

Summary
Symbiosis is one of the most important evolutionary processes shaping biodiversity on Earth. Symbiotic associations often bring together organisms from different domains of life, which can provide an unparalleled route to evolutionary innovation1–4. The phylum Apicomplexa encompasses 6000 ubiquitous animal parasites, however, species in the recently described apicomplexan family, Nephromycidae, are reportedly non-virulent5,6. Members of the genus Nephromyces live within a specialized organ of tunicates, called the renal sac, in which they use concentrated uric acid as a primary nitrogen source7,8. Here we report genomic and transcriptomic data from the diverse genus Nephromyces, as well as the three bacterial symbionts that live within this species complex. We show that the diversity of Nephromyces is unexpectedly high within each renal sac, with as many as 20 different species inhabiting renal sacs in wild populations. The many species of Nephromyces can host three different types of bacterial endosymbionts, however, FISH microscopy allowed us to demonstrate that each individual Nephromyces cell only hosts a single bacterial type. Through the reconstruction and analyses of the endosymbiont bacterial genomes, we infer that each bacterial type supplies its host with different metabolites. No individual species of Nephromyces, in combination with its endosymbiont, can produce a complete set of essential amino acids and culture experiments demonstrate that individual Nephromyces species cannot form a viable infection. Therefore, we hypothesize that Nephromyces spp. depend on co-infection with congeners containing different bacterial symbionts in order to exchange metabolites to meet their needs.
eTOC:
Paight et al. demonstrate that members of the apicomplexan genus Nephromyces can host three different types of highly reduced bacterial endosymbionts, however none produce all necessary metabolites. Therefore, metabolite exchange between congeners harboring different endosymbionts likely explains universal multispecies Nephromyces infections.
Results and Discussion
The exploitation of extreme or low resource environments is often accomplished via symbioses1–4. One such extreme environment is the renal sac of the marine invertebrate genus Molgula. These marine tunicates (sea squirts) produce a liquid-filled organ, which contains high enough concentrations of uric and oxalic acids to form crystal structures analogous to human kidney stones7. Surprisingly, within this renal sac environment live multiple apicomplexan species of Nephromyces (Figure 1), with vertically transmitted bacterial endosymbionts living inside them9. Fluorescence in situ hybridization (FISH) of wild Molgula manhattensis tunicates revealed persistent infections of alphaproteobacterial, Bacteroides (Figure 1) and betaproteobacteria (Figure S1). Unlike the ~6000 described species of “core apicomplexans” (Hematozoa, Coccidia, Marosporida) in which they are nested5, Nephromyces spp. do not have an intracellular stage and are not predicted to be parasitic6. Instead, they maintain a likely commensal relationship with molgulid tunicates, using a combination of retained metabolic pathways and their bacterial endosymbionts8.
Figure 1: A multi-layered endosymbiosis between a tunicate, apicomplexan and bacterial endosymbionts.
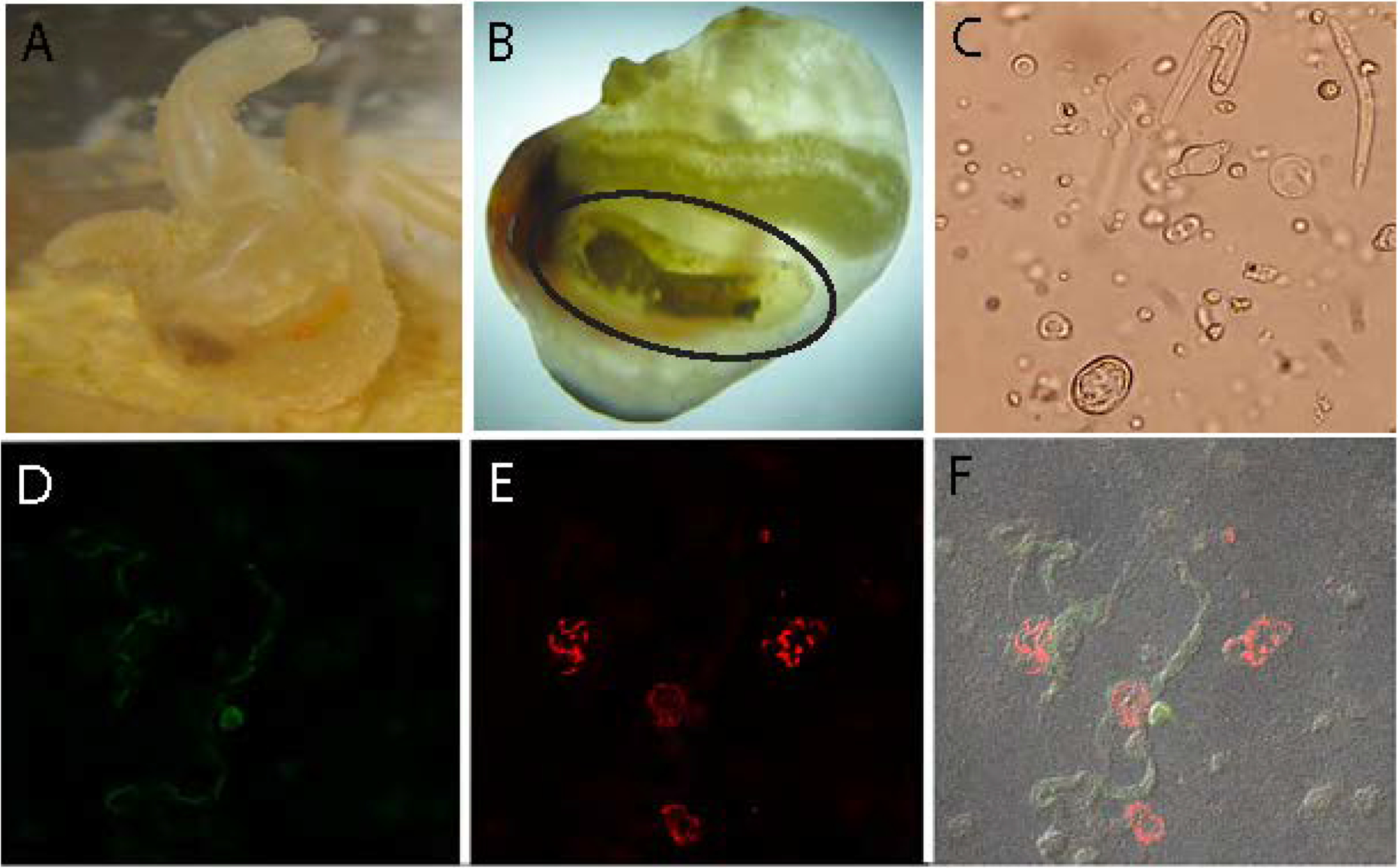
A) Laboratory cultured Molgula manhattensis examined in this study. B) Molgula manhattensis with tunic removed to show renal sac (circled), where Nephromyces spp. spend their entire life cycle. The brown substance inside the renals sac are concretions of crystallized uric acid and calcium oxalate. C) Light microscopy photo of several Nephromyces life stages. Bacteriodes (D) and alphaprotebacterial (E) endosymbionts labeled with 16S rRNA class specific probes, using fluorescence in situ hybridization. F) The composite image of (D) and (E) showing these endosymbionts localized to different Nephromyces cells. Additional images can be seen in Figure S1.
A single species of Nephromyces was described with a complex lifecycle and bacterial endosymbionts9. Based on this information, high-quality RNA was extracted from the contents of a single renal sac, with the goal of clarifying the metabolic contributions of Nephromyces and its symbionts. Our data revealed that the renal sac contained ~6 species of Nephromyces and associated endosymbionts, resulting in 195,694 transcripts from the host Molgula manhattensis, different species of Nephromyces, and bacteria. Whereas the multispecies co-infection was surprising, the genome-scale data provided the impetuous to investigate whether this is a critical feature of Nephromyces biology (see below). After binning by organism, 60,223 transcripts were attributed to Nephromyces, and 6,589 were attributed to the bacterial endosymbionts. The large number of transcripts attributed to Nephromyces was due to multiple species infecting a single host (estimated 69% gene duplication). Orthologous sequences were collapsed by protein sequence identity with CD-HIT, reducing the number of assembled transcripts to estimate the number of genes for a single Nephromyces species; 26,938 transcripts at 90%, 23,850 at 80%, 21,762 at 15270%, 19,540 at 60%, 16,668 at 50%. When accounting for the multi-species community, we estimate that there are between 8,000 and 12,000 unique transcripts produced by an individual Nephromyces. The KEGG database predicted functionality for 6,987 transcripts and BUSCO estimated 81.8% complete transcripts and 6.3% partial.
To better interpret the complexity of the transcriptome dataset, we used short and long read sequencing technologies to produce a pan-genome. Over 150 tunicates were dissected to amass sufficient high molecular weight DNA, which was pooled for Illumina and PacBio sequencing. We estimate the resulting data captures ~20 Nephromyces species and their endosymbionts. Due to the multispecies nature of the system, we cannot rule out that some of our assembled contigs and transcripts are potentially chimeric. However, even with these limitations we are able to characterize the metabolic contribution of Nephromyces. The Nephromyces pan-genome assembled into 2,156 contigs, has an N50 of 50,747 bp, with a 36% GC content. The sum genome size is 60,134,441 bp, and BUSCO estimated completeness of 51.1% and duplication of 13.5%.
There are 12,391 predicted protein-coding genes after dereplication in the Nephromyces dataset, with 5,482 annotated open reading frames (ORFs) and 6,909 without functional annotations. Of these putative protein-coding genes, 11,664 were recovered in the transcriptome and 727 are unique to the genome, demonstrating active transcription of nearly all of the detected ORFs. The fragmentation of the genome makes determining the true absence of genes impossible but the combination of the genomic data, with the more complete transcriptome, allows a better prediction of the metabolic capabilities and contributions of Nephromyces to the system. The genome also provides support for the Nephromyces transcript bins that were formed based on phylogenetic analysis.
The genomes of three bacterial endosymbionts were retrieved from the metagenome of Nephromyces. These genomes were small, gene dense, and AT-rich. The Bacteroidetes (NBe) genome found within Nephromyces is circular, 494,352 nucleotides long and extremely AT rich (22% GC content; Figures S2 & S3). The genome encodes many amino acid pathways, including two that are partial but complement partial pathways in Nephromyces (Figure 2). The NBe genome has lost almost all complete pathways for carbon and lipid metabolism (Figure 3). The betaproteobacterial (Nβe) genome also circularized, is 866,396 bp long, with 30% GC content (Figures S2 & S4). Like the NBe genome, Nβe encodes partial amino acid pathways that perfectly complement losses to these pathways in Nephromyces (Figure 2). The presence of at least two closely related alphaproteobacterial (Nαe) genomes, with high AT bias (25% GC content) and regions of low complexity, limited our ability to assemble these genomes completely. The fragmented Nαe pan genome assembled into 11 mosaic contigs, ranging in size from 13 kb to 312 kb, for a minimum length of 995,540 and an average of 90,503 (Figure S2). Genomic re-arrangements between the different Nαe species are likely preventing independent assemblies, but the Nαe have substantially reduced coding capacity (Figures 2 & 3). The Microbial Gene Atlas (MiGA) predicted this assembly is 85.6% complete with 0% contamination. The KEGG Automatic Annotation Server (KAAS) annotations for the endosymbint genomes are described in the STAR methods.
Figure 2: Reconstruction of the key metabolic exchanges within the renal sac of Molgula manhattensis.
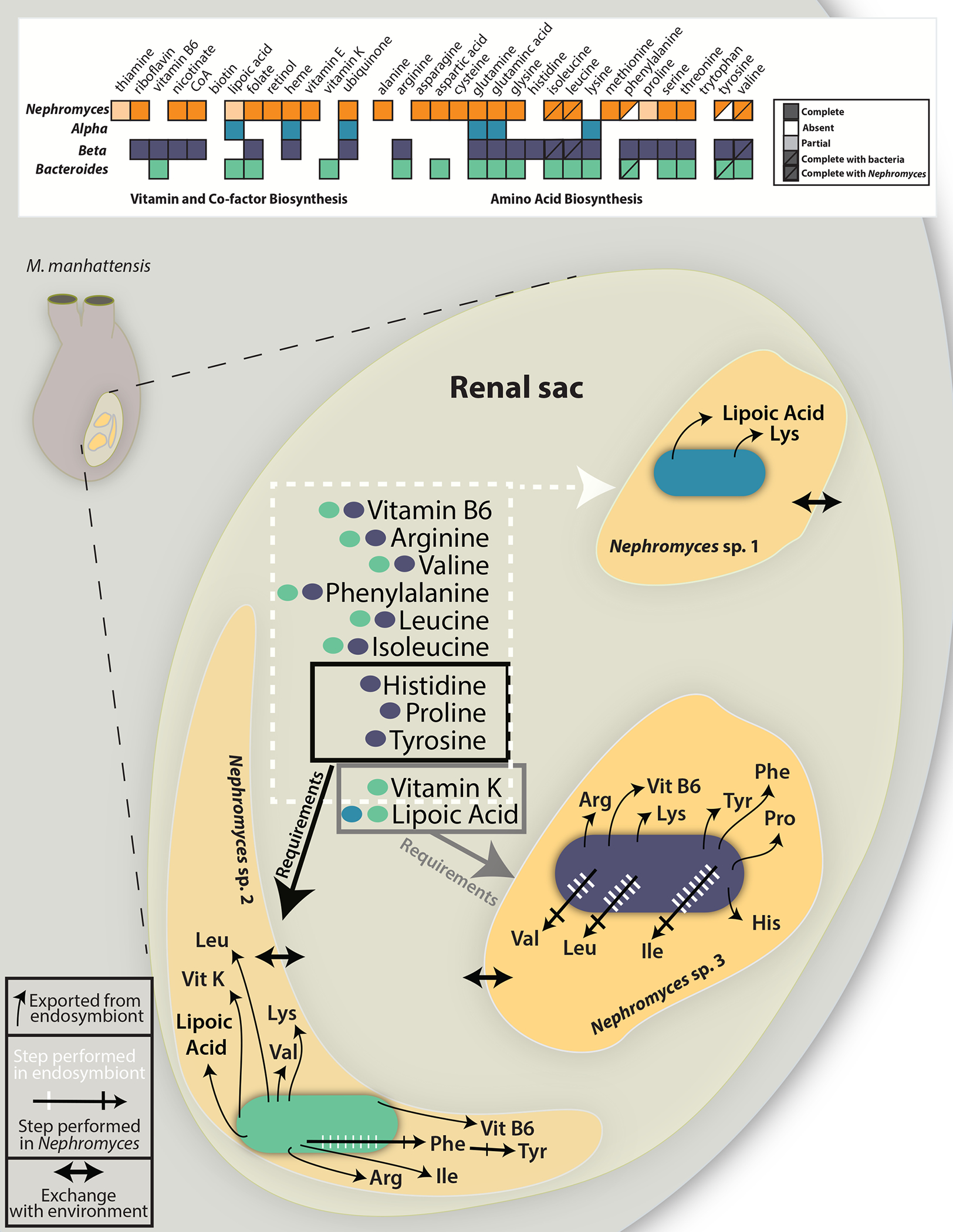
A) Graphical representation of amino acid, vitamin and co-factor biosenthesis pathways in Nephromyces and the three types of bacterial endosymbionts. Filled boxes represent complete pathways, empty boxes indicate no genes in the pathway are encoded, light shading represent partially complete pathways, and half-filled boxes indicate pathways that require genes encoded by Nephromyces and a bacterial endosymbiont. B) The renal sac of M. manhattensis (inset) with each bacterial type represented within a different Nephromyces. Black arrows from bacteria, into each Nephromyces, represent metabolites each endosymbiont provides its host. In shared pathways, hashed arrows show the number of steps and location where the biosynthetic reactions occur (white for endosymbiont, black for Nephromyces), predicted from the genomes of each organism. In all cases, Nephromyces only encodes genes for the final biosynthetic reaction in the amino acid pathways. Arrows between the Nephromyces and the lumen of the renal sac represent the predicted exchange of metabolites. Boxes around metabolites predicted to exist in the renal sac show the requirements of the different Nephromyces with endosymbionts. Genome maps of the endosymbionts can be found in Figure S2.
Figure 3: Carbon, lipid and nucleotide metabolism.
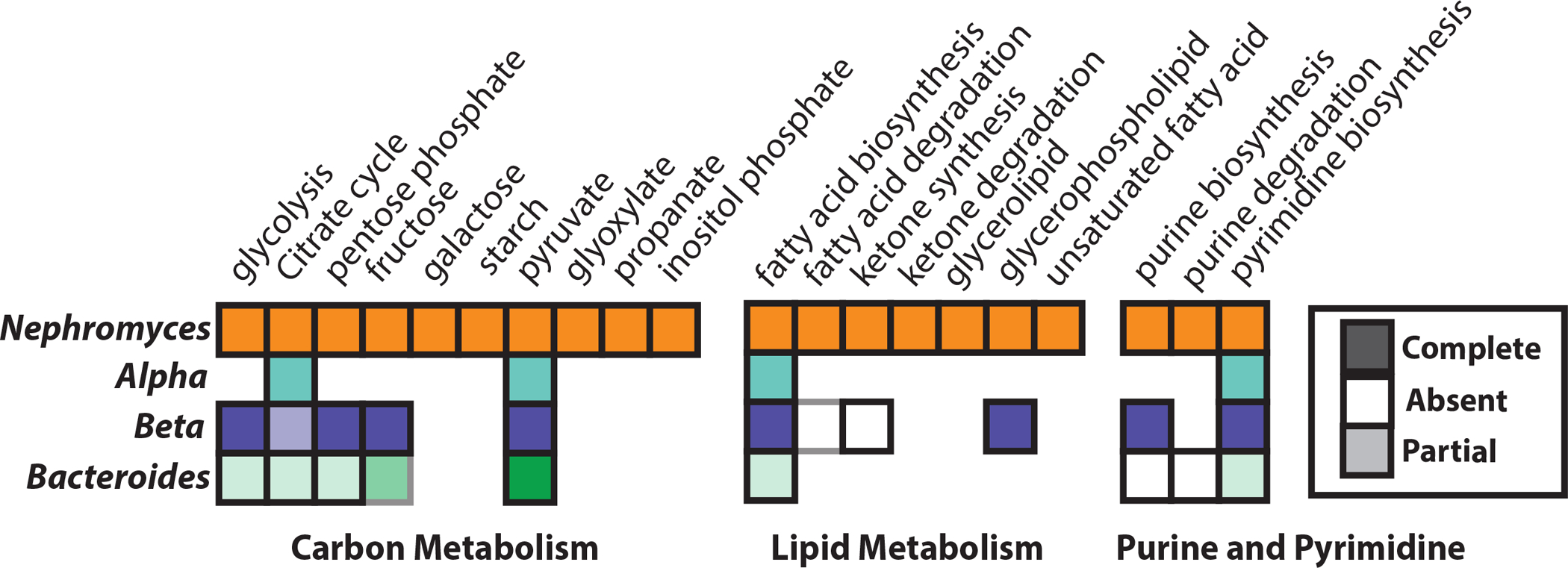
Predicted carbon, lipid and nucleotide metabolism of Nephromyces and hosted endosymbionts. All three endosymbiont types have greatly reduced metabolic capabilities. Additional coding capacity can be seen in Figure S2.
Metabolic Pathways Essential for the partnership between Nephromyces and Endosymbionts
Amino Acids
The genome data revealed the metabolic interplay between Nephromyces and its bacterial partners. The genomes of the three bacterial endosymbiont types are highly reduced, but encode several key functions for the partnership. Additionally, the bacteria complete pathways partially encoded by Nephromyces, which is predicted to synthesize only 10 amino acids on its own (Figure 2). Nephromyces has partial pathways for the synthesis of phenylalanine from phenylpyruvate and can synthesize tyrosine from phenylalanine. These pathways are complemented in two of the bacterial endosymbionts (Figure 2).
Only Nβe is capable of synthesizing a nearly full complement of amino acids (with the exception of tryptophan) with Nephromyces. Nαe is only capable of synthesizing the amino acids glutamine, glutamic acid, and lysine. Of these three amino acids, lysine is the only amino acid that Nephromyces is unable to synthesize (Figure 2). Nbe is capable of synthesizing 11 amino acids and is able to synthesis phenylpyruvate, but lacks the ability to synthesize phenylalanine (Figure 2). Genes encoding biosynthesis of arginine, isoleucine, leucine, lysine, and valine are not present in the Nephromyces transcriptome and may represent the Nbe contribution to this community. Nβe also encodes the genes for 11 amino acids (Figure 2) and encodes all the genes for synthesis of isoleucine, leucine, and valine, but lacks the last gene in the pathway branched-chain amino acid aminotransferase. However, Nephromyces encodes branched-chain amino acid aminotransferase and may be able to complete isoleucine, leucine, and valine by adding the final amine group (Figure 2).
Nucleotides
Both Nαe and Nbe endosymbionts lack all genes in the inosine monophosphate (IMP) biosynthesis pathway, as well as the ability to convert IMP to guanine or adenine (Figure 3). Nephromyces is unique among core apicomplexans because it contains a complete pathway for the biosynthesis of IMP, suggesting it supplies guanine or adenine to Nαe and Nbe. De novo biosynthesis of purines has been lost in all other sequenced apicomplexans, which scavenge precursor molecules to IMP and convert them into IMP. Purine biosynthesis in Nβe is complete, encoding adenylosuccinate lyase (purB) and IMP cyclohydrolase (purH) (Figure 3). Additionally, Nβe is able to synthesize IMP from 5-Phosphoribosyl diphosphate (PRPP) with the histidine synthesis pathway, and also encodes the genes required to convert IMP to both guanine and adenine.
Purine degradation and purine synthesis could be critical to the unusual epidemiology of Nephromyces. Tunicates lack the enzymatic ability to degrade purines past uric acid and none of the three endosymbionts encode any genes involved in purine degradation. In addition to the biosynthesis of purines, Nephromyces are the only known apicomplexans capable of purine degradation (Figure 3). By obtaining the bulk of the required carbon, nitrogen, and energy from a tunicate metabolic waste product, Nephromyces is able to limit impact on its host, while still reaching high cellular densities. De novo synthesis of purines means Nephromyces is not dependent on the host for IMP, also reducing virulence. The retention of purine degradation and purine biosynthesis may have been the critical factors, which allowed Nephromyces to leave the intracellular environment and colonize the renal sac. Both Nαe and Nβe encode the necessary genes for pyrimidine biosynthesis (Figure 3). However, because Nbe is missing several genes involved in pyrimidine biosynthesis it seems likely that Nbe is dependent on Nephromyces for both purines and pyrimidines.
Other small molecules
Nephromyces has the predicted biosynthetic capabilities to produce riboflavin, acetyl CoA, nicotinate, folate, retinol, vitamin E, heme, and ubiquinone. It also encodes a copy of lipoyl synthase (lipA) but lacks lipoyl (octanoyl) transferase (lipB) in the lipoic acid synthesis pathway. Lipoic acid is an essential cofactor involved in the citric acid cycle, pyruvate dehydrogenase complex, 2-oxoglutarate dehydrogenase complex, branched-chain oxoacid dehydrogenase, and acetoin dehydrogenase10–13. The Nαe genome only encodes genes for the biosynthesis of three vitamins and co-factors; heme, ubiquinone, and Lipoic acid (Figures 2 & 3). As lipoic acid can only be partially synthesized in Nephromyces, it may be an important product produced by the alphaproteobacteria. In contrast Nbe is capable of biosynthesizing vitamin B6, lipoic acid, folate and vitamin K (Figures 2 & 3). The Nβe genome encodes most cofactors including riboflavin, vitamin B6, nicotinate, coenzyme-A, folate, heme, and ubiquinone (Figure 3).
Nephromyces Diversity in wild populations
To test our hypothesis that coinfection is necessary for the long term survival of Nephromyces, wild populations of two tunicate species M. occidentalis (n= 54) and M. manhattensis (n= 50), were surveyed for infection. A total of 335 distinct Nephromyces amplicon sequence variants (ASVs) from amplified mitochondrial cytochrome oxidase I (COI) were recovered from samples collected from M. manhattensis, and 182 from M. occidentalis (Figure 4). Only one wild tunicate out of 104 we examined contained just a single Nephromyces species. The average population was three to five Nephromyces species in M. manhattensis and seven to eight in the larger M. occidentalis (Figure 4). Whereas single genus, multispecies, apicomplexan infections are relatively common10–13 the extreme diversity and universal multispecies infections found in Molgula tunicates is striking. All three bacterial endosymbionts were found in 40% of M. manhattensis and Molgula occidentalis samples, and at least two types were in 90% of M. manhattensis, and 80% of Molgula occidentalis (Figure 4).
Figure 4: Biodiversity data across Nephromyces and their bacterial endosymbionts.
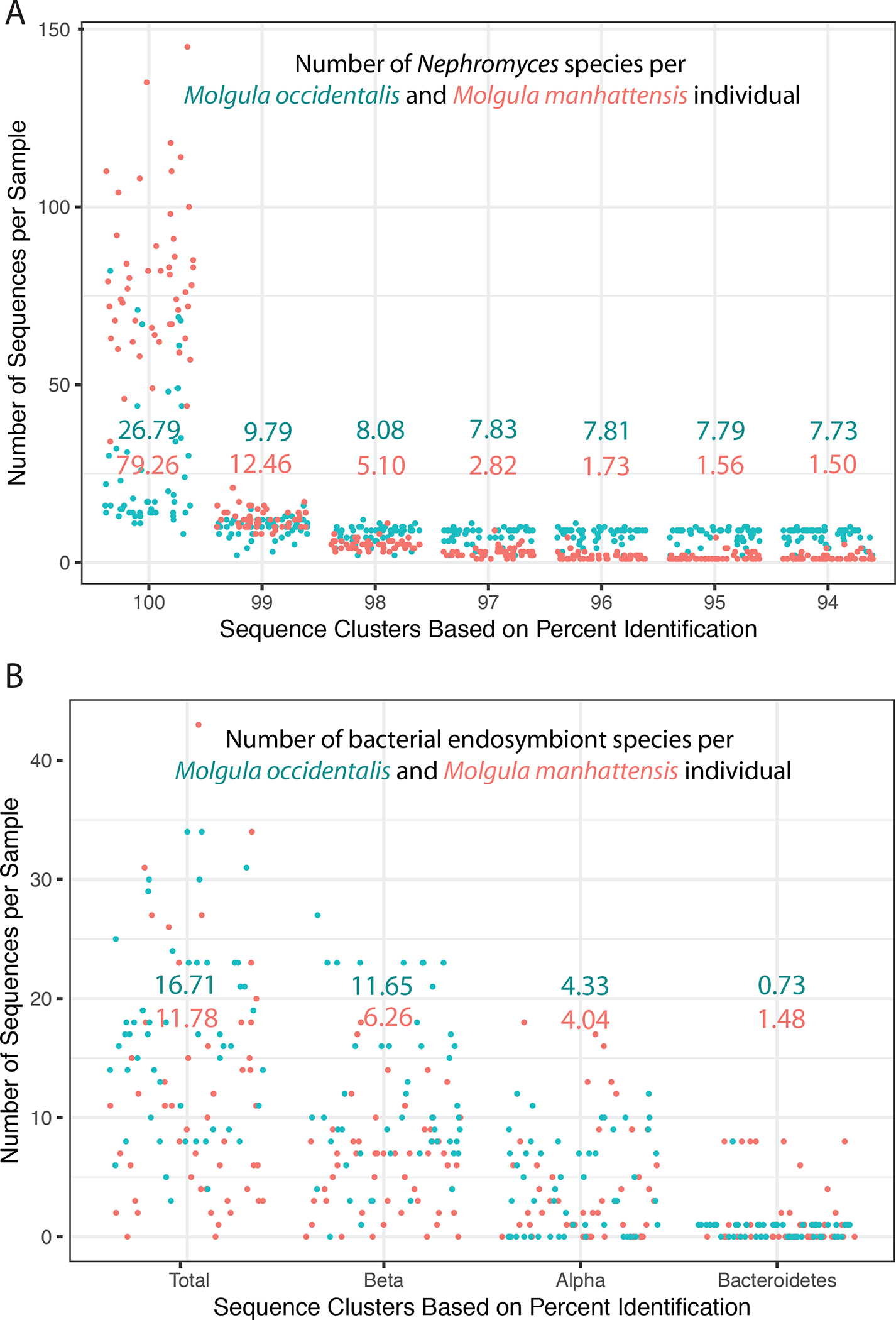
A) Number of Nephromyces ASVs per tunicate individual (blue = M. occidentalis, red = M. manhattensis). The x-axis shows the different percent identity levels that the ASVs were clustered at. The mean at every clustering level for each host species is shown. B) The number and type of bacterial endosymbionts found from each tunicate host. Bacterial ASV’s were not clustered by percent identity. The mean for each host is shown. Phylogenetic placement of Nβe and Nbe is shown in Figures S3 & S4.
The most common endosymbiont type in the wild populations we sampled was Nβe (Figure 4), which has the most complete metabolism among the three types (Figures 2 & 3). Almost counter-intuitively, the second most commonly found endosymbiont is Nαe, which is the most reduced endosymbiont. However, Nαe is the oldest of the endosymbionts14 and produces lipoic acid, which is not encoded in Nβe and we hypothesize is an essential cofactor. Nephromyces harboring Nβe and Nbe in the same renal sac would only be missing vitamin K among all of the metabolites the endosymbionts produce but Nephromyces cannot (Figure 2).
Metabolic mosaicism across endosymbiont systems
There are several stunning parallels between the Nephromyces system and the well-characterized relationship between sap-feeding insects and their bacterial endosymbionts. We hypothesize that the relationship between Nephromyces and the different endosymbionts is collaborative, as defined as when “hosts supply endosymbionts with metabolic precursors that endosymbionts metabolically transform into host-required dietary components” in4 Like the pea aphid15,16 and the citrus mealbug3,17, branched chain amino acids (Valine, Leucine and Isoleucine) are synthesized cooperatively between Nephromyces and Nβe (Figure 2) due to the loss of ilvE in Nβe. Additionally, phenylalanine is widely collaboratively synthesized between insects and endosymbionts18as well as between Nephromyces and Nbe (Figure 2). Whereas synthesis of vitamin B5 is a critical component on many insect / bacterial symbioses, Nephromyces requires vitamins B6 and K, as well as lipoic acid from its endosymbionts.
Unlike insects, which can host multiple bacterial symbionts to complete their metabolic needs19, we do not have direct evidence that Nephromyces hosts multiple endosymbiont types (Figure 1, Figure S1). As a unicellular organism, it is unclear whether the lack of specialized cells to sequester and separate the bacteria9 is responsible for individuals only hosting one bacterial type, or whether the cost of two endosymbionts outweighs the benefit in this particular system. The likelihood of a single Nephromyces species colonizing a tunicate appears low, as wild tunicates are either infected by multiple species of Nephromyces or have not yet been infected (Figure 4). In experiments with laboratory-raised tunicates, limiting the number of Nephromyces species per host affected cell densities and transmission efficiency. Single-species Nephromyces inoculations in laboratory-raised tunicates grew poorly and were lost in subsequent generations of tunicates. Limited infections with two Nephromyces species were transmitted to other tunicates but contained an order of magnitude fewer cells than tunicates colonized by four or more species with undiluted renal fluid from wild tunicates.
Vertical transmission of bacterial endosymbionts relies on provisioning gametes (often the macrogamete) with at least one bacterial endosymbiont cell20. In systems where the bacterial endosymbiont is essential for survival of the host, as in phloem feeding insects, failure to provision gametes with a bacterial endosymbiont is lethal21. In contrast, our culturing experiments indicate that Nephromyces are able to survive and transmit to other tunicates in the short term, without bacterial endosymbionts, even if there are no other Nephromyces species present in the renal sac. Therefore, there may be a low abundance of the required metabolites that Nephromyces cannot biosynthesize present in the renal sac, or the infective merozoites can persist long enough to infect a second tunicate. If the presence of multiple Nephromyces species with multiple types of bacterial endosymbionts increases the availability of essential metabolites in the renal sac, then Nephromyces strains without bacterial endosymbionts could persist long term with comparable fitness to co-infecting Nephromyces strains with endosymbionts. This may explain why we have not observed any Nephromyces cell with two types of bacterial endosymbionts.
Even in some phloem feeding insects that host two endosymbionts, the endosymbionts are maintained in separate cells19. As single-celled organisms, Nephromyces appear to have evolved a parallel mechanism to host multiple endosymbionts, without individual cells maintaining more than one type. However, unlike the dual endosymbiont systems seen in insects, Nephromyces strains appear able to lose their bacterial endosymbiont and persist long enough to acquire a new bacterial endosymbiont. The Nαe found in Nephromyces and its parasitic sister taxon, Cardiosporidium cionae, are monophyletic, indicating that they have been maintained and vertically transmitted since the divergence of taxa14. In contrast, Nβe and Nbe have been acquired within the Nephromyces clade, indicating novel acquisition has occurred after the original endosymbiosis was established. Even if a individual Nephromyces did, at one time, have multiple types of bacterial endosymbionts, the problem of provisioning gametes with bacterial cells remains, and the fitness advantage of having both may not have been enough to prevent subsequent loss of one of the bacterial types.
One possible mechanism for the transport of molecules between individual Nephromyces may be clathrin-dependant endocytosis. An additional 25 genes related to endocytosis are predicted in Nephromyces compared with Plasmodium falciparum. Many of the additional genes found in Nephromyces are in the VPS and CHMP protein families, which form part of the ESCRT machinery and are important in the biogenesis of multivesicular bodies22. Multivesicular bodies transport ubiquitinated proteins to lysosomes for degradation. Other proteins not found in P. falciparum include a number of genes in the AP2 complex, which are accessory proteins in clathrin-mediated endocytosis23. The AP2 complex plays an important role in the regulation of the assembly of clathrin-coated vesicles24. Endocytosis may play a critical role in the exchange of metabolites between Nephromyces spp in the renal sac.
Taken together, these data show that Nephromyces spp. form complex species swarm communities inside the renal sacs of their hosts, where closely related conspecifics and their functionally distinct bacterial endosymbionts are in immediate proximity with each other. Based on the genomic data from the bacterial endosymbionts, no single bacterial endosymbiont is capable of producing all of the metabolites and precursor molecules that are predicted to be essential for Nephromyces. As of yet, we have no support for a mechanism of direct metabolite exchange between Nephromyces species, but propose that these metabolites may become available in the renal sac indirectly by leaky membranes, cell lysis25 or via an expanded repertoire of 25 additional endocytosis genes, compared with Plasmodium. This hypothesis does not preclude that these metabolites may also come from the tunicate host, but it is unlikely the expense of hosting an endosymbiont within Nephromyces individuals would be tolerated if these metabolites were available in sufficient quantities within the renal sac. Instead, these data suggest that the presence of multiple endosymbionts, with different types of bacterial endosymbionts, increases the availability of critical metabolites inside the renal sac. In this way, Nephromyces appear to have evolved a complex symbiosis with their host, sister species, and bacterial endosymbionts.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher Lane (clane@uri.edu).
Materials availability
Lab cultured tunicates are available upon request.
Data and Code Availability
All raw nucleotide data is available as an NCBI Bioproject (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA666913)
All R and bash code used in this project can be found on Github (https://github.com/cpaight/Nephromyces)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
A scientific collection permit was obtained from Rhode Island Department of Environmental Management. We generated genomes and transcriptomes, from several Nephromyces species, and their bacterial endosymbionts. Genetic material was collected from 250+ Molgula Manhattensis individuals collected from Greenwich Bay, RI (41.653N, −71.452W). Live tunicates were housed in 5-gallon buckets with an air stone for no longer than 36 hours. Additional DNA was collected for ampicon amplification of Nephromyces and bacterial endosymbionts from 50 Molgula Manhattenensis were collected from a single floating dock located in Greenwich Bay, RI (41.653N, −71.452W) and 54 Molgula occidentalis were collected from Alligator Harbor, FL (29.899N, −84.381W) by Gulf Specimens Marine Laboratories, Inc. (https://gulfspecimen.org/).
Laboratory cultures of uninfected Molgula manhattensis were started by collecting M. manhattensis tunicates from a dock in Greenwich Bay, Rhode Island (41.653N, −71.452W) during the summer of 2014. Gonads were dissected from sexually mature, M. manhattensis, mixed with sterile seawater and split between eight cell culture dishes. Plates were incubated at 18° C with a 24 hr dark cycle to limit growth of contaminants. Tunicates were fed by 100% water exchange with cultures of the diatoms from the National Center for Marine Algae and Microbiota, Isochrysis galbana (CCMP1323) and Chaetoceros sp. (CCMP209), three days a week.
Method details
DNA Extraction & Genome Assembly &Annotation
The renal sacs from eight lab grown M. manhattensis individuals were dissected and their renal fluid was pooled for DNA extraction, using a CTAB + proteinase K buffer and phenol / chloroform extraction. Following DNA quality control, an Illumina library was constructed with an Illumina MiSeq 2×250 v3 kit and sequenced at the URI Genomics and Sequencing Center on the Illumina MiSeq platform and with HiSeq 2500 2×125 HiSeq SBS v4 kit on the HiSeq platform at the University of Maryland Baltimore sequencing center, on three lanes.
One MiSeq run and three lanes of HiSeq, all from the same library, were trimmed using Trimmomatic v0.39 flags (SLIDINGWINDOW:4:5 TRAILING:5 MINLEN:36)26 then assembled using SPAdes v3.13.0 --careful27 assembler on the URI server BlueWaves.
Using the contents of 150 (done in batches of 10 then pooled) M. manhattensis renal sacs, the same DNA extraction protocol was performed as for Illumina sequencing. The resulting DNA was sequenced using three SMRT cells on the Pacific Biosciences platform at the University of Baltimore sequencing center.
Pacific Biosciences reads were error corrected using PBSuite v15.8.2428 on the Brown University server, Oscar. Reads were then assembled using Canu v1.6 (genomeSize=200, useGrid=false, corOutCoverage=10000, corMhapSensitivity=high)29. Contigs generated by Canu were combined with Illumina MiSeq/HiSeq short reads with ABySS v2.02 k=9630. Nephromyces contigs were identified by mapping Nephromyces transcriptome reads to the genomic assembly using Bowtie2 v1.2.2. Contigs with greater than 90x coverage assessed by bedtools31 were binned as Nephromyces additional screening of binned contigs was done using VizBin32. Contigs were additionally binned using CAT33, and those that were classified as apicomplexan were added to the existing assembly). Scaffolded contigs were annotated with Maker version 3.01.03. Using a training set of Nephromyces genes assembled from RNA data and Agustus ab initio predictions.
Bacterial Endosymbiont Genome Assembly and Annotation
Using the contigs from the ABySS assembly bacterial contigs were initially identified by hexamers using VizBin, transcriptomic reads that were identified as bacterial were mapped using Bowtie234. Bacterial contigs were separated based on a 90x coverage threshold with bedtools31. Binned bacterial contigs were preliminarily annotated with Prokka35. Resulting annotations were run through KEGG GhostKoala to assign and separate by taxonomy. Taxon separated contig bins were merged and scaffolded using PBJelly from the PBsuite v15.8.24 of tools36. Illumina MiSeq and HiSeq reads were remapped to resulting contigs to ensure accurate assembly using Bowtie2 (-p 20 --very-sensitive --al-conc, --no-unal). Final assembled bacterial genomes were re-annotated with Prokka using a genus-specific database.
RNA Extraction & Sequencing
Renal fluid was centrifuged at 8,000g for 4 minutes, liquid was removed and tissue was flash frozen in liquid nitrogen. RNA extraction buffer (Zymo Research LLC. Irvine, CA) was added to frozen samples of an individual M. manhattensis and ground with a pestle. Following grinding, the Zymo Quick-RNA kit (Zymo Research LLC. Irvine, CA) was used and the manufacturer’s protocol was followed. RNA was converted to cDNA with Illumina TruSeq RNA kit with polyA enrichment and sequenced at the School of Medicine Genome Resource Center, University of Maryland. One paired-end RNA library was run on one lane of the Illumina HiSeq platform.
RNA Assembly & Binning
Transcriptome data was assembled and proteins were predicted with Trinity/Trinotate pipeline v2.4.0 with the --normalize_reads flag set run on the server at Brown University Center for Computation and Visualization37. Protein sequences were predicted using Transdecoder with universal genetic code38. BLASTp v 2.8.1 was used to identify bacterial sequences from assembled transcripts against NCBI’s refseq database and binned. Orthofinder was run on the remaining Eukaryotic sequences with a custom database of alveolate and ascidian transcriptomes downloaded from VEuPathDB and Aniseed. Orthologous genes were binned using closest phylogeny in R.
Trimmed reads were mapped back to each of the three bins (Nephromyces, M. manhattensis, bacteria) with BBmap flags slow k=12 and then reassembled independently in Spades --rna27. The Nephromyces transcriptome from a single renal sac was composed of multiple co-infecting Nephromyces species and CDhit v4.6.8 was used to cluster transcripts based on percent identity39. Transcriptome completeness was assessed with Busco v3 against the Eukaryotic and bacterial reference data sets. Transcripts were annotated using Interproscan v71.040.
Amplicon Sequencing
Molgula Manhattenensis (n=50) and Molgula occidentalis (n=54) renal sacs were dissected and DNA extracted (same DNA extraction method as above). A section of COI and 16S rRNA were PCR amplified (COI primers GYGGWGTAGGWSCWGGWTGGA, GACTTCWGGATGWCCAAARAA) (16S primers GCCTACGGGNGGCWGCAG, GGACTACHVGGGTATCTAATCC). The addition of well specific adaptors, library preparation, and sequencing was done at the URI genome sequencing center on the Illumina MiSeq platform using the Illumina MiSeq reagent kit V2 2X250. Adaptors and primer sequences were removed with cutadapt v2.1041. Cleaned read sets were processed in R using dada2 with the pool=”pseudo”42. CO1 sequences were assigned taxonomy using blastx against NCBI’s refseq protein database. All CO1 sequences that did not return an apicomplexan as top hit were removed from the count table, taxonomy table, and sequence files. The 16S sequences were aligned with MAFFT43 to 16S rRNA sequences from the three known bacterial endosymbionts found in Nephromyces, an alphaproteobacteria, betaproteobacteria and Bacteroides.
Fluorescence in situ Hybridization (FISH) Microscopy
Three 16S targeted bacterial probes with attached fluorophores were used in each fluorescence in situ hybridization (FISH) reaction. The alpha and bacteroides were designed by Seah44 to be specific to the classes of bacterial endosymbionts present in Nephromyces. The Nbe probe from was replaced with a probe from45 based on better sequence complementary and probe exposure.
Renal sacks from wild Molgula manhattensis collected from Greenwich Bay, Rhode Island, USA, were dissected and emptied onto a positively charged slide. The sample was dried on the slide, and then subjected to a dehydration time series in sterile seawater: 50% ethanol (5 mins), 70% ethanol (8 mins), 95% ethanol (10 mins), and finally 100% ethanol (15 mins). A hybridization solution consisting of 0.9M NaCl, 0.02M Tris-Cl (pH 7.4), 0.01% SDS, 20% HiDi Formamide, and 5% dextran sulfate was mixed with 2pmol/μL of probe, and 200μL applied to the slide44. The slide was then incubated in a humidified dark chamber at 46C overnight. Two washes were conducted by draining the remaining hybridization buffer from the slide and applying 200μL of wash buffer (0.215M NaCl, 0.02M Tris-Cl (pH 7.4), 0.01% SDS) for 30 minutes, with one change of solution44. Samples were then mounted with Prolong Gold w/ DAPI, cured, and imaged using a Zeiss Axioimager M2 imaging system at the URI Genomics and Sequencing Center (URIGSC) and at the NRI-MCDB Microscopy Facility at the University of California Santa Barbara.
Molgula manhattensis laboratory innoculation with Nephromyces
After ten days of tunicate growth, a single wild collected M. manhattensis was dissected, and the renal fluid was extracted with a syringe. From the raw fluid, 2μL was added to the seawater in two petri dishes of growing tunicates. Two microliters of a dilution of 1/100 renal fluid was added to two of the tunicate petri dishes, and a single oocyst was hand picked and added to two petri dishes. The final two petri dishes were not infected with Nephromyces. After several weeks tunicates were moved to aerated 1L beakers to meet their increased nutrient and gas exchange requirements. Feeding regimen remained the same except that food volume was increased with tunicate growth. Tunicates were grown for six months until they were ~10mm across, after which they were periodically sacrificed to check infection status.
Quantification and Statistical Analysis
Amplicon Analysis
Reference sequences were trimmed to the amplicon sequence length and CDhit was used to cluster sequences with 85% sequence identity. All bacterial sequences, which did not cluster were deemed contamination and removed from the count table, taxonomy table, and sequences file. Samples with less than 5% of the mean number of reads were considered failed and were removed (three samples). Any ASV represented by less than 20 reads from a single sample was set to 0. Clustering thresholds for species assignment from ASVs, all available Plasmodium COI sequences were downloaded from NCBI’s Genbank. Sequences were aligned with MAFFT and trimmed to the region amplified by our COI primers. Sequences were then clustered with CD-hit at 99, 98, 97, 96% sequence identity. Clustered at 98% sequence identity collapsed almost all of the sequences to assigned species levels without collapsing multiple species. At 97% all sequences collapse to species level and a few species are collapsed. We clustered Nephromyces along the same gradient (Figure 2), and based on the Plasmodium analysis, the 98% and 97% sequence identity ASV cluster for Nephromyces.
Supplementary Material
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Deposited data | ||
| DNA and RNA sequences | This Paper | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA666913 |
| VEuPathDB | VeuPathDB | https://veupathdb.org/veupathdb/app |
| Aniseed | aniseed | https://www.aniseed.cnrs.fr/aniseed/species/ |
| Busco v3 | Swiss Institute of Bioinformatics | https://busco.ezlab.org/ |
| Interproscan v71.0 | InterPro | https://www.ebi.ac.uk/interpro/about/interproscan/ |
| KEGG | Kanehisa Laboratories | https://www.kegg.jp/ |
| Software and algorithms | ||
| Trimmomatic v0.3 | USADELL Lab | http://www.usadellab.org/cms/?page=trimmomatic |
| SPAdes v3.13. | Saint Petersburg State University | https://github.com/ablab/spades |
| PBSuite v15.8.2 | Pacific Biosciences | https://sourceforge.net/projects/pb-jelly/ |
| Canu v1.6 | Maryland Bioinformatics Labs | https://github.com/marbl/canu/releases/tag/v2.2 |
| ABySS v2.02 | BC Cancer Canada’s Michael Smith Genome Sciences Centre | https://github.com/bcgsc/abyss |
| Bowtie2 v1.2.2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bedtools | University of Utah | https://bedtools.readthedocs.io/en/latest/index.html |
| VizBin | university du luxembourg | https://claczny.github.io/VizBin/ |
| CAT | Universiteit Utrecht | https://github.com/dutilh/CAT |
| Trinity/Trinotate pipeline v2.4.0 | The Broad Institute | https://github.com/trinityrnaseq/trinityrnaseq |
| BLASTp | National Center for Biotechnology Information | ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ |
| Transdecoder | The Broad Institute | https://github.com/TransDecoder/TransDecoder |
| BBmap | Brian Bushnell | https://sourceforge.net/projects/bbmap/ |
| CDhit v4.6.8 | UCSD | https://sourceforge.net/projects/cdhit/ |
| Prokka | The University of Melbourne | https://github.com/tseemann/prokka |
| Maker | University of Utah | http://www.yandell-lab.org/software/maker.html |
| R and bash code | This paper | https://github.com/cpaight/Nephromyces |
| Critical commercial assays | ||
| MiSeq Reagent Kit v3 | Illumina | Product Number MS-102–3003 |
| HiSeq 2500 2×250 HiSeq SBS v4 | Illumina | Product Number FC-401–4003 |
| SMRT cells | Pacific Biosciences | Product Number 102-281-700 |
| Zymo Quick-RNA kit | Zymo | Catalogue number R1054 |
| Illumina TruSeq RNA kit | Illumina | Product Number RS-122–2001 |
| Prolong Gold with DAPI | ThermoFisher | Product Number P-36931 |
| Experimental models: Organisms/strains | ||
| M. manhattensis | This paper | Live collection, Greenwich Bay, RI (41.653N, −71.452W) |
| M. occidentalis | This paper | Live collection by Gulf Specimens Marine Laboratories, Inc. from Alligator Harbor, FL (29.899N, −84.381W) |
| Isochrysis galbana | National Center for Marine Algae and Microbiota | CCMP1323 |
| Chaetoceras sp. | National Center for Marine Algae and Microbiota | CCMP209 |
Highlights:
Nephromyces is a diverse apicomplexan that can harbor three bacterial endosymbionts
Each individual Nephromyces only contains a single bacterial species.
No bacterial endosymbiont completes the metabolic needs of Nephromyces.
Nephromyces with different endosymbionts likely must exchange metabolites.
Acknowledgements
The authors would like to acknowledge the comments of anonymous reviewers and Dr. John Archibald in helping to improve this manuscript. Support for this project was provided by National Institute of Health, grant AI124092, as well as National Science Foundation award 1541510. Sequencing library prep was conducted at a Rhode Island NSF EPSCoR research facility, the Genomics and Sequencing Center (GSC), supported in part by the National Science Foundation EPSCoR Cooperative Agreement #OIA-1655221.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Cavanaugh CM, Gardiner SL, Jones ML, Jannasch HW, and Waterbury JB (1981). Prokaryotic Cells in the Hydrothermal Vent Tube Worm Riftia pachyptila Jones: Possible Chemoautotrophic Symbionts. Science 213, 340–342. [DOI] [PubMed] [Google Scholar]
- 2.Brune A, Frenzel P, and Cypionka H (2000). Life at the oxic–anoxic interface: microbial activities and adaptations. FEMS Microbiology Reviews 24, 691–710. [DOI] [PubMed] [Google Scholar]
- 3.Husnik F, and McCutcheon JP (2016). Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proceedings of the National Academy of Sciences 113, E5416–E5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson ACC, and Duncan RP (2015). Signatures of host/symbiont genome coevolution in insect nutritional endosymbioses. Proceedings of the National Academy of Sciences 112, 10255–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz-Gómez SA, Durnin K, Eme L, Paight C, Lane CE, Saffo MB, and Slamovits CH (2019). Nephromyces Represents a Diverse and Novel Lineage of the Apicomplexa That Has Retained Apicoplasts. Genome Biol Evol 11, 2727–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saffo MB, McCoy AM, Rieken C, and Slamovits CH (2010). Nephromyces, a beneficial apicomplexan symbiont in marine animals. Proceedings of the National Academy of Sciences 107, 16190–16195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saffo MB (1988). Nitrogen Waste or Nitrogen Source? Urate Degradation in the Renal Sac of Molgulid Tunicates. The Biological Bulletin 175, 403–409. [Google Scholar]
- 8.Paight C, Slamovits CH, Saffo MB, and Lane CE (2019). Nephromyces Encodes a Urate Metabolism Pathway and Predicted Peroxisomes, Demonstrating That These Are Not Ancient Losses of Apicomplexans. Genome Biology and Evolution 11, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saffo MB (1990). Symbiosis within a symbiosis: Intracellular bacteria within the endosymbiotic protistNephromyces. Marine Biology 107, 291–296. [Google Scholar]
- 10.Anderson TJC, Haubold B, Williams JT, Estrada-Franco§ JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, et al. (2000). Microsatellite Markers Reveal a Spectrum of Population Structures in the Malaria Parasite Plasmodium falciparum. Molecular Biology and Evolution 17, 1467–1482. [DOI] [PubMed] [Google Scholar]
- 11.Lee K-S, Divis PCS, Zakaria SK, Matusop A, Julin RA, Conway DJ, Cox-Singh J, and Singh B (2011). Plasmodium knowlesi: Reservoir Hosts and Tracking the Emergence in Humans and Macaques. PLoS Pathogens 7, e1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnott A, Barry AE, and Reeder JC (2012). Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malaria Journal 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lalremruata A, Jeyaraj S, Engleitner T, Joanny F, Lang A, Bélard S, Mombo-Ngoma G, Ramharter M, Kremsner PG, Mordmüller B, et al. (2017). Species and genotype diversity of Plasmodium in malaria patients from Gabon analysed by next generation sequencing. Malaria Journal 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter ME, Ferrante JA, Meigs-Friend G, and Ulmer A (2019). Improving eDNA yield and inhibitor reduction through increased water volumes and multi-filter isolation techniques. Scientific Reports 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson ACC, Ashton PD, Calevro F, Charles H, Colella S, Febvay G, Jander G, Kushlan PF, Macdonald SJ, Schwartz JF, et al. (2010). Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect Mol Biol 19 Suppl 2, 249–258. [DOI] [PubMed] [Google Scholar]
- 16.Russell CW, Bouvaine S, Newell PD, and Douglas AE (2013). Shared Metabolic Pathways in a Coevolved Insect-Bacterial Symbiosis. Appl. Environ. Microbiol 79, 6117–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCutcheon JP, and von Dohlen CD (2011). An Interdependent Metabolic Patchwork in the Nested Symbiosis of Mealybugs. Current Biology 21, 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen AK, and Moran NA (2011). Aphid genome expression reveals host–symbiont cooperation in the production of amino acids. PNAS 108, 2849–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCutcheon JP, McDonald BR, and Moran NA (2009). Convergent evolution of metabolic roles in bacterial co-symbionts of insects. PNAS 106, 15394–15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bright M, and Bulgheresi S (2010). A complex journey: transmission of microbial symbionts. Nat Rev Microbiol 8, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa H (2001). Symbiotic microoiganisms in aphids (Homoptera, Insecta): A secret of one thriving insect group. Korean Journal of Biological Sciences 5, 163–177. [Google Scholar]
- 22.Hurley JH (2008). ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol 20, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, et al. (2011). Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol 13, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mettlen M, Chen P-H, Srinivasan S, Danuser G, and Schmid SL (2018). Regulation of Clathrin-Mediated Endocytosis. Annu Rev Biochem 87, 871–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamagishi JF, Saito N, and Kaneko K (2020). The advantage of leakage of essential metabolites and resultant symbiosis of diverse species. Phys. Rev. Lett 124, 048101. [DOI] [PubMed] [Google Scholar]
- 26.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, et al. (2012). Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS ONE 7, e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Research 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackman SD, Vandervalk BP, Mohamadi H, Chu J, Yeo S, Hammond SA, Jahesh G, Khan H, Coombe L, Warren RL, et al. (2017). ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Research 27, 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laczny CC, Sternal T, Plugaru V, Gawron P, Atashpendar A, Margossian H, Coronado S, der Maaten L, Vlassis N, and Wilmes P (2015). VizBin - an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Meijenfeldt FAB, Arkhipova K, Cambuy DD, Coutinho FH, and Dutilh BE (2019). Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- 36.Bushell B (2014). BBMap. SourceForge. https://sourceforge.net/projects/bbmap/. [Google Scholar]
- 37.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.TransDecoder (2021). (TransDecoder).
- 39.Li W, and Godzik A (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- 40.Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang H-Y, Dosztányi Z, El-Gebali S, Fraser M, et al. (2017). InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Research 45, D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- 42.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K, and Standley DM (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seah B (2011). A Tripartite Animal-Protist-Bacteria Symbiosis: Culture-Independent and Phylogenetic Characterization.
- 45.Batani G, Bayer K, Böge J, Hentschel U, and Thomas T (2019). Fluorescence in situ hybridization (FISH) and cell sorting of living bacteria. Sci Rep 9, 18618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw nucleotide data is available as an NCBI Bioproject (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA666913)
All R and bash code used in this project can be found on Github (https://github.com/cpaight/Nephromyces)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.