

Abstract
The incorporation of a secondary network into traditional single network hydrogels can enhance mechanical properties, such as toughness and loading to failure. These features are important for many applications, including as biomedical materials; however, the processing of interpenetrating polymer network (IPN) hydrogels is often limited by their multi-step fabrication procedures. Here, we report a one-pot scheme for the synthesis of biopolymer IPN hydrogels mediated by the simultaneous crosslinking of two independent networks with light, namely (i) free-radical crosslinking of methacrylate-modified hyaluronic acid (HA) to form the primary network and (ii) thiol-ene crosslinking of norbornene-modified HA with thiolated guest-host assemblies of adamantane and β-cyclodextrin to form the secondary network. The mechanical properties of the IPN hydrogels are tuned by changing the network composition, with high water content (~94%) hydrogels exhibiting excellent work of fracture, tensile strength, and low hysteresis. As proof-of-concept, the IPN hydrogels are implemented as low viscosity digital light processing resins to fabricate complex structures that recover shape upon loading, as well as in microfluidic devices to form deformable microparticles. Further, the IPNs are cytocompatible with cell adhesion dependent on the inclusion of adhesive peptides. Overall, the enhanced processing of these IPN hydrogels will expand their utility across applications.
Keywords: interpenetrating polymer network, hydrogels, photocrosslinking, digital light processing, microparticles
Graphical Abstract
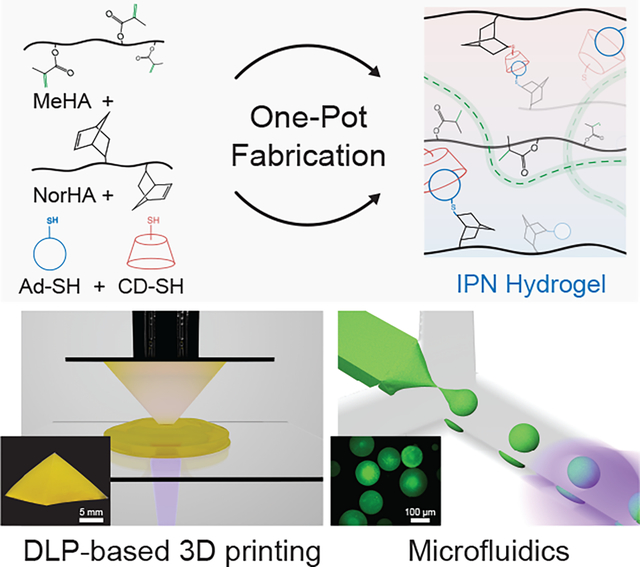
A one-pot photo-initiated reaction strategy is utilized to create tough IPN hydrogels that are amenable to processing modalities such as digital light processing (DLP)-based 3D printing and microfluidic fabrication of microparticles. The mechanical properties of these IPN hydrogels are highly tunable with optimal formulations exhibiting high toughness and low hysteresis despite the high water content.
1. Introduction
Conventional single network (SN) hydrogels generally possess weak mechanical properties, which limits their use in many applications. Interpenetrating polymer network (IPN) hydrogels have emerged to address this limitation, particularly in applications where high mechanical strength and work of fracture are desired.[1] IPN hydrogels consist of two or more independent polymer networks that are held together through inter-network entanglement. These hydrogels are typically fabricated via multi-step approaches wherein the networks are formed sequentially (e.g., swelling of additional monomer or crosslinker into the first network);[2–4] however, this strategy is often time-consuming, results in network heterogeneities, and is not compatible with many processing techniques. To address this, efforts have been made towards the one-pot synthesis of IPNs. Approaches have included thermoreversible sol-gel transitions that require heating (> 60 °C),[5] orthogonal click chemistries that require long reaction times,[6,7] multi-mode crosslinking strategies,[8] or harsh reaction conditions (e.g., toxic monomer, pH).[9] However, these techniques are not directly translatable towards advanced fabrication modalities that require rapid photocrosslinking, such as with stereolithography. Further, there is interest in the processing of hydrogels with high water content and from biopolymers, particularly to introduce cytocompatible materials useful for biomedical applications. Thus, there is a need for one-pot IPN formulations that are rapid, controllable, light-mediated and leverage biopolymers such as hyaluronic acid (HA) in their design.
Many new processing modalities have been developed where single-step reactions are useful, particularly for the fabrication of constructs for tissue repair. As an example, digital light processing (DLP) based 3D printing relies on the crosslinking of non-viscous photoreactive resins in a layer-by-layer fashion.[10–12] The design criteria for DLP resins are that they must (i) have low viscosity to facilitate recoating of the build stage with uncured resin between successive layers and (ii) undergo rapid photo-crosslinking to minimize print times. Although significant progress has been made in the development of IPN inks for alternate 3D printing techniques (e.g., extrusion printing),[13,14] little attention has been paid towards IPN resins for light-based fabrication. Recent studies on DLP-based 3D printing of semi-IPN hydrogels or elastomers have employed synthetic polymers at high concentration (> 20 wt.%) or have utilized multi-step crosslinking.[15–18] As an example, an acrylamide network was DLP printed with alginate present, followed by submersion into a calcium bath to induce ionic crosslinking, which resulted in significant swelling that affected print resolution and fidelity.[17] Sequential crosslinking of IPN hydrogels also introduces challenges related to gradients in mechanical properties or the inability to preserve complex, negative features due to swelling and material crosslinking within these regions. Although there are numerous commercial resins available related to elastomers, most of these contain cytotoxic crosslinkers or solvents that render them unfit for applications that require encapsulating proteins or growth factors and they result in non-degradable structures that are not compatible with many biomedical applications.
Another processing modality that has gained significant interest is the microfluidic fabrication of hydrogel microparticles, which relies on precursor solutions having (i) low viscosity for droplet formation and (ii) rapid crosslinking to prevent droplet coalescence. The flow of viscous precursors in microfluidic channels is often met with challenges such as device failure due to a rise in pressure or increase in droplet polydispersity,[19] while alternate strategies rely on multi-step crosslinking.[20,21] Hydrogel microparticles are useful in a range of biomedical applications, including in the formation of granular hydrogels that are injectable and can be processed with 3D printing.[22] The expansion of microparticle properties, such as with deformable IPN-based properties, would be of great interest to increase their applicability to tissues and constructs where such mechanics are important.
With these examples in mind, new IPN hydrogel strategies are needed to merge the benefits of IPN hydrogel properties with processing compatibility. Thus, the objective of this study is to develop a one-pot IPN fabrication approach to fabricate high water content hydrogels from cytocompatible biopolymers and illustrate the processing of these IPNs across numerous modalities. We first focus on optimizing IPN formulations based on the iteration of various inputs such as network concentrations and compositions, particularly to illustrate superior mechanical properties when compared to traditional SN hydrogels. Next, we show the ease of their processability via DLP or microfluidics and establish their utility through proof-of-concept biomedical applications. Further, we illustrate the cytocompatibility of the IPNs by assessing cellular interactions and show control over cell adhesion through the introduction of adhesive peptides.
2. Results and Discussion
2.1. Design and synthesis of IPN hydrogels via a one-pot reaction
We implement a one-pot scheme with a single photo-initiating step that enables the simultaneous formation of networks via orthogonal chain-growth (i.e., radical crosslinking) and step-growth (i.e., thiol-ene) chemistries (Figure 1). The primary network is composed of methacrylate-modified HA (MeHA) and formed via radical crosslinking (Figure 1a), whereas the secondary network is formed via light-mediated guest-host (GH) crosslinking of norbornene modified HA (NorHA) (Figure 1b, Figure S1). GH supramolecular chemistry (e.g., the interaction of host β-cyclodextrin, CD with guest adamantane, Ad) is used within the secondary network due to the rapid timescales of bond formation that allow the network to adapt to applied mechanical strains without rupture, making it suitable as a ductile network.[23] To overcome the issues of viscosity that occur in traditional supramolecular networks and to meet our design criteria, we introduce an innovative approach for GH network formation. Specifically, monofunctional thiols, namely Ad thiol (Ad-SH) and CD thiol (CD-SH), are reacted with pendant norbornene functional groups via a photo-initiated thiol-ene step-growth reaction with crosslinking due to the dynamic interaction of Ad and CD. Ad-SH and CD-SH, alone or in combination have been utilized to introduce GH functionality to hydrogels, but in different contexts to our approach.[24–26] Supramolecular hydrogels have also been formed by radical copolymerization of inclusion complexes with acrylamide; however, to keep the two networks within IPNs distinct and prevent reactions with MeHA, radical polymerizations of guest-host monomers were avoided.[27] In our design, all reagents (MeHA, NorHA, Ad-SH, CD-SH) are mixed together along with photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate, LAP) and reacted via visible light (400 – 500 nm) exposure to transition a non-viscous solution into an IPN hydrogel (Figure 1c). Additionally, the total polymer concentration is kept low (5 wt.%) to avoid issues with viscosity and to retain the benefits of high water content hydrogels.
Figure 1.
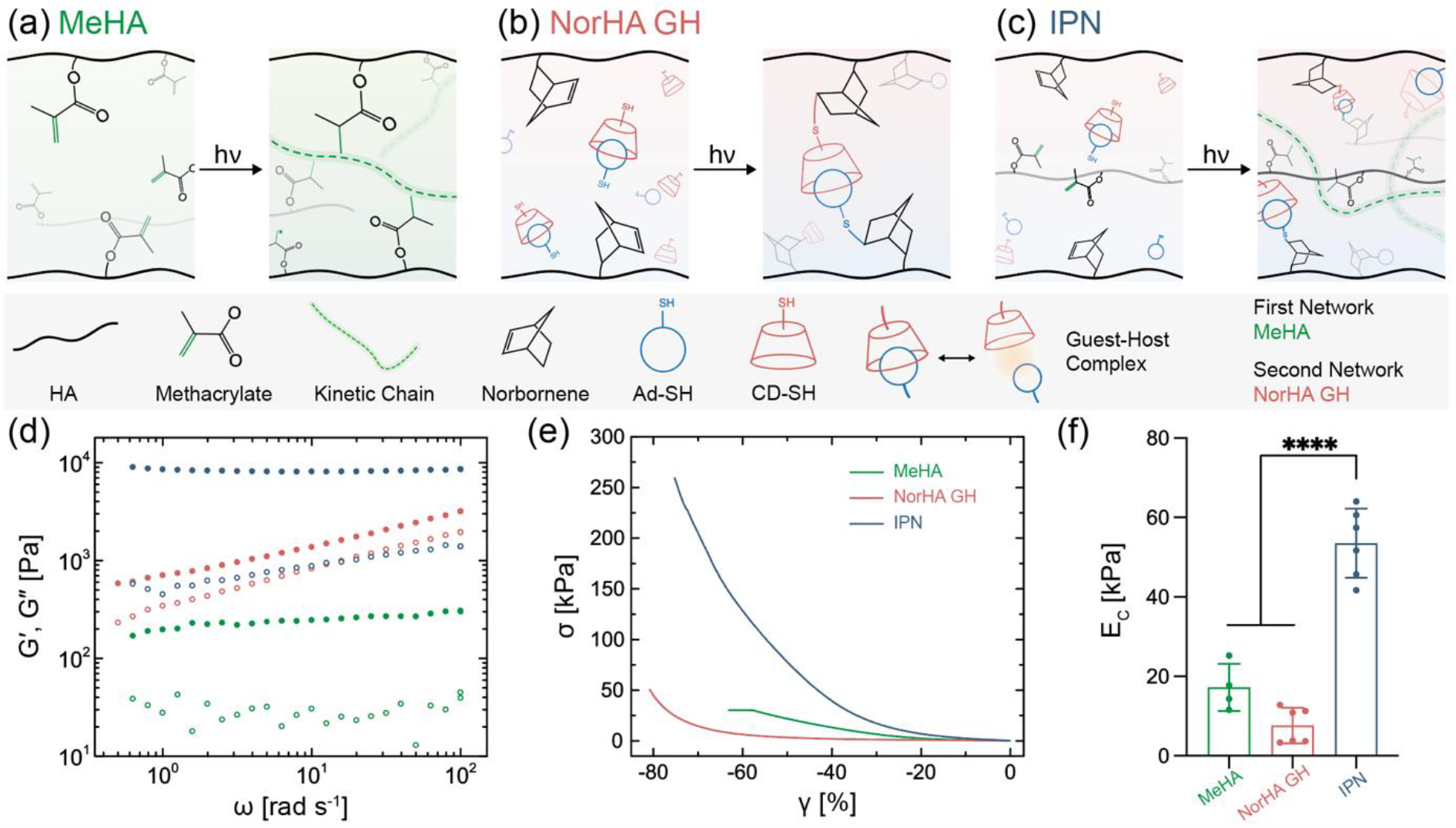
Interpenetrating polymer network (IPN) hydrogels formed via one-pot, light triggered orthogonal reactions. Schematic illustration of chemistry involved in (a) free radical crosslinking of methacrylate modified hyaluronic acid (MeHA) single network (SN), (b) photo-initiated thiol-ene reaction between norbornene modified HA (NorHA) and adamantane thiol (Ad-SH) and β-cyclodextrin thiol (CD-SH) to form a network with guest-host (GH) crosslinks, resulting in a NorHA GH SN, and (c) IPN consisting of simultaneous formation of MeHA (first, brittle) and NorHA GH (second, ductile) networks. Representative profiles of (d) G′ (closed) and G″ (open) as a function of angular frequency and (e) compressive stress-strain response, as well as (f) quantified compressive modulus (EC) for SNs of MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%), and an IPN comprised of these same SN formulations. Data are reported as mean ± SD; n ≥ 4; ****p < 0.0001 (one-way ANOVA).
To better understand IPN hydrogel formation in our design, it is important to understand each crosslinking reaction independently. The influence of alkene (e.g., norbornene, methacrylate) on the thiol-ene reaction rate has been well-studied and it has been reported that norbornenes (ring-strained alkenes) react much more rapidly with thiols than electron poor alkenes such as methacrylates.[28] Additionally, the reaction of methacrylate groups in the presence of radicals is dominated by homopolymerization and therefore, in the presence of both norbornene and methacrylate groups, we expect thiols to preferentially react with norbornenes.[29,30] We illustrate this via the complete consumption of methacrylates and norbornenes within the crosslinked IPN hydrogels, which would show unreacted norbornenes if thiols were undergoing an alternate reaction (Figure S2a). To further corroborate this finding, we performed Ellman’s assay to quantify the conversion of free thiols (Ad-CD) and found a near stoichiometric consumption of thiol:norbornene, indicating minimal thiol-methacrylate side-reaction (Figure S2b). Taken together, we expect the MeHA and NorHA GH networks within the IPN to be primarily independent of each other; however, given the nature of simultaneous light-mediated reactions of methacrylates and norbornenes, chain transfer could occur and result in loose interconnection between the two networks. Also, to avoid any undesired Michael addition between thiols and methacrylate groups, the crosslinker is mixed into the solution immediately before use.
The reaction of Ad-CD crosslinker with NorHA proceeds rapidly (within six seconds) upon light exposure, whereas photo-crosslinking of MeHA occurs relatively slower (on the order of tens of seconds) (Figure S2c). This is likely due to the less oxygen-sensitive thiol-ene reaction and the well understood kinetics of methacrylates via radical polymerization.[31] Despite the differences in the reaction kinetics of the networks, we refer to these reactions as “simultaneous” as there is overlap in their reaction profiles and they form from a single step of radical formation with light exposure. It also remains unclear if the crosslinking of the methacrylate network perturbs the dynamic guest-host bonds within the secondary network. To assess the compatibility of the two networks, we visualize IPN hydrogels formed with fluorescently labelled MeHA. IPNs and MeHA SNs appear fairly homogeneous with no evidence of macroscale phase separation or disruption of the hydrogel microstructure (Figure S3). Future studies can further investigate hydrogel microstructures as a result of the interpenetration of two networks, particularly through the development of theoretical and computational models.
Additionally, our approach of leveraging light to decorate HA with small molecule GH moieties via thiol-ene reaction circumvents the prior challenges with mixing during gelation of previously investigated GH hydrogels.[32,33] Moreover, the use of Ad-SH and CD-SH results in rapid conversions in part due to their high mobility during reaction. Consistent with previous studies,[34] we expect that supramolecular networks formed with preorganized Ad-CD complexes result in improved homogeneity and mechanics when compared to those formed by the mixing of guest- and host-modified polymers.
The interpenetration of covalent and physical networks endows the IPN hydrogel with tunable properties that are distinct from the SN hydrogels. Oscillatory shear rheology of the NorHA GH network displays a frequency dependent response, whereas the MeHA and IPN show largely constant storage moduli (G′) across the frequency sweep, confirming the elastic nature of the hydrogels (Figure 1d). Further, based on the differences in crosslinking chemistry, both MeHA and NorHA GH SN hydrogels exhibit distinct properties under compression (Figure 1e,f). MeHA hydrogels are relatively strong but display brittle behavior under compression (EC = 17.9 ± 5.9 kPa), whereas NorHA GH hydrogels exhibit lower moduli (EC = 7.6 ± 4.5 kPa) and are able to sustain compressive strains up to ~80% without failure. IPN hydrogels have compressive properties that are far higher than the constituent networks, with a compressive modulus (53.5 ± 8.7 kPa) and ultimate compressive strength that are greater than an additive increase of the two SNs. In addition to the mechanical properties, the mass swelling ratio and equilibrium water content of MeHA hydrogels, NorHA GH hydrogels, and IPN hydrogels are 47.9 and 98%, 14.7 and 93%, and 15.3 and 94%, respectively (Figure S4). Furthermore, the IPN hydrogels are stable under physiological conditions with no significant reduction in compressive modulus over 35 days (Figure S5). Macroscopically, IPN hydrogels are strong and flexible to withstand large deformations such as bending, compression, and molding into twisted knots (Figure S6).
2.2. Influence of IPN formulation on mechanical properties
To elucidate the influence of network composition on IPN hydrogel properties, tensile testing is performed across variations in input network formulations (Figure 2). There are numerous mechanical properties of interest, however, we focus on work of fracture (Wf), tensile modulus (ET), stretch at failure (λf), and stress at failure (σf) to provide a holistic picture of material properties. Wf is defined by the area under the stress-stretch curve and is an important property of IPN hydrogels and correlates to the energy required to stretch a sample until failure. Although it is sometimes referred to as work of extension, we chose Wf to emphasize that the value is related to when the sample fails. Additionally, λf is also denoted as stretchability and σf as tensile strength.
Figure 2.

Influence of network composition and chemistry on mechanical properties of IPN hydrogels. (a) Various parameters within IPNs that are important to overall material properties. (b) Increasing MeHA (brittle network) concentration at a fixed NorHA GH concentration. (i) Stress (σ)-stretch (λ) curves of samples stretched uniaxially to rupture, (ii) work of fracture (Wf), (iii) tensile modulus (ET), and (iv) stretch at failure (λf) for IPN hydrogels with varying MeHA concentration and constant NorHA GH concentration (4.5 wt.%). Statistical comparisons of all groups to only the MeHA (0.6 wt.%) group shown on graphs. (c) Increasing NorHA GH (ductile network) concentration at a fixed MeHA concentration. (i) Stress-stretch curves of samples stretched uniaxially to rupture, (ii) Wf, (iii) ET, and (iv) λf for IPN hydrogels with varying NorHA GH concentrations and constant MeHA concentration (0.6 wt.%). Statistical comparisons of all groups to only the NorHA GH (4.5 wt.%) group shown on graphs. (d) Varying crosslinker chemistry (DTT) and crosslinker ratio (Ad:CD) at a fixed MeHA and NorHA GH concentration. (i) Stress-stretch curves of samples stretched uniaxially to rupture, (ii) Wf, (iii) ET, and (iv) λf for IPN hydrogels at constant MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%) concentration. Statistical comparisons of all groups to only Ad:CD (1:1) group shown on graphs. Data are reported as mean ± SD; n ≥ 4; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (one-way ANOVA).
With respect to SNs, NorHA GH hydrogels are able to sustain high degrees of stretch before failure but display low tensile moduli and tensile strengths. Conversely, MeHA SN hydrogels are extremely brittle and fail at low stretch. Both SN hydrogels exhibit low work of fracture, which greatly increase with all IPN hydrogel formulations, including the highest being 175- and 9-fold higher than those of MeHA and NorHA GH SNs, respectively. Similar improvements are observed in the measured stress at failure between the SNs and the IPN hydrogels (Figure S7). The fold change difference in tensile properties between our SNs and IPN hydrogels are on par with those reported for other double network hydrogels.[35]
To systematically investigate the contribution of each network to overall IPN hydrogel properties (Figure 2), we first alter the MeHA network concentration (i.e., brittle component) while keeping the NorHA GH concentration fixed (Figure 2b, Figure S7). As expected, tensile moduli of IPN hydrogels increase and consequently, the stretch at failure values decrease significantly, even with a small increase in MeHA concentration. Increasing the MeHA concentration relative to the NorHA GH results in IPN hydrogels with more brittle properties, which is consistent with changes in compressive moduli from 6.5 kPa to 106 kPa with small changes in MeHA concentration from 0.25 wt.% to 1.00 wt.% (Figure S8). For IPN hydrogels, the work of fracture reaches a maximum of 35.2 ± 11.1 kJ m−3 at 0.6 wt.% MeHA. Next, we alter the NorHA GH concentration (i.e., ductile component) while keeping the MeHA concentration constant (Figure 2c, Figure S7). Similar to trends with the MeHA concentration, work of fracture reaches a peak value at 4.5 wt.% NorHA GH concentration. Tensile moduli and stretch at failure both increase with an initial increase in NorHA GH concentration but do not change significantly at higher concentrations. This can be attributed to the fact that at high NorHA GH concentrations, the relative contribution of MeHA becomes less prominent and the IPN behaves as a ductile NorHA GH SN. Hence, there exists a delicate balance between the MeHA and NorHA GH concentrations at which synergistic enhancement in mechanical properties are realized. This data also highlights the tunability of our IPN system, where IPN properties can be easily tailored for applications that require a high tensile modulus or those that might prefer a high tensile stretch, by simply changing the network concentrations.
Beyond the concentrations of the individual networks, the degrees of modification (mod.) of MeHA (Figure S9) and NorHA (Figure S10) also strongly affect the mechanical behavior of IPN hydrogels. While keeping the concentration of MeHA and NorHA GH within the IPN constant at 0.6 wt.% and 4.5 wt.%, respectively, the work of fracture and stress at fracture increase with increasing MeHA and NorHA mod. While stretch at failure does not change significantly with changes in MeHA mod., it increases significantly with NorHA mod., highlighting the increased number of GH bonds that need to be broken for the sample to fail. Due to challenges associated with synthesis of NorHA at high mod., the highest degree of modification studied is 45% mod. To better understand the significance of GH crosslinking within the secondary network, we replace it with dithiothreitol (DTT) as the crosslinker (Figure 2d). This covalent crosslinking within the secondary network results in brittle IPN hydrogels with a much higher tensile modulus (93.4 ± 24.9 kPa) but significantly lower work of fracture when compared to IPN hydrogels with GH crosslinking. The use of thiol-ene chemistry to crosslink norbornenes in our approach can be extended to a wide range of multifunctional thiol crosslinkers beyond guest-host and DTT illustrated here.
Since the light mediated GH crosslinking used in this study provides tunability in terms of functionalization of Ad and CD, we also investigate the influence of the molar ratios of Ad to CD on the tensile properties of resulting IPN hydrogels. IPN hydrogels that are solely crosslinked with either Ad or CD (i.e., Ad:CD ratio of 1:0 and 0:1, respectively) exhibit lower work of fracture and stretch at failure when compared to IPNs that are crosslinked with Ad:CD at a ratio of 1:1. In general, perturbing the ratio of Ad:CD away from the stoichiometric amount results in guest or host moieties that do not find binding partners, thereby reducing crosslinking and resulting in IPNs with lower tensile properties. To further illustrate this, we soak IPN hydrogels crosslinked with 1:1 Ad:CD ratio in a solution of free host molecule (β-cyclodextrin) to disrupt the GH crosslinking. Displacing the Ad molecules from crosslinking with added host molecules decreases the work of fracture by 21.4 kJ m−3, likely due to a reduction in the effective crosslinking of the secondary network (Figure S11).
When MeHA SN (0.6 wt.%) and IPN containing the same amount of MeHA network are stretched until rupture, their failure properties are very distinct from one another. In the case of MeHA SN, an initiated crack propagates rapidly and causes the entire sample to break at low stretch (λ = 1.26 ± 0.05). In contrast, catastrophic crack propagation in an IPN hydrogel is suppressed by the presence of the NorHA GH network, which allows the sample to sustain much higher stretch (λ = 2.43 ± 0.25). Taken together, this supports that the toughening mechanism for the IPN hydrogel is similar to that observed in traditional double-network hydrogels.[36] When the IPN hydrogels are tested in tension, the (sacrificial) MeHA network breaks to dissipate stress, while the NorHA GH network extends to withstand elongation. The level of elongation exhibited by IPN hydrogels is relevant to the physiological stretch experienced by musculoskeletal tissues such as cartilage and tendons.[37]
Although the toughness of IPN hydrogels (166 ± 52 J m−2) reported in the current study is lower than the conventional tough double network hydrogels previously reported, it is important to note that here the polymer concentration is kept relatively low (5 wt.%) to enable a low-viscosity solution compared to the high polymer concentrations (> 20 wt.%) widely used in previous studies (Table S1). Moreover, the reactive group (methacrylate or norbornene) concentrations within our IPN precursors are orders of magnitude lower than the concentrations of monomer typically employed. Further, when compared to other IPN hydrogels with similar water contents, the work of fracture, toughness, and tensile modulus are similar to those that we observe for our IPNs, now with the benefit of a one-pot light-mediated fabrication that allows processing that is not feasible with many other IPN hydrogel fabrication procedures.
2.3. IPN hydrogel response to cyclic loading
In addition to high tensile strength, IPN hydrogels designed for loading must be able to sustain cyclic stretch (Figure 3). Based on the peak in work of fracture highlighted in Figure 2, an optimal IPN formulation comprised of MeHA (0.6 wt.%, 100% mod.) and NorHA (4.5 wt.%, 45% mod.) is employed for further studies. Samples are first subjected to loading-unloading cycles of varying maximum stretch (Figure 3a). IPN hydrogels effectively dissipate energy as evidenced by pronounced hysteresis at higher stretch, where the hysteresis energy or energy dissipated per unit volume – defined as the area between the loading and unloading cycles – increases with imposed stretch (Figure S12). Under small deformation, IPN hydrogels undergo negligible hysteresis and recover their original length after unloading; however, when the sample is subjected to higher stretch, covalent bonds within the MeHA network irreversibly break and result in relatively higher hysteresis energy. Interestingly, there is no macroscopic crack propagation, and the samples exhibit negligible residual stretch upon unloading, potentially due to the intact NorHA GH network and any remaining MeHA network.
Figure 3.
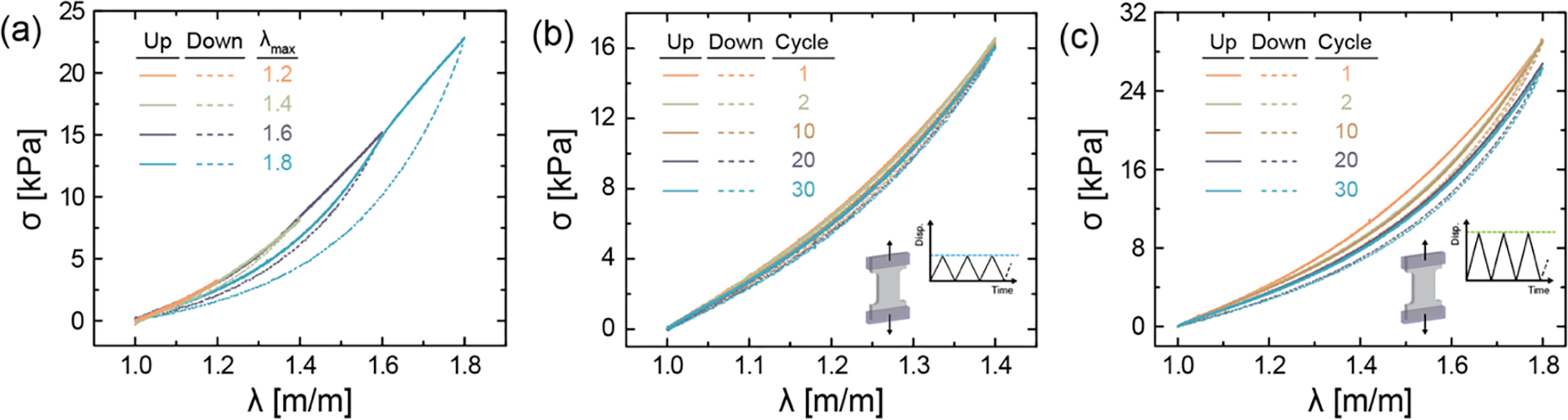
IPN hydrogels subjected to loading and unloading. Representative (a) stress-stretch curves of the same sample subjected to increasing stretch with each cycle (λ = 1.2 to 1.8). Stress-stretch curves of IPN hydrogels subjected to cyclic loading (up to 30 cycles) at constant stretch of (b) λ = 1.4 (λ < λc) and (c) λ = 1.8 (λ > λc) to assess damage. IPN hydrogel composition is kept fixed at MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%) for all studies. Data are reported as mean ± SD; n ≥ 3.
In order to determine the critical stretch at which crack propagation begins, notched IPN samples are subjected to uniaxial tension (Figure S13). IPN hydrogels subjected to cyclic loading at maximum stretch (i) lower than (Figure 3b) and (ii) higher than (Figure 3c) the critical stretch are able to sustain multiple cycles without failure. Minimal change in hysteresis, stress at maximum stretch, and tensile modulus are observed over cycles when the samples are subjected to λ = 1.4; however, for samples cycled at λ = 1.8, hysteresis energy in the first cycle is relatively larger than subsequent cycles and the maximum stress at imposed stretch decreases minimally from 29.1 kPa (cycle 1) to 26.4 kPa (cycle 30). Similarly, with repeated loading at high stretch, softening of the IPN hydrogel occurs as the tensile modulus of the sample decreases by 4.8 kPa over 30 cycles. These trends are in agreement with those previously reported for other double-network hydrogels.[38,39]
2.4. DLP-based 3D processing of IPN hydrogels
To demonstrate the utility of tough, one-pot IPN hydrogels, we explore their processability through advanced fabrication techniques (Figure 4). For example, light-based printing has emerged as a promising fabrication technique for biomedical applications.[40,41] DLP-based 3D printers utilize (i) a micromirror device to control spatial light illumination and (ii) a build stage that moves vertically to allow fabrication of constructs in a layer-by-layer manner (Figure 4a). The xy resolution is determined by the light path, whereas z resolution is dictated by light-attenuating additives that confine the polymerization to the desired layer thickness, thereby improving pattern fidelity.[12] Hence, this technique can achieve higher resolution structures with faster print speeds over other 3D printing techniques (e.g., extrusion-based methods). Owing to the light-initiated simultaneous formation of networks, our non-viscous resin is readily processed into IPN hydrogel constructs directly on a DLP printer. To control resolution in the z-direction and to print structures with negative or over-hanging features, photoabsorber (in this case, tartrazine) is added to the resin. The amount of photoabsorber added is a balance between the resolution desired and construct mechanical properties as these additives can affect the crosslinking kinetics and storage moduli of IPN hydrogels in a dose-dependent manner (Figure S14). Working curves for the IPN resin with different concentrations of photoabsorber are used to minimize exposure times for gelation. To this end, constructs could be printed with tartrazine (1 mM), exposure times of 2.5 s per layer, and a voxel step size of 100 μm. Post-printing, the IPN constructs retain their resolution and do not undergo significant swelling over seven days (Figure S14). Printability assessment of IPN in comparison to SN hydrogels reveals the feasibility of printing and resolution of IPN is similar to commercial resins (Figure S15).
Figure 4.
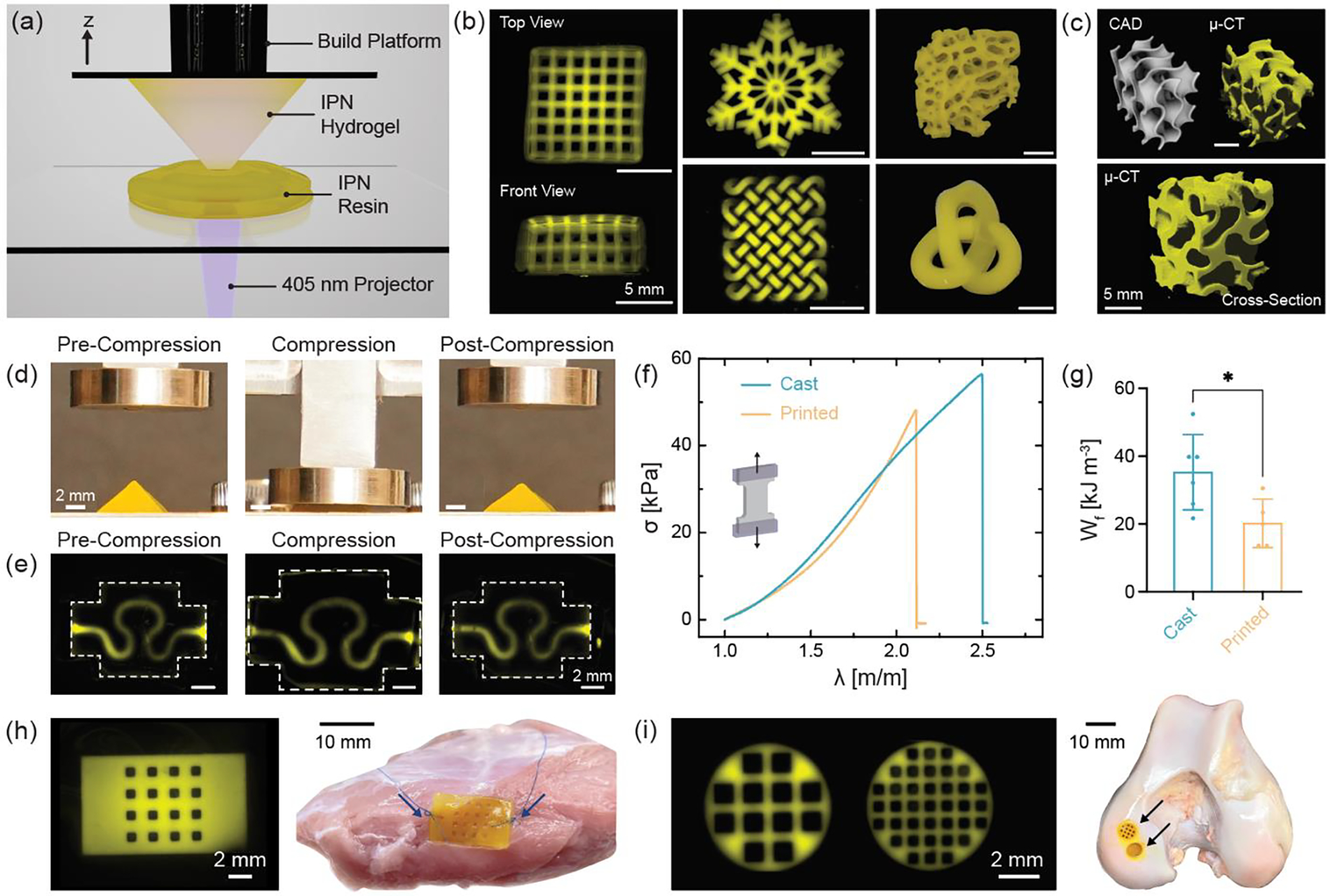
3D processing of IPN resin into complex shapes using one-step light triggered orthogonal reactions. (a) Schematic representation of digital light processing (DLP) stereolithography fabrication of IPN hydrogel structures. (b) Fluorescence images of macroporous lattice (top and front views), snowflake, and knotted mesh structures and photographs of trabecular bone and 3D knot printed with IPN resin. Scale bar: 5 mm. (c) CAD and μ-CT rendering of gyroid 3D printed with IPN resin. Scale bars: 5 mm. Shape-recovery ability of (d) pyramid and (e) serpentine microfluidic device before, during, and after compression to ~65% strain. Scale bars: 2 mm. (f) Stress-stretch curves until failure and (g) work of fracture (Wf) for casted and 3D printed dog-bone specimens. Data are reported as mean ± SD; n ≥ 5; *p < 0.05 (two-tailed t-test). (h) 3D printed porous patch sutured to bovine skeletal muscle. Arrows indicate suture location. Scale bars: 2 mm and 10 mm. (i) 3D printed macroporous discs implanted into a bovine femoral condyle defect. Arrows indicate site of implantation. Scale bars: 2 mm and 10 mm. IPN hydrogel composition is kept fixed at MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%) for all studies.
To illustrate the capability of our IPN resin, we 3D print microporous structures such as a lattice (1 mm square channels in all directions and 0.5 mm distance between struts), as well as constructs that mimic trabecular bone (Figure 4b). To demonstrate the advantageous precision and sophistication of DLP, we fabricate geometries such as a snowflake and those with intertwined loops such as the knotted mesh and 3D (continuous) knot. The capacity of the IPN resin to form structures with highly curved features and ordered interconnected pores is further validated with 3D printing of a gyroid structure and visualized using micro-computed tomography (μ-CT). The capability of the IPN resin to form complex scaffolds with high fidelity despite the high (~94%) water content is notable and structures as tall as 12.5 mm (z-height) can be 3D printed in under 30 minutes without the need for any support.
Beyond being processed with DLP printing, the printed hydrogel structures are highly deformable and tough. IPN hydrogels exhibit shape-recovery as qualitatively evidenced from the ability of 3D printed (i) pyramid, (ii) gyroid, and (iii) brain structures to regain their original shape without permanent damage even after large deformation (Figure 4d, Figure S16). In addition to compression, 3D printed rings can be stretched without rupture and recover to their original shape upon removal of load (Figure S16). Next, we print perfusable microfluidic devices with shape-recovery capabilities that could be useful to mimic the dynamic loading of tissues in vitro.[42,43] The serpentine channel (~1 mm diameter) within the device is able to sustain high compressive strain without constriction, collapse, or macroscopic cracks, as evidenced by the perfusion of fluorescently labelled dextran through the channel before, during, and after compression (Figure 4e). We also investigate if the mechanical properties of IPN hydrogels are affected by the printing process by comparing printed hydrogels to those fabricated via simple casting into a mold. Dog-bone specimens and cylindrical discs are 3D printed and subjected to tension or compression testing, respectively (Figure S17). The work of fracture for the 3D printed samples (20.2 ± 7.1 kJ m−3) is lower than the cast specimens; however, there is no significant difference between the tensile moduli and stress at failure values of the two groups (Figure 4f). The drop in work of fracture of the printed samples can be attributed to the use of photoabsorber and the low light exposure time used for the fabrication of each layer. While addition of photoabsorber allows control over light penetration depth, it also creates a gradient in crosslinking density through each layer, often causing a reduction in mechanical properties, as has been reported previously.[44]
Importantly, the mechanical properties (work of fracture, tensile and compressive moduli, stress at failure) of 3D printed IPN hydrogels are superior to other commerically available and widely used SN hydrogel resins such as poly(ethylene glycol) diacrylate (PEGDA) and gelatin methacrylamide (GelMA) (Figure S18). 3D processing via DLP also allows fabrication of complex, porous constructs that retain the properties of IPN hydrogels. For example, we leverage the enhanced ability of the IPNs to resist crack propagation toward applications that involve suturing. IPNs exhibit significantly increased suture retention strengths when compared to MeHA and NorHA GH SNs (Figure S19). Like bulk IPN hydrogels, 3D printed IPNs can be sutured directly to tissue (e.g., skeletal muscle) (Figure 4h). Such a property greatly increases the utility of hydrogels for applications such as biomaterial patches or to secure cell-laden hydrogel constructs to tissues. Further, we show that porous 3D printed IPNs can be readily press-fit within defects of the femoral condyle (Figure 4i). In order for these IPNs to be suitable as acellular scaffolds within cartilage defects, it is important that they recapitulate native tissue properties such as low friction coefficient (Figure S19). The measured coefficient of friction of IPN hydrogels (0.13 ± 0.02) is similar to that measured for native bovine cartilage as well as that reported previously for other double network hydrogels.[45]
In addition to mechanical strength, cytocompatibility is an important requirement for many biomedical applications. We first compare cell behaviors when interacting with SN and IPN hydrogels modified with adhesion peptide, particularly through the visualization of cytoskeletal organization and nucleus. No significant differences are observed in the number of cells (per unit area) attached across SN and IPN hydrogels after one day of culture in growth media. Cell spreading is also seen across all groups; however, cells seeded on NorHA GH SN and IPN spread to a lower extent to those on MeHA SN hydrogels (Figure S20). These results agree with previous reports where such differences are attributed to the higher viscous modulus (G″) for NorHA GH and IPN hydrogels, whereas MeHA SN hydrogels are primarily elastic.[46]
Beyond processing, the control over cellular interactions is important towards their use, as some applications require limited cell adhesion whereas cell adhesion is desired in other applications (Figure 5). IPNs by themselves do not support cell adhesion or spreading due to the lack of integrin binding motifs; however, with the incorporation of thiolated RGD (GCGYGRGDSPG, cell adhesion sites), IPN hydrogels do support cell attachment and spreading (Figure 5a–c). Cell proliferation as evidenced by an increase in the number of cells per unit area is seen over seven days of culture on IPN hydrogels (Figure S21). To highlight applications of DLP-processed IPN hydrogels, we first leverage their resistance to cell adhesion to our advantage. Engineered 3D microtissues are useful for in vitro disease modeling but are often produced in PDMS molds that require multi-step fabrication steps.[47,48] With the help of DLP and our one-pot IPN resin we fabricate complex molds in one-step (Figure 5d). Collagen gels seeded with 3T3 fibroblasts fill the molds and undergo compaction to form microtissues, where the posts are flexible and can bend to accommodate tissue contraction. In other applications, cell adhesion is desired, such as for tissue engineering scaffolds. IPN hydrogels fabricated into microporous scaffolds with adhesive ligands incorporated can support cell colonization throughout the scaffold (Figure 5e). Due to the properties of IPN, such scaffolds can also be mechanically stimulated without failure to influence the behavior of interacting cells.
Figure 5.
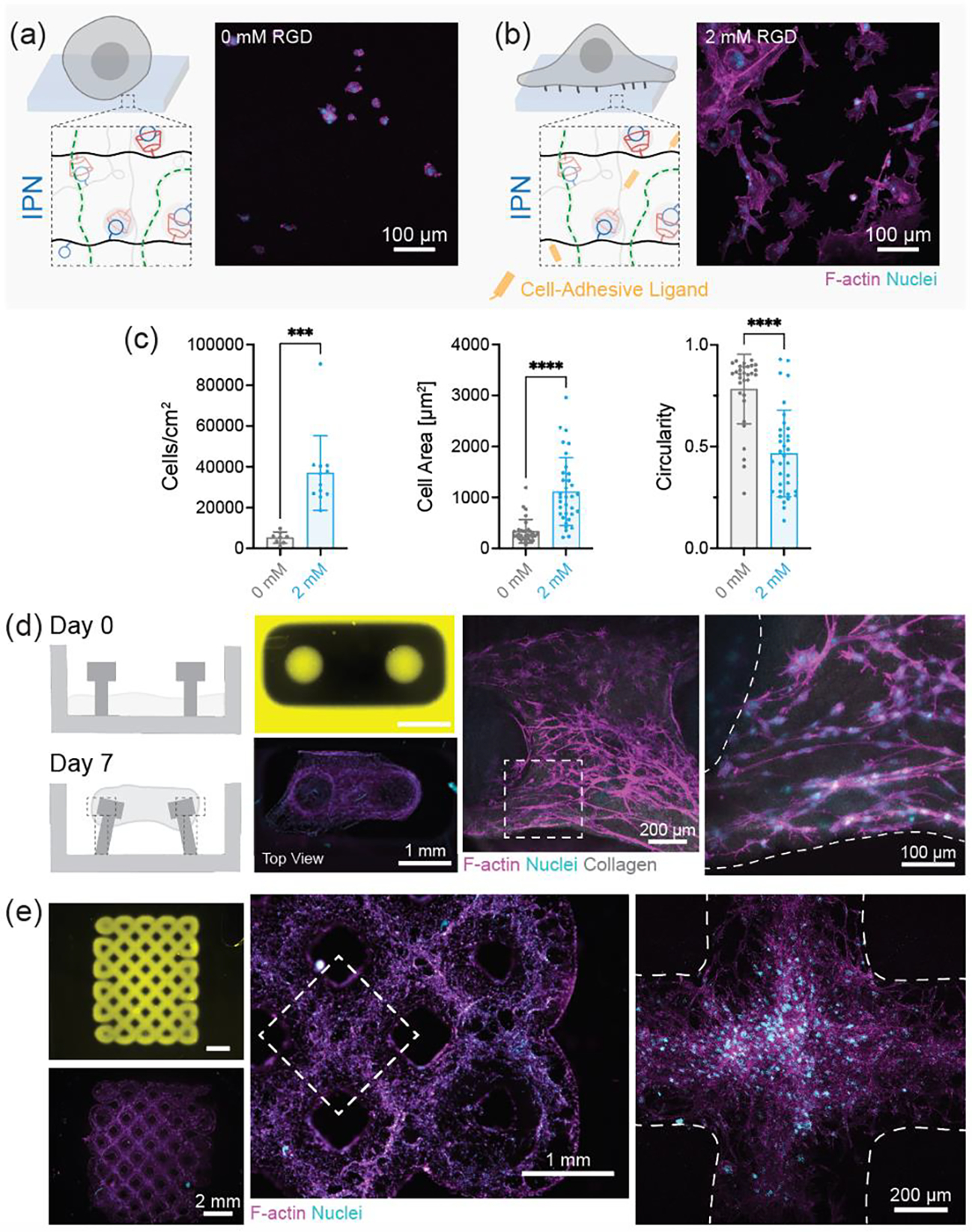
Cell interactions with 3D printed IPN hydrogels. Schematic representation and maximum projection image of 3T3 fibroblasts on IPN hydrogels with (a) 0 mM RGD (absence of cell adhesive ligands) and (b) 2 mM RGD (presence of cell adhesive ligands). Scale bars: 100 μm. (c) Quantification of the number of cells per unit area, cell spread area, and circularity of cells from (a) and (b). Data are reported as mean ± SD; n ≥ 8; ***p < 0.001; ****p < 0.0001 (two-tailed t-test). (d) Schematic representation of 3D printed mold for formation of collagen microtissue. Collagen gels seeded with fibroblasts at day 0 undergo contraction to form a dense tissue by day 7. Fluorescence images of 3D printed mold (yellow) at day 0 and tissue at day 7. Maximum projection images of microtissue to visualize cell morphology. Scale bars (left to right): 1 mm, 200 μm, and 100 μm. (e) Fluorescence images of 3D printed macroporous lattice (yellow) at day 0 and cellularization at day 1. Maximum projection images of lattice to visualize cell morphology. Scale bars (left to right): 2 mm, 1 mm, and 200 μm. F-actin (magenta), nuclei (cyan). IPN hydrogel composition was kept fixed at MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%) for all studies
2.5. 3D processing of IPNs into microparticles and granular hydrogels
Characteristics such as low viscosity and rapid photo-crosslinking of the IPN precursor also allows for the formation of IPN microparticles via droplet microfluidics (Figure 6). Droplets are generated in oil through a flow-focusing device and photocrosslinked off-chip to form IPN microgels with an average diameter of 167 ± 27 μm (Figure 6a,b). As above, we investigate if the fabrication process affects IPN hydrogel mechanical properties; nanoindentation of IPN microgels and cast IPN hydrogel films reveal no significant difference in the modulus of the two groups (Figure 6c–d, Figure S22). Since each individual microgel consists of interpenetrating MeHA and NorHA GH networks, they should be endowed with shape-recovery abilities similar to bulk hydrogels. 3D reconstruction of fluorescent z-slices revealed that IPN microgels readily withstand compressive loading (0.2 kg, 1.96 N) without rupture or significant permanent deformation (Figure 6e, Figure S23).
Figure 6.
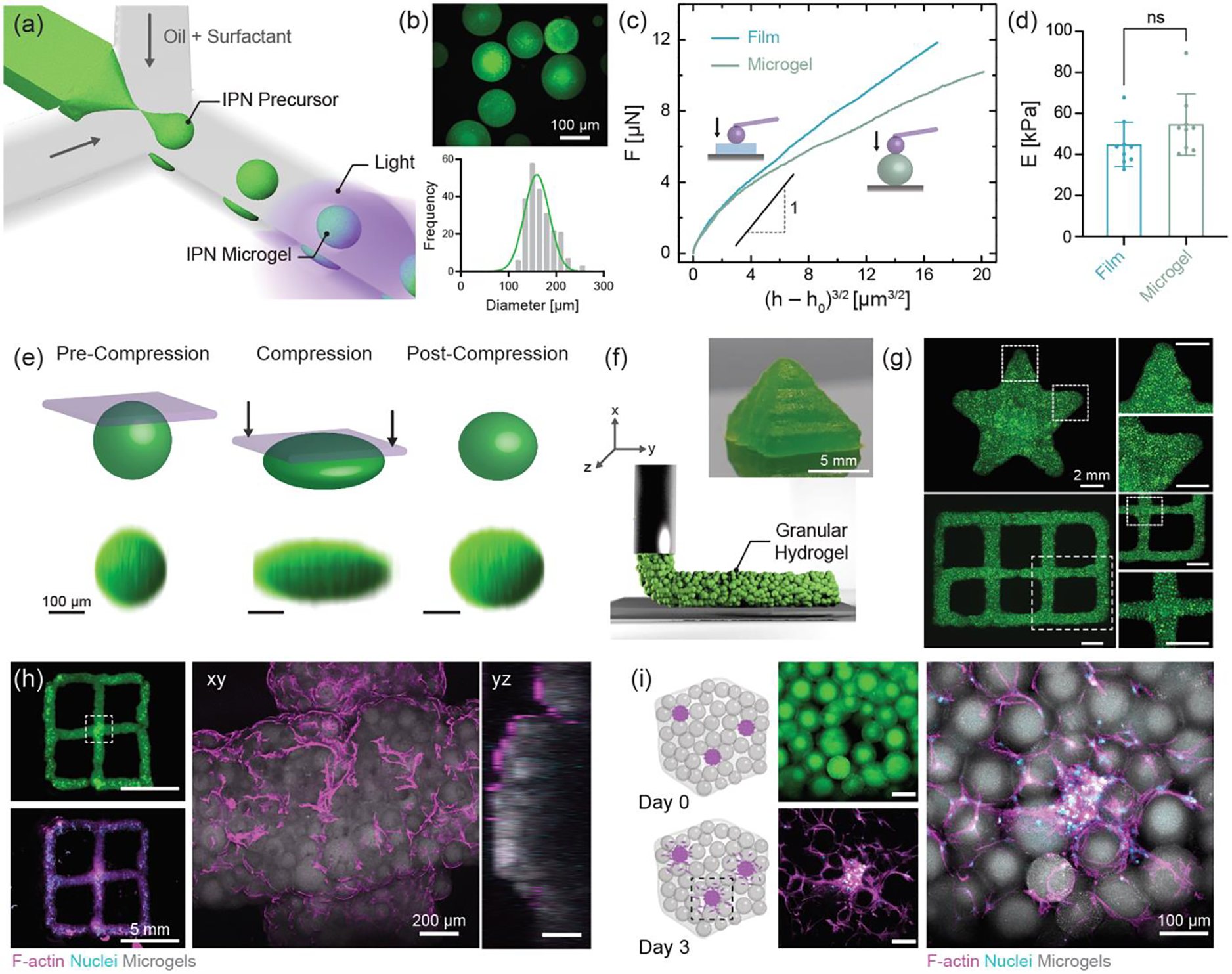
3D processing of IPN precursor into microparticles using one-step light triggered orthogonal reactions (a) Schematic representation of microfluidic fabrication of IPN microgels. (b) Fluorescence image and size distribution of IPN microgels. Scale bar: 100 μm. (c) Force (F) as a function of effective indentation, (h − h0)3/2 and (d) Young’s modulus (E) for casted IPN hydrogel films and IPN microgels. Data are reported as mean ± SD; n ≥ 8; ns = not significant (two-tailed t-test). Representative (e) shape-recovery of IPN microgel shown before, during (Force = 1.96 N), and after compression. Scale bar: 100 μm. (f) Schematic illustration of extrusion printing of jammed IPN microgel ink. Photograph of 3D printed pyramid from IPN microgel ink. Scale bar: 5 mm. (g) Fluorescence images of 3D printed star and lattice structures. Scale bar: 2 mm. (h) 3D printed lattice seeded with fibroblasts. Representative maximum projection image showing cell attachment and spreading at day 1. F-actin (magenta), nuclei (cyan), and microgels (gray). Scale bars: 5 mm and 200 μm. (i) Schematic illustration of hMSC/HUVEC spheroids encapsulated in a 3D granular hydrogel. Cell outgrowth seen from spheroids at day 3. Scale bar: 100 μm. IPN hydrogel composition is kept fixed at MeHA (0.6 wt.%) and NorHA GH (4.5 wt.%) for all studies.
To extend the utility of IPN microgels, they are assembled into granular hydrogels through vacuum jamming. Granular hydrogels are formed as a result of particle-particle interaction[22] and exhibit porosity due to interstitial voids between microgels that support cell infiltration and migration.[49] Such hydrogels formed with IPN microgels undergo yielding (G″ > G′), albeit at higher strains (~200%) than those typically reported for granular hydrogels formed from SN microgels (Figure S24).[50–52] We expect that this difference arises due to inter-particle GH bond formation when the microgels are packed closely. Next, we leverage the shear-thinning property of these granular systems to process them into 3D microporous shapes such as a pyramid and star, as well as hierarchical structures like a two-layered lattice via extrusion printing (Figure 6f,g). 3D printed constructs are stable enough to hold their shape without any need for post-crosslinking.
In order to investigate the cell-material interactions of IPN microgels we conduct experiments with cells seeded atop granular hydrogels in 2D and cells as spheroids embedded within granular hydrogels in 3D. 3T3 fibroblasts seeded on granular hydrogel discs are seen to attach and spread along the microgel periphery (Figure S25) at day 1. 3D printed granular hydrogel lattices are also seeded with fibroblasts to visualize cell morphology after one day of culture in growth media (Figure 6h). The cells are seen to not only adhere and proliferate on the surface of microgels but also to invade into the voids between microgels. Taking advantage of the porosity of granular hydrogels, we encapsulate human mesenchymal stromal cell/human umbilical vein endothelial cells (hMSC/HUVEC) spheroids within interstitial voids. IPN granular hydrogels could support cell outgrowth from the spheroids as seen at day 3 (Figure 6i). These examples indicate that granular hydrogels from IPN-fabricated microgels support not only cytocompatibility but the growth of cells, which is particularly important for applications as injectable and 3D printed biomaterials towards tissue repair and regeneration.
3. Conclusions
In summary, we establish a one-pot scheme for the fabrication of IPN hydrogels via orthogonal photo-initiated reactions. Both networks within the IPN hydrogel are formed from a biopolymer and avoid the use of toxic monomers or harsh crosslinking conditions. Despite their high water content (~94%), IPN hydrogels are obtained with high work of fracture (~24 to 46 kJ m−3) that can sustain multiple cycles of loading-unloading without significant damage. The ability to tune mechanical properties by small changes in the concentrations of each network establishes these IPN hydrogels as modular and versatile candidates for a wide variety of biomedical applications. There is room for improvement in the mechanical properties of the IPN hydrogels described here which can be addressed in future studies of one-pot reaction schemes; however, the low viscosity and rapid photo-crosslinking of these hydrogels addresses a major challenge in the field of IPN fabrication, making them amenable to high resolution, high-throughput DLP-based 3D printing and processing into microparticles and granular hydrogels. Further, through the use of adhesive ligands, processed structures from IPN hydrogels are shown to be either non-adhesive or adhesive, depending on the desired application.
Experimental Section
Polymer synthesis and characterization:
All chemicals were obtained from Millipore-Sigma unless stated otherwise. Sodium hyaluronate (HA, mol. wt. = 88 kDa) was obtained from Lifecore Biomedical (USA). Methacrylate modified hyaluronic acid (MeHA) was synthesized as previously described.[53] Briefly, HA (5 g) was dissolved in deionized water (0.2 L) under ice-cold conditions. Methacrylic anhydride (38.5 g, 20 equivalent) was added dropwise to the solution and pH was maintained at 8.5–9.5 throughout the 8 h reaction. The solution was transferred to a dialysis tubing (Spectra Por, 6–8 kDa cutoff) and dialyzed against deionized water for 10 d, frozen at −80 °C, and lyophilized. Dry polymer was stored under inert nitrogen at −20 °C. Norbornene modified hyaluronic acid (NorHA) was synthesized following benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) coupling protocol as described previously.[54] First, HA was converted into its tetrabutylammonium salt form (HA-TBA) by dissolving HA (5 g) in deionized water (0.3 L) along with Dowex 50Wx200 proton exchange resin (15 g), mixing for 30 minutes, recovering the resin via vacuum filtration, and titrating the filtrate with tetrabutylammonium hydroxide (0.2 M) solution to a pH of 7.02–7.05. The HA-TBA solution was then frozen and lyophilized. Next, HA-TBA (4.5 g) was dissolved in anhydrous dimethyl sulfoxide (DMSO, ~0.3 L) and 5-norbornene-2-methylamine (0.48 mL, TCI Chemicals) under inert nitrogen at room temperature. After complete dissolution, BOP was added via cannulation and the reaction was allowed to proceed for 2 h. The reaction was quenched with cold deionized water (20 mL) and the solution was dialyzed for 14 d. The final product was frozen, lyophilized, and stored under nitrogen at −20 °C. In order to determine the degree of modification of HA, polymer (10 mg) was dissolved in deuterium oxide (D2O, 1 mL) and characterized using 1H-NMR spectroscopy (Bruker Neo400 360 MHz) (Figure S1). To obtain MeHA and NorHA with varying degrees of modification, the amounts of methacrylic anhydride and 5-norbornene-2-methylamine were adjusted accordingly. Methacrylate modification (~100%) was determined by integration of the vinyl protons (1H, δ ~ 5.5 to 5.8 ppm) relative to the methyl groups on both HA and pendant methacrylates (δ ~ 1.6 to 2.2 ppm, 6H), while the modification of HA with pendant norbornene group (~45%) was determined by integration of vinyl protons (2H, δ ~ 5.8 to 6.2 ppm) relative to the methyl group on HA (δ ~ 1.8 to 2.2 ppm, 3H).
Hydrogel Fabrication:
MeHA (0.25–1.00 wt.%) was dissolved in Dulbecco’s (1×) phosphate buffered saline (PBS) along with photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate, LAP, Colorado Photopolymer Solutions, USA) to form single network MeHA hydrogels. A stock solution of Ad-CD crosslinker was prepared by combining Ad-SH (200 mM, 1-adamantanethiol) and CD-SH (200 mM, 6-Mercapto-6-deoxy-β-cyclodextrin, Crysdot LLC, USA) dissolved in DMSO as the solvent. NorHA (3.0–7.5 wt.%) was dissolved in PBS along with LAP (17 mM) and Ad-CD (16–42 mM, 1:1 thiol:norbornene ratio) to form single network NorHA GH hydrogels. Precursor solutions for IPN hydrogels consisted of MeHA (0.25–1.00 wt.%), NorHA (3.0–7.5 wt.%), LAP (17 mM), and Ad-CD (1:1 thiol:norbornene ratio). All precursor solutions were vortexed for 2–3 minutes and crosslinked in the presence of visible light (5 min., 10 mW cm−2, Exfo Omnicure Vis S1000 lamp, 400–500 nm filter). To quantify the consumption of functional groups (methacrylate and norbornene), hydrogels were degraded in sodium hydroxide solution (2 N NaOH) for 3 days at 37 °C. Final solutions were then frozen, lyophilized, and resuspended in deuterium oxide (D2O, 1 mL) to determine the presence of functional groups using 1H-NMR spectroscopy (Bruker Neo400 360 MHz). Ellman’s assay was performed according to manufacturer’s instructions to quantify the consumption of free thiols for NorHA GH and IPN hydrogels. Hydrogel films (8 mm diameter, 30 μL) were prepared as described and soaked in reaction buffer [2 mL, containing 0.1 M sodium phosphate, 1 mM EDTA, and Ellman’s reagent (5,5’-Dithio-bis-2-nitrobenzoic acid, Thermo-Scientific, USA)]. The samples were placed on an orbital shaker at room temperature for 90 minutes and the absorbance was recorded at 412 nm using a Tecan Infinite M200 spectrometer. To assess compatibility between networks in IPNs, hydrogels were formed with fluorescently (FITC)-labeled MeHA and allowed to reach equilibrium swelling overnight at 37 °C. Fluorescence intensity was then visualized using maximum projection images acquired on confocal microscope (Leica SP5 equipped with 10× objective). Randomly selected regions of interest across samples were first thresholded and % fluorescent area was quantified using ImageJ. IPN hydrogels were immersed in PBS (1 mL) and placed on an orbital shaker at 37 °C overnight, the swelling ratio was calculated as the ratio of mass of swollen gel (ms) to that of dry polymer after lyophilization (md), while the equilibrium water content was defined as (ms − md)/ms.
Mechanical Characterization:
Rheological characterization was performed on an AR2000 stress-controlled rheometer (TA Instruments) fitted with a 20 mm diameter parallel plate geometry, and 100 μm gap. Rheological properties were measured by oscillatory frequency sweeps (0.1–100 rad s−1, 0.1% strain) and oscillatory time sweeps (10 rad s−1, 0.1% strain). Photorheological characterization was conducted to obtain the gelation profile through oscillatory shear time sweeps (time to gel point defined as the crossover between storage, G′ and loss modulus, G″) in the presence of visible light (10 mW cm−2, Exfo Omnicure Vis S1000 lamp, 400–500 nm filter) or 405 nm light (15 mW cm−2, Alpha Prototype Projector, Volumetric Inc.). Hydrogels were cast into 5 mm diameter cylindrical discs for compression testing (Q800 DMA, TA Instruments, 0.5 N min−1 force ramp). Compressive moduli were determined from the linear elastic region (10–20% strain) of the stress-strain curves. To assess degradation, IPN hydrogels were immersed in PBS (1 mL), placed on an orbital shaker at 37 °C, and their compressive modulus was measured over 35 days. For tensile testing, hydrogels were cast into dog-bone shaped samples using custom poly(dimethyl siloxane) PDMS molds (5 mm width; 10 mm gauge length; and 1 mm thickness). Samples were secured with sand-blasted clamps and loaded under uniaxial tension (Instron 5542, 5 N load cell, 3 mm min−1). Nominal stress was defined as the force on the deformed gel divided by the cross-sectional area of the undeformed sample, while stretch was defined as the ratio of the length of deformed to the length of undeformed samples. Tensile modulus (slope from 5–15% strain), stretch, and stress at failure were determined using a custom MATLAB (MathWorks, USA) script. Work of fracture was quantified as the area under the stress-stretch curves for samples without pre-cut crack.[55] Hysteresis energy (Uhys) was determined by the area between the loading and unloading curve. Hysteresis (Hys = Uhys/W) was defined as the ratio of irreversible work to total work done by extension. Tensile testing of notched samples was performed using hydrogels with a 1 mm (20% of the specimen width) crack (i.e., pre-cut with a razor blade). Toughness was estimated as Γ = W(λc) h, where λc is the critical stretch at which crack begins to propagate, W(λc) is the strain energy per unit volume of an unnotched sample measured up to the critical stretch, and h is the length of undeformed sample, while G is estimated as , where c is the initial crack length as described previously.[56] For suture retention measurements, hydrogels were cast into rectangular samples in a PDMS mold (10 mm width and 1.5 mm thickness). 4–0 absorbable sutures (Chromic gut, 45 cm, Syneture Inc.) were employed using the needle provided by the manufacturer and passed through the hydrogel specimen (suture bite centered with respect to the sample width at a distance of 4.5 mm from the free end) into a suture holder and closed into a loop by multiple knots. The samples were pre-loaded until the suture thread was taut and then subjected to uniaxial tension (Instron 5542, 5 N load cell, 3 mm min−1) and stretched until suture pullout. Suture retention strength was quantified as the ratio of suture load to the product of suture diameter and sample thickness as described previously.[57] For surface friction measurements, hydrogels were cast on methacrylated glass coverslips (18 mm diameter, treated with 3-(trimethoxysilyl)propyl methacrylate, Millipore sigma) and covered by glass slide (treated with Sigmacote™, Millipore Sigma) to ensure a flat surface. The friction measurement tests were performed on a custom micro-tribometer setup with custom hemispherical PDMS probe (sliding velocity: 0.5 mm s−1, sliding distance: 5 mm and pre-load force: 5 mN)
DLP-based 3D printing of IPN hydrogels:
The IPN resin used for printing consisted of MeHA (0.6 wt.%), NorHA (4.5 wt.%), Ad-CD (25 mM), and LAP (17 mM) along with photoabsorber (Tartrazine, 0–2 mM). IPN resins were exposed to a range of irradiation doses (varying exposure time, intensity = 15 mW cm−2) and the thickness of the crosslinked layer was measured to generate a working curve. Fabrication of 3D hydrogel constructs was conducted on a Lumen Alpha DLP bioprinter (Volumetric Inc., USA). IPN resin was dispensed into the PDMS vat, the build platform was slowly lowered (30 mm min−1) into the resin bath and the layers were crosslinked sequentially with light exposure (2.5–7.5 s, 15 mW cm−2, 100 μm step size). After printing was completed, the 3D fabricated hydrogel was removed from the build platform with a razor and washed with PBS to remove excess or uncured resin. CAD models for solid shapes or complex geometries used in the study were either designed in Fusion 360 (Autodesk, USA) or downloaded from online repositories (NIH 3D print exchange and MakerBot Thingiverse). Polyethylene glycol diacrylate (PEGDA Photoink™) and gelatin methacrylate (GelMA Photoink™) resins were obtained from Volumetric Inc. (distributed by Cellink) and used as per manufacturer’s protocol.
Fabrication and characterization of IPN microgels:
IPN hydrogel microparticles (or microgels) were produced using a mixture of MeHA (0.6 wt.%), NorHA (4.5 wt.%), Ad-CD (25 mM), and LAP (17 mM) with or without fluorescein isothiocyanate (FITC)-labelled dextran (0.2 wt.%, 2 MDa) in PBS. A PDMS-based microfluidic device was fabricated using a custom-designed mold (Microfine green; ProtoLabs). Briefly, PDMS (Sylgard 184, Dow Corning) was cast onto the mold, cured, cleaned, and plasma-bonded to a glass slide. Silicone tubing (Tygon; ABW00001, Saint-Gobain) was connected to the inlets and outlet. Light mineral oil (Fisher Scientific, USA) containing Span 80 (2 wt.%) was used as the continuous phase, and IPN precursor solution was used as the dispersed phase. An optimized oil flow rate of 75 μL min−1 and precursor flow rate of 4 μL min−1 was employed to create ~160 μm diameter spherical droplets. Downstream, droplets were crosslinked by exposure to visible light (20 mW cm−2, 2.5 min residence time, Exfo Omnicure Vis S1000 lamp, 400–500 nm filter) off-chip to form IPN microgels. The microgel suspension was centrifuged at 10000 ×g for 3 minutes, the oil phase decanted, and the microgels were then washed multiple times with PBS and an aqueous solution of Tween-20 (1 wt.%) to remove any remaining oil and surfactant. Granular hydrogels were assembled through vacuum jamming of IPN microgels on a PVDF membrane (0.22 μm pores, Steriflip, Millipore). Shear-dependent yielding of granular hydrogels was characterized through an oscillatory strain sweeps (10 rad s−1, 0.05–500% strain) on the rheometer (20 mm parallel plate geometry, 1 mm gap height). Young’s moduli of IPN microgels and cast hydrogel films were determined in the wet state (using PVDF membrane soaked in PBS as substrate) using a Piuma nanoindenter (Optics 11, Netherlands). A spherical tip indenter (tip radius 51.5 μm, stiffness 4.33 N m−1) was used to probe the samples to an indentation depth of 10 μm. The force-indentation depth data was fit to a modified Hertzian contact model as described previously.[58] The moduli were determined at low strains using the relationship in Equations 1 and 2.
| (1) |
| (2) |
where, F is the force, E is the Young’s modulus, h is the indentation depth, h0 is the indentation at first contact between particle and tip (characterized by an initial increase in measured force), υ is the Poisson ratio (assumed to be 0.5), Reff is the effective radius, R is the radius of the sample (= 84.5 μm for microgel and ∞ for flat hydrogel film), and Rtip is the indenter tip radius.
Extrusion-based 3D printing of granular hydrogels:
A granular hydrogel ink composed of IPN microgels was loaded into a 1 mL syringe (BD) equipped with a blunt-end 18-gauge needle (McMaster-Carr). Constructs were printed using a customized 3D FDM printer (Velleman K8200). CAD models of interest were first sliced using Slic3r software. The deposition of microgel ink onto the print-bed was then controlled through Repetier-Host software.
Imaging and visualization:
0.5 mg mL−1 of rhodamine-labelled dextran (500 kDa) or FITC-labelled dextran (2 MDa) was added to the IPN resin before DLP-based 3D printing. For visualization of lumen topology in the IPN microfluidic device, 2-hydroxyethyl agarose (0.5 wt.%, 37 °C) containing fluorescent dextran was perfused into the open channel and the agarose allowed to cure before imaging. Fluorescent images of 3D printed IPN bulk and granular hydrogels were captured on a wide-field microscope Axio Zoom V.16 (Zeiss, Germany). Z-stack (5–20 μm step size) fluorescence scans of IPN microgels were conducted on confocal microscopes (Leica SP5 and Nikon A1plus). The raw images of microgels pre-compression, during compression, and post-compression were reconstructed in 3D. Images were analyzed and the brightness/contrast was adjusted using ImageJ (NIH). Macroscopic photographs of hydrogel constructs (in air or immersed in PBS) were taken with a Sony Alpha 7R3 camera. The background was manually segmented using Photoshop editor (Adobe Inc., USA). Pattern fidelity of printed constructs was evaluated using micro-computed tomography (μ-CT). Hydrogels (gyroid) were incubated in Lugol’s solution overnight and scanned using a MicroCT 35 system (Scanco Medical, USA; exposure: 300 ms, voltage: 55 kVp, isotropic voxel size: 6 μm). For volumetric visualization of interstitial porosity and curved features of the gyroid, μ-CT reconstructions were created using DragonFly (Object Research Systems, Canada) software. The images were filtered to improve the signal to noise ratio for subsequent threshold-based segmentation of high intensity signal (gyroid; upper Otsu) from low intensity signal (background; lower Otsu). Following reconstruction, images were pseudo-colored and custom rendering settings (lighting, specularity, opacity) were adjusted for 3D visualization.
Cell culture, seeding, and visualization:
NIH 3T3 fibroblasts were used for all experiments unless otherwise stated. The culture media contained high glucose Dulbecco’s modified Eagle’s Medium (DMEM, 10% v/v fetal bovine serum, 1% v/v penicillin–streptomycin). Hydrogel specimens were prepared (cast or 3D printed) as described earlier (with 0 or 2 mM thiolated RGD, GCGYGRGDSPG, Genscript), washed with PBS, and sterilized with germicidal UV lamp for 1 h. Cells were seeded (density: 7500 cells/cm2) on hydrogels and cultured for specified number of days with media replaced every two days. For microtissue formation, acid-solubilized type I bovine telocollagen (Advanced BioMatrix) was mixed with prechilled 10× α-MEM and neutralized with 1 N NaOH to achieve a final concentration of 2.5 mg mL−1. Cell suspension (density: 1 million cells mL−1) was mixed with collagen precursor, the resultant solution was transferred to IPN micropillar molds to allow collagen gel formation for 20 min at 37 °C in a 5% CO2 incubator and then hydrated with cell culture medium. For encapsulation within IPN granular hydrogels, multicellular spheroids consisting of human umbilical vein endothelial cells (HUVEC, Lonza) and human mesenchymal stromal cells (hMSC) in 2:1 ratio were prepared by seeding cells on AggreWell 400 templated agarose wells as described previously.[59] Cell concentrations were adjusted to achieve ~1000 cells per spheroid. IPN microgels (containing 2 mM RGD) were sterilized, washed with PBS, and incubated with DTT and LAP for 15 minutes. The microgel suspension was packed via centrifugation at 15000 ×g and spheroids were gently mixed into microgel pellet. Mixture was transferred to cylindrical molds and exposed to light (2 min., 20 mW cm2) to stabilize the microgels together. The constructs were washed and cultured in media (endothelial basal medium, Lonza) supplemented with 100 ng mL−1 rhVEGF with media replaced every day. At terminal time points, cells were fixed with 10% v/v buffered formalin in PBS, permeabilized with 0.1% v/v TritonX-100, blocked with 3% v/v horse serum (in case of 3D cell seeding) and stained with Alexa Fluor™ 647 or rhodamine-conjugated phalloidin (1:250; Thermo Fisher Scientific) and Hoechst 33342 (1:500; Thermo Fisher Scientific) to visualize F-actin and nucleus, respectively. Z-stack images were acquired on Leica SP5 confocal microscope equipped with a 10× or 25× immersion objective lens. Cell descriptors were obtained from maximum projection images analyzed on ImageJ (NIH). Cell number was calculated based on total number of nuclei (binary mask, Otsu threshold). Cell area and circularity were determined based on manual thresholding of isolated cells that did not exhibit any cell-cell contacts. Femoral condyle and skeletal muscle were isolated from juvenile bovine joints (obtained from Research 87 Inc., USA)
Statistical Analysis:
All data are reported as mean ± standard deviation with n ≥ 3. All statistics were conducted using Prism 9 (GraphPad Inc., USA). Comparisons between two experimental groups were performed using two-tailed t-tests (α = 0.05, significance determined at p < 0.05) and comparisons between more than two groups were performed using a one-way analysis of variance (ANOVA) with post hoc Tukey’s multiple comparison test.
Supplementary Material
Acknowledgements
This work was supported by the AO Foundation, the National Science Foundation (graduate research fellowship to J.H.G., UPenn MRSEC: DMR-1720530), the National Institutes of Health (R01AR056624, F32DK117568 to M.D.D.), and the German Research Foundation (QA 58/1-1 to T.H.Q.). The authors would like to acknowledge Dr. Claudia Loebel for intellectual input and helpful discussions, Selen Uman for assistance with hydrogel suturing, Victoria Muir for help with 1H-NMR, and Nikolas Di Caprio and Tian Ren for assistance with photography. The authors are grateful to Gokulanand Iyer and Dr. Kevin Turner for assistance with surface friction measurements. The authors would also like to thank the BIOLines laboratory (Dr. Dan Huh) for use of their wide-field fluorescence microscope and the Penn Center for Musculoskeletal Disorders (PCMD, P30AR069619) for tensile testing equipment. The NSF Major Research Instrumentation Program (award NSF CHE-1827457) and Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study.
Contributor Information
Abhishek P. Dhand, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA
Matthew D. Davidson, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA BioFrontiers Institute and Department of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80309, USA.
Jonathan H. Galarraga, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA
Taimoor H. Qazi, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA
Ryan C. Locke, Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, PA 19104, USA Department of Orthopaedic Surgery, McKay Orthopaedic Research Laboratory, University of Pennsylvania, Philadelphia, PA 19104, USA.
Robert L. Mauck, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, PA 19104, USA; Department of Orthopaedic Surgery, McKay Orthopaedic Research Laboratory, University of Pennsylvania, Philadelphia, PA 19104, USA.
Jason A. Burdick, Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, PA 19104, USA; Department of Orthopaedic Surgery, McKay Orthopaedic Research Laboratory, University of Pennsylvania, Philadelphia, PA 19104, USA; BioFrontiers Institute and Department of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80309, USA.
References
- [1].Dhand AP, Galarraga JH, Burdick JA, Trends Biotechnol. 2021, 39, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gong JP, Katsuyama Y, Kurokawa T, Osada Y, Adv. Mater. 2003, 15, 1155. [Google Scholar]
- [3].Sun JY, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z, Nature 2012, 489, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shin H, Olsen BD, Khademhosseini A, Biomaterials 2012, 33, 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Q, Zhu L, Zhao C, Wang Q, Zheng J, Adv. Mater. 2013, 25, 4171. [DOI] [PubMed] [Google Scholar]
- [6].Truong VX, Ablett MP, Richardson SM, Hoyland JA, Dove AP, J. Am. Chem. Soc. 2015, 137, 1618. [DOI] [PubMed] [Google Scholar]
- [7].Chen F, Le P, Lai K, Fernandes-Cunha GM, Myung D, Chem. Mater. 2020, 32, 5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Daniele MA, Adams AA, Naciri J, North SH, Ligler FS, Biomaterials 2014, 35, 1845. [DOI] [PubMed] [Google Scholar]
- [9].Shete AU, Kloxin CJ, Polym. Chem. 2017, 8, 3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ma X, Qu X, Zhu W, Li Y-S, Yuan S, Zhang H, Liu J, Wang P, Lai CSE, Zanella F, Feng G-S, Sheikh F, Chien S, Chen S, Proc. Natl. Acad. Sci. 2016, 113, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lim KS, Levato R, Costa PF, Castilho MD, Alcala-Orozco CR, Van Dorenmalen KMA, Melchels FPW, Gawlitta D, Hooper GJ, Malda J, Woodfield TBF, Biofabrication 2018, 10, 0. [DOI] [PubMed] [Google Scholar]
- [12].Grigoryan B, Paulsen SJ, Corbett DC, Sazer DW, Fortin CL, Zaita AJ, Greenfield PT, Calafat NJ, Gounley JP, Ta AH, Johansson F, Randles A, Rosenkrantz JE, Louis-Rosenberg JD, Galie PA, Stevens KR, Miller JS, Science 2019, 364, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hong S, Sycks D, Chan HF, Lin S, Lopez GP, Guilak F, Leong KW, Zhao X, Adv. Mater. 2015, 27, 4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guo Z, Dong L, Xia J, Mi S, Sun W, Adv. Healthc. Mater. 2021, 10, 2100036. [DOI] [PubMed] [Google Scholar]
- [15].Kuang X, Zhao Z, Chen K, Fang D, Kang G, Qi HJ, Macromol. Rapid Commun. 2018, 39, 1700809. [DOI] [PubMed] [Google Scholar]
- [16].Wang P, Berry DB, Song Z, Kiratitanaporn W, Schimelman J, Moran A, He F, Xi B, Cai S, Chen S, Adv. Funct. Mater. 2020, 30, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kunwar P, Jannini AVS, Xiong Z, Ransbottom MJ, Perkins JS, Henderson JH, Hasenwinkel JM, Soman P, ACS Appl. Mater. Interfaces 2020, 12, 1640. [DOI] [PubMed] [Google Scholar]
- [18].Caprioli M, Roppolo I, Chiappone A, Larush L, Pirri CF, Magdassi S, Nat. Commun. 2021, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kesselman LRB, Shinwary S, Selvaganapathy PR, Hoare T, Small 2012, 8, 1092. [DOI] [PubMed] [Google Scholar]
- [20].Raz N, Li JK, Fiddes LK, Tumarkin E, Walker GC, Kumacheva E, Macromolecules 2010, 43, 7277. [Google Scholar]
- [21].Mahou R, Vlahos AE, Shulman A, Sefton MV, ACS Biomater. Sci. Eng. 2017, 4, 3704. [DOI] [PubMed] [Google Scholar]
- [22].Daly AC, Riley L, Segura T, Burdick JA, Nat. Rev. Mater. 2019, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mantooth SM, Munoz-Robles BG, Webber MJ, Mantooth SM, Munoz-Robles BG, Webber MJ, Macromol. Biosci. 2019, 19, 1800281. [DOI] [PubMed] [Google Scholar]
- [24].Hui E, Gimeno KI, Guan G, Caliari SR, Biomacromolecules 2019, 20, 4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jing Y, Yang B, Yuan W, Han S, Song L, Ye M, Zhang ZY, Bian L, Appl. Mater. Today 2021, 22, 100892. [Google Scholar]
- [26].Widener AE, Bhatta M, Angelini TE, Phelps EA, Biomater. Sci. 2021, 9, 2480. [DOI] [PubMed] [Google Scholar]
- [27].Kakuta T, Takashima Y, Nakahata M, Otsubo M, Yamaguchi H, Harada A, Adv. Mater. 2013, 25, 2849. [DOI] [PubMed] [Google Scholar]
- [28].Northrop BH, Coffey RN, J. Am. Chem. Soc. 2012, 134, 13804. [DOI] [PubMed] [Google Scholar]
- [29].Lecamp L, Houllier F, Youssef B, Bunel C, Polymer 2001, 42, 2727. [Google Scholar]
- [30].Fairbanks BD, Macdougall LJ, Mavila S, Sinha J, Kirkpatrick BE, Anseth KS, Bowman CN, Chem. Rev. 2021, 121, 6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Blasco E, Wegener M, Barner-Kowollik C, Adv. Mater. 2017, 29, 1604005. [DOI] [PubMed] [Google Scholar]
- [32].Rodell CB, Dusaj NN, Highley CB, Burdick JA, Adv. Mater. 2016, 28, 8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Loebel C, Ayoub A, Galarraga JH, Kossover O, Simaan-Yameen H, Seliktar D, Burdick JA, J. Mater. Chem. B 2019, 7, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Harada A, Takashima Y, Nakahata M, Acc. Chem. Res. 2014, 47, 2128. [DOI] [PubMed] [Google Scholar]
- [35].Xin H, Saricilar SZ, Brown HR, Whitten PG, Spinks GM, Macromolecules 2013, 46, 6613. [Google Scholar]
- [36].Nakajima T, Kurokawa T, Ahmed S, Wu W, Gong JP, Soft Matter 2013, 9, 1955. [Google Scholar]
- [37].Cook RF, Oyen ML, J. Phys. Mater. 2021, 4, 021001. [Google Scholar]
- [38].Webber RE, Creton C, Brown HR, Gong JP, Macromolecules 2007, 40, 2919. [Google Scholar]
- [39].Zhang W, Liu X, Wang J, Tang J, Hu J, Lu T, Suo Z, Eng. Fract. Mech. 2018, 187, 74. [Google Scholar]
- [40].Yu C, Schimelman J, Wang P, Miller KL, Ma X, You S, Guan J, Sun B, Zhu W, Chen S, Chem. Rev. 2020, 120, 10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee M, Rizzo R, Surman F, Zenobi-Wong M, Chem. Rev. 2020, 120, 10950. [DOI] [PubMed] [Google Scholar]
- [42].Qiu Y, Ahn B, Sakurai Y, Hansen CE, Tran R, Mimche PN, Mannino RG, Ciciliano JC, Lamb TJ, Joiner CH, Ofori-Acquah SF, Lam WA, Nat. Biomed. Eng. 2018, 2, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Paek J, Song JW, Ban E, Morimitsu Y, Osuji CO, Shenoy VB, Huh DD, Sci. Rep. 2021, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Uzcategui AC, Muralidharan A, Ferguson VL, Bryant SJ, McLeod RR, Adv. Eng. Mater. 2018, 20, 1800876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yasuda K, Gong JP, Katsuyama Y, Nakayama A, Tanabe Y, Kondo E, Ueno M, Osada Y, Biomaterials 2005, 26, 4468. [DOI] [PubMed] [Google Scholar]
- [46].Charrier EE, Pogoda K, Wells RG, Janmey PA, Nat. Commun. 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB, Chen CS, Tissue Eng. - Part A 2012, 18, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen Z, Zhao R, ACS Biomater. Sci. Eng. 2019, 5, 3688. [DOI] [PubMed] [Google Scholar]
- [49].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat. Mater. 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Highley CB, Song KH, Daly AC, Burdick JA, Adv. Sci. 2019, 6, 1801076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Seymour AJ, Shin S, Heilshorn SC, Adv. Healthc. Mater. 2021, 2100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hirsch M, Charlet A, Amstad E, Adv. Funct. Mater. 2021, 31, 2005929. [Google Scholar]
- [53].Loebel C, Rodell CB, Chen MH, Burdick JA, Nat. Protoc. 2017, 12, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vega SL, Kwon MY, Song KH, Wang C, Mauck RL, Han L, Burdick JA, Nat. Commun. 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bai R, Yang J, Suo Z, Eur. J. Mech. - A/Solids 2019, 74, 337. [Google Scholar]
- [56].Creton C, Ciccotti M, Reports Prog. Phys. 2016, 79, 046601. [DOI] [PubMed] [Google Scholar]
- [57].Hong Y, Huber A, Takanari K, Amoroso NJ, Hashizume R, Badylak SF, Wagner WR, Biomaterials 2011, 32, 3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Aufderhorst-Roberts A, Baker D, Foster RJ, Cayre O, Mattsson J, Connell SD, Nanoscale 2018, 10, 16050. [DOI] [PubMed] [Google Scholar]
- [59].Qazi TH, Wu J, Muir VG, Weintraub S, Gullbrand SE, Lee D, Issadore D, Burdick JA, Adv. Mater. 2022, 2109194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.