardial oxygen consumption both through increased contraction rate and the stress of dyssynchrony. While showing promising short-term success, this approach is potentially limited by the risk of acute adverse symptomatic and haemodynamic effects, and its long-term effectiveness may be blunted by the known predisposition of repetitive ventricular dyssynchrony towards pathological ventricular remodelling with adverse outcomes. An intriguing question thus becomes: what aspects of preconditioning stress are essential vs. what parts are unnecessary or even inadvertently detrimental to an optimal outcome?
‘Natural’ forms of myocardial preconditioning may provide insight to this question. The most effective non-clinical intervention known to enhance myocardial stress resistance is exercise. Voluminous data indicate that exercise in moderation clearly promotes cardiac conditioning, improves cardiovascular outcomes and does so without creating myocardial dyssynchrony. There is also a solid body of evidence indicating that the degree and duration of heart rate (HR) acceleration are reliable indicators of both the intensity of physical activity and its cardiovascular conditioning potential. Specifically, exercise with a HR increase to 75–85% maximum predicted by age for 75–150 min per week, divided over 3–5 days per week, is a typical evidence-based recommendation to promote cardiovascular conditioning in humans and in mice.2 Conversely, an impaired HR response to exercise has been associated with adverse cardiac outcomes in healthy adults and those with established heart disease.3
While exercise is a complex process that impacts the cardiovascular system in numerous ways, we hypothesize that since HR magnitude and duration are tightly correlated with beneficial exercise outcomes, the HR envelope may be more than simply a readout, but rather also act as a significant trigger of myocardial conditioning and stress resistance independent of other components of exercise. We also hypothesize that the HR envelope of exercise is sufficient to induce this favourable response without dyssynchronous atrioventricular (AV) and/or interventricular (VV) activation. To test this theory, we employed a pacing approach to isolate and study the exercise HR acceleration envelope while preserving synchronous myocardial activation. Specifically, we delivered atrial pacing, with intrinsic, intact AV conduction and intrinsic, intact synchronous VV activation, to gradually increase the HR from resting to a physiologic rate and duration similar to that typically identified as a marker for exercise-derived beneficial myocardial conditioning2 followed by a gradual return to baseline. Similar to exercise, average daily HR is thus minimally affected since episodes were delivered at most once daily such that any adverse effects of sustained, elevated HR are avoided. Furthermore, atrial-only pacing, with careful titration of maximum target HR to avoid AV Wenckebach and aberrant distal conduction, can prevent the harmful haemodynamic and remodelling effects of AV and VV dyssynchrony that are known to provoke or worsen left ventricular dysfunction.4 Indeed, no long-term adverse effects are associated with atrial pacing at physiologic rates such as those proposed here.5 Furthermore, the maximum rates under the studied protocol are those reflective of a normal exercise response, typically well-tolerated by patients with heart disease undergoing cardiac rehabilitation programs and thus would not be expected to result in limiting anginal or other symptoms, exacerbation of heart failure, or risk of myocardial injury in patients with or without coronary artery disease who are otherwise in stable condition.
Here we establish that an exercise-similar intermittent HR acceleration stimulus elicits a myocardial adaptation and stress resistance cardioprotective response by triggering a spectrum of molecular changes involving calcium handling and transcriptional remodelling. There is significant overlap in adaptations with those of exercise and traditional IPC. These findings may support the repurposing of atrial pacing from the treatment or prevention of bradycardia to the novel purpose of reconstituting a healthy, physiologic, exercise-similar HR acceleration envelope for myocardial conditioning, thereby offering a practical clinical approach to injury prevention in ischaemic heart disease.
Methods
Transoesophageal pacing
Wild-type male C57-Bl6 mice (Charles River), aged 8–10 weeks, were anaesthetized with isoflurane with maintenance of a constant respiratory rate. Body temperature was maintained at 37°C using a heating pad (PhysioSuite, Kent Scientific). An octapolar pacing catheter (Cybermouse catheter, NuMED for Children, Inc.) was inserted through the mouth into the oesophagus and positioned for pacing capture of the atria. Pacing output was twice the diastolic pacing threshold and continuous observation of mice during pacing confirmed that no skeletal muscle or diaphragm capture occurred. Atrial pacing capture, AV, and VV synchrony were also continuously monitored during delivery of the protocol through use of limb-lead electrocardiogram (ECG) recorded using subcutaneous needle electrodes (Grass), amplified using a standard 12-lead ECG amplifier (Data Sciences International), digitized, recorded, and displayed in real time (pClamp, Molecular Devices). Pacing started at a cycle length of 120 ms (corresponding to a typical murine resting HR of ∼500 beats/min), which was then gradually reduced in 10 ms decrements every 3 min to a target cycle length of 80 ms (or the shortest cycle length longer than 80 ms for which 1:1 AV synchrony was stably maintained) for 45 min. Subsequently, the pacing cycle length was gradually increased by 10 ms every 3 min until the pacing interval was longer than the intrinsic sinus cycle length. The oesophageal catheter and ECG electrodes were then removed, and mice were recovered from anaesthesia. Since the cardioprotective effects of exercise are similar in response to short-term (3–5 days) and long-term (weeks–months) exercise,6 in the present study the pacing was performed once daily for five consecutive days (Monday to Friday), followed by a 2-day rest and then paced once daily for three more days (Monday, Tuesday, Wednesday) for a total of 8 days. Sham-treated mice were anaesthetized and had placement of ECG electrodes and the oesophageal pacing catheter but without pacing. Pacing- and sham-treated mice underwent the protocol simultaneously and on an identical schedule to minimize confounding variables. Mice were sacrificed for subsequent experiments 2 h after the last episode of pacing or sham treatment.
Cardiomyocyte isolation
Ventricular cardiomyocytes were isolated from mouse hearts using standard methods as described in the Supplementary material online.
Isolated heart studies and myocardial ischaemia–reperfusion injury
Hearts were extracted from anaesthetized mice and retrogradely perfused at 90 mmHg with Krebs–Henseleit buffer bubbled with 95% O2/5% CO2, at 37°C and pH 7.4. Myocardial IR injury was created as follows: after allowing 20 min for stabilization, the hearts were subjected to 20 min of global ischaemia and 45 min reperfusion. Some hearts were first subjected to IPC consisting of two 2 min cycles of global ischaemia followed by 5 min of reperfusion each before undergoing the IR injury protocol. At the end of this treatment hearts were removed from the Langendorff perfusion apparatus and immediately frozen at −20°C. The frozen hearts were then cut into transverse slices of approximately equal thickness (∼0.8 mm) from apex to base. The slices were placed into a small cell culture dish and then incubated in 1% triphenyltetrazolium chloride (TTC) in phosphate buffer (Na2HPO4 88 mmol/L, NaH2PO4 1.8 mmol/L, pH 7.8) at 37°C for 20 min while gently shaking the dish. The development of the red formazan pigment in living tissues relies on the presence of lactate dehydrogenase or NADH, while failure to stain red indicates a loss of these constituents from necrotic tissue. After staining, the TTC buffer was replaced by 10% formaldehyde. The slices were fixed for the next 4–6 h, then the areas of infarct tissue were quantified using ImageJ software. The risk area is taken as the total ventricular cross-sectional area of all slices. The infarct size is presented as infarcted percentage of at-risk area.
Treadmill exercise training
Three days before exercise, mice were acclimated for 45 min daily on a non-moving treadmill (Columbus Instruments, Columbus, OH) followed by 15 min at a velocity of 3.5 m/min at 15° incline. Mice were then exercised daily for five consecutive days (Monday to Friday), followed by a 2-day rest and then exercised once daily for three more days (Monday, Tuesday, Wednesday; total 8 days) at a speed of 15 m/min and inclination of 15° for 45 min. Mice were sacrificed for further studies 2 h after the last exercise session.
Single-cell calcium imaging
For confocal imaging of sarcoplasmic reticulum (SR) calcium content, isolated mouse ventricular myocytes were loaded with Fluo-4 AM (5 μM) for 30 min at room temperature. Fluo-4 AM was used for its rapid kinetics. After 10 min of de-esterification, the cells were placed in a recording chamber and perfused with normal Tyrode’s solution (1.8 mM Ca2+) at 36 ± 1°C (Temperature Controller, TC2BIP, Cell MicroControls). Confocal Ca2+ imaging was performed in line-scan mode with a laser scanning confocal microscope (510, Carl Zeiss) equipped with a numerical aperture 1.35, 63× lenses. Images of Ca2+ transients and caffeine-induced Ca2+ were acquired at a sampling rate of 1.93 ms per line along the longitudinal axis of the cells. Steady-state Ca2+ transients were achieved by 30 s field stimulation at 1 Hz, 8 V, 4 ms pulse width. SR Ca2+ content was determined by measuring the amplitude of Ca2+ release induced by local delivery of 20 mM caffeine. All digital images were processed with IDL 6.0 program (Research System Inc.).
For ratiometric calcium imaging, cardiac myocytes were placed into Tyrode’s buffer with 2 mM Ca2+ for 1 h to isolate cells resistant to physiological Ca2+ levels. High vacuum grease (Dow Corning Corporation, Midland, MI, USA) was used to make a border along the centre of a Geltrex-coated glass-bottom microwell dish (MatTec Corporation). The grease border prevented mixing between cell types. A single drop of cells from a sham-paced or paced mouse was placed onto each half of the dish, allowing simultaneous loading of both populations with 1 μM Fura-2 AM (Invitrogen) for 20 min at room temperature. Fura-2 AM was used for its sensitivity for detection of low-level diastolic Ca2+. Cells were then washed twice with normal Tyrode’s buffer and incubated for another 10 min at 37°C to allow for de-esterification of Fura-2 AM. Cells were excited alternatively at 340 and 380 nm. Fluorescence signal intensity was acquired at 510 nm. Real-time shifts in Fura-2AM fluorescence ratio were recorded in 30 s intervals on each half of the plate using a Nikon Eclipse Ti2 inverted light microscope.
Transcription analysis
Analysis of transcription from freshly isolated murine ventricular tissue was performed using standard methods as described in the Supplementary material online.
Western blot
Standard methods and reagents were used as described in the Supplementary material online.
Statistics
Data are presented as individual data points with means ± the standard deviation. For Ca2+ handling data, because different numbers of cardiomyocytes were studied from each heart, a mixed-effects statistical model was used. Statistical significance was evaluated by ANOVA with post hoc testing or two-sided Student’s t-test with tests for normality/lognormality as appropriate (GraphPad Prism version 9.2).
Study approval
Animal experiments were performed according to protocols approved by the University of Iowa Institutional Animal Care and Use Committee.
Results
Minimally invasive atrial pacing to maintain atrioventricular and interventricular synchrony
An octapolar pacing catheter (Cybermouse catheter, NuMED for Children, Inc.) was inserted into the oesophagus of healthy, sedated mice positioned supine on a warmed pad. Pacing (n = 50 mice) was delivered by selectively capturing the atria as monitored by ECG (Figure 1). Because pacing was bipolar, with the stimulating current set just above the atrial capture threshold, no skeletal muscle or diaphragmatic capture occurred. Sham-paced mice (n = 50 mice) underwent sedation and placement of the pacing catheter but without delivery of any pacing stimulation. In the pacing group, during a 15-min HR ramp-up period, atrial pacing was used to gradually increase the HR from the resting intrinsic HR (471 ± 41 b.p.m.) to the maximum HR achievable while maintaining 1:1 AV synchrony (710 ± 29 b.p.m.). These HRs are consistent with typical average HRs reported during rest in a home cage (∼480 b.p.m.) and during vigorous treadmill exercise (∼760 b.p.m.) in awake adult mice when studied by implanted telemetry monitors7 with the typical peak HR during pacing slightly exceeding 85% of the maximum HR induced by cholinolytics during peak treadmill exercise in that study (∼800 b.p.m.). The maximum HR was maintained by atrial pacing for 45 min, then gradually reduced over 15 min to baseline (Figure 1). The resting cycle length for sham group mice was 128.8 ± 11.9 ms (466 ± 40 b.p.m.), not significantly different from that of pacing group mice (P = 0.54) and was not changed significantly during the experiment. Pacing did not significantly affect the PR interval (35.1 ± 2.4 at the nadir vs. 34.3 ± 3.8 ms at the longest paced cycle length, P = 0.21), QRS duration (the rapid component of the ventricular electrogram, 15.8 ± 3.4 vs.16.0 ± 2.7 ms, P = 0.75), or QRS amplitude (0.31±0.08 vs. 0.29±0.09 mV, P = 0.24). Furthermore, no animal developed aberrant ventricular conduction during pacing or sham treatments as evidenced by a narrow and consistent QRS morphology throughout. During paced acceleration of HR, any observation of AV block resulted in immediately lowering the pacing rate until 1:1 AV conduction was restored, then maintaining that rate as the maximum, so that by design no sustained AV Wenckebach occurred in any animal. No atrial or ventricular arrhythmias, other than rare, isolated ectopic beats, were seen in any animal during the study. The protocol was repeated once daily, 5 days/week, for a total of eight pacing days (Monday to Friday, Monday to Wednesday). At the end of the pacing protocol, echocardiographic assessment of LV ejection fraction showed no difference in paced mice compared to the sham group (0.68±0.06 vs. 0.70±0.06%, n = 4 each, P = 0.65). Two hours after the final episode, mice were sacrificed, their hearts extracted and freeze-clamped, or the entire heart retrogradely perfused for the IR protocol.
Figure 1.

Transoesophageal atrial pacing protocol. (A) Schematic of the transoesophageal atrial pacing protocol from a typical resting HR (∼470 b.p.m.), accelerated to the maximum HR obtainable while maintaining 1:1 AV and VV synchrony (∼710 b.p.m.), sustained for 45 min, then gradually restored to baseline. (B) Representative baseline NSR (top) and an AP rate at 600 b.p.m. (100 ms cycle length, bottom). Note: 1:1 AV synchrony and a normal VV activation pattern, identical to that during NSR, are preserved during atrial pacing. Horizontal scale bar represents 100 ms. AP, atrial-paced; AV, atrioventricular; HR, heart rate; NSR, normal sinus rhythm; p, atrial waveform; qrst, ventricular waveform; VV, interventricular.
An exercise-similar atrial-paced heart rate envelope triggers protection from ischaemia–reperfusion injury with an effect size similar to that of ischaemic preconditioning and treadmill exercise
Rapid ventricular pacing with AV and/or VV dyssynchrony is known to induce protection from IR. However, the cardioprotective effect of maximally physiological atrial pacing without dyssynchrony is unknown. Here (Figure 2), the atrial pacing protocol resulted in augmented stress resistance as evidenced by smaller myocardial infarct size in response to IR (47.54 ± 4.49% of total ventricular area, n = 8) than that for sham-paced mice (59.94 ± 4.87%, n = 7, P = 0.02) or for sedentary control mice that did not undergo any sedation or pacing catheter placement (60.19 ± 6.03%, n = 7, P = 0.01). Furthermore, when compared to other interventions known to provide preconditioning, the exercise-similar atrial pacing produced equivalent protection from IR injury. Specifically, infarct sizes following IPC (46.89 ± 10.00%, n = 8) and preconditioning by exercise (50.17 ± 9.20%, n = 10) were no smaller than that achieved in the atrial-paced group (atrial-paced group: 47.54 ± 4.49%, n = 8, P = 0.86 vs. IPC and 0.46 vs. exercise). One-way ANOVA values for all comparisons between groups, including comparisons to sham pacing, are shown in Supplementary material online, Table S1.
Figure 2.
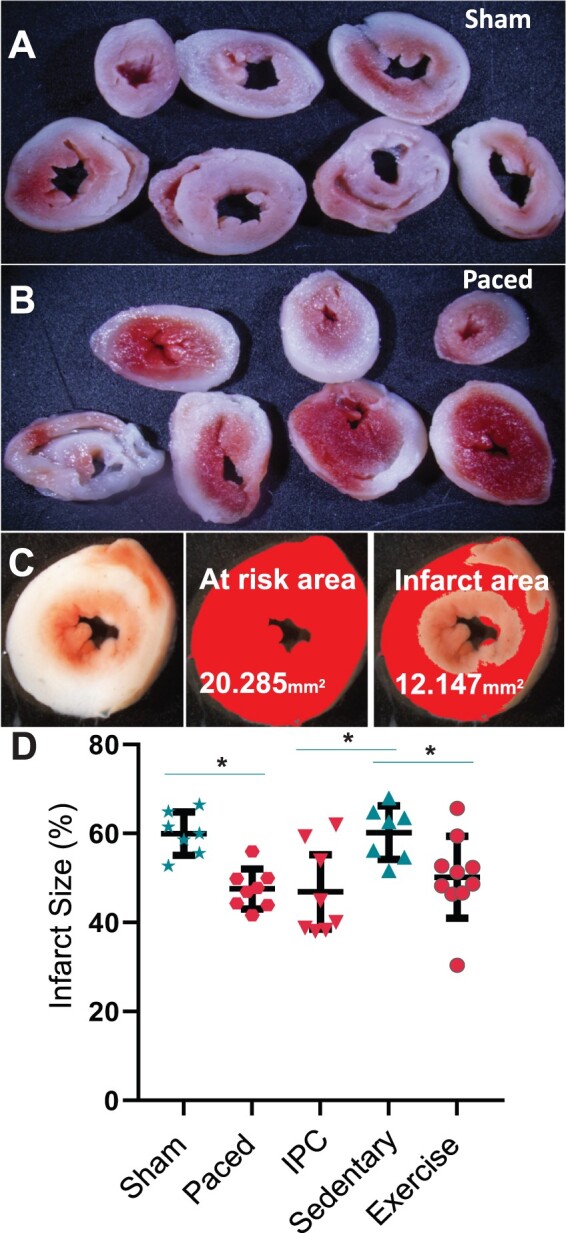
Cardioprotection from ischaemia–reperfusion (IR) injury. Representative ventricle slices after IR from (A) a sham-treated and (B) an atrial-paced mouse. (C) Example of infarct size calculation: the degree of IR injury is measured as the infarct area of ventricles as a percentage of total ventricular area in tetrazolium-stained tissue sections. Infarcted tissue is white, while healthy tissue is red in the left-most panel. The middle panel shows the at-risk area in magenta while the right-most panel shows the infarct area in magenta. (D) Summary statistics of infarct size (sham 59.94 ± 4.87 n = 7, paced 47.54 ± 4.49 n = 8, sedentary control 60.19 ± 6.03 n = 7, ischaemic preconditioning 46.89 ± 10.0 n = 8, exercise preconditioned 50.17 ± 9.20 n = 10; one-way ANOVA, *P < 0.05). IPC, ischaemic preconditioning.
An exercise-similar atrial-paced heart rate envelope alters Ca2+ handling
The dependence of Ca2+ transients on HR is a fundamental property of cardiac myocytes. Since remodelling of Ca2+ handling has been reported in response to both exercise and pacing,8 we investigated the effect on ventricular cardiomyocyte Ca2+ handling of the atrial pacing approach that mimics exercise-related HR acceleration (Figure 3). Ventricular myocytes isolated from mice exposed to 8 days of the atrial pacing protocol, compared to cells from sham-treated mice, demonstrated greater SR Ca2+ content (F/F0 ratio: 6.1 ± 1.9 vs. 5.4 ± 1.4 for sham, n = 8 mice both groups, with n = 124 and 154 cells, respectively, P = 0.01, Figure 3). Ratiometric measurement of diastolic Ca2+ revealed significantly reduced levels for the paced group (FURA 340/380 ratio: 0.32±0.10, n = 84 cells, n = 3 mice) vs. sham (0.37±0.11, n = 101 cells, 3 mice, P < 0.0001). Confocal Ca2+ imaging demonstrated that the atrial pacing protocol also increased the Ca2+ transient duration at 90% of the maximum amplitude (T90 decay time: 264.7 ± 77.2 vs. 203.8 ± 74.1 ms for sham, n = 8 mice both groups, 150 and 213 cells, respectively, P < 0.02). Time to peak amplitude (25.29 ± 8.25, vs. 25.78 ± 8.78 ms), time of decay to 50% of peak amplitude (T50: 92.24 ± 20.92 vs. 80.37 ± 18.16 ms), and 75% of peak amplitude (T75: 159.84 ± 41.83 vs. 131.88 ± 37.52 ms) were not significantly different (n = 8 mice both groups, 150 and 219 cells in each group, respectively). In agreement with previously published data9 there was a trend towards increased Ca2+ transient peak amplitude but it did not reach statistical significance (F1/F0 ratio: 4.71 ± 1.32 vs. 4.40 ± 1.25, n = 8 mice both groups, 150 and 219 cells in each group, respectively P = 0.31).
Figure 3.

Exercise-similar atrial pacing alters Ca2+ dynamics in cardiomyocytes. (A) Representative Ca2+ fluorescence in isolated cardiomyocytes. (B) Summary statistics for sarcoplasmic reticulum Ca2+; diastolic Ca2+; rate of Ca2+ decay at 90% of the transient amplitude; and peak of Ca2+ transients. Mixed-effects model analysis results expressed as individual data points with mean ± standard deviation: *P < 0.01. Ca2+ transient data are from 150 (paced) and 219 (sham) cells, 8 mice for each of paced and sham. Sarcoplasmic reticulum content from 154 (paced) and 124 (sham) cells, 8 mice for each of paced and sham. Diastolic Ca2+ from 84 (paced) and 101 (sham) cells, 3 mice for each of paced and sham. SR, sarcoplasmic reticulum.
Post-translational regulation of Ca2+ handling proteins
To better understand molecular changes underlying the altered Ca2+ handling, we evaluated expression of principal proteins known to be responsible for the regulation of Ca2+ cycling (Figure 4):10 calcium/calmodulin-dependent protein kinase II (CaMKII); sodium-calcium exchanger (NCX), SR Ca2+ ATPase (SERCA 2A), ryanodine receptor Ca2+ release channel (RyR2), protein kinase A (PKA), and phospholamban (PLN).
Figure 4.

Post-translational regulation of Ca2+ handling proteins. Whole cell protein extracts, from the hearts of mice collected 2 h after last pacing or sham intervention, were investigated for expression of total of phosphorylated proteins involved in Ca2+ handling. (A). Representative western blots of NCX; SERC 2A; RyR2 phosphorylated at serine residue 2814, 2030, 2808, and total (t); CaMKII phosphorylated at threonine residue 286 and total (t); PLN phosphorylated at serine residue 16 and threonine residue 17 as well as total (t); and GAPDH. (B). Summary statistics for phosphorylated protein with statistically different expression in paced vs. sham hearts (labelled with orange background on A). Results expressed as individual data points with mean ± standard deviation: *P ≤ 0.05 vs. sham. CaMKII, calcium/calmodulin-dependent protein kinase II; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NCX, sodium-calcium exchanger; PLN, phospholamban; RyR2, Ryanodine receptor 2; SERC 2A, sarcoplasmic reticulum Ca2+-ATPase.
In tissue collected 2 h after the last pacing intervention the expression of SERCA 2A, total RyR2, p-RyR(S2814), p-RyR(S2030), PLN, p-PLN(T17), NCX, PKA, and total CaMKII were not significantly changed by pacing vs. sham (Figure 4 and Supplementary material online, Table S2). However, increases were seen in the expression of RyR2 phosphorylated at S2808, normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (0.12±0.04 vs. 0.07±0.02, n = 7 each, P = 0.008) or to total RyR2 (0.35±0.12 vs. 0.20±0.10, n = 7 each, P = 0.02) and p-PLN (S16 and T17 PKA and CaMKII sites, respectively) over GAPDH (1.11±.25 vs. 0.32±0.29, n = 7 each, P = 0.006) and over total PLN (0.79±.18 vs. 0.49±0.28, n = 7 each, P = 0.036). Auto-phosphorylated (active) CaMKII at T286 normalized to GAPDH (0.60±0.52 vs. 1.16±0.29, n = 8 each, P = 0.02) and normalized to total CaMKII (0.28±0.14 vs. 1.02±0.66, n = 8 each, P = 0.008) was reduced.
Transcriptional changes
We assessed the transcriptional changes that occur in the heart in response to the atrial pacing protocol. RNAseq analysis revealed that 242 genes are differentially expressed (DE) in the ventricles of mice in the paced vs. sham groups with a Log2 fold change >1.5 and at a threshold of more than one fragment per kilobase per million mapped reads (FDR > 0.05) (Figure 5A). Interestingly, the majority of significant DE genes (93.8%) are upregulated. We performed Ingenuity Pathway Analysis (IPA) to provide insight into the pathways and processes that are potentially altered at the transcriptional level by the pacing protocol. Ingenuity Pathway Analysis results suggest that the atrial pacing protocol elicits a cardioprotective expression profile (Figure 5B and C). Potential causal upstream master regulators that are predicted to drive atrial pacing protocol-dependent transcriptional output primarily fall into IP3/Ca2+ signalling, metabolic regulation, and lipid signalling pathways. Many of the observed changes have been previously linked to cardioprotective responses. For instance, within the IP3/Ca2+ signalling group, Ppp3r1 is a calcium-binding regulatory subunit of calcineurin and cardiomyocyte-specific Ppp3r1 knockout hearts are dysfunctional with impaired contractility and excitation–contraction coupling. Plcl2, an inactive phospholipase C gene recently found to be a susceptibility locus for myocardial infarction and is negatively implicated as a master regulator by the pacing protocol. Another example that is also negatively predicted is the growth regulator Sik1. Its deletion destabilizes HDAC7 and blunts pressure overload-dependent cardiac remodelling and heart failure.11 Furthermore, Pde4d activity is also predicted to be inhibited. Genetic deletion of Pde4d has been found to promote progressive cardiomyopathy and reduced RyR2 function. At the same time pharmacological inhibition of Pde4d results in improved cardiac function and enhanced SERCA2 activity.
Figure 5.

Exercise-similar atrial pacing cardioprotective transcriptional response. (A) RNAseq profile of ventricles from sham vs. atrial-paced mice showing significant differentially expressed (DE) upregulated (red) and downregulated (blue) genes (Log2 fold change > 1.5; FDR < 0.05). (B and C) Ingenuity Pathway Analysis of DE genes >1.5-fold for candidate upstream Master Regulators (B) and for enriched Diseases and Functions annotation categories (C). An activation Z-score threshold of ±2 is used to identify likely hits. FPKM, fragments per kilobase per million mapped reads.
KEGG analyses indicate that our pacing protocol results in predicted enhanced activity of the PPAR signalling pathway involved in cardiac energetics (FDR = 0.023), while transcription factors that generally antagonize adult cardiac metabolism are predicted be repressed by the pacing protocol (e.g. Med13, Plin1, Thrap3).12 Meanwhile, transcription-independent metabolic regulator Fgf21, a secreted hormone that protects against oxidative damage and cardiac hypertrophy as well as promotes mitochondrial biogenesis and function through CREB activation13 is also predicted to be magnified. Thus, the atrial pacing-induced exercise HR envelope with preserved cardiac synchrony promotes transcriptional remodelling associated with a cardioprotective paradigm.
Discussion
In this study, atrial pacing was used to uncover the ability of an exercise-similar episodic HR acceleration envelope, with a physiologic myocardial activation pattern (preserved AV and VV synchrony), to enhance myocardial resistance to ischaemic insult. Exercise-similar pacing-dependent reduction in IR-induced infarct size is coupled with changes in calcium handling and mRNA expression patterns to promote cardioprotection. These findings suggest the potential use of such pacing for prevention of the myocardial injury that often underlies heart failure initiation and/or progression in the presence of coronary artery disease.
The mechanisms of cardioprotection induced by IPC and exercise are very complex and still under investigation. However, the present work indicates that HR acceleration may be an important trigger for the molecular and cellular changes of exercise conditioning. Interestingly, the effect size of the exercise HR envelope on cardiac resistance to injury, under the tested pacing protocol, is close to that provided by the exposure to short treadmill exercise training. This suggests that the role of the HR envelope is not marginal, but rather is a key factor driving the phenomenon of cardiac conditioning by exercise. This is somewhat surprising given the complexity of exercise and the numerous potential pathways by which it may exert beneficial cardiac conditioning effects. However, although HR acceleration may seem like a small part of ‘whole exercise’, it is worth noting that the integrated physiologic effects of HR acceleration by pacing are far from trivial. For instance, it is known that HR acceleration augments the force of contraction in healthy hearts through the ‘force-frequency’ effect. Systemically, HR acceleration may have autonomic, endocrine, and vascular consequences. For instance, by altering cardiac chamber filling and stretch, HR acceleration may impact the release of natriuretic peptides. The impact of increased arterial volume and pulsation rate may alter baroreceptor activity, vascular shear, and vascular tone. The overall increase in cardiac output may have far-reaching consequences due to enhanced blood flow to other organ systems including kidneys, skeletal muscle, and brain. Thus, while pacing can dissociate HR acceleration from other components of exercise and thus help to deconstruct the complex physiology of exercise, the actual physiologic mechanisms by which paced HR acceleration can exert beneficial stress-tolerance effects may extend well beyond the presented myocardial adaptations.
An apparent conundrum here is that beneficial heart disease outcomes, particularly in ischaemic heart disease and heart failure, have been strongly associated with slow average HR which may discourage consideration of the role of episodic HR acceleration in cardioprotection. The apparent paradox presented by the association of positive myocardial outcomes with both slow HR and with exercise that accelerates HR (and the data here on paced HR acceleration) may be resolved by considering that the relatively brief HR acceleration of exercise (or the studied pacing protocol) provides a signal for myocardial adaptation, while the longer intervening periods of bradycardia for the remainder of a day may provide adequate time and surplus resources needed for the actual work of cellular adaptation. Since episodic exercise-related or exercise-similar HR acceleration minimally impact daily average HR, they could be expected to augment, not counter, the known beneficial myocardial effects of a slow average or resting HR. In other words, the two concepts are not mutually exclusive—both slow average HR and brief episodic HR acceleration could exert beneficial effects via very different mechanisms, as well as have synergistic effects.
The concept of pacing as a stimulus for myocardial functional, structural, and/or metabolic adaptation has precedence. On one hand, biventricular (ventricular resynchronization) pacing, which corrects abnormal VV synchrony, results in significant gains in quality of life, left ventricular contractile function, exercise capacity, and all-cause mortality in patients with left ventricular dysfunction. In contrast, pacing that impairs AV and/or VV synchrony creates abnormal and inefficient myocardial contraction, haemodynamics, and energetics that can lead to a pacing-induced cardiomyopathy.4,5 Importantly, while any pacing that causes a sustained high HR can cause adverse myocardial remodelling (tachycardia-induced cardiomyopathy), physiologic rate pacing-induced cardiomyopathy is caused only by right ventricular, but not atrial, pacing.4,5 Some previous studies have purposely used non-physiologic pacing manoeuvers to induce molecular and gross structural or functional changes in diseased, dysfunctional hearts.14 For instance, rapid atrial pacing (significantly higher than normal physiologic rates) has been used to promote preconditioning or cardiac remodelling.8 The substantial differences in approach between the current study and others make it difficult to compare them directly, but they all indicate an influence of pacing-induced HR acceleration and/or associated myocardial activation patterns on healthy and pathological myocardial adaptations. The pacing approach in the present study, which specifies the paced HR acceleration be episodic, within a normal exercise-induced range, atrium driven with 1:1 AV and normal VV activation patterns, is designed to be maximally physiologic, thereby minimizing negative stimuli that could limit the overall beneficial impact. Future studies may quantify the relative merits of physiologic rates, durations, and activation patterns vs. stress rebound responses on the balance between beneficial cardiac conditioning and maladaptive changes.
At the myocardial molecular level, the pathways underlying HR or pacing-driven cardiac remodelling are still not fully understood. One possibility is that changes in Ca2+ homeostasis are a primary trigger for downstream remodelling effects. Ca2+ is a central element of excitation–contraction coupling, but also impacts diverse signalling cascades and influences the regulation of gene expression, cell proliferation, growth, and death. Thus, the HR acceleration of exercise may act through such Ca2+ signalling cascades to initiate the witnessed protective adaptations. This assumption is supported by the present data indicating that induction of an exercise HR pattern by atrial pacing results in changes in Ca2+ handling, exhibited as reduction in diastolic Ca2+, increase in the SR Ca2+ content, and T90 prolongation. The hypothesis is further supported by the similarity between the changes in diastolic Ca2+ and SR content with those observed after exercise training.15 We also note a trend towards increased amplitude of Ca2+ transients after pacing, although it did not reach statistical significance. This finding is not surprising as it has previously been shown that both exercise and pacing can induce an increase in the Ca2+ transient amplitude. However, our results do not match previously reported exercise training-related changes exactly. Specifically, in contrast to previous reports, which linked exercise and increased paced HR to a decrease in Ca2+ transient duration,6,9 our atrial pacing protocol increased the Ca2+ transient duration. This may be explained by a reported reduction in the potassium Ito current after short episodes of rapid atrial pacing. This could also be due to variation in the effect of different HR acceleration intensities, durations, and quiescent times between the final conditioning episode and the cell isolation/study. It is important to note, especially when comparing the present data to other studies, that Ca2+ transients were measured here in cardiomyocytes isolated 2 h after the last pacing intervention, so the changes indicate a new steady state, rather than the acute changes that occur during the period of HR acceleration.
With respect to a potential mechanistic explanation of the observed changes of Ca2+ handling, we found three major changes in the phosphorylation of key proteins involved in Ca2+ homeostasis: CaMKII, PLN, and RyR2 at site S2808.10 Calcium/calmodulin-dependent protein kinase II is a multifunctional kinase, densely expressed in cardiomyocytes, that targets numerous proteins involved in excitation–contraction coupling and excitability to support short-term enhancement of cardiac performance, while persistent activation results in adverse cardiac remodelling and dysfunction.16 Inhibition of CaMKII has been shown to increase cardioprotection17 but the effect of exercise training on CaMKII activity is unclear, as some studies report an increase, vs. some reduction, in the enzyme activity. The data here indicate a reduction in the presence of phosphorylated CaMKII which is interpreted to reflect a reduction in basal CaMKII activity.16 RyR2 is central to cytoplasmic Ca2+ signalling in the heart. Intracellular Ca2+ release via RyR2 is modulated by phosphorylation. The location and physiological importance of the different phosphorylation target sites at RyR2 are controversial. Serine at 2808 and 2030 were implicated as the major PKA phosphorylation targets, at the same time other groups identified serine at 2808 and 2814 as the primary targets of CaMKII. Some studies indicate that PKA phosphorylation of RyR2 S2808 reduces the binding affinity of the channel-stabilizing subunit calstabin2, resulting in leaky RyR2 channels, while others confront this finding. This leakiness of RyR2 can explain the increased T90 observed in myocytes after the pacing intervention. This prolonged Ca2+ re-uptake time was observed in spite of increased phosphorylated PLN. Phospholamban is a small phosphoprotein closely associated with the cardiac SR. Dephosphorylated PLN tonically inhibits SERCA 2A, while phosphorylation at S16 by PKA and T17 by CaMKII relieves the inhibition, thus increasing SR Ca2+ uptake.18 We postulate that an increase in SR Ca2+ and a reduction in diastolic Ca2+ can be linked to an elevated presence of phosphorylated PLN, while the prolonged T90 reflects the ‘new balance’ between SERCA2 and RyR2 activities. It is also possible that the pacing intervention affected NCX activity without altering its expression and thus promoted Ca2+ time.
The present data also indicate that the pacing protocol increased expression of S16 and T17 phosphorylated PLN. These changes occurred despite unchanged expression of the PKA alpha isoform and reduced expression of phosphorylated (active) CaMKII. This discrepancy can be explained by the dynamic balance between the activity of kinases and phosphatases, as well as time-related differences in the presence of active/inactive states of specific proteins. We speculate that since post-translational co-modification of different proteins can have different ‘life spans’, our one-time snapshot of post-translational protein modification may not represent the full spectrum of potential functional effects on Ca2+ regulatory cascades.
To identify molecular pathways remodelled by pacing-accelerated HR, we performed RNA sequencing of ventricles. Although we observe changes in intracellular Ca2+ dynamics, the expression of prototypical Ca2+ handling genes remained unchanged except for Cacna1h and Camk1d. The changes in intracellular Ca2+ handling may therefore be primarily regulated by the observed post-transcriptional modifications discussed in the paragraph above, among others. Enriched IPA disease and biological functional annotations further underscore the likelihood that the exercise-similar atrial pacing is cardioprotective. While detrimental fibrosis, oxidative stress, organismal death, and cancer-related phenotypes are negatively associated with the pacing protocol; angiogenic, survival, and cell movement pathways are positively correlated. Several transcription factors are candidate drivers of the metabolic regulation master regulator group and suggest exercise-similar atrial pacing enhances the expression of genes involved in cardiac energetics. The predicted activation of Ppars would enhance the expression of genes involved in the metabolism of fatty acids especially if complexed with PGC1α.19 It is noteworthy that KEGG analysis significantly identified only the PPAR signalling pathway (FDR = 0.023). Other candidate activating transcription factors involved in lipid metabolism are Ncoa2, Nr1h3, and Srebf2. On the other hand, transcription factors that generally antagonize adult cardiac metabolism are predicted be repressed by atrial pacing (e.g. Med13 and Thrap3).12 Again, these data represent just one time point 2 h after the last exposure to pacing, to indicate a sample of the potential spectrum of transcriptional changes.
Furthermore, it is well accepted that cardiac remodelling from physiological exercise and pathological insults such as ischaemia can lead to similar degrees of hypertrophy but generate distinct transcriptional responses. As our proposed pacing protocol generates repetitive episodes of tachycardia yet mimics some of the beneficial cardiac outcomes observed with ex vivo IPC, we further wondered how the transcriptional profile of pacing-treated hearts aligns with consensus pathological vs. physiological datasets. We compared genes that are consistently altered in human idiopathic and ischaemic heart failure, or in rodent models of pathological and physiological cardiac hypertrophy20 to those we found DE in response to the atrial pacing protocol. We found a correlation between the pacing group profile and consensus human heart failure or pathological hypertrophic genes,20 particularly for extracellular matrix-associated transcripts. In addition, the pacing protocol and exercised datasets were both enriched for organismal survival and PPAR signalling annotations.20 A limitation is that these data are obtained on homogenized flash-frozen ventricular tissue to avoid the time-delay inherent in cardiomyocyte isolation that could affect expression profile, as well as to allow for more direct comparison to published data in hypertrophy and heart failure. Future assessments in isolated cardiomyocytes could produce valuable cell-specific information.
Additional investigation, including direct comparison of the pacing protocol with HR- and exposure time-matched exercise interventions, are needed to fully understand the range of adaptations in underlying molecular machinery that underlie the beneficial remodelling effects. Indeed, the mechanisms of cardioprotection induced by IPC and exercise are very complex and still under investigation. However, the current work indicates that HR acceleration may be an important trigger of the molecular and cellular changes of exercise conditioning.
Thus, the atrial pacing-induced exercise HR envelope with preserved cardiac synchrony promotes transcriptional remodelling associated with a cardioprotective paradigm. Beyond advancing the understanding of HR effects and IPC mechanisms, this study has translational implications: traditionally, cardiac pacing is used to prevent bradycardia. More recently, there has been a trend towards using pacing to enact myocardial structural, functional, and/or metabolic changes to treat heart disease such as via the resynchronization therapy mentioned above and via cardiac contractility modulation. The current pacing approach follows on this important trend while extending the concept to preconditioning of hearts to improve their tolerance of future ischaemic insults.
Supplementary material
Supplementary material is available at Europace online.
Supplementary Material
Contributor Information
Zhiyong Zhu, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA; Department of Medicine, Veterans Affairs Medical Center, 601 Hwy 6 West, Iowa City, IA 52246, USA.
Zhan Gao, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Biyi Chen, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Duane D Hall, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Rachel Minerath, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Olha Koval, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Ana Sierra, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Ekaterina Subbotina, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Xiaoyi Zhu, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Young Rae Kim, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Jun Yang, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Isabella Grumbach, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Kaikobad Irani, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Chad Grueter, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Long Sheng Song, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Denice M Hodgson-Zingman, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA.
Leonid V Zingman, Department of Medicine, University of Iowa, 200 Hawkins Drive, CBRB 2270B, Iowa City, IA 52242, USA; Department of Medicine, Veterans Affairs Medical Center, 601 Hwy 6 West, Iowa City, IA 52246, USA.
Funding
There are no relationships with industry involved in this work. This work was supported by funding from the National Institutes of Health (HL144199 and HL113089 to D.M.H.-Z., HL108932 to I.G., DK092412 to L.V.Z.), the Veterans Affairs Merit Review Program (I01BX000163 to I.G., 1I0BX004840 to L.V.Z.), and the Carver Trust (Medical Research Initiative Grant to D.M.H.-Z.).
Conflict of interest: none declared.
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files). Gene sequencing data is available at https://www.ncbi.nlm.nih.gov/geo/; access # 186604.
References
- 1. Murry CE, Jennings RB, Reimer KA.. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124–36. [DOI] [PubMed] [Google Scholar]
- 2. Ferreira R, Moreira-Goncalves D, Azevedo AL, Duarte JA, Amado F, Vitorino R.. Unraveling the exercise-related proteome signature in heart. Basic Res Cardiol 2015;110:454. [DOI] [PubMed] [Google Scholar]
- 3. Girotra S, Kitzman DW, Kop WJ, Stein PK, Gottdiener JS, Mukamal KJ.. Heart rate response to a timed walk and cardiovascular outcomes in older adults: the cardiovascular health study. Cardiology 2012;122:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spragg DD, Kass DA.. Pathobiology of left ventricular dyssynchrony and resynchronization. Prog Cardiovasc Dis 2006;49:26–41. [DOI] [PubMed] [Google Scholar]
- 5. Nielsen JC, Kristensen L, Andersen HR, Mortensen PT, Pedersen OL, Pedersen AK.. A randomized comparison of atrial and dual-chamber pacing in 177 consecutive patients with sick sinus syndrome: echocardiographic and clinical outcome. J Am Coll Cardiol 2003;42:614–23. [DOI] [PubMed] [Google Scholar]
- 6. Gielen S, Schuler G, Adams V.. Cardiovascular effects of exercise training: molecular mechanisms. Circulation 2010;122:1221–38. [DOI] [PubMed] [Google Scholar]
- 7. Andreev-Andrievskiy AA, Popova AS, Borovik AS, Dolgov ON, Tsvirkun DV, Custaud Met al. Stress-associated cardiovascular reaction masks heart rate dependence on physical load in mice. Physiol Behav 2014;132:1–9. [DOI] [PubMed] [Google Scholar]
- 8. Kneller J, Sun H, Leblanc N, Nattel S.. Remodeling of Ca(2+)-handling by atrial tachycardia: evidence for a role in loss of rate-adaptation. Cardiovasc Res 2002;54:416–26. [DOI] [PubMed] [Google Scholar]
- 9. Varian KD, Janssen PM.. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol 2007;292:H2212–9. [DOI] [PubMed] [Google Scholar]
- 10. Dewenter M, von der Lieth A, Katus HA, Backs J.. Calcium signaling and transcriptional regulation in cardiomyocytes. Circ Res 2017;121:1000–20. [DOI] [PubMed] [Google Scholar]
- 11. Hsu A, Duan Q, McMahon S, Huang Y, Wood SA, Gray NSet al. Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling. J Clin Invest 2020;130:2966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi Xet al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell 2012;149:671–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Planavila A, Redondo-Angulo I, Villarroya F.. FGF21 and cardiac physiopathology. Front Endocrinol (Lausanne) 2015;6:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uitterdijk A, Springeling T, Hermans KCM, Merkus D, de Beer VJ, Gorsse-Bakker Cet al. Intermittent pacing therapy favorably modulates infarct remodeling. Basic Res Cardiol 2017;112:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu L, Mei DF, Gu AG, Wang S, Lentzner B, Gutstein DEet al. Exercise training normalizes altered calcium-handling proteins during development of heart failure. J Appl Physiol 2002;92:1524–30. [DOI] [PubMed] [Google Scholar]
- 16. Anderson ME, Brown JH, Bers DM.. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 2011;51:468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni Get al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 2005;11:409–17. [DOI] [PubMed] [Google Scholar]
- 18. Cerra MC, Imbrogno S.. Phospholamban and cardiac function: a comparative perspective in vertebrates. Acta Physiol (Oxf) 2012;205:9–25. [DOI] [PubMed] [Google Scholar]
- 19. Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka Oet al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 2005;1:259–71. [DOI] [PubMed] [Google Scholar]
- 20. Galindo CL, Skinner MA, Errami M, Olson LD, Watson DA, Li Jet al. Transcriptional profile of isoproterenol-induced cardiomyopathy and comparison to exercise-induced cardiac hypertrophy and human cardiac failure. BMC Physiol 2009;9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files). Gene sequencing data is available at https://www.ncbi.nlm.nih.gov/geo/; access # 186604.