

Abstract
The effect of inhaled corticosteroids (ICS) on the airway microbiome requires longitudinal research for corroboration. Asthma patients, not undergoing ICS treatment (baseline), were enrolled and prescribed ICS; all these patients were followed up with regular visits at 3 months (visit 1) and 9 months (visit 2). Induced sputum was collected, and fungal microbiota (mycobiome) and bacterial microbiota (bacteriome) were estimated using 16S rRNA and internal transcribed spacer (ITS) sequencing. Bacterial α diversity indices were not significantly different between baseline, visit 1, and visit 2. Visit 1 showed lower fungal evenness than the baseline, and visit 2 showed lower fungal diversity and evenness than the baseline. Fungal, but not bacterial, community compositions differed significantly between the baseline, visit 1, and visit 2. The most abundant bacterial phyla and genera did not differ significantly between the baseline, visit 1, and visit 2. Compared with the baseline, visit 1 showed significantly increased frequency of the fungal phylum Ascomycota and lower frequency of Basidiomycota. We found sharply decreased fungal genera Wallemia, Cladosporium, Penicillium, and Alternaria at visit 1 and visit 2 compared with the baseline, although the differences were not statistically significant. We also found the proportion of Basidiomycota was positively correlated with percentages of sputum eosinophils and neutrophils. The proportions of Saccharomyces, Wallemia, and Aplosporella were positively correlated with percentage of sputum eosinophils. Moreover, we identified distinct inter‐ and intra‐kingdom interactions in baseline, visit 1, and visit 2. Therefore, ICS use altered the airway microbial diversity, evenness, community composition, and microbial connections.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Previous studies have reported on the associations of bacteriome and asthma; however, no longitudinal studies have examined the correlations between inhaled corticosteroid (ICS) use and airway bacteriome, mycobiome, and microbial interactions.
WHAT QUESTION DID THIS STUDY ADDRESS?
We aimed to investigate the effect of ICS prescriptions on fungal microbiota (mycobiome) and bacterial microbiota (bacteriome) in the airways of asthma patients.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study refines the list of bacterial and fungal groups associated with asthmatic therapies. ICS use mainly altered the airway mycobiome diversity, evenness, community composition, and microbial connections.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Our preliminary results regarding the changes in airway microbiome based on ICS treatment over time might provide additional information on the role of ICS in asthma, which merits further research.
INTRODUCTION
Asthma is one of the most frequent causes of chronic airway diseases. Recent evidence has demonstrated that an abnormal airway microbiome may be involved in the pathogenesis of asthma. 1 The human airway harbors thousands of bacterial, fungal, and viral species, together making up a population dominated by the phyla Bacteroidetes, Firmicutes, Ascomycota, and Basidiomycota; however, these communities are easily affected by a multitude of factors including host genetics, lifestyle, and the environment. 2
Increasing evidence suggests associations between airway bacterial dysbiosis and airway inflammation, disease severity, and disease control in asthma, indicating that microbe–host interactions have the potential to influence or serve as biomarkers of asthmatic development. 3 , 4 , 5 , 6 , 7 For instance, previous research demonstrated that airway bacteriome was associated with hyperresponsiveness, disease control, 8 eosinophilic phenotype, 4 disease severity, 9 and steroid sensitivity. 10 In one of these studies, severe patients were found to be significantly enriched in Actinobacteria and a Klebsiella genus member compared with healthy controls or patients with mild‐to‐moderate asthma. 9 Studies of fungal dysbiosis have also attracted increasing attention with useful insight into its correlation with airway inflammation phenotypes in asthma 11 , 12 ; however, the understanding regarding the influence of fungal imbalance is still lacking. Recent observations (from sputum, bronchoalveolar lavage fluid [BALF], and endobronchial brush [EB]) have provided insights into the airway mycobiome in asthma patients. Studies have revealed lower fungal diversity in asthma patients with a T2‐high phenotype compared to those with a T2‐low phenotype, and have shown significant correlations between specific fungal exact sequence variants and the fraction of exhaled nitric oxide (FeNO), FEV1, and corticosteroid therapy. 11
Pharmacotherapy based on inhaled corticosteroids (ICS) is the first‐line therapy for asthma. A previous study investigated the association between ICS usage and the characteristics of the bacteriome of asthma patients. 10 , 13 Actinobacteria was correlated with indicators of steroid responsiveness and bronchial epithelial gene expression of the FK506 binding protein (FKBP5). 9 Durack et al. reported that 6‐week ICS treatment increased the proportions of Microbacteriaceae, Neisseria, and Moraxella species, but reduced that of the Fusobacterium members. 14 Different inhaled ICS showed different effects on the lung microbiome, although different inhaled ICS may have similar effects on the phagocytic function of macrophages in animal experiments. Hartmann et al. performed a systematic review and reported that corticosteroids may have an important effect on the composition of the respiratory microbiome, but there was no clear shared direction as a result of the heterogeneous nature of the methods used and the populations investigated. 15 The effect of ICS on the microbiome, especially mycobiome and bacterial–fungal, fungal–fungal, and bacterial–bacterial networks, remains to be elucidated. In this study we aimed to investigate the effect of ICS on the airway microbiome and microbial connections in asthma patients over a 9‐month period.
METHODS
Study population
The study was carried out at Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Asthma patients without ICS treatment (baseline) were enrolled and prescribed budesonide; all patients were followed up with regular visits at 3 months (visit 1) and 9 months (visit 2). Patients with asthma were enrolled based on the diagnosis of respiratory symptoms and bronchodilator responsiveness (an improvement of FEV1 (forced expiratory volume in the first second) by at least 12% and 200 ml after albuterol inhalation). The Asthma Control Questionnaire (ACQ) was used to assess asthma control. Exclusion criteria were smoking history, acute respiratory infection, or antibiotic use within 1 month before enrollment. The demographic characteristics including age, sex, body mass index (BMI), ICS types, FEV1% pre, FEV1/FVC (forced vital capacity) percentage, and Asthma Control Questionnaire 7 score (ACQ 7 score) were recorded.
Experiments were carried out in accordance with the principles of the Declaration of Helsinki of 1975. Written informed consent was obtained from all participants and the study was approved by the Institutional Medical Ethics Review Board of Ruijin Hospital, Shanghai Jiao Tong University, School of Medicine.
Specimen collection
First, patients were asked to rinse their mouth with saline. Subsequently, sputum was induced by hypertonic saline nebulization. 16 Samples for cell differential counts were stored at room temperature, the percentages of eosinophils (EOS%) and neutrophils (NEU%) were calculated. Samples intended for microbial analysis were frozen in dry ice and then stored at −80°C prior to analyses.
DNA extraction, 16S rRNA, and ITS sequencing
Bacterial and fungal DNA were isolated and purified using the QIAamp DNA Microbiome Kit (Qiagen) according to the manufacturer's instructions. Briefly, mixed host/microbe samples were exposed to lysis buffer to deplete host nucleic acid, then bacteria and fungi cells were effectively lyzed by chemical and mechanical disruption. After the removal of residual contaminants affecting DNA binding, microbial DNA was purified by elution. 16S rRNA (338F: 5′‐ACTCCTACGGGAGGCAGCAG‐3′; 806R: 5′‐GGACTACHVGGGTWTCTAAT‐3′), and ITS (1F: 5′‐CTTGGTCATTTAGAGGAAGTAA‐3′; 2R: 5′‐GCTGCGTTCTTCATCGATGC‐3′) were amplified, and the reaction occurred under the following conditions: 95°C for 3 min (1 cycle); 95°C for 30 s, 53°C for 60 s, 72°C for 45 s (37 cycles), and a final step of 72°C for 10 min. Then, polymerase chain reaction (PCR) products were used to construct a library for microbial sequencing using the Illumina Miseq system.
Data analysis
Raw data were processed as previously described, 17 including the removal of low‐quality reads and those shorter than 200 base pairs (bp). Unique reads of each sample were clustered into an operational taxonomic unit (OTU) based on 97% identity using the UPARSE algorithm and chimeric sequences were identified and removed. The representative sequence of each OTU was screened for further annotation by assigning to the SILVA (version 138, http://www.arb‐silva.de) and UNITE (version 8.0, http://unite.ut.ee/index.php) databases using RDP Classifier (version 2.11, http://sourceforge.net/projects/rdp‐classifier/), which provides taxonomic assignments from domain to genus.
Core species analysis was used to judge whether the sample size was sufficient to evaluate the total core species in sputum (Figure S1). α diversity was estimated by calculating community richness (Chao index), community evenness (Shannoneven index), and community diversity (Shannon). Faith phylogenetic diversity (PD index), which weighs not only diversity and evenness, but additionally weighs phylogenetic relationships, was also measured to reflect microbial phylogenetic diversity. Principal coordinate analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA) were used for observation of the difference or similarity in mycobiome and bacteriome between different groups. Bacterial and fungal correlation networks (Spearman test) between different taxa were performed by Networkx (version.2.4, http://networkx.github.io), and only significant correlations (false discovery rate [FDR]‐corrected p < 0.05) with an absolute correlation coefficient > 0.5 were displayed. Microbial modularity analyses were performed using the modularity function in Gephi software, and inter‐kingdom and intra‐kingdom networks in modules were generated by calculating Spearman correlations with the FDR‐corrected p value. Statistical analysis was performed using R package or Prism. Data are presented as mean ± SD. A normality test was conducted to assess the distribution of the variables. If the data were normally distributed, a paired t‐test was used for comparisons, otherwise a nonparametric test (Wilcoxon signed‐rank test) was used for comparisons of paired samples. A FDR‐corrected p < 0.05 was considered significant.
RESULTS
Characteristics of subjects
For the bacteriome and mycobiome analyses, 14 and 13 asthma patients were enrolled (baseline) and followed up at 3 months (visit 1) and 9 months (visit 2), respectively. Half the patients were male, and most had a history of rhinosinusitis. FEV1%pre were 79.2 ± 24.8 and 78.97 ± 23.1, respectively. ACQ 7 scores were 1.05 ± 0.72 and 1.04 ± 0.75, respectively (Table S1). We compared the ACQ 7 score in patients before and after treatment and found that the ACQ 7 score at visit 1 and visit 2 were significantly lower than that at baseline (Figure S2).
Effect of ICS on airway bacteriome
After filtering for low‐quality reads, the qualified sequence reads in each sample were normalized. Finally, 27,828 sequence reads per sample were used for the subsequent analyses, resulting in 18 phyla, 116 families, 222 genera, and 648 OTUs. Rarefaction curves (Shannon and Sobs indices) of all samples at the OTU level reached a plateau, indicating that the sequencing data were large enough to reflect the microbial information of all the samples in our study (Figure 1a).
FIGURE 1.
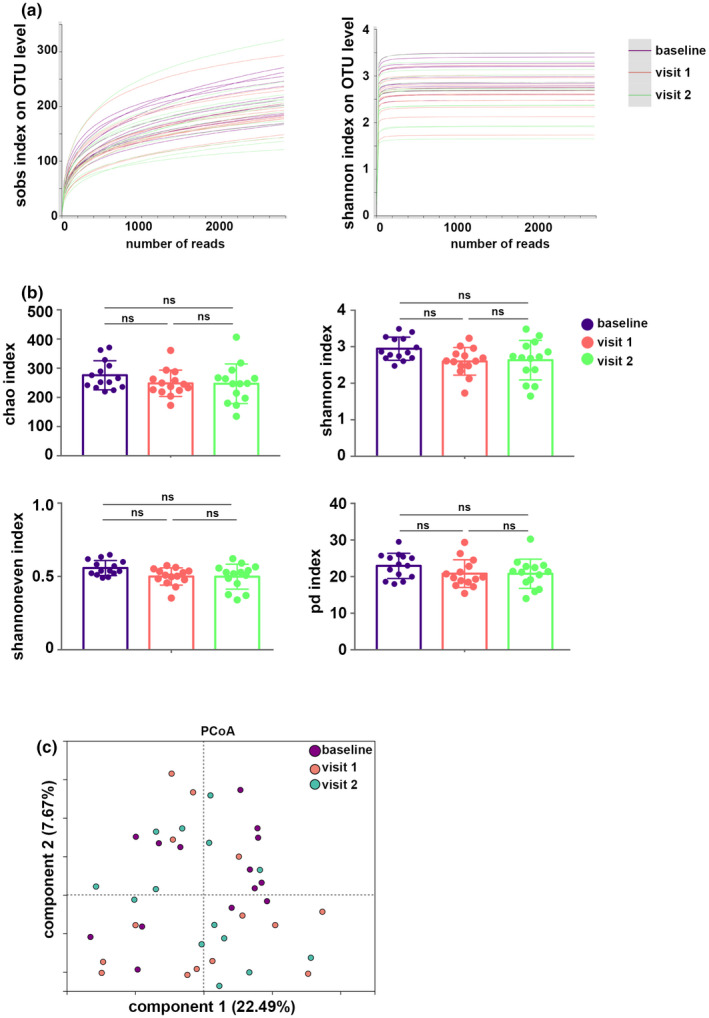
Basic estimations of airway bacteriome. (a) Rarefaction curves (Shannon and Sobs indices) on operational taxonomic unit (OTU) level. (b) Chao, Shannon, Shannoneven, and phylogenetic diversity (PD) indices of the airway bacteriome. (c) Principal coordinate analysis (PCoA) based on Bray–Curtis distances. NS, not significant.
The Chao (community richness) index, Shannoneven (community evenness) index, and Shannon (community diversity) index were used to describe the α diversity features of our bacterial community results. The PD index was also calculated. We found that these indices were not significantly different between the baseline, visit 1, and visit 2 (Figure 1b). PCoA showed a similarity of bacterial community composition, and it was supported by PERMANOVA analysis based on Bray–Curtis distances (R 2 = 0.047, p = 0.492; Figure 1c, Table S2).
Detection of the OTUs revealed 545 OTUs in the baseline, 503 OTUs in the visit 1, and 488 OTUs in the visit 2; a total of 400 OTUs were shared by all of them; unique OTUs in baseline, visit 1, and visit 2 were 92, 36, and 32, respectively (Figure 2a). The taxonomic assignment for the sequencing data was carried out and the relative abundance of the airway microbiome was calculated; the percentages of phyla and genera are shown in Figure 2b and c. The airway bacteriome of all the samples was dominated by four major phyla: Firmicutes, Actinobacteriota, Fusobacteria, and Proteobacteria (Figure 2b). The genera Streptococcus, Rothia, Actinomyces, Leptotrichia, and Neisseria were the most frequent in all samples (Figure 2c).
FIGURE 2.
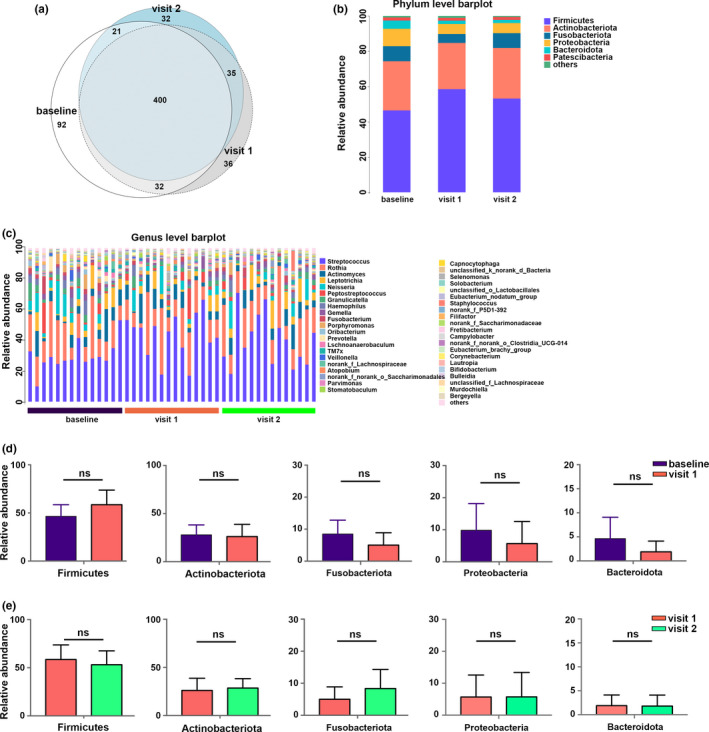
Taxa distribution and compositional comparisons of airway bacteriome. (a) Venn diagram illustrating the bacterial operational taxonomic units (OTUs) in sputum for the baseline, visit 1, and visit 2. (b, c) Bar charts of the airway bacterial composition at the phylum and genus levels. (d) Comparisons of the most abundant phyla between the baseline and visit 1. (e) Comparisons of the most abundant phyla between visit 1 and visit 2. NS, not significant.
Next, we investigated the effects of ICS treatment on the bacterial composition of asthma patients. At the phylum level, the abundant phyla Firmicutes, Actinobacteriota, Proteobacteria, Fusobacteriota, and Bacteroidota did not show any difference between the baseline and visit 1, or between visit 1 and visit 2 (Figure 2d and e). At the genus level, no significant differences in the relative abundances of the top seven genera Streptococcus, Rothia, Actinomyces, Leptotrichia, Neisseria, Peptostreptococcus, and Granulicatella were observed between the baseline and visit 1, or between visit 1 and visit 2 (Figure S3).
Effect of ICS on airway mycobiome
Qualified sequence reads in each sample were normalized, and 30,370 sequence reads per sample were used for further analyses, resulting in 9 phyla, 201 families, 359 genera, and 789 OTUs. Rarefaction curves (Shannon and Sobs indices) of all samples at the OTU level reached a plateau, indicating that the sequencing data were large enough to reflect the microbial information of all the samples in our study (Figure 3a).
FIGURE 3.
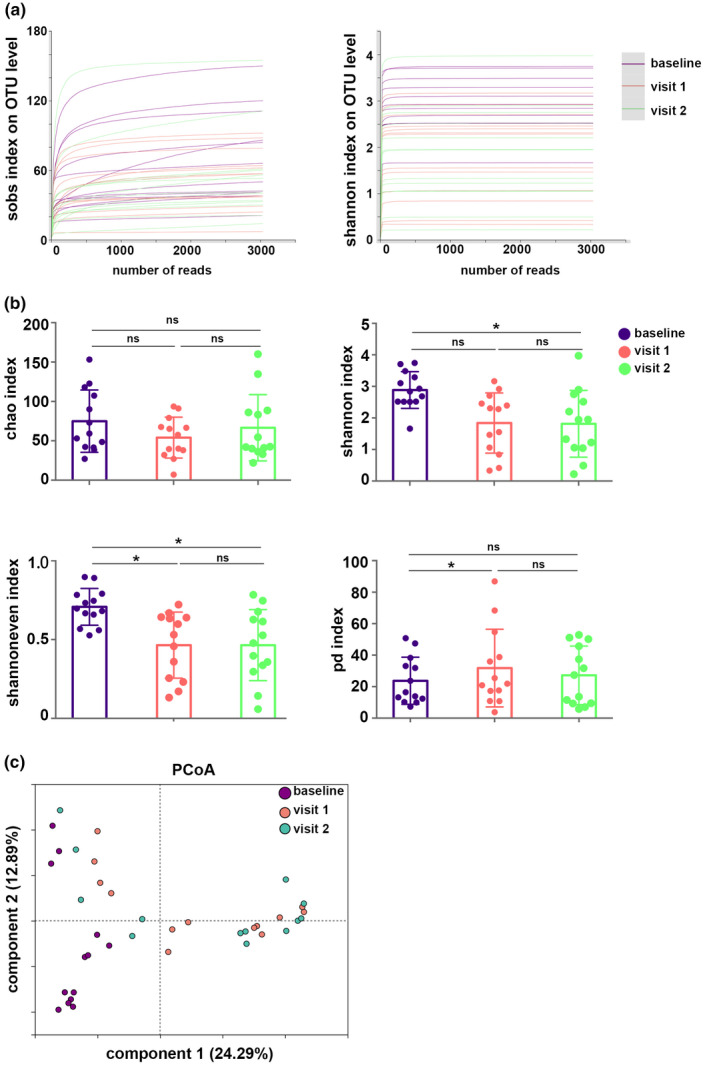
Basic estimations of airway mycobiome. (a) Rarefaction curves (Shannon and Sobs indices) on operational taxonomic unit (OTU) level. (b) Chao, Shannon, Shannoneven, and phylogenetic diversity (PD) indices of the airway mycobiome. Corrected *p < 0.05. (c) Principal coordinate analysis (PCoA) based on Bray–Curtis distances. NS, not significant.
The analysis revealed lower fungal diversity and lower fungal evenness in visit 2 compared with the baseline, as evidenced by the lower Shannon and Shannoneven indices. Visit 1 showed lower fungal evenness (Shannoneven index) and higher phylogenetic diversity than the baseline. Microbial richness (Chao) did not show statistical difference between the baseline and visit 1, or between the baseline and visit 2. Moreover, there were no significant differences in all of these indices between visit 1 and visit 2, indicating that ICS could alter fungal diversity or evenness, and 9‐month ICS therapy may not further affect the alterations of the mycobiome induced by 3 months of therapy (Figure 3b). PCoA showed differences in the fungal community composition between baseline, visit 1, and visit 2, and this was confirmed by PERMANOVA analysis based on Bray–Curtis distances (R 2 = 0.16, p = 0.001; Figure 3c, Table S3).
The comparison of the OTUs revealed 437 OTUs in the baseline, 317 OTUs in visit 1, and 375 OTUs in visit 2; a total of 98 OTUs were shared by them; unique OTUs in baseline, visit 1, and visit 2 were 237, 128, and 182, respectively (Figure 4a). The airway mycobiome of all the samples was dominated by four major phyla (Ascomycota, Basidiomycota, unclassified_k_Fungi, and Mortierellomycota) and seven genera (Aspergillus, Candida, Wallemia, Cladosporium, Saccharomyces, Penicillium, and Alternaria) (Figure 4b and c).
FIGURE 4.
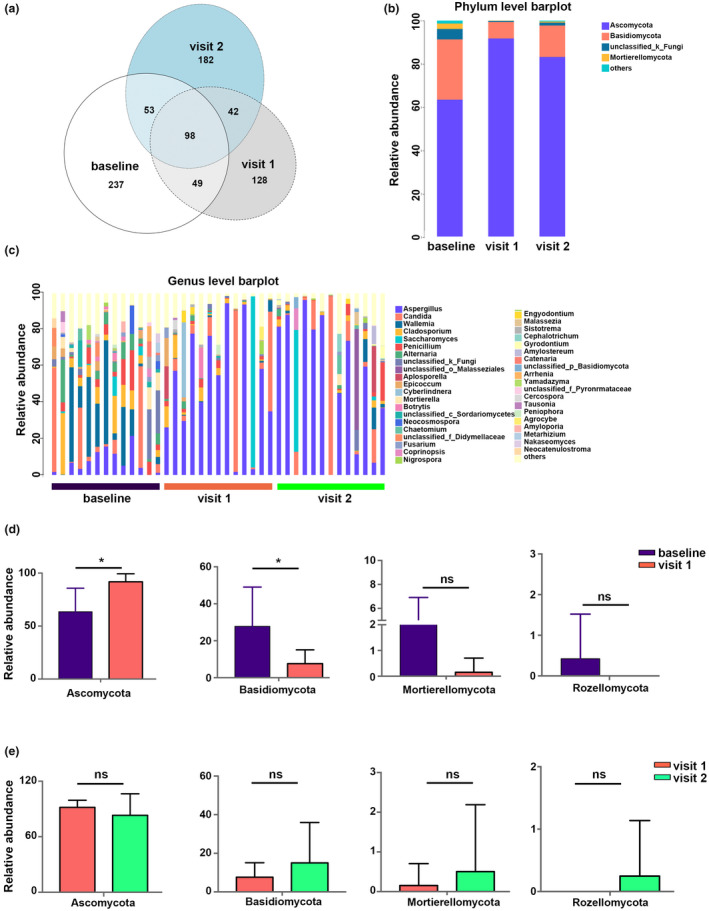
Taxa distribution and compositional comparisons of airway mycobiome. (a) Venn diagram illustrating the fungal operational taxonomic units (OTUs) in sputum for the baseline, visit 1, and visit 2. (b, c) Bar charts of the airway fungal composition at the phylum and genus levels. (d) Comparisons of the most abundant phyla between the baseline and visit 1. Corrected *p < 0.05. (e) Comparisons of abundant phyla between visit 1 and visit 2. NS, not significant.
Comparisons of the community composition at the phylum level revealed an enhanced frequency of Ascomycota and lower abundances of Basidiomycota in visit 1 compared with the baseline (Figure 4d); however, the relative abundances of these phyla did not show significant differences between visit 1 and visit 2 (Figure 4e). Among the top seven genera, we showed that compared with the baseline, the percentages of Wallemia, Cladosporium, Penicillum, and Alternaria tended to show a more than two‐fold decrease in visit 1 and visit 2, although the difference was not statistically significant (Figure S4).
Associations between the bacteriome, mycobiome, and clinical characteristics of asthma patients
We further investigated the association between the bacteriome and clinical characteristics of the patients. At the phylum level, the relative abundance of Proteobacteria was negatively correlated with sputum EOS%. The relative abundance of Pastescibacteria was negatively correlated with ACQ 7 score, and positively correlated with sputum EOS% and NEU% (Figure 5a, left). At the genus level, the frequency of Haemophilus and Fusobacterium was negatively associated with sputum EOS% (Figure 5a, right). With respect to the airway mycobiome, the relative abundance of the phylum Basidiomycota was positively correlated with sputum EOS% and NEU% (Figure 5b, left). The relative abundances of the genera Saccharomyces, Wallemia, and Aplosporella were positively correlated with sputum EOS% (Figure 5b, right).
FIGURE 5.
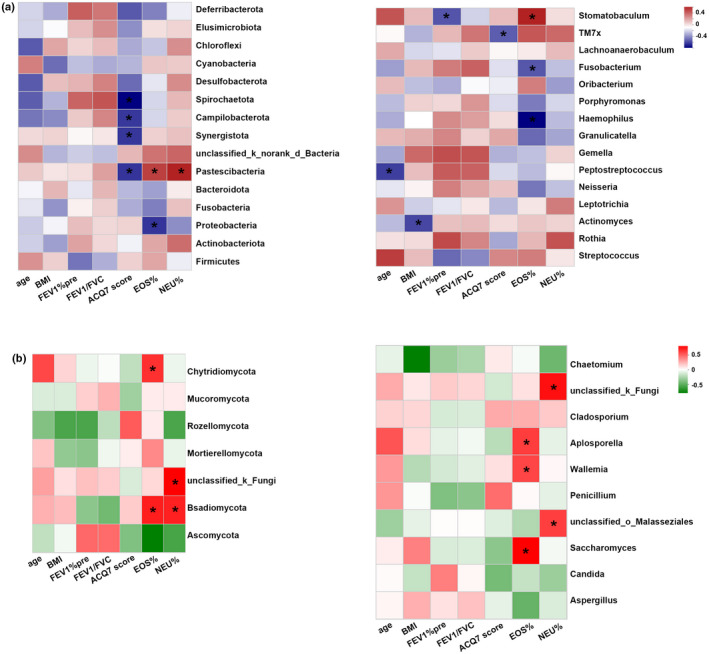
Heatmap showing the correlation between airway bacteriome (a), mycobiome (b), and clinical indices of asthma at the phylum (left) and genus (right) levels. Correlations were performed based on the Spearman correlation with false discovery rate (FDR) correction. Each row indicates a phylum or genus, and each column corresponds to a trait. The table is color coded by correlation according to the color legend. Corrected *p < 0.05. ACQ, Asthma Control Questionnaire; BMI, body mass index; EOS%, percentage of eosinophils; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; NEU%, percentage of neutrophils.
Bacterial and fungal networks in baseline, visit 1, and visit 2
Bacterial networks
We built a bacterial correlation network at the OTU level to assess the bacterial ecosystem structure in the baseline, visit 1, and visit 2. Globally, compared with visit 1 and visit 2, the baseline showed a rich and complex network of correlations between bacteria as indicated by increased node degree and closeness centrality. In the baseline, the network comprised one large‐connected component containing 50 OTUs with an average node degree of 11.84 (SD 7.19) and closeness centrality value of 0.46 (SD 0.08; Figure 6a). In visit 1, the network was comprised of one large‐connected component containing 49 OTUs with an average node degree of 6.89 (SD 3.5) and closeness centrality value of 0.39 (SD 0.06; Figure 6b). In visit 2, the network was comprised of 49 OTUs with an average node degree of 5.63 (SD 3.1) and closeness centrality value of 0.36 (SD 0.06; Figure 6c).
FIGURE 6.
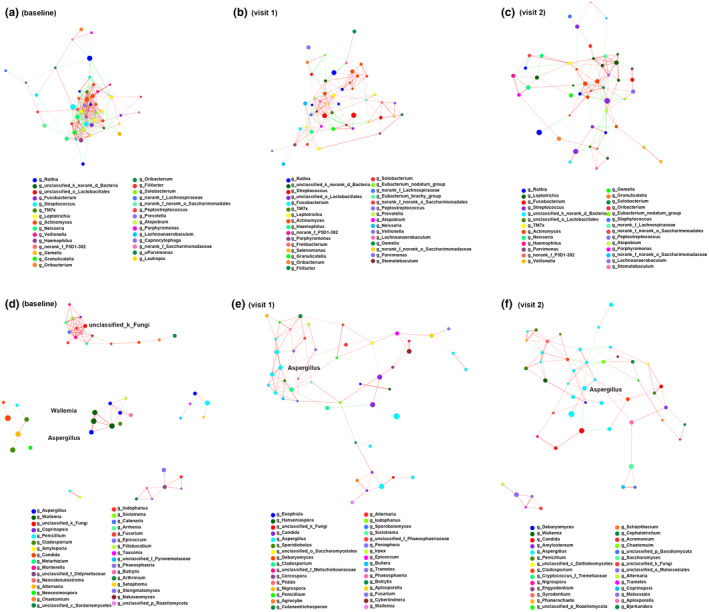
Bacterial and fungal networks from baseline, visit 1, and visit 2 using Networkx. (a–c) Bacterial networks. (d–f) Fungal networks. Each colored edge shows a statistical correlation to indicate positive correlation (red) or negative correlation (green) (determined using the Spearman test). Same node color indicates a species from the same genus. Only significant correlations (corrected p value < 0.05) with an absolute correlation coefficient > 0.5 are displayed.
Fungal networks
Fungal networks in baseline consisted of one large‐connected component, four small connections, and one dyad. In detail, an unclassified_k_Fungi was the core genus in the large connection, while members from Wallemia and Aspergillus were positively correlated in the small connections (Figure 6d). Visit 1 and visit 2 showed similar fungal networks, in which members from Aspergillus showed extensive correlations with several genera such as Penicillium, Cladosporium, Candida, and Wallemia (Figure 6e and f).
These results suggested that the fungal correlations were less complex and interconnected in asthma patients treated with ICS, and highlighted a distinct fungal topology between baseline, visit 1, and visit 2.
Effect of ICS on inter‐kingdom and intra‐kingdom connectivity in modules
To investigate inter‐ and intra‐kingdom connectivity of the baseline, visit 1, and visit 2, modularity‐based co‐occurrence networks between fungal and bacterial taxa were parsed at a Spearman correlation cutoff of 0.6 and a corrected p value of less than 0.05. Modules that maintained highly connected sets of nodes with few connections to other modules were identified.
Overall, the baseline and visit 1 had a greater density of connections between nodes compared with visit 2. As shown in Figure 7a (baseline), extensive bacteria–fungi, bacteria–bacteria, and fungi–fungi connections were revealed. We observed one major module of co‐occurring bacterial networks associated with fungal nodes such as Candida, Cladosporium, Alternaria, Penicillium, and Aspergillus. Other trans‐kingdom networks showed associations between Fusarium and Granulicatella, Porphyromonas, Neisseria (orange module), between Aspergillus and Bulleidia (green module), between Wallemia and Peptostreptococcus (pink module), and between Penicillium and Stomatobaculum (purple module). There was also a major bacterial module consisting of Lachnoanaerobaculum, Provella, Actinomyces, Selenomonas, Rothia, Fusobacterium, Streptococcus, and Leptotripia (yellow module). With respect to fungal networks, Candida significantly correlated with Penicillium and Motierella. Four additional fungal networks are also shown in Figure 7a.
FIGURE 7.
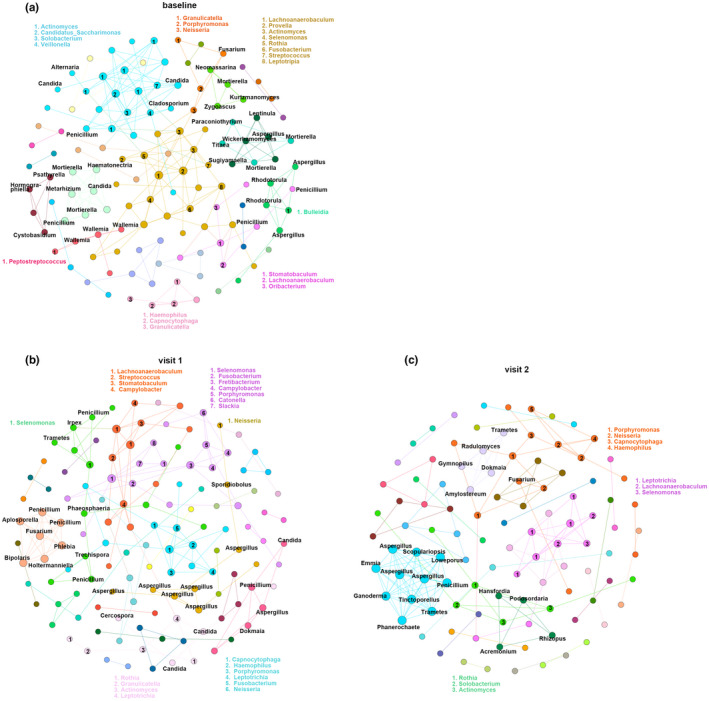
Effect of inhaled corticosteroids on inter‐kingdom and intra‐kingdom connectivity between different modules in asthma patients. Networks represent statistically significant correlations (corrected p < 0.05). Fungal nodes are labeled inside the networks (black), and bacterial nodes are labeled outside the network in different colors according to the modules. Nodes are colored by modules or microbial communities at a resolution of 0.8 using the modularity function in Gephi software.
In visit 1, we observed only small bacteria–fungi co‐occurring networks (green and light pink modules). In the bacteria–bacteria networks, Haemophilus showed interactions with Capnocytophaga, Porphyromonas, Leptotrichia, Fusobacterium, and Neisseria (blue module). Streptococcus showed associations with Lachnoanaerobaculum, Stomatobaculum, and Campylobacter (orange module) (Figure 7b). Specifically, the fungi–fungi networks showed that Fusarium was significantly associated with the fungal genera Penicillium, Phlebia, Aplosporella, Bipolaris, and Holtermanniella (light orange module). Penicillium was significantly correlated with Aspergillus and Candida in the pink module. In addition, we found several correlations between Aspergillus species (Figure 7b).
However, unlike the baseline and visit 1, there were fewer modules in networks in visit 2. Only one bacteria–fungi association between Fusarium and Neisseria was observed in the orange module. Bacterial genus Lachnoanaerobaculum showed interactions with bacterial genera Leptotrichia and Selenomonas (purple module). Similarly, Actinomyces was correlated with Rothia and Solobacterium (green module). Notably, we found distinct fungi–fungi correlations. For instance, Aspergillus significantly correlated with Penicillium, Tinctoporellus, Trametes, Phanerochaete, Ganoderma, Emmia, Scopulariopsis, and Loweporus (blue module) (Figure 7c).
DISCUSSION
In this study, we compared the mycobiome and bacteriome communities in the airways of asthma patients with regular visits (3 months, 9 months). We investigated the effect of ICS on airway microbiome and cross‐domain connections. There were no differences in bacterial richness, diversity, and evenness between the baseline, visit 1, and visit 2. Visit 2 showed lower fungal diversity and evenness than the baseline, and visit 1 showed lower fungal evenness than the baseline. The fungal, but not bacterial, community composition differed significantly between the baseline, visit 1, and visit 2. The relative abundances of abundant bacterial phyla and genera did not differ significantly between them. With regard to mycobiome, compared with baseline, visit 1 showed increased frequency of the phyla Ascomycota and lower abundance of Basidiomycota; however, these differences were not observed between visit 1 and visit 2. Compared with baseline, the percentage of abundant genera Wallemia, Cladosporium, Penicillum, and Alternaria sharply decreased in visit 1 and visit 2, although they did not reach statistical difference. We also investigated the associations between microbiome and clinical factors. In addition, we reported the distinct inter‐kingdom and intra‐kingdom connections in baseline, visit 1, and visit 2.
Maintenance treatment with ICS is the basic therapy for asthma. The effect of ICS on airway microbiome dysbiosis has not yet been fully delineated. In the present study we demonstrated no differences in bacterial richness, diversity, and evenness in asthma patients with different states of ICS treatment. No significant differences were observed in the bacterial community structure (PCoA based on Bray–Curtis distances) between the baseline, visit 1, and visit 2, which is consistent with the findings of Durack et al. 14 Community compositional comparisons of frequent phyla and genera showed no differences, which did not suggest the bacterial alterations to be as previously considered. This is inconsistent with the observations of ICS or oral corticosteroid (OCS)‐associated differences in microbial compositions noted in previous studies. 10 , 18 For instance, Denner et al. reported corticosteroid‐associated bacterial differences, especially following combination therapy of ICS and OCS. They found an increase in the abundance of Proteobacteria and Pseudomonas, and a decrease in that of Bacteroidetes, Fusobacteria, Prevotella, and Veillonella in endobronchial brushing (EB) samples of asthma patients receiving ICS compared with those without ICS use. Moreover, the generalized linear regression model (GLM) predicted OCS as having a significant influence on the proportions of genera Pseudomonas, Rickettsia, Prevotella, Lactobacillus, and Streptococcus species. 10 This discrepancy could be attributable to different sample types (EB vs. induced sputum) and different research methodologies. We also noted that Denner et al. demonstrated the effect of ICS on the bacteriome based on the stratification of patients with or without ICS treatment. Therefore, their study might not be directly comparable in explaining the impact of ICS on the airway microbiome; instead, further longitudinal research will be more powerful in helping to explain this issue. Despite the insignificant differences in frequent compositional comparisons, we found correlations between bacteria and clinical indicators. For instance, the phylum Proteobacteria, genera Haemophilus, and Fusobacterium were negatively associated with sputum EOS%, which is consistent with previous studies. Genera Haemophilus, Moraxella, and Neisseria belong to the phylum Proteobacteria. Taylor et al. demonstrated that the relative abundance of Moraxella taxon positively correlated with sputum NEU%, Haemophilus taxon negatively correlated with EOS%. 3 Sverrild et al. reported a significantly higher proportion of Neisseria in ‘eosinophil low’ compared to ‘eosinophil high’ asthma. 4 The persistence of Haemophilus colonization in the airways, an important pathogenic entity in asthma, 19 was associated with switches from eosinophilic to neutrophilic inflammation. 20 Haemophilus‐predominant asthma patients were shown to be ICS non‐responders who showed enhancement of xenobiotic degradation capacity. 14 However, the mechanisms of the switches of inflammatory state induced by Haemophilus still remain to be delineated.
With respect to airway mycobiome, we demonstrated the most abundant mycobiome phyla (Ascomycota, Basidiomycota, unclassified_k_Fungi, and Mortierellomycota) and genera (Aspergillus, Candida, Wallemia, Cladosporium, Saccharomyces, Penicillium, Alternaria). The result showed decreased fungal diversity and evenness in patients receiving ICS therapy compared with those not receiving ICS treatment (baseline). ICS could affect microbial communities (mycobiome), as evidenced by the significantly different community composition between the baseline and visit 1, and visit 2. Moreover, we found that asthma patients had an increased abundance of phyla Ascomycota, and a lower abundance of Basidiomycota, after 3 months of ICS treatment. The relative abundance of phylum Basidiomycota was positively correlated with sputum EOS% and NEU%. At the genus level, compared with the baseline, the percentages of abundant genera Wallemia, Cladosporium, Penicillium, and Alternaria sharply decreased in visit 1 and visit 2, although the differences were not statistically significant. Wallemia, Cladosporium, Penicillium, and Alternaria have been suggested to be associated with asthma in pioneering research, 11 , 21 and we found that the relative abundances of the genera Wallemia, Saccharomyces, and Aplosporella were positively correlated with sputum EOS%. A previous study showed that species from the genus Wallemia could induce immediate positive reactions of skin prick tests and radioallergosorbent tests (RAST) in asthma patients, which indicates that this fungal species may be important as a causative agent in atopic diseases. 22 These results indicated that ICS not only affects the mycobiome diversity, but also influences the relative abundances of specific taxa associated with asthma. These results are partly consistent with those of Sharma et al., who compared mycobiome composition between asthmatic patients with and without ICS use. They identified that the abundance of the taxa belonging to the genus Penicillium decreased in patients using ICS. 11 Of note, our study may be a more direct way of observing the altered fungal signature caused by ICS use than those results performed by GLMs. Our results indicated that ICS treatment can influence airway mycobiome. However, there have been no large‐scale longitudinal studies that attempted to discern the effect of ICS on the airway microbiome, especially the mycobiome. Therefore, more extensive research to investigate the direct influence of ICS on airway bacteriome and mycobiome is warranted.
In addition, we investigated the bacterial or fungal correlations in different disease states. We found that the bacterial correlations in visit 1 and visit 2 were less complex and interconnected than those in the baseline, indicating that ICS treatment affected the associations between airway bacterial species. Moreover, we also highlighted the distinct fungal topology in the three states, especially the discriminate correlations of airway mycobiome in ICS‐treated patients when compared with patients not treated with ICS.
Fungi and bacteria coexist in the gut and airway, where they can exhibit a regulatory role in the immune system by interacting with each other and the human microenvironment. 23 To gain insights into the interactions of bacteria–bacteria, bacteria–fungi, and fungi–fungi, global intra‐ and inter‐kingdom equilibria according to disease phenotype at the genus level involving both bacteria and fungi were established. Globally, more dense significant correlations were observed in the baseline and visit 1 compared with visit 2. Modularity‐based correlation networks observed distinct patterns in the baseline, visit 1, and visit 2. Specifically, in the baseline, we identified fungi−fungi correlations between species from Candida, Mortierella, and Penicillium, and between Wallemia species. In visit 1, we observed correlations between Candida, Penicillium, and Aspergillus, between Penicillium and Fusarium, and associations within species from Aspergillus. However, in visit 2, we noted the correlations between Penicillium and Aspergillus. These fungi were potential pathogens of asthma, 11 , 24 and our study suggested the potential effect of asthmatic treatment (ICS) on microbial interactions in the airway, which is worthy of continued investigation.
There are some limitations with our study. Inclusion of an untreated control group would be more rigorous for the present study. Although the induced sputum could be used to assess the airway microbiome, additional microbial analysis in BALF and lungs is needed to compare the similarities and differences between different sample types. Further longitudinal studies are also required to investigate how the microbial diversity and community composition change with the time of ICS treatment, given the relatively short follow‐up time. Besides, we did not assess the compliance with ICS regimens, which may exert an effect on airway microbiome. We also noted that the altered microbial correlations in our analysis are preliminary results; the microbe–microbe relationships suggested are only correlative, and further experimentation on isolated microbes will be needed to validate these relationships. Given that these microbial interactions may alter the host immune response in asthma, the clinical consequences of altered microbial interactions in ICS‐treated patients are our further research avenues to pursue, especially the biological consequences of interactions between Aspergillus and other taxa in our study.
This study refines the list of airway microbiome associated with asthma therapies. ICS use altered the airway microbial diversity, evenness, community composition, and microbial connections. Our preliminary results regarding changes in the airway microbiome based on ICS treatment over time might provide additional information on the role of ICS in asthma. Consequently, large‐scale longitudinal studies that attempt to discern the effect of ICS on the airway microbiome and microbial cross‐domain connections merit further research.
CONFLICT OF INTEREST
The authors have declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
C.H. wrote the manuscript. G.S. designed the research. C.H. and Y.N. performed the research. C.H analyzed the data. W.D. contributed analytical tools.
Supporting information
Appendix S1
Huang C, Ni Y, Du W, Shi G. Effect of inhaled corticosteroids on microbiome and microbial correlations in asthma over a 9‐month period. Clin Transl Sci. 2022;15:1723‐1736. doi: 10.1111/cts.13288
Chunrong Huang and Yingmeng Ni contributed equally to this work.
Funding information
This study was supported by the following grants: National Natural Science Foundation of China (82170023, 81970020), Shanghai Municipal Health Commission (2019SY006), Shanghai Key Laboratory of Emergency Prevention, Diagnosis and Treatment of Respiratory Infectious Diseases (20dz2261100), Shanghai Municipal Key Clinical Specialty (shslczdzk02202), Cultivation Project of Shanghai Major Infectious Disease Research Base (20dz2210500), Shanghai Key Discipline for Respiratory Diseases (2017ZZ02014), and Shanghai Shenkang Hospital Development Center Clinical Science and Technology Innovation Project (SHDC12018102).
REFERENCES
- 1. Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The role of lung and gut microbiota in the pathology of asthma. Immunity. 2020;52:241‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hufnagl K, Pali‐Schöll I, Roth‐Walter F, Jensen‐Jarolim E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin Immunopathol. 2020;42:75‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor SL, Leong LEX, Choo JM, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141:94‐103.e15. [DOI] [PubMed] [Google Scholar]
- 4. Sverrild A, Kiilerich P, Brejnrod A, et al. Eosinophilic airway inflammation in asthmatic patients is associated with an altered airway microbiome. J Allergy Clin Immunol. 2017;140:407‐417.e11. [DOI] [PubMed] [Google Scholar]
- 5. Li N, Qiu R, Yang Z, et al. Sputum microbiota in severe asthma patients: relationship to eosinophilic inflammation. Respir Med. 2017;131:192‐198. [DOI] [PubMed] [Google Scholar]
- 6. Zhang Q, Cox M, Liang Z, et al. Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS One. 2016;11:e0152724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simpson JL, Daly J, Baines KJ, et al. Airway dysbiosis: haemophilus influenzae and tropheryma in poorly controlled asthma. Eur Respir J. 2016;47:792‐800. [DOI] [PubMed] [Google Scholar]
- 8. Huang YJ, Nelson CE, Brodie EL, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 2011;127:372‐381.e1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang YJ, Nariya S, Harris JM, et al. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136:874‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Denner DR, Sangwan N, Becker JB, et al. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J Allergy Clin Immunol. 2016;137:1398‐1405.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma A, Laxman B, Naureckas ET, et al. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol. 2019;144:1214‐1227.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rick E‐M, Woolnough KF, Seear PJ, et al. The airway fungal microbiome in asthma. Clin Exp Allergy. 2020;50:1325‐1341. [DOI] [PubMed] [Google Scholar]
- 13. Martin MJ, Zain NMM, Hearson G, et al. The airways microbiome of individuals with asthma treated with high and low doses of inhaled corticosteroids. PLoS One. 2020;15:e0244681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durack J, Lynch SV, Nariya S, et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140:63‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartmann JE, Albrich WC, Dmitrijeva M, Kahlert CR. The effects of corticosteroids on the respiratory microbiome: a systematic review. Front Med (Lausanne). 2021;8:588584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sohn S‐W, Lee H‐S, Park H‐W, et al. Evaluation of cytokine mRNA in induced sputum from patients with allergic rhinitis: relationship to airway hyperresponsiveness. Allergy. 2008;63:268‐273. [DOI] [PubMed] [Google Scholar]
- 17. Huang C, Yu Y, Du W, et al. Fungal and bacterial microbiome dysbiosis and imbalance of trans‐kingdom network in asthma. Clin Transl Allergy. 2020;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goleva E, Jackson LP, Harris JK, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188:1193‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diver S, Richardson M, Haldar K, et al. Sputum microbiomic clustering in asthma and chronic obstructive pulmonary disease reveals a Haemophilus‐predominant subgroup. Allergy. 2020;75:808‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z, Locantore N, Haldar K, et al. Inflammatory endotype‐associated airway microbiome in chronic obstructive pulmonary disease clinical stability and exacerbations: a multicohort longitudinal analysis. Am J Respir Crit Care Med. 2021;203:1488‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soffer N, Green BJ, Acosta L, et al. Alternaria is associated with asthma symptoms and exhaled NO among NYC children. J Allergy Clin Immunol. 2018;142:1366‐1368.e10. [DOI] [PubMed] [Google Scholar]
- 22. Sakamoto T, Torii S, Yamada M, et al. Allergenic and antigenic activities of the osmophilic fungus Wallemia sebi asthmatic patients. Arerugi. 1989;38:352‐359. [PubMed] [Google Scholar]
- 23. Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khosravi AR, Fatahinia M, Shokri H, Yadegari MH. Allergens from Fusarium solani identified by immunoblotting in asthma patients in Iran. Arh Hig Rada Toksikol. 2012;63:1‐6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1