

SUMMARY
Mutational signatures defined by single base substitution (SBS) patterns in cancer have elucidated potential mutagenic processes that contribute to malignancy. Two prevalent mutational patterns in human cancers are attributed to the APOBEC3 cytidine deaminase enzymes. Among the seven human APOBEC3 proteins, APO-BEC3A is a potent deaminase and proposed driver of cancer mutagenesis. In this study, we prospectively examine genome-wide aberrations by expressing human APOBEC3A in avian DT40 cells. From whole-genome sequencing, we detect hundreds to thousands of base substitutions per genome. The APOBEC3A signature includes widespread cytidine mutations and a unique insertion-deletion (indel) signature consisting largely of cytidine deletions. This multi-dimensional APOBEC3A signature is prevalent in human cancer genomes. Our data further reveal replication-associated mutations, the rate of stem-loop and clustered mutations, and deamination of methylated cytidines. This comprehensive signature of APOBEC3A mutagenesis is a tool for future studies and a potential biomarker for APOBEC3 activity in cancer.
In brief
APOBEC3 cytidine deaminases are putative cancer mutagens. DeWeerd et al. experimentally define the genome-wide spectrum of mutagenesis caused by APOBEC3A deamination activity, which includes base substitutions, deletions, and mutations of 5-methylcytidines at CpG motifs. This mutational signature is prevalent in human cancers and provides a biomarker for APOBEC3A activity.
Graphical Abstract
INTRODUCTION
Characterization of mutational patterns in cancer genomes has indicated processes responsible for somatic mutagenesis that may contribute to initiation or progression of malignancy. These patterns consist of single base substitutions (SBS) and are defined by the nucleotide context in which they occur (Alexan-drov et al., 2013; Nik-Zainal et al., 2012). Mutational patterns identified in human cancer genomes are designated as mutational signatures, many of which have proposed or experimentally defined etiologies (Alexandrov et al., 2013, 2020; Tate et al., 2019). Two signatures, SBS2 and SBS13, are identified frequently in human tumors and have been attributed to the enzymatic activity of the APOBEC3 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3) family of cytidine deaminases (Burns et al., 2013b; Chen et al., 2017, 2020; Cortez et al., 2019; Nik-Zainal et al., 2012; Roberts et al., 2013; Robertson et al., 2017; Wang et al., 2017). The APOBEC3 family consists of seven members (APOBEC3A–APOBEC3H) that catalyze conversion of cytidine to uracil (C > U) in single-stranded DNA (ssDNA) (Chen et al., 2006; Conticello, 2012; Jarmuz et al., 2002; Yu et al., 2004). SBS2 and SBS13 consist predominantly of cytidine mutations within a TC dinucleotide context, the preferred context for activity of most APOBEC3 enzymes in vitro (Bogerd et al., 2007; Chan et al., 2015). SBS2 consists primarily of C > T mutations, thought to be caused by replication across a uracil base, whereas SBS13 consists of C > G and C > A mutations, possibly caused by error-prone polymerase activity at abasic sites (Chan et al., 2013). The prevalence of SBS2 and SBS13 across cancer genomes suggests that deamination by APOBEC3 enzymes is a frequent source of somatic mutation in human tumors (Burns et al., 2013b; Nik-Zainal et al., 2012; Petljak and Alexandrov, 2016; Roberts et al., 2013).
The APOBEC3 enzymes function as a part of the innate immune response by deaminating cytidine bases in viral genomes and retroelements to restrict infection and limit retrotransposition (Chen et al., 2006; Harris et al., 2003; Jarmuz et al., 2002; Man-geat et al., 2003; Narvaiza et al., 2009). Several APOBEC3 family members, specifically APOBEC3A and APOBEC3B, can localize to the nucleus and act on genomic DNA, causing mutations, breaks, and DNA damage signaling (Burns et al., 2013a; Green et al., 2016; Landry et al., 2011; Mussil et al., 2013). The finding of APOBEC3-induced DNA damage in experimental systems, combined with attribution of APOBEC3 activity to prevalent mutational patterns in cancer, suggests that these enzymes are important cancer mutagens (Green and Weitzman, 2019). Studies in 293T cell lines, yeast, and mouse carcinoma models have evaluated the spectrum of SBS mutations caused by APOBEC3 activity, yielding patterns similar to SBS2 and SBS13 (Akre et al., 2016; Law et al., 2020; Taylor et al., 2013). Although the specific APOBEC3 family member responsible for cancer mutagenesis has not been defined, evidence in favor of APOBEC3A and APOBEC3B as mutagenic drivers exists. APOBEC3A is a more potent enzyme, and recent studies indicate that mutational patterns distinctly associated with APOBEC3A activity are more prevalent in cancer cells and human tumors (Cortez et al., 2019; Jalili et al., 2020; Petljak et al., 2021). Although several studies to date have examined the SBS signature of APOBEC3 activity, we sought to establish a comprehensive view of the various genomic components that contribute to and result from APOBEC3 deamination in a single experimental system.
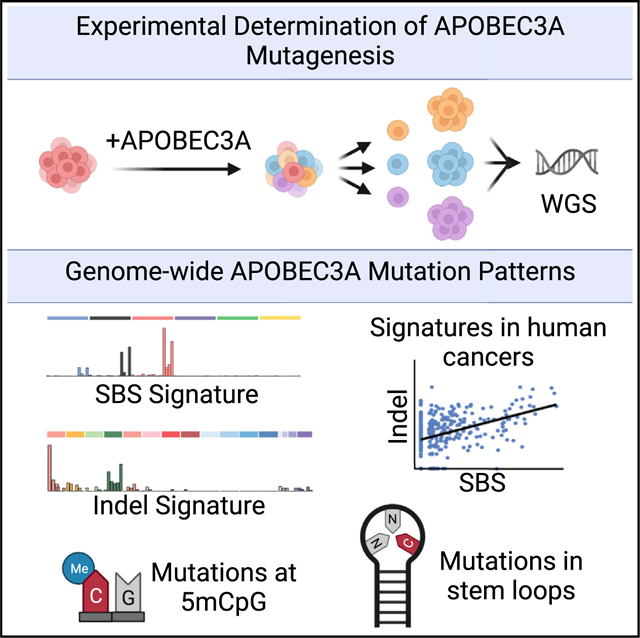
In this prospective study, we utilize the avian DT40 cell line to experimentally define the genome-wide consequences of APO-BEC3A mutagenesis. The DT40 line, which does not encode APOBEC3 orthologs, acquires few mutations in long-term culture and, thus, is a suitable system in which to examine human APO-BEC3A activity. By whole-genome sequencing (WGS), we demonstrate that the experimentally defined APOBEC3A mutational signature includes SBSs, insertions-deletions (indels), and clustered mutations. The SBS signature generated by APOBEC3A is a combination of COSMIC (Catalogue of Somatic Mutations in Cancer) signatures SBS2 and SBS13 and is prevalent in human cancers. Surprisingly, we find that APOBEC3A generates genomic indels that arise independent of base substitutions and comprise a unique indel signature which is also evident in human cancer genomes that harbor the APOBEC3A SBS signature. Our data show that APOBEC3A generates clustered and dispersed mutations across the genome in an apparently stochastic manner. Consistent with prior reports, we find that cytidine deamination by APOBEC3A occurs primarily on the lagging strand during DNA replication. Interestingly, WGS of DT40 genomes demonstrates that in vivo APOBEC3A activity results in deamination of 5-methylcytidines (5mC) at CpG sites with a frequency similar to deamination of unmodified cytidines. Our findings present a multi-dimensional mutational signature of APO-BEC3A activity that is a potential biomarker in human cancers and implicates APOBEC3A activity in previously undefined consequences on the genome.
RESULTS
Modeling human APOBEC3A activity in avian cells
To evaluate APOBEC3A mutational patterns, we sought a system with minimal endogenous mutational processes. The avian DT40 cell line, which has a diploid genome, acquires notably few mutations in culture (Szikriszt et al., 2016) and has remarkable cloning efficiency, making it suitable for experiments requiring single-cell clone expansion (Harris et al., 2003; Yamazoe et al., 2004). DT40 cells, which express uracil DNA glycosylase, are used frequently to study the activities of activation-induced deaminase (AID), an enzyme that mutates cytidines in a sequence context distinct from that of APOBEC3A (Rogozin et al., 2016). However, DT40 cells do not encode an APOBEC3 ortholog, making them an ideal system to study APOBEC3A mutagenesis. Through lentiviral integration, we introduced a doxycycline (dox)-inducible human APO-BEC3A transgene to DT40 cells (DT40-A3A). We subsequently evaluated a single DT40-A3A ancestral clone. Expression of the APOBEC3A transcript and protein in the clone was detected upon treatment with dox (Figures 1A and S1A). Enzymatic activity of human APOBEC3A was verified by deamination assay (Figure 1B). Using a low dose of dox, we observed a moderate proliferativedefectinDT40-A3Acellsthatdidnotoccurinadox-treated wild-type DT40 clone (Figure S1B). Consistent with prior studies, we observed an increase in the phosphorylated histone variant H2AX (γH2AX), a marker of DNA breaks, in DT40-A3A cells (Figure S1C; Burns et al., 2013a; Green et al., 2016; Landry et al., 2011; Mussil et al., 2013). We additionally observed a modest cell cycle arrest in G2, a consequence of APOBEC3A activity during replication (Figure S1D; Green et al., 2016). Our findings demonstrate that human APOBEC3A acts similarly in avian cells as in human cells, resulting in deamination, DNA damage, and cell cycle arrest.
Figure 1. The genome-wide spectrum of mutations generated by human APOBEC3A in DT40 cells.

(A) Human APOBEC3A (A3A) was expressed inavian DT40 cells. Doxycycline (dox)-induced A3A expression was evaluated by immunoblot. A3A is detected by a hemagglutinin (HA) tag. The image is representative of two biological replicates.
(B) Deaminase activity in DT40-A3A cells. Lysates were incubated with a ssDNA oligonucleotide containing a single cytosine. Cytosine deamination followed by addition of uracil-DNA glycosylase (UDG) results in an abasic site; incubation with NaOH results in oligonucleotide cleavage. Substrate (S) and product (P) bands are visualized by gel electrophoresis. Oligonucleotides that contain a single uracil or cytosine (TU and TC), incubated in the absence of cell lysate, are controls. The image is representative of three biological replicates.
(C) Experimental schematic for evaluating A3A mutagenesis. A3A expression was induced in a DT40-A3A ancestral clone for 30 days. Subsequent single cell selection yielded descendant clones (n = 16), which were evaluated by whole-genome sequencing (WGS). In parallel, three control populations were cultured, sequenced, and analyzed: DT40 wild type (WT) untreated (n = 3), WT dox treated (n = 3), and DT40-A3AC106S (catalytically inactive A3A mutant, n = 3).
(D) Number of SBS mutations per genome. Each dot represents the base substitution burden within an individual descendant clone. Statistical analysis was performed by two-tailed t test; the bar indicates the median. ***p < 0.001.
(E) Total cytidine mutations. Left: cumulative cytidine mutations shown as a percentage of all SBSs in descendant clone genomes from WT and DT40-A3A cells. Right: dinucleotide contexts in which C base substitutions are shown as fractions of all mutated cytidines.
APOBEC3A deamination results in an increased burden of genomic cytidine mutations
To study mutations resulting from APOBEC3A activity, we designed a 50-day experiment that included initial expansion of a DT40-A3A ancestral clone, treatment with dox for 30 days, and isolation of descendant clones (n = 16). WGS was utilized to identify mutations acquiredin descendant clones (Figure 1C). Intermittent dox dosing resulted insustained APOBEC3A expression (Figure S1E). Controls included wild-type DT40 clones cultured in the presence or absence of dox in parallel to examine dox-induced and spontaneous mutagenesis, respectively, as well as transgenic expression of a catalytically inactive APOBEC3A mutant, A3AC106S (Figures S1F–S1H). Ancestral and descendant clones were subjected to WGS and analysis by IsoMut, which enables streamlined identification of mutations across isogenic samples (Pipek et al., 2017). The overall mutation burden among DT40-A3A descendant clones was significantly higher and notably variable in comparison with controls (Figures 1D and 1E). Mutations acquired in DT40-A3A descendant clones consisted largely of substitutions at cytidine bases in the TC context, characteristic of in vitro APOBEC3A activity and consistent with prior studies of APOBEC3A mutagenesis in mouse and yeast models (Figure 1E; Bogerd et al., 2007; Chan et al., 2015; Chen et al., 2006; Hoopes et al., 2016; Law et al., 2020; Taylor et al., 2013).
Analysis of 16 descendant DT40-A3A clones revealed the genome-wide spectrum of all mutations acquired during the 50-day experiment (Figure S2A, top). The observed base substitutions included mostly C > T and C > G substitutions, both of which are associated with APOBEC3 activity (Akre et al., 2016; Alexandrov et al., 2013; Burns et al., 2013b; Law et al., 2020; Nik-Zainal et al., 2012; Petljak et al., 2021; Roberts et al., 2013; Tate et al., 2019). However, we observed an unremarkable mutational spectrum among DT40-A3AC106S descendant clones with few base substitutions that resembled the mutational spectrum in DT40 wild-type genomes (Figure S2A). These data demonstrate that enzymatically active APOBEC3A generates substantial genomic mutagenesis.
The experimentally defined SBS signature of APOBEC3A activity
We sought to determine the de novo APOBEC3A SBS signature by removing SBS caused by background mutagenesis in the DT40 system. By applying non-negative matrix factorization (NMF), a mathematical algorithm that was used in the original determination of somatic mutational signatures from cancer genomes (Alexan-drov et al., 2013; Lee and Seung, 1999; Nik-Zainal et al., 2012), to the DT40 descendant clone genomes, we extracted two distinct mutational signatures (Figures 2A and S2B). The first (Figure 2A, top) is comprised of mostly TC context mutations and represents the experimentally defined APOBEC3A mutational signature, hereafter referred to as the A3A SBS Signature. The second (Figure 2A, bottom) has a non-specific spectrum of mutations and is attributed to background mutational processes in cultured DT40 cells. Among individual clones, we found that the majority of mutations in DT40-A3A clones are comprised of the A3A SBS Signature, whereas mutations in control clones consist almost exclusively of the background signature (Figure 2B).
Figure 2. The mutational signature of APOBEC3A activity is prevalent in human cancers.

(A) From the entire spectrum of mutations found in DT40-A3A descendant clones and controls, two SBS mutational signatures were derived using non-negative matrix factorization (NMF). The relative contribution of each SBS (top, x axis) within a trinucleotide context (bottom, x axis) is shown. The mutational signature consistent with APOBEC3A deaminase activity is denoted the A3A SBS Signature.
(B) The contribution of each experimentally derived mutational signature for individual descendant clones.
(C) The pentanucleotide context in which cytidine mutations occur in DT40-A3A descendant clones. Letter size represents relative frequency of each base flanking mutated cytidines. Shown is the pentanucleotide context of all TC > TN mutations, where N is any nucleotide (left), and TCA > TTA mutations (right). p < 0.0001 by Fisher’s exact test for the overrepresentation of G.
(D) The two experimentally generated mutational signatures derived from DT40-A3A genomes are compared with the SBS signatures defined in COSMIC v.3. Pearson’s correlation was used to generate the heatmap. Arrows mark SBS2 and SBS13, attributed previously to APOBEC3 activity.
(E) Cancer genomes from tissues associated previously with APOBEC3 were evaluated for A3A SBS Signature mutations. The fraction of A3A SBS Signature mutations in individual tumors from PCAWG is shown for bladder cancer (BLCA), breast adenocarcinoma (BRCA), cervical squamous cell carcinoma (CESC), head and neck squamous cell carcinoma (HNSC), and uterine corpus endometrioid carcinoma (UCEC). The bar indicates the median.
(F) All cancer genomes from PCAWG were evaluated for SBS2 and SBS13 mutations compared with A3A SBS Signature mutations. The slope, R value, andsignificance (p value) of the correlation were determined using linear regression.
Prior in silico and yeast studies have aimed to refine the APO-BEC3 mutational signature by analyzing the extended nucleotide context in which deamination-induced mutations occur. These studies demonstrated a tetranucleotide context preference of YTCA (Y = pyrimidine) for APOBEC3A (Chan et al., 2015; Cortez et al., 2019). Consistent with this, we found TC mutations mostly in the YTCA nucleotide context (Figure 2C, left) and also observed a preference for G in the +2 position, which was enhanced when evaluating C > T mutations within a TCA motif (Figures 2C, right, and S2C). The YTCAG pentanucleotide context offers a more granular definition of the APOBEC3A deamination context and may further distinguish APOBEC3A mutagenesis from that of other deaminases.
The A3A SBS Signature is similar to COSMIC signatures SBS2 and SBS13
Prior studies have relied on the attribution of APOBEC3 activity as the mutagenic source of SBS2 and SBS13, and important inferences have been drawn from this association. For example, the tissues in which APOBEC3 enzymes are thought to act were derived from enrichment of SBS2 and SBS13 in specific cancer genomes (Alexandrov et al., 2013; Burns et al., 2013b; Cortez et al., 2019; Leonard et al., 2013; Nik-Zainal et al., 2012). We compared the A3A SBS Signature with all published SBS signatures in COSMIC v.3 and found substantial similarity to SBS2 and SBS13 (Figure 2D, arrows). Deconstruction of the A3A SBS Signature revealed that SBS2 and SBS13 together comprise more than 85% of the experimentally defined A3A SBS Signature (Figure S2D). To assess the prevalence of the A3A SBS Signature in human cancers, we examined tumor genomes from the Pan-Cancer Analysis of Whole Genomes (PCAWG) study (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020). Among tumor types that have been associated previously with SBS2 and/or SBS13 (Burns et al., 2013b; Chan et al., 2015; Cortez et al., 2019; Leonard et al., 2013; Roberts et al., 2013; Shi et al., 2020), we found that most individual tumorsin this group harbor a high proportion of mutations from the A3A SBS Signature (Figure 2E). We also found a significant preference for G in the +2 position at TCA context mutations, as observed in DT40 cells (Figure S2E). We next evaluated all PCAWG tumors and found a striking correlation between the contribution of the A3A SBS Signature and SBS2 and/or SBS13 (Figures 2F, S2F, and S2G). The correlation was strongest when the A3A SBS Signature was compared with a combination of SBS2 and SBS13 (R = 0.97, Figure 2F). These data demonstrate that the A3A SBS Signature is prevalent in human cancers and encompasses SBS2 and SBS13 as a singular measure of APOBEC3A activity.
APOBEC3 deamination has been associated with mutations in specific genes (Burns et al., 2013a; Henderson et al., 2014; Mas-Ponte and Supek, 2020; Shi et al., 2019). A recent study delineated several oncogenes and tumor suppressor genes that were affected by putative APOBEC3 signature mutations in human cancers (Mas-Ponte and Supek, 2020). We assessed whether specific genes were mutated by APOBEC3A in DT40 genomes and found remarkably few recurrent mutations across all 16 DT40-A3A descendant clones (Figure S3A). Given the lack of mutational hotspots, we evaluated how frequently A3A SBS Signature mutations occur within genes compared with intergenic regions. We found a minority of mutations within genes and even fewer within transcribed regions of the genome (Figure S3B). We evaluated the frequency of mutations within specific genes among all descendant clones and found only seven genes that acquired more than one mutation per 20 kb in at least three DT40-A3A clones (Figure S3C). These avian genes did not cluster in biologically functional groups, nor did they overlap with the human genes in tumors with a high contribution of A3A SBS Signature mutations. Importantly, our system does not enable evaluation of clonal selection that may result from a specific APOBEC-induced oncogene mutation, as has been reported within PIK3CA in cervical cancer (Henderson et al., 2014). The heterogeneity in recurrent gene mutations reported to be caused by APOBEC3, our finding of few recurrently mutated genes, and the relative paucity of mutations within genes across DT40 genomes demonstrate that mutagenesis by APOBEC3A is stochastic and does not occur at hotspots.
APOBEC3A deamination generates a unique indel signature
In addition to the SBS spectrum, we observed an unexpected increase in the number of indels in DT40-A3A genomes (Figures 3A, S4A, and S4B; Table S1). Among DT40-A3A descendant clones, the frequency of indels correlated significantly with the SBS burden (R = 0.76; Figure S4C). Indels in DT40-A3A descendant clones were mostly 1-bp indels and some longer, microho-mology-associated deletions (Figure S4D, top). The difference between the DT40-A3A indel spectrum (Figure S4D, top) and that of control samples (Figure S4D, bottom) comprises a unique indel (ID) signature (Figure 3B). This signature shows few deletions with microhomology, which would indicate scars of double-stranded break (DSB) repair (Figure 3B). Interestingly, the predominant component of the A3A ID Signature was single cytidine deletions that occurred in a TC context (Figure 3C), suggesting that replicative or translesion synthesis (TLS) polymerase slippage at abasic sites created after uracil excision generate a significant portion of the observed indels.
Figure 3. APOBEC3A-related deletions in cancer genomes.

(A) Indels are increased in genomes exposed to APOBEC3A. The numbers of insertions and deletions per genome are displayed as an average of all descendant clone genomes evaluated for each cell type. Statistical analysis was performed using a two-tailed t test. ***p < 0.005, **p < 0.01; error bars indicate SD.
(B) The indel signature generated by APOBEC3A. Indels unique to the DT40-A3A genomes are characterized by indel length (top, x axis) and homology/repeat regions (bottom, x axis).
(C) The context of cytidine deletions in cultured DT40 genomes. The base preceding all C deletions in DT40 descendant clones is shown. DT40 controls include untreated WT, dox-treated WT, and DT40-A3AC106S.
(D) Indel signature deconstruction into COSMIC v.3 ID signatures. The contribution of each COSMIC ID signature to the A3A ID Signature is shown. An empty gray bar denotes combined remaining ID signature contributions.
(E) Correlation between A3A ID and SBS Signature contribution in PCAWG whole genomes. Tumor genomes from PCAWG were analyzed for the presence of theA3A SBS Signature (x axis) and the A3A ID Signature (y axis). The slope, confidence interval, and R and p values of linear regression for each dataset are shown. Shown are BLCA, BRCA, HNSC, and UCEC.
(F) The context of cytidine deletions in cancer. The base preceding cytidine deletions was analyzed in breast cancers with more than 15% A3A SBS Signature contribution (black dots) or less than 15% (gray dots). Each dot represents an individual tumor sample, the bar indicates the mean, and error bars indicate SEM. Statistical analysis was performed using Wilcoxon rank-sum test. ***p < 0.005; ns, non-significant.
We assessed the similarity of the A3A ID Signature to the ID signatures in COSMIC and identified substantial contributions from several previously detected ID signatures, three of which have no proposed etiology (Figure 3D). We found that ID9, of unknown etiology, comprises the most significant fraction of ID signature deconstruction (Figure 3D). The ID9 signature, which also contains primarily single cytidine deletions, is notably more frequent in breast, bladder, uterine, and lung cancer, all tumors in which the APOBEC-associated SBS signatures are detected (Alexan-drov et al., 2013, 2020; Tate et al., 2019). We applied the A3A ID Signature to human cancers and found a highly significant correlation with the A3A SBS Signature (R = 0.4, p < 2.6 3 10–14; Figures 3E and S4E). Addition of the A3A ID Signature to the COSMIC ID signature set significantly increased the accuracy of ID signature deconstruction in samples that bore evidence of A3A SBS mutagenesis (Figures S4F and S4G). We assessed the nucleotide context of single cytidine deletions in breast cancer genomes with a substantial burden (>15%) of the A3A SBS Signature and found a TC deletion preference, which was not present in genomes without A3A SBS Signature mutations (Figure 3F). The TC deletion preference was noted across all cancer types in PCAWG with substantial burdens of A3A SBS Signature mutations (Figure S4H). These data indicate that deamination by APOBEC3A generates indels that occur in a unique, previously unidentified pattern and that this indel pattern co-occurs with the A3A SBS Signature in human cancers.
APOBEC3A activity generates small omikli clusters more frequently than kataegis
An additional facet of mutational patterns previously attributed to APOBEC activity is that of clustered mutagenes is (Nik-Zainaletal., 2012; Taylor et al., 2013). Two patterns of mutation clusters have been observed: kataegis (thunderstorm), consisting of more than 5 mutations per cluster, and omikli (fog), consisting of 2–4 mutations per cluster (Mas-Ponte and Supek, 2020; Nik-Zainal et al., 2012; Petljak et al., 2021). We examined the DT40 descendant clones for evidence of clustered mutations. No mutation clusters were observed in DT40 wild-type clones, and only 5 omikli events occurred across all DT40-A3AC106S clones examined. In contrast, DT40-A3A clones harbored 688 omikli and 10 kataegis events across all 16 genomes sequenced (Figures 4A–4C), consistent with the contribution of clustered patterns reported previously in cancer genomes (Mas-Ponte and Supek, 2020; Seplyarskiy et al., 2016). The average number of omikli clusters per genome was 42 with a range of 4–161 (Figure 4A), which was similar to the frequency of omikli events reported recently in human cancer genomes(Mas-Ponte and Supek, 2020).With in individual descendant clones, a strong correlation between the frequency of mutation clusters and SBS burden was observed (Figure 4D; R = 0.97), indicating that APOBEC3A activity generates a relatively fixed ratio of clustered and dispersed mutations. We assessed the spectrum of mutations within clusters in all DT40-A3A genomes (Figure 4E) and found a spectrum indistinguishable from the total DT40-A3A spectrum (Figure S2A), suggesting that nucleotide context does not influence whether mutations occur in clusters. We additionally analyzed strand coordination of mutation clusters, in which series of only mutated guanines or only mutated cytosines appear on a single strand, as reported previously in yeast (Roberts et al., 2012). Among clustered mutations in DT40-A3A clones, we observed significant strand coordination (Figure 4F), which highly suggests that mutagenesis resulting in clusters occurs during a single replication cycle by a common mechanism.
Figure 4. APOBEC3A activity generates clustered mutations.

(A and B) The number of mutation clusters per descendant clone genome are quantified as (A) omikli, defined as 2–4 mutations, or (B) kataegis, defined as 5 or more mutations within 20 kb.
(C) Rainfall plots from representative descendant clones. All SBSs within a single genome are plotted by genome position (x axis) and distance betweenneighboring mutations (y axis). Mutations in close proximity appear toward the low end of the y axis. Dot color indicates a specific base substitution (shown in the legend).
(D) The number of mutation clusters is correlated with the number of total base substitutions in DT40-A3A descendant clones. The slope and R and p values oflinear regression are shown.
(E) The averaged spectrum of mutations located within kataegis and omikli clusters from all DT40-A3A descendant clones.
(F) Mutation clusters are quantified according to number and type of bases altered within each cluster; for example, only C mutations, only G mutations, orcombinations of base mutations. Mutated bases within each cluster are shown on the x axis. C-only and G-only clusters, which indicate strand coordination of deaminase activity, are highlighted in purple and green, respectively.
Genomic substrates of APOBEC3A mutagenesis
Clustered mutations have been proposed to occur through processive deamination of the lagging strand during replication (Bhagwat et al., 2016; Haradhvala et al., 2016; Hoopes et al., 2016; Seplyarskiy et al., 2016), the exposed ssDNA remaining after resection of DSB ends (Lei et al., 2018; Roberts et al., 2012; Taylor et al., 2013), opposite R-loops during transcription (Hamperl and Cimprich, 2014; Love et al., 2012), and, most recently, ssDNA tracts formed during mismatch repair (Mas-Ponte and Supek, 2020). To assess the substrates on which APOBEC3 activity results in DNA mutations, we first evaluated the frequency of mutations on the leading and lagging replication strands (Figure 5A, diagram). We determined the probability of the direction of replication at each point of the DT40 genome by sequencing highly purified Okazaki fragment DNA (Ok-seq) (Petryk et al., 2016). We found that mutations of C and G in the reference genome correlated with replication fork directionality (Figure 5A). Specifically, TC mutations occurred more frequently on the parental lagging-strand template (Figure 5B). We evaluated mutations at transcribed regions of the genome and found no increase in mutated cytidines at transcribed (template) strands compared with nontranscribed (coding) strands (Figure 5C), consistent with prior in silico studies (Haradhvala et al., 2016; Hoopes et al., 2016; MasPonte and Supek, 2020).
Figure 5. APOBEC3A deamination occurs at specific genomic substrates.

(A) Density plot of mutated C and G bases relative to replication fork directionality (RFD). Positive RFD indicates a rightward-oriented fork, in which case the sense strand is the lagging strand. Negative RFD indicates a leftward-oriented fork. RFD is calculated by the difference between rightward- and leftward-oriented forks; thus, a value of 0 means that, at that location, equal numbers of forks go to the right and to the left. A replication fork diagram depicting leading and lagging strand directions is shown below.
(B) SBSs found to be associated with replication forks from all DT40-A3A descendant clone genomes are quantified as leading-strand (blue) or lagging-strand(red) mutations. Base substitution in the indicated trinucleotide context is shown on the x axis.
(C) The numbers of SBSs in transcribed and non-transcribed strands from DT40-A3A genomes are quantified and categorized by base substitution (x axis).
(D) The DT40 genome was divided into deciles by timing of replication from early (left) to late (right). Mutated cytidines in a TCN context are shown, where N is any base.
(E) Clustered mutations by the timing of replication. Top: number of mutation clusters (omikli and kataegis) per replication decile. Bottom: number of kataegis events per replication decile.
(F) APOBEC3A mutates DNA stem loops. Cytidine mutations in DT40-A3A genomes were compared with 38,325 control positions that represent the same trinucleotide spectrum. The ratio of APOBEC3A-induced mutations and control positions found in putative stem loops is categorized based on stem strength. The ratio was normalized to 1 in the “no stem loop” column (<3 bases in the stem).
(G) The difference between APOBEC3A and random stem loop mutation frequency is shown as a heatmap. The fraction of APOBEC3A mutations in each type ofstem loop (based on stem strength and loop length) relative to that of the control positions shows that the putative loops mutated by APOBEC3A in the DT40 genome have a stem strength of 5–25 and a loop length of 3–5.
(H) Sequence preference of APOBEC3A reflected in 3 nucleotide stem-loop mutations. TpC sites located entirely within the loop are mutated preferentially compared with loops where T is part of the stem structure.
(I) Bisulfite sequencing revealed the frequency with which each cytidine base within a CpG dinucleotide context in the DT40 genome was methylated (x axis). The proportion of cytidines mutated by APOBEC3A (red) and all other cytidines (gray) are indicated with respect to the likelihood that each cytidine base is methylated.
From in silico evaluation of cancer genomes, APOBEC3-associated mutagenesis is proposed to correlate with replication timing. However, different analyses have found SBS2/13 to be enriched in early-replicating regions (Kazanov et al., 2015) or late-replicating regions (Morganella et al., 2016). This variability is perhaps dependent on whether mutations occur in clusters or independently and the mechanisms by which deaminated bases are repaired (Mas-Ponte and Supek, 2020). It is also possible that the expression and/or activity levels of APOBEC3A in tumors or experimental systems influence the promiscuity of mutagenesis (e.g., during early replication or throughout the entirety of replication). We investigated APOBEC3A mutagenesis relative to replication timing by dividing replication of the DT40 genome into chronological deciles (Shang et al., 2013). When all TC mutations were assessed, they appeared to occur with equal frequency throughout replication (Figure 5D). Similarly, we found no correlation with replication timing when all mutation clusters were analyzed together (Figure 5E, top). However, independent analysis of kataegis events demonstrated enrichment in the earliest-replicating regions (Figure 5E, bottom). Although the total number of kataegis events was low (n = 10), this suggests distinct molecular mechanisms driving omikli and kataegis clusters (Mas-Ponte and Supek, 2020) and may provide insights into mechanisms of kataegis formation.
An additional substrate on which APOBEC3A has been shown to act is putative DNA stem loops, formed by palindromic sequences in close proximity in the genome (Buisson et al., 2019; Langenbucher et al., 2021; Shi et al., 2020). We assessed APOBEC3A activity at predicted stem loops in DT40 genomes by analyzing cytidines mutated by APOBEC3A in comparison with 38,325 randomly selected control positions throughout the genome with a trinucleotide spectrum matching that of APOBEC3A target sites. We found that APOBEC3A-mediated mutations were enriched in putative stem loops with a strong base pairing palindrome stem sequence compared with the control positions (Figures 5F, 5G, and S5A). An examination of loop lengths revealed that stem loops mutated by APOBEC3A were only enriched in the case of shorter loops (Figures 5G, S5A, and S5B) and that APOBEC3A activity occurred preferentially at cytidines in the final but not the initial loop position (Figure 5H), consistent with prior biochemical and in silico studies (Buisson et al., 2019; Langenbucher et al., 2021). The spectrum of APOBEC3A-mediated mutations in loops differs slightly from the global A3A SBS Signature (Figures S5C and S5D), consistent with a previously reported tendency for APOBEC3A to mutate VpC sites (V = not T) within optimal stem-loop structures (Lan-genbucher et al., 2021). However, we found that the altered spectrum of mutations caused by APOBEC3A in stem loops reflects the composition of the loops (Figures S5C and S5D), which are likely to be GC rich.
Deamination of methylated and unmodified cytidines by APOBEC3A in vivo
Based on the apparent alteration of mutation spectra in stem loops, we evaluated the genome-wide A3A SBS Signature normalized to trinucleotide occurrence. Surprisingly, after trinucleotide normalization, we found that TCG > TTG is the most frequent event resulting from APOBEC3A activity (Figure S6A). CpG dinucleotides are infrequent in vertebrate genomes, and the majority contain 5mC (Bird, 2002), an epigenetic modification critical for regulation of gene expression and numerous other cellular processes. The A3A SBS Signature includes predominantly C > T and only rarely C > A and C > G at CpG motifs (Figure 2A). Deamination of 5mC catalyzes a direct C > T transition without variability in mutations resulting from processing of a uracil base. We hypothesized that the frequency of TCG > TTG mutations was due to deamination of 5mC by APOBEC3A. Consistent with this hypothesis, in the absence of APOBEC3A, the trinucleotide occurrence normalized mutational spectrum demonstrates a C > T mutation pattern consistent with that of the previously defined SBS1, which represents spontaneous 5mC deamination (Figure S6B). Previous in vitro studies have shown that 5mCs are not deaminated as efficiently by APOBEC3A as unmodified cytidines (Ito et al., 2017; Schutsky et al., 2017). To assess 5mC deamination by APOBEC3A in vivo, we mapped 5mC sites in the DT40 genome by bisulfite sequencing. Distinct from prior studies, we found that methylated and unmodified cytidines were mutated at similar rates by APOBEC3A (Figure 5I). The preference of APOBEC3A for TCG trinucleotides is not necessarily influenced by CpG methylation but reflects a previously undocumented high affinity for this sequence context.
DISCUSSION
The inherent genomic stability of the DT40 genome, along with the depth of previous characterization of this cell line, has enabled comprehensive and granular characterization of genomic aberrations elicited by APOBEC3A deamination. The human APOBEC3A enzyme acts similarly in DT40 cells as it does in human cancer cells based on deamination activity, DNA damage responses, and effect on proliferation (Figures 1 and S1). Whole-genome analysis of APOBEC3A activity in DT40 cells yielded a SBS signature comprised almost entirely of a combination of SBS2 and SBS13 and resembles SBS spectra generated recently in other model systems in which APOBEC3 activity has been analyzed prospectively (Akre et al., 2016; Law et al., 2020; Petljak et al., 2021). Although APOBEC3B has been proposed as a source of mutagenesis and DNA damage in cancer (Burns et al., 2013a, 2013b), our data show that APOBEC3A is sufficient to generate a SBS spectrum consistent with those found in human cancer genomes. However, a limitation of this model is that only APOBEC3A, and not the other APOBEC3 family members that are potential cancer mutagens, is assessed. We rely on ectopic expression of APOBEC3A, which may not accurately reflect the variable and presumably fluctuating levels of expression or activity reported in human cancers. Among descendant clones, we find a large range of mutational burden resulting from APOBEC3A, which may reflect the stochastic nature of enzyme activity or intermittent interactions between APOBEC3A and the genome. Analysis of DT40-A3A clones reveals a multidimensional mutational pattern generated by APOBEC3A deamination activity, including a distinct indel signature, characteristic clustered mutations, replication-associated mutagenesis, and deamination of methylated cytidines. We show that this extended mutational pattern is evident in cancer genomes, bolstering the long-standing hypothesis that APOBEC3A contributes to somatic mutagenesis in human malignancies (Alexandrov et al., 2013; Burns et al., 2013a; Harris et al., 2002; Nik-Zainal et al., 2012).
Indels have been reported previously as a result of APOBEC3 activity near Cas9-generated DSBs (Lei et al., 2018), although genome-wide studies have not reported APOBEC3-mediated indels (Akre et al., 2016; Chan et al., 2015; Law et al., 2020; Taylor et al., 2013). Here we show that APOBEC3A generates an indel pattern composed of primarily single cytidine deletions arising in the canonical TC context as well as longer deletions with microhomology. This unique indel signature is also found in cancer genomes that harbor A3A SBS Signature mutations. By deconstructing the A3A ID Signature, we find components of several previously noted ID patterns, most of which have unknown etiologies (ID4, ID9, and ID11). In our studies, we have shown that APOBEC3A alone is sufficient to generate a combination of SBS2 and SBS13. The same paradigm may apply to ID signatures; APOBEC3A generates a unique ID signature that may encompass several COSMIC ID patterns. Although the mechanism by which APOBEC3A-mediated indels has not been determined, characterization of the indels reveals a likely contribution of strand-slippage errors. Uracils resulting from deamination events are excised by uracil-DNA glycosylase (UDG), leaving abasic sites. In the context of DNA break repair, indels induced by APOBEC3 deamination are dependent on UDG and promoted by the activity of endonucleases such as MRE11 (Lei et al., 2018). Outside of DNA break repair, slippage of TLS or replicative polymerases acting at abasic sites can generate addition or deletion of a nucleotide (Taylor et al., 2004).
Recent studies have demonstrated the importance of the TLS polymerase REV1 in APOBEC3-mediated mutations (Petljak et al., 2021; Taylor et al., 2013), and REV1 may also function in APOBEC3-mediated small indels. In addition to small indels, TLS is proposed to be the mechanism by which APOBEC3 mutagenesis converts C > A and C > G, as in SBS13. In contrast, SBS2, which is comprised of C > T mutations, is thought to be caused by replicative polymerases pairing U with A following deamination. In subsequent replication cycles, A is paired with T, ultimately resulting in a C > T transition. We observed both patterns of mutations resulting from APOBEC3A activity, which may represent various methods by which uracils are processed in a repair-competent cell. These patterns may be skewed in cancer cells with deficiencies in base excision repair, TLS, or other DNA repair pathways.
Longer deletions that display microhomology are likely to represent sites of DSB repair by non-homologous end joining or microhomology-mediated end joining, suggesting that APOBEC3A generates DSBs. This has been proposed previously based on observation of pan-nuclear γH2AX staining upon ectopic APOBEC3A expression (Burns et al., 2013a; Landry et al., 2011). Generation of DSBs occurs during deaminase-mediated class-switch recombination because of activity of the related AID on opposite strands of DNA in close proximity (Di Noia and Neuberger, 2007). Subsequent abasic sites on opposing strands result in DSBs (Daniel and Nussenzweig, 2013). Deamination by APOBEC3A may result in a similar phenomenon of DSB generation. However, based on our data and prior studies, APOBEC3A does not appear to act during transcription, which is the modus operandi of AID (Chaudhuri et al., 2003; Ramiro et al., 2003). Experimental and in silico data have shown that replication forks are a substrate for APOBEC3 deamination (Bhagwat et al., 2016; Green et al., 2016; Haradhvala et al., 2016; Hoopes et al., 2016; Seplyarskiy et al., 2016). Deamination of cytidines, followed by uracil excision and subsequent apurinic/apyrimidinic (AP) endonuclease activity on one strand of the replication fork, may result in collapse of the fork to a DSB.
Here we demonstrate that APOBEC3A generates SBSs primarily on the lagging strand, which is consistent with prior studies of SBS2 and SBS13 prevalence on the lagging strand in cancer genomes (Haradhvala et al., 2016; Seplyarskiy et al., 2016). We show that putative stem loops with strong stems and small loops are frequent substrates for APOBEC3A mutagenesis. Among stem loops, the spectrum of APOBEC3A-induced mutations is altered relative to non-loop regions of the genome but appears to reflect the base composition of genomic regions likely to form stem loops. Across the entire genome, we demonstrate that TCG motifs have the highest mutation rate. Deamination of a 5mC would result in T rather than U, leading to a direct C > T mutation, and we find that essentially only C > T (but rarely C > A and C > G) mutations are generated by APOBEC3A at CpG motifs. Thus, the propensity for APOBEC3A to generate CG > TG mutations may result from deamination of methylated CpG, which occurs as frequently as deamination of unmodified cytidines in our analysis. Methylated cytidines are prone to spontaneous deamination, and a common mutational signature in cancer, SBS1, is attributed to attrition of 5mCpG by this mechanism throughout aging (Alexandrov et al., 2015). We found that the most frequently mutated motif in control genomes is also CpG (Figure S6B), suggesting that some degree of 5mC demethylation is occurring regardless of APOBEC3A activity. However, the burden of CpG mutations in control genomes is far lower than that in DT40-A3A genomes (Figure S6). We found not just a predominance of CpG mutations in DT40-A3A genomes but of TCG mutations, which are more specific to APOBEC3A activity. Our data show that APOBEC3A deaminates methylated cytidines in vivo and support the hypothesis that APOBEC3A may contribute to mutagenesis observed in signatures other than SBS2 and SBS13 (Langenbucher et al., 2021).
In vitro biochemical studies have demonstrated the capacity of APOBEC3A to deaminate methylated cytidines, although with reduced efficiency compared with deamination of unmodified cytidines (Carpenter et al., 2012; Ito et al., 2017; Schutsky et al., 2017; Suspene et al., 2013; Wijesinghe and Bhagwat, 2012). Thus, a biochemical affinity for 5mC as a substrate is unlikely to be the reason for substantial deamination of methylated cytidines. Our finding of mutations at 5mC is consistent with a recent observation that APOBEC-dependent C > T mutations in cancer cell lines are not dependent on UDG (Petljak et al., 2021). It is possible that, because 5mC > T mutations occur independent of BER, these events more frequently result in base substitution compared with deamination of unmodified cytidines, which are subject to uracil processing and may be accurately repaired. Methylation of CpG sites can be associated with increased chromatin accessibility (Pott, 2017), which may enable deamination events by APOBEC3A. Although a relationship between APOBEC3 deamination sites and chromatin architecture has not been established, evaluation of cancer genomes identified increased APOBEC3-associated mutations in regions of accessible chromatin (Kazanov et al., 2015). Genome-wide loss of methylation is a hallmark of cancer (Shen and Laird, 2013), and our data suggest that APOBEC3A deamination may contribute to alteration of individual gene expression or general hypomethylation of cancer genomes.
APOBEC3 activity has been suggested as a therapeutic target either by limiting clonal progression of cancer via inhibition of mutagenesis or by promoting genotoxicity and cancer cell death through inhibition of DNA repair pathways. The multi-dimensional APOBEC3A mutational signature from DT40 cells provides a comprehensive genomic biomarker of APOBEC3A activity that can potentially be applied to human cancers and may provide opportunities for targeting APOBEC3 in affected individuals. We define novel roles of APOBEC3A in genome instability through generation of mutagenic indels that enable further potential for synthetic lethality. Our data suggest that APOBEC3A mutagenesis results in demethylation and implicates mutagenic deaminases in epigenetic reprogramming, which extends the potential for APOBEC3 activity to affect cancer development and progression.
Limitations of the study
Indel calling and validation are technical challenges. Here we primarily relied on IsoMut, which can detect indels with reduced sensitivity but apparently higher specificity compared with other variant calling methods (see Table S1 for comparisons). It is likely, therefore, that APOBEC3A generates more indels than observed in this study. In tumor genomes, our results do not specifically tie the APOBEC3 spectrum indels to the activity of APOBEC3A or APOBEC3B beyond demonstrating their correlation with the A3A SBS Signature.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Abby Green (abby.green@wustl.edu).
Materials availability
Cell lines, plasmids, and other reagents used in this study are available upon request.
Data and code availability
Genome sequencing data have been deposited and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H2AX-p-S139 antibody | BD Biosciences | clone N1-431, cat 560443, RRID: AB_1645592 |
| HA antibody | Biolegend | clone HA.11, cat 901514, RRID: AB_2565336 |
| Tubulin | Santa Cruz | clone 6A204, cat sc-69969, RRID: AB_1118882 |
| Bacterial and virus strains | ||
| pSLIK-A3A lentivirus | Vector obtained from addgene, cloned in Green Lab, lentivirus generated in Green Lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline | Sigma | D9891-1G |
| PowerSYBR Green PCR Master Mix | AppliedBiosystems | 43-676-59 |
| Uracil DNA glycosylase | NEB | M0280 |
| Critical commercial assays | ||
| RNeasy kit | Qiagen | N/A |
| RNA-to-cDNA kit | Invitrogen | N/A |
| Deposited data | ||
| OK-Seq raw data from DT40 cells | GEO | Accession number GSE196761 |
| DT40 genome sequencing raw data | European Nucleotide Archive | Accession number PRJEB50626 |
| Replication timing information | Replication Domain database www.replicationdomain.com | Int98808223 data set |
| Replication fork direction processing data from DT40 cells | https://github.com/CL-CHEN-Lab/OK-Seq. | N/A |
| Experimental models: Cell lines | ||
| DT40 cells | Szuts lab | N/A |
| Oligonucleotides | ||
| 5’-FAM TGAGGAATGAAGTTGATTUAA ATGTGATGAGGTGA | IDT | TU Control oligo for deaminase reaction |
| 5’-FAM TGAGGAATGAAGTTGATTCAAA TGTGATGAGGTGA |
IDT | TC oligo for deaminase reaction |
| Recombinant DNA | ||
| pSLIK-A3A lentivector | Addgene | 25735 |
| Software and algorithms | ||
| Prism | GraphPad | version 9 |
| Via7 | Invitrogen | N/A |
| FlowJo | version 10.7.1 | N/A |
| IsoMut | (Pipek et al. 2017) | N/A |
| MuTect2 | GATC Toolkit https://gatk.broadinstitute.org | N/A |
| Bismark | (Krueger and Andrews 2011) | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
All DT40 lines were maintained in RPMI supplemented with 7% heat-inactivated, tetracycline-free fetal bovine serum, 3% chicken serum, 1% penicillin/streptomycin, and 5 μL/L beta-mercaptoethanol. Cells were cultured at 37C with 5% CO2. The creation of the DT40-A3A and DT40-A3AC106S cell lines was achieved through lentiviral transduction with the pSLIK-A3A lentivector with neomycin resistance.
METHOD DETAILS
Generation of ancestral and descendent clones
DT40 ancestral and descendent clones were obtained by single-cell sorting on the MoFlo FACS sorter. Cells were sorted into media, as above, supplemented with 10% additional fetal bovine serum. Single-cell ancestral clones were expanded prior to treatment with doxycycline. Cells were treated with 0.5 μg/mL doxycycline every three days during the 30-day expression period. The pool of treated ancestral clones was again sorted to generate individual descendent clones, which were expanded prior to genomic DNA extraction. Total experimental time was ∼50 days, resulting in approximately 150–200 generations in controls and 100 generations in DT40-A3A cells treated with dox.
Immunoblotting
Cells were lysed by boiling in 1X LDS (Invitrogen) for 15 minutes, followed by the addition of beta-mercaptoethanol (20% of lysate volume). Samples were run on bis-acrylamide gels in MOPS buffer (Invitrogen), then transferred to a nitrocellulose membrane (GE) using a Bio-Rad semi-dry transfer machine (BIO-RAD). Blots were blocked in 5% milk and probed with HA (Biolegend) and tubulin (Santa Cruz Biotechnology) antibodies overnight. Immunoblots were visualized using ECL (Invitrogen) and analyzed on a Bio-Rad ChemiDoc MP (BIO-RAD).
Quantitative PCR
RNA was harvested from cell pellets using the RNeasy kit (Qiagen). cDNA was produced using the Invitrogen RNA-to-cDNA kit. Quantitative PCR was performed using PowerSYBR Green PCR Master Mix (appliedbiosystems) on a Quant Studio 6 qPCR machine (appliedbiosystems) and analyzed by Via7 software.
Cell proliferation, cell cycle, and DNA damage assays
Cell proliferation curves were determined by daily total cell counts for six days using the Countess II (Invitrogen). Fifty thousand cells were plated on the first day of the experiment and treated with doxycycline on days 1 and 4. To analyze cell cycle changes, DT40 cells were treated with doxycycline for 24 to 72 hours prior to FACS analysis. Cells were fixed with 70% ethanol, stained with propidium iodide, and analyzed by FACS. Intracellular gH2AX staining was performed using the anti-H2AX-p-S139 antibody (BD Biosciences) per manufacturer’s protocol. DT40 cells were treated with doxycycline 72 hours prior to FACS analysis performed on a Fortessa cytometer (BD Biosciences) and analyzed by FlowJo (v10.7.1).
Deaminase assay
DT40 cells were plated and treated with doxycycline for 72 hours. Cells were harvested in a lysis buffer containing 50 mM Tris HCl pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 0.5% sodium deoxycholate, 1 mM sodium orthovanodate, 20 mM sodium fluoride, and freshly added protease inhibitors and incubated on ice for ten minutes followed by sonication. Protein concentration was determined by Bradford assay. 2 mg of cell lysates were incubated with incubation buffer (20 mM MES and 0.1% Tween 20), 50 mM EDTA pH 8, water, and 2.5mM of an oligonucleotide containing a single cytosine and a 5’ FAM tag as previously described. Control reactions in the absence of lysate included all of the components above with the addition of lysis buffer. The negative full-length substrate control reaction contained the oligonucleotide with a single cytosine, the positive product control reaction contained the oligonucleotide with a uracil in the place of that cytosine. All sample and control reactions were incubated at 37°C for two hours, followed by the addition of 2.5 units of uracil-DNA glycosylase (NEB) and again incubated at 37°C for 15 minutes. A loading dye solution containing formamide, sodium hydroxide, and EDTA with bromophenol blue was added and reactions were boiled at 95°C for 15 minutes. Reactions were run out on a urea-acrylamide gel in 1X TBE buffer and the gel was imaged using the fluorescein channel on a Bio-Rad ChemiDoc MP imager.
Sequencing
Genomic DNA was extracted using the PureLink Genomic DNA Mini Kit (Invitrogen). Library preparation and whole genome sequencing (WGS) was performed at Beijing Genome Institute (BGI) using 150 bp paired end reads on the DNB-seq platform. Bisulfite conversion of genomic DNA prior to library preparation was also performed by BGI.
Alignment of WGS reads and mutation calling
The sequencing reads were aligned to the chicken (Gallus gallus) reference sequence Galgal4.73 as previously described (Zamborszky et al., 2017). SBS and short indels (<50 bp) were identified using the IsoMut method (Pipek et al., 2017; Poti et al., 2019; Szikriszt et al., 2016; Zamborszky et al., 2017) developed for multiple isogenic samples. In brief, after applying a base quality filter of 30, data from all samples were compared at each genomic position and filtered using optimized parameters of minimum mutated allele frequency (0.2), minimum coverage of the mutated sample (5) and minimum reference allele frequency of all the other samples (0.93). IsoMut assumes independent samples, therefore the independence of samples was checked with preliminary runs of sample subsets. The final input set for IsoMut is listed in Table S1. Hits were also filtered using a probability-based quality score calculated from the mutated sample and one other sample with the lowest reference allele frequency (Pipek et al., 2017), which was 3.2 in case of SBS, 1.2 for insertions and 1.7 for deletions. We validated the indel calling on a representative set of sequenced samples using MuTect2 of the GATK Toolkit (https://gatk.broadinstitute.org/) as well as manual checking of each event on aligned reads and found that approximately 90% of indels identified by IsoMut were correct, whereas the sensitivity of detection was no more than 60% (Table S1).
Analysis of WGS data
De novo signature extraction
SBS triplet spectra were determined for each sample and averaged for parallel clones. De novo APOBEC and background spectra were extracted using NMF in the R package MutationalPatterns (Blokzijl et al., 2018). An optimal component number of two was chosen based on the cophenetic correlation coefficient and the residual sum of squares values (Figure S2B). The obtained experimental signatures were corrected for the differences in the human and chicken triplet frequencies before comparison to COSMIC v3.1 SBS signatures.
Spectrum deconstruction
Human cancer data collected in the PCAWG database were obtained from Alexandrov LB, et al. (Alexandrov et al., 2020). Using the DeconstructSigs R package (Rosenthal et al., 2016), SBS spectra were deconvoluted to the COSMIC v3.1 reference signature set as well as to a modified signature set excluding SBS2 and SBS13 but supplemented with the de novo extracted experimental APOBEC3A signature corrected for human triplet frequency. A sample was considered APOBEC positive if the sum of SBS2 and SBS13 contributions was greater than 15%.
Indel spectra
Indel spectra were determined according to the COSMIC indel categories (Alexandrov et al., 2020). Deconstruction of indel spectra to indel signatures was done with the DeconstructSigs R package (Rosenthal et al., 2016). Indel deconstruction was performed using the COSMIC indel signature reference set supplemented with the experimental A3A indel signature. The deconstruction of indel spectra to an unlimited number of COSMIC signatures resulted in a weak correlation of APOBEC3A-mediated SBSs and indels. The reason may be the lower number of indels as compared to SNVs, making the procedure less robust. To avoid the overfitting of data, we limited the number of reference indel signature components in the deconstruction to no more than four components picked from the whole set, which resulted in a significant improvement in the deconstruction of samples with A3A SBS contribution as compared to samples with no A3A SBS contribution.
Sequence context
The sequence context of SBS was visualized on normalized seqlogo plots generated using the R package seqLogo.
Transcriptional strand bias
The transcriptional strand for each mutated position was determined based on the Gallus gallus 4.73 annotation database downloaded from ensembl.org, using the mutstrand function of MutationalPatterns.
Replicational strand bias
OK-seq was performed as described previously (Blin et al., 2021; Petryk et al., 2016). Replication fork directionality (RFD) was computed from mapped, trimmed, and de-duplicated OK-seq reads in book-ended windows of 1 kb according to RFD = (R-F)/(F+R). F and R correspond to reads mapped on forward and reverse strand respectively (Petryk et al., 2016). RFD value reflects the proportions of forks moving rightward and leftward within each window in the cell population. The raw OK-seq data of DT40 cells are available at GEO (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE196761, and the processed RFD data are available at https://github.com/CL-CHEN-Lab/OK-Seq within the folder for the published results.
Replicational timing
Replication timing information was downloaded from the Replication Domain database (www.replicationdomain.com), based on the Int98808223 data set (Shang et al., 2013). The replication timing score at the position of mutations was determined by linear interpolation between the nearest data points. For bar plots, replication timing scores of the whole reference set were divided into deciles.
Cluster analysis
A set of mutations was considered to belong to a cluster if the distances between neighbouring mutations was no greater than 20000 base pairs, a distance chosen empirically to ensure the detection of all strand-coordinated kataegis mutation clusters. Clusters containing 5 or more mutations were termed kataegis, while smaller clusters (2–4 mutations) of diffuse hypermutation were termed omikli events (Mas-Ponte and Supek, 2020).
Trinucleotide normalization
Normalization of trinucleotide spectra was performed by dividing the mutation counts with the occurrence of the given triplet category in the reference genome.
DNA stem loops
Putative stem loops were identified based on the presence of flanking palindrome sequences that are expected to form the stem structure. Putative loops were accepted based on the criteria in Buisson R, et al. (Buisson et al., 2019) with a loop length of 3–11 bases and a palindrome sequence with minimal length of 3. In case there was more than one putative loop at a mutation site, the stronger loop was used for analysis. Loop strength was calculated as 3 × GC + 1 × AT. Mutations within the loop were identified for each stem loop category based on loop length and stem strength. For comparison, 38,235 control positions were selected that are randomly distributed throughout the genome and match the trinucleotide spectrum of APOBEC3A-mutated bases. The difference between APOBEC3A and random stem loop hit frequency was calculated as (nAcat/nAall) – (nRcat/nRall), where nAcat and nRcat are the number of positions found in a given stem loop category of mutations from APOBEC3A and control positions respectively, and nAall and nRall are the total number of mutations from APOBEC3A and control positions, respectively.
DNA methylation
Alignment of reads from bisulfite sequencing and the analysis of CpG methylation levels was done using Bismark (Krueger and Andrews, 2011) with default parameter settings of 1 = 20, n = 0. A minimum coverage of 15 was required at the CpG sites.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics for individual experiments were performed as described in figure legends. P-values were determined by two tailed t-tests with Welch’s correlation or Wilcoxon Rank Sum test. Statistical analysis was performed using GraphPad Prism v9. Specific quantitative and statistical details of genome analysis are included in the previous section “analysis of WGS data”.
Data availability
Raw sequence data are available from the European Nucleotide Archive under study accession number PRJEB50626.
Supplementary Material
Highlights
The comprehensive APOBEC3A mutational signature is experimentally defined
Deamination of methylated and unmodified cytidines by APOBEC3A occurs at similar rates
APOBEC3A generates a unique genome-wide signature of deletions
APOBEC3A base substitution and deletion signatures are prevalent in human cancers
ACKNOWLEDGMENTS
The authors thank all members of the Green and Szüts labs for critical evaluation of experimental data and thoughtful review of the manuscript. We are thankful to colleagues and collaborators for experimental discussions and manuscript editing, especially Drs. Matthew Weitzman, Rahul Kohli, Sebastien Landry, Jeffrey Bednarski, and members of the Bednarski lab. We acknowledge the high-throughput sequencing facility of I2BC for its sequencing and bioinformatics expertise. BioRender was used to generate schematic figures and the graphical abstract. This work was supported by funding from the American Cancer Society (to A.M.G.), the Cancer Research Foundation (to A.M.G.), the National Institutes of Health (K08 CA212299 to A.M.G.), the Department of Defense (CA200867 to A.M.G.), the Children’s Discovery Institute and the Washington University School of Medicine (to A.M.G.), and the National Research Development and Innovation Office of Hungary (K_134779 and VEKOP-2.3.3-15-2017-00014 to D.S. and PD_134818 to E.N.). The C.-L.C. lab is supported by the YPI program of I. Curie, the ATIP-Avenir program from Centre National de la Recherche Scientifique and Plan Cancer (N° 18CT014-00), the Agence Nationale de la Recherche (ReDeFINe 19-CE12-0016-02), and Institut National Du Cancer (PLBIO19-076). The O.H. lab is supported by Ligue Nationale Contre le Cancer (Comité de Paris), Agence Nationale de la Recherche (ANR 2010 BLAN 161501), Association pour la Recherche sur le Cancer, the Fondation pour la Recherche Médicale (FRM DEI201512344404), and the France Génomique national infrastructure “Investissements d’Avenir” program managed by the ANR (ANR-10-INBS-09).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of the genomic datasets. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110555.
REFERENCES
- Akre MK, Starrett GJ, Quist JS, Temiz NA, Carpenter MA, Tutt AN, Grigoriadis A, and Harris RS (2016). Mutation processes in 293-based clones overexpressing the DNA cytosine deaminase APOBEC3B. PLoS One 11, e0155391. 10.1371/journal.pone.0155391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, and Stratton MR (2015). Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407. 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat AS, Hao W, Townes JP, Lee H, Tang H, and Foster PL (2016). Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. U S A 113, 2176–2181. 10.1073/pnas.1522325113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Blin M, Lacroix L, Petryk N, Jaszczyszyn Y, Chen CL, Hyrien O, and Le Tallec B (2021). DNA molecular combing-based replication fork directionality profiling. Nucleic Acids Res. 49, e69. 10.1093/nar/gkab219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, Janssen R, van Boxtel R, and Cuppen E (2018). MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 10, 33. 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, and Cullen BR (2007). The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology 364, 486–493. 10.1016/j.virol.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, and Lawrence MS (2019). Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 364, eaaw2872. 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. (2013a). APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, and Harris RS (2013b). Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SM, Bohn MF, Schiffer CA, et al. (2012). Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 287, 34801–34808. 10.1074/jbc.M112.385161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Resnick MA, and Gordenin DA (2013). The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair (Amst) 12, 878–889. 10.1016/j.dnarep.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, et al. (2015). An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 47, 1067–1072. 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, and Alt FW (2003). Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 422, 726–730. 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, and Weitzman MD (2006). APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16, 480–485. 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chen TW, Lee CC, Liu H, Wu CS, Pickering CR, Huang PJ, Wang J, Chang IY, Yeh YM, Chen CD, et al. (2017). APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat. Commun. 8, 465. 10.1038/s41467-017-00493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Roumeliotis TI, Chang YH, Chen CT, Han CL, Lin MH, Chen HW, Chang GC, Chang YL, Wu CT, et al. (2020). Proteogenomics of non-smoking lung cancer in east Asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244.e17. 10.1016/j.cell.2020.06.012. [DOI] [PubMed] [Google Scholar]
- Conticello SG (2012). Creative deaminases, self-inflicted damage, and genome evolution. Ann. N. Y Acad. Sci. 1267, 79–85. 10.1111/j.1749-6632.2012.06614.x. [DOI] [PubMed] [Google Scholar]
- Cortez LM, Brown AL, Dennis MA, Collins CD, Brown AJ, Mitchell D, Mertz TM, and Roberts SA (2019). APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 15, e1008545. 10.1371/journal.pgen.1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, and Nussenzweig A (2013). The AID-induced DNA damage response in chromatin. Mol. Cell 50, 309–321. 10.1016/j.mol-cel.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22. 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Green AM, Landry S, Budagyan K, Avgousti DC, Shalhout S, Bhagwat AS, and Weitzman MD (2016). APOBEC3A damages the cellular genome during DNA replication. Cell Cycle 15, 998–1008. 10.1080/15384101.2016.1152426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AM, and Weitzman MD (2019). The spectrum of APOBEC3 activity: from anti-viral agents to anti-cancer opportunities. DNA Repair (Amst) 83, 102700. 10.1016/j.dnarep.2019.102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2014). The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 19, 84–94. 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. (2016). Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 164, 538–549. 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, and Malim MH (2003). DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809. 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Harris RS, Petersen-Mahrt SK, and Neuberger MS (2002). RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10, 1247–1253. 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- Henderson S, Chakravarthy A, Su X, Boshoff C, and Fenton TR (2014). APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 7, 1833–1841. 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, and Roberts SA (2016). APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 14, 1273–1282. 10.1016/j.celrep.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito F, Fu Y, Kao SA, Yang H, and Chen XS (2017). Family-wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J. Mol. Biol. 429, 1787–1799. 10.1016/j.jmb.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili P, Bowen D, Langenbucher A, Park S, Aguirre K, Corcoran RB, Fleischman AG, Lawrence MS, Zou L, and Buisson R (2020). Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat. Commun. 11, 2971. 10.1038/s41467-02016802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, and Navaratnam N (2002). An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79, 285–296. 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Kazanov MD, Roberts SA, Polak P, Stamatoyannopoulos J, Klimczak LJ, Gordenin DA, and Sunyaev SR (2015). APOBEC-induced cancer mutations are uniquely enriched in early-replicating, gene-dense, and active chromatin regions. Cell Rep. 13, 1103–1109. 10.1016/j.celrep.2015.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry S, Narvaiza I, Linfesty DC, and Weitzman MD (2011). APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 12, 444–450. 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbucher A, Bowen D, Sakhtemani R, Bournique E, Wise JF, Zou L, Bhagwat AS, Buisson R, and Lawrence MS (2021). An extended APO-BEC3A mutation signature in cancer. Nat. Commun. 12, 1602. 10.1038/s41467-021-21891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, Starrett GJ, Temiz NA, Larson LK, Durfee C, et al. (2020). APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 217, e20200261. 10.1084/jem.20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DD, and Seung HS (1999). Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791. 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- Lei L, Chen H, Xue W, Yang B, Hu B, Wei J, Wang L, Cui Y, Li W, Wang J, et al. (2018). APOBEC3 induces mutations during repair of CRISPR-Cas9-generated DNA breaks. Nat. Struct. Mol. Biol. 25, 45–52. 10.1038/s41594-017-0004-6. [DOI] [PubMed] [Google Scholar]
- Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, et al. (2013). APOBEC3B up-regulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231. 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love RP, Xu H, and Chelico L (2012). Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 287, 30812–30822. 10.1074/jbc.M112.393181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, and Trono D (2003). Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103. 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mas-Ponte D, and Supek F (2020). DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nat. Genet. 52, 958–968. 10.1038/s41588-020-0674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, et al. (2016). The topography of mutational processes in breast cancer genomes. Nat. Commun. 7, 11383. 10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussil B, Suspene R, Aynaud MM, Gauvrit A, Vartanian JP, and Wain-Hobson S (2013). Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS One 8, e73641. 10.1371/journal.pone.0073641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, Landau NR, and Weitzman MD (2009). Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 5, e1000439. 10.1371/journal.ppat.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, and Alexandrov LB (2016). Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 37, 531–540. 10.1093/carcin/bgw055. [DOI] [PubMed] [Google Scholar]
- Petljak M, Chu K, Dananberg A, Bergstrom EN, Morgen P.v., Alexandrov LB, Stratton MR, and Maciejowski J (2021). The APOBEC3A deaminase drives episodic mutagenesis in cancer cells. Preprint at bioRxiv. 10.1101/2021.02.14.431145. [DOI] [Google Scholar]
- Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen CL, and Hyrien O (2016). Replication landscape of the human genome. Nat. Commun. 7, 10208. 10.1038/ncomms10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipek O, Ribli D, Molnar J, Poti A, Krzystanek M, Bodor A, Tusnady GE, Szallasi Z, Csabai I, and Szuts D (2017). Fast and accurate mutation detection in whole genome sequences of multiple isogenic samples with Iso-Mut. BMC Bioinformatics 18, 73. 10.1186/s12859-017-1492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poti A, Gyergyak H, Nemeth E, Rusz O, Toth S, Kovacshazi C, Chen D, Szikriszt B, Spisak S, Takeda S, et al. (2019). Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 20, 240. 10.1186/s13059-019-1867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott S (2017). Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife 6, e23203. 10.7554/eLife.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, and Nussenzweig MC (2003). Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4, 452–456. 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. (2013). An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976. 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. (2012). Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 46, 424–435. 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. (2017). Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540–556.e25. 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Lada AG, Goncearenco A, Green MR, De S, Nudelman G, Panchenko AR, Koonin EV, and Pavlov YI (2016). Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci. Rep. 6, 38133. 10.1038/srep38133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, and Swanton C (2016). DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31. 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutsky EK, Nabel CS, Davis AKF, DeNizio JE, and Kohli RM (2017). APOBEC3A efficiently deaminates methylated, but not TET-oxidized, cytosine bases in DNA. Nucleic Acids Res. 45, 7655–7665. 10.1093/nar/gkx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, and Nikolaev SI (2016). APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 26, 174–182. 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang WH, Hori T, Martins NM, Toyoda A, Misu S, Monma N, Hiratani I, Maeshima K, Ikeo K, Fujiyama A, et al. (2013). Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev. Cell 24, 635–648. 10.1016/j.devcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi MJ, Meng XY, Fontugne J, Chen CL, Radvanyi F, and BernardPierrot I (2020). Identification of new driver and passenger mutations within APOBEC-induced hotspot mutations in bladder cancer. Genome Med. 12, 85. 10.1186/s13073-020-00781-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi MJ, Meng XY, Lamy P, Banday AR, Yang J, Moreno-Vega A, Chen CL, Dyrskjot L, Bernard-Pierrot I, Prokunina-Olsson L, et al. (2019). APOBEC-mediated mutagenesis as a likely cause of FGFR3 S249C mutation over-representation in bladder cancer. Eur. Urol. 76, 9–13. 10.1016/j.eururo.2019.03.032. [DOI] [PubMed] [Google Scholar]
- Suspene R, Aynaud MM, Vartanian JP, and Wain-Hobson S (2013). Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One 8, e63461. 10.1371/journal.pone.0063461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szikriszt B, Poti A, Pipek O, Krzystanek M, Kanu N, Molnar J, Ribli D, Szeltner Z, Tusnady GE, Csabai I, et al. (2016). A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 17, 99. 10.1186/s13059-016-0963-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941– D947. 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, and Neuberger MS (2013). DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2, e00534. 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Ponting CP, and Copley RR (2004). Occurrence and consequences of coding sequence insertions and deletions in Mammalian genomes. Genome Res. 14, 555–566. 10.1101/gr.1977804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, McPherson A, Horlings HM, Senz J, Prentice LM, et al. (2017). Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 49, 856–865. 10.1038/ng.3849. [DOI] [PubMed] [Google Scholar]
- Wijesinghe P, and Bhagwat AS (2012). Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 40, 9206–9217. 10.1093/nar/gks685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe M, Sonoda E, Hochegger H, and Takeda S (2004). Reverse genetic studies of the DNA damage response in the chicken B lymphocyte line DT40. DNA Repair (Amst) 3, 1175–1185. 10.1016/j.dnarep.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, and Landau NR (2004). Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11, 435–442. 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Zamborszky J, Szikriszt B, Gervai JZ, Pipek O, Poti A, Krzystanek M, Ribli D, Szalai-Gindl JM, Csabai I, Szallasi Z, et al. (2017). Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 36, 746–755. 10.1038/onc.2016.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome sequencing data have been deposited and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H2AX-p-S139 antibody | BD Biosciences | clone N1-431, cat 560443, RRID: AB_1645592 |
| HA antibody | Biolegend | clone HA.11, cat 901514, RRID: AB_2565336 |
| Tubulin | Santa Cruz | clone 6A204, cat sc-69969, RRID: AB_1118882 |
| Bacterial and virus strains | ||
| pSLIK-A3A lentivirus | Vector obtained from addgene, cloned in Green Lab, lentivirus generated in Green Lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline | Sigma | D9891-1G |
| PowerSYBR Green PCR Master Mix | AppliedBiosystems | 43-676-59 |
| Uracil DNA glycosylase | NEB | M0280 |
| Critical commercial assays | ||
| RNeasy kit | Qiagen | N/A |
| RNA-to-cDNA kit | Invitrogen | N/A |
| Deposited data | ||
| OK-Seq raw data from DT40 cells | GEO | Accession number GSE196761 |
| DT40 genome sequencing raw data | European Nucleotide Archive | Accession number PRJEB50626 |
| Replication timing information | Replication Domain database www.replicationdomain.com | Int98808223 data set |
| Replication fork direction processing data from DT40 cells | https://github.com/CL-CHEN-Lab/OK-Seq. | N/A |
| Experimental models: Cell lines | ||
| DT40 cells | Szuts lab | N/A |
| Oligonucleotides | ||
| 5’-FAM TGAGGAATGAAGTTGATTUAA ATGTGATGAGGTGA | IDT | TU Control oligo for deaminase reaction |
| 5’-FAM TGAGGAATGAAGTTGATTCAAA TGTGATGAGGTGA |
IDT | TC oligo for deaminase reaction |
| Recombinant DNA | ||
| pSLIK-A3A lentivector | Addgene | 25735 |
| Software and algorithms | ||
| Prism | GraphPad | version 9 |
| Via7 | Invitrogen | N/A |
| FlowJo | version 10.7.1 | N/A |
| IsoMut | (Pipek et al. 2017) | N/A |
| MuTect2 | GATC Toolkit https://gatk.broadinstitute.org | N/A |
| Bismark | (Krueger and Andrews 2011) | N/A |
Raw sequence data are available from the European Nucleotide Archive under study accession number PRJEB50626.