

Abstract
The pancreas and liver arise from a common pool of progenitors. However, the underlying mechanisms driving their lineage diversification from the foregut endoderm are not fully understood. To tackle this question, we took a multifactorial approach integrating human pluripotent stem cell guided differentiation, genome-scale CRISPR-Cas9 screening, single-cell analysis, genomics and proteomics. We discovered that HHEX, a transcription factor (TF) widely recognized as a key regulator of liver development, acts as a gatekeeper of pancreatic lineage specification. HHEX deletion impaired pancreatic commitment and unleashed a surprising degree of cellular plasticity towards the liver and duodenum fates. Mechanistically, HHEX cooperates with pioneer TFs FOXA1/2 and GATA4 shared by both pancreas and liver differentiation programs to promote pancreas commitment, and this cooperation also restrains the shared TFs from activating alternative lineages. These findings provide a generalizable model for how gatekeeper TFs like HHEX orchestrate lineage commitment and plasticity restriction in broad developmental contexts.
A central question in developmental biology concerns the demarcation of organ-specific progenitor domains during organogenesis. For instance, the pancreas, liver and duodenum (the first segment of the small intestine) arise from adjacent progenitor domains that share a common lineage history1. However, little is known about the intrinsic control that ensures proper lineage choices among these neighboring embryonic cells. A recent study discovered a high degree of cellular plasticity in the hepato-pancreato-biliary territory2, which is likely critical for both normal development and tissue regeneration. On the other hand, uncontrolled plasticity can also lead to aberrant organogenesis and sometimes predispose tissues to malignant transformation3,4, and the loss of lineage-specifying transcription factors (TFs) can lead to the acquisition of tumor cell plasticity that parallels the cell’s developmental history5. However, it remains unclear how transcriptional programs orchestrate lineage commitment and plasticity restriction and what are the roles of lineage-specifying TFs. CRISPR-Cas9 mediated gene editing and human pluripotent stem cell (hPSC) directed differentiation have opened a new avenue to explore lineage segregation. We and others have demonstrated a critical role for the “master” TF PDX1 in hPSC pancreatic differentiation6,7, which is consistent with its established roles in mouse and human pancreas development8–10. In addition, FOXA2, GATA4 and GATA6 are required for activating PDX1 expression and initiating the pancreatic program11–17. Since these upstream TFs are broadly expressed in the gut tube and are required for the formation of additional organ domains such as the liver18–21, the mechanisms that control the segregation of early PDX1+ pancreatic progenitors from the closely related liver and intestine domains remain incompletely understood.
To identify genes specifically required for endoderm differentiation towards the pancreas, we devised a sequential, genome-scale CRISPR-Cas9 screening strategy and successfully identified known regulators of pancreatic differentiation such as PDX1 and GATA4 as well as previously unknown regulators. Hematopoietically-expressed homeobox protein (HHEX) is a top hit identified from the screen and is well known for its roles in the development of the liver, thyroid, and forebrain22–24. However, Hhex is not intrinsically required for mouse pancreatic differentiation, as Hhex deficient endoderm cells are competent to activate the pancreatic program in explant cultures25. Here, through analyzing HHEX knockout (KO) phenotypes in hPSC guided differentiation, we show a dual cell-intrinsic requirement of HHEX for both specifying human pancreatic progenitors and restricting endoderm differentiation to liver and duodenum fates. Further proteomic and genomic investigations show that HHEX drives pancreatic differentiation through cooperating with FOXA1/2 and GATA4, TFs with pioneer factor activity and critical for both pancreas and liver development15–20,26. In the absence of HHEX, these shared TFs become unrestrained and activate liver and duodenum lineages instead. HHEX typifies a class of lineage TFs with the gatekeeper function capable of simultaneously promoting cell fate commitment and restricting plasticity through engaging pioneer TFs that are broadly expressed.
Results
Genome-scale screens identify regulators of human pancreatic differentiation
The initiation of pancreatic development can be modeled using well-established hPSC differentiation protocols that converts hPSCs first to definitive endoderm (DE), followed by primitive gut tube (GT), and subsequently pancreatic progenitors that first express PDX1 (PP1) and then co-express PDX1 and NKX6.1 (PP2) (Fig. 1a)12,27–31. Here we focus on the induction of PDX1, which marks the onset of pancreatic specification32. Using our established knock-in strategy33, we generated a PDX1GFP/+ reporter in H1 iCas9 human embryonic stem cells (hESCs), which express Cas9 upon doxycycline treatment12,34 (Fig. 1b). After induction of DE differentiation16, cells were exposed to FGF7 and vitamin C for four days, with retinoic acid added after the GT stage, resulting in most cells co-expressing PDX1 and GFP at the PP1 stage (Fig. 1c). PDX1/GFP+ cells emerge around 24 hours after the GT stage, and increased to ~90% of the culture in the following 24 hours (Fig. 1d).
Fig. 1: CRISPR-Cas9 screens identify regulators of pancreatic differentiation.
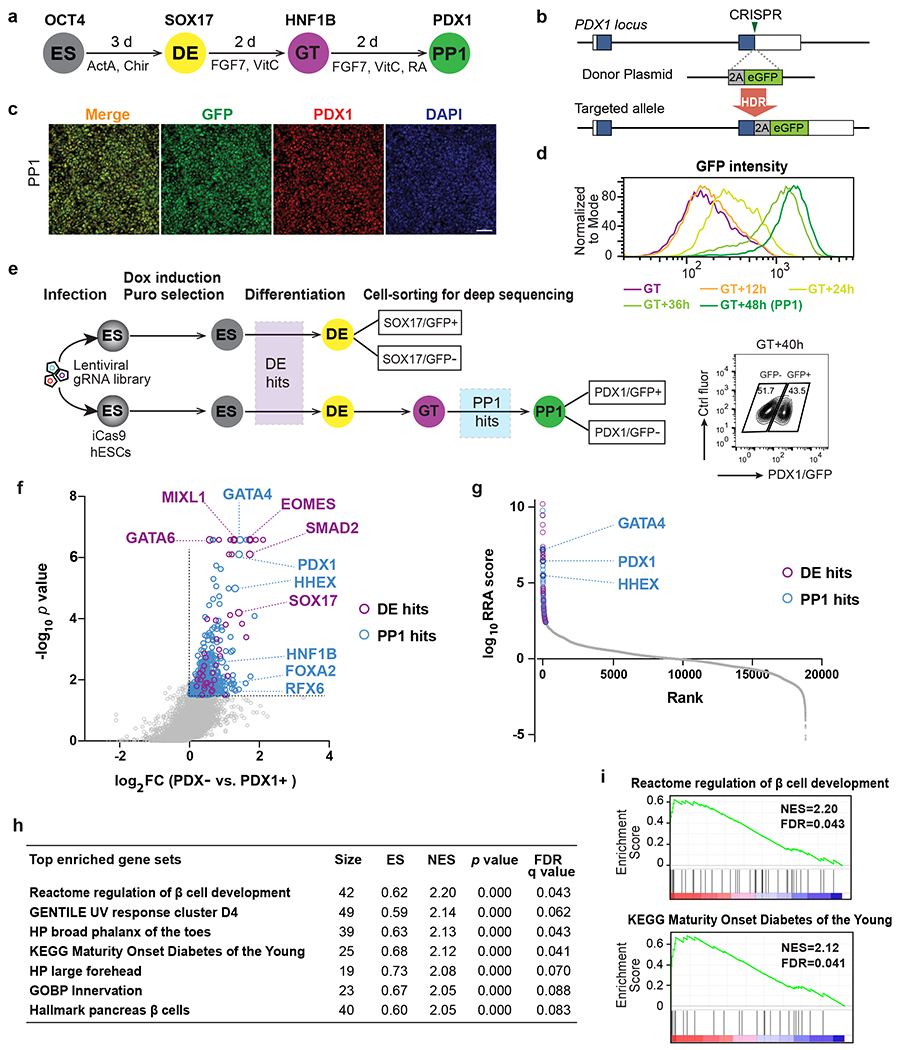
a, Schematic of stepwise pancreatic differentiation protocol from hESCs. ES, embryonic stem cells; DE, definitive endoderm; GT, gut tube; PP1, early pancreatic progenitors. ActA (Actvin A); VitC (vitamin C); RA (retinoic acid). Lineage-specific markers were indicated.
b, Strategy for generating the PDX1GFP/+ reporter cell line. In the presence of the donor plasmid, HDR results in the replacement of the PDX1 stop codon with 2A-eGFP. Boxes indicate PDX1 exons, and filled blue boxes indicate the coding sequence of PDX1.
c, Immunofluorescence staining shows the co-expression of PDX1 and GFP at PP1 stage. Images shown represent three independent experiments. Scale bar, 50 μm.
d, Histogram plots for live GFP expression from GT to PP1 stage.
e, Schematic of sequential screening approach for regulators in pancreatic differentiation. Upper panel, screen schematic for DE formation using SOX17 GFP/+ reporter iCas9hESCs; Lower panel, screen schematic for pancreas induction using PDX1GFP/+ reporter iCas9hESCs. FACS plot shows the sorted information for PDX1/GFP+ and PDX1/GFP− sub-populations. DOX, doxycycline; puro, puromycin. Ctrl fluor, control fluorescence. Each screen was conducted once with two technical repeats.
f, Scatter plot of −log10 p value versus log2 fold change (FC) for all gRNA targeted genes in the PP1 screen. Each circle represents an individual gene. −log10 p >1.5 and log2 FC > 0 were used to identify PP1 hits (in blue), except that the purple circles indicate genes also identified as DE hits (p < 0.01 and log2 FC > 0 based on DE screening results). Selected DE and PP1 positive regulator hits are indicated.
g, Each gene target in the screen ranked based on the MAGeCK robust ranking aggregation (RRA) score at the PP1 stage. Y axis represents log10 (PP1 positive score) – log10 (PP1 negative score). The top 200 genes are labeled with blue (PP1 specific) and purple circles (also identified as DE hits), respectively. Selected top PP1 hits are indicated.
h, GSEA analysis showing the top gene sets that are associated with PP1 screening results. ES, enrichment score; NES, normalized enrichment score.
i, GSEA enrichment plots of selected gene sets.
To identify regulators of pancreatic specification, we infected H1 iCas9 PDX1GFP/+ hESCs with a genome-scale CRISPR library35 using a pooled screening strategy36,37 (Fig. 1e). We targeted ~40 hours after GT when PDX1/GFP+ cells are steeply upregulated (Fig. 1e) to isolate PDX1/GFP+ and PDX1/GFP− cells through fluorescence-activated cell sorting (FACS). Next-generation sequencing was conducted to determine the abundance of individual gRNAs within each population (Fig. 1e and Supplementary Table 1). To distinguish genes specifically required for pancreatic differentiation from those that regulate earlier DE specification, we performed another screen using a SOX17GFP/+ DE reporter36 (Fig. 1e and Supplementary Table 1). Overlapping hits from both screens identified genes essential for DE formation, which included genes such as EOMES, GATA6, SMAD2, and SOX17 that were also identified in our previous DE screens36. Furthermore, the hits unique to the pancreatic screen, referred to as PP1 hits, included known pancreatic regulators such as PDX1, GATA4, and RFX6 (Fig. 1f,g). Gene set enrichment analysis (GSEA) uncovered genes related to maturity onset diabetes of the young (MODY) and regulation of β cell development (Fig. 1h,i), highlighting the utility of unbiased genetic screens focused on the initiation of pancreatic differentiation for discovery of genes relevant to β cell biology and diabetes risk.
Characterization of HHEX expression in pancreatic development
We focused on one of the top hits HHEX for further investigation. HHEX expression was first detected at the DE stage and maintained through the GT, PP1 and PP2 stages and co-expressed with stage-specific markers (Fig. 2a–c). We next investigated human tissues that correspond to the hPSC PP1-to-PP2 transition by analyzing published pancreatic cell RNA sequencing (RNA-seq) data from late CS12 (29–31 days post-conception [dpc]), when PDX1 expression first becomes apparent, to early CS14 (33-35 dpc)38. HHEX was detected in the dorsal pancreatic bud along with archetypal pancreatic progenitor genes such as PDX1 and NKX6-1 (Extended Data Fig. 1a). HHEX expression in pancreatic progenitors was also confirmed based on scRNA-seq data from CS12-16 human embryos39. We further examined human tissues at 22 and 33 weeks post-conception (wpc). In line with a previous study in adult human pancreas40, HHEX was expressed in somatostatin-expressing δ cell but not in insulin-expressing β cells (Fig. 2d). Beyond the islets, HHEX expression was detected in endo-ductal progenitors (also known as “trunk progenitors”) marked by PDX1 and CD13341,42 (Fig. 2e,f). The 33 wpc sample also had positional information that allowed us to identify HHEX expression in progenitor cells in both ventrally derived “head” tissue and dorsally derived “body/tail” pancreas (Fig. 2f). Therefore, HHEX is present in multipotent pancreatic progenitors in the early stages of human pancreas organogenesis and maintained in pancreatic progenitors later restricted to the ductal and endocrine fates (that is, in the bipotent pool) in both ventral and dorsal pancreas. In mouse embryos, expression of Hhex is detected in the ventral-most cells in the anterior intestinal portal at E8.5 and the ventral pancreas at E9.525, and expression levels increase in the dorsal pancreas at E10.5 based on scRNA-seq data43 (Extended Data Fig. 1b). Possibly reflecting a delay in protein production or ineffective antibody labeling, we were unable to reliably detect Hhex protein expression in the dorsal pancreas at E9.5 and E10.5, but at E11.5, Hhex showed co-production with Pdx1 in both dorsal and ventral pancreatic buds (Fig. 2g,h). In addition, the neighboring liver bud expressed Hhex but not Pdx1 (Fig. 2g), whereas the duodenum expressed low levels of Pdx1 but not Hhex (Fig. 2h). Thus, while neither Hhex nor Pdx1 is exclusively expressed in the pancreatic buds, their combined expression uniquely defines the multipotent pancreatic progenitor domain.
Fig. 2: HHEX expression in early pancreas development.
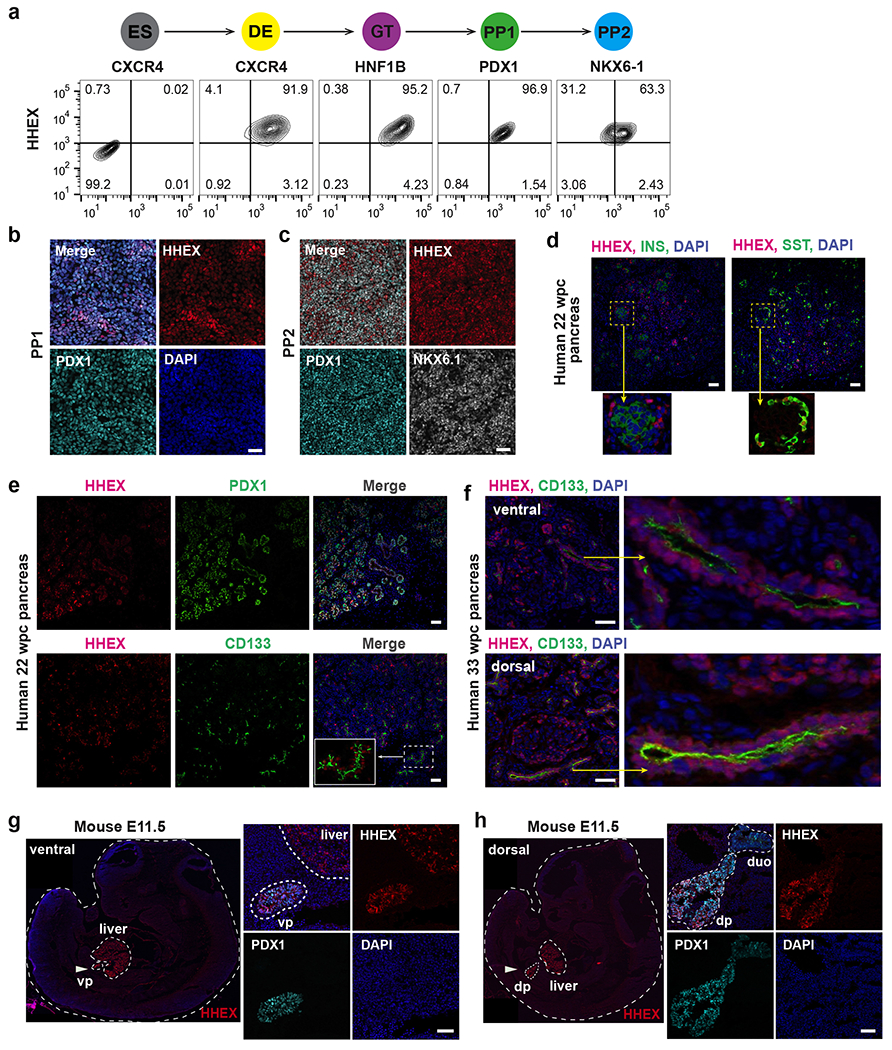
a, Schematic of stepwise pancreatic differentiation from hESCs and flow cytometry analysis of HHEX and lineage-specific markers CXCR4 (DE), HNF1B (GT), PDX1 (PP1), and NKX6-1 (PP2) expression during WT hESCs pancreatic differentiation. ES cells are used as the negative control for HHEX expression.
b,c, Immunofluorescence staining of HHEX, PDX1 (b,c), and NKX6-1 (c) at the PP1 and PP2 stage. Scale bar, 50 μm.
d, Immunofluorescence staining of HHEX, INSULIN (INS), and SOMATOSTAIN (SST) in human pancreas at 22 wpc. Staining was performed in adjacent tissue sections. Inset represents a close-up view of HHEX, INS, and SST expression. Scale bar, 50 μm.
e, Immunofluorescence staining of HHEX, PDX1, and CD133 in human pancreas at 22 wpc. Inset represents a close-up view showing co-expression of HHEX and CD133. Scale bar, 50 μm. Images shown in b-e represent three independent experiments.
f, Immunofluorescence staining of HHEX and CD133 in ventrally derived “head” and dorsally derived “body/tail” pancreas from human pancreas at 33 wpc. Scale bar, 50 μm.
g,h, Immunofluorescence staining of Hhex and Pdx1 in mice embryos at E11.5. vp, ventral pancreas; dp, dorsal pancreas, duo, duodenum; White arrow indicates the ventral pancreas (g) and dorsal pancreas (h). Scale bar, 50 μm. Images shown in f-h represent two independent experiments.
HHEX deletion impairs pancreatic differentiation and causes ectopic liver differentiation
We generated clonal HHEX KO hESC lines to characterize the roles of HHEX in human pancreatic differentiation (Fig. 3a–c and Extended Data Fig. 1c,d). All five KO hESC lines could form DE cells and GT cells as efficiently as the parental wildtype (WT) hESCs (Extended Data Fig. 1e–h). Supporting a critical role for HHEX in initiating pancreatic differentiation, the KO lines formed significantly reduced numbers of PDX1+ cells compared to WT cells at the PP1 stage (Fig. 3d,e) with no significant increase in apoptosis based on cleaved caspase-3 expression (Extended Data Fig. 1i,j). Principal component analysis (PCA) of RNA-seq results showed that KO cells clustered closely with WT cells at the DE and GT stages but diverged from WT cells at the PP1 stage (Extended Data Fig. 1k). 2,154 differentially expressed genes (DEGs) were identified between WT and KO at the PP1 stage, compared to negligible numbers of DEGs at the earlier stages (Fig. 3f and Supplementary Table 2). Focusing on the PP1 stage, we found that KO cells showed a significant reduction in the expression of early pancreatic genes, including PDX1, ONECUT1 and PROX1 (Fig. 3g and Supplementary Table 2). GSEA analysis showed significant enrichment of the gene signatures representing the liver program as well as targets of the liver TF HNF4A in PP1 KO versus WT cells (Fig. 3h). Indeed, many liver genes such as HNF4A, AFP, and APOA2 were significantly upregulated in KO cells (Fig. 3g,i and Supplementary Table 2). These RNA-seq results were confirmed by immunostaining showing that the KO cells formed predominantly HNF4A+ and AFP+ cells that resemble liver progenitors instead of forming PDX1+ pancreatic progenitors (Fig. 3j,k), indicating that HHEX restricts the liver fate in pancreatic differentiation conditions.
Fig. 3: Deletion of HHEX impairs human pancreatic differentiation.
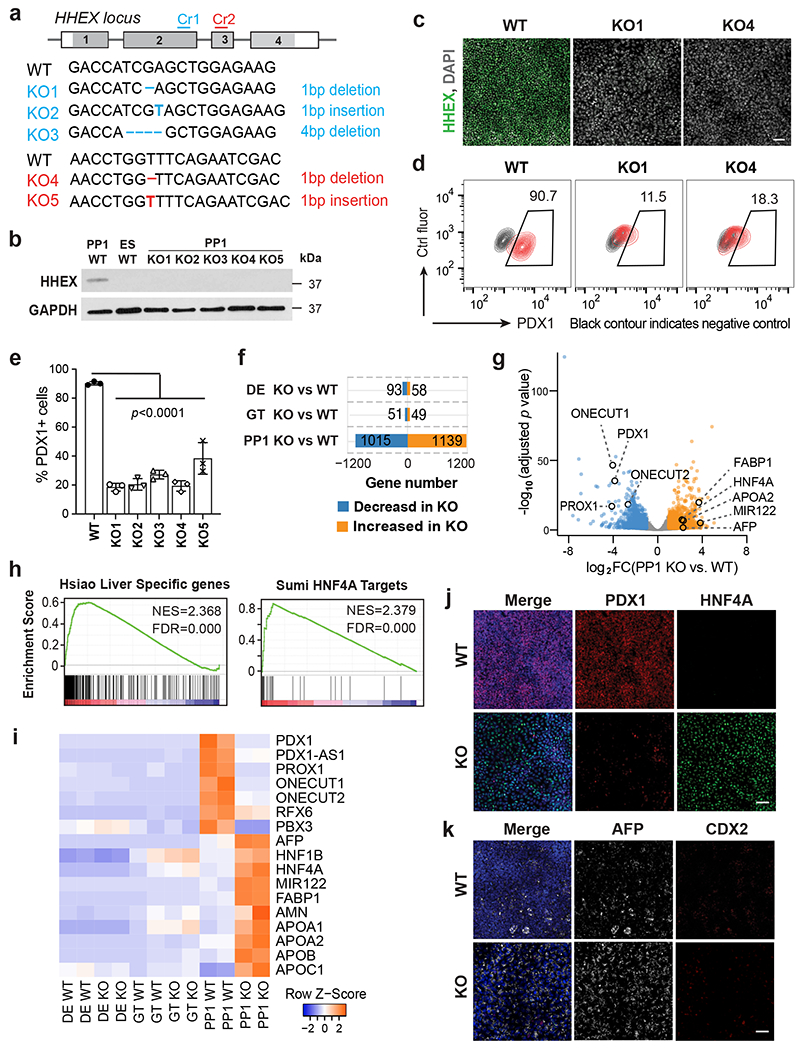
a, Schematic illustrating HHEX targeting. CRISPR gRNA 1 and gRNA 2 (Cr1, Cr2) were designed and targeted at HHEX exon 2 and exon 3, respectively. The consequential homozygous mutations are summarized. Boxes indicate HHEX exons, and filled gray boxes indicate the coding sequence of HHEX.
b,c, The loss of HHEX protein was verified by western blot (b) at the PP1 stage and immunofluorescence staining (c) at the DE stage. GAPDH was used as a loading control. Scale bar, 50 μm.
d,e, Flow cytometry analysis (d) and quantification (e) for PDX1 expression at the PP1 stage. Each symbol represents one independent experiment (n = 3 independent experiments) and data are presented as mean ± SD. One-way ANOVA followed by Dunnett multiple comparisons test vs. WT control.
f,g, Bar graph (f) and volcano plot (g) showing differentially expressed genes identified in KO cells vs. WT during pancreatic differentiation. Genes with log2 FC >1 and FDR <0 .05 are counted as one significant hit. Significantly up-regulated and down-regulated genes in the PP1 KO cells were labeled by orange and blue color, respectively.
h, GSEA shows the enrichment of liver genes and HNF4A targets in PP1 KO cells vs. WT.
i, Heatmap showing the expression of pancreatic and liver genes in WT and KO cells during pancreatic differentiation.
j,k, Immunofluorescence staining of PDX1 and HNF4A (i), AFP and CDX2 (j) at the PP1 stage WT and KO cells. Scale bar, 50 μm. Images shown in b,c, j and k represent three independent experiments, and RNA-seq data shown in f-i are from two independent experiments.
We further explored chromatin accessibility changes at the GT and PP1 stages. Consistent with the RNA-seq analysis, PCA (Fig. 4a) and analysis of differential peaks (Extended Data Fig. 2a,b and Supplementary Table 3) showed that WT and KO cells became divergent at the PP1 stage. Hierarchical clustering identified three main clusters (Fig. 4b,c and Supplementary Table 3). WT and KO cells both showed decreased accessibility during the GT-to-PP1 transition in Cluster I (1,680 regions), and increased accessibility in Cluster II (977 regions). We identified SOX and PBX family members, respectively, as the top enriched TF motifs in Cluster I and Cluster II (Extended Data Fig. 2c,d), suggesting that HHEX deletion did not have a major impact on the developmental transition driven by these transcriptional programs. We focused on Cluster III (3,705 regions), which showed increased accessibility specifically in KO cells at the PP1 stage (Fig. 4b,c). Consistent with the RNA-seq results, this cluster included regions near liver genes such as AFP and FABP1 (Extended Data Fig. 2e). Genomic Regions Enrichment of Annotations Tool (GREAT) analysis on Cluster III regions showed that the top enriched mouse phenotypes were related to abnormal liver physiology and morphology (Extended Data Fig. 2f). Motif analysis on Cluster III regions identified HNF4A and HNF1B as the top enriched TF motifs (Fig. 4d and Supplementary Table 3), implicating the corresponding TFs as drivers of the liver-like chromatin landscape in HHEX KO cells.
Fig. 4: HHEX deletion results in early induction of a liver-like transcriptional program.
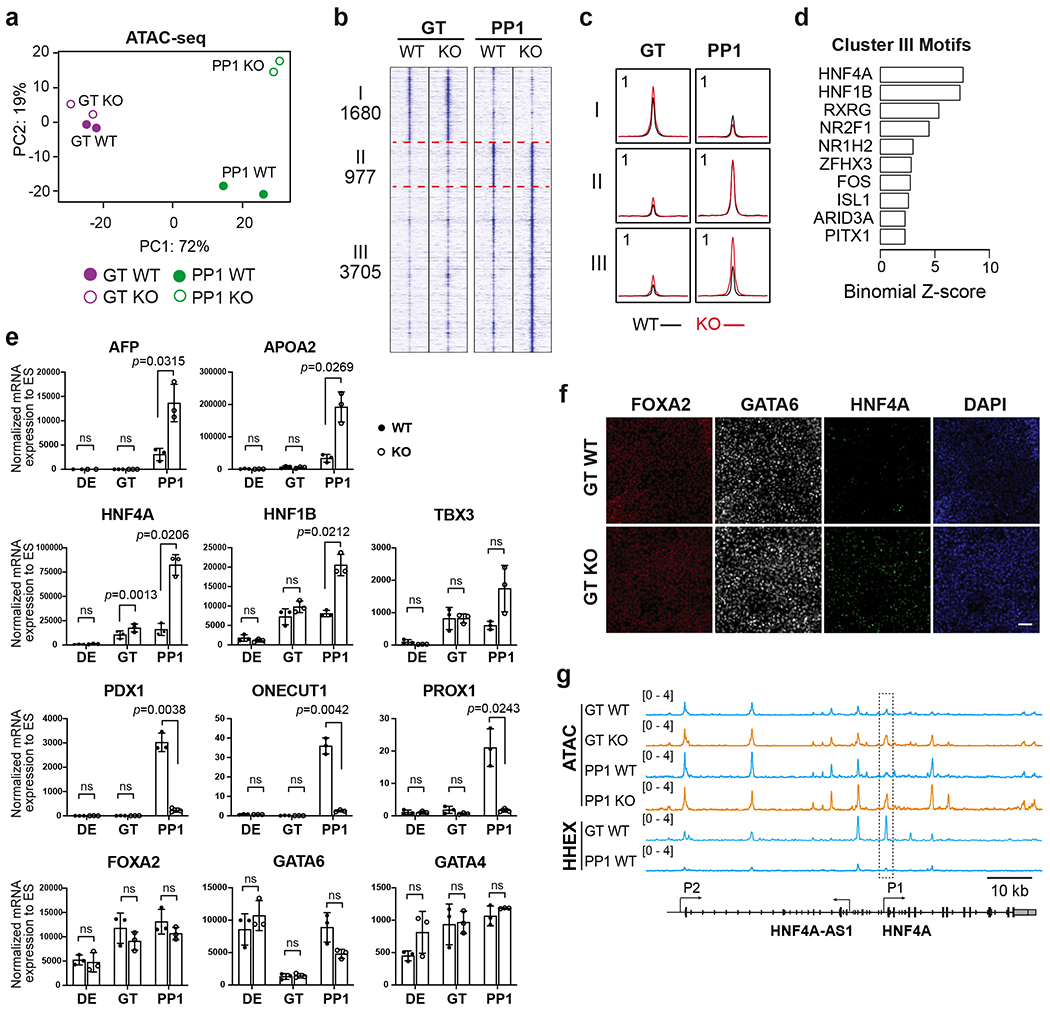
a, PCA of top 3000 most variable genes in WT and HHEX KO cells at the GT and PP1 stages.
b,c, Visualization of ATAC-seq profile at the GT and PP1 stages, patterned by hierarchical clustering of signal tracks (average of two independent experiments) around ATAC-seq peak summits ± 3 kb. (b), tornado plots. (c), metapeaks defined from the column average of the signal. The maximum value of each y axis is annotated in tags per million (TPM).
d, Enrichment of top 10 TF motifs in the regulatory regions III. One-sided hypergeometric test was used to compare the enrichment of proportions of TF motifs for each cluster (foreground ratio) versus those for total atlas (background ratio). The horizontal axis shows the binomial Z-score, representing the number of standard deviations between the observed count of each cluster peaks containing a TF motif and the expected count based on the background ratio. The p values are provided in the Supplementary Table 3.
e, The expression levels of pancreatic and liver genes in WT and KO cells were measured at the DE, GT, and PP1 stages. Each symbol represents one independent experiment (n = 3 independent experiments) and data are presented as mean ± SD. Statistical analysis was performed by paired two-tailed Student’s t-test KO vs. WT control. ns, not significant (p ≥ 0.05).
f, Immunofluorescence staining of FOXA2, GATA6 and HNF4A at the GT stage WT and KO cells. Scale bar, 50 μm. Images shown represent three independent experiments.
g, Integrative Genomics Viewer (IGV) tracks (average of two independent experiments) to show chromatin accessibility and HHEX binding activities at the HNF4A locus. The P1 promoter region of HNF4A showing significantly increased chromatin accessibility upon HHEX deletion was indicated. Scale bar, 10 kb. Data shown in a-d and g are from two independent experiments.
Corresponding to motif analysis results, a significant increase of HNF4A expression in KO cells was first detected by RT-qPCR at the GT stage, and by the PP1 stage both HNF4A and HNF1B were significantly upregulated (Fig. 4e). Differences in the numbers of HNF4A+ cells were further confirmed by immunostaining and flow cytometry at the GT and PP1 stages (Fig. 4f and Extended Data Fig. 2g,h). In contrast, differences in downstream liver and pancreatic genes became significant only at the PP1 stage, and key upstream TFs shared between liver and pancreas such as GATA4/6 and FOXA2 were not significantly affected by HHEX deletion (Fig. 4e,f). Chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) showed binding of HHEX to the HNF4A P1 promoter, which gained chromatin accessibility upon HHEX deletion (Fig. 4g), suggesting that HHEX directly represses HNF4A expression. Given HNF4A’s critical role in liver differentiation44, our results suggest that HHEX restricts the HNF4A-driven liver program in pancreatic differentiation conditions.
HHEX restricts alternative endoderm-derived lineages
The small yet significant increase of HNF4A expression at the GT stage was the earliest detectable phenotype in HHEX KO (Fig. 4e,f). This defect preceded the impaired induction of pancreatic markers detected at the PP1 stage, indicating that the ectopic liver fate was not a consequence of failed pancreatic differentiation. However, it was unclear whether the impaired pancreatic differentiation observed in HHEX KO was a consequence of ectopic liver differentiation, or alternatively, due to a direct role of HHEX in initiating pancreatic differentiation. To distinguish between these possibilities, we attempted to inhibit the liver fate by applying a cocktail of chemicals that includes LDN-193189 to selectively block the liver-inducing BMP signaling45–48 for two or six days (Conditions 1 and 2, Fig. 5a,b). The KO cells formed more AFP+ liver progenitor cells compared to WT at the PP1 stage (Extended Data Fig. 3a–c), but by the PP2 stage, no significant difference of AFP+ cells were detected between the WT and KO cells in either condition (Fig. 5a–c). Despite the suppression of the liver phenotype by BMP inhibition, the KO cells failed to form any bona fide PDX1+NKX6–1+ pancreatic progenitors in either condition (Fig. 5a–d). HHEX KO cells were able to form substantial numbers of PDX1+ cells at the PP2 stage, but the expression levels were much reduced compared to the WT (Fig 5a–c, 5e, Extended Data Fig. 3). The low PDX1 expression was accompanied by CDX2 expression (Fig. 5a–c,e, Extended Data Fig. 3d), suggesting that most HHEX KO cells differentiated into duodenum-like cells. A significant increase of CDX2+ cells was also detected in HHEX KO at the PP1 stage (Extended Data Fig. 3a–c). In summary, while WT cells predominantly form pancreatic progenitor cells in both differentiation conditions, HHEX KO cells fail to form PDX1+NKX6-1+ pancreatic progenitor cells regardless of the differentiation conditions. Therefore, HHEX is intrinsically required for pancreatic differentiation, and it also restricts alternative liver and duodenum differentiation programs.
Fig. 5: HHEX KO cells acquire duodenum-like cell state upon inhibition of liver differentiation.
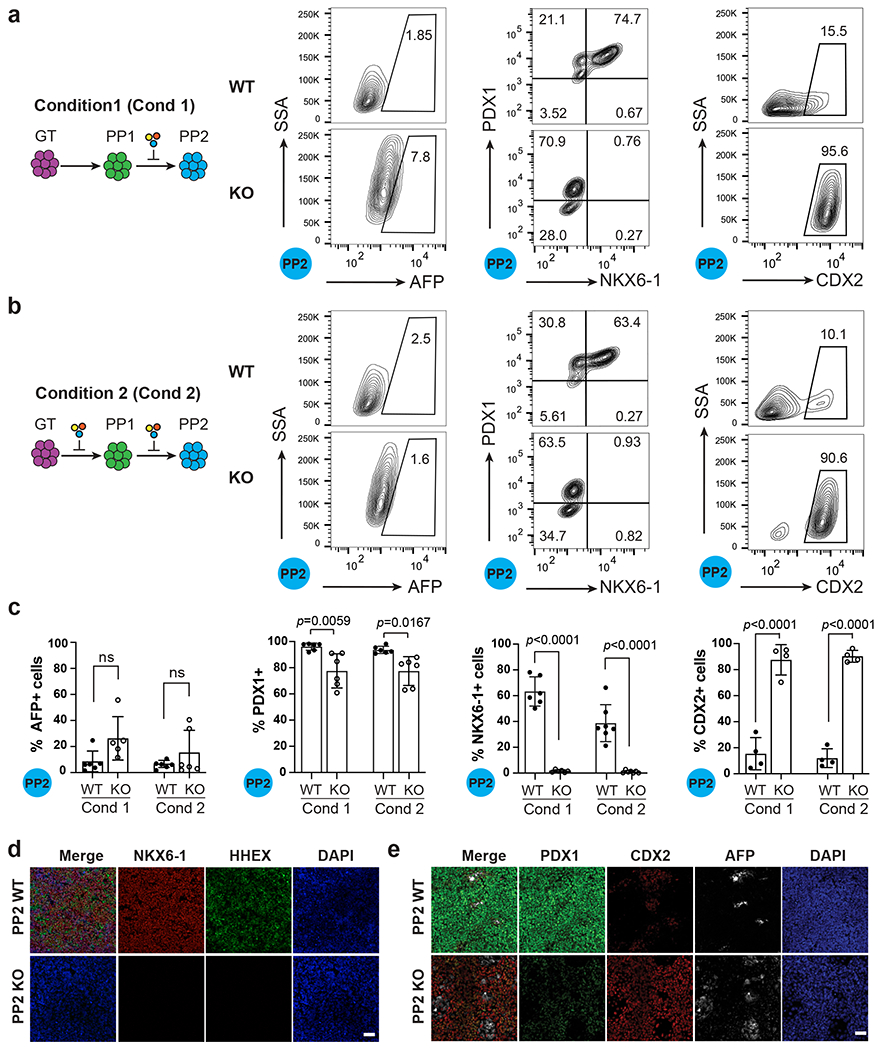
a, Schematic showing the strategy of differentiation using Condition 1 (Cond 1) and flow cytometry analysis for AFP, PDX1, NKX6.1 and CDX2 expression under Condition 1 at the PP2 stage. Additional chemical cocktail was induced after the PP1 stage, a stage displaying ectopic liver genes expression in HHEX KO cells.
b, Schematic showing the strategy of differentiation using Condition 2 (Cond 2) and flow cytometry analysis for AFP, PDX1, NKX6.1 and CDX2 expression under Condition 2 at the PP2 stage. Additional chemical cocktail was induced during both the PP1 and PP2 stage.
c, Quantification of flow cytometry analysis of AFP+, PDX1+, NKX6-1+, and CDX2+ cells at the PP2 stage in both conditions. Each symbol represents one independent experiment (n = 6 independent experiments, except for CDX2 staining, where n = 4 independent experiments) and data are presented as mean ± SD. Statistical analysis was performed using two-way ANOVA followed by multiple comparisons with Tukey correction.
d, Immunostaining images for HHEX, PDX1 and NKX6-1 expression at the PP2 stage WT/KO cells using differentiation Condition 1. Scale bar, 50 μm.
e, Immunostaining images for PDX1, CDX2, AFP expression at the PP2 stage WT/KO cells using differentiation Condition 1. Images shown in d and e represent three independent experiments. Scale bar, 50 μm.
HHEX safeguards pancreatic differentiation trajectory
We performed scRNA-seq to further characterize lineage diversification during WT and HHEX KO cell differentiation. Focusing on cells at the PP1 and PP2 stages derived from the two differentiation conditions, we identified a total of 15 clusters (Fig. 6a–c, Extended Data Fig. 4a). We next grouped these clusters based on their transcriptional profiles, and signature gene markers were identified for each group (Fig. 6d and Supplementary Table 4). These analyses led to the annotation of four main groups: (1) posterior foregut (PFG) containing the emerging PDX1+ cells with high FOXA2, HHEX, and PBX1 expression; (2) pancreatic cells (PAN) expressing progenitor markers such as NKX6-1, PDX1, and SOX9; (3) liver-like cells (LV) expressing AFP, APOA2, and FABP1; and (4) duodenum-like cells (DUO) expressing CDX2, KLF5, and SOX4. Two minor groups, labeled as “OTH” (for “other”), likely representing a small number of heterogeneous stomach and pharynx cells in the differentiation culture, were not overtly affected by HHEX deletion. The dominant groups identified from the WT cells in both differentiation conditions were the PFG group at the PP1 stage, and the PAN group at the PP2 stage (Fig. 6b,c). In contrast, KO cells in both differentiation conditions contained predominantly cells in the LV group at the PP1 stage and then cells in the DUO group at the PP2 stage (Fig. 6b,c). We confirmed the annotations by mapping published scRNA-seq profiles from E9.5 mouse embryonic foregut populations49 to our scRNA-seq data based on their transcriptional similarity with orthologous genes. The PAN, LV and DUO groups each showed high transcriptional similarity to their corresponding organ domains in mouse embryos, whereas the PFG group showed modest similarity to multiple developing organ domains (Fig. 6e, and Supplementary Table 4), consistent with its broader differentiation potential compared to the three organ-specific groups.
Fig. 6: scRNA-seq reveals differentiation trajectories of WT and HHEX KO cells.
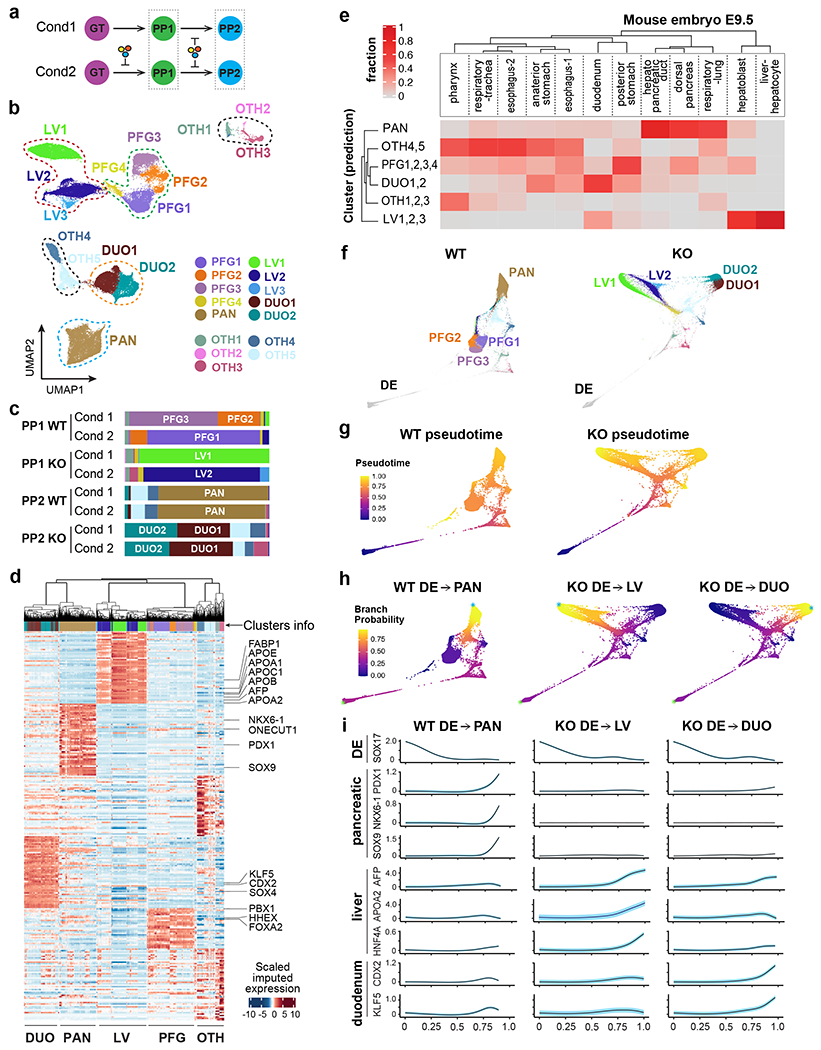
a, Schematic of single cell transcriptome profiling of WT and KO cells at PP1/PP2 stages from two differentiation conditions (one biological replicate per condition).
b, UMAP visualization of the main PP1/PP2 populations. Seurat clusters 1-15, shown with distinct colors, were annotated and grouped based on known markers.
c, Bar plots illustrating the composition of each sample across the clusters in (b). The stacks show fractions per sample.
d, Heatmap reporting MAGIC-imputed expression values (standardized per gene) of the top 50 differentially expressed genes for each group, as annotated in (b). Significant genes (FC > 1.5 and adjusted p < 0.05) were selected by comparing each group to the rest using MAST.
e, Heatmap reporting the fraction of E9.5 mouse endoderm-derived populations49 mapping to in vitro-derived human populations from (b). The sum of values in each column equals 1.
f, WT and KO lineages visualized by forced-directed layouts of the integrated data from the DE, GT, PP1, and PP2 stages. PP1/PP2 populations were annotated as in (b).
g, h, The pseudotime ordering (g) and branch probabilities (h) of selected WT or KO differentiation trajectories. A DE cell was selected as the start of each trajectory (indicated by green asterisks), and the terminal points (indicated by blue asterisks) were identified by Palantir.
i, The trends of gene expression along the pseudotime of trajectories in (g,h). The highlights show ± standard error.
To investigate the differentiation trajectories of WT and KO cells, we integrated the data from earlier DE and GT stages (Extended Data Fig. 4b). Consistent with the bulk RNA-seq results, KO cells were indistinguishable from WT cells at these earlier stages, while there were evident distinctions at the later stages (PP1, PP2) (Extended Data Fig. 4b). Palantir pseudotime ordering50 showed similar patterns between WT and KO cells at the earlier DE and GT stages, but the trajectories diverged at the PP1 and PP2 stages (Fig. 6f–h and Extended Data Fig. 4c). In WT cells, the major branch started from PFG clusters and ended at the PAN cluster. On the other hand, two distinct branches were observed in KO cells, one ending at the LV clusters and the other one ending at the DUO clusters (Fig. 6f–h). Gene expression trends in each trajectory show the changes of known markers versus the pseudotime (Fig. 6i). The expression of endoderm marker SOX17 decreased with pseudotime in all three trajectories, but the expression of pancreatic genes PDX1, SOX9, NKX6-1 only increased in the WT pancreatic trajectory (Fig. 6i). In contrast, the expression of liver (AFP, APOA2, HNF4A) and duodenum markers (CDX2 and KLF5) was dramatically increased in the KO liver and KO duodenum trajectories, respectively (Fig. 6i). Altogether, these results demonstrate that HHEX is essential to safeguard the pancreatic differentiation trajectory from diverging to the closely related liver and duodenum lineages.
HHEX safeguards the pancreatic gene regulatory network
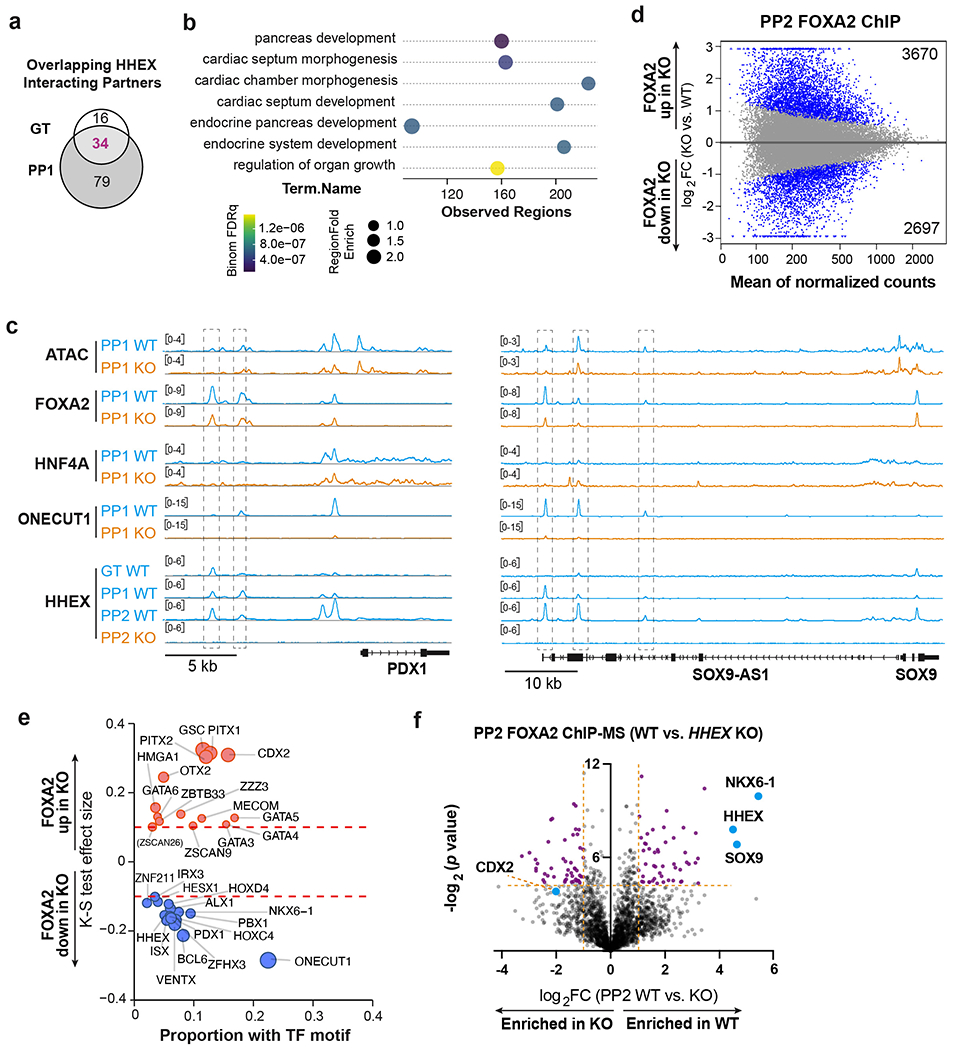
To further the mechanistic study, we investigated chromatin associated partners for HHEX through ChIP followed by mass spectrometry (ChIP-MS) (Fig. 7a). We identified 50 and 113 significantly enriched proteins at the GT and PP1 stages, respectively (Extended Data Fig. 5a and Supplementary Table 5). 34 proteins were enriched at both stages (Extended Data Fig. 5a), including four TFs: FOXA1, FOXA2, GATA4 and SALL4 (Fig. 7b,c). For further investigation, we focused on FOXA2, a well-known pioneer factor in both pancreas and liver induction26. A strong overlap was observed between the top enriched proteins in FOXA2 and HHEX ChIP-MS at the PP1 stage (Fig. 7d,e and Supplementary Table 5), suggesting that the two TFs cooperate with common partners such as FOXA1, PBX1 and TLE3 to promote pancreatic development. Comparing FOXA2 ChIP-MS data in WT versus HHEX KO cells showed differential enrichment of pancreatic TFs PBX1 and PBX2 in WT, and liver TF HNF4A in HHEX KO cells (Fig. 7f). These findings suggest that HHEX mediates selective interaction of FOXA2 with lineage-specifying TFs. FOXA2 cooperates with HHEX and PBX factors to drive pancreatic differentiation, and in the absence of HHEX, FOXA2 cooperates with HNF4A to drive liver differentiation.
Fig. 7: HHEX cooperates with FOXA2 to safeguard pancreatic differentiation.
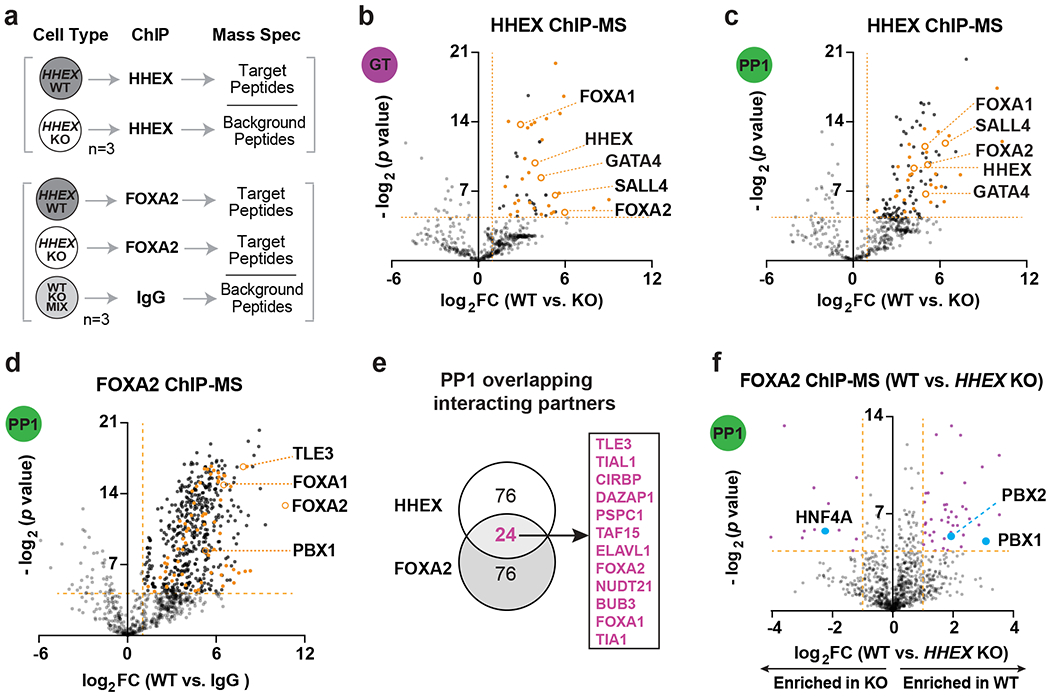
a, Schematic of ChIP-MS experiments.
b,c, Volcano plots of identified and overlapping proteins (orange labeled) for HHEX ChIP-MS at the GT(b) and PP1 (c) stages. Dotted lines indicate the cutoffs (log2 FC > 1, −log2 p > 4.32) for significantly enriched proteins. TFs that are significantly enriched at both stages are indicated with orange circles.
d, Volcano plots of significantly enriched proteins for FOXA2 ChIP-MS at the PP1 WT cells. Dotted lines indicate the significance cutoffs (log2 FC > 1, −log2 p > 4.32). Overlapping proteins that are enriched in both HHEX and FOXA2 ChIP-MS are orange labeled. TFs that are enriched in both HHEX and FOXA2 ChIP-MS are indicated.
e, Venn diagram of top 100 significantly enriched proteins (−log2 p > 4.32, top 100 hits were ranked based on log2 FC) at the PP1 stage in HHEX and FOXA2 ChIP-MS. Representative overlapping interacting hits were indicated in the box.
f, Volcano plots of significantly enriched proteins (purple labeled) for FOXA2 ChIP-MS at the PP1 stage WT or KO cells. Dotted lines indicate the significance cutoffs (log2 FC > 1, −log2 p > 4.32) for significantly enriched proteins. ChIP-MS data shown in b-f are generated from three independent experiments.
Further supporting the cooperation of HHEX and FOXA2 during pancreatic differentiation, we observed a substantial number of overlapping HHEX and FOXA2 ChIP-seq peaks at both GT and PP1 stages in WT cells (Fig. 8a and Supplementary Table 6). In addition, we identified 11,311 differential FOXA2 ChIP-seq peaks between WT and HHEX KO at the PP1 stage, whereas few differential peaks were found at the GT stage (Fig. 8b and Supplementary Table 6). Consistent with the pioneer factor activity of FOXA2, regions with decreased or increased FOXA2 occupancies in KO, referred to as “FOXA2-down” and “FOXA2-up”, respectively, showed a corresponding decrease or increase in chromatin accessibility (Fig. 8c,d). Further motif analysis showed that FOXA2-down regions were enriched for ONECUT1 and PBX1 motifs, whereas FOXA2-up regions were enriched for HNF4A and HNF1B motifs (Fig. 8e). Consistent with the motif analysis, higher overall HNF4A ChIP-seq signals were observed in the FOXA2-up regions in WT cells, while higher ONECUT1 were observed in the FOXA2-down regions (Fig. 8c,d). In HHEX KO, HNF4A occupancies increased in the FOXA2-up regions compared to WT, and ONECUT1 decreased in the FOXA2-down regions. GREAT analysis with Gene Ontology terms showed that genes in FOXA2-down regions were associated with pancreas and endocrine system development (Extended Data Fig. 5b). Consistent with a requirement for HHEX in activating the pancreatic differentiation program, we also observed a progressive increase of HHEX binding in the FOXA2-down regions from GT to PP2 in WT cells (Fig. 8c,d) as evident at the PDX1 and SOX9 loci (Extended Data Fig. 5c).
Fig. 8: HHEX protects FOXA2 binding to pancreatic regulatory regions.
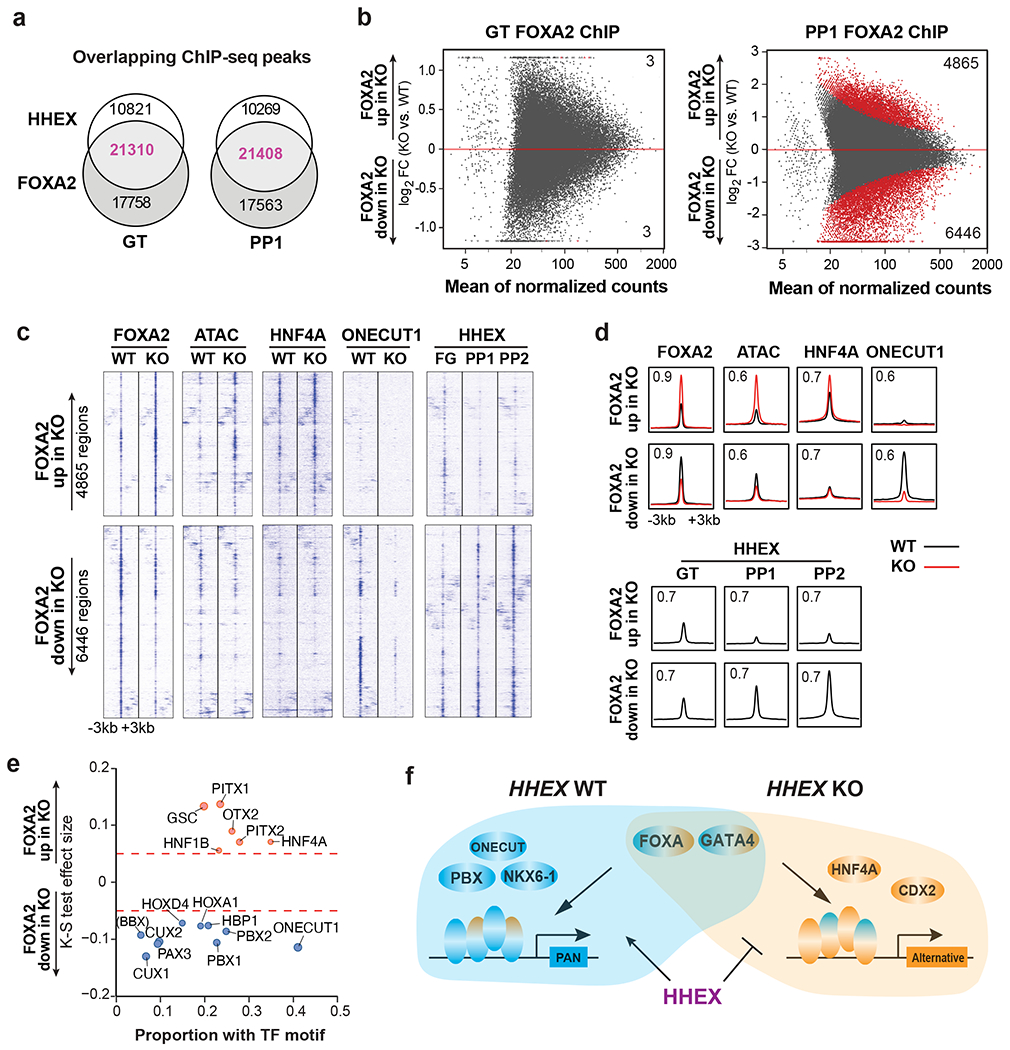
a, Venn diagram representing the number of co-bound regions identified in HHEX and FOXA2 ChIP-seq at the GT and PP1 stage WT cells.
b, MA plot of significantly increased and decreased FOXA2 binding sites (red color) at the GT and PP1 stages upon HHEX deletion. The number of significantly increased and decreased FOXA2 binding sites is indicated.
c,d, Genomic visualization of loci associated with the significantly increased and decreased activity of FOXA2 binding in KO cells versus WT. ChIP-seq of FOXA2, HNF4A, and ONECUT1 at the PP1 stage were shown, and ATAC-seq was visualized at the same regions. HHEX ChIP-seq was shown for GT, PP1, and PP2 WT. (c), tornado plots. (d), metapeaks, from the column average of the signal. The maximum value of each y axis is annotated in TPM. Plots represent the average of two independent experiments.
e, TF motifs enriched in the differential FOXA2 binding regions upon HHEX deletion. Significantly increased/decreased FOXA2 binding peaks were compared with the total atlas to examine the TF motif enrichments using the one-sided Kolmogorov-Smirnov (KS) test. The KS test effect size is shown on the y axis, and the proportion of peaks associated with the TF motif is plotted on the x axis. The size of each circle represents the odds ratio, which was defined as the frequency of the TF in an opened or closed group divided by its frequency in the entire atlas. TF motifs with a KS test effect size ≥ 0.05 (indicated by the dashed lines) and odds ratio ≥ 1.2 are shown.
f, Schematic illustrating HHEX interaction with FOXA2 and other TFs in pancreatic differentiation conditions. Data shown in a-e are from two independent experiments.
We conducted similar FOXA2 ChIP-seq analysis under conditions when the liver differentiation was inhibited (Condition 2, PP2) (Supplementary Table 6). Motif analysis showed that FOXA2-down regions were enriched for NKX6-1, ONECUT1, and PBX1 motifs, whereas FOXA2-up regions were enriched for CDX2 motifs (Extended Data Fig. 5d,e). Similarly, FOXA2 ChIP-MS experiments also showed a significant enrichment of NKX6-1 and SOX9 in WT compared to HHEX KO, and a trend towards enrichment for duodenum TF CDX2 in HHEX KO cells (Extended Data Fig. 5f and Supplementary Table 5). Together, our findings support a continuous requirement for HHEX to cooperate with FOXA2 and GATA4, and additional pancreatic TFs such as PBX1, ONECUT1 and NKX6-1 to activate pancreatic regulatory regions (Fig. 8f). Furthermore, HHEX safeguards the pancreatic gene regulatory network through restricting the cooperation of pioneer factor FOXA2 with TFs (HNF4A and CDX2) that drive liver and duodenum differentiation programs.
Discussion
Advances in CRISPR-Cas9 loss-of-function screening have enabled systematic discovery of intrinsic gene requirements for hPSCs pluripotency regulation51. We and others recently conducted genome-wide screens to interrogate the segregation of the endoderm, mesoderm, and ectoderm germ layer identities36,52,53. Here, our sequential screening strategy enabled high-throughput discovery of genetic regulators required for initiating human pancreas development. The lead hits identified from our screens are enriched for molecular signatures related to MODY and β cell development, highlighting the utility for discovery of disease-relevant genes. Notably, single-nucleotide polymorphisms at the HHEX locus have been associated with impaired insulin secretion and type 2 diabetes in genome-wide association studies54, but HHEX is not produced by adult human or mouse β cells40. We speculate that reduced HHEX expression during development could decrease the number of pancreatic progenitors, thereby the total number of endocrine cells produced, and increase diabetes susceptibility later in life. Broadly, organogenesis forms the basis for understanding human health, and studies of organogenesis can be greatly assisted through expanding the sequential screening strategy to appropriate differentiation stages that match the emergence of organ-specific progenitors or tissue subtypes therein.
There are notable similarities and distinctions between our findings in hPSCs and previous studies in mice. Hhex null mouse embryos fail to form a ventral pancreas25 and have ectopic duodenal cells55. The impaired morphogenetic movement of the endoderm causes the prospective ventral pancreatic domain to be incorrectly positioned next to cardiac mesoderm cells, thus indirectly affecting ventral pancreas specification25. Our investigation of HHEX function builds upon an hPSC differentiation platform which closely mimics human pancreas organogenesis. Some studies suggest that current methods favor a dorsal-like program38. The fact that HHEX emerged from a pooled CRISPR-Cas9 screen strongly supports a cell-autonomous role for HHEX within the pancreatic anlagen in establishing the early pancreatic differentiation program, and one that is distinct from the indirect tissue-positioning role described above for mice. It remains to be determined if HHEX might have additional indirect effects through regulating tissue appositions in human embryogenesis. Conversely, revisiting the Hhex KO mouse model may uncover ectopic liver phenotypes and intrinsic requirements for Hhex in pancreas development as predicted by the findings in our hPSC model.
The separation of the pancreatic domain from progenitors of the neighboring organs, the liver and duodenum, marks an important fate-allocation branchpoint in organogenesis and could help us understand generalizable principles that govern organ-domain demarcation. Histone acetyltransferase P300 (an activator influence) and histone methyltransferase Ezh2 (a repressor influence) have been shown to promote the “liver not pancreas” program choice56. In addition to chromatin factors, a gatekeeper function has been proposed for specific TFs. Some studies suggested that such gatekeeper TFs directly repress the transcriptional programs of alternative lineages57–60. It is not yet clear to what extent HHEX directly suppresses alternative lineages. We show that HHEX promotes pancreatic differentiation through cooperation with pancreatic TFs as well as endodermal TFs such as FOXA2 and GATA4 that are broadly required for both pancreas and liver specification. Eliminating HHEX promotes the FOXA2-HNF4A interaction, leading to liver differentiation. Therefore, our findings suggest a model that a gatekeeper TF, through cooperation with broadly expressed pioneer TFs, can efficiently direct pioneer TFs to activate one lineage-specific programs and at the same time restrict the activation of alternative programs (Fig. 8f). During lineage segregation, multiple gatekeeper TFs may compete for the shared TFs to fine-tune the balance of lineage-specific transcriptional programs in diverse developmental contexts, as has been proposed for hematopoietic development61. Identifying gatekeeper TFs and their mechanisms of function especially in early stages of organogenesis for multiple organ domains – and the period for which fate-allocation states might remain intrinsically pliable – is fundamental to our understanding of lineage commitment and plasticity with implications for stem cell biology, regenerative medicine, and tumor cell plasticity.
Methods
Culture of hESCs
Experiments in this study were performed using H1 (NIHhESC-10-0043) and HUES8 (NIHhESC-09–0021), which were regularly confirmed to be mycoplasma-free by the Memorial Sloan Kettering Cancer Center (MSKCC) Antibody & Bioresource Core Facility. All experiments were conducted per NIH guidelines and approved by the Tri-SCI Embryonic Stem Cell Research Oversight (ESCRO) Committee. hESCs were maintained in Essential 8 (E8) medium (Thermo Fisher Scientific, A1517001) on vitronectin (Thermo Fisher Scientific, A14700) pre-coated plates at 37 °C with 5% CO2. 5 μM Rho-associated protein kinase (ROCK) inhibitor Y-27632 (Selleck Chemicals, S1049) was added into the E8 medium the first day after passaging or thawing hESCs.
Mouse and human tissues
Male and female C57BL/6N wildtype mice of 8-12 weeks of age were bred to generate timed pregnancies in accordance with the animal protocol approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). Mice were housed with a 12h-12h light-dark cycle at 18-23 °C temperature and under 40-60% humidity. Gender information was not determined for the embryos, and no embryos were excluded based on gender. Deidentified human 33 wpc pancreatic tissue from neonatal organ donors were obtained through the National Disease Research Interchange (http://ndriresource.org) or the International Institute for the Advancement of Medicine (http://www.iiam.org) as part of studies led by Alvin C. Powers and Marcela Brissova at Vanderbilt University Medical Center. The Vanderbilt University Institutional Review Board (IRB) has declared that studies on de-identified human pancreatic specimens do not qualify as human subject research. Human 22 wpc fetal pancreas tissue was obtained from secondary sources (Advanced Biosystems Resources [ABR], Inc.) under approved Material Transfer Agreements and with protocols approved by the University of Wisconsin’s IACUC and IRB (Study #2013-141). ABR, Inc. obtained consent in accordance with Uniform Anatomical Gift Act and National Organ Transplant Act guidelines. ABR, Inc. warrants that appropriate consent for tissue donation is obtained and adequate records of such consents are maintained. In addition, these tissues were obtained with local, state, and federal laws and regulations governing the procurement of human tissue.
hESC-directed pancreatic differentiation
All differentiation experiments were performed on hESCs grown on vitronectin. hESCs were maintained in E8 medium for 2 days to reach ~80% confluence. Cells were washed with PBS and differentiated to DE, GT, PP1, and PP2 stage following the protocols previously described27,37. Briefly, hESCs were rinsed with PBS and first differentiated into DE using S1/2 media supplemented with 100 ng/ml Activin A (Bon Opus Biosciences) for three days and CHIR99021 (Stemgent, 04-0004-10) for two days (1st day, 5 μM; 2nd day, 0.5 μM). DE cells were rinsed with PBS and then exposed to S1/2 media supplemented with 50 ng/ml of KGF (FGF7) (PeproTech, 100-19) and 0.25mM vitamin C (VitC) (Sigma-Aldrich, A4544) for 2 days to reach GT stage. For PP1 stage, two conditions were used. For Condition 1, cells were switched to S3/4 media37 supplemented with 50 ng/ml of FGF7, 0.25 mM VitC, and 1μM retinoic acid (RA) (Sigma-Aldrich, R2625) for two days to reach PP1 stage. For Condition 2, cells were exposed to S3/4 media supplemented with 50 ng/ml of FGF7, 0.25 mM VitC, 1μM RA, 100 nM LDN (Stemgent, 04-0019), 0.25 μM SANT-1 (Sigma, S4572), 200 nM TPB (EMD Millipore, 565740), and 1:200 ITS-X for two days. The PP1 cells derived from both conditions were then differentiated to the PP2 stage using S3/4 media supplemented with 2 ng/ml of FGF7, 0.25 mM VitC, 0.1 μM RA, 200 nM LDN, 0.25 μM SANT-1, 100 nM TPB, and 1:200 ITS-X for 4 days.
Generation of the iCas9 H1 PDX1GFP/+ reporter hESC line
The reporter hESC line was generated with a previously established knockin strategy33. Briefly, iCas9 H1 hESC were treated with Y-27632 and doxycycline one day before transfection. Transfection of the gRNAs and donor plasmid (Plasmid #66964, Addgene) into iCas9hESCs was performed using Lipofectamine 3000 (Thermo Fisher Scientific, L3000001) following manufacturer’s guidelines. Correct targeting was verified by Southern blotting. gRNA target sequence is listed in Supplementary Table 7.
Genome-Wide CRISPR-Cas9 screens
The lentiviral CRISPR libraries were produced and tested as previously described36,37. An ~1,000-fold library coverage is targeted to maximize sensitivity. For the PP1 screen, ~200 million H1 PDX1GFP/+ iCas9 hESCs were harvested and infected with the lentiviral human GeCKO v2 library35 at a low multiplicity of infection ~0.35. 6 μg/ml protamine sulfate was added concurrently with the virus infection to enhance the infection efficiency. Infected cells were treated with 2 μg/ml doxycycline at day 1-6 and 1 μg/ml puromycin at day 2-6. On day 6, cells were dissociated with TrypLE Select, and in total ~108 million cells were plated into twelve 150 mm plates for pancreatic differentiation (Condition 1). PDX1/GFP+ and PDX1/GFP− cells were collected through FACS, and genomic DNA was extracted using the Qiagen blood & cell culture DNA maxi kit (Qiagen, 13362). For the DE screen, ~200 million HUES8 SOX17GFP/+ iCas9 hESCs were harvested and infected with the lentiviral human Brunello library62. Hi-seq was performed as previously described36,37. Data was analyzed with MAGeCK 0.5.9.4 default RRA parameters63 (Supplementary Table 1). After removal of DE hits (log2 FC > 0 [GFP− versus GFP+], p < 0.01), all genes were ranked by log10 (PP1 positive score) – log10 (PP1 negative score) for GSEA analysis with MSigDB v6 using the pre-ranked option. The direction of positive or negative score was based on the enrichment of gRNAs in GFP+ versus GFP− cells, or vice versa.
Generation of clonal KO hESC lines
To control for potential CRISPR off-target effects, we generated five homozygous KO lines carrying frameshift mutations using two separate gRNAs targeting exon 2 and exon 3 of HHEX, respectively (Fig. 3a). Mutant lines were generated as previously described with some modifications37. gRNAs and tracer RNA were ordered from IDT (Alt-R® CRISPR-Cas9 crRNA and #1072532) and added at a 15 nM final concentration. Briefly, gRNA/tracer RNA and Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778030) were diluted separately in Opti-MEM (Invitrogen, 31985070), mixed together, and incubated for 15 min at room temperature (RT), and added dropwise to freshly seeded iCas9 hESCs in a 24-well plate. 2 μg/ml doxycycline was added the day prior to transfection, the day of transfection, and one day after transfection to induce the Cas9 expression. Three days after transfection, hESCs were dissociated into single cells and ~1000 cells were plated into one 100 mm tissue culture dish for colony formation. After ~10 days of expansion, single colonies were picked. Genomic DNA from crude cell lysate was used for PCR genotyping. gRNA target sequences and primers used for PCR and sequencing are listed in Supplementary Table 7.
Immunofluorescence staining
In vitro pancreatic differentiated cells were fixed in 4% paraformaldehyde (Thermo Fisher Scientific, 50980495) for 10 minutes at RT. After washing with PBST (PBS with 0.1% Triton X-100) three times, cells were blocked in 5% donkey serum in PBST buffer for 30 minutes at RT. Primary and second antibodies were diluted in the blocking solution. Cells were incubated with primary antibodies 1 hour at RT or overnight at 4 °C, followed by 1 hour staining for secondary antibodies at RT. The cells were stained with DAPI for ~15 min RT. Images were taken using the Confocal Laser Scanning Platform Leica TCS SP8.
Human Pancreas samples from 22 wpc tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Five-micron sections were cut and deparaffinized with xylene and ethanol. Antigen retrieval was performed for 2 h at 90 °C in 10 mM sodium citrate buffer. Following washing with PBST (1× PBS/0.05% Triton X-100) and blocking for 35 min (1× PBS/10% BSA) at RT, antibody-specific staining was performed. Images were taken by Nikon A1RS HD confocal microscope. Human Pancreas samples from 33 wpc tissues and mouse E11.5 samples were fixed with 4% paraformaldehyde in 1x PBS, equilibrated in 30% sucrose overnight and embedded in optimal cutting temperature compound (Tissue-Tek, Sakura). Briefly, slides were air dried for 10 min and washed in PBS RT for 5 min. After a further fixation (1% PFA/PBS for 10 minutes RT, human embryo sample only), slides were permeabilized in 0.1% Triton X-100 for 10 min. Slides were then blocked and stained. Signal was enhanced with VECTASTAIN ABC TSA kit (Vector lab, PK6100) and Cy3-Avidin TSA reagent (Akoya Biosciences, NEL744001KT) and mounted with Prolong Gold with DAPI (Invitrogen, P36931). Images were taken using the Zeiss Apotome. All primary antibodies are listed in Supplementary Table 8.
Flow cytometry
Cells were dissociated using TrypLE Select and resuspended in FACS buffer (5% FBS in PBS). LIVE-DEAD Fixable Violet Dead Cell Stain (Invitrogen, L34955) was used to discriminate dead cells from live cells. Cell surface marker (CXCR4-APC from R&D, FAB170A) staining and live/dead staining were then performed for 15 min RT in FACS buffer. Intracellular staining was performed with Foxp3 Staining Buffer Set (eBioscience, 00-5523-00) following the manufacturer’s instructions. Permeabilization/fixation was performed at RT for 1 hour. Antibody staining was performed in permeabilization buffer. Antibodies for this study are listed in Supplementary Table 8. Cells were then analyzed using BD LSRFortessa. Flow cytometry analysis and figures were generated in FlowJo v10.
Western blot
Cells were harvested at PP1 stage and lysed using cell lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor (Roche, #05892791001). Sample preparation follows NuPAGE Novex protocol. The lysate (mixed with loading buffer and reducing agent) was loaded into a Bis-Tris 10% gel (Novex, NP0301BOX) and transferred to nitrocellulose membranes (Novex, LC2001). Membranes were blocked with 5% milk in Tris-based saline with Tween 20 (0.1% TBST) buffer for 1 hour at RT. The membrane was incubated with primary antibody HHEX (R&D Systems, MAB83771-100) overnight at 4°C, followed by incubation with HRP conjugated secondary antibodies at RT for 1 hour. ECL western blotting detection reagent (Amersham, RPN2236) was used to visualize the protein bands.
RNA Isolation, quantitative real-time PCR, RNA-seq and analysis
RNA samples from HHEX KO and WT cells were collected at the DE, GT, and PP1 stage and isolated with the miRNeasy Mini Kit (Qiagen, 217004). 1 μg of RNA per sample was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, 4368814). Quantitative real-time PCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25742) on the ABI PRISM® 7500 Real Time PCR System (Applied Biosystems) using manufacturer recommended PCR settings. Quantitative real-time PCR primers are listed in Supplementary Table 7.
RNA-seq was performed by the MSKCC Integrated Genomics Operation (IGO) Core as previously described16,37. RNA-seq reads were trimmed for quality and adapter sequences using TrimGalore (v0.4.5) and aligned to the human genome (hg19) by STAR (v2.6) with default parameters64. Read counts per gene were created using HTSeq (v0.9.1)65. Normalization, principal component analysis, and differentially expressed genes were generated by DESeq266. Analyzed RNA-seq data (normalized counts) can be found in Supplementary Table 2. Differentially expressed genes were identified based on cutoff log2 FC > 1 and FDR < 0.05. GSEA was performed with MSigDB v6 using the pre-ranked option and log2 FC for pairwise comparisons. Heat map was generated using the online analysis tool Heatmapper67.
Single-cell RNA-seq
For single-cell RNA-seq analysis, two experiments were performed using WT and HHEX KO cells at different stages of differentiation. In one experiment, we profiled 4 samples (#1-4) that consist of WT and KO cells at the DE and GT stages. In the second experiment, we profiled 8 samples (#5-12) that consist of WT and KO cells grown under two differentiation conditions at the PP1 and PP2 stages (Fig. 6a,b), as well as two DE WT samples (#13-14). The last two DE samples were included to evaluate and correct the potential batch effect.
To dissociate the cells into a single cell suspension, cells were incubated in 0.5 mM EDTA 3 mins at RT, followed by TrypLE Select for 10 mins at 37°C. Cells were filtered through CellTrics™ (20μm, Sysmex) and centrifuged at 200 rcf for 2 mins to remove the debris in the supernatant. To further remove the debris, the pellets were further washed in PBS containing 0.04%BSA and centrifuged at 200 rcf for 90s three times. Cells were resuspended with PBS containing 0.04%BSA and filtered through CellTrics™ twice to obtain single cells. Cells were then loaded into Single Cell Master Mix (10x Genomics). Cellular suspensions were loaded on a Chromium Controller targeting a 5,000–7,000 cell range collection. Single-cell 3′ RNA-seq libraries were generated following the manufacturer’s instructions (10x Genomics Chromium Single Cell 3′ Reagent Kit v3 User Guide). cDNA libraries were then quantified on the Agilent Bioanalyzer with a high-sensitivity chip (Agilent), and Kapa DNA quantification kit for Illumina platforms (Roche). Sequencing setup is following the manufacturer’s instructions.
Single-cell RNA-seq computational analysis
Pre-processing the 10x single-cell RNA-seq data.
FASTQ files of 14 hESC-derived samples from two experiments (Supplementary Table 4) were pre-processed with 10x Genomics Cell Ranger v6.0.068. Each sample was individually aligned to GRCh38 (Cell Ranger human reference 2020-A) and counted by ‘cellranger count’. The outputs were combined, without any sequencing depth normalization, by ‘cellranger aggr --normalize=none’ to obtain the filtered gene-barcode count matrix. This matrix was further filtered based on the number of transcripts (>3000), the number of detected genes (>1500), and the fraction of mitochondrial transcripts (<12%). Then, any genes detected in fewer than 3 cells were discarded, leaving 29012 genes for downstream analyses. Finally, the count matrix was split into experiment one (samples 1-4) with 14391 cells retained, and experiment two (samples 5-14) with 47703 cells retained.
Data normalization and dimensionality reduction.
The experiments were treated as two separate batches and downstream analyses on each experiment were separately performed using standard functions from Seurat v4.0.069,70. Each filtered count matrix was library-size normalized and log-transformed to obtain the ‘log-normalized’ expression values, and the top 5000 highly variable genes were identified. To remove the cell cycle effect, S phase and G2/M phase scores71,72 were computed from and regressed out of the gene expression values, and the residuals were scaled to obtain the ‘scaled’ expression values. PCA was performed on the scaled expression values of the top variable genes.
Cell clustering.
For the data from experiment two, a Shared Nearest Neighbor (SNN) graph was built from its first 20 PCs, computed as described in the previous section, with k=50 nearest neighbors and used to cluster the cells and find their UMAP embedding, by standard Seurat functions (Fig. 6b–c, Extended Data Fig. 4a, and Supplementary Table 4). Clusters 1-15 consist of the main PP1/PP2 populations, clusters 16-18 consist of DE WT cells, and clusters 19-20 are small outlying clusters with mostly low-quality cells.
Differential gene expression tests.
Differentially expressed genes were identified by Seurat functions performing MAST73 on the log-normalized expression values. All tests were restricted to non-mitochondrial and non-ribosomal genes that were detected in at least 10% of the cells in at least one of the groups that are compared. Signature genes were selected based on their fold change >1.5 and adjusted p <0.05. Top genes with the highest fold change were subsequently selected per group (Fig. 6d and Supplementary Table 4).
Mapping mouse endoderm derived cells to human PP1/PP2 populations
Single-cell RNA-seq data from dissected mouse foreguts at embryonic days 8.5-9.5 were downloaded from GEO GSE13668949. The raw counts of cells labeled as ‘Endoderm’ from clusters that were annotated as definitive endoderm (DE) and splanchnic mesoderm (SM) were used for this analysis. The name of any gene that was uniquely mapped to an orthologous gene, by gprofiler2 v0.2.074, was replaced by its corresponding human gene ID. Any genes detected in fewer than 3 cells were discarded, and the count matrix was split into stages E8.5, E9.0, and E9.5. Stage E9.5 was used as the ‘query’ data because we expected it to best fit the human PP1/PP2 stages. The counts were log-normalized, top 5000 highly variable genes were identified, cell cycle effect was regressed out, and PCA was performed on the scaled data from the top variable genes. As for the human ‘reference’ data, the analysis was restricted to the main PP1/PP2 populations (clusters 1-15) from experiment two. This data was already analyzed (see ‘Data normalization and dimensionality reduction’), but the top variable genes were identified for this subset of cells, and PCA was performed again. Seurat functions were used to identify ‘transfer anchors’ by Canonical Correlation Analysis (CCA) and predict the most likely cluster in the reference human data for each query mouse cell. The fraction of cells in each mouse cluster that mapped to each human cluster was computed as a measure of similarity between human and mouse populations (Fig. 6e and Supplementary Table 4).
Data integration.
The data from both experiments were integrated, by standard Seurat functions, after the initial analyses (see ‘Data normalization and dimensionality reduction’). 5000 genes were selected as integration features and used to identify ‘integration anchors’ by CCA. Log-normalized, batch-corrected expression values were computed for the integration features. PCA was performed on the integrated data after removing the cell cycle effect and scaling. A UMAP embedding was computed for the integrated data from a k=50 SNN graph built using the first 20 PCs (Extended Data Fig. 4 b and Supplementary Table 4). The overlapping embedding of samples 1-2 (DE WT/KO in experiment 1) and samples 13-14 (DE WT 60h/72h in experiment 2) suggests that any potential batch effects were removed.
Data imputation.
The log-normalized expression values were imputed by MAGIC75 to de-noise gene expression. The imputed values were not used in the analyses unless stated.
Pseudotime analysis.
Pseudotime analysis was performed on the integrated data of both experiments to study cell state transitions. The overlapping embedding of samples #1-2 (DE WT/KO in experiment 1) and samples #13-14 (DE WT 60h/72h in experiment 2) suggests that any potential batch effects were removed. In the following analyses, the data from experiment two was restricted to the main PP1/PP2 populations (clusters 1-15). The following steps were repeated, once for the WT cells (samples 1, 3, 5, 6, 9, 10), and once for the HHEX KO cells (samples 2, 4, 7, 8, 11, 12). The first 20 PCs of the integrated data (see ‘Data integration’) were used to build an affinity kernel (k=100, ka=⌊k/3⌋), from which a force-directed layout (FDL) and 15 diffusion map (DM) components were computed. The FDL embedding was computed using ForceAtlas2 v0.3.576 and used to visualize the cells (Fig. 6f, Extended Data Fig. 4c and Supplementary Table 4). The ‘multi-scale space’ was determined from the DM components and used to perform pseudotime analysis (k=100), by standard functions from Palantir v1.0.050. A DE cell was manually selected as the starting point, but the potential trajectories and their terminal cells were identified by Palantir (Fig. 6g,h). Every cell’s fate is modeled with an estimated pseudotime, the probability of reaching each terminal state (Branch Probability), and its differentiation potential (Entropy). Gene expression trends for selected trajectories (Fig. 6i) were calculated by fitting generalized additive models, implemented in Palantir, on imputed log-normalized expression values of the integrated data (see ‘Data integration’ and ‘Data imputation’).
ATAC-seq and Analysis
ATAC-seq was performed as previously described16,36. Two biological repeats per genotype were used for ATAC-seq experiment. Libraries were prepared using the NEBNext Q5 Hot Start HiFi PCR Master Mix (NEB, M0543L) and Nextera primers77. Samples were sent to MSKCC IGO for PE50 sequencing using a HiSeq 2500. Sequencing data was aligned to the hg19 reference genome using bowtie2 version 2.3.3.178. Ends of the aligned reads were shifted to remove Tn5 transposase artifacts as described79,80. Macs2 version 2.1.1.2016030981 was used to remove duplicate reads and to call peaks. Peaks were filtered using Irreproducible Discovery Rate (IDR) version 2.0.382 using the two replicates of each cell type with the threshold of 0.01. Filtered peaks showing reproducibility in any cell type were combined to create the atlas used in subsequent analyses.
PCA plots.
Among the ATAC-seq atlas, the top 3000 peaks with high variance among cell types were selected and were used to generate a PCA plot.
Identification of clusters.
Using the Wald test with the adjusted p value cutoff of 0.01, we defined 6362 peaks that showed variability in the cell types of GT and PP1 cell types, in both HHEX WT and KO. The quantification of ATAC-seq of all cell types in the loci was combined and ordered by hierarchical clustering, with the agglomeration method of ward.D2.3 clusters at the top of the dendrogram were used.
Motif enrichment of clusters by hypergeometric test.
The hypergeometric test was used to investigate the motifs enriched in each cluster (foreground ratio) compared with the total atlas (background ratio). The binomial Z-score was used to visualize the motif enrichment as previously described16. The expected count was calculated from the size of the foreground group and the background ratio, and the standard deviation was estimated based on the binomial distribution. Then the observed count was transformed to a Z-score, showing the number of standard deviations away from the expected count. For reference, a dotted vertical line was added at the point where the motif generated the Bonferroni-corrected hypergeometric p closest to 1 × 10−10.
ChIP-seq and Analysis
ChIP-seq was performed as previously described16,37. Antibodies used for immunoprecipitation are listed in Supplementary Table 8. One-half of a confluent 15 cm plate cells and two biological repeats per genotype were used for ChIP-seq experiment. Libraries were prepared using the NEBNext® Ultra II DNA Library Prep Kit (NEB, E7103S) and NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1; NEB, E7335S). Samples were pooled and submitted to MSKCC IGO for sequencing. Sequencing data was aligned to the hg19 reference genome using bowtie2 version 2.3.3.1. Peak calling was performed by MACS2 with the paired input and the choice of default extension size81. ChIP-seq signal was quantified by two independent sets of domains: one based on ATAC-seq and the other based on FOXA2 ChIP-seq. To show co-binding of transcription factors at ATAC-seq peaks, ChIP-seq signal was quantified on the same ATAC-seq-based atlas (Fig. 8c and Supplementary Table 6). Then the coverage of ChIP-seq on those peaks was quantified by RPKM, with the library size defined as the number of reads mapped to any genomic location within 3 kb of summits in the ChIP-seq atlas. Finally, the RPKM cutoffs were chosen as shown in Supplementary Table 6 to determine whether a transcription factor was co-bound with the ATAC-seq. Additionally, to show the effect of FOXA2 binding in the ChIP-seq of other transcription factors, the FOXA2-based atlas was also created, with the same peak calling and reproducibility filtering steps83. The differentially bound group of FOXA2 ChIP-seq between WT and KO was defined with the adjusted p value cutoff of 0.05.
Motif enrichment by Kolmogorov-Smirnov test.
One-sided Kolmogorov-Smirnov test was used to quantify and visualize the effect of motif enrichment in the significantly differential ChIP-seq groups. The cumulative distribution function of logFCs associated with the differential group was compared with that in the entire atlas. The effect size and significance were used to show the top enriched motifs, and the odds ratio was defined as the foreground ratio divided by the background ratio.
Visualization of ATAC-seq and ChIP-seq signal tracks
To reduce the effect of outliers with very high peak activity compared with the rest of the plot, all signal in the tornado plot was capped at the 99th percentile. The relevant groups of ATAC-seq or ChIP-seq peaks were ordered by hierarchical clustering. The column average of the signal was plotted as the metapeak. The y axis was annotated at the unit of tags per million.
ChIP Mass Spectrometry and Analysis
ChIP Mass Spectrometry (ChIP-MS) was performed as previously described37. Antibodies used for immunoprecipitation are listed in Supplementary Table 8. Briefly, ChIP-MS was performed using the same protocol as ChIP-seq through washing of the magnetic beads that contained the chromatin/protein immuno-complex. Proteins were digested using the S-trap (Protifi, C02-micro-80) sample preparation followed by LC-MS/MS analysis with an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific), as previously described37.
Raw files were searched using Proteome Discoverer software (v2.4, Thermo Scientific) using SEQUEST search engine and the SwissProt human database (updated February 2020). Each analysis was performed with three biological replicates. Prior to statistics, proteins were log2 transformed, normalized by the average value of each sample and missing values were imputed using a normal distribution 2 standard deviations lower than the mean as previously described84. The normalized data was shown in Supplementary Table 5.
Statistical analysis
All data points refer to the biological repeats. No statistical method was used to predetermine sample size. The Investigators were not blinded to allocation during experiments and outcome assessment. No data were excluded from the analyses unless the differentiation experiment itself failed. The number of biological and technical replicates are reported in the legend of each figure. Flow cytometry analysis and immunofluorescence staining as well as RT-qPCR experiments were derived from at least three independent experiments unless specified in the legends. For ATAC-seq, ChIP-seq, and bulk RNA-seq, quantification and statistics were derived from two independent experiments. CRISPR-Cas9 screening and scRNA-seq experiments were performed once. All ChIP-MS data are from three independent experiments. All the statistical analysis methods are indicated in the figure legends and methods parts. Quantification of flow cytometry and RT-qPCR data are shown as mean ± SD. Student’s t-test was used for comparison between two groups. ANOVA was used for multiple comparisons. Statistical significance (the exact p value) was indicated in each figure.
Data availability
Sequencing data is available at the Gene Expression Omnibus (GEO) under the accession code of GSE181480. Previously published data that were re-analyzed here are available at GEO under the accession codes of GSE136689 and GSE86225. Source data are provided with this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1. Analysis of HHEX expression and examination of HHEX KO cells through flow cytometry and RNA-seq.
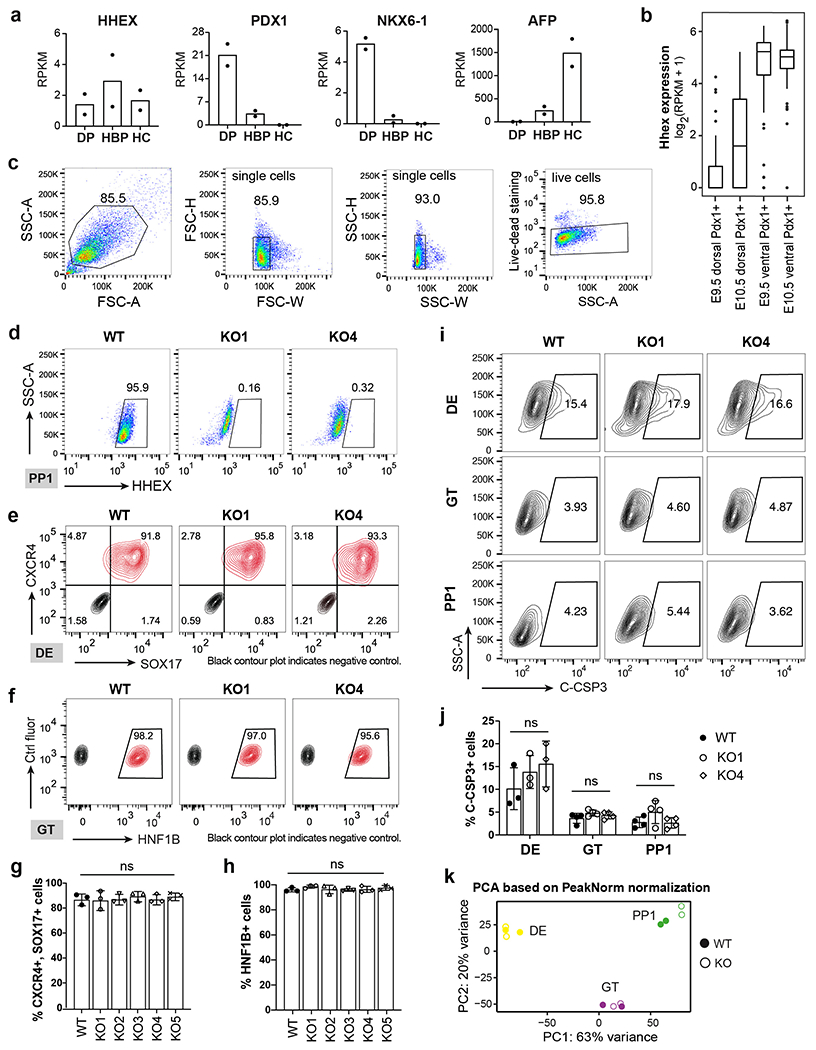
a, Bar plots for HHEX, PDX1, NKX6-1, and AFP expression in human CS12-14 stages dorsal pancreatic bud (DP), hepatobiliary primordium (HBP), and hepatic cords (HC) based on a published study38. n = 2 independent experiments. The y-axes represent the expression levels RPKM.
b, Box plots for Hhex expression in Pdx1 cells sorted from ventral and dorsal pancreatic buds in E9.5 and E10.5 Pdx1-GFP mouse embryos based on a published study43. The y-axe of the box plots represent the expression levels (log2(RPKM+1)). In each boxplot, the rectangle shows the inter-quartile range (IQR), with the bottom and top hinges representing the 25 and 75 percentiles, respectively. The middle line represents the median. The whiskers extend to the most extreme value within 1.5*IQR above or below the hinges. E9.5 dorsal Pdx1+ cells, n=31; E10.5 dorsal Pdx1+ cells, n=27; E9.5 ventral Pdx1+ cells, n=44; E10.5 ventral Pdx1+ cells, n=59. E9.5 and E10.5 data are generated from two and three independent mice, respectively.
c,d, Flow cytometry gating strategy for HHEX expression. The SSC-A/FSC-A gate identifies cells based on the size and granularity. The FSC-H/FSC-W and SSC-H/SSC-W gates identify single cells. Live-dead staining distinguishes live cells from dead cells (c). HHEX KO cells were used as negative control for WT HHEX expression at the PP1 stage (d).
e,f, Flow cytometry analysis of SOX17 and CXCR4 expression at the DE stage (e) and HNF1B expression at the GT stage (f).
g,h, Quantification of flow cytometry analysis for SOX17, CXCR4 expression at the DE stage (g) and HNF1B expression at the GT stage (h). n = 3 independent experiments and data are presented as mean ± SD.
i,j, Flow cytometry analysis (i) and quantification (j) of cleaved caspase-3 (C-CSP3) expression at the DE, GT and PP1 stage. n = 3 independent experiments and data are presented as mean ± SD. k, PCA based on PeakNorm normalization for all WT and KO samples during pancreatic differentiation. Two independent experiments were performed at each stage. Statistical analysis of g, h and j was performed using one-way ANOVA followed by Dunnett multiple comparisons test vs. the WT control.
Extended Data Fig. 2. Chromatin accessibility and transcriptional changes upon HHEX deletion.
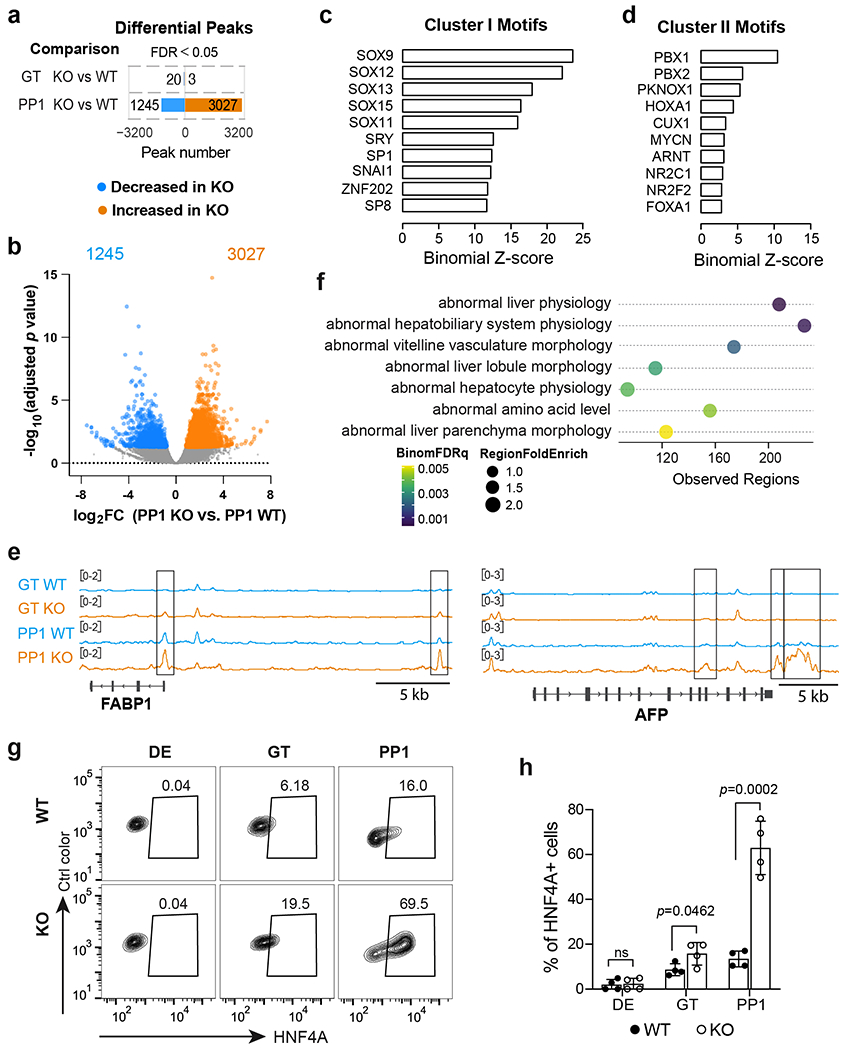
a,b, Bar graph and volcano plot showing the number (a) and adjusted p value distribution (b) of differential peaks in KO cells compared to WT. Differential ATAC peaks were identified by DESeq2 using default parameters. FDR<0.05 are counted as one significant peak. Less accessible peaks in KO are marked in blue and more accessible peaks in KO in orange. The number of differential peaks is indicated.
c,d, TF motif enrichment in cluster I (c) and II (d) regions. One-sided hypergeometric test was used to compare the enrichment of proportions of TF motifs for each cluster (foreground ratio) versus those for total atlas (background ratio). The horizontal axis shows the binomial Z-score, representing the number of standard deviations between the observed count of each cluster peaks containing a TF motif and the expected count based on the background ratio. The p values are provided in the Supplementary Table 3.
e, IGV tracks (average of two independent experiments) show chromatin accessibility at representative liver genes loci identified in cluster III. Scale bar, 5 kb.
f, Top 7 mouse phenotypes associated with the regulatory regions identified in cluster III. The term of mouse phenotypes was selected based on the binom rank and cutoffs of region fold enrich >1.4 and observed regions >80.
g,h, Flow cytometry analysis (g) and quantification (h) of HNF4A+ cells at DE, GT, and PP1 stage. Each symbol represents one independent experiment (n = 4 independent experiments) and data are presented as mean ± SD. Statistical analysis was performed using unpaired two-tailed Student’s t-test. Data shown in a-f are from two independent experiments.
Extended Data Fig. 3. The effects of inhibiting liver differentiation on WT and HHEX KO cells.
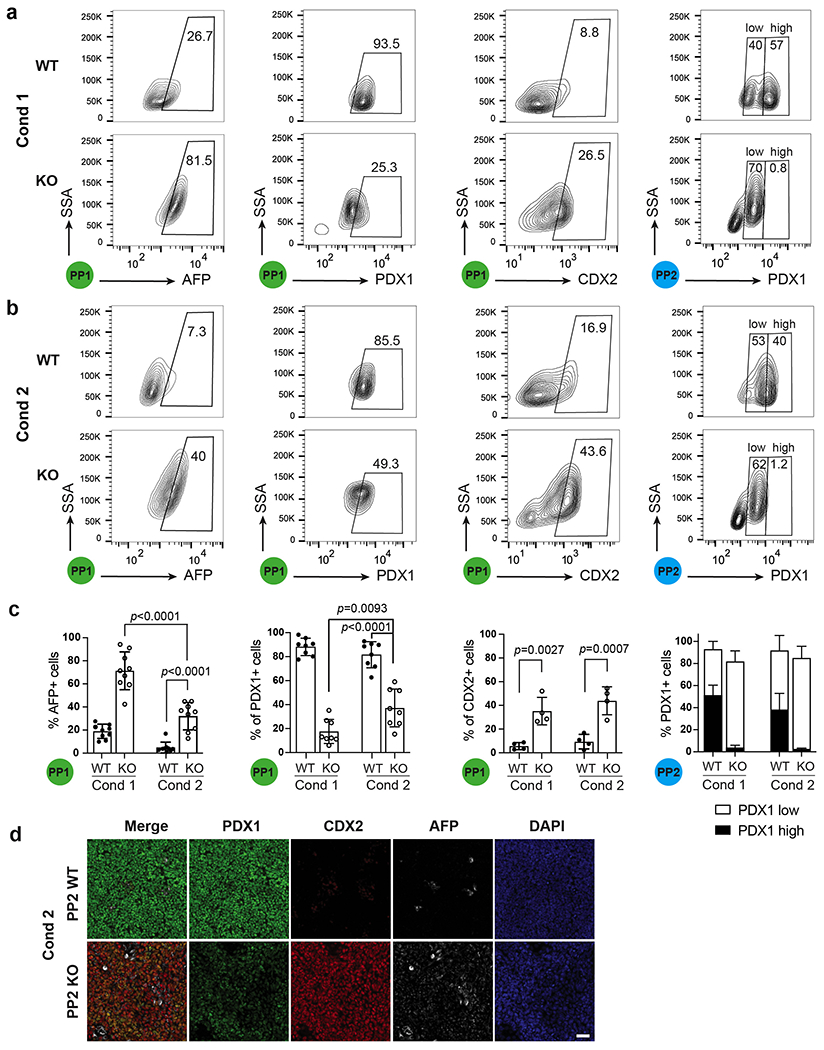
a,b, Flow cytometry analysis of AFP, PDX1 and CDX2 expression at the PP1 and PP2 stage WT/KO cells using differentiation Condition 1 (a) and Condition 2 (b).
c, Quantification of flow cytometry analysis of AFP+, PDX1+ and CDX2+ cells at the PP1 and PP2 stages in both conditions. Each symbol represents one independent experiment (n = 8 independent experiments, except for CDX2 staining, where n = 4 independent experiments) and data are presented as mean ± SD. Statistical analysis was performed using two-way ANOVA followed by multiple comparisons with Tukey correction.
d, Immunostaining images for PDX1, AFP and CDX2 expression at the PP2 stage WT/KO cells using differentiation Condition 2. Images shown represent three independent experiments. Scale bar, 50 μm.
Extended Data Fig. 4. Investigation of differentiation trajectories in WT and HHEX KO cells through scRNA-seq analysis.
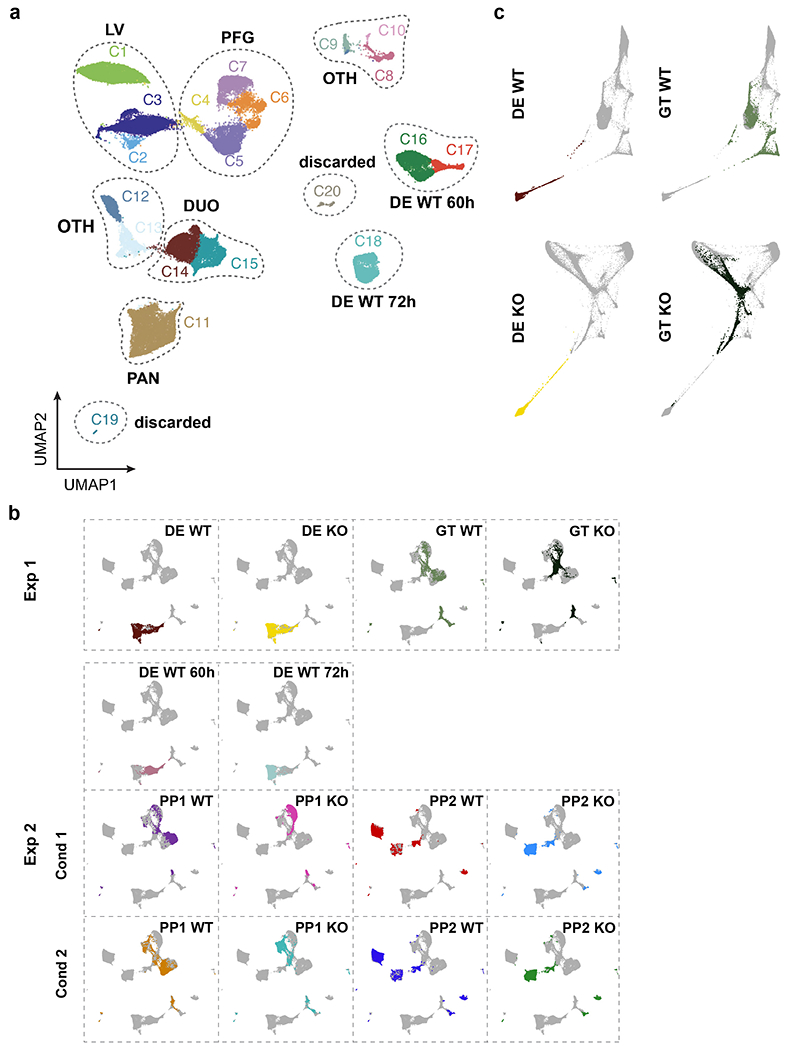
a, UMAP visualization of all Seurat clusters from experiment 2, shown with distinct colors. Clusters 1–15 were annotated as in Fig. 6b.
b, UMAP visualization of the integrated data from all samples of both experiments at the DE, GT, PP1, and PP2 stages. The overlapping embedding of DE cells shows that the batch effect was removed.
c, WT and KO lineages visualized by forced-directed layouts of the integrated data from the DE, GT, PP1, and PP2 stages. Cells at DE and GT stages are shown here. Data shown in a-c represent one independent experiments.
Extended Data Fig. 5: HHEX and FOXA2 ChIP-MS and ChIP-seq analysis.
a, Venn diagram of significantly enriched proteins at the GT and PP1 stages for HHEX ChIP-MS.
b, GREAT Gene Ontology showing the top 7 biological process associated with the FOXA2-down regions. The term of mouse phenotypes was selected based on the binom rank and cutoffs of region fold enrich >1.5 and observed regions>80.
c, IGV tracks (average of two independent experiments) to show chromatin accessibility and TFs binding activities at the PDX1 (left panel) and SOX9 (right panel) loci in WT and KO cells. Tracks are generated from two independent experiments. Scale bars was indicated. The regions showed significant decreasing of FOXA2 binding upon HHEX deletion were indicated.
d, MA plot of significantly increased and decreased FOXA2 binding sites (blue color) at the PP2 stage upon HHEX deletion. The number of significantly increased and decreased FOXA2 binding sites is indicated.
e, TF motifs enriched in the differential FOXA2 binding regions upon HHEX deletion. Significantly increased/decreased FOXA2 binding peaks were compared with the total atlas to examine the TF motif enrichments using the one-sided KS test. The KS test effect size is shown on the y axis, and the proportion of peaks associated with the TF motif is plotted on the x axis. The size of each circle represents the odds ratio, which was defined as the frequency of the TF in an opened or closed group divided by its frequency in the entire atlas. TF motifs with a KS test effect size ≥ 0.1 (indicated by the dashed lines) and odds ratio ≥ 1.2 are shown.
f, Volcano plots of significantly enriched proteins (purple labeled) for FOXA2 ChIP-MS at the PP2 stage WT or KO cells. Dotted lines indicate the fold change and significance cutoffs. CDX2 (log2 FC = −2.01, p = 0.067) is also indicated. Data shown in a-e are from two independent experiments, and data shown in f represent three independent experiments.
Supplementary Material
Supplementary Table 1 MAGeCK analysis of the DE and PP1 screens. Sheet 1, results from MAGeCK analysis of the DE and PP1 screens. Data was analyzed with MAGeCK 0.5.9.4 default RRA parameters; Sheet 2, DE screen gRNA raw counts; Sheet 3, PP1 screen gRNA raw counts.
Supplementary Table 2 Results from transcriptome analysis. Sheet 1, all normalized counts; Sheet2, significant hits at the DE stage; Sheet3, significant hits at the GT stage; Sheet4, significant hits at the PP1 stage. A two-tailed Wald test was used for statistical significance and a Benjamini-Hochberg procedure was to correct for multiple hypothesis testing.
Supplementary Table 3 Results from chromatin accessibility analysis. Sheet 1, differential peak analysis in KO cells vs. WT at the GT and PP1 stage. Differential peaks were identified by DESeq2 using default parameters; Sheet 2-4, cluster I, II and III enriched peaks information. Differential peaks were identified by DESeq2 using default parameters, and adjusted p value cutoff is 0.01; Sheet 5-7, hypergeometric test analysis of cluster I, II and III. One-sided hypergeometric test was used to compare the enrichment of proportions of TF motifs for each cluster (foreground ratio) versus those for total atlas (background ratio). Multiple testing correction using Bonferroni. The horizontal axis shows the binomial Z-score, representing the number of standard deviations between the observed count of each cluster peaks containing a TF motif and the expected count based on the background ratio. Analysis is from two independent experiments. FG: foreground. BG: background.
Supplementary Table 4 Results from scRNA-seq analysis. Sheet 1, sample information; Sheet2, cell meta-data; Sheet3, differential expression analysis for the annotated PP1/PP2 group cells; Sheet 4, mapping data from mouse endoderm derived cells to human PP1/PP2 populations. Differential expression tests, comparing log-normalized gene expression values of each group to the rest, were performed by MAST, and p values were adjusted with Bonferroni multiple testing correction.
Supplementary Table 5 Results from HHEX and FOXA2 ChIP-MS analysis. Sheet1, HHEX ChIP-MS analysis. HHEX KO cells stained with HHEX antibody was used as negative control. Sheet2, FOXA2 ChIP-MS analysis on PP1 WT cells. Cells stained with IgG was used as negative control. Sheet3, FOXA2 ChIP-MS analysis on PP1 WT vs. KO cells. Cells stained with IgG was used as negative control. Sheet4, FOXA2 ChIP-MS analysis on PP2 WT vs. KO cells. Cells stained with IgG was used as negative control. Two-tails heteroscedastic t-test were used for all mass spec experiments. Sheet5-7, raw data of HHEX and FOXA2 ChIP-MS experiments.
Supplementary Table 6 Results from FOXA2 and HHEX ChIP-seq analysis. Sheet1, PP1 FOXA2 Atlas analysis; Sheet2, Identification of FOXA2 and HHEX co-bind ATAC peaks at the GT and PP1 stages; Sheet3, PP2 FOXA2 Atlas analysis. To show co-binding of FOXA2 and HHEX at ATAC-seq peaks, ChIP-seq signal was quantified on the same ATAC-seq-based atlas. Differential ChIP-seq analysis using DESeq2’s default parameters.
Supplementary Table 7 List of gRNA target sequences, and primer sequences for PCR genotyping and RT-qPCR.
Supplementary Table 8 List of antibodies.
Acknowledgments
We thank H. Lickert, K. Yang, S.S. Kim, J. Kazakov, and N. Zhang for assisting with additional experiments not included in the manuscript, Z. Bao, S. Chen, and L. Studer for insightful discussions, N. Verma and F.C. Pan for critical reading and editing of the manuscript, M. Ray and B. Jarvis for technical support. This study was supported in part by grants to D.H. from DoD PRMRP (W81XWH-20-1-0298), NIH (R01DK096239, U01HG012051), American Diabetes Association (1-19-IBS-125), and grants to C.V.W. from the NIH (UC4DK104211, UC4DK108120) and the Leona M. and Harry B. Helmsley Charitable Trust, grants to J.S.O. and D.H. from JDRF (3-SRA-2021-1060-S-B) and DoD PRMRP (W81XWH-20-1-0670), and the University of Wisconsin-Madison, Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. S.St. and S.Si. were supported by the Leukemia Research Foundation (Hollis Brownstein New Investigator Research Grant), AFAR (Sagol Network GerOmics award), Deerfield (Xseed award) and the NIH Office of the Director (1S10OD030286-01). This study was also supported by MSKCC Cancer Center Support Grant from NIH (P30CA008748), a postdoctoral fellowship from a NYSTEM training grant (DOH01-TRAIN3-2015-2016-00006 to D.Y.), NIH T32 Training Grants (T32HD060600 to G.D. and T32GM008539 to S.J.K. and B.P.R.), and a Frank Lappin Horsfall Jr Fellowship (to G.D.).
Footnotes
Competing interests
The authors declare no competing interests.
Reference
- 1.Zaret KS & Grompe M Generation and regeneration of cells of the liver and pancreas. Science 322, 1490–4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willnow D et al. Quantitative lineage analysis identifies a hepato-pancreato-biliary progenitor niche. Nature 597, 87–91 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Puri S, Folias AE & Hebrok M Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell 16, 18–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan S, Norgard RJ & Stanger BZ Cellular Plasticity in Cancer. Cancer Discov 9, 837–851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tata PR et al. Developmental History Provides a Roadmap for the Emergence of Tumor Plasticity. Dev Cell 44, 679–693 e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Z et al. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell 18, 755–768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X et al. Point mutations in the PDX1 transactivation domain impair human beta-cell development and function. Mol Metab 24, 80–97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonsson J, Carlsson L, Edlund T & Edlund H Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371, 606–609 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Offield MF et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983–995 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL & Habener JF Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 15, 106–10 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Carrasco M, Delgado I, Soria B, Martin F & Rojas A GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest 122, 3504–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi ZD et al. Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 20, 675–688 e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiyaboonchai A et al. GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic beta Cells. Stem Cell Reports 8, 589–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xuan S et al. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest 122, 3516–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao N et al. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22, 3435–48 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee K et al. FOXA2 Is Required for Enhancer Priming during Pancreatic Differentiation. Cell Rep 28, 382–393 e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geusz RJ et al. Sequence logic at enhancers governs a dual mechanism of endodermal organ fate induction by FOXA pioneer factors. Nat Commun 12, 6636 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Genga RMJ et al. Single-Cell RNA-Sequencing-Based CRISPRi Screening Resolves Molecular Drivers of Early Human Endoderm Development. Cell Rep 27, 708–718 e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CS, Friedman JR, Fulmer JT & Kaestner KH The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–7 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Watt AJ, Zhao R, Li J & Duncan SA Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev Biol 7, 37 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao R et al. GATA6 is essential for embryonic development of the liver but dispensable for early heart formation. Mol Cell Biol 25, 2622–31 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keng VW et al. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem Biophys Res Commun 276, 1155–61 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Hunter MP et al. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol 308, 355–367 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez Barbera JP et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127, 2433–45 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Bort R, Martinez-Barbera JP, Beddington RS & Zaret KS Hex homeobox gene-dependent tissue positioning is required for organogenesis of the ventral pancreas. Development 131, 797–806 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Zaret KS et al. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol 73, 119–26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezania A et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32, 1121–33 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Nostro MC et al. Efficient generation of NKX6–1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports 4, 591–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagliuca FW et al. Generation of functional human pancreatic beta cells in vitro. Cell 159, 428–39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russ HA et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J 34, 1759–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L & Millman JR Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat Biotechnol 38, 460–470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan FC & Wright C Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240, 530–65 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Zhu Z, Verma N, Gonzalez F, Shi ZD & Huangfu D A CRISPR/Cas-Mediated Selection-free Knockin Strategy in Human Embryonic Stem Cells. Stem Cell Reports 4, 1103–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez F et al. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 15, 215–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li QV et al. Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat Genet 51, 999–1010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dixon G et al. QSER1 protects DNA methylation valleys from de novo methylation. Science 372(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jennings RE et al. Laser Capture and Deep Sequencing Reveals the Transcriptomic Programmes Regulating the Onset of Pancreas and Liver Differentiation in Human Embryos. Stem Cell Reports 9, 1387–1394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y et al. A single-cell transcriptome atlas of human early embryogenesis. bioRxiv, 2021.11.30.470583 (2021). [Google Scholar]
- 40.Zhang J, McKenna LB, Bogue CW & Kaestner KH The diabetes gene Hhex maintains delta-cell differentiation and islet function. Genes Dev 28, 829–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugiyama T, Rodriguez RT, McLean GW & Kim SK Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci U S A 104, 175–80 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oshima Y et al. Isolation of mouse pancreatic ductal progenitor cells expressing CD133 and c-Met by flow cytometric cell sorting. Gastroenterology 132, 720–32 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Li LC et al. Single-cell transcriptomic analyses reveal distinct dorsal/ventral pancreatic programs. EMBO Rep 19(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Odom DT et al. Control of Pancreas and Liver Gene Expression by HNF Transcription Factors. Science 303, 1378–1381 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossi JM, Dunn NR, Hogan BL & Zaret KS Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev 15, 1998–2009 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin D et al. Bmp and Fgf signaling are essential for liver specification in zebrafish. Development 134, 2041–50 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Ang LT et al. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep 22, 2190–2205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouon-Evans V et al. BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat Biotechnol 24, 1402–11 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Han L et al. Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nat Commun 11, 4158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Setty M et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol 37, 451–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li QV, Rosen BP & Huangfu D Decoding pluripotency: Genetic screens to interrogate the acquisition, maintenance, and exit of pluripotency. Wiley Interdiscip Rev Syst Biol Med 12, e1464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yilmaz A, Braverman-Gross C, Bialer-Tsypin A, Peretz M & Benvenisty N Mapping Gene Circuits Essential for Germ Layer Differentiation via Loss-of-Function Screens in Haploid Human Embryonic Stem Cells. Cell Stem Cell 27, 679–691 e6 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Naxerova K et al. Integrated loss- and gain-of-function screens define a core network governing human embryonic stem cell behavior. Genes Dev 35, 1527–1547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai Y, Yi J, Ma Y & Fu D Meta-analysis of the effect of HHEX gene polymorphism on the risk of type 2 diabetes. Mutagenesis 26, 309–14 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Bort R, Signore M, Tremblay K, Martinez Barbera JP & Zaret KS Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev Biol 290, 44–56 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Xu CR et al. Chromatin “prepattern” and histone modifiers in a fate choice for liver and pancreas. Science 332, 963–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trizzino M et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci Adv 7(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shalom-Feuerstein R et al. DeltaNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ 18, 887–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Markov GJ et al. AP-1 is a temporally regulated dual gatekeeper of reprogramming to pluripotency. Proc Natl Acad Sci U S A 118(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mall M et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orkin SH & Zon LI Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–44 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Babicki S et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kowalczyk MS et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res 25, 1860–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finak G et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Dijk D et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729 e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jacomy M, Venturini T, Heymann S & Bastian M ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One 9, e98679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 1–21 29 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Veeken J et al. Memory of Inflammation in Regulatory T Cells. Cell 166, 977–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li QH, Brown JB, Huang HY & Bickel PJ Measuring Reproducibility of High-Throughput Experiments. Annals of Applied Statistics 5, 1752–1779 (2011). [Google Scholar]
- 83.Liu T Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150, 81–95 (2014). [DOI] [PubMed] [Google Scholar]
- 84.Aguilan JT, Kulej K & Sidoli S Guide for protein fold change and p-value calculation for non-experts in proteomics. Mol Omics 16, 573–582 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 MAGeCK analysis of the DE and PP1 screens. Sheet 1, results from MAGeCK analysis of the DE and PP1 screens. Data was analyzed with MAGeCK 0.5.9.4 default RRA parameters; Sheet 2, DE screen gRNA raw counts; Sheet 3, PP1 screen gRNA raw counts.
Supplementary Table 2 Results from transcriptome analysis. Sheet 1, all normalized counts; Sheet2, significant hits at the DE stage; Sheet3, significant hits at the GT stage; Sheet4, significant hits at the PP1 stage. A two-tailed Wald test was used for statistical significance and a Benjamini-Hochberg procedure was to correct for multiple hypothesis testing.
Supplementary Table 3 Results from chromatin accessibility analysis. Sheet 1, differential peak analysis in KO cells vs. WT at the GT and PP1 stage. Differential peaks were identified by DESeq2 using default parameters; Sheet 2-4, cluster I, II and III enriched peaks information. Differential peaks were identified by DESeq2 using default parameters, and adjusted p value cutoff is 0.01; Sheet 5-7, hypergeometric test analysis of cluster I, II and III. One-sided hypergeometric test was used to compare the enrichment of proportions of TF motifs for each cluster (foreground ratio) versus those for total atlas (background ratio). Multiple testing correction using Bonferroni. The horizontal axis shows the binomial Z-score, representing the number of standard deviations between the observed count of each cluster peaks containing a TF motif and the expected count based on the background ratio. Analysis is from two independent experiments. FG: foreground. BG: background.
Supplementary Table 4 Results from scRNA-seq analysis. Sheet 1, sample information; Sheet2, cell meta-data; Sheet3, differential expression analysis for the annotated PP1/PP2 group cells; Sheet 4, mapping data from mouse endoderm derived cells to human PP1/PP2 populations. Differential expression tests, comparing log-normalized gene expression values of each group to the rest, were performed by MAST, and p values were adjusted with Bonferroni multiple testing correction.
Supplementary Table 5 Results from HHEX and FOXA2 ChIP-MS analysis. Sheet1, HHEX ChIP-MS analysis. HHEX KO cells stained with HHEX antibody was used as negative control. Sheet2, FOXA2 ChIP-MS analysis on PP1 WT cells. Cells stained with IgG was used as negative control. Sheet3, FOXA2 ChIP-MS analysis on PP1 WT vs. KO cells. Cells stained with IgG was used as negative control. Sheet4, FOXA2 ChIP-MS analysis on PP2 WT vs. KO cells. Cells stained with IgG was used as negative control. Two-tails heteroscedastic t-test were used for all mass spec experiments. Sheet5-7, raw data of HHEX and FOXA2 ChIP-MS experiments.
Supplementary Table 6 Results from FOXA2 and HHEX ChIP-seq analysis. Sheet1, PP1 FOXA2 Atlas analysis; Sheet2, Identification of FOXA2 and HHEX co-bind ATAC peaks at the GT and PP1 stages; Sheet3, PP2 FOXA2 Atlas analysis. To show co-binding of FOXA2 and HHEX at ATAC-seq peaks, ChIP-seq signal was quantified on the same ATAC-seq-based atlas. Differential ChIP-seq analysis using DESeq2’s default parameters.
Supplementary Table 7 List of gRNA target sequences, and primer sequences for PCR genotyping and RT-qPCR.
Supplementary Table 8 List of antibodies.
Data Availability Statement
Sequencing data is available at the Gene Expression Omnibus (GEO) under the accession code of GSE181480. Previously published data that were re-analyzed here are available at GEO under the accession codes of GSE136689 and GSE86225. Source data are provided with this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.