

Abstract
Asciminib is a first‐in‐class inhibitor of BCR::ABL1, specifically targeting the ABL myristoyl pocket. Asciminib is a substrate of CYP3A4 and P‐glycoprotein (P‐gp) and possesses pH‐dependent solubility in aqueous solution. This report summarizes the results of two phase I studies in healthy subjects aimed at assessing the impact of CYP3A and P‐gp inhibitors, CYP3A inducers and acid‐reducing agents (ARAs) on the pharmacokinetics (PK) of asciminib (single dose of 40 mg). Asciminib exposure (area under the curve [AUC]) unexpectedly decreased by ~40% when administered concomitantly with the strong CYP3A inhibitor itraconazole oral solution, whereas maximum plasma concentration (Cmax) decreased by ~50%. However, asciminib exposure was slightly increased in subjects receiving an itraconazole capsule (~3%) or clarithromycin (~35%), another strong CYP3A inhibitor. Macroflux studies showed that cyclodextrin (present in high quantities as excipient [40‐fold excess to itraconazole] in the oral solution formulation of itraconazole) decreased asciminib flux through a lipid membrane by ~80%. The AUC of asciminib was marginally decreased by concomitant administration with the strong CYP3A inducer rifampicin (by ~13–15%) and the strong P‐gp inhibitor quinidine (by ~13–16%). Concomitant administration of the ARA rabeprazole had little or no effect on asciminib AUC, with a 9% decrease in Cmax. The treatments were generally well tolerated. Taking into account the large therapeutic window of asciminib, the observed changes in asciminib PK following multiple doses of P‐gp, CYP3A inhibitors, CYP3A inducers, or ARAs are not considered to be clinically meaningful. Care should be exercised when administering asciminib concomitantly with cyclodextrin‐containing drug formulations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Asciminib is a first‐in‐class BCR::ABL1 inhibitor, specifically targeting the ABL myristoyl pocket, and a substrate of CYP3A4 and P‐gp. Asciminib displays pH‐dependent solubility in aqueous solution.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study explored the drug–drug interaction risk of asciminib as a victim with CYP3A inhibitors, CYP3A inducers, P‐gp inhibitors, and acid‐reducing agents (ARAs).
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Asciminib as a victim was weakly affected by concomitantly administered P‐gp inhibitors, strong CYP3A inhibitors, strong CYP3A inducers, or ARAs. However, a substantial effect of cyclodextrin (as an excipient in itraconazole oral solution) was observed; indirect evidence showed that cyclodextrin markedly decreased asciminib bioavailability.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These results support the concomitant use of CYP3A and P‐gp inhibitors, CYP3A inducers and ARAs in patients treated with asciminib. Care should be exercised when using itraconazole oral solution or other cyclodextrin‐containing formulations in clinical studies due to their potential impact on absorption of orally co‐administered compounds.
INTRODUCTION
Asciminib (ABL001) is an oral BCR::ABL1 inhibitor with a mechanism of action distinct from currently approved ATP‐competitive tyrosine kinase inhibitors (TKIs) used for the treatment of chronic myeloid leukemia (CML). Asciminib is the first‐in‐class BCR::ABL1 inhibitor specifically targeting the BCR::ABL1 myristoyl pocket (STAMP inhibitor). 1 , 2 , 3 , 4 The unique mechanism of action of asciminib means that this drug can maintain activity against forms of BCR::ABL1 carrying resistance mutations (including the T315I mutation) in the ATP‐binding site, thereby becoming a novel treatment option for patients who no longer respond to currently available TKIs. 1 , 2 , 3
The efficacy of asciminib is currently being evaluated in the ASCEMBL study (NCT03106779), a multicenter, open‐label, phase III study of asciminib versus the ATP‐competitive BCR::ABL1 inhibitor bosutinib in patients with CML in chronic phase (CML‐CP) previously treated with greater than or equal to two TKIs. 5 Asciminib 40 mg twice daily (b.i.d.) demonstrated statistically significant and clinically meaningful superiority in efficacy compared with bosutinib 500 mg once daily (q.d.), as well as a favorable safety profile.
Asciminib has a molecular weight of 449.84 Da. The measured log P for asciminib is 3.9, whereas permeability in CaCo‐2 cells is in the medium range (P app [B − A] and P app [A − B] were estimated as 6.8 × 10−6 cm/s and 2.3 × 10−6 cm/s, respectively). 1 A recent study assessing the absorption, distribution, metabolism, and excretion (ADME) of asciminib in healthy male subjects showed that the drug was rapidly absorbed following oral administration, reaching maximum plasma concentration at ~2 h postdose. 6 Asciminib was eliminated mainly through feces, and direct glucuronidation and oxidation were identified as major metabolic pathways; these were catalyzed predominantly by UDP‐glucuronosyltransferase (UGT)2B7 and cytochrome P450 (CYP)3A4, respectively. Based on in vitro data and the human ADME, the relative contribution of CYP3A4 to asciminib clearance was estimated to be 36.0%. 7
Asciminib has been identified as a substrate of P‐glycoprotein (P‐gp), 8 hence, inhibitors of P‐gp may increase asciminib plasma concentration, warranting further studies.
Acid reducing agents (ARAs), such as proton pump inhibitors (PPIs) and H2‐blockers induce significant increases in gastric pH, which may decrease the bioavailability of certain drugs that act as weak bases. A review of clinical literature data suggests that the magnitude of this drug–drug interaction (DDI) is largest for compounds in which in vitro solubility decreases over the pH range 1–4. 9 This is the case for asciminib, a weak base (pK a = 4.0) exhibiting pH‐dependent solubility, which is high at acidic pH and decreases with increasing pH. 1 Patients with cancer frequently take ARAs to alleviate dyspeptic symptoms; analysis of two large US healthcare databases revealed that prevalence of ARA use was 20%–33% among patients with cancer. 10 There is thus potential for significant DDIs between ARAs and asciminib, which could lead to decreased therapeutic benefit via the impact on drug absorption.
In this report, we describe the results from two phase I studies undertaken to assess the pharmacokinetics (PK) of asciminib in healthy individuals when administered alone or in combination with CYP3A or P‐gp inhibitors, CYP3A inducers, or ARAs.
METHODS
Study designs
The inhibitor/inducer study was a phase I, single center, open‐label, six‐cohort, two‐period, single‐sequence, crossover study to assess the effect of multiple doses of itraconazole and clarithromycin (strong CYP3A inhibitors), quinidine (P‐gp inhibitor), and rifampicin (strong CYP3A inducer) on the PK of a single 40 mg oral dose of asciminib in healthy subjects.
All subjects received asciminib alone in the first treatment period and asciminib together with the potentially interacting drug in the second treatment period. This study comprised six cohorts, which followed a similar single‐sequence design (Figure S1A); there was no randomization, but sequential cohort enrollment.
Cohort 1: Asciminib 40 mg with itraconazole 200 mg q.d. oral solution formulation. 11
Cohort 2: Asciminib 40 mg with quinidine 300 mg three times a day.
Cohort 3: Asciminib 40 mg with rifampicin 600 mg q.d.
Cohort 4: Asciminib 200 mg with rifampicin 600 mg q.d. (optional based on interim results from cohort 3).
Cohort 5: Asciminib 40 mg with clarithromycin 500 mg b.i.d.
Cohort 6: Asciminib 40 mg with itraconazole 200 mg q.d. capsule formulation.
The PK analysis of cohort 1 showed an unexpected decrease in asciminib area under the curve (AUC) and maximum plasma concentration (Cmax), which warranted further investigation. The original protocol was therefore amended to introduce two additional cohorts (cohorts 5 and 6) in order to further investigate the effect of strong CYP3A inhibitors on the PK of asciminib. Cohort 4 was to be initiated if a significant effect on asciminib PK was observed from asciminib 40 mg co‐administration with rifampicin (cohort 3). Based on predefined criteria in the protocol, cohort 4 (asciminib 200 mg) was not initiated.
The ARA study was an open‐label, adaptive three‐period, single‐sequence, crossover study to assess the effect of multiple doses of ARAs (rabeprazole and famotidine) on the PK of a single 40 mg oral dose of asciminib in healthy subjects (Figure S1B). A staggered approach was undertaken. First the effect of a PPI (rabeprazole 20 mg q.d.), the strongest ARA class, was tested in period 2. Period 3 (treatment with famotidine 20 mg q.d.) was conditioned on the outcome of data from the first two periods, and was to be initiated if the effect of rabeprazole on the PK of asciminib 40 mg was considered as clinically meaningful. Based on the results of the interim PK analysis, period 3 was not initiated.
Compound dosing and administration for both studies are described in detail in Appendix S1.
Study participants
Eligible subjects included male and/or sterile or postmenopausal female subjects between 18 and 55 years of age for the inhibitor/inducer study, and between 20 and 55 years of age for the ARA study. The ARA study was performed in Japanese healthy volunteers. Other eligibility criteria were body weight between 50 and 120 kg, as well as a body mass index (BMI) of 18.5–29.9 kg/m2 for the inhibitor/inducer study and 18.0–29.9 kg/m2 for the ARA study. Subjects were in good health as determined by lack of clinically significant findings from medical history, physical examination, vital signs, electrocardiogram (ECG), and laboratory data. Inclusion and exclusion criteria are described in Appendix S1.
PK analyses
The primary end points for both studies were the following PK parameters: area under the plasma concentration–time curve from time zero to the time of last quantifiable concentration point (AUClast), area under the plasma concentration–time curve from time zero to infinity (AUCinf), Cmax of drug in blood plasma, time to reach Cmax (Tmax), terminal half‐life (t 1/2), and apparent plasma clearance (CL/F) of asciminib. For the inhibitor/inducer study, trough concentrations of itraconazole, clarithromycin, quinidine, and rifampicin were also secondary end points, whereas the assessment of asciminib PK parameters in cohort 6 (itraconazole capsule formulation) was an exploratory end point.
Approximately 88 healthy subjects were to be enrolled in all six cohorts in the inhibitor/inducer study (18 each in cohort 1, cohort 2, and cohort 3; 10 in cohort 4; 14 in cohort 5, and 10 in cohort 6). The reason for the reduced sample size in cohort 6 is that this cohort was exploratory in nature and implemented to provide supportive data to separate out the effect of itraconazole and cyclodextrin (excipient of the itraconazole oral solution) on the PK of asciminib. For the ARA study, 23 healthy subjects were to be enrolled. Sample size estimates were based on an intrasubject coefficient of variation for the primary asciminib PK parameters of 30% (for the inhibitor/inducer study) and 28.9% (for the ARA study), as determined in a previous phase I study. 12 Enrollment numbers for both studies were determined by an acceptable precision of the 90% confidence intervals (CIs) for the comparison of test and reference parameters on a log scale, and took into account the potential dropout rate.
Across both studies, PK analyses were carried out in all subjects who provided greater than or equal to one evaluable PK profile for asciminib. PK parameters were derived with noncompartmental methods using Phoenix WinNonlin (Pharsight) software version 8.0 (for the inhibitor/inducer study) and version 6.4 (for the ARA study).
Safety assessments
Secondary end points for both studies included the incidence of adverse events (AEs) and serious AEs (SAEs), changes in hematology and blood chemistry values, vital signs, and ECG. Subjects underwent a physical and vital signs examination, clinical laboratory testing (biochemistry and hematology) and cardiac assessments at regular intervals throughout the study period. AE data were collected on an ongoing basis throughout the study and were coded and graded using MedDRA version 23.0 and Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 (for the inhibitor/inducer study), and MedDRA version 19.1 and CTCAE version 4.03 (for the ARA study).
Statistical analysis
Descriptive statistics were used for all PK parameters for analyses of asciminib and trough concentration values of itraconazole, quinidine, rifampicin, and clarithromycin; for Tmax, only the median and range were determined.
PK parameters for asciminib were calculated from individual plasma concentration–time profiles. To estimate the effect of inhibitors/inducers or multiple doses of rabeprazole on asciminib PK, a linear mixed effect model was fitted to the log‐transformed PK parameters (AUCinf, AUClast, and Cmax). For both studies, the model included treatment as a fixed factor and participant as a random factor. The difference between test (asciminib plus other drug) and reference (asciminib) means were calculated. Outputs were back‐transformed to obtain values for adjusted G mean, G mean ratio, and 90% CIs. G mean ratios were not calculated for cohort 6 due to the exploratory nature of this cohort; furthermore, the small sample size of this cohort would limit any conclusions that can be made from the results. Plasma concentrations below the lower limit of quantification (LLOQ) were set to zero, and treated as missing in calculations of G mean and geometric coefficients of variation percentage (GCV%). All analyses were performed using Statistical Analysis System (SAS) version 9.4 (for the inhibitor/inducer study) and version 9.2 (for the ARA study).
Macroflux studies
In order to assess the impact of cyclodextrin on asciminib absorption, a macroflux study was carried out, evaluating the dissolution and simultaneous flux of a 40 mg dose of asciminib through a gastrointestinal tract (GIT)‐Lipid (Pampa) membrane situated on top of a polyvinylidene difluoride (PVDF) membrane, mimicking an enterocyte cell layer. Fasted state simulated intestinal fluid (FaSSIF) media version 2 (V2) and FaSSIF V2 media with the addition of cyclodextrin (900 ml each) were added to a USP II apparatus (Distek Corporation, North Brunswick, NJ) maintained at a constant stirring of 100 rpm and a temperature of 37°C during the study. Two film‐coated tablets containing 20 mg of asciminib per tablet were introduced into the dissolution apparatus for the studies, with two replicates per condition. To mimic the exact total amount of cyclodextrin in each itraconazole solution dose as that used in the clinical study, 11 8 g of hydroxypropyl‐β‐cyclodextrin (as a 40% solution in water) was added to the FaSSIF V2 media prior to introduction of the two asciminib tablets; this setup constituted the donor compartment. The receiver compartment consisted of a miniaturized USP II paddle set‐up with a 0.45 μm PVDF membrane at the bottom of the vessel, coated with the biomimetic membrane (GIT‐Lipid). This compartment was filled with 12 ml of pION acceptor buffer to ensure sink conditions. Concentrations in the donor and receiver compartments were monitored over time using fiber optic probes, collecting data every 30 s over a 2‐h period. Dissolution profiles in the donor compartment were collected as the drug dissolved per time. Additionally, flux profiles in the receiver compartment were also collected in the same manner.
The fiber optic probes were calibrated for both the donor and acceptor compartments based on the media in the compartment. For both studies, the donor compartment was calibrated in FaSSIF V2 media, whereas the acceptor compartment was calibrated in pION acceptor buffer. The calibration curve was constructed from 0, 1, 5, 10, 25, 45, and 50 μg/ml target concentrations. The R 2 is reported as 0.9938 for the FaSSIF V2 donor, 0.9942 for the FaSSIF V2 plus cyclodextrin donor, and 0.9927 for the pION acceptor chambers. The wavelength range used for calibration analysis was a second derivative treatment of the UV data from 350 to 390 nm. As the area of the membrane is known, the linear portion of the flux curve (R 2 > 0.995) from ~70 to 120 min was utilized to calculate the slope in both conditions.
Ethics
All studies were conducted in accordance with the principles of the Declaration of Helsinki, as well as local laws and regulations. All subjects provided written informed consent before any study procedures took place. The study protocol and all amendments were reviewed by the independent ethics committee and/or institutional review board for each study center.
RESULTS
Subject disposition and baseline characteristics
Subject disposition is shown in Table S1. A total of 79 subjects participated in the inhibitor/inducer study (cohort 1, n = 18; cohort 2, n = 19; cohort 3, n = 18; cohort 5, n = 14; and cohort 6, n = 10); 10 subjects were planned to be enrolled in cohort 4, but this cohort was not initiated based on predefined criteria in the protocol. For the ARA study, a total of 23 subjects were enrolled.
All subjects completed treatment in cohorts 1, 3, and 5 of the inhibitor/inducer study, and in the ARA study. Four subjects in cohort 2 and one subject in cohort 6 discontinued the study; reasons for discontinuation were grade 3 QT corrected Fridericia's formula (QTcF) interval prolongation (cohort 2, n = 3) and subject/guardian decision (cohort 2, n = 1; and cohort 6, n = 1).
Subject demographics and baseline characteristics are shown in Table S2. The median age of subjects in the different cohorts of the inhibitor/inducer study ranged between 36.0 and 53.5 years (range 21–55), and the majority of subjects were White (one subject was African American, one Asian, one Native American, and one reported ethnicity as “other”). Subjects in the ARA study were younger (median age 27.0 years; range 22–39) and all were of Japanese origin. Five subjects required concomitant medication during the inhibitor/inducer study: one subject in cohort 1 required posiformin opthalmix salve 2% and dexamytrex ophthalmic salve for external hordeolum, whereas three subjects in cohort 2 and one subject in cohort 3 required paracetamol for headaches. We believe that these medications were unlikely to have affected the study results. No concomitant medications were used in the ARA study.
PK analyses: Inhibitor/inducer study
Overall, PK data from administration of asciminib alone were comparable across cohorts. Plasma concentration–time profiles of asciminib alone and in combination with inhibitors/inducers are shown in Figure 1.
FIGURE 1.
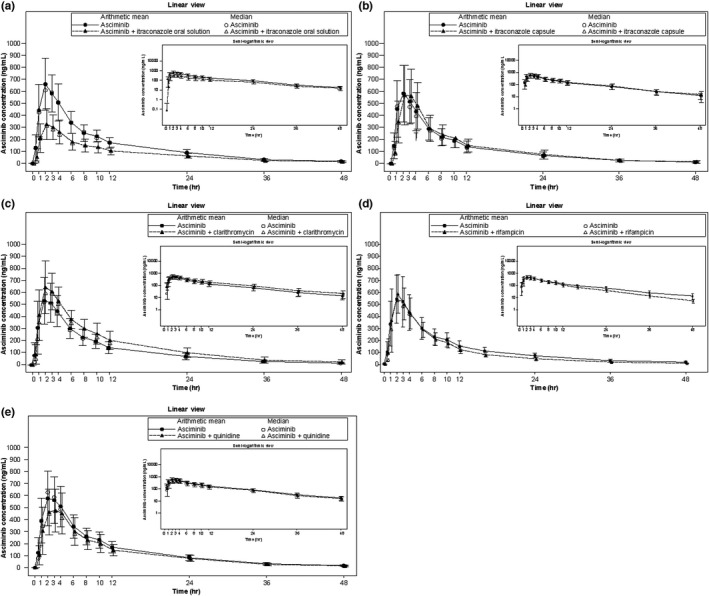
Arithmetic mean (SD) and median plasma concentration‐time profiles for asciminib in cohort 1 (itraconazole oral solution, a), cohort 6 (itraconazole capsule, b), cohort 5 (clarithromycin, c), cohort 3 (rifampicin, d), and cohort 2 (quinidine, e). Linear views are shown in the main panel, with semilogarithmic views in the inset
Interaction with strong CYP3A inhibitors (itraconazole/clarithromycin)
Concentration‐time profiles from subjects in cohort 1 revealed an unexpected decrease in the plasma concentration of asciminib when it was administered concomitantly with the strong CYP3A inhibitor itraconazole (oral solution) compared with administration of asciminib alone (Figure 1a). This was reflected by a lower AUCinf, AUClast, and Cmax and increased CL/F (Table 1), whereas Tmax and t 1/2 for asciminib were similar for both treatments. The intersubject variability increased when asciminib was administered together with itraconazole (oral solution), as shown by the increase in GCV% compared with administration of asciminib alone. When administered in combination with itraconazole (oral solution), the G mean of AUCinf and AUClast decreased by 40.2% and 41.2%, respectively, whereas Cmax decreased by 50.1% compared with asciminib administered alone (Table 2). The median Tmax was similar, and the elimination rate was also similar for both treatments, suggesting that the observed decrease in exposure was related to a decrease in absorption rather than a change in clearance.
TABLE 1.
PK parameters for asciminib in the inhibitor/inducer study
| Parameters | Asciminib (n = 18) | Asciminib + itraconazole (oral solution) (n = 17) | Asciminib (n = 10) | Asciminib + itraconazole (capsule) (n = 10) | Asciminib (n = 14) | Asciminib + clarithromycin (n = 14) | Asciminib (n = 18) | Asciminib + rifampicin (n = 17) | Asciminib (n = 19) | Asciminib + quinidine (n = 18) |
|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 6 | Cohort 5 | Cohort 3 | Cohort 2 | ||||||
| AUCinf (ng*h/ml), G mean (GCV%) | 7000 (25.1) | 4200 (32.8) | 5630 (39.8) | 5830 (45.4) | 5740 (34.0) | 7820 (31.0) | 5630 (31.3) | 4740 (24.2) | 6750 (30.0) | 5800 (32.7) |
| AUClast (ng*h/ml), G mean (GCV%) | 6950 (25.2) | 4090 (33.4) | 5590 (40.2) | 5780 (46.0) | 5670 (34.4) | 7750 (30.4) | 5400 (30.6) | 4670 (24.3) | 6700 (30.0) | 5550 (29.9) |
| Cmax (ng/ml), G mean (GCV%) | 679 (27.4) | 338 (35.2) | 554 (42.1) | 578 (40.4) | 537 (35.6) | 642 (30.8) | 559 (38.8) | 599 (25.1) | 618 (35.8) | 539 (32.5) |
| Tmax (h), median (range) | 2.00 (1.00–4.02) | 2.00 (0.97–3.98) | 2.01 (1.93–3.00) | 2.03 (1.98–3.00) | 2.02 (1.00–3.00) | 2.02 (1.98–3.03) | 2.00 (1.98–4.00) | 2.00 (0.98–3.00) | 2.00 (1.00–4.00) | 2.00 (0.98–3.98) |
| t½ (h), G mean (GCV%) | 12.6 (18.8) | 13.9 (41.1) | 11.5 (17.6) | 10.6 (18.3) | 12.5 (14.9) | 14.5 (18.0) | 10.5 (15.1) | 8.35 (9.5) | 12.4 (17.4) | 11.3 (14.3) |
| CL/F (L/h), G mean (GCV%) | 5.71 (25.1) | 9.53 (32.8) | 7.11 (39.8) | 6.86 (45.4) | 6.97 (34.0) | 5.11 (31.0) | 7.11 (31.3) | 8.43 (24.2) | 5.92 (30.0) | 6.90 (32.7) |
Abbreviations: AUC, area under the curve; AUCinf, AUC from zero to infinity; AUClast, AUC from zero to the last quantifiable concentration; CL/F, apparent plasma clearance; Cmax, maximum concentration of drug in plasma; GCV%, geometric coefficient of variation; G mean, geometric mean; PK, pharmacokinetic; t 1/2, terminal half‐life; Tmax, time to reach maximum concentration of drug in plasma.
TABLE 2.
Statistical analysis of PK parameters for asciminib in the inhibitor/inducer study
| PK parameter (unit) | Treatment | n* | Adjusted G mean | Comparison(s) | Treatment comparison | |
|---|---|---|---|---|---|---|
| G mean ratio | 90% CI | |||||
| Asciminib ± itraconazole (oral solution) ‐ cohort 1 | ||||||
| AUCinf (ng*h/ml) | Asciminib | 18 | 7000 | Asciminib + itraconazole (oral solution) / asciminib | 0.598 | 0.51–0.70 |
| Asciminib + itraconazole (oral solution) | 17 | 4190 | ||||
| AUClast (ng*h/ml) | Asciminib | 18 | 6950 | Asciminib + itraconazole (oral solution) / asciminib | 0.588 | 0.50–0.69 |
| Asciminib + itraconazole (oral solution) | 17 | 4090 | ||||
| Cmax (ng/ml) | Asciminib | 18 | 679 | Asciminib + itraconazole (oral solution) / asciminib | 0.499 | 0.42–0.59 |
| Asciminib + itraconazole (oral solution) | 17 | 338 | ||||
| Asciminib ± clarithromycin ‐ cohort 5 | ||||||
| AUCinf (ng*h/ml) | Asciminib | 14 | 5740 | Asciminib + clarithromycin / asciminib | 1.36 | 1.27–1.46 |
| Asciminib + clarithromycin | 14 | 7820 | ||||
| AUClast (ng*h/ml) | Asciminib | 14 | 5670 | Asciminib + clarithromycin / asciminib | 1.37 | 1.27–1.47 |
| Asciminib + clarithromycin | 14 | 7750 | ||||
| Cmax (ng/ml) | Asciminib | 14 | 537 | Asciminib + clarithromycin / asciminib | 1.19 | 1.10–1.30 |
| Asciminib + clarithromycin | 14 | 642 | ||||
| Asciminib ± rifampicin ‐ cohort 3 | ||||||
| AUCinf (ng*h/ml) | Asciminib | 18 | 5630 | Asciminib + rifampicin / asciminib | 0.851 | 0.80–0.90 |
| Asciminib + rifampicin | 17 | 4790 | ||||
| AUClast (ng*h/ml) | Asciminib | 18 | 5400 | Asciminib + rifampicin / asciminib | 0.873 | 0.83–0.92 |
| Asciminib + rifampicin | 17 | 4720 | ||||
| Cmax (ng/ml) | Asciminib | 18 | 559 | Asciminib + rifampicin / asciminib | 1.09 | 1.00–1.20 |
| Asciminib + rifampicin | 17 | 611 | ||||
| Asciminib ± quinidine ‐ Cohort 2 | ||||||
| AUCinf (ng*h/ml) | Asciminib | 19 | 6750 | Asciminib + quinidine / asciminib | 0.871 | 0.79–0.97 |
| Asciminib + quinidine | 18 | 5880 | ||||
| AUClast (ng*h/ml) | Asciminib | 19 | 6700 | Asciminib + quinidine / asciminib | 0.840 | 0.76–0.93 |
| Asciminib + quinidine | 18 | 5630 | ||||
| Cmax (ng/ml) | Asciminib | 19 | 618 | Asciminib + quinidine / asciminib | 0.887 | 0.78–1.01 |
| Asciminib + quinidine | 18 | 548 | ||||
Note: Model is a linear mixed effects model of the log‐transformed PK parameters. The model includes treatment as a fixed factor and subject as a random factor. The results are back transformed to get adjusted geometric mean, geometric mean ratio, and 90% CI.
n* = number of observations used for the analysis.
Abbreviations: AUC, area under the curve; AUCinf, AUC from zero to infinity; AUClast, AUC from zero to the last quantifiable concentration; CI, confidence interval; Cmax, maximum concentration of drug in plasma; GCV%, geometric coefficient of variation; G mean, geometric mean; PK, pharmacokinetic.
To investigate these findings, an additional smaller cohort was included in the study (cohort 6) to assess the effect of itraconazole in capsule formulation (not containing any cyclodextrin, an excipient in the oral solution formulation added in 40‐fold excess to itraconazole; i.e., 8 g cyclodextrin per 200 mg of itraconazole). In contrast with the results obtained when asciminib was administered with itraconazole oral solution, the plasma concentration–time profile of asciminib showed a similar absorption phase when co‐administered with an itraconazole capsule compared with that of asciminib administered alone (Table 1 and Figure 1b). Exposure of asciminib was slightly increased when administered in combination with itraconazole capsule compared with asciminib administered alone (G mean [GCV%]: AUCinf 5830 ng*h/ml [45.4] vs. 5630 ng*h/ml [39.8]; AUClast 5780 ng*h/ml [46.0] vs. 5590 ng*h/ml [40.2]; and Cmax 578 ng/ml [40.4] vs. 554 ng/ml [42.1]). This is in stark contrast with the results obtained when asciminib was co‐administered with itraconazole oral solution compared with asciminib alone (G mean [GCV%]: AUCinf 4200 ng*h/ml [32.8] vs. 7000 ng*h/ml [25.1]; AUClast 4090 ng*h/ml [33.4] vs. 6950 ng*h/ml [25.2]; and Cmax 338 ng/ml [35.2] vs. 679 ng/ml [27.4]). Consistent with exposure results, administration of asciminib with an itraconazole capsule did not affect Tmax compared with asciminib alone, whereas the CL/F decreased slightly (Table 1).
In order to further investigate these unexpected results with the itraconazole oral solution, asciminib dissolution and flux were assessed in vitro in the presence of a final amount of cyclodextrin mimicking that contained in each dose of itraconazole oral solution. Macroflux results showed that the dissolution rate was higher in fasted‐state simulated intestinal fluid (FaSSIF V2) media with added cyclodextrin than in FaSSIF V2 media alone (105% vs. 87%), whereas the rate of flux was 4.6‐fold lower in FaSSIF V2 media containing cyclodextrin compared with FaSSIF V2 media alone, reflecting limited crossing of asciminib through the lipid membrane due to interaction with cyclodextrin (Figure 2a and b). The calculated slope in FaSSIF V2 media alone was 0.107 μg/ml/min, with a membrane area of 3.88 cm2 and acceptor volume of 12 ml, whereas the flux was 1.29 μg/min (for a calculated flux of 0.332 μg/min*cm2). The calculated slope in FaSSIF V2 plus cyclodextrin was 0.02 μg/ml/min, which corresponds to a flux rate of 0.28 μg/min (for a calculated flux of 0.071 μg/min*cm2).
FIGURE 2.
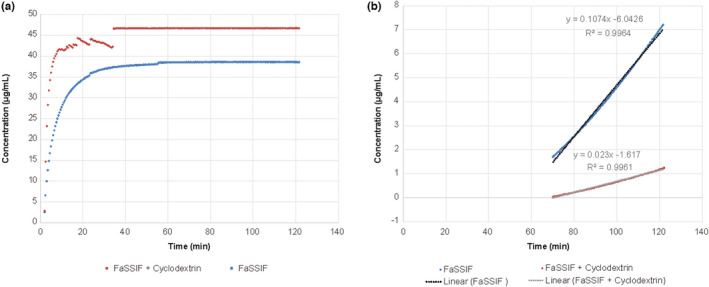
Dissolution (a) and flux (b) of 40 mg asciminib in FaSSIF version 2 media (900 ml) alone (blue) and containing cyclodextrin (red). For both experiments, total asciminib concentrations were measured in the receiver compartment. FaSSIF, Fasted State Simulated Intestinal Fluid
In vitro binding constant data showed a strong complex formation between asciminib and cyclodextrin, which decreased with increasing concentrations of bile acids and also at lower pH (Table S3). The binding constant in buffer was determined to be ~243,046/M. Additionally, we tested asciminib binding in media with increasing bile acid concentrations and found a substantial decrease in the binding constant in the presence of bile components. An ~65‐fold drop was observed in FaSSIF V2 and 0.3× Fed State Simulated Intestinal Fluid (FeSSIF) V2, whereas there was further decrease to practically no binding in the acidic pH of Fasted State Simulated Gastric Fluid (FaSSGF) and the high bile component concentration of FeSSIF V2.
The R 2 for asciminib binding was 0.94 in FaSSGF media; 0.99 in FeSSIF V2 media; 0.95 in 0.3× FeSSIF V2 media; 0.86 in FaSSIF V2 media, and 0.94 in blank buffer media. Saturation observed at the highest concentration in FaSSIF V2 media led to lower R 2 value. Removal of the highest concentration, 40% cyclodextrin, led to an R 2 value of 0.99 and a calculated binding constant of 7.12/M. As this was within two‐fold of the 3.75/M value determined leaving in the 40% value, it was decided to keep consistency among the calculations and report the lower value (3.75).
These in vitro results, coupled with the clinical results from cohort 6 (itraconazole capsule formulation) provide strong evidence that the results from cohort 1 (itraconazole oral solution) were caused by a formulation effect, most probably due to the cyclodextrin present in large quantities in the itraconazole oral solution as excipient to enhance the solubility of itraconazole.
The effect of clarithromycin—another strong CYP3A inhibitor—on the PK of asciminib was also assessed. The plasma concentration–time profile of asciminib in subjects from cohort 5 showed slightly increased plasma concentrations of asciminib when administered with clarithromycin compared to administration of asciminib alone (Figure 1c). The asciminib plasma elimination rate was slightly lower when asciminib was administered with clarithromycin, whereas Tmax was unchanged. AUCinf, AUClast, and Cmax increased when asciminib was administered in combination with clarithromycin compared with asciminib administered alone, whereas CL/F decreased (Table 1); median Tmax was unchanged, whereas t 1/2 was longer when asciminib was administered with clarithromycin. On average, administration of asciminib with clarithromycin increased AUCinf and AUClast of asciminib by 36% and 37%, respectively, whereas Cmax showed an average increase of 19% (Table 2).
Interaction with a strong CYP3A inducer (rifampicin)
Concentration profiles from subjects in cohort 3 revealed slightly decreased plasma concentrations of asciminib when administered with rifampicin compared with administration of asciminib alone (Figure 1d); however, the Cmax was similar for the two treatments. The asciminib plasma elimination rate was slightly higher when asciminib was administered with rifampicin, whereas Tmax was unchanged. Asciminib exposure was slightly decreased by concomitant administration of rifampicin, as shown by decreases in AUCinf and AUClast, although Cmax remained similar between treatments (Table 1). Administration of asciminib with rifampicin decreased AUCinf and AUClast of asciminib on average by 14.9% and 12.7%, respectively, whereas Cmax increased by 9% (Table 2).
Interaction with a P‐gp inhibitor (quinidine)
The plasma concentration profiles from subjects in cohort 2 showed marginally decreased plasma concentrations of asciminib when administered with the P‐gp inhibitor quinidine compared with administration of asciminib alone, with a similar elimination rate between the two treatments (Figure 1e). Administration of asciminib with quinidine decreased exposure, with lower AUCinf, AUClast, and Cmax coupled with slightly higher CL/F for asciminib administered with quinidine compared with administration of asciminib alone (Table 1). Median Tmax for asciminib was similar when administered either in combination with quinidine or alone, whereas median t 1/2 was slightly shorter. The average decrease in AUCinf, AUClast, and Cmax of asciminib was 12.9%, 16.0%, and 11.3%, respectively, when asciminib was administered with quinidine (Table 2).
Trough concentrations of itraconazole, quinidine, rifampicin, and clarithromycin are shown in Figure S2.
PK analyses: ARA study
Plasma concentration‐time profiles of asciminib alone and in combination with the ARA rabeprazole are shown in Figure 3. Concentration profiles were similar for a single oral dose of 40 mg asciminib when administered following multiple doses of rabeprazole compared with asciminib alone, although Cmax was slightly decreased when administered with rabeprazole. Overall, AUCinf, AUClast, and Cmax were similar when asciminib was administered alone or in combination with rabeprazole (Table 3). Administration of asciminib with rabeprazole reduced Cmax of asciminib by ~9%, but did not meaningfully change AUCinf, AUClast, or Tmax (Table 4).
FIGURE 3.
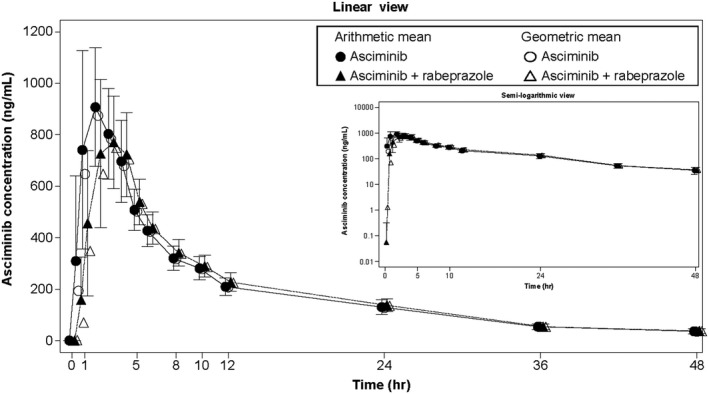
Arithmetic mean (SD) and geometric mean plasma concentration‐time profiles for asciminib alone or in combination with rabeprazole. Linear views are shown in the main panel, with semilogarithmic views on the right hand side
TABLE 3.
PK parameters for asciminib in the ARA study
| Parameters | Asciminib (n = 23) | Asciminib + rabeprazole (n = 23) |
|---|---|---|
| AUCinf (ng*h/ml), G mean (GCV%) | 9850 (18.7) | 9710 (18.1) |
| AUClast (ng*h/ml), G mean (GCV%) | 9200 (18.5) | 9060 (17.9) |
| Cmax (ng/ml), G mean (GCV%) | 943 (27.4) | 856 (21.7) |
| Tmax (h), median (range) | 2.00 (1.00–4.00) | 2.00 (2.00–5.00) |
| t ½ (h), G mean (GCV%) | 12.6 (10.0) | 12.2 (10.1) |
| CL/F (L/h), G mean (GCV%) | 4.06 (18.7) | 4.12 (18.1) |
Abbreviations: ARA, acid‐reducing agent; AUC, area under the curve; AUCinf, AUC from zero to infinity; AUClast, AUC from zero to the last quantifiable concentration; CL/F, apparent plasma clearance; Cmax, maximum concentration of drug in plasma; GCV%, geometric coefficient of variation; G mean, geometric mean; PK, pharmacokinetic; t 1/2, terminal half‐life; Tmax, time to reach maximum concentration of drug in plasma.
TABLE 4.
Statistical analysis of PK parameters for the ARA study
| PK parameter (unit) | Treatment | n* | Adjusted G mean | Comparison(s) | Treatment comparison | |
|---|---|---|---|---|---|---|
| G mean ratio | 90% CI | |||||
| AUCinf (ng*h/ml) | Asciminib | 23 | 9850 | Asciminib + rabeprazole / asciminib | 0.986 | 0.959–1.01 |
| Asciminib + rabeprazole | 23 | 9710 | ||||
| AUClast (ng*h/ml) | Asciminib | 23 | 9200 | Asciminib + rabeprazole / asciminib | 0.985 | 0.957–1.01 |
| Asciminib + rabeprazole | 23 | 9060 | ||||
| Cmax (ng/ml) | Asciminib | 23 | 943 | Asciminib + rabeprazole / asciminib | 0.908 | 0.849–0.972 |
| Asciminib + rabeprazole | 23 | 856 | ||||
| Tmax (h) | Asciminib | 23 | 2.00 | Asciminib + rabeprazole / asciminib | 1.00 | −1.00 to 3.00 |
| Asciminib + rabeprazole | 23 | 2.00 | ||||
Note: Model is a linear mixed effects model of the log‐transformed PK parameters. The model includes treatment as a fixed factor and subject as a random factor. The results are back transformed to get adjusted geometric mean, geometric mean ratio, and 90% CI.
n* = number of observations used for the analysis.
For Tmax, median is presented under “Adjusted geo‐mean,” median difference under “Geo‐mean ratio,” and minimum and maximum differences under 90% CI.
Abbreviations: ARA, acid‐reducing agent; AUC, area under the curve; AUCinf, AUC from zero to infinity; AUClast, AUC from zero to the last quantifiable concentration; CI, confidence interval; Cmax, maximum concentration of drug in plasma; G mean, geometric mean; PK, pharmacokinetic; Tmax, time to reach maximum concentration of drug in plasma.
Safety
In the inhibitor/inducer study, overall, 33 of 79 subjects (41.8%) experienced at least one AE during the study; no AEs were reported for subjects in the ARA study (Table S4). Three AEs of ECG QT prolongation (3.8%) reported in subjects from cohort 2 of the inhibitor/inducer study were grade ≥3 and led to study discontinuation. Increases in QTcF greater than 60 ms were observed in 6 (31.6%) subjects. No incidences of QTc over 500 ms occurred during the study. The three AEs of ECG QT prolongation occurred on day 7 (the first day that subjects were administered asciminib together with quinidine) and were assessed as related to the study drug. All events were asymptomatic ECG findings and recovered or resolved on the same or next day. Similar events were not observed following administration of asciminib alone in this cohort or in any of the other cohorts.
No greater than or equal to grade three AEs were reported for subjects in cohorts 1, 3, 5, or 6 of the inhibitor/inducer study (Table S5). The most common AEs reported across cohorts were flatulence (40.0% of subjects in cohort 6), fatigue (20.0% of subjects in cohort 6), headache (26.3% of subjects in cohort 2 and 5.6% subjects in cohort 3), diarrhea (15.8% of subjects in cohort 2 and 7.1% of subjects in cohort 3), and dysgeusia (14.3% of subjects in cohort 5). No clinically significant abnormalities in laboratory evaluations or vital signs were detected during the inhibitor/inducer study. No AEs were reported during the ARA study, although two subjects had alterations in laboratory values, one with a grade two increase in total bilirubin and one with a grade two increase in creatine kinase.
DISCUSSION
In vitro data indicated the involvement of multiple metabolic enzymes and transporters in asciminib human clearance, suggesting that the risk for a clinically significant DDI is low or moderate for this drug; however, further investigation was warranted. This report summarizes the results of two clinical studies aimed at assessing the impact of CYP3A and P‐gp inhibitors, CYP3A inducers and ARAs on the PK of a single dose of asciminib. Overall, the results show that none of these compounds appear to affect exposure of asciminib in a clinically meaningful way.
As itraconazole is a strong CYP3A inhibitor, based on in vitro data it was expected that asciminib exposure would increase with concomitant administration. However, asciminib exposure unexpectedly decreased when administered concomitantly with itraconazole oral solution. The observed decrease in Cmax was not accompanied by a decrease in Tmax, suggesting that the observed reduction in exposure was due to altered absorption. As the decrease in exposure appeared to be a phenomenon occurring at the absorption level in the gut, it was speculated that there might be an interaction between asciminib and an excipient present in the oral solution.
Cyclodextrin is an excipient in the itraconazole oral solution formulation, where it is present at a 40‐fold higher concentration than itraconazole (8 g of cyclodextrin per 200 mg itraconazole). 11 Cyclodextrins are present in pharmaceutical formulations to help improve compound solubility by forming strong complexes. 13 It was hypothesized that asciminib might form a strong complex with the cyclodextrin present in the oral solution at a very high concentration, leading to decreased free asciminib concentration available in the gut and hence reduced absorption. Sequestration by cyclodextrin has been reported for drugs such as warfarin, 14 as well as for proteins, growth factors, and other compounds. Macroflux experiments revealed that flux of a 40 mg dose of asciminib through an artificial lipid membrane was reduced ~4.6‐fold in FaSSIF media containing an equivalent amount of cyclodextrin as that present in the itraconazole oral solution than in media alone. The observed negative food effect on asciminib exposure (where administration with food decreased asciminib AUCinf) is thought to be caused by a similar type of interaction. 4 Briefly, asciminib is sequestered by bile acids when these are present at high levels in the GIT following food intake; the food effect is stronger after consuming high‐fat meals, which cause higher levels of bile acid secretion into the gut compared with low‐fat meals.
To assess the impact of itraconazole in the absence of cyclodextrin in a clinical setting, an additional smaller cohort of subjects (cohort 6) was recruited. These subjects received asciminib in combination with itraconazole in capsule formulation, which does not contain any cyclodextrin: the expected increase in asciminib exposure, although small, was observed in this cohort. Quantitative measurement of itraconazole concentrations in both formulations was comparable and consistent with historical data, indicating that the full inhibition potential of itraconazole was reached with both formulations. Furthermore, the expected increase in asciminib exposure was also observed following co‐administration with clarithromycin, another strong CYP3A inhibitor. Overall, our findings provide strong evidence that the observed decrease in asciminib exposure in combination with the itraconazole oral solution was caused by sequestration of asciminib by cyclodextrin contained in the formulation.
A similar unexpected decrease in exposure was reported for the Bruton’s TKI fenebrutinib when administered concomitantly with itraconazole oral solution. 15 Authors reported that fenebrutinib permeability across a Madin‐Darby canine kidney cell monolayer decreased in a cyclodextrin concentration‐dependent manner. Indeed, the in vitro binding constant of asciminib and cyclodextrin had a similar value to that reported for fenebrutinib (200,000/M), 15 further confirming that asciminib has a strong binding affinity to hydroxypropyl‐β‐cyclodextrin. Consistent with literature reports that bile components have a strong binding affinity to cyclodextrins, 16 we found that asciminib binding to cyclodextrin was dramatically reduced in the presence of high concentrations of bile acids. Our results further demonstrate the impact of cyclodextrin on asciminib absorption using core evidence data, including a macroflux study applied for the first time in this context, as well as the compilation of detailed clinical results from different subjects exposed to itraconazole oral solution and capsule formulations, as well as clarithromycin, another strong CYP3A4 inhibitor. Before selecting itraconazole oral solution as CYP3A4 perpetrator in a DDI study, researchers should assess whether cyclodextrin may form a complex with the compound of interest. As shown by our results, macroflux studies and binding constant data can inform study design, helping to predict the negative impact of excipients present in the formulation of concomitantly administered drugs.
Our results and those of others show the unexpected effects of itraconazole on drug metabolism, and highlight that care should be exercised when using it in DDI studies. Both formulations of itraconazole are widely used in this type of studies to assess the effect of strong CYP3A4 inhibition, with the oral solution reported to provide higher systemic exposure and reduced variability. 17 In addition to its strong inhibitory effect on CYP3A4/5, itraconazole is described as an in vitro inducer of CYP1A1 and as an inhibitor of CYP1A1, P‐gp, BCRP, OATP1B1, OATP1B3, OATP2B1, OATP4C1, UGT1A1, and UGT1A4. 18 , 19 , 20 , 21 , 22 , 23 As expected from a compound with multiple perpetrator characteristics, similar unexpected DDIs between itraconazole and other compounds have been published previously. For example, an unexpected 10%–24% decrease in exposure has been reported for the CYP3A4 substrate siponimod when administered concomitantly with an itraconazole capsule formulation. 18 This effect was proposed to be a consequence of itraconazole‐mediated induction of CYP1A1, but the authors concluded the reasons behind the decrease in exposure were not fully understood.
Given the unexpected effects of itraconazole on drug metabolism, alternative strong CYP3A4/5 inhibitors should be considered: several are available for clinical DDI studies, each one having its advantages and limitations. Ritonavir has the fastest onset of complete inhibition and shows the strongest CYP3A4/5 inhibition, achieved already at the second day of administration. 24 However, inhibition is not very specific to CYP3A4/5, as ritonavir has been reported to also induce CYP1A2, CYP2B6, and CYP2C9. 25 , 26 Moreover, ritonavir inhibits multiple other enzymes and transporters based on clinical or in vitro data (i.e., CYP2D6, P‐gp, OATP1B1, OATP1B3, OAT3, OCT1/2, MATE1, BCRP, and MRP1). 27 , 28 , 29 On the other hand, clarithromycin is a time‐dependent CYP3A4/5 inhibitor which also inhibits P‐gp and OATP1B1/1B3, whereas voriconazole is a reversible inhibitor of CYP3A4/5 and P‐gp, OATP1B3, and OAT1. 30 , 31 , 32 , 33 All of these factors should be carefully taken into account by the researchers when selecting the perpetrator for this type of DDI study.
As expected from the weak DDI effect with the CYP3A inhibitor clarithromycin, administration of asciminib following multiple doses of the strong CYP3A inducer rifampicin modestly decreased asciminib exposure. A slight decrease in exposure was observed following multiple doses of the P‐gp inhibitor quinidine. These results suggest that asciminib as a victim is weakly affected by concomitantly administered drugs that are strong CYP3A inducers or inhibitors of P‐gp.
For both formulations of itraconazole (oral solution and capsule), it is assumed that almost complete CYP3A4 inhibition was reached, although the trough concentrations increased up to the day when asciminib was co‐administered. The trough itraconazole concentrations for the capsule formulation tended to be higher and showed higher variability compared with those for the itraconazole oral solution. For quinidine, rifampicin, and clarithromycin, steady‐state conditions appear to have been reached when asciminib was co‐administered with these drugs. Of note, for rifampicin the maximum CYP3A4 induction may not have been reached by day 5 of treatment. However, based on physiologically‐based pharmacokinetic (PBPK) simulation studies, 34 , 35 ~90% and almost 100% of the maximal CYP3A4 induction effect in the liver and intestines, respectively, are reached at day 5. Hence, it is believed that the observed reduction in asciminib exposure of 15% is close to the maximum induction effect of rifampicin.
Taking into account the large therapeutic window of asciminib, the observed changes in asciminib exposure following multiple doses of P‐gp, CYP3A inhibitors, or CYP3A inducers are not considered to be clinically meaningful. The findings on CYP3A inhibitors and inducers are supported by previous studies showing that the relative contribution of CYP3A4 metabolism to asciminib clearance is ~36.0%. 7
The primary purpose of the ARA study was to investigate the effect of a PPI (rabeprazole) and a H2‐blocker (famotidine) on the PK of asciminib in healthy subjects. Although both medications are used to treat gastroesophageal reflux disease, PPIs induce a more profound and longer lasting reduction in gastric acid secretion than H2‐blockers, which have a limited ability to reduce postprandial gastric acid secretion. 36 The results of the study showed little to no effect of multiple doses of 20 mg rabeprazole on asciminib exposure, suggesting that asciminib bioavailability was not affected by co‐administration with rabeprazole. Given that rabeprazole would have a more potent effect on gastric acid secretion (and therefore on asciminib absorption) than famotidine, assessment of asciminib PK in combination with famotidine was not carried out. Among currently available PPIs, rabeprazole is considered the most potent 37 ; this means that results obtained with rabeprazole can be extrapolated to other ARAs. In line with clinical results, GastroPlus PBPK predictive modeling showed little to no effect of elevated gastric pH on the rate and extent of asciminib absorption (data not shown). These data demonstrated that the asciminib hydrochloride salt contained in the tablet is expected to fully dissolve in the stomach due to high solubility and will not precipitate in intestinal medium as it forms a supersaturated solution in basic conditions (data not shown). The amount of drug substance in solution and available for absorption is therefore greater than predicted by the low saturation solubility values within the pH range of 4.5–6.8.
The ARA study exclusively enrolled Japanese healthy volunteers; however, the PK characteristics of asciminib in Japanese and non‐Japanese subjects are not expected to differ significantly. This is further supported by clinical results: based on limited data on asciminib PK in the pivotal phase III study (ASCEMBL) of asciminib in patients with CML, 38 no relevant differences in PK parameters were apparent between Japanese and non‐Japanese subjects. The G mean (GCV%) AUC0–12 h at steady‐state following administration of 40 mg asciminib b.i.d. were 6450 ng*h/ml (22.8%) for Japanese subjects (n = 5) and 5370 ng*h/ml (39.4%) for non‐Japanese subjects (n = 8). Hence, these results and their interpretation can be extrapolated to non‐Japanese subjects.
The treatments were generally well‐tolerated. In the inhibitor/inducer study, asciminib in combination with itraconazole (oral solution and capsule formulations), clarithromycin, or rifampicin was well‐tolerated, with generally mild AEs reported and no laboratory abnormalities or changes in vital signs observed. A greater than or equal to grade three QTc prolongation event was reported in 3 of 19 (15.8%) subjects receiving asciminib in combination with quinidine. These events were considered as related to the study drug, and the three subjects discontinued the study; all events resolved on the same or next day. QTc prolongation was only observed when subjects were treated with asciminib in combination with quinidine, which is known to cause QTc prolongation due to its ability to block Na+ and K+ channel function. 39 No QTc prolongation events were observed in any of the other cohorts, including the cohort with clarithromycin, which is also known to prolong the QTc interval, 40 and none were reported in the phase I study, including 141 heavily pretreated patients with CML who received doses of asciminib up to 200 mg b.i.d. 41 No other greater than or equal to grade three AEs were reported during the inhibitor/inducer study. No AEs were reported in the ARA study.
In conclusion, asciminib as a victim was weakly affected by concomitantly administered strong CYP3A inhibitors, strong CYP3A inducers, P‐gp inhibitors, or ARAs. Overall, these results support the concomitant use of CYP3A and P‐gp inhibitors, CYP3A inducers, and ARAs in patients treated with asciminib, given that the negligible effect on asciminib PK is not considered clinically relevant. Care should be exercised when administering asciminib concomitantly with cyclodextrin‐containing drug formulations.
CONFLICT OF INTEREST
All authors are employees of Novartis Pharmaceuticals Corporation. M.H., F.H., M.Q., S.D., S.K., and F.H.‐P. are also Novartis stockholders.
AUTHOR CONTRIBUTIONS
M.H., F.H., M.S., T.S., M.Q., S.D., S.K., and F.H.‐P. wrote the manuscript. F.H., M.S., and M.Q. designed the research. S.D. performed the research. M.H., F.H., M.S., T.S., M.Q., S.D., S.K., and F.H.‐P. analyzed the data.
Supporting information
Figure S1
Figure S2
Table S1
Table S2
Table S3
Table S4
Table S5
Appendix S1
ACKNOWLEDGEMENTS
The authors would like to thank Angela Sinn, MD, of Parexel, Germany, principal investigator of the inhibitor/inducer study, and Harumi Murakami, MD, of Sumida Hospital, Japan, principal investigator of the ARA study, for their contribution to the execution of these studies; and Yunlin Fu, Birk Poller, Paulo G. Santos, and Hilmar Schiller, all of Novartis, for helping to elucidate the role of cyclodextrin. The authors would also like to thank Vanesa Martinez Lopez, PhD, of Novartis Ireland Ltd., for medical writing support.
Hoch M, Huth F, Sato M, et al. Pharmacokinetics of asciminib in the presence of CYP3A or P‐gp inhibitors, CYP3A inducers, and acid‐reducing agents. Clin Transl Sci. 2022;15:1698‐1712. doi: 10.1111/cts.13285
Funding information
This study was sponsored and funded by Novartis Pharmaceuticals Corporation.
REFERENCES
- 1. Schoepfer J, Jahnke W, Berellini G, et al. Discovery of Asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR‐ABL1. J Med Chem. 2018;61:8120‐8135. [DOI] [PubMed] [Google Scholar]
- 2. Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR‐ABL1. Nature. 2017;543:733‐737. [DOI] [PubMed] [Google Scholar]
- 3. Manley PW, Barys L, Cowan‐Jacob SW. The specificity of asciminib, a potential treatment for chronic myeloid leukemia, as a myristate‐pocket binding ABL inhibitor and analysis of its interactions with mutant forms of BCR‐ABL1 kinase. Leuk Res. 2020;98:106458. [DOI] [PubMed] [Google Scholar]
- 4. Hoch M, Zack J, Quinlan M, et al. Pharmacokinetics of Asciminib when Taken with Imatinib or with Food. Clin Pharmacol Drug Dev. 2021;11:207‐219. [DOI] [PubMed] [Google Scholar]
- 5. Rea D, Mauro MJ, Boquimpani C, et al. A phase 3, open‐label, randomized study of Asciminib, a STAMP inhibitor, vs bosutinib in CML after ≥2 prior TKIs. Blood. 2021;138:2031‐2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tran P, Hanna I, Eggimann FK, et al. Disposition of asciminib, a potent BCR‐ABL1 tyrosine kinase inhibitor, in healthy male subjects. Xenobiotica. 2020;50:150‐169. [DOI] [PubMed] [Google Scholar]
- 7. Novartis, data on file.
- 8. Eadie LN, Saunders VA, Branford S, White DL, Hughes TP. The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro. Oncotarget. 2018;9:13423‐13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH‐dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92:203‐213. [DOI] [PubMed] [Google Scholar]
- 10. Smelick GS, Heffron TP, Chu L, et al. Prevalence of acid‐reducing agents (ARA) in cancer populations and ARA drug–drug interaction potential for molecular targeted agents in clinical development. Mol Pharm. 2013;10:4055‐4062. [DOI] [PubMed] [Google Scholar]
- 11. Sporanox SMPC. 2021. https://www.medicines.org.uk/emc/medicine/7514/SPC/Sporanox+10+mg+ml+Oral+Solution/#gref.
- 12. Menssen HD, Quinlan M, Kemp C, Tian X. Relative bioavailability and food effect evaluation for 2 tablet formulations of Asciminib in a 2‐arm, crossover, randomized, open‐label study in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8:385‐394. [DOI] [PubMed] [Google Scholar]
- 13. Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998;98:2045‐2076. [DOI] [PubMed] [Google Scholar]
- 14. Al‐Dubaili N, Saleh N. Sequestration effect on the open‐cyclic switchable property of warfarin induced by cyclodextrin: time‐resolved fluorescence study. Molecules. 2017;22:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durk MR, Jones NS, Liu J, et al. Understanding the effect of hydroxypropyl‐β‐cyclodextrin on Fenebrutinib absorption in an itraconazole‐Fenebrutinib drug–drug interaction study. Clin Pharmacol Ther. 2020;108:1224‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schönbeck C. Complexation kinetics of cyclodextrins with bile salt anions: energy barriers for the threading of ionic groups. J Phys Chem B. 2019;123:9831‐9838. [DOI] [PubMed] [Google Scholar]
- 17. Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug–drug interaction studies. J Clin Pharmacol. 2016;56:143‐151. [DOI] [PubMed] [Google Scholar]
- 18. Gardin A, Shakeri‐Nejad K, Feller A, Huth F, Neelakantham S, Dumitras S. Siponimod pharmacokinetics, safety, and tolerability in combination with the potent CYP3A4 inhibitor itraconazole in healthy subjects with different CYP2C9 genotypes. Eur J Clin Pharmacol. 2019;75:1565‐1574. [DOI] [PubMed] [Google Scholar]
- 19. Maeda K, Ikeda Y, Fujita T, et al. Identification of the rate‐determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther. 2011;90:575‐581. [DOI] [PubMed] [Google Scholar]
- 20. Prueksaritanont T, Tatosian DA, Chu X, et al. Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin Pharmacol Ther. 2017;101:519‐530. [DOI] [PubMed] [Google Scholar]
- 21. Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609‐613. [DOI] [PubMed] [Google Scholar]
- 22. Walsky RL, Bauman JN, Bourcier K, et al. Optimized assays for human UDP‐glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab Dispos. 2012;40:1051‐1065. [DOI] [PubMed] [Google Scholar]
- 23. Sato T, Mishima E, Mano N, Abe T, Yamaguchi H. Potential drug interactions mediated by renal organic anion transporter OATP4C1. J Pharmacol Exp Ther. 2017;362:271‐277. [DOI] [PubMed] [Google Scholar]
- 24. Umehara KI, Huth F, Won CS, Heimbach T, He H. Verification of a physiologically based pharmacokinetic model of ritonavir to estimate drug–drug interaction potential of CYP3A4 substrates. Biopharm Drug Dispos. 2018;39:152‐163. [DOI] [PubMed] [Google Scholar]
- 25. Kirby BJ, Collier AC, Kharasch ED, et al. Complex drug interactions of HIV protease inhibitors 2: in vivo induction and in vitro to in vivo correlation of induction of cytochrome P450 1A2, 2B6, and 2C9 by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39:2329‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greenblatt DJ, Peters DE, Oleson LE, et al. Inhibition of oral midazolam clearance by boosting doses of ritonavir, and by 4,4‐dimethyl‐benziso‐(2H)‐selenazine (ALT‐2074), an experimental catalytic mimic of glutathione oxidase. Br J Clin Pharmacol. 2009;68:920‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. FDA. Center for Drug Evaluation and Research . Application Number: 203100Orig1s000 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203100Orig1s000ClinPharmR.pdf> (2012).
- 28. Shebley M, Liu J, Kavetskaia O, et al. Mechanisms and predictions of drug–drug interactions of the hepatitis C virus three direct‐acting antiviral regimen: Paritaprevir/ritonavir, Ombitasvir, and dasabuvir. Drug Metab Dispos. 2017;45:755‐764. [DOI] [PubMed] [Google Scholar]
- 29. Vermeer LM, Isringhausen CD, Ogilvie BW, Buckley DB. Evaluation of ketoconazole and its alternative clinical CYP3A4/5 inhibitors as inhibitors of drug transporters: the in vitro effects of ketoconazole, ritonavir, clarithromycin, and itraconazole on 13 clinically‐relevant drug transporters. Drug Metab Dispos. 2016;44:453‐459. [DOI] [PubMed] [Google Scholar]
- 30. Nakakariya M, Goto A, Amano N. Appropriate risk criteria for OATP inhibition at the drug discovery stage based on the clinical relevancy between OATP inhibitors and drug‐induced adverse effect. Drug Metab Pharmacokinet. 2016;31:333‐339. [DOI] [PubMed] [Google Scholar]
- 31. Zapater P, Reus S, Tello A, Torrús D, Pérez‐Mateo M, Horga JF. A prospective study of the clarithromycin–digoxin interaction in elderly patients. J Antimicrob Chemother. 2002;50:601‐606. [DOI] [PubMed] [Google Scholar]
- 32. Li J, Olaleye OE, Yu X, et al. High degree of pharmacokinetic compatibility exists between the five‐herb medicine XueBiJing and antibiotics comedicated in sepsis care. Acta Pharm Sin B. 2019;9:1035‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park SJ, Song IS, Kang SW, et al. Pharmacokinetic effect of voriconazole on cyclosporine in the treatment of aspergillosis after renal transplantation. Clin Nephrol. 2012;78:412‐418. [DOI] [PubMed] [Google Scholar]
- 34. Baneyx G, Parrott N, Meille C, Iliadis A, Lavé T. Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: influence of time between substrate and inducer administration. Eur J Pharm Sci. 2014;56:1‐15. [DOI] [PubMed] [Google Scholar]
- 35. Kapetas AJ, Sorich MJ, Rodrigues AD, Rowland A. Guidance for rifampin and midazolam dosing protocols to study intestinal and hepatic cytochrome P450 (CYP) 3A4 induction and De‐induction. AAPS J. 2019;21:78. [DOI] [PubMed] [Google Scholar]
- 36. Wang WH, Huang JQ, Zheng GF, et al. Head‐to‐head comparison of H2‐receptor antagonists and proton pump inhibitors in the treatment of erosive esophagitis: a meta‐analysis. World J Gastroenterol. 2005;11:4067‐4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Graham DY, Tansel A. Interchangeable use of proton pump inhibitors based on relative potency. Clin Gastroenterol Hepatol. 2018;16:800‐808.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Novartis, data on file.
- 39. Nachimuthu S, Assar MD, Schussler JM. Drug‐induced QT interval prolongation: mechanisms and clinical management. Ther Adv Drug Saf. 2012;3:241‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Albert RK, Schuller JL. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med. 2014;189:1173‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hughes TP, Mauro MJ, Cortes JE, et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019;381:2315‐2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Table S1
Table S2
Table S3
Table S4
Table S5
Appendix S1