

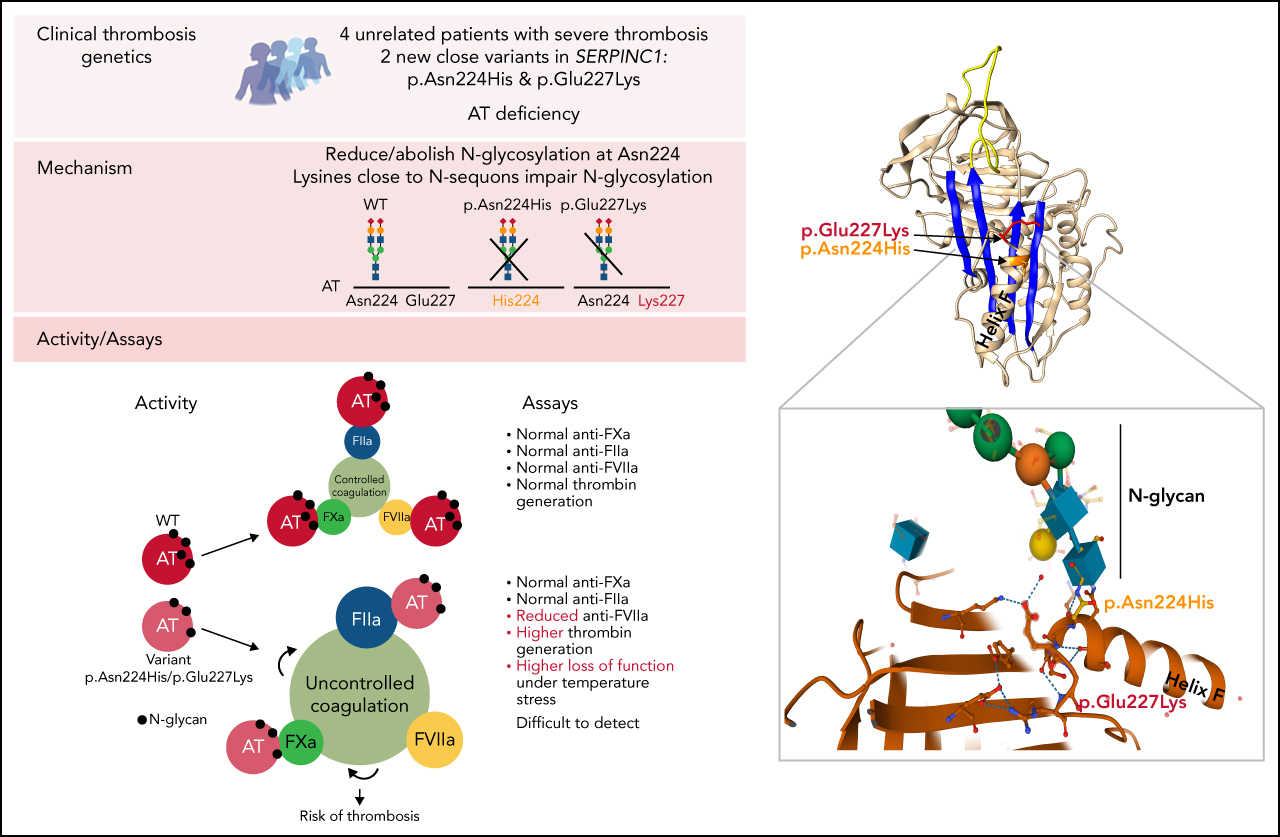
Antithrombin (AT), encoded by the gene SERPINC1, is a serine protease inhibitor whose deficiency causes a severe form of dominantly inherited thrombophilia. de la Morena-Barrio et al describe 2 novel variants of AT with altered glycosylation profiles that were identified in 4 unrelated families with thrombophilia. This study finds that although these mutations are clearly associated with abnormal thrombosis, carriers are not identified using routine testing for AT deficiency. These discoveries provide insight into the regulation of AT function, and they have implications for the investigation of severe thrombophilia, particularly in families.
Key Points
Severe thrombophilia caused by antithrombin deficiency is detected only by impaired anti-FVIIa activity and increased thrombin generation.
This antithrombin deficiency is caused by 2 new SERPINC1 variants affecting N-glycosylation.
Visual Abstract

Abstract
Antithrombin deficiency, the most severe congenital thrombophilia, might be underestimated, as some pathogenic variants are not detected by routine functional methods. We have identified 2 new SERPINC1 variants, p.Glu227Lys and p.Asn224His, in 4 unrelated thrombophilic patients with early and recurrent thrombosis that had normal antithrombin activity. In one case, the mutation was identified by whole genome sequencing, while in the 3 remaining cases, the mutation was identified by sequencing SERPINC1 based on a single functional positive finding supporting deficiency. The 2 variants shared a common functional defect, an impaired or null N-glycosylation of Asn224 according to a eukaryotic expression model. Carriers had normal anti-FXa or anti-FIIa activities but impaired anti-FVIIa activity and a detectable loss of inhibitory function when incubating the plasma for 1 hour at 41°C. Moreover, the β glycoform of the variants, lacking 2 N-glycans, had reduced secretion, increased heparin affinity, no inhibitory activity, and a potential dominant–negative effect. These results explain the increased thrombin generation observed in carriers. Mutation experiments reflected the role that Lysine residues close to the N-glycosylation sequon have in impairing the efficacy of N-glycosylation. Our study shows new elements involved in the regulation of N-glycosylation, a key posttranslational modification that, according to our results, affects folding, secretion, and function, providing new evidence of the pathogenic consequence of an incorrect N-glycosylation of antithrombin. This study supports that antithrombin deficiency is underestimated and encourages the development of new functional and genetic tests to diagnose this severe thrombophilia.
Introduction
Antithrombin is the most important endogenous anticoagulant. It inhibits not only thrombin but also all other procoagulant serine proteases.1 Additionally, as a member of the serpin superfamily, antithrombin inhibits target proteases by an efficient suicide mechanism that is enhanced by its cofactor, heparin.1 Thus, even mild antithrombin deficiencies significantly increase the risk of thrombosis.2 Congenital antithrombin deficiency is a dominant disorder. The defect of 1 allele of SERPINC1, the gene encoding antithrombin, significantly increases the risk of venous thrombosis, while complete or very severe deficiency causes embryonic lethality.3,4 Antithrombin deficiency was the first inherited thrombophilia identified and the most severe.5 Diagnosis of antithrombin deficiency has clinical implications for providing extended anticoagulant treatment in symptomatic carriers6 and for using thromboprophylaxis under predisposing situations in asymptomatic carriers.7 In addition, patients with antithrombin deficiency may benefit from a specific anticoagulant therapy by using concentrates of antithrombin.8 All these data support the screening of antithrombin deficiency among patients with thrombophilia, which is currently done in most hospitals by functional methods.9 Unfortunately, these methods may fail to detect some pathogenic variants causing type II deficiencies.10 This may also contribute to explaining the contradiction between the relatively high prevalence of antithrombin deficiency in the general population (around 1 out of 500),11 the high risk of thrombosis (odds ratio, >10) associated with this disorder,12 and the low incidence of antithrombin deficiency in consecutive cases with venous thrombosis (1% to 5%).13
Methods
Antithrombin assays in plasma samples
After informed consent following ethical guidelines, blood samples were collected, and plasma and DNA were obtained (supplemental Methods).
Anti-FXa and anti-FIIa assays were performed using chromogenic methods (supplemental Methods). Antigen levels were measured by Rocket immunoelectrophoresis, enzyme-linked immunoassay (ELISA), or immunonephelometry. Analysis of plasma antithrombin included crossed immunoelectrophoresis (CIE) in the presence of heparin and polyacrylamide gel electrophoresis. Western blot immunostaining and CIE was done using specific antihuman antithrombin polyclonal antibodies (A9522 from Sigma-Aldrich and OSAY09 from Siemens, respectively).
Thrombin–antithrombin complexes were evaluated by Western blotting in the presence and absence of thrombin.
FVIIa-antithrombin complexes were determined by ELISA (00491, Stago, Valencia, Spain) following the manufacturer’s procedures after the incubation of plasma with recombinant activated FVII (FVIIa) (Novoseven, Novo Nordisk, Madrid, Spain) and unfractionated heparin (UFH, Hospira, Spain) for 30 minutes at 37°C. FVIIa antithrombin complexes were also detected by Western blotting in basal plasma samples as well as in samples supplemented with FVIIa and unfractionated heparin.
Densitometry analyses of Western blots were done with the Image J software (ImageJ 1.49n).
Thrombin generation potential
Thrombin generation triggered by 5 pM tissue factor was measured in platelet-poor plasma using calibrated automated thrombogram (CAT) method.14 The main output parameter of this assay was the area under the thrombin generation curve, which represents the endogenous thrombin potential (ETP). ETP was calculated in nM.min by the Thrombinoscope software (Stago, Valencia, Spain).
Genetic analysis
For the French thrombophilic family, the proband was included in a whole-genome sequencing (WGS) program including 200 unrelated individuals with unprovoked venous thrombosis, family history of venous thrombosis, or multiple unprovoked venous thrombotic events, and no evidence of classical biological thrombophilia recruited in the CEHT (Centre d’Exploration des Pathologies Hémorragiques et Thrombiques) of La Timone Hospital, Marseille, France. WGS was done as indicated in the supplemental Methods. Variant calling was performed using the GATK Haplotype Caller (GenomeAnalysisTK-v3.3-0, https://software.broadinstitute.org/gatk/documentation/article.php?id=4148) tool, followed by recalibration. Single nucleotide variants that succeeded the “PASS” filter were then annotated using Annovar.15 Prediction of pathogenicity was made using a Mutation taster.16 The entire coding and flanking intron regions of SERPINC1 were sequenced by NGS on a Miseq sequencer (Illumina, France). Raw data analysis was done on Workbench (Qiagen, France) pipeline. The alignment was made using a variant studio (Illumina).
For all remaining cases, Sanger’s sequencing of the 7 exons and intron and exon boundaries of the SERPINC1 gene was done using primers and conditions previously described.17
Multiplex ligation-dependent probe amplification (Holland, SERPINC1) was performed in all patients following the manufacturer’s protocol.
Recombinant antithrombins
Antithrombin has 4 N-glycosylation sites, Asn128, Asn167, Asn187, and Asn224. Since Asn167 is inefficiently glycosylated, 2 glycoforms may be generated: α and β with 4 and 3 N-glycans, respectively.18,19 Expression of the human antithrombin complementary DNA (cDNA) in eukaryotic cells generates these 2 glycoforms at a 60/40 proportion (supplemental Figure 1). However, the higher heparin affinity of the β glycoform increases its clearance from circulation, and the proportions of antithrombin glycoforms in plasma are 90/10. To reduce the heterogeneity of antithrombin N-glycoforms and facilitate its purification by heparin affinity chromatography, most models of recombinant expression of human antithrombin usually introduce a mutation in the cDNA, Ser169Ala. Cells transfected with a plasmid containing this cDNA only generate β antithrombin (supplemental Figure 1). The β glycoform with this mutation (p.Ser169Ala) has full anticoagulant activity.17
For this study, recombinant antithrombins in wild-type (Ser169) or β (Ala169) backgrounds were generated by site-directed mutagenesis in the pCEP4-human antithrombin plasmid, generously donated by J. Huntington, and they were expressed in HEK293T-EBNA cells. Recombinant antithrombins were purified as described elsewhere.17
Glycosylation of recombinant antithrombins was studied by Western blotting/silver staining after PNGase F treatment (Sigma-Aldrich, Madrid, Spain) (supplemental Methods).
Functional analysis of recombinant antithrombins included chromogenic assays (anti-FXa and anti-FIIa) and identification of thrombin–antithrombin complexes by Western blotting, following the same procedures used in plasma.
Heparin affinity was evaluated by endogenous fluorescence titration as described elsewhere.20
For all studies, at least 3 independent experiments were done.
In silico studies and statistical analysis
Structural representation of antithrombins was performed by using SWISS-MODEL and the Swiss-PdbViewer programs21 using native (protein data bank, PDB accession number: 1t1fA) conformational states as a template. Electrostatic potential was studied using both Poisson-Boltzmann Solver program22 and a Coulomb computation method. Python Molecule Viewer23 was used as the graphic interface for the calculated Poisson-Boltzmann electrostatic potential.
Values were shown as mean (standard deviation) or median (interquartile range) for normally and nonnormally distributed data. Student t test (ANOVA for more than 2 groups) or Mann-Whitney U (nonparametric K samples analysis for more than 2 groups) tests were used for group comparisons of normally and nonnormally distributed data, respectively. A P value <0.05 was considered statistically significant. These studies were performed using IBM SPSS software v21.
Results
SERPINC1 p.Glu227Lys
Figure 1A and supplemental Table 1 summarize the clinical characteristics and main laboratory data from the index French thrombophilic family. The proband (patient 1) is a 44-year-old man presenting an unprovoked thrombosis of the portal vein. He had no acquired risk factors for vascular complications other than a history of tobacco in the previous 5 years. During the acute episode, under therapeutic doses of heparin, laboratory findings demonstrated low functional antithrombin (0.52 IU/mL [reference values 0.80 to 1.20 IU/mL]). A few months later, after stopping anticoagulant treatment, both functional (0.83 IU/mL) and immunologic (0.27 g/L; reference values 0.24 to 0.36 g/L) values of antithrombin were normal. Complete thrombophilia workup was negative.
Figure 1.

Thrombophilic families carrying new SERPINC1 variants affecting N-glycosylation of Asn224. Clinical information, including type and age of the first thrombotic event (between brackets) and recurrence, functional values (anti-FXa activity), antigen levels, and molecular data (including the electropherogram of exon 4 in a symptomatic patient and a healthy control patient) are shown. Symbols half filled with red represent heterozygous subjects, and a red border indicates a patient who had a thrombotic event. The proband is pointed by an arrow. (A) p.Glu227Lys. Pedigree tree of the index French thrombophilic family. Thrombin generation data in available subjects are also shown. The c.679G>A (p.Glu227Lys) mutation is pointed by a red arrow. (B) p.Asn224His. Pedigree 3 of 2 unrelated Norwegian families. The c.670A>C (p.Asn224His) mutation is pointed by a red arrow. DVT, deep venous thrombosis; PVT, portal venous thrombosis; PE, pulmonary embolism; ND, not determined; AT Ag, antithrombin antigen; ETP, endogenous thrombin potential; OC, oral contraceptives; R, recurrence; F1, family 1; F2, family 2.
His mother had a deep vein thrombosis (DVT) at the age of 70. Moreover, his 2 daughters presented venous thrombosis. The older one had an unprovoked proximal DVT and a massive pulmonary embolism (PE) at the age of 18 while on a combined oral contraceptive for 2 years. She had been smoking for 1 year. During the acute event and while receiving therapeutic heparin, both functional and immunological antithrombin levels were normal (0.80 IU/mL and 0.25 g/L, respectively); 3 days later, the functional dosage was low (0.66 IU/mL), apparently due to hemolysis. The younger daughter had a proximal DVT at the age of 19 when a plaster cast was applied for a sprained knee and while receiving a combined oral contraceptive for 4 years. Her antithrombin was normal (0.98 IU/mL of functional antithrombin).
Plasma antithrombin of 2 symptomatic members of this family (III-2 and III-3) was evaluated. CIE revealed normal antithrombin levels without a significant increase in forms with low heparin affinity (Figure 2A). However, an aberrant form of antithrombin with reduced electrophoretic mobility in native polyacrylamide gel electrophoresis (PAGE) was detected in both subjects (Figure 2B). Moreover, an aberrant form of antithrombin was also observed in the 2 sisters after native PAGE with 5 M urea (Figure 2B). The anticoagulant activity of these samples was nearly normal, as verified by anti-FXa and anti-FIIa activity (both progressive and heparin cofactor activity), and the levels of thrombin–antithrombin complexes formed after the addition of thrombin were also normal (Figure 2C).
Figure 2.

Analysis of plasma antithrombin (AT) in symptomatic members of the French thrombophilic family (II-3, III-2, and III-3, p.Glu227Lys+/−) and in a control plasma generated with a pool of 100 healthy blood donors. (A) Crossed immunoelectrophoresis in the presence of heparin. Plasma from a patient heterozygous carrier of the p.Arg79Cys variant (AT Toyama), a genetic defect that causes a type II deficiency with heparin-binding site (HBS) defect, was also included. (B) Western blotting of native PAGE in the presence and absence of 5M urea. The aberrant forms of antithrombin detected in the sisters are pointed by red arrows. (C) Western blotting after SDS-PAGE in the presence or absence of thrombin and unfractionated heparin (UFH). Thrombin-antithrombin complexes (T-AT) and antithrombin are pointed by arrows. The anti-FXa and anti-FIIa (heparin cofactor and progressive) activity of the samples evaluated by chromogenic assays is also indicated. (D) Thrombin generated in platelet-poor plasma of 3 symptomatic members of the French thrombophilic family (II-3, III-2, and III-3) and 1 healthy control after triggering the coagulation by 5 pM tissue factor. Thrombin was measured using the CAT method.
The thrombin generation potential of platelet-poor plasma from all symptomatic members of the family was evaluated using the 5 pM tissue factor. Supplemental Table 1 shows ETP results as well as other coagulation parameters. The first study was done on the proband (II-3) and youngest daughter (III-2) as they were not under anticoagulant treatment. Both subjects showed a high ETP (2759 and 3095 nM.min; normal range <2268 nM.min) when anti-FXa antithrombin activity of the same sample was 0.82 IU/mL and 0.98 IU/mL, respectively. A few months later, a CAT assay was done for patient III-3 showing a high ETP of 2254 nM.min (normal range <1616 nM.min) when her antithrombin activity was 0.85 IU/mL. Figure 2D shows the profile of thrombin generation observed in the 3 symptomatic members of the family and a healthy control subject.
The severe clinical phenotype of thrombosis in this family and increased thrombin generation capacity strongly suggested a dominant thrombophilia. However, as all thrombophilic studies rendered negative or conflictive results, the search for this thrombophilia was done by WGS in the proband. The selection of potentially pathogenic variants, considering a dominant effect, was firstly focused on genes involved in coagulation and inflammation. This analysis revealed an interesting heterozygous mutation in SERPINC1: c.679G>A, potentially responsible for a missense change p.Glu227Lys. This mutation is not included in the SERPINC1 mutation database of the HGMD, ExAC, or Gnomad. The mutation was confirmed by NGS and Sanger sequencing and verified in the 2 symptomatic daughters (Figure 1A).
The mutation changes an electronegative glutamic acid residue by a positively charged lysine. The glutamic acid 227 is not conserved among the serpin superfamily but is located at the end of helix F in a region containing a high proportion of conserved residues in the serpin superfamily (supplemental Figure 2). Moreover, it is highly remarkable the close position of Glu227 to the Asn224 N-glycosylation sequon (supplemental Figure 2).
In silico studies suggested a minor effect of this mutation on the molecular weight (1 Da less for the mutant) but a relevant impact on the pI; 6.32 for the wild-type and 6.97 for the mutant (supplemental Figure 3). This may explain the aberrant electrophoretic mobility in native PAGE of the variant antithrombin, as we have documented for other variants removing or inserting electropositive residues (supplemental Figure 4).
The significance of this mutation increased when we identified an unrelated patient carrying the same genetic variant that also shared other clinical, biochemical, and functional characteristics with the French family. The patient, a 43-year-old woman originally from Peru, had a severe clinical history of thrombosis, with a first early DVT event (31 years old) after immobilization due to trauma and a recurrent DVT after a transoceanic flight. The anti-FXa activity was low (0.60 IU/mL) in a sample drawn during the second event, but the sample collected 6 months later showed normal activity (0.82 IU/mL). The anti-FIIa heparin cofactor and progressive activity were normal (0.90 and 0.88 IU/mL, respectively). Antigen levels were also in the normal range (0.29 g/L). The aberrant antithrombin detected in the French family was also observed in this patient (supplemental Figure 5). The patient was also heterozygous for the prothrombin G20210A polymorphism (rs1799963).
The consequences of the p.Glu227Lys mutation were evaluated in a recombinant system. Site-directed mutagenesis to generate the p.Glu227Lys mutation was done in a plasmid containing the cDNA of human antithrombin in the β context (ie, with a mutation p.Ser169Ala). Expression of the Lys227 variant resulted in a twofold reduction of the secretion of the variant compared with the wild-type molecule. The mutation generated similar amounts of 2 antithrombin forms, one with the wild-type size and one smaller (Figure 3A). As the mutation was close to an N-glycosylation site (Asn224), we speculated a potential hypoglycosylation. This hypothesis was confirmed by PNGase F treatment, as both forms uniformed their size, which was identical to that of the wild-type molecule treated with this glycosidase (Figure 3A). To demonstrate that the Lys227 variant impaired N-glycosylation, Lys227 was mutated to Ala227, and the resulting antithrombin was fully glycosylated at Asn224 (Figure 3B).
Figure 3.

Recombinant antithrombin Lys227 variant expressed in HEK-EBNA cells. (A) Glycoforms generated in the β context (Ala169) revealed an impaired N-glycosylation in Lys227 antithrombin compared with Glu227 antithrombin (residue present in the wild-type molecule). The study was done with (+) or without (−) treatment with PNGase F. (B) Forms secreted to the conditioned medium of cells transfected with the Glu227 (residue present in the wild-type molecule) plasmid and the mutated plasmids Lys227 and Ala227 in the β context (Ala169). The proposed number of N-glycans of each form of antithrombin is indicated. (C) Antithrombin glycoforms secreted to the conditioned medium of HEK-EBNA cells transfected with plasmids in the wild-type context (Ser169). A more complex heterogeneity of forms was observed for both Glu227 (residue present in the wild-type molecule) and mutated Lys227 plasmids, the last one generating increased levels of 3N-hypoglycosylated form and a new 2N-glycan variant form. The proposed number of N-glycans of each form of antithrombin is indicated. (D) Anticoagulant activity of the β AT (Glu227, residue present in the wild-type molecule) and mutant (Lys227) recombinant antithrombins. Anti-FXa activity was determined by a chromogenic assay. The activity of a pool of plasma of 100 healthy blood donors was taken as a reference value of 100%. (E) T-AT complexes were detected by Western blotting after adding thrombin (T) with or without UFH. The abnormal antithrombin is pointed by a red arrow.
Expression of the Lys227 variant in the wild-type background (Ser169) generated 3 glycoforms with 4, 3, and 2 N-glycans (Figure 3C).
Both anti-FXa activity, analyzed by chromogenic assays, and anti-FIIa activity, evaluated by Western blotting detection of thrombin–antithrombin complexes, revealed significant impairment of the anticoagulant activity of the mutated Lys227 antithrombin in the β context (Ala169) (Figure 3D-E).
Modulation of N-glycosylation efficiency by lysine (Lys) residues
In order to validate if a Lys electropositive residue close to the N-glycosylation sequon might affect the efficacy of the glycosylation, we performed a set of variations in the N-glycosylation sequons of antithrombin by introducing or deleting Lys residues and evaluating the effect on the N-glycosylation efficacy (supplemental Figure 6). The presence of a new Lys up to 4 positions before or after an Asn significantly impaired the N-glycosylation of the sequon. In contrast, the mutation of any of the 3 Lys residues surrounding Asn167 (at positions −2, +1, and +4 of this sequon) in the wild-type background, with Ser169, that produces similar amounts of α and β glycoforms, resulted in nearly full occupancy of Asn167 by the N-glycan, and thus, all secreted antithrombin had 4 N-glycans. Similarly, the mutation of any of the 2 Lys residues present in the Asn224 sequon restored a full occupancy of N-glycosylation in the p.Glu227Lys background (supplemental Figure 6).
SERPINC1 p.Asn224His
The interest in knowing the pathogenic consequences of a genetic variant disturbing the N-glycosylation at Asn224 significantly increased when we identified 2 unrelated Norwegian patients with early and severe thrombosis having a conflictive antithrombin deficiency that shared the same heterozygous SERPINC1 mutation affecting the Asn224 sequon: c.670 A>C (p.Asn224His) (Figure 1B). This mutation is not described in HGMD, ExAC, or Gnomad, although the p.Asn224Asp was found in 3 out 282 864 alleles. Mutation Taster and PROVEAN (Protein Variation Effect Analyzer) predicted a potentially damaging effect for the p.Asn224His and p.Asn224Asp mutations.
In the first family with p.Asn224His, the proband is a male who got a proximal DVT at the age of 53 complicated with PE. He suffered a recurrent DVT a few weeks after discontinuing anticoagulation. Since then, he remains with lifelong anticoagulation. His antithrombin anti-FXa activity and antigen levels were normal (97% and 87%, respectively; reference, 80% to 120%). The proband has 2 asymptomatic children, both carrying the mutation. In the second family (Figure 1B), the proband is a young man who was diagnosed with venous thrombosis in the inferior cava vein at the age of 11. He was anticoagulated for 1 year, but at the age of 14 years, he was again admitted with pelvic vein thrombosis. Since that time, he has been permanently anticoagulated. Anti-FXa activity measured outside the thrombotic episodes and while on warfarin treatment was between 67% and 87%. The proband had 2 siblings, a younger healthy brother and a sister who died abruptly at 18 years old after a short period of mild infectious symptoms. She was on oral contraceptives at the time. The autopsy showed massive PE. The healthy father also carried the mutation.
Analysis of plasma antithrombin in probands from both families revealed nearly normal antigen levels with no increase of forms with low heparin affinity, but the presence of smaller antithrombin with slower mobility than the wild-type antithrombin in native PAGE and faster mobility in SDS-PAGE (Figure 4).
Figure 4.

Analysis of plasma antithrombin in 2 symptomatic unrelated Norwegian probands carrying the p.Asn224His mutation in the heterozygous state and 2 healthy control samples. (A) Crossed immunoelectrophoresis in the presence of heparin. (B) Western blot of native PAGE and SDS-PAGE. The aberrant forms of antithrombin detected in the probands are pointed by red arrows. The anti-FXa activity of the sample evaluated by Western blot is also indicated.
Recombinant expression of the p.Asn224His variant in the wild-type background (Ser169) showed slighter reduced secretion than the wild-type molecule (Figure 5). As for the wild-type, 2 glycoforms were also observed for the mutant antithrombin, but with 1 glycan less (Figure 5). Interestingly, the variant antithrombin showed similar anticoagulant activity as the wild-type by chromogenic assays (anti-FXa: 1.38 IU/mL and 1.39 IU/mL, respectively) and Western blotting after adding thrombin (Figure 5). The expression of the variant in the β background (Ala169), with no glycosylation at Asn167, resulted in a significant reduction of the secretion of the variant antithrombin that only presented 2 N-glycans. Interestingly, this 2 N-glycan variant had no anticoagulant activity and seemed to be cleaved by thrombin (Figure 5).
Figure 5.

Recombinant expression of the His224 variant in HEK-EBNA cells. Supernatant of HEK-EBNA cells transfected with the human SERPINC1 Asn224 (residue present in the wild-type molecule) or the mutant His224 cDNAs in the wild-type (Ser169) or β (Ala169) backgrounds were analyzed by Western blotting. The number of N-glycans of each form is indicated. The formation of T-AT, when incubated with thrombin (T) with or without UFH, is pointed by arrows. Cleaved antithrombin (cAT) is also indicated.
Recombinant antithrombins in the β background were purified, and further biochemical analysis was done. Heparin affinity determined by intrinsic fluorescence analysis revealed higher heparin affinity than wild-type antithrombin (Kd: 22.1 ± 2.9 nM vs 36.8 ± 1.2 nM, respectively) (supplemental Figure 7). Moreover, the variant had negligible anti-FXa activity (0.05 ± 0.07 IU/mL). Interestingly, the β glycoform of variant His224 impaired the anticoagulant activity of the wild-type molecule when mixed at equimolecular concentrations (0.53 ± 0.29 IU/mL) (supplemental Figure 8).
Anti-FVII activity of variants affecting the glycosylation of Asn224
The severe clinical phenotype, together with the increased thrombin generation but the nearly normal anti-FXa and anti-FIIa observed in carriers of these mutations, suggested that the variant antithrombins might have a defect not detected by current functional methods. We evaluated the anti-FVIIa activity of variants impairing or abolishing the N-glycosylation of Asn224. Carriers of both p.Glu227Lys and p.Asn224His variants had reduced levels of FVIIa-antithrombin complexes than healthy control samples (Figure 6A-B) detected by Western blotting. Although a small reduction of free antithrombin was observed by Western blotting in carriers of p.Asn224His and p.Glu227Lys, total antithrombin in basal samples without treatment of FVIIa was similar in controls and patients according to ELISA, Western blotting, and Rocket immunoelectrophoresis. Thus, the ratio FVIIa-antithrombin/total antithrombin was significantly lower in carriers of these mutations (Figure 6B). Moreover, the quantification of FVIIa-antithrombin complexes by ELISA also revealed reduced levels in carriers of these mutations (Figure 6C). The normalization of these values by total antithrombin also maintained these differences (Figure 6C).
Figure 6.

Anti-FVIIa activity of plasma antithrombin (AT) in healthy control samples and carriers of mutations affecting the glycosylation at Asn224. (A) Formation of FVIIa-antithrombin complexes (FVIIa-AT) in vitro by incubation of plasma from carriers of both variants and a control subject, with recombinant FVIIa and UFH. FVIIa-AT complexes (C) and free antithrombin were detected by Western blotting. The image shows representative cases. Values from densitometry analysis of FVIIa-AT, free AT, and total AT are indicated. *arbitrary units. (B) Violin plots of FVIIa-AT complexes and ratio FVIIa-AT/total AT determined by densitometry in 3 carriers of the p.Asn224His and p.Glu227Lys variants and 4 healthy control samples after incubation with FVIIa and UFH. a.u., arbitrary units. *P < .05. (C) Violin plots of FVIIa-AT complexes determined by a specific ELISA in the same samples. The ratio FVIIa-AT/total AT is also shown. *P < .05.
Anticoagulant activity after moderate heating
The plasma anti-FXa activity of carriers of p.Asn224His, p.Glu227Lys, and healthy control samples was evaluated in basal conditions and after heating for 1 hour at 41°C. Although the loss of activity was small, it was higher in carriers of mutations impairing the glycosylation at Asn224, and differences reached statistical significance for p.Asn224His carriers (Figure 7).
Figure 7.

Anti-FXa activity of basal plasma samples from patients and control samples, and after incubation for 1 hour at 41°C. (A) Global values in each group of patients and control samples. (B) Ratio basal/41°C of anti-FXa activities in each group of patients and control samples. *P > .05.
Discussion
The relevance of genetic factors in thrombosis and their high heritability have been proven to a great degree by the description of thrombophilic families.24,25 Since the discovery of antithrombin deficiency in 1965, hundreds of studies have tried to identify new gene defects increasing the risk of thrombosis. In the 1980s, the study of families with thrombophilia and intermediate phenotypes in the hemostatic system allowed the identification of congenital protein C and protein S deficiencies as another class of strong dominant thrombophilias.12 In the 1990s, after the discovery of FV Leiden,26 genetic risk factors of thrombosis also included certain common prothrombotic polymorphisms. However, a recent meta-analysis of GWAS evaluating more than 34 million SNPs comprising more than 20 000 patients and 400 000 control samples revealed that only a few common polymorphisms significantly increase the risk of thrombosis, most of them with a very mild effect.27,28 Currently, the search for new thrombophilias returned to the first strategy: the recruitment of thrombophilic families with new intermediate hemostatic phenotypes such as increased FIX activity, antithrombin resistance, or increased FVIII levels. The genetic defects in these families were identified by sequencing the candidate genes (F9, F2, and F8, respectively).29-31 However, the results obtained by massive sequencing (whole exome or whole genome) of thrombophilic families or patients with thrombosis have not been very gratifying, as only a few novel (and conflicting) thrombophilias have been identified.32,33 We speculated that genetic defects affecting key hemostatic proteins previously involved in classical thrombophilias might not have been detected because of the limitation of current diagnostic functional tests. This study underscores that classical thrombophilia may still go missing with current diagnostic approaches and provides new concepts for the recognition of antithrombin deficiency, the congenital thrombophilia with the most severe consequences.
Here, we describe 4 unrelated patients with thrombosis and antithrombin deficiency not identifiable by current functional methods that shared a common pathogenic mechanism. Detection of the gene defect was done by a nontargeted strategy (WGS) or sequencing of the candidate gene, not following current diagnostic algorithms since probands had no family history of antithrombin deficiency, and samples drawn at basal conditions exhibited normal anticoagulant activity. Our findings support the limitation of current functional tests, which have been designed to identify strong functional defects and fail to recognize milder forms or, as in this study, genetic variants that impair other hemostatic roles. The p.Asn224His and p.Glu227Lys variants have a negligible effect on the anti-FXa or anti-FIIa activity, which are the common assays used to diagnose antithrombin deficiency,9 but might impair an effect of the anticoagulant which has been barely studied: the anti-FVIIa activity.34-38 It is a fact that FVIIa–antithrombin complexes are considered good biomarkers of prothrombotic states.39 In the cascade of proteolytic reactions leading to the generation of thrombin, any element that can reduce, even moderately, the inhibition of the first procoagulant protease, FVIIa, will probably result in a significantly higher thrombin generation, as our study also found. Thus, new functional assays (or modifications of current ones), such as thrombin generation assays, might improve the diagnosis and prognosis of cases with SERPINC1 defects.
Our study also emphasizes the relevance of minority forms of hemostatic elements, such as the β glycoform, which is significantly impaired in its anti-FXa and anti-FIIa anticoagulant activity by these mutations. In addition, the higher heparin affinity of the β variants explains a dominant–negative effect on the wild-type antithrombin that contributes to the severe clinical phenotype, as it has been previously suggested for antithrombin London (p.Arg425del).40
We aimed to further identify and characterize the underlying pathogenic mechanism of these 2 variants. N-glycosylation is a key PTM involved in the folding, secretion, stability, specificity, and function of most secreted proteins,41 and its results are crucial for the hemostatic system.42 There are few data supporting the consequences that PTM might have in the functional activity of antithrombin. The physiological inefficient glycosylation of Asn167 causes the β form,18 a hypoglycosylated antithrombin with higher heparin affinity; that, despite being minor in circulation, could have a relevant anticoagulant role.19 A more pronounced defect of glycosylation, such as the one caused by congenital disorders of glycosylation, a heterogeneous group of disorders with reduced capacity to generate the glycan precursor, is usually associated with antithrombin deficiency.43 We have demonstrated that up to 20% of cases with antithrombin deficiency but no SERPINC1 defect have hypoglycosylation.44 However, few papers have reported specific clues on a potential pathogenic effect of an aberrant N-glycosylation specific to antithrombin caused by mutations in SERPINC1. As far as we know, 3 mutations introducing new N-glycans cause antithrombin deficiency, 1 in the heparin-binding domain, p.Ile71Asn, causing a type II HBS deficiency,45 and 2 in the shutter of antithrombin, p.Ser146Asn and p.Ile283Thr, which cause type I deficiencies probably by disturbing the correct folding of antithrombin.46,47 In contrast, only 1 report found a genetic variant disturbing an N-glycosylation site of antithrombin, p.Asn167Thr, in a family with mild antithrombin deficiency, increased β glycoform concentrations, and a nondefinitive higher risk of thrombosis48 but increased antimicrobial protection.49 Our study identified for the first time 2 genetic variants disturbing another N-glycosylation site with thrombotic consequences. It remains to be determined whether this impaired N-glycosylation may cause a transient deficiency of antithrombin and the triggering factors exacerbating the pathological consequences of these mutations that cause antithrombin deficiency and a high risk of thrombosis.
The biological bonus of our study was the identification of a new key residue that determines the N-glycosylation efficacy, which, when corroborated, should be incorporated into in silico predictions. Classically, the consensus sequence for N-glycosylation only considers 3 residues: Asn-X-Ser/Thr.41 However, data suggest that other residues close to this sequence must modulate, positively or negatively, the incorporation of the N-glycan precursor to the Asn.50 Our previous report demonstrated that an aromatic residue close to Asn167 in antithrombin increased the efficacy of glycosylation.51 In silico studies showed the involvement of charge and polarity environment of amino acids in the N-glycosylation process.52,53 In this study, we demonstrated that the presence of the electropositive Lys close to an N-glycosylation sequon negatively modulates the efficiency of this PTM, probably by impairing the binding of the oligosaccharyltransferase (OST EC 2.4.1.119). The final consequence is a reduced efficacy of glycosylation and the presence of secreted hypoglycosylated forms. This finding might also be useful for glycoengineered recombinant proteins.
In addition, this work also highlights the concept of transitory deficiency of antithrombin and the possibility that the deleterious effect of pathological alterations is manifested only in certain conditions at certain times.54 The precise function of antithrombin is sensitively dependent on the maintenance of its metastable structure. Glycosylation protects this fragile stability by forming an encompassing surrounding to the folded protein. The loss of a major glycosyl sidechain renders it vulnerable to perturbations that may affect its inhibitory activity. Such perturbations favor the transient loss of function and can result from a number of challenges, including a mild increase in body temperature, as observed during incidental viral infections.55,56 Strong indications that this may indeed contribute to transient and episodic thrombotic episodes associated with the identified mutations in the submitted paper come from family 2 with the sudden death of an 18-year-old female due to a massive thrombotic episode during an infectious bout. The increased loss of anticoagulant activity after moderate heating observed in carriers of these mutations supports this protective role of N-glycosylation and points toward new conditions that may cause a transient antithrombin deficiency with an increased risk of thrombosis.
In conclusion, we identified 2 new SERPINC1 defects that cause hypoglycosylation of Asn224. These variants have minor, if any, functional consequences when using routine methods to diagnose antithrombin deficiency but increase thrombin generation and reduce the inhibition of FVIIa. Moreover, these gene defects might significantly increase the risk of thrombosis according to the segregation of genetic and clinical phenotypes in affected families. The severity of these mutations may be explained by multiple prothrombotic mechanisms: a loss of function for the 2N-glycan variant that also has a dominant–negative effect, an impaired anti-FVIIa activity, and an overexposed sensitivity to stress factors caused by the hypoglycosylation. Finally, this study reinforces the usefulness of molecular analysis to identify thrombophilic defects, supports the notion that antithrombin deficiency may be an underestimated disorder, and encourages the development of new functional assays to diagnose antithrombin deficiency to unveil additional pathogenic defects affecting this key anticoagulant.
Supplementary Material
The online version of this article contains a data supplement.
{kind=link}
Acknowledgments
The authors thank the participants involved in this study and their families. They thank Sonia Aguila and Julia Peñas for helpful assistance in fluorescence analysis and Marit Sletten for sequencing analysis.
This work was supported by PI18/00598 & PI21/00174 (ISCIII & FEDER); 19873/GERM/15 project (Fundación Séneca). This work partially benefited from the support of the GENMED Laboratory of Excellence on Medical Genomics [ANR-10-LABX-0013], a research program managed by the National Research Agency (ANR) as part of the French Investment for the Future. This work also benefited from the financial support from the «EPIDEMIOM-VTE» Senior Chair (D.A.T) from the Initiative of Excellence of the University of Bordeaux.
Footnotes
For original data, please contact javiercorraldelacalle@gmail.com. All participants provided written informed consent to participate in the study. The study was approved by the Hospital Morales Meseguer Ethic committee. The research conforms with the principles of the Declaration of Helsinki.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.E.d.l.M.-B., D.A.T., P.M.S., and P.E.M. designed research; M.E.d.l.M.-B., A.M., B.d.l.M.-B., C.B.-P., J.P., and R.C. performed research; S.A., P.S., J.F.D., P.M.S., N.I., and E.M.J. contributed with patients and clinical data; D.A.T., M.E.d.l.M.-B., P.S., J.C., and E.M.J. analyzed data; and M.E.d.l.M.-B., J.C., M.L.L., V.V., D.A.T., P.M.S., and P.E.M. wrote and reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pierre Emmanuel Morange, Aix Marseille Univ, INSERM, INRAE, C2VN, Marseille, France; e-mail: Pierre.MORANGE@ap-hm.fr; Per Morten Sandset, Department of Haematology, Oslo University Hospital; Department of Haematology, Oslo University Hospital, Box 4950 Nydalen, 0424 Oslo, Norway; e-mail: p.m.sandset@medisin.uio.no; and Javier Corral, Servicio de Hematología y Oncología Médica, Hospital Universitario Morales Meseguer, Centro Regional de Hemodonación, Universidad de Murcia, IMIB, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Centro de Hemodonación, Ronda de Garay s/n, 30003, Murcia, Spain; email: javiercorraldelacalle@carm.es.
REFERENCES
- 1.Björk I, Olson ST. Antithrombin. A bloody important serpin. Adv Exp Med Biol. 1997;425:17-33. [PubMed] [Google Scholar]
- 2.Bucciarelli P, Passamonti SM, Biguzzi E, et al. Low borderline plasma levels of antithrombin, protein C and protein S are risk factors for venous thromboembolism. J Thromb Haemost. 2012;10(9): 1783-1791. [DOI] [PubMed] [Google Scholar]
- 3.Ishiguro K, Kojima T, Kadomatsu K, et al. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. 2000;106(7):873-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bravo‐Pérez C, Morena‐Barrio ME, Palomo A, et al. Genotype–phenotype gradient of SERPINC1 variants in a single family reveals a severe compound antithrombin deficiency in a dead embryo. Br. J. Haematol. 2020;191(1):e.32-e.35. [DOI] [PubMed] [Google Scholar]
- 5.Corral J, de la Morena-Barrio ME, Vicente V. The genetics of antithrombin. Thromb Res. 2018;169:23-29. [DOI] [PubMed] [Google Scholar]
- 6.Lijfering WM, Brouwer J-LP, Veeger NJGM, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood. 2009;113(21):5314-5322. [DOI] [PubMed] [Google Scholar]
- 7.Mahmoodi BK, Brouwer J-LP, Ten Kate MK, et al. A prospective cohort study on the absolute risks of venous thromboembolism and predictive value of screening asymptomatic relatives of patients with hereditary deficiencies of protein S, protein C or antithrombin. J Thromb Haemost. 2010;8(6):1193-1200. [DOI] [PubMed] [Google Scholar]
- 8.Bravo-Pérez C, Vicente V, Corral J. Management of antithrombin deficiency: an update for clinicians. Expert Rev Hematol. 2019;12(6):397-405. [DOI] [PubMed] [Google Scholar]
- 9.Van Cott EM, Orlando C, Moore GW, Cooper PC, Meijer P, Marlar R; Subcommittee on Plasma Coagulation Inhibitors . Recommendations for clinical laboratory testing for antithrombin deficiency; communication from the SSC of the ISTH. J Thromb Haemost. 2020;18(1): 17-22. [DOI] [PubMed] [Google Scholar]
- 10.Corral J, Vicente V. Puzzling questions on antithrombin: diagnostic limitations and real incidence in venous and arterial thrombosis. Thromb Res. 2015;135(6):1047-1048. [DOI] [PubMed] [Google Scholar]
- 11.Tait RC, Walker ID, Perry DJ, et al. Prevalence of antithrombin deficiency in the healthy population. Br J Haematol. 1994; 87(1):106-112. [DOI] [PubMed] [Google Scholar]
- 12.Martinelli I, De Stefano V, Mannucci PM. Inherited risk factors for venous thromboembolism. Nat Rev Cardiol. 2014;11(3):140-156. [DOI] [PubMed] [Google Scholar]
- 13.Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism--results of the Spanish multicentric study on thrombophilia (EMET-study). Thromb Haemost. 1997;77(3): 444-451. [PubMed] [Google Scholar]
- 14.Hemker HC, Giesen P, AlDieri R, et al. The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol Haemost Thromb. 2002;32(5-6):249-253. [DOI] [PubMed] [Google Scholar]
- 15.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362. [DOI] [PubMed] [Google Scholar]
- 17.Martínez-Martínez I, Ordóñez A, Navarro-Fernández J, et al. Antithrombin Murcia (K241E) causing antithrombin deficiency: a possible role for altered glycosylation. Haematologica. 2010;95(8):1358-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picard V, Ersdal-Badju E, Bock SC. Partial glycosylation of antithrombin III asparagine-135 is caused by the serine in the third position of its N-glycosylation consensus sequence and is responsible for production of the beta-antithrombin III isoform with enhanced heparin affinity. Biochemistry. 1995;34(26):8433-8440. [DOI] [PubMed] [Google Scholar]
- 19.McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. 2003;326(3):823-833. [DOI] [PubMed] [Google Scholar]
- 20.Langdown J, Belzar KJ, Savory WJ, Baglin TP, Huntington JA. The critical role of hinge-region expulsion in the induced-fit heparin binding mechanism of antithrombin. J Mol Biol. 2009;386(5):1278-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714-2723. [DOI] [PubMed] [Google Scholar]
- 22.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98(18):10037-10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999; 17(1):57-61. [PubMed] [Google Scholar]
- 24.Souto JC, Almasy L, Borrell M, et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic analysis of idiopathic thrombophilia. Am J Hum Genet. 2000; 67(6):1452-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desch KC. Dissecting the genetic determinants of hemostasis and thrombosis. Curr Opin Hematol. 2015;22(5):428-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369(6475):64-67. [DOI] [PubMed] [Google Scholar]
- 27.Lindström S, Brody JA, Turman C, et al. ; INVENT Consortium . A large-scale exome array analysis of venous thromboembolism. Genet Epidemiol. 2019;43(4):449-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klarin D, Busenkell E, Judy R, et al. ; Veterans Affairs’ Million Veteran Program . Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51(11):1574-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simioni P, Tormene D, Tognin G, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009; 361(17):1671-1675. [DOI] [PubMed] [Google Scholar]
- 30.Simioni P, Cagnin S, Sartorello F, et al. Partial F8 gene duplication (factor VIII Padua) associated with high factor VIII levels and familial thrombophilia. Blood. 2021; 137(17):2383-2393. [DOI] [PubMed] [Google Scholar]
- 31.Miyawaki Y, Suzuki A, Fujita J, et al. Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med. 2012;366(25):2390-2396. [DOI] [PubMed] [Google Scholar]
- 32.Lee E-J, Dykas DJ, Leavitt AD, et al. Whole-exome sequencing in evaluation of patients with venous thromboembolism. Blood Adv. 2017;1(16):1224-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desch KC, Ozel AB, Halvorsen M, et al. Whole-exome sequencing identifies rare variants in STAB2 associated with venous thromboembolic disease. Blood. 2020; 136(5):533-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo S, Kisiel W. Regulation of factor VIIa activity in plasma: evidence that antithrombin III is the sole plasma protease inhibitor of human factor VIIa. Thromb Res. 1987;46(2):325-335. [DOI] [PubMed] [Google Scholar]
- 35.Rao LV, Rapaport SI, Hoang AD. Binding of factor VIIa to tissue factor permits rapid antithrombin III/heparin inhibition of factor VIIa. Blood. 1993;81(10):2600-2607. [PubMed] [Google Scholar]
- 36.Lawson JH, Butenas S, Ribarik N, Mann KG. Complex-dependent inhibition of factor VIIa by antithrombin III and heparin. J Biol Chem. 1993;268(2):767-770. [PubMed] [Google Scholar]
- 37.Jesty J, Lorenz A, Rodriguez J, Wun TC. Initiation of the tissue factor pathway of coagulation in the presence of heparin: control by antithrombin III and tissue factor pathway inhibitor. Blood. 1996;87(6): 2301-2307. [PubMed] [Google Scholar]
- 38.Broze GJ Jr, Likert K, Higuchi D. Inhibition of factor VIIa/tissue factor by antithrombin III and tissue factor pathway inhibitor. Blood. 1993;82(5):1679-1681. [PubMed] [Google Scholar]
- 39.Spiezia L, Campello E, Valle FD, Woodhams B, Simioni P. Factor VIIa-antithrombin complex: a possible new biomarker for activated coagulation. Clin Chem Lab Med. 2017; 55(4):484-488. [DOI] [PubMed] [Google Scholar]
- 40.Raja SM, Chhablani N, Swanson R, et al. Deletion of P1 arginine in a novel antithrombin variant (antithrombin London) abolishes inhibitory activity but enhances heparin affinity and is associated with early onset thrombosis. J Biol Chem. 2003; 278(16):13688-13695. [DOI] [PubMed] [Google Scholar]
- 41.Clerc F, Reiding KR, Jansen BC, Kammeijer GS, Bondt A, Wuhrer M. Human plasma protein N-glycosylation. Glycoconj J. 2016;33(3):309-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansson K, Stenflo J. Post-translational modifications in proteins involved in blood coagulation. J Thromb Haemost. 2005;3(12):2633-2648. [DOI] [PubMed] [Google Scholar]
- 43.Linssen M, Mohamed M, Wevers RA, Lefeber DJ, Morava E. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab. 2013;109(1):107-111. [DOI] [PubMed] [Google Scholar]
- 44.de la Morena-Barrio ME, Martínez-Martínez I, de Cos C, et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absence of a SERPINC1 gene defect. J Thromb Haemost. 2016;14(8):1549-1560. [DOI] [PubMed] [Google Scholar]
- 45.Brennan SO, Borg JY, George PM, et al. New carbohydrate site in mutant antithrombin (7 Ile----Asn) with decreased heparin affinity. FEBS Lett. 1988;237(1-2):118-122. [DOI] [PubMed] [Google Scholar]
- 46.Fitches AC, Lewandowski K, Olds RJ. Creation of an additional glycosylation site as a mechanism for type I antithrombin deficiency. Thromb Haemost. 2001;86(4):1023-1027. [PubMed] [Google Scholar]
- 47.Picard V, Bura A, Emmerich J, et al. Molecular bases of antithrombin deficiency in French families: identification of seven novel mutations in the antithrombin gene. Br J Haematol. 2000;110(3):731-734. [DOI] [PubMed] [Google Scholar]
- 48.Bayston TA, Tripodi A, Mannucci PM, et al. Familial overexpression of beta antithrombin caused by an Asn135Thr substitution. Blood. 1999;93(12):4242-4247. [PubMed] [Google Scholar]
- 49.Papareddy P, Rossnagel M, Doreen Hollwedel F, et al. A human antithrombin isoform dampens inflammatory responses and protects from organ damage during bacterial infection. Nat Microbiol. 2019; 4(12):2442-2455. [DOI] [PubMed] [Google Scholar]
- 50.Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH. The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry. 1998;37(19): 6833-6837. [DOI] [PubMed] [Google Scholar]
- 51.Aguila S, Martínez-Martínez I, Dichiara G, et al. Increased N-glycosylation efficiency by generation of an aromatic sequon on N135 of antithrombin [published correction appears in PloS One. 2015;10(3):e0122177]. PLoS One. 2014;9(12):e114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manwar Hussain MR, Iqbal Z, Qazi WM, Hoessli DC. Charge and polarity preferences for N-glycosylation: a genome-wide in silico study and its implications regarding constitutive proliferation and adhesion of carcinoma cells. Front Oncol. 2018;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Senger RS, Karim MN. Variable site-occupancy classification of N-linked glycosylation using artificial neural networks. Biotechnol Prog. 2005;21(6):1653-1662. [DOI] [PubMed] [Google Scholar]
- 54.Bravo-Pérez C, de la Morena-Barrio ME, de la Morena-Barrio B, et al. Molecular and clinical characterization of transient antithrombin deficiency: a new concept in congenital thrombophilia. Am J Hematol. 2022;97(2):216-225. [DOI] [PubMed] [Google Scholar]
- 55.Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin rouen-VI (187 Asn-->Asp). J Clin Invest. 1994;94(6):2265-2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beauchamp NJ, Pike RN, Daly M, et al. Antithrombins wibble and wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood. 1998;92(8): 2696-2706. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.