

Abstract
Background:
Nonalcoholic fatty liver disease (NAFLD) is highly prevalent in young adults with obesity. Obesity is associated with relative growth hormone (GH) deficiency, and data from animal studies and from humans with pituitary GH deficiency suggest a role for GH deficiency in the pathogenesis of NAFLD. The effects of GH on NAFLD in those with obesity are unknown, however, prompting this pilot study to assess effects of GH administration on measures of NAFLD in young adults.
Methods:
Twenty-four men and women ages 18–29 years with BMI ≥ 30kg/m2, hepatic fat fraction (HFF) ≥ 5% on proton magnetic resonance spectroscopy (1H-MRS), and insulin-like growth factor 1 (IGF-1) z-score ≤ 0 were randomized to treatment with recombinant human GH (rhGH) versus no treatment for 24 weeks. The primary endpoint was change in HFF.
Results:
Compared to no treatment, the effect size of rhGH on absolute HFF over 24 weeks was −3.3% (95% confidence interval −7.8%, 1.2%; p=0.14). At 24 weeks, HFF < 5% was achieved in 5 of 9 individuals receiving rhGH versus 1 of 9 individuals receiving no treatment (p=0.04). rhGH did not significantly reduce ALT, AST, or GGT. Serum IGF-1 increased as expected with rhGH treatment, and there were no changes in fasting lipids, c-reactive protein, fasting glucose, or 2-hour glucose following an oral glucose tolerance test.
Conclusion:
Data from this pilot study suggest that rhGH treatment in young adults with obesity and NAFLD may have benefits to reduce liver fat content, although larger studies are needed to confirm this effect.
Clincialtrials.gov registration:
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of liver disease in adolescents and young adults. Over the past 3 decades, in conjunction with rising obesity, the prevalence of NAFLD in adolescents and young adults has more than doubled (1, 2), such that the current prevalence of NAFLD among individuals with obesity in this age group is estimated at up to 38–45% (3, 4). More concerningly, 20% of adolescents with NAFLD show signs of nonalcoholic steatohepatitis (NASH), characterized by inflammation, hepatocellular injury and fibrosis (5, 6). In many, NASH will progress to advanced fibrosis and cirrhosis, causing significant morbidity. Effective treatment options for NAFLD in adolescents and young adults are limited. Lifestyle modification is effective in reducing hepatic lipid and biomarkers of liver injury (7, 8), but these efforts are difficult to maintain over the long-term. Thus, there is significant need to better understand the pathophysiology of NAFLD in adolescents and young adults and to explore possible pharmacologic strategies.
We and others have reported significant reductions in growth hormone (GH) in adolescents and adults with obesity (9–11). Furthermore, human studies and animal models implicate reduced GH and insulin-like growth factor-1 (IGF-1) signaling in the pathogenesis of NAFLD (12, 13). In humans, pituitary GH deficiency is associated with increased risk for NAFLD and NASH, and GH replacement improves histological findings of NAFLD (14–16). In an adult population with HIV-associated NAFLD, we have demonstrated that augmentation of GH using a GH releasing hormone (GHRH) analog significantly reduces liver fat (17). However, to our knowledge, the effect of GH treatment in reducing liver fat in young adults with NAFLD has never been tested. Our objective in this pilot study was to determine whether treatment with recombinant human GH (rhGH), somatropin, may reduce liver fat in young adults with obesity and NAFLD and below-average IGF-1 levels (suggesting relative baseline reductions in endogenous GH). We hypothesized that, compared to no treatment, rhGH therapy would decrease hepatic fat fraction as measured by 1H-Magnetic Resonance Spectroscopy (1H-MRS).
Materials and Methods
Study design and participants
We conducted a randomized, open-label, 6-month trial of 24 participants at the Massachusetts General Hospital (MGH) in Boston, MA, USA. Eligible participants were young adults 18 to 29 years of age with BMI ≥30kg/m2 and hepatic fat fraction (HFF) of ≥5% on 1H-MRS (18). Participants were required to have IGF-1 z-scores less than average (IGF-1 z-score ≤0), suggesting relative reductions in endogenous GH, but could not have a known clinical diagnosis of pituitary GH deficiency or another condition known to affect the GH axis. Participants with heavy alcohol use (>14 drinks per week for women or >21 drinks per week for men) were excluded, as were participants with significant liver disease as indicated by alanine aminotransferase (ALT) or aspartate aminotransferase (AST) concentrations greater than 2.5 times the upper limit of normal (ULN), total bilirubin greater than ULN, hemoglobin less than 11.0g/dL (6.89mmol/L), positive hepatitis B surface antigen, positive hepatitis C antibodies, or known diagnoses of α1 antitrypsin deficiency, Wilson’s disease, hemochromatosis, or autoimmune hepatitis. Other exclusion criteria were as follows: known cirrhosis, history of or active malignancy, use of chronic systemic corticosteroids, use of growth hormone in the past year, use of weight-loss medications or previous weight loss surgery, diagnosis of diabetes or use of antidiabetic medications. Participants reporting use of any antihypertensives or lipid-lowering medications within the 3 months prior to study entry were required to have had stable use of such medications during that time. Participants reporting use of vitamin E or ursodiol within the 6 months prior to study entry were required to have had stable use of these medications during that time. Female participants who were pregnant or breastfeeding were excluded, and those reporting use of oral estrogen/progesterone contraceptives or depot progesterone formulations within the 12 months prior to study entry were required to have had stable use of these medications during that time. Hormone-releasing intrauterine devices were permitted. Potentially eligible participants were recruited from the greater Boston area through advertisements at ambulatory care practices including pediatric and adult gastroenterology and obesity clinics as well as website advertisements. All participants provided written informed consent. The study was approved by the MGH Institutional Review Board.
Randomization and Treatment
Participants were randomized in a 1:1 ratio to somatropin (Norditropin®) or no treatment. The study was open-label, and placebo was not used to avoid daily injections without benefit for participants randomized to no treatment. The randomization list was prepared by the study statistician, stratified by sex and vitamin E use ≥400 IU/day. Somatropin was started at a dose of 0.5mg daily and adjusted according to IGF-1 z-score at 2, 4, 6, 12, and 18 weeks in order to reach a target z-score between 0–2. A 20% dose increase was performed for any rhGH-treated subject with IGF-1 z-score ≤0. Conversely, a 20% decrease was performed for any rhGH-treated subject with IGF-1 z-score >2. Participants received somatropin (or no treatment) for 24 weeks.
Procedures
The study was 24 weeks in duration, with visits at baseline, 12 weeks, and 24 weeks for all subjects, and additional safety visits at 2, 4, 6, and 18 weeks for those randomized to somatropin. All participants received nutritional counseling from clinical research nutritionists at baseline and 24 weeks. Visits were conducted in the fasting state. The screening visit included history and physical examination, laboratory investigations for eligibility, magnetic resonance imaging (MRI) to assess cross-sectional area of visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT), and 1H-MRS of the liver for quantification of HFF. For quantification of VAT and SAT, a T1 water-suppressed axial single slice was obtained at the level of the L4 vertebra, and cross-sectional area of VAT and SAT was measured by semi-automated tracing with manual adjustment (ViTrak/AccuImage, EFilm, Inc.). The radiologist performing measures of VAT, SAT, and liver fat was not aware of treatment assignment. VAT, SAT, and HFF from the screening visit were used as the baseline measures. The baseline assessment included the following: standard 75g oral glucose tolerance test with glucose assessment at 0 and 120 minutes; clonidine-arginine GH stimulation test; whole body dual energy x-ray absorptiometry (DXA); fasting assessment of liver function tests, lipids, serum inflammatory markers, and IGF-1; bionutrition assessment including 24-hour food recall and anthropometric measures done in triplicate; and questionnaires derived from the Centers for Disease Control and Prevention 2013 Standard High School Youth Risk Behavior Survey. These assessments were repeated at 24 weeks along with repeat MRI and 1H-MRS.
GH stimulation testing with clonidine and arginine was performed with administration of clonidine 100mcg/m2 (maximum 250mcg) orally at time 0 and continuous intravenous infusion of 30g arginine between 0–30 minutes, with measurement of GH values at 0, 30, 60, 90, and 120 minutes. Biochemical analyses were performed at the MGH clinical laboratory, LabCorp, and Quest Laboratories. IGF-1 was measured centrally at Quest Laboratories.
Outcomes and Statistical Analysis
The prespecified primary endpoint was change in HFF between baseline and 24 weeks. HFF was measured with 1H-MRS in the morning, fasting, and was calculated as the area under the spectroscopic lipid peak divided by the total area under the water and lipid peaks. Image acquisition followed a standard protocol and liver fat content was quantified by a radiologist who was not aware of treatment assignment. Prespecified secondary endpoints were changes after 24 weeks in ALT, AST, γ-glutamyl transferase (GGT), VAT area, fasting lipids, CRP, and fasting and 2-hour glucose and insulin. Prespecified endpoints related to safety included measures of glucose homeostasis, namely fasting glucose and HbA1c.
A sample size of 24, assuming a discontinuation rate up to 20%, had 80% power to detect an absolute reduction of 4.2 percentage points or more in HFF over 24 weeks in the rhGH group compared to the no treatment group by using a two-sample two-sided t-test at 5% significance level. For all endpoints, an intention-to-treat analysis was done, using all available data, with missing data mechanism was assumed to be missing at random (MAR). Per the prespecified analysis plan, differences between groups in change in variables measured only at baseline and 24 weeks, including the primary endpoint of HFF, were assessed using a two-sample Student’s t-test. Differences between groups in categorical variables were assessed using a likelihood ratio chi-square test. One individual moved out of state prior to completing the study and had an early final assessment, including MRI/MRS, at his 18-week visit. This individual was included in the primary analysis but was excluded in a secondary sensitivity analysis. GH stimulation test results were available for 19 participants. Statistical analyses were performed with JMP Pro, version 15 (SAS Institute Inc., Cary, NC). This study was registered on ClinicalTrials.gov, NCT02726542.
Results
Participant flow is shown in the Consort Diagram in Figure 1. Of 103 participants screened, 24 were eligible and randomized between May 19, 2017 and August 14, 2019. Two participants in the rhGH group discontinued before the baseline visit and never had drug initiation. One participant in the rhGH group and one participant in the no treatment group were lost to follow up prior to their final visit. One participant in the rhGH group was unable to undergo the 24-week MRI assessment due to anxiety, and one participant in the no treatment group was unable to undergo their 24-week MRI due to the COVID-19 pandemic. The overall discontinuation rate was not significantly different between the two groups (p=0.36).
Figure 1:
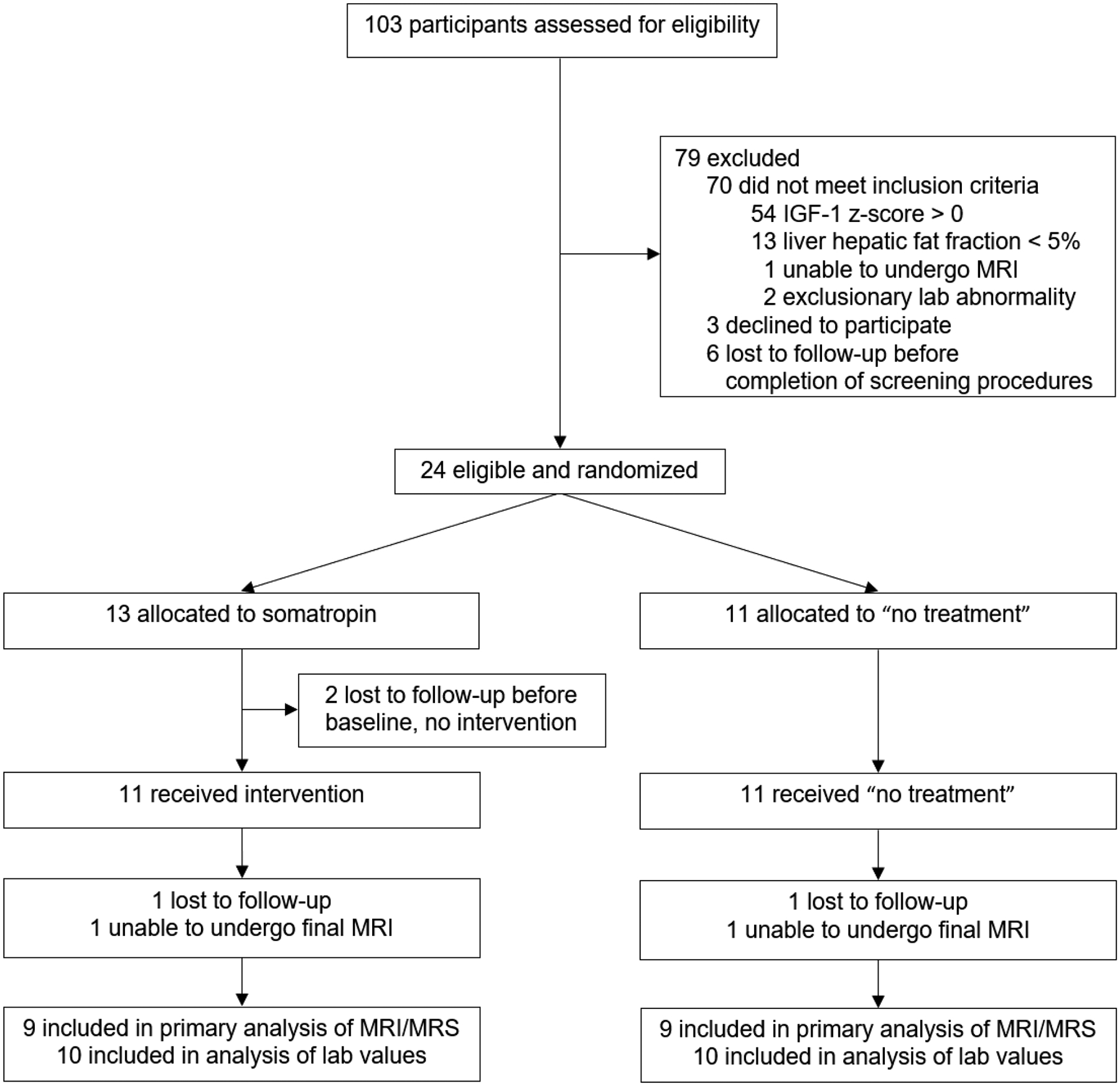
CONSORT Diagram outlining the flow of subjects through the study.
Baseline clinical characteristics and measures of body composition and metabolism were similar between groups (Table 1, Table 2), except that baseline serum IGF-1 was lower in the GH group compared to the no treatment group (p = 0.01); however, IGF-1 z-score did not differ between groups. Fifty-four percent of the cohort was comprised of females, and no participants were taking Vitamin E. Mean BMI among the entire cohort at baseline was 39.9 ± 6.8 kg/m2, and mean HFF was 11.3 ± 7.2%. Thirteen of 24 participants (7 in the rhGH group and 6 in the no treatment group) had elevated ALT (≥30 U/L for males or ≥19 U/L for females) at the screening visit (19).
Table 1:
Baseline Demographic and Clinical Characteristics
| GH (N = 13) |
No treatment (N = 11) |
|
|---|---|---|
| Sex [N (%) Female] | 7 (53.9%) | 6 (54.6%) |
| Age (years) | 25 (3) | 23 (4) |
| Race [N (%)] | ||
| American Indian or Alaskan Native | 1 (7.7%) | 0 (0%) |
| Asian | 1 (7.7%) | 1 (9.1%) |
| White | 9 (69.2%) | 6 (54.6%) |
| Other or more than 1 race | 2 (15.4%) | 4 (36.4%) |
| Ethnicity [N (%) Hispanic] | 4 (30.8%) | 4 (36.4%) |
| Smoking status | ||
| Current | 1 (7.7%) | 1 (9.1%) |
| Former | 0 | 1 (9.1%) |
| Never | 12 (92.3%) | 9 (81.8%) |
| Current lipid-lowering medications | 0 | 1 (9.1%) |
| Hepatic fat fraction | 12.1% (7.2%) | 10.4% (7.6%) |
Continuous data are presented as mean (Standard Deviation). There are no statistically significant differences between GH and No Treatment groups.
Table 2:
Baseline and Final Hepatic and Metabolic Endpoints
| Baseline | Change at 24 weeks | Treatment effect (95% CI) |
P-value | |||
|---|---|---|---|---|---|---|
| GH (N = 13) |
No treatment (N=11) |
GH (N = 10) |
No treatment (N = 10) |
|||
| Liver endpoints | ||||||
| Hepatic fat fraction (%)* | 12.1 (7.2) | 10.4 (7.6) | −3.0 (5.2) | 0.3 (3.5) | −3.3 (−7.8, 1.2) | 0.14 |
| ALT (U/L) | 29 (22) | 27 (13) | −2 (9) | 1 (8) | −3 (−11, 5) | 0.46 |
| AST (U/L) | 24 (15) | 21 (3) | −3 (9) | −1 (4) | −1 (−8, 5) | 0.67 |
| GGT (U/L) | 43 (50) | 30 (18) | −9 (30) | 5 (9) | −14 (−35, 8) | 0.19 |
| Body Composition | ||||||
| VAT (cm2) | 141 (72) | 145 (47) | −6 (50) | 6 (24) | −12 (−52, 29) | 0.53 |
| SAT (cm2) | 636 (209) | 674 (250) | −17 (81) | 8 (147) | −25 (−146, 96) | 0.66 |
| BMI (kg/m2) | 40.5 (6.6) | 39.3 (7.3) | −0.8 (1.7) | 1.1 (1.6) | −1.9 (−3.5, −0.3) | 0.02 |
| Waist (cm) | 120 (14) | 119 (14) | −3 (11) | 7 (10) | −10 (−21, 1) | 0.08 |
| Total body fat (kg) | 52.3 (14.2) | 52.8 (17.3) | −2.6 (3.0) | 0.4 (4.0) | −3.1 (−6.8, 0.7) | 0.10 |
| Total lean body mass (kg) | 61.5 (7.6) | 59.7 (10.5) | −0.6 (3.3) | 2.0 (1.4) | −2.6 (−5.3, 0.0) | 0.0498 |
| Metabolic indices | ||||||
| IGF-1 (nmol/L) [ng/mL] |
18.7 (4.3) [143 (33)] |
24.7 (5.5) [189 (42)] |
11.5 (9.6) [88.3 (73.7)] |
−3.5 (5.5) [−26.9 (42.1)] |
15.0 (7.6, 22.6) [115 (58, 173)] |
0.001 |
| IGF-1 z-score | −0.7 (0.6) | −0.3 (0.4) | 1.1 (0.8) | −0.3 (0.6) | 1.4 (0.7, 2.0) | 0.001 |
| Triglycerides (mmol/L) [mg/dL] |
1.2 (0.6) [110 (50)] |
1.1 (0.4) [98 (31)] |
0.2 (0.5) [14 (47)] |
0.1 (0.3) [−7 (27)] |
0.2 (−0.2, 0.7) [21 (−16, 58)] |
0.24 |
| HDL cholesterol (mmol/L) [mg/dL] |
1.1 (0.2) [42 (7)] |
1.0 (0.2) [40 (6)] |
0.1 (0.1) [2 (4)] |
0.1 (0.1) [3 (4)] |
0.0 (−0.1, 0.1) [−1 (−5, 3)] |
0.57 |
| LDL cholesterol (mmol/L) [mg/dL] |
2.7 (0.9) [103 (33)] |
2.5 (1.0) [96 (39)] |
0.2 (0.5) [6 (21)] |
0.2 (0.3) [6 (12)] |
0 (−0.4, 0.4) [0 (−16, 16)] |
0.98 |
| CRP (mg/L) | 6.1 (4.5) | 7.0 (7.7) | −0.2 (3.5) | −0.4 (7.6) | 0.2 (−5.6, 6.0) | 0.95 |
| Fasting glucose (mmol/L) [mg/dL] |
4.8 (0.6) [87 (10)] |
4.7 (0.4) [84 (8)] |
−0.2 (0.6) [−4 (11)] |
0.1 (0.5) [1 (9)] |
−0.3 (−0.8, 0.2) [−5 (−15, 4)] |
0.24 |
| 2-hour glucose (mmol/L) [mg/dL] |
6.0 (1.6) [108 (29)] |
6.1 (1.1) [110 (20)] |
0.7 (2.4) [12 (44)] |
−0.1 (1.3) [−2 (24)] |
0.8 (−1.2, 2.7) [14 (−22, 49)] |
0.42 |
Data are presented as mean (Standard Deviation). There are no statistically significant differences at baseline between GH and No Treatment groups except for serum IGF-1 (p = 0.01); IGF-1 Z-scores were not different.
One individual in each treatment group was unable to undergo 24-week MRS/MRI; sample size is 9 individuals in each group for this endpoint. Treatment effect and p-value using Student’s T-test.
Hepatic Endpoints
Compared to no treatment, the effect size of rhGH on absolute HFF over 24 weeks was −3.3% (95% confidence interval −7.8%, 1.2%; p = 0.14; Table 2, Figure 2A), corresponding to a relative reduction in HFF of −36% (95% CI −88.4%, 15.9%; p = 0.16). Figure 2B shows individual changes in HFF for each participant. At 24 weeks, 5 of 9 individuals in the rhGH group (55.6%) versus 1 of 9 individuals in the no treatment group (11.1%) had achieved HFF < 5%, below the threshold for NAFLD (p = 0.04). In a secondary analysis excluding the individual in the rhGH group who had assessment of HFF early due to moving out of state, the effect size of rhGH on absolute HFF was −4.6% (95% CI −8.3%, −0.9%; p = 0.02), corresponding to a relative reduction in HFF of −51.5% (95% CI −94.6%, −8.4%; p = 0.02). rhGH did not significantly reduce ALT AST, or GGT over the treatment period, although effect sizes suggest the possibility of modest reductions, particularly in GGT (Table 2). Adjusting for changes in BMI or VAT during the study attenuated the effect of GH on liver fat content (absolute effect size adjusting for BMI −2.7% [95% CI −7.9, 2.5], p = 0.29; absolute effect size adjusting for VAT −2.7% [95% CI −6.8, 1.4], p = 0.19). Among the entire study cohort, changes in HFF were strongly associated with changes in VAT (r = 0.49, p = 0.04) but not changes in SAT (r = 0.24, p = 0.34), total fat (r = 0.37, p = 0.16), or BMI (r = 0.29, p = 0.25).
Figure 2:
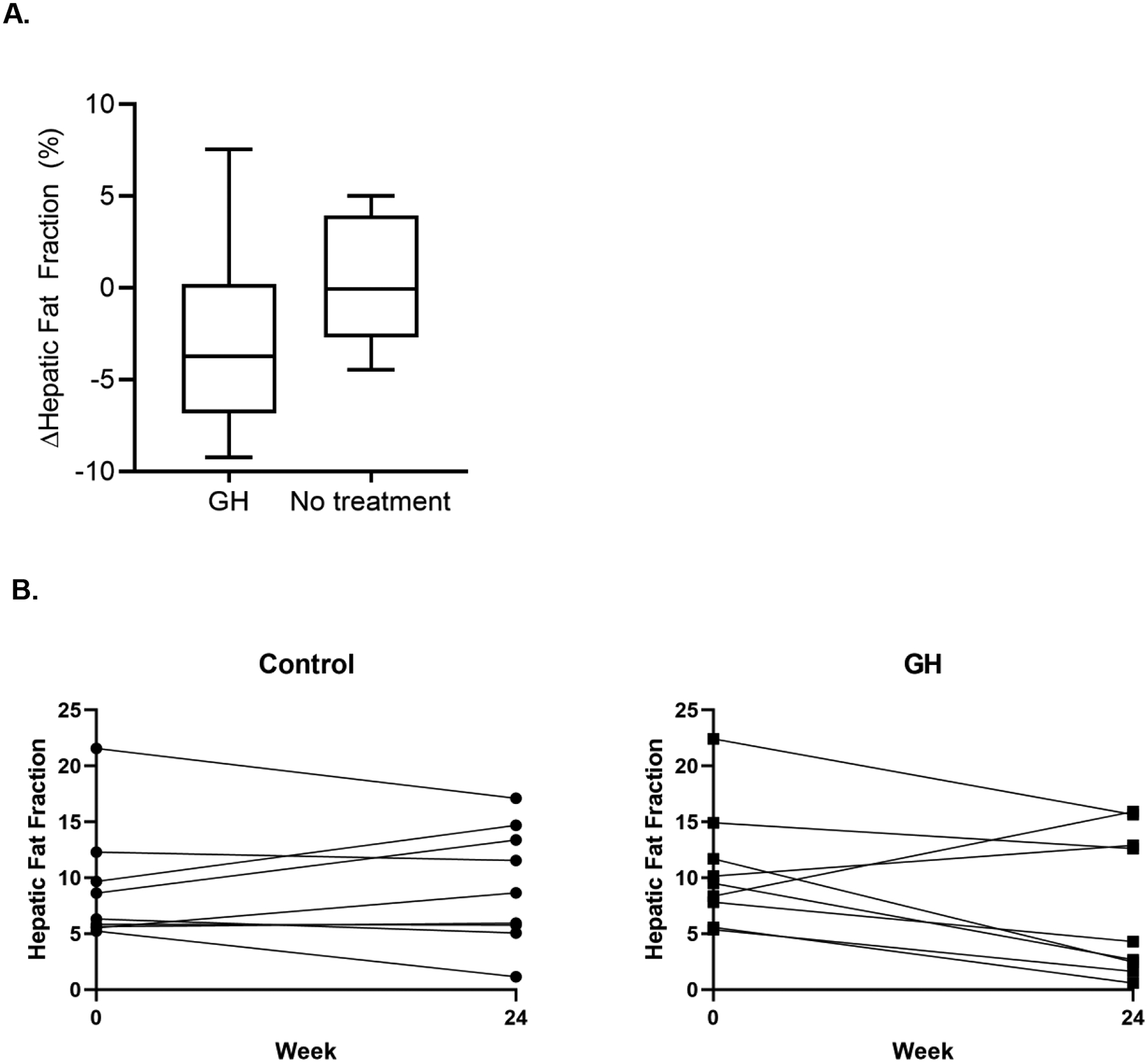
(A) Box-plot representation of the change in hepatic fat fraction in each treatment group between baseline and 24 weeks, with the middle bar representing the median, boxes representing interquartile range, and whiskers representing 95th percentile. (B) Baseline and 24 week HFF for each participant by treatment group.
Body Composition and Metabolic Endpoints
Changes in body composition and metabolic endpoints are shown in Table 2. At 24 weeks, participants in the rhGH group had a decrease in BMI compared to those receiving no treatment. This represents a combination of modest reductions in both fat mass and lean mass in the rhGH group. rhGH did not significantly reduce VAT, SAT, or waist circumference, although effect sizes suggest possible modest reductions. There was no effect of rhGH on LDL-cholesterol, HDL-cholesterol, or triglycerides. rhGH also did not significantly affect fasting glucose or 2-hour glucose following 75g glucose challenge.
Changes in Nutritional Intake and Activity
There were no significant differences between groups in changes in estimates of daily caloric intake by 24-hour food recall over 24 weeks (419.9 ± 2643.8 in the rhGH group versus −127.1 ± 687.4 kcal/day in the no treatment group, p = 0.59) nor in macronutrient intake (data not shown). There were also no significant differences between groups in changes in grams of alcohol consumed per week (−0.02 ± 0.15 in the rhGH group versus −0.02 ± 0.08 g/day in the no treatment group, p = 0.94) reported by 24-hour food recall.
Safety and Adherence
One serious adverse event, a hospitalization for psychiatric reasons in a patient with history of previous hospitalizations for psychiatric reasons, was seen in the rhGH group and was judged unrelated to study drug. No participants had fasting glucose above 7mmol/L (126mg/dL) at any point in the study, nor did any subject have a 2-hour glucose level >11.1mmol/L (200mg/dL) following oral glucose load at the baseline or 24-week oral glucose tolerance test. Five subjects in the rhGH group compared to one subject in the no treatment group complained of mild headaches during the study. Four subjects in the rhGH group experienced injection-site bruising. One subject in the rhGH group complained of jaw stiffness and pain. No subjects complained of edema or extremity swelling. There were no drug-related reasons for discontinuation from the study. Other reasons for study discontinuation are outlined in Figure 1.
Adherence to daily injections was measured by change in IGF-1 z-score. Participants in the rhGH group had an average increase of 1.1 ± 0.8 in IGF-1 z-scores, and participants in the no treatment group had an average decrease of −0.3 ± 0.6 in IGF-1 z-score (Table 2, Figure 3). The average IGF-1 z-score in participants at the 24-week visit was 0.4 ± 0.9 in the rhGH group versus −0.5 ± 0.7 in the no treatment group. Within the rhGH group, one participant had an IGF-1 z-score over the prespecified dose-reduction threshold during the study and required a dose reduction to 0.4mg. Three participants had IGF-1 z-scores below the prespecified dose-increase threshold: one required 2 dose increases to 0.7mg, one required 3 dose increases to 0.8mg, and one required 4 dose increases to 0.9mg daily.
Figure 3:
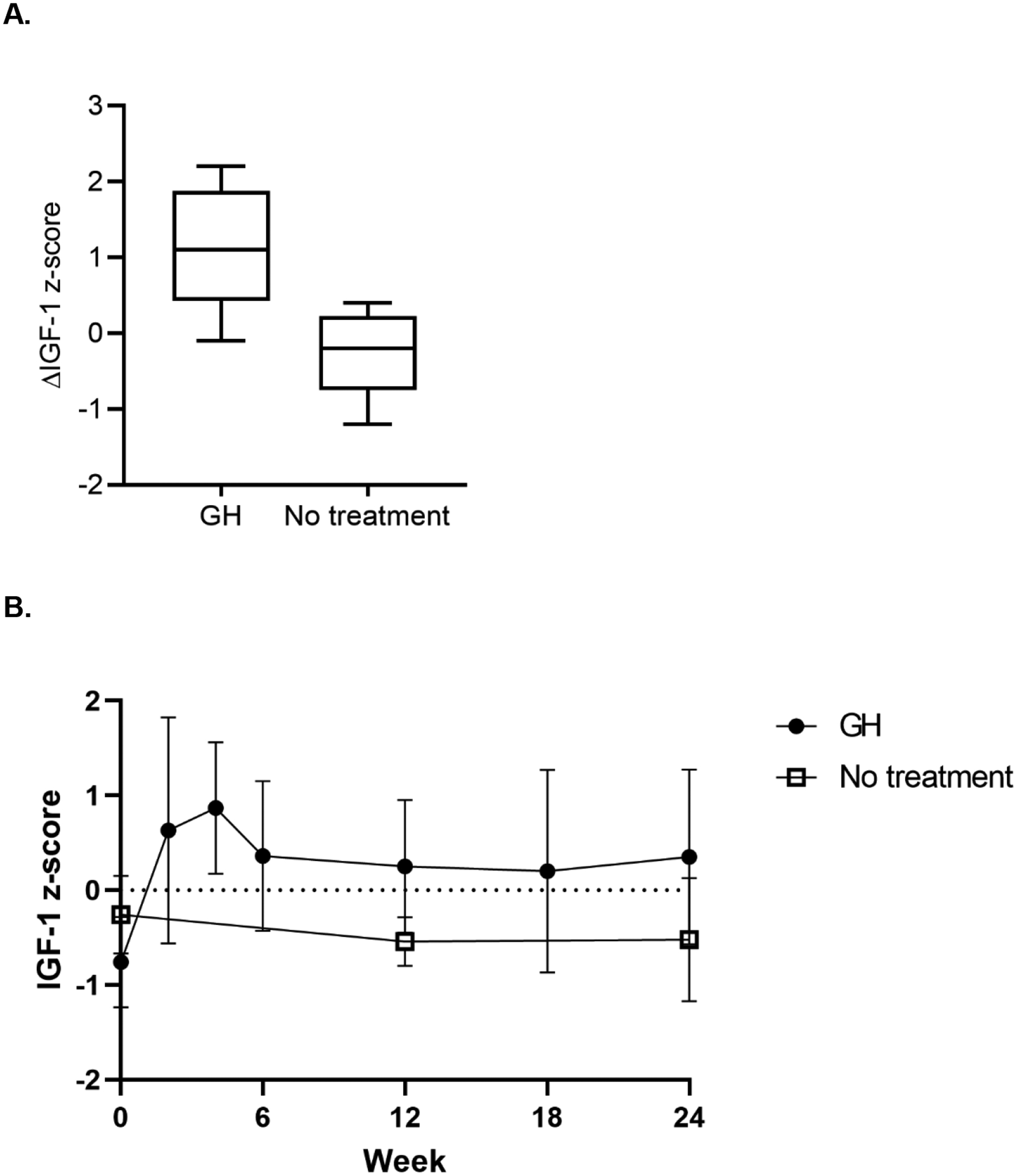
(A) Box-plot representation of the change in IGF-1 z-score, with the middle bar representing the median, the boxes representing interquartile range, and the whiskers representing 95th percentile. (B) IGF-1 z-score (mean with standard deviation) at each timepoint in each treatment group.
Relationships between GH, IGF-1, Hepatic and Metabolic Endpoints, and Body Composition at Baseline
Among the entire cohort at baseline, higher peak GH on arginine-clonidine stimulation testing was associated with a trend toward lower VAT (r = −0.40, p = 0.09) and was not associated with HFF (r = −0.11, p = 0.64) or BMI (r = −0.30, p = 0.21). There was a significant inverse relationship between peak GH and CRP (r = −0.52, p = 0.02) and a significant positive association with HDL (r = 0.57, p = 0.01), with no relationships with LDL or triglycerides. There were no associations between peak GH and ALT (r = 0.12, p = 0.63), AST (r = 0.19, p = 0.44), or GGT (r = 0.33, p = 0.17). In subgroup analyses by sex, females demonstrated associations between peak GH and body composition parameters (r = −0.65, p = 0.03 for VAT, r = −0.79, p = 0.004 for BMI, and r = −0.51, p = 0.11 for HFF), as well as a possible relationship between peak GH and ALT (r = −0.49, p = 0.13), whereas none of these relationships was present in males.
Differential relationships by sex were also seen in baseline relationships between IGF-1 z-score and hepatic measures. Among the entire cohort, there were no associations at baseline between IGF-1 or IGF-1 z-score and HFF, ALT, AST, or GGT. In subgroup analyses by sex, however, males demonstrated associations between IGF-1 z-score and ALT (r = −0.71, p = 0.02), GGT (r = −0.75, p = 0.01), and AST (r = −0.59, p = 0.07), whereas these associations were not present in females.
Among the entire cohort at baseline, HFF was strongly and positively correlated with VAT (r = 0.49, p = 0.01) but not SAT (r = 0.17, p = 0.42), waist circumference (r = 0.22, p = 0.33), or BMI (r = 0.14, p = 0.53). As expected HFF was strongly and positively associated with ALT (r = 0.71, p = 0.000), AST (r = 0.66, p = 0.0008), and GGT (r = 0.59, p = 0.004) at baseline.
Discussion
This pilot study administered rhGH titrated to IGF-1 z-score between 0 and 2 to young adults with obesity and IGF-1 z-scores below 0 at study entry, over 24 weeks, with the hypothesis that rhGH would reduce liver fat content in this group. Although there was not a statistically significant reduction in HFF, the rhGH effect size (−3.3% [95% CI −7.8 to 1.2%] absolute and −36% [−88.4 to 15.9%] relative change in HFF) suggests a magnitude of change that may be clinically significant and may warrant investigation in larger studies. Further, a significantly greater number of individuals in the rhGH group had “resolution” of steatosis to < 5% on 1H-MRS compared to those in the no treatment group. These changes may have been attributable in part to a reduction in BMI during the study in participants receiving rhGH. We did not note any significant effects of rhGH on ALT, AST, or GGT.
The known physiologic actions of GH, combined with evidence from multiple animal studies, strongly suggest a role for GH in mediating hepatic lipid content. Stable isotope studies demonstrate that approximately 60% of stored hepatic triglyceride is derived from uptake of circulating free fatty acid, 25% from hepatic de novo lipogenesis (DNL), and 15% from dietary fat intake (20). Relative reductions in GH are associated with increased VAT (10, 11), a highly lipolytic depot providing a substantial source of free fatty acid to the portal vein. Further, GH inhibits hepatic DNL, and treatment with rhGH has been shown to reduce DNL in both animal (21–25), and human (26) models. Liver-specific GH-receptor knock-out (LGHRKO) mice have significantly increased hepatic lipogenesis and severe hepatic steatosis, both of which are reversed with adenovirus-mediated rescue of GHR expression (12). Similarly, the spontaneous dwarf rat, which has a GH gene mutation, also demonstrates hepatic steatosis and fibrosis that can be reversed by treatment with exogenous GH (13). Consistent with these animal data, human studies show a higher prevalence of NAFLD and NASH in patients with pituitary GHD, with improvements in NAFLD and NASH following GH replacement (14, 15, 27).
To date, studies of the effects of rhGH on NAFLD are largely limited to individuals with pituitary GH deficiency (14, 15, 27, 28). Obesity is associated with relative reductions in endogenous GH secretion (9–11, 29), such that many individuals with obesity have a relative physiologic GH deficiency that is reversible with weight loss (30). Physiologic rhGH replacement in individuals with obesity has previously been shown to reduce visceral fat and/or overall body fat mass (31–33), and use of a GHRH analog to increase endogenous pulsatile GH has also been shown to reduce visceral fat and systemic inflammation in men and women with abdominal obesity and relative reductions in endogenous GH secretion (34). In individuals with HIV-infection, GHRH analog is used to reduce visceral fat and also has recently been shown to reduce liver fat and decrease the rate of fibrosis progression in individuals with HIV-infection and NAFLD (17, 35).
To our knowledge, the current data represent the first reported effects of rhGH on liver fat content in a randomized trial in individuals with obesity and NAFLD. These pilot data are not definitive but suggest a possible effect of rhGH to reduce hepatic fat content. This effect was partly attenuated when adjusting for changes in BMI or VAT, such that our data do not necessarily support an effect of GH to reduce liver fat independent of changes in body composition. Further, we saw a strong association between changes in VAT and changes in liver fat over the 24 weeks of the study. Existing literature suggests that rhGH achieves reductions in VAT, a modest decrease in body fat mass, and a modest increase in lean mass (31, 32). Significant reductions in overall BMI are usually not reported. We cannot be sure if the changes in BMI seen in our rhGH group are attributable to GH or to other factors independent of treatment assignment, and these results should be interpreted with caution in this pilot study.
The 36% relative liver fat reduction seen in our study is less than that reported with pioglitazone and FGF19 agonist, but relatively similar to effects reported with many other agents for which 1H-MRS or proton density fat fraction data from MRI are available. Pioglitazone has repeatedly shown benefit to decrease liver fat, with relative reductions of roughly 45–55% by MRI or MRS (36–38). More recently, glucagon-like peptide-1 (GLP-1) agonists have shown relative liver fat reductions of 30–45% (39–41); the GHRH analog tesamorelin has shown 37% relative reduction (17); resmetirom, a liver-directed selective thyroid hormone receptor-β agonist has shown a relative reduction of 29% (42); the FGF21 analog pegbelfermin has shown approximately 30–35% relative reduction over only 16 weeks (43); and an FGF19 agonist achieved 48–60% relative reduction over only 12 weeks (44). Importantly, current United States Food and Drug Administration guidance on trials for NAFLD and NASH does not support reduction in liver fat content as a meaningful clinical endpoint; rather, reductions in inflammation and/or fibrosis by serial biopsy are required to demonstrate efficacy in reducing liver-related morbidity and mortality (45). Accordingly, the comparison of liver fat reduction above does not necessarily correlate with clinical efficacy against NAFLD/NASH. Additionally, the comparison does not include Phase 2b/3 trials with biopsy-based outcomes for agents including liraglutide (46) and obeticholic acid (47). Our data are not intended to suggest rhGH as a treatment for NAFLD/NASH, but rather to further support meaningful effects of GH in regulating liver fat content.
Our study has multiple limitations of note, primarily the small sample size in this pilot investigation. Our study was powered for complete data on 19 participants, and we had complete data on 18 for HFF. Secondary endpoints are likely further underpowered, such that lack of effect in this study cannot be interpreted as definitive evidence of no effect. Additionally, our participants were chosen based on ≥5% steatosis by 1H-MRS; we did not perform liver biopsies or surrogate tests for fibrosis, and many of our participants likely had simple steatosis rather than NASH. Thus, while we did not see effects on ALT, AST, or GGT, these results might have differed in a cohort chosen specifically for NASH. Additionally, the use of IGF-1 z-score ≤0 as an inclusion criterion is not a perfect means to select individuals with GH deficiency, as some individuals with low responses to provocative stimulation of GH have normal IGF-1 levels, and some people with lower IGF-1 levels have normal GH response to provocative stimulation. Both arginine and clonidine are provocative agents used to stimulate GH secretion (48, 49), but this is not a standard clinical testing protocol, and we did not aim to diagnose GH deficiency in our participants, who were otherwise healthy adults who likely but not definitively had relative reductions in GH secretion due to obesity based on below-average serum IGF-1. Provocative GH testing protocols have relatively high intra-individual variability when performed in individuals without pituitary GH deficiency (50), and we consider our results regarding associations of peak stimulated GH with body composition and metabolic variables to be exploratory in nature. Duration of GH treatment may have affected results. Although many studies have shown reductions in liver fat after 6 months or less, it is possible that continuing reductions may take place with further treatment or, less likely, that rebound effect may occur with longer duration. We also chose not to use an identical placebo, such that we are comparing the effects of rhGH to the natural history of NAFLD but may not be capturing placebo effect. BMI was reduced in the treatment group, and adjustment for BMI attenuated the effect of rhGH on liver fat. BMI reduction is not a commonly observed effect of rhGH administration and may have been attributable to lifestyle changes that subjects made related to their knowledge of treatment assignment. Finally, as discussed above, hepatic lipid content is of physiological interest, and is likely to be relevant as the first step in development of NASH, but changes in inflammation and fibrosis are more relevant endpoints with regard to liver-specific morbidity and mortality. Strengths of our study include a focus on young adults, a population that is relatively understudied in investigations of NAFLD and dose titration to maintain IGF-1 levels between 0 and 2 standard deviations above the mean for age and sex.
In summary, our data demonstrate that treatment with rhGH may have the potential to reduce liver fat content, and, in this small cohort, did not disturb glycemia or result in significant adverse events. Additional larger studies, as well as studies with endpoints based on liver biopsy, would be necessary to support the clinical relevance of this finding.
Acknowledgements:
We would like to thank the research volunteers for their participation and dedication to the study, as well as the Massachusetts General Hospital Clinical Research Center research nurses, bionutritionists, and staff.
Funding Statement:
This work was supported by an investigator-initiated research grant to Dr. Stanley from Novo Nordisk, Inc. Novo Nordisk had no role in the performance of the study, the analysis of data, or the preparation or approval of this manuscript. This work was also supported in part by P30DK040561 (T.L.S.) and 1K23HD100266 (L.T.F.).
Conflict of Interest:
CSP and JJW have nothing to report. LTF has served as a consultant for Theratechnologies, Inc. CB, KLB, HL and MT have nothing to report. MM has served as a consultant for Abbvie and Sanofi. TLS received investigator-initiated grant funding for this work to her institution from Novo Nordisk, Inc.
Data Sharing Statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr. 2013;162(3):496–500 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doycheva I, Watt KD, Rifai G, Abou Mrad R, Lopez R, Zein NN, et al. Increasing Burden of Chronic Liver Disease Among Adolescents and Young Adults in the USA: A Silent Epidemic. Dig Dis Sci. 2017;62(5):1373–80. [DOI] [PubMed] [Google Scholar]
- 3.Goldner D, Lavine JE. Nonalcoholic Fatty Liver Disease in Children: Unique Considerations and Challenges. Gastroenterology. 2020;158(7):1967–83 e1. [DOI] [PubMed] [Google Scholar]
- 4.Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J Pediatr Gastroenterol Nutr. 2017;64(2):319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118(4):1388–93. [DOI] [PubMed] [Google Scholar]
- 6.Xanthakos S, Miles L, Bucuvalas J, Daniels S, Garcia V, Inge T. Histologic spectrum of nonalcoholic fatty liver disease in morbidly obese adolescents. Clin Gastroenterol Hepatol. 2006;4(2):226–32. [DOI] [PubMed] [Google Scholar]
- 7.Nseir W, Hellou E, Assy N. Role of diet and lifestyle changes in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(28):9338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koutoukidis DA, Astbury NM, Tudor KE, Morris E, Henry JA, Noreik M, et al. Association of Weight Loss Interventions With Changes in Biomarkers of Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. JAMA Intern Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanley TL, Levitsky LL, Grinspoon SK, Misra M. Effect of body mass index on peak growth hormone response to provocative testing in children with short stature. J Clin Endocrinol Metab. 2009;94(12):4875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makimura H, Stanley T, Mun D, You SM, Grinspoon S. The effects of central adiposity on growth hormone (GH) response to GH-releasing hormone-arginine stimulation testing in men. J Clin Endocrinol Metab. 2008;93(11):4254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misra M, Bredella MA, Tsai P, Mendes N, Miller KK, Klibanski A. Lower growth hormone and higher cortisol are associated with greater visceral adiposity, intramyocellular lipids, and insulin resistance in overweight girls. Am J Physiol Endocrinol Metab. 2008;295(2):E385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, et al. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284(30):19937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishizawa H, Takahashi M, Fukuoka H, Iguchi G, Kitazawa R, Takahashi Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem Biophys Res Commun. 2012;423(2):295–300. [DOI] [PubMed] [Google Scholar]
- 14.Nishizawa H, Iguchi G, Murawaki A, Fukuoka H, Hayashi Y, Kaji H, et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur J Endocrinol. 2012;167(1):67–74. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Iida K, Takahashi K, Yoshioka S, Fukuoka H, Takeno R, et al. Growth hormone reverses nonalcoholic steatohepatitis in a patient with adult growth hormone deficiency. Gastroenterology. 2007;132(3):938–43. [DOI] [PubMed] [Google Scholar]
- 16.Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology. 2004;39(4):909–14. [DOI] [PubMed] [Google Scholar]
- 17.Stanley TL, Fourman LT, Feldpausch MN, Purdy J, Zheng I, Pan CS, et al. Effects of tesamorelin on non-alcoholic fatty liver disease in HIV: a randomised, double-blind, multicentre trial. Lancet HIV. 2019;6(12):e821–e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeder SB, Sirlin CB. Quantification of liver fat with magnetic resonance imaging. Magn Reson Imaging Clin N Am. 2010;18(3):337–57, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prati D, Taioli E, Zanella A, Della Torre E, Butelli S, Del Vecchio E, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med. 2002;137(1):1–10. [DOI] [PubMed] [Google Scholar]
- 20.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman HM. Effects of chronic growth hormone treatment on lipogenesis by rat adipose tissue. Endocrinology. 1963;72:95–9. [DOI] [PubMed] [Google Scholar]
- 22.Adamafio NA, Ng FM. Effects of growth hormone on lipogenesis and glucose oxidation in genetically GH-deficient mice. Mol Cell Endocrinol. 1984;37(2):241–4. [DOI] [PubMed] [Google Scholar]
- 23.Perry WF, Bowen HF. The effect of growth hormone on lipogenesis in intact and adrenalectomized rats. Endocrinology. 1955;56(5):579–83. [DOI] [PubMed] [Google Scholar]
- 24.Ng FM, Adamafio NA, Graystone JE. Effects of exogenous growth hormone on lipid metabolism in the isolated epididymal fat pad of the growth hormone-deficient little mouse. J Mol Endocrinol. 1990;4(1):43–9. [DOI] [PubMed] [Google Scholar]
- 25.Vernon RG. GH inhibition of lipogenesis and stimulation of lipolysis in sheep adipose tissue: involvement of protein serine phosphorylation and dephosphorylation and phospholipase C. J Endocrinol. 1996;150(1):129–40. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz JM, Mulligan K, Lee J, Lo JC, Wen M, Noor MA, et al. Effects of recombinant human growth hormone on hepatic lipid and carbohydrate metabolism in HIV-infected patients with fat accumulation. J Clin Endocrinol Metab. 2002;87(2):942. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto R, Fukuoka H, Iguchi G, Nishizawa H, Bando H, Suda K, et al. Long-term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Horm IGF Res. 2014;24(5):174–9. [DOI] [PubMed] [Google Scholar]
- 28.Gilliland T, Dufour S, Shulman GI, Petersen KF, Emre SH. Resolution of non-alcoholic steatohepatitis after growth hormone replacement in a pediatric liver transplant patient with panhypopituitarism. Pediatr Transplant. 2016;20(8):1157–63. [DOI] [PubMed] [Google Scholar]
- 29.Miller KK, Biller BM, Lipman JG, Bradwin G, Rifai N, Klibanski A. Truncal adiposity, relative growth hormone deficiency, and cardiovascular risk. J Clin Endocrinol Metab. 2005;90(2):768–74. [DOI] [PubMed] [Google Scholar]
- 30.Rasmussen MH, Hvidberg A, Juul A, Main KM, Gotfredsen A, Skakkebaek NE, et al. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J Clin Endocrinol Metab. 1995;80(4):1407–15. [DOI] [PubMed] [Google Scholar]
- 31.Johannsson G, Marin P, Lonn L, Ottosson M, Stenlof K, Bjorntorp P, et al. Growth hormone treatment of abdominally obese men reduces abdominal fat mass, improves glucose and lipoprotein metabolism, and reduces diastolic blood pressure. J Clin Endocrinol Metab. 1997;82(3):727–34. [DOI] [PubMed] [Google Scholar]
- 32.Pasarica M, Zachwieja JJ, Dejonge L, Redman S, Smith SR. Effect of growth hormone on body composition and visceral adiposity in middle-aged men with visceral obesity. J Clin Endocrinol Metab. 2007;92(11):4265–70. [DOI] [PubMed] [Google Scholar]
- 33.Albert SG, Mooradian AD. Low-dose recombinant human growth hormone as adjuvant therapy to lifestyle modifications in the management of obesity. J Clin Endocrinol Metab. 2004;89(2):695–701. [DOI] [PubMed] [Google Scholar]
- 34.Makimura H, Feldpausch MN, Rope AM, Hemphill LC, Torriani M, Lee H, et al. Metabolic effects of a growth hormone-releasing factor in obese subjects with reduced growth hormone secretion: a randomized controlled trial. J Clin Endocrinol Metab. 2012;97(12):4769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falutz J, Allas S, Blot K, Potvin D, Kotler D, Somero M, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359–70. [DOI] [PubMed] [Google Scholar]
- 36.Bajaj M, Suraamornkul S, Pratipanawatr T, Hardies LJ, Pratipanawatr W, Glass L, et al. Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes. 2003;52:1364–70. [DOI] [PubMed] [Google Scholar]
- 37.Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39(1):188–96. [DOI] [PubMed] [Google Scholar]
- 38.Belfort R A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307. [DOI] [PubMed] [Google Scholar]
- 39.Frossing S, Nylander M, Chabanova E, Frystyk J, Holst JJ, Kistorp C, et al. Effect of liraglutide on ectopic fat in polycystic ovary syndrome: A randomized clinical trial. Diabetes Obes Metab. 2018;20(1):215–8. [DOI] [PubMed] [Google Scholar]
- 40.Dutour A, Abdesselam I, Ancel P, Kober F, Mrad G, Darmon P, et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: a prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes Metab. 2016;18(9):882–91. [DOI] [PubMed] [Google Scholar]
- 41.Matikainen N, Soderlund S, Bjornson E, Pietilainen K, Hakkarainen A, Lundbom N, et al. Liraglutide treatment improves postprandial lipid metabolism and cardiometabolic risk factors in humans with adequately controlled type 2 diabetes: A single-centre randomized controlled study. Diabetes Obes Metab. 2019;21(1):84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394(10213):2012–24. [DOI] [PubMed] [Google Scholar]
- 43.Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392(10165):2705–17. [DOI] [PubMed] [Google Scholar]
- 44.Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2018;391(10126):1174–85. [DOI] [PubMed] [Google Scholar]
- 45.FDA. Noncirrhotic Nonalcoholic Steatohepatitis With Liver Fibrosis: Developing Drugs for Treatment Guidance for Industry from the United States Food and Drug Administration 2018. [Available from: https://www.fda.gov/media/119044/download.
- 46.Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–90. [DOI] [PubMed] [Google Scholar]
- 47.Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394(10215):2184–96. [DOI] [PubMed] [Google Scholar]
- 48.Aimaretti G, Baffoni C, DiVito L, Bellone S, Grottoli S, Maccario M, et al. Comparisons among old and new provocative tests of GH secretion in 178 normal adults. Eur J Endocrinol. 2000;142(4):347–52. [DOI] [PubMed] [Google Scholar]
- 49.Webb SM, Strasburger CJ, Mo D, Hartman ML, Melmed S, Jung H, et al. Changing patterns of the adult growth hormone deficiency diagnosis documented in a decade-long global surveillance database. J Clin Endocrinol Metab. 2009;94(2):392–9. [DOI] [PubMed] [Google Scholar]
- 50.Hoeck HC, Jakobsen PE, Vestergaard P, Falhof J, Laurberg P. Differences in reproducibility and peak growth hormone responses to repeated testing with various stimulators in healthy adults. Growth Horm IGF Res. 1999;9(1):18–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.