

Abstract
Background
Hyperventilation resulting in hypocapnic alkalosis (HA) is frequently encountered in spontaneously breathing patients with acute cerebrovascular conditions. The underlying mechanisms of this respiratory response have not been fully elucidated. The present study describes, applying the physical–chemical approach, the acid-base characteristics of cerebrospinal fluid (CSF) and arterial plasma of spontaneously breathing patients with aneurismal subarachnoid hemorrhage (SAH) and compares these results with those of control patients. Moreover, it investigates the pathophysiologic mechanisms leading to HA in SAH.
Methods
Patients with SAH admitted to the neurological intensive care unit and patients (American Society of Anesthesiologists physical status of 1 and 2) undergoing elective surgery under spinal anesthesia were enrolled. CSF and arterial samples were collected simultaneously. Electrolytes, strong ion difference (SID), partial pressure of carbon dioxide (PCO2), weak noncarbonic acids (ATOT), and pH were measured in CSF and arterial blood samples.
Results
Twenty spontaneously breathing patients with SAH and 25 controls were enrolled. The CSF of patients with SAH, as compared with controls, was characterized by a lower SID (23.1 ± 2.3 vs. 26.5 ± 1.4 mmol/L, p < 0.001) and PCO2 (40 ± 4 vs. 46 ± 3 mm Hg, p < 0.001), whereas no differences in ATOT (1.2 ± 0.5 vs. 1.2 ± 0.2 mmol/L, p = 0.95) and pH (7.34 ± 0.06 vs. 7.35 ± 0.02, p = 0.69) were observed. The reduced CSF SID was mainly caused by a higher lactate concentration (3.3 ± 1.3 vs. 1.4 ± 0.2 mmol/L, p < 0.001). A linear association (r = 0.71, p < 0.001) was found between CSF SID and arterial PCO2. A higher proportion of patients with SAH were characterized by arterial HA, as compared with controls (40 vs. 4%, p = 0.003). A reduced CSF-to-plasma difference in PCO2 was observed in nonhyperventilating patients with SAH (0.4 ± 3.8 vs. 7.8 ± 3.7 mm Hg, p < 0.001).
Conclusions
Patients with SAH have a reduction of CSF SID due to an increased lactate concentration. The resulting localized acidifying effect is compensated by CSF hypocapnia, yielding normal CSF pH values and resulting in a higher incidence of arterial HA.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12028-022-01450-1.
Keywords: Acid–base equilibrium, Cerebrospinal fluid, Blood, Subarachnoid hemorrhage, Electrolyte disturbances
Introduction
According to Stewart’s physical–chemical approach [1], the acid–base equilibrium of biological fluids is regulated independently by three variables: the partial pressure of carbon dioxide (PCO2), the strong ion difference (SID), and the total amount of weak nonvolatile buffers (ATOT). Cerebrospinal fluid (CSF) in normal conditions is a clear fluid surrounding the brain, characterized by a very low concentration of proteins. It is produced mainly by the choroid plexus and reabsorbed by the arachnoid villi [2]. Given its negligible amount of proteins, only two variables regulate the acid–base equilibrium of CSF, namely PCO2 and SID [3]. The acid–base equilibrium of this small and very particular extracellular fluid compartment, i.e., CSF, is of particular importance because it contributes significantly to the regulation of cerebral blood flow [4] and spontaneous breathing activity [5].
Hyperventilation resulting in hypocapnic alkalosis (HA) is frequently encountered in spontaneously breathing patients with acute cerebrovascular conditions [6–8] or severe brain injuries [9]. The pathophysiologic mechanisms underlying this abnormal respiratory response have not been fully elucidated. On one hand, the existence of a neurogenic stimulus causing a pathologic activation of the respiratory centers has been hypothesized [8, 10, 11]. On the other hand, the mixing of CSF with exogenous blood could determine an acidification of the extracellular cerebral milieu, fostering an increase in minute ventilation to restore normal pH values in proximity of the respiratory centers [7].
The aim of the present study was to (1) describe, applying Stewart’s physical–chemical approach, the acid–base characteristics of CSF and arterial plasma of spontaneously breathing patients with subarachnoid hemorrhage (SAH); (2) compare these results with those of healthy controls; and (3) investigate the pathophysiologic mechanisms leading to HA in SAH. We hypothesized that patients with SAH would be characterized by a local CSF metabolic acidosis (lower CSF SID), which would favor the development of hypocapnia.
Methods
Institutional Review Board and Patient Consent
The study protocol and the informed consent process were approved by the Ethical Committee Milano Area B (protocol number: 25_2017bis), and written informed or deferred consent was obtained from patients with SAH and controls.
Patients with Spontaneous SAH
Patients with SAH admitted to the neurological intensive care unit (ICU) of our hospital, in whom an external ventricular drain was placed for clinical reasons, with preserved spontaneous respiratory activity were enrolled. Details about our clinical practice regarding SAH treatment can be found elsewhere [12]. Both nonintubated patients and intubated patients on pressure support ventilation were studied. Age younger than 18 years and pregnancy constituted exclusion criteria. In each patient, CSF and arterial blood samples were collected simultaneously as close as possible to the day of SAH onset. A CSF sample of 1.5 mL was drawn anaerobically and under strict asepsis from the external ventricular drain after the dead space of the catheter was discarded. Simultaneously, an arterial blood sample was collected from an indwelling arterial catheter. In addition, the following clinical data were recorded: medical history, the severity of bleeding (modified Fisher scale), day from bleeding, neurological assessment at ICU admission (World Federation Neurological Surgeons Scale), and Glasgow Coma Scale score on the study day [13].
Control Population
Patients undergoing spinal anesthesia for elective surgery constituted the control group. Exclusion criteria were American Society of Anesthesia (ASA) physical status above 2, age younger than 18, pregnancy, and known neurological, renal, or respiratory disease. Before the local anesthetic injection, 1.5 mL of CSF was collected anaerobically from the needle used for spinal anesthesia. The radial artery was punctured to collect an arterial blood sample. Anesthesiologists were asked to limit intravenous fluids prior to spinal anesthesia and to choose balanced solutions to avoid intravenous-fluids-related acid–base derangements [14, 15].
Analysis of CSF and Blood
Acid–base and electrolytes of CSF and arterial blood samples were immediately measured (ABL800 FLEX, Radiometer, Copenhagen, Denmark). Magnesium, phosphate, and albumin levels (COBAS 8000, Roche Diagnostics GmbH, Mannheim, Germany); hemoglobin concentration; and red and white blood cell counts (XN-9000 V, Sysmex Corporation, Kobe, Japan) were analyzed for CSF and blood samples. Plasma and CSF osmolarity were measured (Osmometer MIR 300-P, E. Mires, Milan, Italy).
For each CSF and blood sample, the SID was calculated as indicated in Eq. 1 [16]:
| 1 |
where Na+, K+, Ca2+, Cl−, and Lac− refer to plasma sodium, potassium, ionized calcium, chloride, and lactate concentrations expressed as millimolar.
Moreover, the dissociated part of noncarbonic weak acid was computed using Eq. 2 [16]:
| 2 |
where [A−] is expressed in mmol/L, [Alb] is the plasma concentration of albumin expressed in g/L, [Pi] is the plasma concentration of phosphates expressed in mmol/L, and pH denotes arterial or cerebrospinal pH.
The total amount of weak acids [ATOT] was calculated, rearranging the dissociation equation as reported in Eq. 3:
| 3 |
where pH is the arterial or cerebrospinal pH, and pKA is equal to 6.8 and represents the negative logarithm to base 10 of the dissociation constant of noncarbonic buffers.
The difference between CSF and plasma SID and CSF and plasma PCO2 was computed and termed ΔSID and ΔPCO2, respectively.
| 4 |
| 5 |
The ratios between CSF and arterial plasma were calculated as previously described [11].
| 6 |
Bicarbonate ion concentration ([HCO3−]) was calculated from pH and PCO2 according to the Henderson–Hasselbalch equation (), assuming pK = 6.1 and α = 0.0306 for plasma and pK = 6.13 and α = 0.0318 for CSF [17].
| 7 |
Clinical Definition of HA
Patients with an arterial PCO2 < 35 mm Hg and an arterial pH > 7.45 were included in the HA subgroup, whereas patients who did not present these features were included in the subgroup without HA.
Clinical Outcomes
The Glasgow Outcome Scale Extended (GOS-E) [18, 19] was scored at 12 months. A good clinical outcome was defined as a GOS-E score ≥ 6. Information regarding clinical outcome was gathered over the phone, directly, or through the caregiver. Data about the incidence of clinical vasospasm, defined as new neurological symptoms associated with confirmed vasospasm diagnosed by a neuroradiologist [20], were collected from the patients’ medical charts.
Sample Size and Statistical Analysis
Data analysis and statistical plans were written and filed with the institutional review board before data were accessed. We calculated that a sample of 40 patients (20 patients per group) would provide a statistical power of 0.9 with an α error of 0.05 to detect a difference in SID of 3 mEq/L, assuming a standard deviation of 2.5 mEq/L. Given the novelty of the measurement of some CSF variables, we decided to increase the number of control patients to 25.
Comparison between continuous variables was performed via Student’s t-test or the Mann–Whitney rank-sum test, as appropriate. Differences between categorical variables were assessed using the χ2 test. The relationship between quantitative or categorical variables was investigated using linear and multilinear regression, as appropriate. Data are expressed as mean ± standard deviation or median and interquartile range, unless otherwise stated. Statistical significance was defined as p < 0.05. Analysis was performed with SAS v.9.2 (SAS Institute, Inc., Cary, NC) and SigmaPlot v.12.0 (Systat Software, San Jose, CA). The Strengthening the Reporting of Observational Studies in Epidemiology checklist was used.
Results
Description of the Study Population
Patients were enrolled between February 2017 and October 2018. Twenty spontaneously breathing patients with aneurismal SAH (aged 57 ± 11 years, 16 women, body mass index 25 [23–29]) were enrolled and studied on day 2 (2–4) after bleeding. The median World Federation Neurological Surgeons Scale score at ICU admission was 3.5 (1–4), the median modified Fisher scale score was 4 (3–4), and the median Glasgow Coma Scale core on the study day was 12 (9–15). Ten patients were not intubated and were spontaneously breathing. The remaining ten were intubated on pressure support ventilation. At the sampling time, no patient exhibited neurogenic breathing patterns [21] or hypoxic respiratory failure (PaO2/FiO2 = 352 ± 121). Twenty-five healthy controls were enrolled (aged 56 ± 16 years, 9 women, body mass index 24 [23–25], 8 patients with an ASA physical status of 1, 17 patients with an ASA physical status of 2). The only statistically significant difference found between controls and patients with SAH was a higher prevalence of female sex in patients with SAH (80% vs. 36%, p = 0.004). Comorbidities of patients with SAH and controls are reported in Table S1.
Description of Cerebrospinal and Arterial Acid–Base Characteristics
Cerebrospinal and arterial acid–base findings are summarized in Tables 1 and 2. CSF of patients with SAH was characterized by an increased presence of red and white blood cells, whereas no red blood cells and only 2 ± 2 white blood cells per microliter were found in controls. The CSF of patients with SAH, as compared with controls, had lower SID and PCO2 and similar ATOT. No difference in CSF pH was observed. The reduced CSF SID was caused mainly by an increased lactate concentration (p < 0.001). On the plasma side (Table 2), similar SID and PCO2 values were recorded. The plasma of patients with SAH was characterized by lower ATOT, as compared with controls, leading to a significantly higher arterial pH. The reduction in ATOT observed in patients with SAH was caused both by lower albumin (4.2 ± 0.4 vs. 3.5 ± 0.4 g/dL, p < 0.001) and phosphate (3.2 ± 0.6 vs. 2.5 ± 0.8 mg/dL, p = 0.003) concentrations.
Table 1.
Acid–base characteristics of cerebrospinal fluid of the study population
| Variables | Controls (n = 25) | SAH (n = 20) | p value |
|---|---|---|---|
| pH | 7.35 ± 0.02 | 7.34 ± 0.06 | 0.69 |
| PCO2 (mm Hg) | 46 ± 3 | 40 ± 4 | < 0.001 |
| ATOT (mmol/L) | 1.2 ± 0.2 | 1.2 ± 0.5 | 0.95 |
| SID (mmol/L) | 26.5 ± 1.4 | 23.1 ± 2.3 | < 0.001 |
| Na+ (mmol/L) | 141 ± 1 | 143 ± 3 | 0.007 |
| K+ (mmol/L) | 2.8 ± 0.1 | 2.6 ± 0.5 | 0.07 |
| Ca2+ (mmol/L) | 0.99 ± 0.02 | 0.98 ± 0.06 | 0.22 |
| Mg2+ (mmol/L) | 1.10 ± 0.04 | 1.15 ± 0.06 | 0.002 |
| Cl− (mmol/L) | 120 ± 2 | 123 ± 4 | 0.001 |
| Lac− (mmol/L) | 1.4 ± 0.2 | 3.3 ± 1.3 | < 0.001 |
| HCO3− (mmol/L) | 24.7 ± 1.5 | 21.7 ± 2.4 | < 0.001 |
| Phosph− (mEq/L) | 0.77 ± 0.08 | 0.66 ± 0.20 | 0.02 |
| Alb− (mEq/L) | 0.19 ± 0.14 | 0.30 ± 0.23 | 0.05 |
| Osmolarity (mOsm/L) | 285 ± 3 | 290 ± 7 | 0.001 |
| Gluc (mg/dL) | 59 ± 7 | 74 ± 14 | < 0.001 |
| Hb (g/dL) | 0.0 ± 0.0 | 0.3 ± 0.3 | < 0.001 |
| RBC (× 106/µL) | 0.00 ± 0.00 | 0.11 ± 0.12 | < 0.001 |
| WBC (cells/µL) | 2 ± 2 | 359 ± 562 | 0.003 |
Data are expressed as mean ± standard deviation
Alb−, ionized albumin concentration, ATOT, total amount of weak noncarbonic acids, Ca2+, ionized calcium concentration, Cl−, chloride concentration, Gluc, glucose concentration, Hb, hemoglobin concentration, HCO3−, bicarbonate concentration, K+, potassium concentration, Lac−, lactate concentration, Mg2+, magnesium concentration, Na+, sodium concentration, PCO2, partial pressure of carbon dioxide, Phosph−, ionized phosphate concentration, RBC, red blood cell count, SAH, aneurismal subarachnoid hemorrhage, SID, strong ion difference, WBC, white blood cell count
Table 2.
Acid–base characteristics of arterial plasma of the study population
| Variables | Controls (n = 25) | SAH (n = 20) | p value |
|---|---|---|---|
| pH | 7.42 ± 0.02 | 7.45 ± 0.04 | < 0.001 |
| PCO2 (mm Hg) | 39 ± 3 | 37 ± 6 | 0.15 |
| ATOT (mmol/L) | 16.3 ± 1.5 | 13.5 ± 1.6 | < 0.001 |
| SID (mmol/L) | 36.2 ± 1.8 | 37.2 ± 3.7 | 0.23 |
| PO2 (mm Hg) | 99 ± 49 | 111 ± 28 | 0.35 |
| Na+ (mmol/L) | 138 ± 2 | 142 ± 4 | < 0.001 |
| K+ (mmol/L) | 4.0 ± 0.3 | 3.5 ± 0.2 | < 0.001 |
| Ca2+ (mmol/L) | 1.22 ± 0.03 | 1.17 ± 0.04 | < 0.001 |
| Mg2+ (mmol/L) | 0.84 ± 0.07 | 0.85 ± 0.07 | 0.88 |
| Cl− (mmol/L) | 109 ± 2 | 111 ± 5 | 0.03 |
| Lac− (mmol/L) | 1.1 ± 0.4 | 1.0 ± 0.5 | 0.25 |
| HCO3− (mmol/L) | 25.0 ± 2.0 | 25.5 ± 2.9 | 0.19 |
| Phosph− (mEq/L) | 1.9 ± 0.3 | 1.5 ± 0.5 | 0.004 |
| Alb− (mEq/L) | 11.3 ± 1.1 | 9.6 ± 1.1 | < 0.001 |
| SBE (mmol/L) | 0.5 ± 2.0 | 1.6 ± 2.6 | 0.09 |
| Osmolarity (mOsm/L) | 281 ± 3 | 291 ± 8 | < 0.001 |
| Gluc (mg/dL) | 100 ± 11 | 129 ± 20 | < 0.001 |
| Gluc ratio (%) | 59 ± 6 | 58 ± 8 | 0.55 |
| Hb (g/dL) | 14.0 ± 1.6 | 11.4 ± 1.6 | < 0.001 |
| RBC (× 106/µL) | 4.5 ± 0.5 | 3.6 ± 0.7 | < 0.001 |
| WBC (× 103/µL) | 5.9 ± 1.0 | 10.2 ± 2.4 | < 0.001 |
Data are expressed as mean ± standard deviation
Alb−, ionized albumin concentration, ATOT, total amount of weak noncarbonic acids, Ca2+, ionized calcium concentration, Cl−, chloride concentration, Gluc, glucose concentration, Hb, hemoglobin concentration, HCO3−, bicarbonate concentration, K+, potassium concentration, Lac−, lactate concentration, Mg2+, magnesium concentration, Na+, sodium concentration, PCO2, partial pressure of carbon dioxide, Phosph−, ionized phosphate concentration, PO2, partial pressure of oxygen, RBC, red blood cell count, SAH, aneurismal subarachnoid hemorrhage, SBE, standard base excess, SID, strong ion difference, WBC, white blood cell count
The ΔSID, i.e., the difference between CSF and plasma SID, differed significantly between the two groups (Fig. 1). Furthermore, ΔPCO2, the CSF-to-plasma difference in PCO2, showed significantly different values between controls and patients with SAH (7.5 ± 2.5 vs. 3.4 ± 5.2 mm Hg, p = 0.001). Measured osmolarity of both plasma (p < 0.001) and CSF (p = 0.001) was significantly higher in patients with SAH.
Fig. 1.
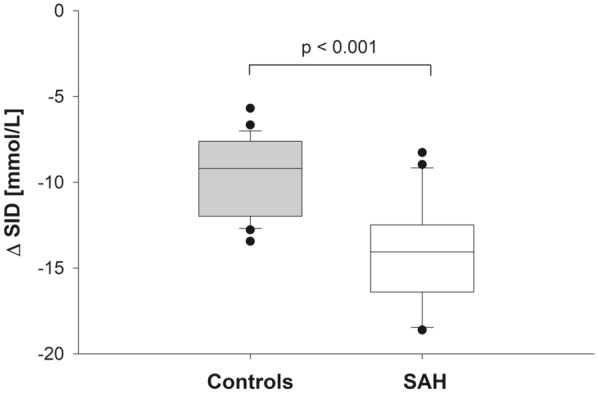
ΔSID in controls and patients with SAH. Box-plot graph representing the CSF-to-plasma SID difference (ΔSID). Whiskers represent 10th and 90th percentiles. CSF cerebrospinal fluid, SAH aneurismal subarachnoid hemorrhage, SID strong ion difference
HA and Underlying Mechanisms
The incidence of HA was higher in spontaneously breathing patients with SAH, as compared with controls (40% vs. 4%, p = 0.003).
When comparing hypocapnic and nonhypocapnic patients with SAH, no differences in demographic and clinical variables were observed (Table S2). Arterial and CSF acid–base characteristics of patients with HA and those without HA are summarized in Table S3 and Table 3, respectively.
Table 3.
Acid–base characteristics of cerebrospinal fluid of the SAH population
| Variables | Nonhypocapnic alkalosis (n = 12) | Hypocapnic alkalosis (n = 8) | p value |
|---|---|---|---|
| pH | 7.36 ± 0.05 | 7.33 ± 0.06 | 0.26 |
| PCO2 (mm Hg) | 41 ± 4 | 39 ± 4 | 0.47 |
| ATOT (mmol/L) | 1.3 ± 0.5 | 1.1 ± 0.3 | 0.18 |
| SID (mmol/L) | 24.2 ± 1.7 | 21.3 ± 1.9 | 0.002 |
| Na+ (mmol/L) | 143 ± 5 | 143 ± 1 | 0.98 |
| K+ (mmol/L) | 2.5 ± 0.4 | 2.6 ± 0.7 | 0.78 |
| Ca2+ (mmol/L) | 1.00 ± 0.04 | 0.96 ± 0.07 | 0.11 |
| Mg2+ (mmol/L) | 1.17 ± 0.07 | 1.13 ± 0.04 | 0.15 |
| Cl− (mmol/L) | 122 ± 5 | 125 ± 3 | 0.13 |
| Lac− (mmol/L) | 3.4 ± 1.4 | 3.1 ± 1.2 | 0.60 |
| HCO3− (mmol/L) | 22.7 ± 2.1 | 20.4 ± 2.3 | 0.03 |
| Phosph− (mEq/L) | 0.68 ± 0.23 | 0.62 ± 0.17 | 0.56 |
| Alb− (mEq/L) | 0.37 ± 0.26 | 0.19 ± 0.12 | 0.09 |
| Osmolarity (mOsm/L) | 290 ± 9 | 290 ± 2 | 0.95 |
| Gluc (mg/dL) | 73 ± 15 | 76 ± 14 | 0.72 |
| Hb (g/dL) | 0.4 ± 0.4 | 0.3 ± 0.3 | 0.70 |
| RBC (× 106/µL) | 0.12 ± 0.14 | 0.09 ± 0.07 | 0.59 |
| WBC (cells/µL) | 326 ± 572 | 408 ± 588 | 0.76 |
| ΔSID (mmol/L) | 14.4 ± 3.7 | 13.8 ± 1.2 | 0.65 |
| ΔPCO2 (mm Hg) | 0.4 ± 3.8 | 7.8 ± 3.7 | < 0.001 |
Data are expressed as mean ± standard deviation
Alb−, ionized albumin concentration, ATOT, total amount of weak noncarbonic acids, Ca2+, ionized calcium concentration, Cl−, chloride concentration, Gluc, glucose concentration, Hb, hemoglobin concentration, HCO3−, bicarbonate concentration, K+, potassium concentration, Mg2+, magnesium concentration, Na+, sodium concentration, PCO2, partial pressure of carbon dioxide, Phosph−, ionized phosphate concentration, RBC, red blood cell count, SAH, aneurismal subarachnoid hemorrhage, SID, strong ion difference, WBC, white blood cell count
Although CSF SID was lower (p = 0.002) in patients with HA, CSF PCO2 and pH did not differ significantly between groups. Notably, CSF electrolytes did not differ significantly between patients with HA and those without HA. Regarding plasma, in addition to the differences in PCO2 and pH due to the selection criteria, arterial SID was significantly lower in patients with HA.
A lower ΔPCO2 was measured in patients without HA compared with those with HA (Table 3). Of note, the ΔPCO2 of patients with HA was similar to values measured in the control group (p = 0.78).
The Interplay Between Variables and pH
Linear and multilinear regression analyses were used to investigate the association between variables both in controls and in patients with SAH. In controls, no association was found at univariate analysis between CSF SID (p = 0.93) or CSF PCO2 (p = 0.13) and CSF pH.
In patients with SAH, both CSF SID and PCO2 showed a significant association with CSF pH (r = 0.54 [p = 0.01] and r = − 0.59 [p = 0.006], respectively). Both variables remained independently associated at the multilinear regression analysis (r2 = 0.79, p < 0.001; Table S4). At univariate analysis, CSF sodium (r = − 0.55, p = 0.01), chloride (r = − 0.62, p = 0.003), and lactate (r = − 0.60, p = 0.005) were significantly associated with CSF pH. However, at multilinear regression analysis, only lactate was independently associated with CSF pH (Table S5). Lastly, in the overall SAH population, a positive correlation between CSF SID and arterial PCO2 was found (Fig. 2). Similarly, a negative correlation between CSF SID and arterial pH was observed (r = − 0.62, p < 0.004; Fig. S1).
Fig. 2.
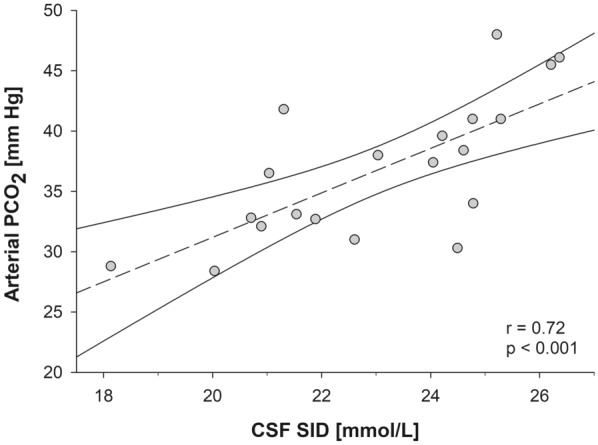
Scatter plot graph of CSF SID and arterial PCO2 in patients with SAH. Scatter Plot representing the association between CSF SID and arterial PCO2 in patients with SAH. Linear regression model (dashed line) is represented with 95% confidence interval. Equation of the linear model: Arterial PCO2 = 1.84 × CSF SID − 5.7. CSF cerebrospinal fluid, PCO2 partial pressure of carbon dioxide, SAH aneurismal subarachnoid hemorrhage, SID strong ion difference
Clinical Outcome
Six patients with SAH experienced an episode of clinical vasospasm; two of these patients died in the ICU, and four patients were lost at follow-up. Plasma and CSF acid–base characteristics of patients who developed vasospasm did not differ significantly from those of patients who did not (Tables S6 and S7). No significant association was observed between CSF SID (or ΔSID) and the modified Fisher scale score by Pearson’s correlation (data not shown). The median 12-month GOS-E score was 6 (3–8). No difference (p = 0.64) was observed in GOS-E scores between patients with HA and those without HA. A good clinical outcome (GOS-E score ≥ 6) was observed in 10 of 16 patients. Patients with worse clinical outcomes had higher CSF lactate concentrations (4.5 ± 1.4 vs. 2.7 ± 0.9, p = 0.006).
Discussion
In the present study, we used the physicochemical approach to acid–base [1] to describe the acid–base characteristics of both arterial blood and CSF of spontaneously breathing patients with SAH and compared these results with those of a control population, i.e., healthy, spontaneously breathing patients undergoing elective surgery under spinal anesthesia. The main finding of our study was that patients with SAH had lower CSF SID, mainly due to an increased concentration of lactate. Arterial SID did not differ between the two populations. Therefore, when blood SID was subtracted from CSF SID (ΔSID), significantly different values were observed in patients with SAH and controls (Fig. 1). This finding clearly points toward a primary metabolic acid–base disorder caused by intracranial bleeding and compartmentalized in the cerebral/CSF environment. Despite the acidifying effect of a reduced CSF SID, CSF pH was similar in the two populations, as CSF PCO2 was significantly reduced in patients with SAH. The respiratory compensation of this local metabolic acidosis led to blood HA in 40% of the SAH population.
Isolated CSF acidosis has been induced in several animal models [22–25] by directly injecting acids into the ventricular system. In these studies, local CSF acidification triggered the respiratory chemoreceptors and caused an increase in minute ventilation to restore CSF pH through a reduction in CSF PCO2 [22]. Given the anatomical and functional separation between blood and CSF, the acids injected locally did not reach the bloodstream, which was therefore unaffected by the acid–base disorder. Consequently, the compensatory hyperventilation corrected CSF pH and led to a systemic HA of the blood.
Although the presence of HA frequently characterizes patients with cerebrovascular conditions, only a few reports [5, 11, 26] investigated the underlying mechanisms. We hypothesized that a local CSF metabolic acidosis, i.e., a reduction in CSF SID, could partially explain the finding. We therefore studied the blood/CSF electrolytes and acid–base of healthy patients undergoing spinal anesthesia and considered these data, for the present study, as baseline values, i.e., normal reference values before the SAH.
Our results from the control population regarding CSF electrolytes and acid–base variables are in line with the available literature [11, 27–29]: normal CSF has a pH around 7.35 with a SID close to 26 mmol/L. In CSF, the protein concentration is negligible, and electrical neutrality is guaranteed by a higher chloride concentration as compared with plasma (120 vs. 109 mmol/L).
The CSF acid–base characteristics of patients with SAH differed significantly as compared with controls. In particular, in patients with SAH, we observed a lower CSF SID, which favors acidification. The causes of the reduced SID are likely multifactorial. First, spontaneous SAH causes the sudden release of blood in the subarachnoid space, frequently including the cerebral ventricles. Although blood has a higher pH and SID, as compared with CSF (Tables 1 and 2), the presence of living red blood cells reaching the CSF could increase CSF lactate concentration because of their anaerobic metabolism [7]. Second, cerebrovascular events frequently lead to cerebral hypoperfusion/ischemia fostering the release of metabolites (mainly lactate) from suffering cerebral tissue [30–33]. Finally, therapies administered in the critical care setting might affect the electrolyte composition of the extracellular fluid, including CSF. Indeed, although the increase in CSF lactate concentration observed in our patients was a consequence of SAH, the alterations of other electrolytes, such as sodium and chloride, could be secondary to the use of isotonic/hypertonic NaCl or other crystalloids. Despite the difference in CSF SID, CSF pH did not differ between the two populations because CSF PCO2 was lower in patients with SAH. This finding might be interpreted as a compensatory hyperventilation triggered by the local CSF acidification.
Similar considerations can be made when analyzing the differences in arterial blood composition (Table 2). Indeed, some differences are likely a consequence of medical treatment. For instance, we observed higher concentrations of sodium and chloride and lower concentrations of albumin, hemoglobin, and phosphates. All these findings can be attributed (at least in part) to intravenous fluids with slightly hypertonic, chloride-rich fluids. In this population, supranormal sodium concentrations and plasma osmolarity are therapeutic targets to reduce the cerebral swelling risk [34]. Overall, the plasma SID of patients with SAH was similar to that of controls. On the other hand, patients with SAH had lower concentrations of weak noncarbonic acids, mainly due to hypoalbuminemia. According to Stewart’s approach, a decrease in weak noncarbonic acids unbalances the acid–base equilibrium toward alkalosis. Of note, hypoalbuminemia is very frequent in critically ill patients [35], in particular in patients with neurologic disorders, given the relative contraindication to its exogenous supplementation [36, 37].
Patients were divided according to the presence/absence of HA (Table 3). This condition was frequent (40%) in patients with SAH, whereas it was present only in one control patient. Per definition, patients with HA had lower arterial PCO2 and higher arterial pH. Moreover, patients with HA had a lower arterial SID, mainly due to a higher chloride concentration. It is conceivable that the renal compensation of HA might have contributed to this finding [38].
On the CSF side, patients with HA had significantly lower CSF SID as compared with nonhypocapnic patients. The reduced SID was not attributable to a single major electrolyte derangement: several nonsignificant electrolyte differences were observed.
Another striking difference between patients with HA and those without HA was ΔPCO2, i.e., the CSF-to-plasma difference in PCO2. In patients with HA, ΔPCO2 had normal values, i.e., values similar to those of controls. On the contrary, ΔPCO2 was significantly reduced in patients without HA (Table 3). Sambrook et al. [11] already described a reduced ΔPCO2 in a case series of patients with SAH, and a reduction of cerebral metabolism was hypothesized to explain this finding [32, 39]. A possible interpretation is the following: a lower cerebral metabolism could characterize patients without HA, leading to a lower local carbon dioxide production and thus a lower difference in PCO2 as compared with arterial blood. Consequently, in patients without HA, CSF pH is close to normal as a result of a lower CSF SID and lower CSF PCO2. Apparently, however, the lower CSF PCO2 is not the result of hyperventilation, because arterial PCO2 is normal, but could be the expression of a reduced local carbon dioxide production. Of note, using positron emission tomography, Carpenter and colleagues [39] documented a reduced cerebral metabolism in patients with SAH.
We used linear and multilinear regression analyses to investigate the association between acid–base variables in the two extracellular compartments. Interestingly, no linear association was found in controls between both CSF PCO2 and SID and CSF pH, suggesting the presence of a stable physiologic equilibrium. On the contrary, in patients with SAH, there was an association between both CSF SID and PCO2 and CSF pH at linear and multilinear regression. Interestingly, only lactate was independently associated with CSF pH at multilinear regression. In the overall SAH population, CSF SID was linearly correlated with both arterial PCO2 (Fig. 2) and pH, suggesting its key role in determining the CSF acid–base disorder and thus favoring the respiratory compensation.
Clinical Implications and Future steps
The presence of hypocapnia in patients with SAH has been hypothesized to be a marker of severity and to be associated with worse outcomes [40]. The idea is that a primary hyperventilation might lead to cerebral hypocapnia and alkalosis, leading to a reduced cerebral blood flow and possibly fostering the development of vasospasm. We did not find any difference regarding clinical outcomes between patients with HA and those without HA. Similarly, we did not find an association between CSF SID and the modified Fisher scale score. This finding might be due to both the limited sample size and the fact that the modified Fisher scale score is based on a computed tomography scan obtained on the day of intensive care admission, whereas the samples are obtained a median of 2 days after bleeding, after the placement of an external ventricular drain.
Our data suggest that the response to a local CSF acidification might differ among patients with SAH and that cerebral metabolism, and therefore local CO2 production, might be an important variable. An additional clinical message of our study could be the following: patients with SAH with an external ventricular drain have an easy and safe access to CSF. This particular extracellular fluid (CSF) contains several physiologic information linked to spontaneous breathing activity and cerebral blood flow. In a patient population characterized by an acute neurologic condition, this fluid might even gain more importance, including, possibly, a prognostic role. Our limited sample size is of course insufficient to draw inferences about the prognostic role of a single value or of its time course. However, we think that it might be a good research question for future studies.
Limitations
We need to acknowledge some limitations of our study. First, a higher presence of men characterized our controls, as compared with the SAH population. Although hormones certainly play a role in respiratory regulation, this is more evident during pregnancy, which was an exclusion criterion for our study. Second, we sampled CSF in different sites, i.e., lumbar in controls and ventricular in patients with SAH. Differences in PCO2 and pH of the two sites are minimal in physiologic conditions [41]. Therefore, it is conceivable to consider the lumbar CSF samples of controls to be similar to their ventricular CSF.
On the contrary, meaningful differences between lumbar and ventricular CSF could be present in patients with SAH [29, 42, 43]. For this reason, we analyzed only ventricular CSF in patients with SAH because this is the extracellular fluid in close contact with the chemosensitive area [44–46]. Moreover, we did not collect and report information regarding the type and amount of intravenous fluids administered to patients with SAH. As discussed above, intravenous fluid therapy might significantly alter the acid–base and electrolyte equilibrium of both blood and CSF [47–51]. Finally, a potential source of variability among CSF samples could derive from the collection timing. The CSF samples of the SAH population were collected as soon as clinically feasible, i.e., reliant on the clinical decision to place an external ventricular drain.
Conclusions
Patients with SAH have a localized CSF acidosis, characterized by a reduced CSF SID and mainly caused by increased lactate concentrations. CSF PCO2 is reduced to restore a normal CSF pH. This compensatory mechanism might explain the high incidence of arterial HA in patients with spontaneous SAH.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
Concept and study design: T.L., F.Z., M.C., R.F., A.P., N.S. Patient recruitment and sample collection: F.Z., A.C., S.B., F.B. Sample analysis: T.L., F.Z., A.C., F.B., C.F.F., A.M. Data analysis and results interpretation: T.L., F.Z., M.C., T.Z., A.Z., E.R.Z., N.S. Drafting and design of the manuscript and figures: T.L., F.Z. Review and editing of the manuscript: All authors. All the authors approved the final version of the manuscript.
Source of Support
Support was provided solely from institutional and/or departmental sources of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy.
Conflict of interest
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or nonfinancial interest in the subject matter discussed in this article.
Ethical approval/informed consent
The study protocol and the informed consent process were approved by the Ethical Committee Milano Area B (protocol number: 25_2017bis), and written informed or deferred consent was obtained from patients according to Italian regulation.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fencl V, Leith DE. Stewart’s quantitative acid-base chemistry: Applications in biology and medicine. Respir Physiol. 1993;91(1):1–16. doi: 10.1016/0034-5687(93)90085-O. [DOI] [PubMed] [Google Scholar]
- 2.Sakka L, Coll G, Chazal J. Anatomy and physiology of cerebrospinal fluid [Internet] Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128(6):309–316. doi: 10.1016/j.anorl.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Langer T, Zanella A, Caironi P. Understanding the role of the cerebrospinal fluid in acid–base disorders. Intensive Care Med. 2016;42(3):436–439. doi: 10.1007/s00134-015-4059-8. [DOI] [PubMed] [Google Scholar]
- 4.Siesjö BK, Kjällquist Å, Pontén U, Zwetnow N. Extracellular pH in the brain and cerebral blood flow. Prog Brain Res. 1968;30:93–98. doi: 10.1016/S0079-6123(08)61444-2. [DOI] [PubMed] [Google Scholar]
- 5.Fencl V, Miller T, Pappenheimer Studies on the respiratory response to disturbances of acid-base balance, with deductions concerning the ionic composition of cerebral interstitial fluid. Am J Physiol Content. 1966;210(3):459–472. doi: 10.1152/ajplegacy.1966.210.3.459. [DOI] [PubMed] [Google Scholar]
- 6.Froman C, Smith AC. Hyperventilation associated with low pH of cerebrospinal fluid after intracranial haemorrhage. Lancet. 1966;1(7441):780–782. doi: 10.1016/S0140-6736(66)91859-9. [DOI] [PubMed] [Google Scholar]
- 7.Froman C, Smith AC. Metabolic acidosis of the cerebrospinal fluid associated with subarachnoid haemorrhage. Lancet. 1967;1(7497):965–967. doi: 10.1016/S0140-6736(67)92354-9. [DOI] [PubMed] [Google Scholar]
- 8.Lane DJ, Rout MW, Williamson DH. Mechanism of hyperventilation in acute cerebrovascular accidents. BMJ. 1971;3(5765):9–12. doi: 10.1136/bmj.3.5765.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Froman C. Alterations of respiratory function in patients with severe head injuries. Br J Anaesth. 1968;40(5):354–360. doi: 10.1093/bja/40.5.354. [DOI] [PubMed] [Google Scholar]
- 10.Plum F, Swanson AG. Central Neurogenic Hyperventilation in Man. Arch Neurol Psychiatry. 1959;81(5):535–549. doi: 10.1001/archneurpsyc.1959.02340170001001. [DOI] [PubMed] [Google Scholar]
- 11.Sambrook MA, Hutchinson EC, Aber GM. Metabolic studies in subarachnoid hæmorrhage and strokes. I: Serial changes in acid-base values in blood and cerebrospinal fluid. Brain. 1973;96(1):171–190. doi: 10.1093/brain/96.1.171. [DOI] [PubMed] [Google Scholar]
- 12.Zoerle T, Lombardo A, Colombo A, et al. Intracranial pressure after subarachnoid hemorrhage. Crit Care Med. 2015;43(1):168–176. doi: 10.1097/CCM.0000000000000670. [DOI] [PubMed] [Google Scholar]
- 13.Rosen DS, Macdonald RL. Subarachnoid hemorrhage grading scales: A systematic review. Neurocrit Care. 2005;2(2):110–118. doi: 10.1385/NCC:2:2:110. [DOI] [PubMed] [Google Scholar]
- 14.Langer T, Carlesso E, Protti A, et al. In vivo conditioning of acid-base equilibrium by crystalloid solutions: An experimental study on pigs. Intens Care Med. 2012;38(4):686–693. doi: 10.1007/s00134-011-2455-2. [DOI] [PubMed] [Google Scholar]
- 15.Langer T, Ferrari M, Zazzeron L, Gattinoni L, Caironi P. Effects of intravenous solutions on acid-base equilibrium: from crystalloids to colloids and blood components. Rev Anaesthesiol Intensive Ther. 2014;46(5):350–360. doi: 10.5603/AIT.2014.0059. [DOI] [PubMed] [Google Scholar]
- 16.Fencl V, Jabor A, Kazda A, Figge J. Diagnosis of metabolic acid-base disturbances in critically III patients. Am J Respir Crit Care Med. 2000;162(6):2246–2251. doi: 10.1164/ajrccm.162.6.9904099. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell RA, Carman CT, Severinghaus JW, Richardson BW, Singer MM, Shnide S. Stability of cerebrospinal fluid pH in chronic acid-base disturbances in blood. J Appl Physiol. 1965;20(3):443–452. doi: 10.1152/jappl.1965.20.3.443. [DOI] [PubMed] [Google Scholar]
- 18.Wilson JTL, Pettigrew LEL, Teasdale GM. Structured interviews for the glasgow outcome scale and the extended glasgow outcome scale: Guidelines for their use. J Neurotrauma. 1998;15(8):573–580. doi: 10.1089/neu.1998.15.573. [DOI] [PubMed] [Google Scholar]
- 19.Lu J, Marmarou A, Lapane K, Turf E, Wilson L. A method for reducing misclassification in the extended Glasgow Outcome Score. J Neurotrauma. 2010;27(5):843–852. doi: 10.1089/neu.2010.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg ED, Gobin YP, Riina H, et al. Role of CT perfusion imaging in the diagnosis and treatment of vasospasm. Imaging Med. 2011;3(3):287–297. doi: 10.2217/iim.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wijdicks EFM. Recording Neurogenic Breathing Patterns in Acute Brain Injury. Neurocrit Care. 2021;34(2):674–676. doi: 10.1007/s12028-020-00922-6. [DOI] [PubMed] [Google Scholar]
- 22.Leusen IR. Chemosensitivity of the respiratory center. Am J Physiol Content. 1953;176(1):39–44. doi: 10.1152/ajplegacy.1953.176.1.39. [DOI] [PubMed] [Google Scholar]
- 23.Pappenheimer JR, Fencl V, Heisey SR, Held D. Role of cerebral fluids in control of respiration as studied in unanesthetized goats. Am J Physiol Content. 1965;208(3):436–450. doi: 10.1152/ajplegacy.1965.208.3.436. [DOI] [PubMed] [Google Scholar]
- 24.Leusen I. Regulation of cerebrospinal fluid composition with reference to breathing. Physiol Rev. 1972;52(1):1–56. doi: 10.1152/physrev.1972.52.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Mascheroni D, Kolobow T, Fumagalli R, Moretti MP, Chen V, Buckhold D. Acute respiratory failure following pharmacologically induced hyperventilation: an experimental animal study. Intens Care Med. 1988;15(1):8–14. doi: 10.1007/BF00255628. [DOI] [PubMed] [Google Scholar]
- 26.Choma L, Kazemi H. Importance of changes in plasma HCO3- on regulation of CSF HCO3- in respiratory alkalosis. Respir Physiol. 1976;26(2):265–278. doi: 10.1016/0034-5687(76)90104-3. [DOI] [PubMed] [Google Scholar]
- 27.Kazemi H, Johnson DC. Regulation of cerebrospinal fluid acid-base balance. Physiol Rev. 1986;66(4):953–1037. doi: 10.1152/physrev.1986.66.4.953. [DOI] [PubMed] [Google Scholar]
- 28.Kazemi H. Regulation of CSF composition- blocking chloride-bicarbonate exchange. 2018; [DOI] [PubMed]
- 29.Rossanda M, Sganzerla EP. Acid-base and gas tension measurements in cerebrospinal fluid. Br J Anaesth. 1976;48(8):753–760. doi: 10.1093/bja/48.8.753. [DOI] [PubMed] [Google Scholar]
- 30.Yatsu FM, Lee LW, Liao CL. Energy metabolism during brain ischemia: stability during reversible and irreversible damage. Stroke. 1975;6(6):678–683. doi: 10.1161/01.STR.6.6.678. [DOI] [PubMed] [Google Scholar]
- 31.Mendelow AD, McCalden TA, Hattingh J, Coull A, Rosendorff C, Eldelman BH. Cerebrovascular reactivity and metabolism after subarachnoid hemorrhage in Baboons. Stroke. 1981;12(1):58–65. doi: 10.1161/01.STR.12.1.58. [DOI] [PubMed] [Google Scholar]
- 32.Fein JM. Cerebral energy metabolism after subarachnoid hemorrhage. Stroke. 1975;6(1):1–8. doi: 10.1161/01.STR.6.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Fujishima M, Sugi T. Cerebrospinal fluid and arterial lactate, pyruvate and acid-base balance in patients with intracranial hemorrhages. Stroke. 1975;6(6):707–714. doi: 10.1161/01.STR.6.6.707. [DOI] [PubMed] [Google Scholar]
- 34.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 35.Langer T, Brusatori S, Carlesso E, et al. Low noncarbonic buffer power amplifies acute respiratory acid-base disorders in patients with sepsis: an in vitro study. J Appl Physiol. 2021;131(2):464–473. doi: 10.1152/japplphysiol.00787.2020. [DOI] [PubMed] [Google Scholar]
- 36.The SAFE Study Investigators Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357(9):874–884. doi: 10.1056/NEJMoa067514. [DOI] [PubMed] [Google Scholar]
- 37.Suarez JI, Martin RH, Calvillo E, et al. The albumin in subarachnoid hemorrhage (ALISAH) multicenter pilot clinical trial. Stroke. 2012;43(3):683–690. doi: 10.1161/STROKEAHA.111.633958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krapf R, Beeler I, Hertner D, Hulter HN. Chronic respiratory alkalosis. N Engl J Med. 1991;324(20):1394–1401. doi: 10.1056/NEJM199105163242003. [DOI] [PubMed] [Google Scholar]
- 39.Carpenter DA, Grubb RL, Tempel LW, Powers WJ. Cerebral oxygen metabolism after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 1991;11(5):837–844. doi: 10.1038/jcbfm.1991.143. [DOI] [PubMed] [Google Scholar]
- 40.Solaiman O, Singh JM. Hypocapnia in aneurysmal subarachnoid hemorrhage: Incidence and association with poor clinical outcomes. J Neurosurg Anesthesiol. 2013;25(3):254–261. doi: 10.1097/ANA.0b013e3182806465. [DOI] [PubMed] [Google Scholar]
- 41.van Heijst ANP, Maas AHJ, Visser BF. Comparison of the acid-base balance in cisternal and lumbar cerebrospinal fluid. Pflugers Arch Gesamte Physiol Menschen Tiere. 1966;287(3):242–246. doi: 10.1007/BF00362485. [DOI] [PubMed] [Google Scholar]
- 42.Shimoda M, Yamada S, Yamamoto I, Tsugane R, Sato O. Time course of csf lactate level in subarachnoid haemorrhage correlation with clinical grading and prognosis. Acta Neurochir (Wien) 1989; [DOI] [PubMed]
- 43.Plum F, Price RW. Acid-Base Balance of Cisternal and Lumbar Cerebrospinal Fluid in Hospital Patients. N Engl J Med. 1973;289(25):1346–1351. doi: 10.1056/NEJM197312202892505. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell RA, Loeschcke HH, Massion WH, Severinghaus JW. Respiratory responses mediated through superficial chemosensitive areas on the medulla. J Appl Physiol. 1963;18(3):523–533. doi: 10.1152/jappl.1963.18.3.523. [DOI] [PubMed] [Google Scholar]
- 45.Kiley JP, Eldridge FL, Millhorn DE. The roles of medullary extracellular and cerebrospinal fluid pH in control of respiration. Respir Physiol. 1985;59(2):117–130. doi: 10.1016/0034-5687(85)90001-5. [DOI] [PubMed] [Google Scholar]
- 46.Nattie E, Li A. Central chemoreceptors: locations and functions. Compr Physiol. 2012;2(1):221–254. doi: 10.1002/cphy.c100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monroe CB, Kazemi H. Effect of changes in plasma bicarbonate level on CSF bicarbonate in respiratory acidosis. Respir Physiol. 1973;17(3):386–393. doi: 10.1016/0034-5687(73)90011-X. [DOI] [PubMed] [Google Scholar]
- 48.Hoorn EJ. Intravenous fluids: balancing solutions. J Nephrol. 2017;30(4):485–492. doi: 10.1007/s40620-016-0363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilic O, Gultekin Y, Yazici S. The impact of intravenous fluid therapy on acidbase status of critically ill adults: A stewart approach-based perspective. Int J Nephrol Renovasc Dis. 2020;13:219–230. doi: 10.2147/IJNRD.S266864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rundgren M, Jonasson H, Hjelmqvist H. Water intake and changes in plasma and CSF composition in response to acute administration of hypertonic NaCl and water deprivation in sheep. Acta Physiol Scand. 1990;138(1):85–92. doi: 10.1111/j.1748-1716.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- 51.Langer T, Santini A, Scotti E, Van Regenmortel N, Malbrain MLNG, Caironi P. Intravenous balanced solutions: From physiology to clinical evidence. Anaesthesiol Intensive Ther. 2015;47(1):s78–88. doi: 10.5603/AIT.a2015.0079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.