

Abstract
Converting solar energy to fuels has attracted substantial interest over the past decades because it has the potential to sustainably meet the increasing global energy demand. However, achieving this potential requires significant technological advances. Polymer photoelectrodes are composed of earth-abundant elements, e.g. carbon, nitrogen, oxygen, hydrogen, which promise to be more economically sustainable than their inorganic counterparts. Furthermore, the electronic structure of polymer photoelectrodes can be more easily tuned to fit the solar spectrum than inorganic counterparts, promising a feasible practical application. As a fast-moving area, in particular, over the past ten years, we have witnessed an explosion of reports on polymer materials, including photoelectrodes, cocatalysts, device architectures, and fundamental understanding experimentally and theoretically, all of which have been detailed in this review. Furthermore, the prospects of this field are discussed to highlight the future development of polymer photoelectrodes.
1. Need for solar fuels
The energy required for the planet is primarily derived from the sun through natural photosynthesis.5 Over time, energy demand has substantially increased due to the industrial revolution, the increasing population, and the increase in quality of life-related human activities such as transportation, electricity generation, and heating.6 At present, fossil fuels such as oil, coal, and natural gas together satisfy ∼80% of the global energy demand largely driven by characteristics such as high energy density, low cost, availability, ease of handling, storage, and transportation.7 Nevertheless, the combustion of fossil fuels releases a massive amount of CO2, a key greenhouse gas, into the atmosphere leading to global warming and climate change.8 On the other hand, worldwide energy consumption is projected to double by 2050 due to the rapid upsurge of the population.9 To address these issues, the development of sustainable energy technologies is essential.
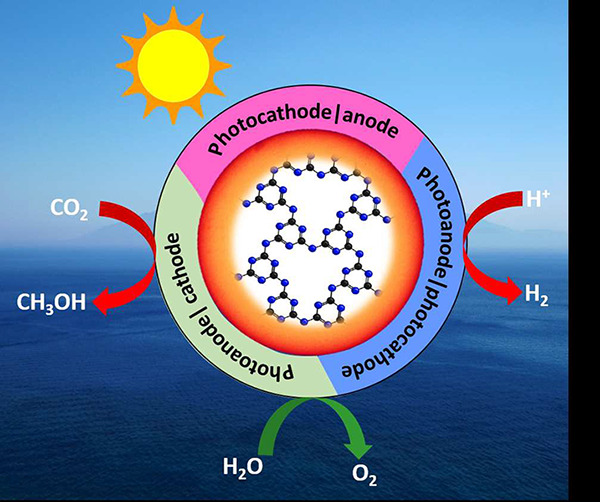
Fascinatingly, the sun provides 1000 times higher energy (1.9 × 108 TWh/yr) than the global energy consumption (1.3 × 105 TWh/yr). To harness this primary energy, remarkable advances have been made to produce sustainable electricity by solar cells.10 Although solar electricity will surely play a key role in future energy infrastructure, there is a significant need to produce high energy density fuels and store them for prolonged time use.11 Thus, conversion of solar energy into chemical fuels, especially via hydrogen (H2) generation from earth-abundant water or high value-added chemical synthesis involving utilization of CO2, is highly desirable. Storing solar energy in the form of chemical bonds, similar to natural photosynthesis, is very attractive as it could be released upon demand. For instance, solar-derived green H2 has the potential to replace gray or blue H2, which has been vastly used for ammonia synthesis and in refineries.12 It is worth noting that the H2 specific mass energy (140 MJ kg–1) is much higher than that of natural gas (55 MJ kg–1), gasoline (34.2 MJ kg–1), diesel (45 MJ kg–1), and coal (24 MJ kg–1).13 Presently, the H2 fuel cell has been demonstrated to power aircraft, rail vehicles, buses, and passenger cars.14 Similarly, high-value chemicals can be produced from CO2 including liquid fuels, e.g., methanol and ethanol, which can be directly used in place of fossil fuels. More importantly, alcohols are easy to store in liquid form for an extended time and can be used right away with the existing energy distribution infrastructure. Therefore, this review mainly covers water splitting and CO2 reduction reactions using polymer photoelectrodes driven by solar energy.
1.1. Solar Fuel Production Techniques
To substitute both gray and blue H2 production technologies, electrocatalysis was proposed decades ago, which is purely driven by an external bias, using the appropriate electrocatalyst-loaded anode and cathode for separate H2 and O2 production.15,16 The faradaic efficiency (FE) and product selectivity depend on the electrocatalyst, electrolyte, and applied potential. Electrocatalytic water splitting and CO2 reduction have been reviewed extensively.17−20 Although this field has progressed, conventional electrolysis has high capital and operational cost. Some other methods such as thermolysis, photolysis, and biomass conversion techniques for green H2 production are also under active development, and the reader can find more details in these reviews.21−24
Photocatalysis (suspension-based, Figure 1a) is highly desirable as it is driven only by sustainable solar energy, providing the potential to produce H2 and high-value chemicals competitively and economically. If the bandgap of the semiconductor is equal to or smaller than the irradiation energy, incoming light irradiation on a photocatalyst powder suspended in an aqueous solution photoexcite the valence band (VB) electrons to the conduction band (CB) of the semiconductor, leaving the corresponding holes in the VB. The photogenerated electrons and holes perform the reduction and oxidation reactions, respectively. Since the first report on photocatalytic water splitting, the single photoabsorber-based system has been vastly studied.25,26 In recent times, dual photoabsorber systems, also known as Z-scheme similar to the natural photosystem, have increased in popularity as they have several advantages such as better charge separation, more efficient solar energy harvesting, and the possibility of onsite separation of evolved gases. In such systems, the dual photoabsorbers are combined using an appropriate shuttle redox mediator to regenerate both photocatalysts in a cyclic process. More details about Z-scheme water splitting can be found in our recent review.27 Similarly, there are several reports28−30 on Z-scheme CO2 conversion into value-added fuels. The various Z-scheme approaches, photoreactor designs, and the discussion on large-scale solar fuel production are also available in the literature.31−34
Figure 1.

Solar fuel production approaches. (a) Suspension-based photocatalysis. (b) Photoanode|dark cathode. (c) Photocathode|dark anode. (d) Photoanode|Photocathode. Note: For CO2 reduction, the same design is applied with the exception of CO2 reduction rather than proton reduction on the cathodes. The bias in Figure 1d could be omitted if two photoelectrodes can form a Z-scheme.
The term photoelectrochemical process (PEC) in itself conveys the advantages that are chalked up to both electrocatalysis and photocatalysis. In PEC, a photocatalyst thin film is deposited on a conducting substrate to make what is called a photoelectrode (either the photoanode or photocathode). This is used to absorb the solar energy to generate electron–hole pairs, which can be separated by the internal electric field at the interface. In most cases, an external bias is applied to increase the electric field and help separate charges, in turn increasing the potential of the charges and promoting reactivity. The separated electrons and holes perform the reduction and oxidation reactions, respectively, either directly on the surface of the photocatalyst or on the surface of a cocatalyst.
1.2. PEC Approaches
On the basis of the photoelectrode configurations, three PEC approaches viz., (i) photoanode|dark cathode, (ii) photocathode|dark anode, and (iii) photoanode|photocathode have been used for solar fuel production. In the photoanode|dark cathode system (Figure 1b), the photoanode acts as the working electrode (WE) for the oxidation half-reaction and a nonphotoactive cathode acts as the counter electrode (CE) for the reduction half-reaction. A stable reference electrode (RE) is also used to control the applied external bias. These electrodes are immersed into the aqueous electrolyte solution, leading to the formation of a semiconductor–electrolyte interface. For the Fermi level of the photoanode to equilibrate with the electron electrochemical potential of the aqueous electrolyte, electrons are transferred from the photoanode (n-type semiconductors) to the electrolyte, which creates a space-charge region also known as an electrical double layer. This built-in potential at the interface induces the upward band bending of the semiconductor. Upon light irradiation, the VB electrons are excited to the CB of the semiconductor and pass to the CE (dark cathode) through an external circuit. These extracted photogenerated electrons perform the reduction reaction at the cathode. The holes left at the VB are transferred to the surface of the photoanode, where an oxidation half-reaction takes place. The n-type inorganic semiconductor-based photoanodes for PEC water splitting and CO2 conversion have been vastly reported.35−41 Compared with the inorganic counterpart, polymer-based photoanodes have not received the same attention yet and will be addressed in Section 2.
In the photocathode|dark anode approach (Figure 1c), the photocathode acts as the WE and the photoinactive anode as the CE. The rest of the conditions are similar to the photoanode|dark cathode configuration. The Fermi-level difference between the semiconductor and the electrolyte induces the electrochemical potential across the interface, known as a built-in electric field, causing the downward band bending of the semiconductor (p-type photocathode). Upon light irradiation of the photon energy higher than or equal to the bandgap, the photocathode semiconductor electrons are excited to the CB, leaving the corresponding holes in the VB. The built-in potential drives the photogenerated electrons to the surface of the semiconductor followed by interfacial charge transfer to molecules on its surface. An external bias can be applied to facilitate this process. Subsequently, the corresponding holes pass through the external circuit to the CE, where the oxidation reaction takes place. Several inorganic semiconductor-based photocathodes for solar fuel production have been studied and many reviews are available.4,42,43 Similarly, polymer-based photocathodes have been little reviewed and will be comprehensively discussed in Section 3.
The photoanode|photocathode configuration (Figure 1d), also known as a tandem cell, consists of an anode and cathode that are both photoactive. Often, n-type and p-type semiconductors are used as the photoanode and photocathode, respectively. If the CB of the photoanode semiconductor is more negative than the VB potential of the photocathode semiconductor, the couple can form a Z-scheme and the external bias is not required. The holes left in the VB of the photoanode and the electrons in the photocathode CB perform oxidation and reduction reactions, respectively. The readers can find more details about the inorganic metal oxide-based tandem-PEC approach for solar water splitting and CO2 conversion in the literature,44−46 which is beyond the scope of this review.
1.3. Polymer-Based Photoelectrodes
One of the main objectives of the polymer (including some examples of organic molecules in this review) based photoelectrodes is to reduce the overall fabrication cost. Standard inorganic materials such as compound semiconductors (groups III–V, Si, etc.),43 transition metal oxides (BiVO4, Fe2O3, TiO2, and Cu2O), sulfides (NiS, CdS), oxynitrides (BaNbO2N), and chalcogenides47−51 can be expensive and composed of less abundant elements. Next, the fabrication of inorganic photoelectrodes often involves complex deposition techniques such as atomic layer deposition (ALD), high-temperature sintering, electron-beam evaporation, and high energy sputtering, which increase the manufacturing cost and are less straightforward for large-scale production. The synthesis and preparation of polymer-based photoelectrodes are less complex and suitable for preparing large-scale devices as it mostly involves synthetic approaches at ambient conditions, followed by coating on a conductive substrate using low-cost methods such as spin coating, doctor blading, and printing.52 Another key advantage of the polymers or organic molecules is that their band positions can be readily tuned, much easier than the inorganic counterparts.53
The stability of the photoelectrodes is one crucial factor to consider, given the cost competition with conventional fossil fuel technologies. For instance, photoelectrodes must have a lifetime of 10 years with 10% solar to hydrogen conversion efficiency (STH) to produce H2 with a commercially competitive cost of 2–4 USD per kg.54 We believe that polymer-based photoelectrodes have the potential to achieve such high stability and efficiency after careful optimization. Organic semiconductors used in photoelectrodes include conducting polymers, oligomers, or self-assembled discrete molecules. Their electronic properties can be tuned more easily than inorganic semiconductors, and hence, diverse materials can be prepared to suit various desired oxidation and reduction reactions. For instance, the photocathode must have the CB potential more negative to the water/CO2 reduction potential to produce H2/carbon-fuel efficiently. Similarly, the photoanode must have the VB potential more positive than the water oxidation potential of +1.23 V vs RHE to produce O2.
In recent years, reports about polymer-based (including organic molecule-based) photoelectrodes for hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and carbon dioxide reduction (CRR) have become more frequent, complementary to the widely used particle suspension system and inorganic counterparts. Many reviews have concentrated on the latter while few of them address the progress, challenges, and prospects of polymer photoelectrodes.1−4 Polymer-based photoelectrodes have the advantage of high optical absorption (absorption coefficient 105 cm–1)55 to harvest solar light efficiently, and their bandgap (absorption spectrum) can be tuned by altering the synthesis conditions to, for example, introduce heteroatoms or change functional groups. Polymer photocatalysts have also shown good charge carrier mobility.56 All these advantages have been successfully demonstrated in organic photovoltaics (OPVs).57 For instance, the conjugated polymers and self-assembled molecules were substantially reported in OPVs and extended to solar fuel production using an OPV-biased-PEC system.58,59
Photoanodes are widely reported for water oxidation reactions, although a limited number of organic semiconductors have been used. This is due to the kinetic complexity of oxidation reactions compared to the water reduction reaction and the typical poor stability of organic photoanodes under oxidation conditions. The use of a polymer-based photoanode for PEC oxidation reaction originated from the modern studies of organic light-harvesting materials for photovoltaic applications. Loading a suitable cocatalyst on the surface of such a polymeric thin film enables photocatalytic water splitting.60 The traditional organic molecules such as phthalocyanine derivatives (Pc), and porphyrin/perylene derivatives (Por) have been reported for water oxidation and share structural similarity to the chlorophyll pigments used in natural photosynthesis. Recently, graphitic carbon nitride (CNx)-based photoanodes have been reported for solar fuel synthesis61 spurred by their stability, low cost, and ease of synthesis. However, sluggish water oxidation kinetics and fast charge-carrier recombination during the PEC reaction have compromised their efficiency, leading to poor photocurrent densities. Therefore, it is crucial to use advanced strategies such as intimate contact between the substrate and the film, maintaining porous structure, and doping as described in Section 2 to improve the photocurrent densities of polymer photoanodes.
Polymer-based photocathodes for solar energy conversion appear to be first reported in 1981.62 Following that, several conjugated polymers such as polythiophene, polypyrrole (PPy), and polyaniline (PANI)63,64 were reported. In particular, an early work on PEC water splitting using a poly(p-phenylene)-based photocathode to produce H2 was reported by Yanagida et al.,65 in which the polymers were prepared by in situ electrochemical polymerization using the respective monomers on conducting substrates. Although utilization of polymer-based photocathodes for solar fuel synthesis was pioneered about four decades ago, interest in this area was low for the next two decades. The progress of OPV, the limitations of inorganic metal oxides, and the urgent need for sustainable fuels have triggered the reconsideration of a wide range of materials for solar-driven catalysis, including polymer photoelectrodes. Recently, the fabrication of polymer photocathodes using CNx,66 covalent organic frameworks (COFs),67 metal–organic frameworks (MOFs),68 conjugated triazine frameworks (CTFs),69 and graphene70 have been explored for solar fuel synthesis. Notably, tuning the optical and catalytic properties by altering the synthesis conditions dramatically enhanced the PEC performances.71 Hence, it is essential to review the latest outcomes on PEC solar fuel synthesis using polymer-based photocathodes and address the challenges, as we do in Section 3.
The PEC activities of the rational polymer-based photoanodes or photocathodes can be enhanced by loading suitable oxidation or reduction electro/molecular catalysts, i.e., cocatalysts.72 The cocatalyst layer also protects the substrate underneath73 and improves the stability of the photoelectrodes together with the selectivity of the reaction.74 Noble metal-based cocatalysts such as Pt,75 Au,76 Ag,77 Pd,78 Rh,79 and Ir80 have been loaded on polymer photoelectrodes for solar fuel synthesis using photodeposition, impregnation, and in situ electrodeposition techniques to incorporate the metals into the polymer mix. These cocatalysts are expensive, and some are rare-earth elements and , hence, have limited use for large-scale practical applications. Recently, molecular-based cocatalysts attracted more attention as a suitable alternative to noble metals. Ruthenium (Ru)- and rhenium (Re)-based molecular catalysts, and scalable transition metals catalysts containing manganese (Mn) and cobalt (Co)-based have been progressively used for fabricating efficient polymer-based photoelectrodes. The latest research findings on molecular-based cocatalyst and transition metal-based cocatalysts toward performance improvement of polymer-based photoelectrodes for H2 production and CO2 reduction reactions have been summarized and presented in Section 4 of this review.
The efficiency of the polymer-based photoelectrodes is mainly determined by the photophysical properties of the polymers and their robustness during the reaction. Specifically, the charge carrier generation, separation, and transfer to the surface, as well as redox reactions at the active sites of the photoelectrodes cumulatively determine the quantum efficiency of the PEC reaction. Hence, a comprehensive understanding of charge carrier dynamics is crucial to the production of solar fuels. Despite several reports on the charge carrier dynamics of inorganic semiconductor-based photoelectrodes for water splitting and CO2 reduction, the mechanistic understanding using polymer-based photoelectrodes is rather limited and challenging because of the complex nature of the organic and aqueous interface, which is illustrated in Section 5. The theoritical modelling on polymer-based photoelectrodes is essential to predict the electronic and charge transfer properties of organic semiconductors, which is discussed in Section 6.
Finally, Section 7 concludes with the current understanding of polymer-based photoelectrodes, what are the leading questions that remain to be addressed, and lists out strategies to overcome long-standing challenges in fabricating efficient photoelectrodes, reducing fabrication costs, and increasing the stability.
2. Polymer-Based Photoanodes
As mentioned above, solar energy conversion using inorganic metal oxide-based photoanodes such as TiO2, SrTiO3, TaON, and ZnO for PEC H2 production81 and BiVO4, Fe2O3, and WO3 for PEC water splitting have been reported and reviewed extensively.82,83 Recently, earth-abundant elements viz. C, N, O, H-based organic semiconductors for solar fuel synthesis have received increasing attention as they are inexpensive. Even though, there are limited examples of polymers used as photoanodes. Here, CNx-based photoanodes followed by the polymers such as 3,4,9,10-perylenetetracarboxylic acid bisbenzimidazole, cobalt(II) phthalocyanine, poly[benzimidazobenzophenanthroline], and fluorinedibenzothiophene-S,S-dioxide-based conjugated polymer are reviewed. Albeit the polymers can be used as a single photoactive component, fabricating a bulk-heterojunction-based photoanode has more benefits including surface roughness, better charge separation, and increased stability as a photoanode for water splitting. The following subsections describe the different polymer materials being used for fabricating photoanodes.
2.1. CNx-Based Photoanode
Quite different from the suspension-based photocatalyst system, the PEC device relies on directional and long-distance charge transfer across the device. To improve the performance of the PEC system, the electrode/substrate contact, grain boundary contact, electrode surface band bending, and surface defects are crucial elements to be considered while preparing photoelectrodes. For example, CNx nanosheets were deposited on FTO substrates via the spray coating method, which simply coated the suspension-like CNx onto the substrate without modifying the contact, leading to a low photocurrent density of 0.0036 mA/cm2 @ 1.23 V vs RHE.84 A similar strategy of exfoliating CNx in methanol with postannealing, photoelectrode fabricated afterward resulted in a low photocurrent density of 0.010 mA/cm2 @ 1.23 V vs Ag/AgCl as well.85 Therefore, a strategy of modifying the contact between the CNx film and the substrate is crucial to obtain high photocurrent density. Thermal vapor condensation is a conventional film fabrication method (Figure 2a), which can grow compact g-CN in situ on an FTO substrate. Bian et al. used this method to fabricate uniform g-CN films at 600 °C using melamine as a single precursor, showing a photocurrent density of 0.12 mA/cm2 @ 1.35 V vs RHE with Na2S as the sacrificial reagent under AM 1.5 illumination.86 Similarly, two-step vapor deposition (Figure 2b) is an appropriate technique to obtain a CNx film. Lv et al. used this process for depositing CNx films with dicyanamide and obtained a photocurrent density of 0.063 mA/cm2 @ 1.23 V vs RHE under AM 1.5 illumination.87 Comparatively, the thermal vapor deposition method also enhances the quality of the CNx film by growing CNx directly on the substrate. All these factors, including contact between the film and the substrate, microstructure, and other parameters are detailed below to represent their influence on the PEC performance.
Figure 2.

Schematic representation of the synthesis of CNx using (a) a thermal vapor condensation method. Adapted with permission from ref (86). Copyright 2015 Elsevier. (b) Two-step vapor deposition method. Adapted with permission from ref (87). Copyright 2017 Elsevier.
2.1.1. Intimate Contact between the Film and the Substrate
The contact between the CNx film and a substrate, usually FTO, is a crucial factor in determining the efficiency of the photoelectrode, and hence, several strategies have been used to obtain a better intimate contact. With the use of a facile solvothermal method with postheating, intimate contact between CNx and the FTO substrate was achieved.88 In a report, the CNx powder was deposited as a compact thin film on the SnO2 flake by electrophoretic deposition using DC power at a constant voltage of 30 V for 3–10 min.89 Prior to this, the SnO2 nanoflakes film was chemically treated in a 0.5 M NaOH solution to add OH– surface functional groups which assisted in obtaining a dense and uniform coating of CNx nanosheets. This led to a photocurrent density of 0.15 mA/cm2 @ 1.23 V vs RHE under AM 1.5 illumination. In another report, the OH– groups on the FTO surface were shown to enable continuous grafting and polymerization of a melamine and cyanuric chloride (1:3 molar ratios) mixture, which was vaporized on FTO at 450 °C for 3 h, and the resultant 200 nm film exhibited a photocurrent density of 0.23 mA/cm2 @ 1.23 V vs RHE under AM 1.5 illumination.90 Apart from the OH– group, Fang et al. showed that sulfur (S) initialized the intimate growth of CNx films on FTO glass.91 The CNx films synthesized via thermal evaporation show that the S existed at the interfaces between the CNx and FTO, which was confirmed by X-ray photoelectron spectroscopy (XPS). The S facilitated charge migration between CNx and the substrate. As a result, it contributed to an improved photocurrent (compared to the absence of S) of ca. 0.1 mA/cm2 at 1.23 V vs RHE under AM 1.5 illumination.
2.1.2. Microstructure Control
The control of the microstructure of the film is another crucial factor to determine the efficiency of the photoelectrodes. In general, the photoelectrode fabrication process gives two types of film, i.e., compact or porous films. The compact film has fewer surface defects, higher crystallinity, controlled thickness, and a large electron transport distance.92 On the other hand, the porous structure has a short electron diffusion distance, large surface area, more active sites, and efficient light absorption.93 We discuss a few examples below to highlight the role of compact and porous CNx films on PEC performance.
Compact Film
Ruan et al. reported a rapid thermal evaporation–condensation method to prepare high-quality, compact CNx films, with controlled thickness from 500–1000 nm as shown in Figure 3a. Due to the reduced deep trap states, the film achieved a high open-circuit photovoltage of 0.3 V, which was much higher than the photovoltage obtained (0.04 V) for a porous CNx film prepared by a traditional thermal evaporation method. Transient photovoltage measurements revealed that the electron diffusion length was nearly 1000 nm for a compact film and obtained a high photocurrent density of 0.180 mA/cm2 at 1.23 V vs RHE with a 150 W xenon lamp.94
Figure 3.

(a) Side view of compact CNx film. Adapted with permission from ref (94). Copyright 2019 Royal Society of Chemistry. (b) Cross-section SEM image of the CNx film. Adapted with permission from ref (95). Copyright 2018 John Wiley & Sons, Inc.
Peng et al. also demonstrated a simple and versatile method to grow crystalline CNx films with a closely packed layered structure on FTO via the seeded crystallization of CN monomers followed by their calcination at high temperature. Upon calcination, a strongly bonded CNx layer on FTO was successfully obtained as shown in Figure 3b, with a thickness of roughly 30 μm.95 The resultant CNx film exhibited impressive PEC performance with a photoanodic photocurrent of 0.116 mA/cm2 at 1.23 V vs RHE and up to a 1 V shift of the onset potential under one sun in 0.1 M KOH aqueous solution. Furthermore, IPCE values at 400 and 420 nm reached 8.5% and 3.6%, respectively. In summary, the method for synthesizing a compact film could improve the contact between the film and the substrate, thus enhancing charge transfer. However, increasing the film thickness beyond 30 μm led to the reduction in the photocurrent density to ∼0.1 mA/cm2 at 1.23 V vs RHE. Hence, it is worth noting that while preparing a compact film, the thickness of the film also must be considered.
Porous Film
Porous photoelectrode usually provides a short electron diffusion distance, large surface area, and a large number of active sites. A simple and versatile doctor blade method could be used to fabricate large-scale and highly porous CNx films with controllable thickness, which can be transferred onto various substrates ranging from FTO, Al foil, porous TiO2, silicon wafer, and glass.96 Upon calcination at 550 °C for 4 h, the uniform CNx layers with the high surface area were obtained according to the electrochemical surface area and dye adsorption measurements. Such a porous film (CNx/FTO) exhibited excellent dye degradation and some PEC performance of ∼0.012 mA/cm2 under white-light illumination at 1.23 V vs RHE in 0.1 M KOH.96 The obtained low photocurrent density may be due to the poor electrode-substrate contact. Similarly, highly porous CNx layer/reduced graphene oxide (rGO) films on FTO substrate were prepared by using a doctor blade method. In this configuration, rGO acted as a support for the CNx layer growth. The obtained film thickness was ∼60 μm. The PEC studies reveal that the rGO layer significantly improved the charge transfer and increased the electrochemically active surface area, leading to a dramatic enhancement of the PEC performance with a photocurrent of 0.072 mA/cm2 at 1.23 V versus RHE in a 0.1 M KOH solution and an external quantum efficiency (EQE) of 5.3% at 400 nm under one-sun illumination.97
The photocurrent density of the porous photoelectrodes can be enhanced by improving the intimate contact between the electrode/substrate and the porous photoactive layer. In a report, a robust method was used to rapidly grow CNx monomers directly from a hot saturated solution of thiourea on various substrates. Upon calcination, a uniform CNx layer with tuned structural and photophysical properties was obtained including the intimate contact with the substrate as shown in Figure 4. The film thickness was ranged from 10 to 50 μm. The detailed PEC and structural studies revealed good photoresponse up to 600 nm, excellent hole extraction efficiency (up to 62%), and strong adhesion of the CNx layer to the substrate. The best CNx photoanode demonstrated a benchmark photocurrent density of 0.353 mA/cm2 (51% faradaic efficiency for oxygen) and an EQE of 12% at 450 nm at 1.23 V versus RHE in an alkaline solution.98 Conclusively, this high performance is benefited from the porous structure and intimate contact between CNx and FTO. A widely reported trend observed with CNx-based photoelectrodes, similar to their inorganic counterparts such as hematite and BiVO4, is that porous films likely possessed better performance than compact films.99 On the other hand, preparing highly crystallized CNx photoelectrodes or single crystals with superior charge transfer ability is very challenging although it is more efficient.
Figure 4.

Top (a,c) and side (b,d) views of porous CNx film. Adapted with permission from ref (98). Copyright 2020 Nature Springer.
2.1.3. Other Strategies
In addition to the above facts, a few other strategies such as doping or structure modification also play a significant role in the optimization of the PEC charge transfer. Poly melamine-formaldehyde resin (PMF) (formed by reacting melamine and formaldehyde) was used as a precursor to dope carbon atoms into the tri-s-triazine units at 500 °C. The layer arrangement disappeared while tri-s-triazine repeating units became prominent in X-ray diffraction (XRD) patterns. The resultant film showed a high photocurrent density of 0.23 mA/cm2 at 1.23 V vs RHE under AM 1.5 illumination, which was believed to be associated with improved charge transfer kinetics in the bulk.100 CNx was also doped with boron (B) using boric acid via a thermal vapor deposition method. The B atom incorporation primarily improved the transport of the minority charge carriers (holes) within the semiconductor, which was four times higher than that of the pristine CNx. In the presence of Na2S as a hole scavenger, the IPCE was nearly 4% at 400 nm with a photocurrent density of 0.055 mA/cm2 at 1.23 V vs RHE under AM 1.5 illumination.101 In another report, Zhang et al. doped CNx with phosphorus (P) using BmimPF6 as the source by polycondensation and achieved 4 orders of magnitude enhanced electrical conductivity and five orders magnitude improved photocurrent generation compared to pristine CNx due to the introduction of intermediate states between VB and CB and more efficient light harvesting.102 Xu et al. demonstrated a comelting strategy by combining molten sulfur and supramolecularly aligned liquid intermediates for in situ fabrication of phenyl-substituted CNx (PhCNx) thin films. Photophysical studies revealed that a sub-band likely formed from oriented phenyl subarrays within the bandgap, which assisted the trapping of photogenerated holes, thus reducing charge recombination and promoting hole transfer to the electrolyte. The sum of improved optical absorption, electronic conductivity, and hole transfer synergistically increased the photocurrent by a factor of 20 under visible light illumination compared to the nonsulfur processed analogue.103 Ruan et al. reported a novel nanojunction architecture that was composed of a B-doped CNx nanolayer and bulk CNx. It was fabricated by a rapid thermal evaporation quenching method. The top layer of the nanojunction had a depth of ca. 100 nm, and the bottom layer was ca. 900 nm. The nanojunction photoanode resulted in a 10-fold higher photocurrent than bulk CNx with an excellent photocurrent density of 0.103 mA/cm2 at 1.23 V vs RHE under one sun condition and an extremely high IPCE of ca. 10% at 400 nm.104 The tightly packed CNx layer prepared by depositing a supramolecular complex comprising melamine-bismuthiol blended with rGO (MSG) on FTO was found to improve the electron diffusion within the CNx and hole extraction to the solution.105 A type-II heterojunction was then formed by depositing a second layer of CNx using the melamine precursor (CNx-M) by thermal vapor condensation. The resulting FTO/CNx-MSG/CNx-M photoanode demonstrated a very high photocurrent density of 0.270 mA/cm2 in 0.1 M KOH solution at 1.23 V vs RHE.
2.2. Other Polymers (Including Organic Molecules)-Based Photoanodes
Decades ago, a thin film of chlorogallium phthalocyanine on a Au electrode was reported for PEC H2 evolution with an efficiency of 0.1%, and the enhanced activity was observed in the presence of Pt cocatalyst.106 Thereafter, a thin film of perylene diimide functionalized with phosphonate groups, N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide (PMPDI), coated on an ITO photoanode was reported for water oxidation, in which CoOx was used as a cocatalyst.107 Under visible-light irradiation, the ITO/PMPDI/CoOx electrode produced a water oxidation photocurrent density of 0.150 mA/cm2 at 1.0 V applied bias vs Ag/AgCl with a FE of 80 ± 15% and internal quantum efficiency of 1% for O2 evolution. Figure 5a shows the working mechanism of the ITO/PMPDI/CoOx photoanode for water oxidation. Recently, an n-type conjugated polymer poly[benzimidazobenzophenanthroline] (BBL)-based photoanode was prepared with exceptional stability and good electron mobility up to 0.1 cm2 V–1 s–1. The BBL polymer film was coated on an FTO substrate using the dispersion-spray method, which showed a morphology-dependent performance. In the presence of the hole acceptor (SO32–), the BBL photoanode displayed photocurrents up to 0.23 ± 0.02 mA/cm2 at 1.23 V vs RHE under standard simulated solar illumination.108 The photocurrent was further enhanced to 0.26 ± 0.02 mA/cm2 by functionalizing the photoanode with 1 nm of TiO2 followed by a nickel–cobalt catalyst.
Figure 5.

(a) Band diagram for the ITO/PMPDI/CoOx system. Adapted from ref (107). Copyright 2014 American Chemical Society. (b) Currents with and without illumination, by PTTh-2/ITO glass in 0.1 M Na2SO4 at 0.9 V vs Ag/AgCl. Stars signify “light on”. Stars with crosses through them indicate, “light off”. Adapted with permission from ref (109). Copyright 2012 Wiley-VCH.
Earlier, porphyrins and metalloporphyrins were used for homogeneous photocatalysis to decompose organic pollutants in the contaminated water and air.110 However, it was limited to short time use because of the poor stability of the porphyrin molecules.111 Later, this issue was resolved by depositing the porphyrin molecules onto solid supports or integrating them with robust nanostructures. One early report on water oxidation using manganese porphyrin (MnPor) came in 1994, in which dimanganese complexes were obtained by linking the triphenylporphyrin (TPP) by an o-phenylene bridge.112 The corresponding photoanode fabricated using MnPor showed PEC water oxidation with a FE of 5–17% at an applied bias of 1.2–1.5 V. MnPor was also used as a cocatalyst for enhancing the PEC water oxidation reaction on polythiophene. The incorporation was achieved by oxidative electrochemical polymerization of terthiophene to poly(terthiophene) (PTTh) on ITO substrates in ethanol–dichloromethane (1:1) solution, in the presence of dissolved 5,10,15,20-tetra(4-sulfonophenyl) porphyrin tetrasodium salt.109 The resulting ITO/PTTh:MnPor photoanode showed a photocurrent of 0.023 mA (not normalized for the surface area) at +0.9 V vs Ag/AgCl (+1.51 V vs RHE) in a 0.1 M Na2SO4 (pH 7) electrolyte solution under the illumination of a halogen lamp (Figure 5b). Furthermore, it was interesting to see seawater oxidation with high selectivity for O2 in this study.
Structurally controlled zinc meso-tetra(4-pyridyl)porphyrin [ZnP(Py)4] nanorods were prepared by encapsulating fullerene derivatives (C60, C60 derivatives, and C70) by a solvent mixture technique in the presence of surfactant molecule cetyltrimethylammonium bromide (CTAB) in a DMF/acetonitrile mixture. Hexagonal nanotubes of ZnP(Py)4 with a large hollow structure was obtained, which became a nanorods shape while combining with fullerenes (Figure 6).113 Time-resolved fluorescence spectra showed efficient fluorescence quenching, suggesting the forward electron-transfer process from the singlet excited state of ZnP(Py)4 to fullerenes. The SnO2/fullerene-ZnP(Py)4 photoanode exhibited a photocurrent density of 1 mA/cm2 at 0 V vs RHE, the solar energy conversion efficiency of 0.63%, and an IPCE of 35%. One organic bilayer composed of 3,4,9,10-perylenetetracarboxylic acid bisbenzimidazole (PTCBI, an n-type semiconductor) and cobalt phthalocyanine (CoPc, a p-type semiconductor) prepared by vapor deposition was also reported for PEC water oxidation. This organic photoanode (ITO/PTCBI/CoPc) exhibited the water oxidation photocurrent density of 0.02 mA/cm2 at 1.2 V vs RHE.114 To improve the stability of the n-type organic fullerene derivative, PC71BM ([6,6]-phenyl C71 butyric acid methyl ester), an ultrathin ZnO (<2 nm) passivation layer was deposited with controlled thickness. The photogenerated holes from the PC71BM were efficiently transferred to the electrolyte through the ZnO passivation layer resulting in the photocurrent density of 0.06 mA/cm2 at 1.23 V vs RHE.115
Figure 6.
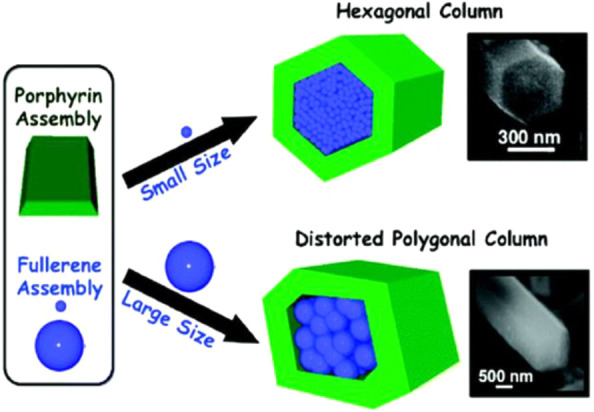
Schematic representation of the formation of the hexagonal C60-ZnP(Py)4 rod and the distorted polygonal C60tBu-ZnP(Py)4 rod. Adapted from ref (113). Copyright 2009 American Chemical Society.
A bulk heterojunction (BHJ) between p-type poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione) (PBDB-T) and n-type 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s indaceno[1,2-b:5,6-b′]dithiophene (ITIC) was reported for PEC water oxidation. This BHJ photoactive layer was preserved by nickel–iron-layered double hydroxides (NiFe-LDHs) to enhance the charge-separation efficiency and reduce the photocorrosion of the organic layer. In addition, GaIn was used as a mediator between BHJ and Ni foil for efficient charge transfer. This photoanode (ITO/PBDB-T/ITIC/GaIn@Ni/NiFe-LDHs) exhibited a record water oxidation photocurrent density of 15.1 mA/cm2 at 1.23 V vs RHE.116 Similarly, a BHJ-based photoanode was prepared using a covalent polymer network (CPN) and SnO2, and the photocurrent density of 3.3 mA/cm2 at 0.54 V vs RHE was obtained at pH 0.117 In addition, a BHJ made up of benzodithiophene-based polymer PBDTTTPD and naphthalenediimide-based polymer PNDITCVT was reported for PEC water oxidation. The BHJ after loading water oxidation catalyst Co3O4 showed a photocurrent density of 2 mA/cm2 at 1.23 V vs RHE at pH 9.0. In this study, the charge accumulation on the photoanode was found to be a big concern for stability. To overcome this issue, a hole extraction layer of poly(triaryl amine) (PTAA) was deposited, which facilitated the charge extraction and reduced the charge accumulation. This FTO/mZnO/PBDTTTPD-PNDITCVT/Co3O4/PTAA/LiO photoanode displayed a slightly improved photocurrent density to 2.3 mA/cm2 at 1.23 V vs RHE, more importantly at a low onset potential of 0.2 V vs RHE and an IPCE of 25%.118
Overall, the limited results on polymer photoanodes (Table 1) were reported, and the highest photocurrent density of 15.1 mA/cm2 at 1.23 V vs RHE was achieved on the ITO/PBDB-T/ITIC/GaIn@Ni/NiFe-LDHs photoanode,116 which is comparable to that reported on inorganic counterpart photoelectrodes.
Table 1. Polymer and Representative Organic Molecules-Based Photoanodes and Their PEC Performances.
| photoanode design | photocurrent density (mA/cm2 vs RHE) | PEC reaction | pH | reference |
|---|---|---|---|---|
| FTO/CNx (TVC) | 0.12 at 1.55 V | water splitting | 11.6 | (86) |
| FTO/SnO2/CNx (EPD) | 0.15 at 1.23 V | water splitting | 7.0 | (89) |
| FTO/2D-CNx | 0.23 at 1.23 V | water splitting | 7.0 | (90) |
| FTO/S-CNx | 0.1 at 1.23 V | water splitting | 13.0 | (91) |
| FTO/CNx (porous) | 0.18 at 1.23 V | water splitting | 7.0 | (94) |
| FTO/CNx | 0.116 at 1.23 V | water splitting | 13.0 | (95) |
| FTO/CNx (porous) | 0.353 at 1.23 V | water splitting | 7.0 | (98) |
| FTO/CNx | 0.23 at 1.23 V | water splitting | 6.0 | (100) |
| FTO/s-BCN | 0.102 at 1.23 V | water splitting | 6.6 | (104) |
| ITO/PMPDI/CoOx | 0.150 at 1.56 | water splitting | 7.0 | (107) |
| OTE/SnO2/C60-ZnP(Py)4 | 1.0 at 1.23 V | water splitting | 7.0 | (113) |
| FTO/CN-MSG/CN-M | 0.270 at 1.23 V | water splitting | 13.5 | (105) |
| ITO/PBDB-T/ITIC/GaIn@Ni/NiFe-LDHs | 15.1 at1.23 V | water splitting | 13.6 | (116) |
| FTO/CPN:SnO2 | 3.3 at 0.54 V | HI splitting | 0 | (117) |
| FTO/mZnO/PBDTTTPD-PNDITCVT/Co3O4 | >2 at 1.23 V | water splitting | 9 | (118) |
| FTO/[Ru(bpy)2(4,4- (PO3H2)2bpy)]Br2/TiO2 | >1.7 at 0.2 V | water splitting | 6.8 | (119) |
3. Polymer-Based Photocathodes
In this section, the emerging polymer-based photocathodes for water and CO2 reduction reactions are reviewed. As explained in the previous section, although single component polymer photocathodes were used for PEC reduction reaction, it is significant to engineer the BHJ to efficiently separate the charge carriers for catalysis. For a polymeric photocathode, it must have good stability when in direct contact with electrolytes. Next, the reduction potential of the polymer, which is approximated by the CB edge, should be more negative than water or CO2 reduction potentials to ensure thermodynamically favored electron transfer. Although polymer semiconductors are good light-absorbing materials, the efficient charge transfer to the surface is challenging because of competitive recombination. The following reviewed the different polymer semiconductors meeting these requirements.
3.1. CNx-Based Photocathode
Although CNx has been used as a photoanode, thermodynamically its VB is aligned around +1.6 V vs RHE, which is not very favorable for water oxidation (+1.23 V vs RHE) due to the small overpotential. This could explain the existing gap in PEC performance between CNx and traditional metal oxide photoanodes (TiO2, BiVO4, WO3, Fe2O3, and TaON)120 which have more positive VB edges. Considering the negative CB (−1.1 V vs RHE) and the great success of CNx for photocatalytic H2 evolution in a suspension system, CNx-based photocathodes are potentially more attractive than photoanodes. Intrinsically, CNx is a n-type semiconductor that generates an upward band bending on the surface and exhibits anodic photocurrent in most studies. To construct a photocathode using an n-type semiconductor, it must overcome the depletion layer filled with holes at the interface, which can be achieved by tuning the electronic properties of CNx by heteroatoms doping. For instance, B doping showed a cathodic current while with a limited current density of 0.01 mA/cm2 at −0.2 V vs RHE for CO2 reduction, due to the positively shifted conduction band (−0.44 V vs RHE) after B doping, therefore mitigating the reduction ability.121 Likewise, Zhang et al. developed phosphorus (P)-doped CNx as a photocathode and observed an accelerated charge transfer in the bulk and an enhanced IPCE of 1.5% at 400 nm at −0.2 V bias vs Ag/AgCl.102 Though the P atoms doping changed the electronic structure of CNx, the PEC efficiency was rather low.
To improve the photoreduction performance of the CNx cathode, it has to be changed from an n-type to a p-type. Ruan et al. substituted NH2 terminals of CNx with −OH groups, which introduced sufficient surface shallow trap states for electrons, extending the lifetime of trapped electrons up to 1 μs for PEC H2 evolution.122 Further by combining the CNx layer with a graphdiyne nanolayer on Cu foil (Figure 7), an efficient photocathode was fabricated to fasten the hole extraction from CNx to graphdiyne/Cu substrate, and the electrons left on the CNx performed the reduction reaction. This photocathode produced a photocurrent density of 0.133 mA/cm2 at a potential of 0 V vs RHE in a neutral aqueous solution.123
Figure 7.

Schematic illustration for the construction of 2D/2D CNx/graphdiyne heterojunction on a 3D GDY nanosheet array. Adapted with permission from ref (123). Copyright 2018 Wiley-Blackwell.
The CNx nanosheets on NiO arrays were reported to form a photocathode with a photocurrent density of 70 μA/cm–2 at 0.42 V vs RHE.124 It was believed that the porous structure of CNx or CNx deposited on aporous conductive substrate would contribute to the photocathode characteristic, similar to minimizing the band bending in a suspension system. The sufficient surface trap states for electrons present in the CNx easily converted it into a photocathode, whereas traditional metal oxide’s porous structure showed photoanode characteristics.125 Similarly, Dong et al. reported a CNx/NiO photocathode for PEC H2 evolution,126 in which the NiO was used to extract the holes from the CNx. The CNx/NiO photocathode showed a photocurrent density of 0.020 mA/cm2 at 0 V vs RHE, which is 10 times higher than the bare NiO and 20 times the bare CNx (Figure 8a).
Figure 8.

(a) Current density vs time of the three electrodes under chopped light illumination in 0.10 M Na2SO4 solution at a bias potential of 0 V vs RHE. (b) The mechanism of PEC H2 generation using the g-C3N4/NiO electrode. Adapted with permission from ref (126). Copyright 2016 Royal Society of Chemistry. (c) Hydrogen production and faradic efficiency of 3D urchin-like ZnO/Au/g-C3N4 and Pt-loaded 3D urchin-like ZnO/Au/g-C3N4 photocathodes. Adapted with permission from ref (127). Copyright 2020 Elsevier. (d) H2 and O2 evolution of the Cu-CN-W photocathode at 0.42 V vs RHE using Pt as the counter electrode. Adapted with permission from ref (128). Copyright 2019 Wiley-VCH.
Interestingly, this photocathode was producing H2 with 100% FE and high stability. A low charge transfer resistance for CNx/NiO photocathode was observed, indicating that it was appropriate for electron transfer to the electrolyte for the proton reduction reaction. The photogenerated holes at the VB of the CNx transferred to the NiO VB, preventing the recombination with CNx CB electrons (Figure 8b). A solid Z-scheme photoelectrode ZnO/Au/CNx was reported for photocathodic performance.127 The addition of the Au layer acted as a solid electron mediator to promote the electron transfer between the ZnO and CNx. With the use of this photocathode, an H2 evolution rate of 3.69 μmol h–1 cm–2 with a FE of 95.2% was obtained under one sun irradiation at an applied bias of 0 V vs RHE in 0.1 M Na2SO4, indicating that the photoelectrons generated by the photocathode were mainly used for the proton reduction reaction. After Pt cocatalyst loading on the surface of the CNx, the H2 evolution rate was increased to 6.75 μmol h–1 cm–2 (Figure 8c) and a FE of 97.5% was achieved. In another report, Basu et al. prepared CNx embedded CoSe2 using a combustion technique followed by a simple hydrothermal route to reduce the charge accumulation on the CoSe2, which led to increased stability.129 CNx-CoSe2 ink was prepared to fabricate the PEC H2 evolution photocathode by placing them on top of the p-Si microwires, in which CNx-CoSe2 was used as an electrocatalyst. It showed a photocurrent density of 4.89 mA/cm2 at 0 V vs RHE and an H2 evolution rate of 1.77 μmol min–1 with a FE of 80%.
Copper (Cu)-modified CNx was reported as an effective photocathode for solar water splitting. The special synthesis method introduced free CuCl and Cu into CNx, which formed the heterojunction between Cu and CNx, similar to a type-II junction leading to enhanced photocurrent density for H2 evolution.128 This work highlighted the molten-salt-based synthesis as an alternative to the liquid-phase synthesis as the molten salt-based approach provides high crystallinity. It produced the cathodic photocurrent density of 0.200 mA/cm2 at 0.42 V vs RHE and the H2 evolution rate of 3 μmol h–1 with a FE of 90.6% (Figure 8d). The mechanistic study revealed that the photogenerated electrons from the CNx transferred to the CB of the CuCl followed by injection into the protons for H2 evolution.
3.2. Covalent Organic Framework (COF) and Metal–Organic Framework (MOF)-Based Photocathodes
COFs are newly developed low-density crystalline polymers that consist of organic units linked via covalent bonds to form porous networks. The selection of appropriate building blocks and linkage motifs provides ways to tailor the optical and electronic properties of COF structures. It has some special potentials for the PEC process such as excellent visible light absorption, fast charge separation and transfer thereby less recombination, and good thermal and chemical stability. Though COFs have been vastly reported as particulate photocatalysts for solar fuel synthesis,130,131 the COFs-based photocathode is also attractive.
An imine-based COF photocathode prepared by using aromatic amine-functionalized tetraphenylethylene (1,1′2,2′-tetra-p-aminophenylethylene, ETTA) and thiophene-based building blocks (benzo[1,2-b:4,5-b′]-dithiophene-2,6-dicarboxaldehyde, BDT) was reported for the PEC H2 evolution. The HOMO and LUMO bandgap measured using the UV–vis and PEC measurements revealed that the LUMO of the COF has higher energy than the water reduction potential. It exhibited a small cathodic photocurrent density of 0.0043 mA/cm2 at +0.3 V vs RHE under one sun irradiation (Figure 9a),132 which was further enhanced by a factor of 4 after loading Pt nanoparticles. In the follow-up study, the same group used a similar COF film but prepared it by an electrophoretic deposition approach (Figure 9b),133 which showed an HER photocurrent density of 0.021 mA/cm2 at 0.1 V vs RHE under the same experimental conditions. By depositing the Pt cocatalyst, the photocurrent density was increased significantly to 0.128 mA/cm2 at 0.1 V vs RHE (Figure 9c). In another report, a new type of COF was synthesized using the Knoevenagel condensation approach in which sp2-carbon-linked triazine core 2D sheets were vertically stacked into high crystalline honeycomb-like structures, forming extended π-delocalization, tunable energy levels, high surface area, regular open channels, and chemical stabilities. The COF-based photocathode showed a photocurrent density of 0.045 mA/cm2 at 0.2 V vs RHE with the average H2 evolution rate of 14.2 μmol h–1 (Figure 9d).134
Figure 9.

(a) Hydrogen evolution on a BDT-ETTA COF electrode was quantified with a hydrogen microsensor (Unisense A/S H2-NPLR) with a selective silicone membrane at a static potential of 0.4 V vs RHE. Illumination of the sample with AM1.5 simulated sunlight results in a photocurrent (black) and the production of hydrogen (red). Adapted from ref (132). Copyright 2018 American Chemical Society. (b) Schematic presentation of the EPD setup with a typical COF film SEM cross-section. (c) Dynamic hydrogen evolution measurement under chopped AM1.5G illumination of a BDT-ETTA COF electrode at 0.2 V vs RHE. Adapted from ref (133). Copyright 2019 American Chemical Society. (d) Time course hydrogen evolution using g-C18N3-COF and g-C33N3-COF as catalysts under visible light (λ > 420 nm) irradiation, monitored over 16 h with evacuation every 4 h (dashed line). Adapted from ref (134). Copyright 2019 American Chemical Society.
2D COF TFBB-TAT (triazine-based) and TFBB-TAB (without triazine) were prepared under solvothermal conditions using a Schiff base type condensation between 1,3,5-tris (4-formylbiphenyl) benzene (TFBB), 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAT), and 2,4,6-tris(4-aminophenyl)-benzene (TAB), and their PEC performances were studied. The triazine-based (ITO/PEDOT:PSS/TFBB-TAT) COF photocathode showed a water reduction photocurrent density of 4.32 mA/cm2 at 0 V vs RHE (Figure 10a),135 which was higher than the nontriazine-based TFBB-TAB COF. In addition to the excellent photoabsorption property, TFBB-TAT showed enhanced charge transfer. Similarly, Dai et al. prepared two 2D COFs (TTA-TTB and TAPB-TTB) based on 2,4,6-triphenyl-1,3,5-triazine by introducing an electron donor triphenylbenzene to reduce the optical bandgap for extended visible light absorption and improved charge transfer. The FTO/TAPB-TTB photocathode showed a photocurrent density of 0.110 mA/cm2 at 0 V vs RHE, at pH 7.0, in the absence of any sacrificial agents, which was higher than the FTO/TTA-TTB (0.035 mA/cm2).136 The extended light absorption and promoted charge transfer were found to be crucial to enhance the PEC water reduction, in accordance with the first-principles calculations. Figure 10b shows the electronic bandgaps of the TAPB-TTB and TTA-TTB COFs modeled using DFT, consistent with their PEC performance. Another COF-based photocathode composed of a bithiophene-bridged donor–acceptor-based 2D sp2 carbon linkages was synthesized using Knoevenagel polymerization between 2,3,8,9,14,15-hexa(4-formylphenyl) diquinoxalino[2,3-a:2′,3′-c]phenazine (HATN-6CHO), an electron-accepting building block, and the first electron-donating linker 2,2′-([2,2′-bithiophene]-5,5′-diyl)diacetonitrile (ThDAN).137 This bithiophene-based COF (2D CCP-Th) photocathode displayed a water reduction photocurrent density of 0.0079 mA/cm2 at 0 V vs RHE (Figure 10c), which was higher than the COF prepared using biphenyl-bridged COF (2D CCP-BD).
Figure 10.

(a) Transient photocurrent responses of TFBB-TAB and TFBB-TAT under dark and light. Adapted with permission from ref (135). Copyright 2020 Zenodo. (b) Hybrid DFT calculated potentials of frontier orbitals and electronic bandgaps in model TAPB-TTB COF and TTA-TTB COF. Adapted with permission from ref (136). Copyright 2021 Wiley-VCH. (c) Photocurrent–time plots for 2D CCP-Th (red line), 2D CCP-BD (blue line), and 2D C=N COF-B (black line) at 0.3 V versus RHE. On: illumination on; off: illumination off. Adapted with permission from ref (137). Copyright 2021 Wiley-VCH.
Besides COF, other known coordinated polymers are metal–organic framework (MOF) materials, which offer the opportunities to develop highly ordered three-dimensional (3D) structures for PEC production of fuels. The pore size control alongside the tunable framework structure offers the appropriate active sites with long-time stability. By choosing suitable central metal atoms, it is possible to create photoresponsive MOF materials. Moreover, the large surface areas and selective porosities in MOF materials can be potentially applied in gas–solid-phase reactions, such as CO2 reduction reactions. Thus, the combination of MOFs with appropriate semiconductors can be a promising approach to design effective solar conversion processes. The MOF-based photocatalyst suspension has been widely reported and reviewed,138 though photocathodes made from MOFs are much less discussed.
The surface coating of MOF material onto a semiconductor would increase the interfacial charge transfer. The porous structure of MOF either ensures high optical absorption or can act as a protective layer to prevent the leaching and aggregation of the photosensitizing particles. For instance, a redox-active MOF was coated on a p-type Si (Figure 11a), which showed a photovoltage of 0.3 V, whereas p-Si functionalized with a naphthalene diimide derivative monolayer exhibited no photoresponse.139 Furthermore, the same MOF coating on a GaP semiconductor shifted the photovoltage to +0.7 V (Figure 11b), which is the highest reported for GaP in a PEC application. This emphasizes the advantage of MOF films regarding enhanced photocathodic operation. In addition, MOF film directly grown on the surface of a substrate improved the diffusion of charges through the film, thereby increasing the photocathodic current.139
Figure 11.

(a) Schematic representation of the MOF surface coating on p-Si. EVB and ECB are the energetic positions of the valence and conduction band, respectively, EF,p, and EF,n are the quasi-Fermi levels of the holes and electrons, respectively, Vph is the semiconductor photovoltage, and Efilm is the electrochemical potential of the MOF film. The molecular structure of the linker is pictured on the right. (b) Cyclic voltammograms of Zr(NDI)@FTO (black dashed) and an illuminated Zr(NDI)|TiO2@GaP working electrode (red) at a scan rate of 100 mV s–1 with 0.5 M LiClO4 in DMF as the supporting electrolyte. The red solid data were collected under AM 1.5 illumination. Adapted with permission from ref (139). Copyright 2020 Nature Springer. (c) Schematic showing the photocathode role of csiMOF-6 in a CO2 photoelectrochemical reduction system employing a rhenium electrocatalyst. A green arrow indicates photoexcitation, the purple arrow indicates CT, and the black arrow signifies CO2 reduction to CO. Adapted with permission from ref (140). Copyright 2021 Royal Society of Chemistry.
Cardoso et al. have reported the MOF-based TiO2 nanotubes photocathode for CO2 reduction, in which the zeolite imidazole framework-8 (ZIF-8) nanoparticles were deposited on TiO2 nanotubes using a layer-by-layer process.141 The prepared polymer photocathode (Ti/TiO2NT-ZIF-8) reduced CO2 to ethanol (up to 10 mM) and methanol (0.7 mM) in a 0.1 M Na2SO4 electrolyte at 0.1 V under UV–vis irradiation for 3 h at room temperature. The utilization of ZIF-8 has the dual advantage that it acted as an active site for CO2 adsorption and activation and also functioned as a cocatalyst to facilitate electron transfer. In a report, Hou et al. used a double solvents approach to synthesize Pt@NH2-MIL-125(Ti) MOF and fabricated the photocathode by drop-casting on the ITO substrate to study the PEC H2 production.142 The optical characterization showed very good absorption in the visible region, precisely at 500 nm. The H2 evolution photocurrent onset was observed at 0.3 V for this MOF-based photocathode. Though the PEC water reduction activity was small, it demonstrated the applicability of the MOFs as photocathode materials for solar fuel conversion. Similarly, a new cofacial photo- and redox-active MOF, i.e., N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide (DPNDI), denoted as csi-MOF-6, was synthesized by Ding et al. and used to fabricate a photocathode by blending it with a photosensitizer into a [Re(bipy-tBu)(CO)3Cl] electrocatalyst (Figure 11c) for PEC CO2 reduction.140 The reaction was performed with a small overpotential under broad visible light irradiation in a CO2-saturated 0.1 M [nBu4N]PF6/MeCN electrolyte, and CO was produced with a FE of 78% and a TON of 7.
3.3. Poly(3-hexylthiophene)-P3HT-Based Photocathodes
P3HT is a commercially available π-conjugated polymer with sufficient solubility in chlorinated organic solvents and has a direct low bandgap of 1.9 eV. The charge carrier mobility is high in P3HT as it has a high degree of intermolecular order. The LUMO of the P3HT is more negative than the water and CO2 reduction potentials; hence, it can be used as a direct photocathode material for solar fuel synthesis. A solution-processed regioregular P3HT photocathode was reported for PEC water reduction, which showed a photocurrent density of 0.020 mA/cm2 under one sun irradiation. In addition, a good stability was observed over a few hours of irradiation.143 The mechanism for proton reduction will be discussed in Section 5. Briefly, the P3HT surface adsorbed H species at the α-site of the thiophene ring. The protonated polymer–electrolyte interface received the photogenerated electrons and produced H2 followed by regeneration of the polymer surface. To enhance the binding of the polymers to the substrate, direct polymerization from the respective monomers has been recommended.
Though P3HT showed direct PEC performance, the charge carrier recombination was presumably a key issue and hence BHJ formation has been introduced to enhance the charge transfer, analogous to the evolution of organic photovoltaics. BHJs are typically obtained by blending a p-type P3HT polymer with an electron acceptor layer. For instance, fullerene derivatives were used as electron acceptors. The BHJ photocathode made up of P3HT:fullerene absorbed light and separated the photogenerated electrons from holes very efficiently. For example, the BHJ photocathode was fabricated by using a P3HT:phenyl-C61-butyric acid (PCBM) blend and sandwiched between the molybdenum sulfide (MoS3) electron selective layer and the PEDOT:PSS hole selective layer. Next, a TiO2 thin film was spin-coated on MoS3 to protect P3HT:PCBM from oxygen and water exposure. The PEC characterization of the BHJ photocathode exhibited a high photocurrent of 0.100 mA/cm2 at 0.16 V vs RHE (Figure 12a).144 After two years, the same group attempted to increase the electron extraction to the catalyst layer MoS3 by adding the metal Al, Ti, and nanocarbon (C60) as an interfacial layer. The ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al/Ti-MoS3 and ITO/PEDOT:PSS/P3HT:PCBM/C60-MoS3 photocathodes showed dramatically enhanced photocurrent densities of 8 mA/cm2 and 1 mA/cm2, respectively, at 0.6 V vs RHE.145 The proposed electron extraction mechanism was shown in Figure 12b.
Figure 12.

(a) Electrolysis at a bias potential of +0.16 V vs RHE, with chopped visible light, in H2SO4 (0.5 M). Photocathode: black, P3HT:PCBM; blue, MoS3/P3HT:PCBM; red, TiO2:MoS3/P3HT:PCBM photocathode. Electrode area: 0.5 cm2. Adapted with permission from ref (144). Copyright 2013 Royal Society of Chemistry. (b) Energy level diagram of the device in contact with the electrolyte. Electrons and holes are represented by black and white dots, respectively. Adapted from ref (145). Copyright 2015 American Chemical Society. (c) Hydrogen evolution of the OPEC measured under continuous 1 sun irradiation at 0 V versus RHE registered experimentally (square points) and theoretically calculated from the measured current by Faraday’s law. (d) Device architecture of the optimized organic photoelectrochemical cell (OPEC), showing the electronic process during device operation. Adapted from ref (146). Copyright 2015 American Chemical Society.
Similarly, Haro et al. fabricated an ITO/PEDOT:PSS/P3HT:PCBM/TiOx-Pt photocathode and obtained a photocurrent density of 1 mA cm–2 at −0.1 V vs RHE and stable H2 evolution of 1.5 μmol h–1 cm–2,146 as shown in Figure 12c. The experimental results agreed with the theoretical value of H2 production, which is represented in the dashed line, indicating nearly 100% FE. The schematic of the photocathode and the mechanism of H2 production are represented in Figure 12d. Fumagalli et al. reported a photocathode made up of P3HT:PCBM sandwiched between the hole separation layer MoO3 and electron separation layer TiO2. The Pt cocatalyst loaded on the TiO2 photocathode (FTO/MoO3/BHJ/TiO2/Pt) showed the photocurrent response of 3 mA cm–2 at 0 V vs RHE with the 100% FE for H2 evolution.147
Rojas et al. used the cuprous iodide (CuI) as a hole selective layer to effectively separate the holes with high performance and stability, along with an electron selective TiO2 layer to fabricate the inorganic–organic hybrid BHJ photocathode.148 After adding a thin Pt catalyst layer, the FTO/CuI/P3HT: PCBM/TiO2-Pt photocathode produced the high photocurrent density of 8 mA at 0 V vs RHE under one sun condition in the pH 1.0 electrolyte. The IPCE value of 50% was obtained with a FE of 100% using the above photocathode. To improve the stability of the photocathode, the polyethylenimine (PEI) layer, due to its good adhesion, hydrophilicity, and proton affinity, was coated on the surface. Such a layer was expected to minimize the Pt loss, and the chronoamperometry results revealed that the photocurrent decrease over time was 2-fold reduced after PEI coating, suggesting improved stability.
Table 2 summarizes representative polymer-based photocathodes reported for water reduction and CO2 conversion reactions. These photocathodes performances were evaluated in terms of photocurrent density or TON under different working conditions. Most of the photocathodes had their PEC reduction activity reported at the aqueous interface. The long-term stability parameters were not discussed in most of the reported polymer-based photocathodes, which should be paid particular attention as organic substances tend to be less stable under strong light irradiation compared with their inorganic counterparts.
Table 2. Representative Polymer-Based Photocathodes and Their PEC Performance.
| photocathode design | cathodic photocurrent (mA/cm2 vs RHE) or TON | PEC reaction | pH | ref |
|---|---|---|---|---|
| CNx | ||||
| FTO/B-C3N4/Rh | 0.01 at –0.2 V | CO2 reduction | 7.3 | (121) |
| ITO/P-C3N4 | 1.5% IPCE at 420 nm | – | 7.0 | (102) |
| FTO/def-C3N4 | 0.010 at 0 V | – | 7.0 | (122) |
| FTO/Pt@g-C3N4/GDY | 0.133 at 0 V | water splitting | 7.0 | (123) |
| FTO/NiO-C3N4 | 0.070 at 0.42 V | water splitting | 6.5 | (124) |
| FTO/NiO-C3N4 | 0.020 at 0 V | water splitting | 6.5 | (126) |
| ITO/ZnO-Au-C3N4/Pt | 0.290 at 0 V | water splitting | 6.8 | (127) |
| FTO/Cu-C3N4 | 0.200 at 0.42 V | water splitting | 6.8 | (128) |
| COF and MOF | ||||
| FTO/BDT-ETTA COF/Pt | 0.129 at 0.1 V | water splitting | 10.0 | (133) |
| FTO/g-C18N3-COF/Pt | 0.045 at 0.2 V | water splitting | 6.8/3.0 | (134) |
| FTO/TAPB-TTB | 0.110 at 0 V | water splitting | 7.0 | (136) |
| FTO/Si@TiO2-Zr(NDI) | +0.3 V (photovoltage) | – | 6.0 | (139) |
| FTO/GaP@TiO2-Zr(NDI) | +0.7 V (photovoltage) | – | 6.0 | (139) |
| FTO/csiMOF-6[Re(bipy-tBu)(CO)3Cl] | 78% FE, TON 5 | CO2 reduction | organic | (140) |
| P3HT | ||||
| FTO/P3HT | 0.020 under one sun | – | 0.1 | (143) |
| ITO/PEDOT:PSS/P3HT:PCBM/MoS3-TiO2 | 0.100 at 0.16 V | – | 0.1 | (144) |
| ITO/PEDOT:PSS/P3HT:PCBM/C60-MoS3 | 1 at 0.6 V | – | 0.1 | (145) |
| ITO/PEDOT:PSS/P3HT:PCBM/TiOx-Pt | 1 at –0.1 V | water splitting | 2.0 | (146) |
| FTO/MoO3/BHJ/TiO2/Pt | 3 at 0 V | water splitting | 1.37 | (147) |
| FTO/CuI/P3HT: PCBM/TiO2-Pt | 8 at 0 V | water splitting | 1.0 | (148) |
| others | ||||
| FTO/PTEB | 0.010 at 0 V | water splitting | 6.8 | (154) |
| FTO/PTEB1.3-co-PDET1 | 0.021 at 0 V | water splitting | 6.8 | (154) |
| FTO/pDEB/p(DEB0.9-co-TEB0.1) | 0.055 at 0.3 V | water splitting | 6.8 | (155) |
3.4. Other Polymer Photocathodes
A direct polymer-based photocathode, for instance, a polyacetylene-based photocathode, was reported for visible-light-driven water reduction. It showed a cathodic photocurrent of 0.005 mA/cm2 at −0.46 V vs RHE at pH 5.7.62 Subsequently, several polymers such as polypyrrole (PPy),149 polyaniline,150 and poly(p-phenylene)151 have been reported for the direct PEC H2 production on the surface of the polymer photoelectrodes, even though poor charge separation efficiency in the absence of a selective charge extraction layer is a major challenge. In addition, stability has been a crucial issue to use the polymers directly in contact with electrolytes. Later, the conjugated acetylenic polymers have been proposed as they have strong electrostatic interaction with water molecules due to the presence of an electron-rich C-to-C triple bond. For instance, the conjugated polymers such as poly(1,3,5-triethynylbenzene) (PTEB) and poly(1,3,5-tris(4-ethynylphenyl)benzene) (PTEPB) nanoflakes were reported for the photocatalytic activity toward H2 and O2 evolutions.152 The PTEB electrode without doping may act as an n-type semiconductor, however, nitrogen doping can convert it into a p-type semiconductor.153 PTEB polymerized on the surface of the Cu or Ti was reported for PEC H2 evolution with the photocurrent density of 0.010 mA/cm2 at 0 V vs RHE. With the use of this photocathode, an H2 evolution rate of 0.253 μmol h–1 cm–1 was achieved with a FE of >90%.154 An increased photocurrent density of 0.021 mA/cm2 was observed upon the incorporation of 2,5-thieno[3,2-b]thiophene into the polymer structure (p(TEB1.3-co-DET1)). Later, the same group prepared a poly(1,4-diethynylbenzene) (pDEB) conjugated polymer by controlled copolymerization of 1,4-diethynylbenzene (DEB) and 1,3,5-triethynylbenzene (TEB) then studied PEC water reduction activity.155 The water reduction photocurrent density of the homojunction FTO/pDEB photocathode was 0.019 mA/cm2, which was enhanced to 0.055 mA/cm2 for the gradient-homojunction pDEB/p(DEB0.9-co-TEB0.1) photocathode at 0.3 V versus RHE at pH 7.0.
The conducting polymer PPy was sometimes used as a supporting substrate to deposit the photoactive complex to fabricate the effective PEC photocathode for solar fuel production. The PPy allows the metal complex to deposit efficiently without modifying the intrinsic properties. For instance, Lattach et al. used PPy to deposit the Ru(II) complex using anodic polymerization onto a carbon electrode,156 which was then incorporated with MoSx by an ion-exchange method followed by electrochemical reduction of MoS42–. The PPy-Ru/MoSx photocathode showed an H2 evolution rate of 0.53 μmol cm–2. PPy has also been used as an electrocatalyst for CO2 reduction reactions by replacing the transition metal electrocatalysts to reduce the energy barrier at the semiconductor-electrolyte junction. Won et al. decorated a p-ZnTe photocathode with PPy to reduce CO2 into formic acid and carbon monoxide (CO).149 Beyond this, organic polymer-based dyes have also been used to improve the performance of the photocathode. Simply coating organic dye molecules via chemisorption or covalently linking organic molecules such as perylene derivatives and donor–acceptor dyes, the visible region of the solar spectrum could be efficiently absorbed. Numerous reports can be found on polymeric dye-sensitized photocathodes,157−160 which is not the focus of this review.
Overall many polymer photocathodes were reported, while the photocurrent is still quite moderate. Except the P3HT-based polymer, which showed a maximum photocurrent density of 8 mA/cm2 at 0 V vs RHE, the highest cathodic photocurrent was 0.29 mA/cm2 observed on the solid Z-scheme ITO/ZnO-Au-C3N4/Pt photocathode, which is nearly 1 order of magnitude lower than that achieved on the benchmark inorganic photocathodes. Therefore, much effort is still required to improve the photocurrent of polymer photocathodes.
3.5. Bias-Free Photoanode|Photocathode System
In addition to the photoanode|dark cathode and photocathode|dark anode systems, a bias-free tandem cell PEC approach has also been recently used for solar fuel production. The term “tandem-cell approach” has been used to represent two types of configurations, (i) a photoanode or photocathode coupled with photovoltaics (PEC|PV) and (ii) a self-powered photoanode|photocathode system. Several PEC|PV systems were reported for solar H2 production44,161 and CO2 reduction,162−164 which is beyond the scope of the present review and the readers can find the related reviews in the literature.4,44,165 The past few years have witnessed the photoanode|photocathode tandem cell approach (Figure 1d) for solar fuel production without the need of any external bias, which potentially reduces the overall cost of the system and increases its efficiency. In this configuration, a n-type semiconductor coated electrode is used as a photoanode for water oxidation to produce O2 and protons and a p-type semiconductor-loaded electrode used as a photocathode for proton reduction to produce H2 or CO2 reduction to produce methanol or high-value chemicals. While both photoelectrodes are connected in series, the light can be harvested by the photoanode and photocathode or two light source can be used. It is also feasible to have both electrodes separated by a membrane to separate the reaction products. So far, the majority of the reported photoanode|photocathode tandem PEC device is made up of inorganic semiconductors166−169 and, hence, limited us to explore further in this review. To the best of our knowledge, the direct use of polymer-based semiconductors to fabricate a photoactive anode and a photoactive cathode in a tandem cell approach is yet to come. But, the organic dye-sensitized photoanode|dye-sensitized photocathode tandem PEC approach has been reported,170,171 in which inorganic metal oxide semiconductors such as TiO2 and NiO were used to extract the charges from the photoexcited dyes followed by the respective oxidation and reduction reactions performed.
4. Molecular and Electro-Cocatalysts
Robust and efficient cocatalysts can be introduced into a conjugated polymer photoelectrode not only to improve the catalytic activity but also to enhance the stability of the polymer. As mentioned above, there were limited polymer photoelectrodes reported, therefore few cocatalysts were tested on polymer photoelectrodes, while there were many cocatalysts loaded on either dyes or organic scaffolds to evidence their efficiency. Due to the similarity of the chemical bonds or affinity between these organic scaffolds and cocatalysts as well as between potential polymer photoelectrodes and cocatalysts, these efficient cocatalysts are reviewed here. To date, ruthenium (Ru)-, cobalt (Co)-, rhenium (Re)-, and manganese (Mn)-based molecular catalysts and inorganic catalysts, e.g. Pt, RuOx, IrOx, NiOx, CoOx, have been extensively grafted on polymer photoelectrodes or organic scaffolds to proceed PEC reactions. To incorporate the catalysts into the matrix, in situ electropolymerization/polymerization, layer-by-Layer (LbL) assembles, etc. have been widely used. Here, the recent advancement of cocatalysts in the matrix, including both polymer photoelectrodes and organic scaffolds is addressed, including for the PEC OER, HER, and CO2 reduction reaction (CRR).
4.1. Molecular OER Cocatalysts
4.1.1. Ru-Based Molecular Cocatalysts
Abundant OER catalysts are based on Ru complexes due to the basis of water oxidation at Ru progressing between four oxidation states: RuII, RuIII, RuIV, and RuV. With RuII and RuIII, an aqua ligand is commonly presented, forming the active site for catalysis. Activation of RuIV spurs the complete proton loss from the aqua ligand and the formation of the Ru-oxo bond. Water oxidation generally occurs at the highest oxidation state, RuV, and the Ru-oxo complex undergoes nucleophilic addition to another water molecule in solution eventually leading to the evolution of O2 from the complex and reformation of the RuII or RuIII aqua complex.172−175 Ru(bda)L2 (bda = 2,2′-bipyridine-6,6′-dicarboxylate; L = 4-picoline, isoquinoline, pyridine, imidazole, or thiophene), a typical class of mononuclear Ru complexes, have been widely exploited both under electrochemical and PEC conditions.176,177 Inspired by the stabilization function of phenolate and carboxylate ligands for high valence states of Mn in natural photosynthesis photosystem II, carboxylate ligands were thereby incorporated into Ru molecular OER catalysts to access high valence Ru = O species at a low oxidation potential,178 which were then utilized in the water oxidation reaction.172,176,177 For instance, Ru(bda)L2 (OER1; L = 4-picoline) was reported to have an onset potential of ca. 0.98 V vs normal hydrogen electrode (NHE) at neutral pH in an electrochemical water oxidation reaction.172 Such low overpotential (0.98 V) renders a homogeneous water oxidation reaction by combing with the photosensitizer [Ru(bpy)3]3+.177 Due to the impressive water oxidation properties in the homogeneous system, developing strategies to further assemble the molecular catalyst in the heterogeneous platform without activity loss attracts much attention. This remains a major challenge holding back molecular artificial photosynthesis.179
To address this issue, OER1 embedded in a Nafion polymer was covered on the dye [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ (RuP) sensitized nanostructured TiO2 photoanode (Figure 13, labeled as TiO2-RuP/Nafion-OER1).180 First, the Nafion polymer has good electrical conducting properties and high chemical/thermal stability, benefiting photoelectrochemical application, due to the existence of numerous sulfonic acid groups.181 Moreover, the oxidation potential E1/2(RuIII/II) of RuP is more positive than the onset potential of OER1 for water oxidation at pH 7.0, thus thermodynamically allowing the photogenerated RuIII in RuP to drive OER1. A negligible photocurrent or fast decay was recorded for the photoanodes without photosensitizer RuP or catalyst OER1 modification. Whereas the TiO2-RuP/Nafion-OER1 showed dramatically enhanced photocurrent very likely due to efficient electron transfer from the catalyst to the photooxidized dye. The photocurrent decay rate of TiO2-RuP/Nafion-OER1 was strongly related to the initial pH of the Nafion membrane, probably due to the rapid proton release during water oxidation thereby affecting the catalytic properties of OER1.177 No O2 could be produced without light illumination for all photoanodes. For TiO2/Nafion-OER1, only 16 nmol mL–1 O2 was generated after 60 min illumination. In the presence of RuP but without OER1 (TiO2-RuP/Nafion), no O2 could be produced as well. In contrast, in the coexistence of RuP and OER1 (TiO2-RuP/Nafion-OER1), 140 nmol mL–1 O2 was obtained after 60 min irradiation. These results clearly proved the light-driven water oxidation by this complete catalytic assembly. Accordingly, assuming all OER1 participating in the water oxidation reaction, TON of 16 was obtained with a corresponding turnover frequency (TOF) of 27 h–1.
Figure 13.

(a) Chemical structure of OER1. (b) Light-driven PEC water splitting device, consisting of TiO2-RuP/Nafion-OER1, a Pt cathode, and an aqueous electrolyte. (c) Transient short-circuit current responses to on–off cycles of illumination. The illumination is provided by a light-emitting diode operated in a 0.1 M Na2SO4 aqueous solution in PEC devices without applying any bias. Nafion pH 7.0 represents the related Nafion film prepared using a pH 7.0 Nafion solution, respectively. (d) Oxygen evolution in PEC devices without applying any bias, operated in a pH 7.0 phosphate buffer solution, detected by a Clark electrode, and illuminated by a 500 W xenon lamp through a 400 nm cut-off filter. Complex 1+ indicates the OER1. Adapted with permission from ref (180). Copyright 2010 Royal Society of Chemistry.
PMMA (poly(methyl methacrylate)) oligomer is another potential support for immobilizing catalysts. Compared to acidic Nafion, PMMA has nearly no influence on the properties of molecular components and displays superior abilities for assembling pH-sensitive devices, thus rendering the utilization in perovskite solar cells as stable hole-transport materials and the immobilization of molecular components for higher stability.182,183 After stabilizing a long carbon chain modified Ru(bda)L2 (OER2; L = 4-picoline) catalyst by PMMA on top of RuP-sensitized TiO2 (Figure 14a,b)184 and upon visible light (λ > 400 nm) illumination, a stable photocurrent density of 1.1 mA/cm2 was obtained at 0.2 V vs NHE in phosphate buffer solution, while nearly no photocurrent was recorded for the only catalyst or photosensitizer-modified TiO2. A maximum IPCE of 9.5% was achieved at 450 nm. A lower photocurrent density with faster decay without long-chain modification indicated that the long carbon chains have a positive effect on immobilizing the catalyst by twining around the polymerized PMMA. After coembedding RuP photosensitizer and OER2 in PMMA, the photocurrent density was further improved to 1.50 mA/cm2.185
Figure 14.

(a) Chemical structure and synthesis route of the OER2. (b) Photocurrent of three photoanodes (TiO2+PS, TiO2+PS+OER2, and TiO2+OER2) with a 0.2 V vs NHE external bias in pH 6.8 phosphate buffer solution upon visible light irradiation (λ > 400 nm, 300 mW cm–2). Adapted with permission from ref (184). Copyright 2015 Wiley-VCH. (c) Chemical structures of OER3, RuP, and RuPdvb2+. (d) Schematic diagram of the surface structure following reductive electropolymerization of OER3 on TiO2-RuPdvb2+. (e) Photocurrents of nTiO2-RuPdvb2+ (dashed) and nTiO2-RuPdvb2+-polyOER3 (solid) at a bias of 0.2 V versus SCE. Adapted with permission from ref (186). Copyright 2015 Wiley-VCH.
Besides polymer encapsulation, the electropolymerization method enables an “on-surface” preparation of assemblies by electrochemically induced C–C bond coupling. This technique exhibits the capacity for polymer film preparation with a vinyl-functionalized complex, Ru(bda)L2 (OER3; L = 4-vinylpyridine), on a metal oxide photoanode (Figure 14c–e).186 The OER3 was prepared by a simple, microwave-assisted, one-pot reaction via self-assembly of three reagents: bda2– (generated in situ by reaction of H2bda and triethylamine), Ru(DMSO)4Cl2, and 4-vinylpyridine. Then, poly-OER3 was obtained by electrochemical polymerization on the RuP-sensitized TiO2 film at −2.0 V vs Ag/AgNO3 for 200 s in an acetonitrile solution containing OER3.187,188 A remarkable initial photocurrent density of ∼3.0 mA/cm2 was obtained by the poly-OER3+RuP@TiO2 photoanode (phosphate buffer solution, 0.2 V vs NHE), while no significant photocurrent was observed for the RuP@TiO2 photoanode. A maximum IPCE of 8.9% was observed at 450 nm, and the FE was measured as 82%. However, the poly-OER3+RuP@TiO2 photoanode was unstable with time, indicated by the photocurrent density decreasing from 1.4 to 0.3 mA/cm2 after 200 s of light illumination, which was due to the decomposition and/or desorption of the photosensitizer. By replacing the RuP@TiO2 with an α-Fe2O3 nanorod array, stable photocurrent density was obtained for a poly-OER3@Fe2O3 photoanode after long-term illumination, indicating that photosensitizer decomposition could be substantially suppressed by using α-Fe2O3 in comparison to RuP.
The results above reveal that molecular catalyst poly-OER3 is an efficient and stable catalyst in PEC devices as well as imply the reliability of the electropolymerization method for fabricating efficient and stable catalyst embedded polymer photoelectrode. In addition, replacing the photosensitizer, RuP, by the phosphonate-derivatized light-harvesting chromophore [Ru(dvb)2((PO3H2)2bpy)]2+ (RuPdvb2+; dvb = 5,5′-divinyl-2,2′-bipyridine; (PO3H2)2bpy = [2,2′-bipyridine]-4,4′-diylbis(phosphonic acid)), more stable photocurrents were obtained under white light illumination compared to poly-OER3+RuP@TiO2.186 The next catalyst OER4 bears a thiophene unit as appended-bridge ligand and a hydrophobic ligand 6-fluoroisoquinoline instead of 4-iodpyridine (Figure 15a,b).189 Under visible light irradiation OER4@Fe2O3 showed a high and stable photocurrent of over 0.3 mA/cm2 at a relatively small bias of 0.8 V vs NHE. This work suggests that the in situ polymerization also has the promise to immobilize molecular catalysts on electrode surfaces to build efficient polymer PEC devices.
Figure 15.

(a) Structure of OER4. (b) Photocurrent densities of the OER4@Fe2O3 and pristine Fe2O3 in the phosphate buffer (pH 7) with a bias of 0.8 V vs NHE, and under AM 1.5 G illumination (100 mW cm–2). Adapted with permission from ref (189). Copyright 2017 Elsevier. (c) Structures of an OER5 (RuOH22+). (d) Schematic diagram of the surface structure following reductive electropolymerization of RuOH22+ on nTiO2-RuPdvb2+. (e) Variation of surface coverage as a function of irradiation time at 475 mW cm–2 at 455 nm over a 16 h photolysis period in aqueous 0.1 M HClO4. Adapted with permission from ref (190). Copyright 2014 American Chemical Society.
Vinyl-functionalized [Ru(Mebimpy)-(dvb)(OH2)]2+ (OER5 = RuOH22+; Mebimpy = 2,6-bis(1-methyl-1H-benzo[d]imidazole-2-yl)pyridine, dvb = 5,5′-divinyl-2,2′-bipyridine) is another OER catalyst polymerized with or without the RuPdvb2+ chromophore on the surface of the metal oxide film by electropolymerization/electrooligomerization (Figure 15c–e).190 In a typical electropolymerization process, propylene carbonate as the solvent rather than CH3CN avoids the displacement of the H2O ligand for RuOH22+. The chromophore/catalyst ratio was controlled by tuning reductive electrochemical cycles. A blue shift in the metal-to-ligand charge transfer absorption maximum from 462 to 453 nm was observed for RuPdvb2+ in the electropolymerized films. This shift is consistent with the conversion of the π* acceptor vinyl substituents in RuPdvb2+ to saturated, electron-donating alkyl substituents in the electropolymerized polymers, suggesting the formation of C–C bonds between RuPdvb2+ and RuOH22+ in the surface assembly.191,192 The surface coverage of the chromophore in nTiO2-RuPdvb2+ decreased by ∼70%, while only ∼10% was lost for nTiO2-RuPdvb2+-polyRuOH22+ after sustaining 16 h of irradiation.
[Ru(tpy)(Mebim-py)(OH2)]2+ (OER6 = RuC; Mebim-py = 2-pyridyl-N-methylbenzimidazole) as another OER catalyst was codeposited with poly(acrylic acid) (PAA), an inert polyanion, to construct chromophore-catalyst assemblies with cationic polystyrene-based Ru polychromophore (PS-Ru) via a LbL self-assembly on the planar SnO2/TiO2 core/shell structure precoated FTO substrate (Figure 16).193 The LbL method enables facile control over the amount and ratio of the chromophore to the catalyst. In the collector-generator (C-G) cell and under irradiation, no measurable cathodic current was detected in the absence of the RuC catalyst layers, while a pronounced anodic photocurrent immediately was produced over FTO//(SnO2/TiO2)//(PAA/PS-Ru)5/(PAA/RuC)5. The photocurrent decayed quickly within the first 30 s and sustained at a stable value over the following 530 s of illumination. Prolonged photoelectrolysis experiments revealed O2 production from the illuminated photoanode with a FE of 22%, equaling the previously investigated systems using Ru(bpy)3-derivatized chromophores with Ru-based OER catalyst stabilized by an atomic layer deposition (ALD) technique.194
Figure 16.

(a) Molecular structures of polystyrene-based PS-Ru and OER6 (RuC). (b) Schematic illustration for fabrication of (b-top) FTO//(SnO2/TiO2)//(PAA/PS-Ru)n and (b-down) FTO//(SnO2/TiO2)//(PAA/PS-Ru)n/(PAA/RuC)m multilayer films. (c) Current–time trace with illumination (1 sun, 100 mW cm–2, 400 nm cutoff filter) of FTO//(SnO2/TiO2)//(PAA/PS-Ru)5/(PAA/RuC)5 photoanode (red) and in the dark (black) in a 0.1 M phosphate buffer at pH 7 with an applied bias of 0.44 V vs NHE. Adapted from ref (193). Copyright 2016 American Chemical Society.
Similarly, a polystyrene-based chromophore-catalyst assembly (poly-2) was obtained on a mesoporous metal oxide photoanode also via the LbL method. [Ru(trpy)(phenq)]2+ (OER7 = RuCat; trpy = 2,2′;6,2″-terpyridine, phenq = 2-(quinol-8′-yl)-1,10-phenanthroline) and [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) derivatives were OER catalyst and chromophores in the assembly, respectively (Figure 17a,b).195 Multilayer photoanodes were constructed with cationic poly-2 and anionic PAA. FTO//(SnO2/TiO2)//PAA/poly-2)5 photoanode had an initial photocurrent (∼0.0185 mA/cm2) partly ascribed to the light-driven water oxidation (0.5 M KNO3 aqueous solution with 0.1 M phosphate buffer (pH 7), 100 μW/cm2, λ > 400 nm). The photocurrent generated from bare FTO//(SnO2/TiO2) was negligible, and the polychromophore-modified photoanode just produced an initial photocurrent density of ∼0.0074 mA/cm2 and finally stabilized at ∼0.006.2 mA/cm2 after three light on–off cycles. In comparison with the previous multicomponent LbL approach, anchoring Ru catalyst molecules via electrostatic LbL self-assembly,196 the LbL films here sustained ∼28% higher photocurrent density after 250 s of continuous illumination. The enhanced photochemical properties of the covalently linked chromophore-catalyst polymer suggested improved charge injection and hole transfer to the catalyst. Polyoxometalate with a tetraruthenium active site, e.g. Na10{Ru4(μ-O)4(μ–OH)2[γ-SiW10O36]2}(OER8 = Ru4POM), has the requisite potential for water oxidation.197−199 Ru4POM was successfully loaded on a KuQ(O)3OH (KuQ(O)3OH = 1-(3-carboxypropyl)KuQuinone) sensitized SnO2 photoanode by soaking in an aqueous solution containing a Nafion polymer and Ru4POM (Figure 17c,d).197 Absorption spectrum and elemental mapping evidenced the successful and uniform loading of Ru4POM over the surface of the SnO2|KuQ(O)3OH|Ru4POM photoanode. Under irradiation, the photocurrent started at a low onset potential of 0.20 V vs NHE, while reaching a net and constant value of 0.020 mA/cm2 in the range 0.4–1 V vs NHE. The attribution of the observed photocurrent to O2 evolution was confirmed by the C-G cell, where the anodic photocurrent produced at the SnO2|KuQ(O)3OH|Ru4POM generator at 0.8 V vs NHE. The FE for O2 evolution was estimated as 70 ± 15%. Conversely, in the case of the catalyst-free SnO2|KuQ(O)3OH electrode (stationary photocurrent of ca. 0.008 mA), no significant cathodic current for O2 reduction was produced, confirming the fundamental role of Ru4POM in driving water oxidation. Evidenced by transient absorption spectroscopy (TAS) results, the absence of long-lived dynamics suggests a fast evolution of the dye excited state in the presence of Ru4POM and likely involving a charge transfer from Ru4POM to *KuQ(O)3OH, forming reduced KuQ(O)2(OH)2* and oxidized Ru4POM, and driving water oxidation reaction. A drop of photocurrent density to 60% of the initial value was observed after 30 min, associated with visible leaching of KuQ(O)3OH and the Ru4POM from the electrode. Furthermore, a remarkably improved PEC water oxidation performance and stability was observed for WO3/PPy:Ru4POM p-n heterojunction by combining PPy doped with Ru4POM (PPy:Ru4POM) with WO3 photoanode.198
Figure 17.

(a) Chemical structure of OER7 (poly-2). (b) Schematic illustration for fabrication of OER7 onto mesoporous substrates. Adapted from ref (195). Copyright 2017 American Chemical Society. (c) Chemical structure of OER8 (Ru4POM) (light blue: W; orange: Ru; gray: Si; and red: O). (d) Schematic representation of the SnO2|KuQ(O)3OH|Ru4POM photoanode for water oxidation (the energy levels are shown for the system at pH = 5.8). Adapted with permission from ref (197). Copyright 2020 Royal Society of Chemistry.
4.1.2. Mn or Co-Based Cubane Catalysts
Tetranuclear Mn or Co cubane clusters render water oxidation due to the inspiration of the O2-evolving complex composed of four Mn atoms in natural photosystem II. Tetranuclear Mn-oxo cluster ([Mn4O4L6]+; L = (MeOPh)2PO2–) OER catalyst has been grafted on [RuII(bipy)2-(bipy(COO)2)]-sensitized TiO2 photoanode by the Nafion polymer (OER9, Figure 18a).200 The photoanode produced a photocurrent due to water oxidation in an almost neutral solution (0.1 M Na2SO4, pH = 6.5) under irradiation (λ > 395 nm) without an external bias. Contemporary excitation of the Mn4O4 catalyst could transfer an electron from the Mn4O4 cluster to the oxidized sensitizer due to their matched electrochemical potentials and then released O2. The RuII sensitizers injected electrons into the CB of TiO2, following transferring electrons to a cathode for H+ reduction to H2, thereby activating the photochemical water splitting cycle (Figure 18b). The photocurrent was measured ca. 100 times that of bare TiO2 (Figure 18c). Later, the Mn-porphyrin monomer (OER10, Figure 18d) was uniformly incorporated into a poly(terthiophene) (PTTh) film on the ITO glass or flexible ITO-coated poly(ethylene terephthalate) (PET) sheet to be a photoanode, which possessed the ability to selectively oxidize seawater instead of producing chloride under irradiation.109 However, interestingly, monomeric Mn-porphyrin is normally catalytically inactive.201
Figure 18.

Chemical structures of (a) OER9 (Mn4O4). (b) Charge transfer route. (c) Photocurrent response of the corresponding photoanode. Representative data from conductive FTO coated glass (black), OER9+-Nafion/TiO2 (gray), Nafion/photosensitizer-TiO2 (red), and OER9+-Nafion/photosensitizer-TiO2 (blue), illuminated at 100 mW/cm2 through a series of long-pass light filters as labeled. Adapted from ref (200). Copyright 2010 American Chemical Society. (d) OER10 (Mn porphyrin; Ar = 4-tBuC6H4, 2,4,6-Me3C6H2, or C6F5). Adapted with permission from ref (109). Copyright 2012 Wiley-VCH. (e) OER11 (Co4O4). Adapted with permission from ref (202). Copyright 2017 Royal Society of Chemistry.
Co4O4(O2CMe)4(py)4 (OER11; py = pyridine derivatives, Figure 18e) with similar merits of Mn cubane catalysts also attracts attention for water oxidation. Several works of cobalt-oxo cubane for photocatalytic water oxidation in homogeneous aqueous solutions consisting of a sacrificial electron acceptor and a photosensitizer have been reported.203,204 It was found that the reactivity of cobalt-oxo cubane was relative to the ligand substitution.205 However, the reactivity of cobalt-oxo cubane on a polymer photoelectrode aiming at heterogeneous PEC water oxidation is rarely explored. After being immobilized on a Nafion film-coated FTO and an α-Fe2O3 photoanode, the incorporation of cobalt-oxo cubane catalysts resulted in a significant cathodic shift of onset potential by 400 mV compared to the Nafion-coated α-Fe2O3 or bare α-Fe2O3. The photocurrent was increased as much as 6-fold to 0.200 mA/cm2 at 0.5 V vs Ag/AgCl with good stability. Furthermore, the vinyl group-modified Co4O4(O2CMe)4(4-vinylpy)4 (py = pyridine) enabled electrochemical polymerization with vinyl phosphate (Vpa) on a RuP-sensitized TiO2 or BiVO4 electrode.202,206 These two kinds of photoanodes showed enhanced photocurrents and cathodic onset shifts compared to the bare metal oxide photoanodes. Moreover, anchoring linkage Vpa allowed better immobilization of the catalyst on the electrode to promote electron transfer between the sensitizer and the catalyst, ultimately, resulting in better performance and excellent stability.
4.2. Inorganic OER Cocatalysts
4.2.1. IrOx Inorganic Cocatalysts
IrOx is a typical noble metal OER catalyst for improving polymer photoanode performance. For instance, an organic bilayer photoanode, composed of 3,4,9,10-perylenetetracarboxyl-bisbenzimidazole (PTCBI, n-type semiconductor) and 29H,31H-phthalocyanine (H2Pc, p-type semiconductor), was capable of O2 evolution in water with the assistance of an IrO2 catalyst that was encapsulated in a Nafion matrix.207 In the electrolyte solution of KOH (pH = 10) and with an applied potential of 0.4 V, O2 evolution was only produced with the IrO2 catalyst under visible light (<750 nm). IrO2 particles were also deposited on a ITO/TiO2-PH (PH = polyheptazine) photoanode by soaking in an IrO2 colloidal solution, deriving from the hydrolysis of Na2IrCl6.208 Basically, the potential of PH valence band maximum (VBM) was estimated to be positive enough to induce water oxidation. Therefore, under light excitation, ITO/TiO2-PH/IrO2 had an O2 evolution with a photocurrent of ca. 0.1 mA/cm2 along with remarkable stability over 90 min (phosphate buffer, +0.5 V vs Ag/AgCl).208,209
4.2.2. Co-Based Inorganic Cocatalysts
Compared to noble metal cocatalysts, earth-abundant Co-based inorganic catalysts are more attractive and have shown great potential both in electrochemical, photoelectrochemical, and photocatalytic water oxidation, as well as over polymer photoanodes for water oxidation.108,210−214 The TiO2-PH photoanode has been modified with cobalt oxide via an in situ photoelectrochemical deposition method in a phosphate buffer containing Co2+ cations (thus, denoted as Co-Pi),210 ensuring the preferential loading of Co-Pi at the active sites with the highest concentration of photogenerated holes. Under monochromatic light irradiation (λ = 450 nm), the ITO/TiO2-PH/Co-Pi photoanode produces a photocurrent density of ∼0.1 mA/cm2, higher than that of the ITO/TiO2-PH photoanode (+0.5 V vs Ag/AgCl). Under visible light (λ > 420 nm), the photocurrent density further increased to 0.19 μA/cm2, capable of producing O2 continuously for 1 h 40 min, while no O2 was detected in the absence of Co-Pi. Substituting Co-Pi particles (∼5 nm) by smaller CoO(OH)x particles (∼1–2 nm) is beneficial for improving activity and stability (Figure 19).211 In detail, the CoO(OH)x cocatalyst was chemically deposited on the FTO/TiO2-PH by successive immersion in a solution of Co(NO3)2 and a weakly basic aqueous ammonia. Due to the smaller size, higher loading of CoO(OH)x than Co-Pi provides more electroactive surface area without decreasing transparency, which would affect the excitation of TiO2-PH and following photocatalytic processes. As a result, the FTO/TiO2-PH/CoO(OH)x (34%) outperformed the FTO/TiO2-PH/Co-Pi (17%) with a photocurrent of higher than 0.11 mA/cm2 after 4 h visible light irradiation.
Figure 19.

(a) Schematic of TiO2-PH hybrid photoanode. (b) Simplified potential scheme illustrating the TiO2-PH photoanode under visible light with a cocatalyst. (c) O2 evolution of the TiO2-PH photoanode without a cocatalyst, with CoO(OH)x or Co-Pi cocatalysts. Adapted from ref (211). Copyright 2017 American Chemical Society.
Ni-Co bimetallic nanoparticles have been developed as an OER catalyst that is directly loaded on an n-type π-conjugated naphthalene benzimidazole polymer photoanode (poly[benzimidazobenzophenanthroline], also labeled as BBL) by the solvothermal method.108 However, no O2 was generated due to the poor catalyst attachment with BBL. After depositing a tunnel junction of an ∼1 nm thin TiO2 layer on the BBL film via ALD to better attach Ni–Co nanoparticles, ultimately, the photocurrent density was improved from ∼0.015 to 0.030 mA/cm2. The O2 evolution was confirmed during the constant illumination with a FE of 82 ± 16%. Recently, Co2+ salt-modified S-gC3N4/BiOCl, prepared by a simple ultrasonically aided hydrothermal method, was reported to have an evident enhancement of the photocurrent density,212 which was measured as 0.393 mA/cm2 for Co-S-gC3N4/BiOCl, ∼3-fold higher than S-gC3N4/BiOCl (1.23 V vs RHE). Similarly, a Ni-based inorganic OER catalyst also has enormous potential to modify polymer photoanodes; the yet reported candidates are just briefly summarized here, including NiOOH,215 Ni(OH)2,216 Ni salt,217 NiFeOx,218 NiOx,219 and the Ni-Co catalyst.108
4.3. Molecular HER Cocatalysts
Up to now, Co complexes are the most widely investigated molecular catalysts to facilitate PEC water reduction. Thereinto, the efficient cobaloxime family contains a coordinated CoIII ion as the redox platform and an −OH group in the second coordination sphere as the proton relay site.220 They have been grafted on the p-type GaP photocathode via the polymerization method and systematically applied for PEC water reduction.221−223 In a typical preparation procedure, the surface oxide of the GaP electrode was first etched by buffered hydrofluoric acid (HF) to create OH-terminated sites for UV-initiated photochemical attachment of linker molecules.221 Then, polymer growth was achieved by reacting 4-vinylpyridine with the hydroxylated surface. The self-initiated photografting and photopolymerization mechanism have been reported on a variety of hydrogen- and OH-terminated materials and occur when hydrogen is abstracted from the surface by a photoactivated monomer to begin surface-initiated free radical polymerization. In this way, polymers bear multiple pyridine binding sites per chain, thereby promising the geometric area loading capacity of catalysts. The catalyst attachment occurs by replacement of one of the axial chloride ligands of Co(dmgH2)(dmgH)Cl2 (HER1) (Figure 20a,c) with a surface-attached pyridine moiety. Compared to bare GaP, the Co-functionalized photocathode showed significantly enhanced PEC performance of 2.4 mA/cm2 photocurrent density at 0.17 V vs RHE (neutral pH, AM 1.5 illumination). By introducing a boron difluoride (BF2) capping group on the glyoximate ligand of cobaloxime (HER2) (Figure 20b,d), the PEC performance could be notably influenced by the change of the ligand environment of molecular catalysts.223 Then, to unveil the effect of the polymeric interface, polyvinylimidazole (PVI) and PVP, providing pyridyl or imidazole ligands, were employed to graft HER2 on GaP(100) electrodes.224 The photocathodes with imidazole and pyridyl immobilized cobaloximes were both capable of achieving a photocurrent density ∼1 mA/cm2 at +0.24 and +0.07 V vs RHE, respectively. The per-cobalt TOF of the Co-PVP-GaP and Co-PVI-GaP photocathodes were estimated as 2.1 s–1 and 2.4 s–1, respectively (0 V vs RHE). Subsequently, the HER2 catalyst was immobilized onto two sides of a p-type GaP substrate via coordination to a surface-grafted PVI brush.225 Through a similar way, cobalt porphyrin catalysts (5,10,15,20-tetra-p-tolylporphyrin cobalt(II)) (CoTTP, HER3) (Figure 20e,f) were assembled on the thin-film polypyridine (4-vinylpyridine as the monomeric unit) surface coatings with a molecular interface for H2 evolution onto a visible-light absorbing p-type GaP semiconductor.226−229
Figure 20.

Chemical structure (marked by red circle) of (a) cobaloxime (HER1, Co(dmgH2)(dmgH)Cl2). (b) Boron difluoride modified HER1 (HER2). (c) Linear sweep voltammograms (LSVs) of GaP (blue dash), HER1 loaded GaP (red), and HER2 loaded GaP (green) recorded at pH 7. (d) Recorded at pH 4.5 under simulated AM 1.5 illumination. Adapted from ref (223). Copyright 2014 American Chemical Society. (e) Cobalt porphyrin (HER3, CoTTP). (f) LSV of HER3 loaded GaP at pH 7 under simulated AM 1.5 illumination. Adapted from ref (226). Copyright 2017 American Chemical Society.
Adopting “click” chemistry for electrode immobilization of a cobaloxime catalyst, diamine-dioxime cobaloxime (HER4, Co-N3) could be successfully grafted to a fullerene derivative (N-methyl-2-(4′-ethynyl)phenyl-3,4-fulleropyrrolidine, C60-ref) on an organic photovoltaic (OPV) electrode (Figure 21).230,231 The LUMO of poly(3-hexylthiophene-2,5-diyl) (P3HT) is more negative than the reduction potential of C60-ref, thus charge transfer between P3HT and C60-ref is thermodynamically feasible. In accordance with the electrochemical measurements, the energy levels of C60-ref0/– and Co-N32+/1+ were estimated to be −4.0 and −4.1 eV, respectively.232 Therefore, an intermolecular charge transfer between the reduced C60-ref and HER4 is expected to reduce protons to H2.230 Compared to ITO/PEDOT:PSS/P3HT:C60-ref, the ITO/PEDOT:PSS/P3HT/C60-Co photocathode had a much higher photocurrent density as shown in Figure 21c (0.1 V vs NHE, 100 mW/cm2), confirming the good catalytic effect of HER4.
Figure 21.

(a, top) Preparation of the OPV-based PEC electrode with a Co catalyst by “click” chemistry. (a, down) The energy level diagram depicts the relevant energy levels under flat band conditions of all materials used in the photocathode. (b) Chemical structure of Co-N3 (HER4). (c) Transient photocurrent response curves of the OPV photocathodes with and without the Co catalyst. Adapted with permission from ref (231). Copyright 2015 Royal Society of Chemistry.
4.4. Inorganic HER Cocatalysts
4.4.1. Pt
Noble metal Pt is the most efficient and extensive inorganic HER catalyst and shows significance for decorating polymer photocathodes. As a special instance, Pt-nanoparticles stabilized by PAA (PAA-Pt) and the anionic poly(isoindigo-co-thiophene) with pendant sodium butylsulfonate groups (PiIT) polyelectrolyte PAA-Pt were codeposited with cationic poly(diallyldimethylammonium) chloride (PDDA) by using LbL self-assembly onto inverse opal (IO) and nanostructured FTO//ITO (nITO) (Figure 22).233 Photoelectrodes with PAA-PT modification showed an enhanced cathodic photocurrent, pointing toward the photoinduced electron transfer from the excited PilT to PAA-Pt and a fast HER rate. Photocatalytic H2 production was confirmed in pH 4.5 acetate buffer with NaClO4 under visible-light irradiation. 0.11 μmol of H2 was produced with 0.047 C of cathodic charge, thus the corresponding FE was ∼45%.
Figure 22.

(a) Molecular structures of PilT, poly(diallyldimethylammonium chloride) (PDDA), and polyacrylatestabilized Pt nanoparticles (PAA-Pt). (b) Schematic illustration of fabrication of FTO//IOnITO//(PDDA/PilT)10//(PDDA/PAAPt)10. (c) Current–time traces with illumination on FTO//IO nITO//(PDDA/PiIT)10 (black) and FTO//IO nITO//(PDDA/PiIT)10//(PDDA/PAA-Pt)10 (red) in 0.1 M acetate buffer, 0.4 M NaClO4, at pH 4.5 with an applied bias of −0.4 V versus Ag/AgCl. (d) H2 production versus time. (e) Proposed mechanism for charge generation/separation in PiIT/PAA-Pt films. Adapted with permission from ref (233). Copyright 2018 American Chemical Society.
In addition, Pt catalyst deposited on a p-type organic photocathode based on P3HT:PCBM BHJ also significantly improved the water reduction performance.146,148,234−238 In detail, after directly depositing a thin layer of Pt onto the top of the P3HT:PCBM (PEDOT:PSS/P3HT:PCBM-Pt), the photocathode represented an increased water reduction onset potential to about +0.69 V vs RHE and the 0.24 mA/cm2 photocurrent density with good stability over 9 h irradiation (0 V vs RHE) (Figure 23a,b). The Pt layer not only improved the charge transport and separation at the Pt/electrolyte interface but also served as electron sinks to effectively extract the electrons from the LUMO of P3HT:PCBM to restrain electron–hole recombination inside the organic semiconductor.234 The photocurrent was further improved via coating a protective TiO2 layer between BHJ and Pt to reduce carrier recombination and to enhance electron collection on Pt (Figure 23c).147,148 In the CdSe and P3HT organic–inorganic photocathode, a thin Pt cocatalyst layer also notably promoted the photocurrent from 0.16 to 1.24 mA/cm2.239 Moreover, obvious enhancement was observed after changing the polymer layer by PTB7:PCBM (PTB7 = poly[(4,8-bis(2-ethylhexyloxy)-benzo(1,2-b:4,5-b’)dithiophene)-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2–6-diyl]),240 or PBDB-T:ITIC (PBDB-T = poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b’]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione))]; ITIC = 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene).241 In addition, there are many other studies relating to the Pt cocatalyst to improve the performance of polymer photocathodes.242−246 Similarly, RuOx is another extensively used noble HER catalyst for decorating the polymer photocathode, and corresponding work has been reviewed here.117,247,248
Figure 23.

(a) Photocurrent density of the photocathodes vs the applied voltage. (b) Prolonged J–t curves of the photocathodes at a bias potential of 0 V vs RHE. Adapted with permission from ref (234). Copyright 2018 Royal Society of Chemistry. (c) Chopped photocurrent density comparison of photoanodes with or without a TiO2 layer. Adapted with permission from ref (148). Copyright 2016 Royal Society of Chemistry.
4.4.2. MoSx
As discussed previously, exploring earth-abundant cocatalysts is of great significance for large-scale solar energy conversion.249,250MoSx, in particular of MoS3, is the most widely investigated catalysts to modify polymer photocathode for water reduction. By adding aqueous HCl into MoO3 and Na2S suspension, MoS3 particles could be successfully synthesized according to the reported method.144,251 Then MoS3 or mixed TiO2:MoS3 were combined with P3HT:PCBM to construct photocathodes for PEC water reduction. Under visible light irradiation, the photocurrent of MoS3/P3HT:PCBM photocathode (0.03 mA/cm2) was only slightly higher than the one without a catalyst layer (0.025 mA/cm2) (0.5 M H2SO4 aqueous solution, −0.05 V vs Ag/AgCl). By contrast, the photocathode with another cocatalyst TiO2:MoS3 layer yielded a higher photocurrent (>0.1 mA/cm2). Similar to the Pt cocatalyst, introducing an electron-collecting layer, routinely used in OPV technology,252 between the P3HT:PCBM and MoS3 layers also contributes to enhancing the overall performance. To prove this concept, an aluminum layer was deposited between the cocatalyst and the polymer layer. Moreover, a metallic LiF/Al/Ti layer or C60 layer was introduced to improve the stability and catalytic performance.145
Besides the surface deposition, the MoSx cocatalyst could also be electrodeposited into a photosensitive Ru complex film by electropolymerization of a pyrrole-functionalized RuII(2,2′-bipyridine)32+.156 The MoS42– was first incorporated into polyRu by ion exchange and then electroreductive transformation in the film to MoSx. For the polyRu photocathodes, no H2 was detected without light irradiation or in the absence of MoSx. In the presence of MoSx and under visible light, the HER process certainly occurred via the oxidative quenching of the excited state of the Ru complex by the MoSx catalyst, thereby exhibiting an enhanced and stable PEC H2 evolution activity.
4.5. CRR Cocatalysts
Ru- and Re-based molecular complexes have been developed as cocatalysts to improve the CRR performance of the polymer photocathode. Only Ru and Re complexes are reviewed in the following, because, to the best of our knowledge, inorganic catalysts have not raised much attention in this field.
4.5.1. Ru-Based Molecular Cocatalysts
Ruthenium-complex polymer [Ru(L-L)(CO)2]n (RCP; L-L = 4,4′-diphosphate ethyl-2,2′-bipyridine) has been developed as a CRR catalyst on the p-type InP:Zn (zinc-doped indium phosphide, p-InP-Zn) photocathode by the in situ photoelectropolymerization of CRR1 (Figure 24a).253 Proper potential positions of p-InP-Zn, RCP, and the CO2 redox reaction ensure the efficient transfer of photoexcited electrons from p-InP-Zn to RCP and the reduction of CO2 to formate (HCOOH) (Figure 24b). The current efficiency for formate formation (EFF) over RCP/p-InP-Zn at pH 4 was measured as 34.3%. Then, three other CRR1 derivatives were designed for polymer RCP/p-InP photocathodes preparation. The selectivity of HCOO– was further improved beyond 70%, and the conversion efficiency of solar energy to chemical energy was achieved to be 0.03–0.04%.254 Finally, a SrTiO3 photoanode was used to combine with this photocathode as a wireless device, which successfully performed solar CO2 reduction and yielded a solar conversion efficiency of 0.08%.255
Figure 24.

(a) Chemical structure of CRR1. (b) Schematic energy diagram of an RCP/p-InP-Zn electrode under visible-light irradiation. Adapted with permission from ref (253). Copyright 2010 Royal Society of Chemistry. (c) Chemical structure of CRR2. Adapted with permission from ref (256). Copyright 2016 Royal Society of Chemistry.
Recently, a new three-step procedure for preparing the highly stable photoelectrochemical CRR photoelectrode comprising a NiO substrate and polymerized complexes of the Ru(II) photosensitizer and a Ru(II) cocatalyst was reported.257 The Ru trisdiimine type photosensitizer (PRuV) contains both methyl phosphonic acid groups and vinyl groups on the diamine ligands and was adsorbed by methyl phosphonic acid groups on the NiO electrode. Another Ru mononuclear complex possessing diimine ligands with a vinyl group and a noncoordinated diimine ligand (VRu-N^N) was connected by electropolymerization of the vinyl groups. These procedures induced the rigid attachment of the polymerized Ru redox photosensitizer with a noncoordinated diimine ligand onto the NiO electrode and increased the amount of the attached Ru photosensitizer units. Finally, a Ru cocatalyst unit of [Ru(CO)2Cl2]n (CRR2) (Figure 24c)256 was introduced into the noncoordinated diimine ligand to form a [(N^N)2-Ru(bpyC2bpy)Ru(CO)2Cl2]2+-type chromophore-catalyst complex on the electrode, which has shown durable CRR ability in homogeneous solutions or hybrid systems with various semiconductor particles.256,258−260 Under visible light irradiation (460 nm < λ < 650 nm, 20 mW cm–2), the NiO/PRu-poly-Ru-RuCAT1 molecular photocathode displayed excellent stability and selectivity for CO and HCOOH production over 100 h in 50 mM NaHCO3 aqueous solution (−0.7 V vs Ag/AgCl). The TON of CRR products exceeded 1200, being the outstanding activity for PEC reactions. Furthermore, a connected device of this polymer photocathode with a CoOx/BiVO4 photoanode, suitable for water oxidation, was developed to facilitate stable solar energy-driven CRR by using water as an electron donor, while a low-energy conversion efficiency of 1.7 × 10–2% was obtained.
4.5.2. Rebased Molecular Cocatalysts
The Re(I) bipyridine tricarbonyl chloride complex (denoted as Re(bpy)(CO)3A; A = Cl or Br) (CRR3,Figure 25a) is one of the extensively studied CO2 reduction catalysts both in electrocatalytic and photocatalytic systems. In particular, the heterogenization of Re(bpy)(CO)3Cl has been successfully applied to a wide range of surfaces through both noncovalent and covalent interactions.261,262 In a heterogeneous polymer system, a stable and efficient molecular photocathode (poly-RuRe/NiO) exists by stabilizing the Ru(II)–Re(I) supramolecular photocatalyst encapsulated CRR3 on the p-type NiO electrode via electrochemical polymerization (Figure 25b–d).159 Due to the coexistence of the vinyl groups in the diimine ligand and methyl phosphonic acid anchors, the new poly-RuRe/NiO photocathode adsorbed more metal complexes and displayed better stability compared to that only using methyl phosphonic acid anchor groups. Compared to the unmodified electrode, the poly-RuRe/NiO photocathode produced approximately 2.5 times more CO (300 W Xe lamp with λ > 460 nm, 50 mM NaHCO3 aqueous solution, −0.7 V vs Ag/AgCl), and the FE was improved from 57 to 85%.
Figure 25.

(a) Chemical structure of CRR3. (b) Preparation of the poly-RuRe/NiO electrode by electropolymerization. (c) Photoelectrochemical CO2 reduction system using H2O as a reductant. (d) Time courses of CO (red ▲), H2 (blue ●), HCOOH (green ■), and half amounts of electrons (black line) passed through the poly-RuRe/NiO (2.5 cm–2) at E = −0.7 V vs Ag/AgCl under irradiation at λex > 460 nm. CO2 purged 50 mM NaHCO3 (aq) (pH 6.6) was used as an electrolyte. Adapted from ref (159). Copyright 2019 American Chemical Society.
Recently, a variety of supporting materials were successfully modified with Re(bpy)(CO)3Cl by a surface-localized electropolymerization method.263 The coordination environments of the rhenium bipyridine tricarbonyl sites are preserved upon immobilization, and the polymerized cocatalyst moieties exhibit long-range structural order with uniform film growth. Though at a low cocatalyst loading, CRR3 modified TiO2 photocathode demonstrates an enhanced activity with TON up to 70 during 5 h. Then, CRR3 and [Ru(bpy)3]2+ (RuL) were coembedded in a polymer Nafion matrix.264 The ternary CRR3-RuL-Nafion system exhibited higher photoconversion of CO2 and photostability than the homogeneous RuL-CRR3 system without Nafion. The Nafion could connect RuL sensitizers and hinder the destructive self-sensitized reaction via enhancing the electron transfer from excited RuL to CRR3. The TON in the CRR3-RuL-Nafion system was 454 for a 20 h reaction, which was ca. 4 times higher than that in the RuL-CRR3 system.
A photocathode assembly (NiO|Si–poly(RuII)–poly(ReI)) by silanization of NiO and a two-step electropolymerization is shown in Figure 26. Vinyl groups functionalized molecular components of silane surface bridge, chromophore, and the catalyst are vinyltrimethoxysilane (Si), [Ru(dvb)2bpy]2+ (RuII; dvb = 5,5′-divinyl-2,2′-bipyridine), and [Re(dvb)(CO)3Cl] (ReI), respectively. NiO|Si–poly(RuII)–poly(ReI) had a stable activity toward CRR over 10 h with a FE of ∼65% and a TON of 58. The long-term stability arises from the silane surface-anchoring groups and the formation of C–C bonds between the three components due to the electropolymerization approach. It was revealed that excitation of the chromophore was followed by rapid hole injection into NiO and the catalyst reduction. The relatively slow interfacial back electron transfer from the reduced catalyst to NiO, in turn, facilitates electron transfer toward CRR.160
Figure 26.

(a) Molecular structures for the surface bridge (Si), the chromophore (RuII), and the catalyst (ReI). (b–d) Possible surface assembly structures on NiO|Si–poly(RuII)–poly(ReI). Adapted from ref (160). Copyright 2019 American Chemical Society.
Overall, the cocatalyst, no matter molecular or inorganic ones, dramatically improved both oxidation and reduction activity of a polymer photoelectrode by about 10-fold in most cases. More importantly, the stability of the polymer photoelectrodes was enhanced greatly. However, one can see that the scale of the photocurrent density of the cocatalyst-loaded polymer photoelectrodes is mainly at a few mA/cm2, still smaller than the inorganic counterparts (about tens of mA/cm2)265 under similar experimental conditions.
5. Charge Carrier Dynamics of Polymer Photoelectrodes
The recent development of polymer-based photoelectrodes for OER, HER, and CRR has dramatically outpaced our fundamental understanding of their inner workings. The synthetic techniques and material discovery strategies have benefitted from a large body of previous work in organic photovoltaics266−268 because of the similarities in the materials targeted. In contrast, the aqueous interface in organic PEC devices adds considerable challenges to spectroscopic investigations. The aqueous environment is vastly different from the solid-state polymer environment, leading to shifts in spectral signatures derived from thin films for photovoltaics and photoinduced protonation changes that are hard to anticipate.269 The collective pathways taken by charges and their kinetics termed charge carrier dynamics ultimately dictate the efficiency and performance of the photochemical systems.270 Knowledge of the charge carrier dynamics provides a pathway to gain key insights into the limitations of a system. As the time scales (kinetics) of different photophysical processes are determined, one can identify the most restrictive limitation. This information could be used to guide efforts on impactful modifications that may be incorporated in the design of more efficient PEC systems.
In this section, we review charge carrier dynamics studies of organic photoelectrodes for HER and OER in the context of designing next-generation systems. Of note, to the best of our knowledge, there are no charge carrier dynamics studies of CO2 reduction on organic photoelectrodes that highlights the limitations of our current understanding, which will be underlined at the end of the section.
Several approaches to the design of semiconductor-based devices for solar fuel generation have been proposed and tested.271 The two most relevant for organic photoelectrodes are PEC cells272 and PV-driven electrolytic cells.273,274 These approaches are distinguished from the source of the asymmetry that separates photogenerated charge carriers and the types of interfaces. PEC photoelectrodes function as monolithic devices where charge carriers are separated at the solid|electrolyte interface. The photovoltage and photocurrent generated by PEC devices under illumination are due to charge separation moderated by differences in electrochemical potentials at the semiconductor|electrolyte interface or by asymmetries in the charge-transfer kinetics for charge carriers across the junction. Typical PV-driven electrolytic cells consist of buried junctions arranged electrically in series with electrocatalysts immersed in an electrolyte. The electrocatalyst can either be in physical contact with the photoelectrode or can be electrically connected by wires, with the PV device completely separated from the electrolyte.275,276 Here the photovoltage and photocurrent produced under illumination originate from charge separation mediated within the PV cell and, hence, are independent of the nature of the electrocatalyst|electrolyte interface. It is generally thought that this working principle is akin to that of PV cells, and consequently, concepts of OPVs are applicable.277 Because of this, we shall not consider PV-driven electrolytic cells in this section and will focus on PEC devices and the understanding of architectural modifications such as heterojunction formation, porosity, and crystallinity, coinciding with the contents addressed in the previous sections. Before discussing these characteristics, we first discuss mechanistic distinctions between inorganic and organic PEC cells.
5.1. Mechanism
The equilibration of charges across the semiconductor|electrolyte interface is fundamental to the function of photoelectrodes and our understanding of them. The electron electrochemical potential of the semiconductor equilibrates with that of the electrolyte solution when they are placed in contact. The dark charge flow across the semiconductor|liquid interface induces a space-charge region in the semiconductor. As mentioned in Section 1.2, the band bending generates a thermodynamic driving force for photogenerated charges to move in opposite directions such that one kind of carrier accumulates at the surface while the other is pushed toward the bulk. The spatial charge separation is key to lengthening the charge carrier lifetimes and making interfacial redox reactions kinetically competitive with recombination.
Our understanding of PEC cells has been built upon knowledge of inorganic photoelectrodes as the charge carrier dynamics of photoelectrodes have been much more studied for inorganic materials, namely metal oxides, compared to organic materials.47,278−281 Being comparatively a recent development, it has not been unambiguously established whether key concepts derived from inorganic photoelectrodes can apply to organic photoelectrodes. For example, Section 4 shows that depositing a cocatalyst on the surface of photoelectrodes is important, both for inorganic and organic materials, to increase their activity. While there is some debate whether the activity improvement arises from passivation of trap states, enhancement of band bending,282−284 improvement of catalytic activity,285,286 or a combination thereof, it is clear that the semiconductor|cocatalyst interface is critical. Structurally ill-defined polymers like CNx have a high density of defects287 and structural disorder can also lead to defect-rich organic materials.288 The defects are associated with localized electronic states, which may reduce the applicability of concepts such as band bending which are derived from a delocalized band structure. Notably, it has been realized that transport models derived from metal-oxide-semiconductor field-effect transistors (MOSFETs) could not accurately describe the behavior of organic field-effect transistors (OFETs).289 Trap states must be explicitly considered to develop models with good agreement. Ionic functionalities have also been shown to lead to doping of organic semiconductors and can influence the energy of electronic states at the interface, which would impact function.290
The level of understanding of OER and HER is much higher for inorganic materials than organic ones. Considering the OER, the inorganic core of the oxygen-evolving complex (OEC) in photosystem II has been intensely studied, more recently with cutting edge X-ray free-electron lasers.291−293 While there is still some debate whether the O–O bond is formed through a radical mechanism or a nucleophilic attack in the OEC,294 it is clear that the successive oxidation of Mn centers occurs in a relatively narrow electrochemical potential window. This has also been observed for inorganic hematite photoanodes and suggests some generality.295 A concept known as redox leveling is invoked, where the loss of electrons is coupled to the loss of protons to maintain a relatively constant redox potential for subsequent oxidations.296 The inorganic semiconductors used in photoanodes, like PSII, have redox-active metal centers with a range of possible oxidation states in close proximity of each other, which appear to determine the OER mechanism.295 It seems doubtful that such mechanisms apply to organic materials considering the more limited accessible oxidation states and that it is unclear whether neighboring repeat units could act cooperatively for catalysis. The limited operando studies to reveal the reaction intermediate formed in organic systems hinder a deep understanding.
For PEC HER, the mechanism is not well understood for conjugated polymer systems compared to inorganic metallic electrodes. Model systems such as Pt and NiPt have been used to understand details such as the catalytic role of adsorbed hydroxyls and alkali metal cations,297 the change in the rate-determining step based on pH and the surface structure,298 and the water structure at the electrode|electrolyte interface.299 In contrast, the identification of active sites in organic polymers from where HER proceeds are not straightforward. Two main active sites have been proposed. The first is metal-centered catalytic active sites,300,301 dominant in conjugated polymers with metal centers, where HER is thought to occur through a homolytic or heterolytic path involving the formation of metal hydride intermediates.302,303 The second suggested active site is an excited state antibonding orbital with localized electrons, in the absence of metal.304,305 This follows recent experimental results by Sun et al. where poly(2,5-diethynylthieno[3,2-b]thiophene grown on copper support (pDET/Cu) displayed similar photocurrents (0.37 mA/cm2 at 0.3 V vs RHE) in both pristine electrolyte and one blended with 10 mM of thiocyanate (SCN–) ions,304 a widely known poisoner of metal-centered catalytic sites.306,307 This presents demonstrable evidence that the Cu species in pDET/Cu may not be the active site for PEC HER. Instead, the acetylenic units, which have excited states with a high electron density antibonding orbital, are more liable to act as highly active centers for HER. This assumption has been investigated via electrochemical-Raman (EC-Raman) spectroscopy.308 Laser lines above (594 nm) and below (647 nm) the bandgap of pDET (2.17 eV, equivalent to 571 nm) were chosen to understand the influence of the photoexcited states. The spectra obtained under 647 nm laser excitation presented bands at 1924 and 2174 cm–1 corresponding to the vibrations of neighboring acetylenic units (C≡C—C≡C) (Figure 27a). The potential dependent current plot under illumination at this wavelength showed an onset potential for HER at 0.2 V vs RHE (Figure 27c). On the other hand, additional bands at 2089 and 2054 cm–1 representing active and transient bands, respectively, were observed under sub-band gap 594 nm laser excitation (Figure 27b). The current onset potential was shifted to 0.5 V vs RHE (Figure 27c). The transient band appeared at a less positive potential (below 0.5 V) in place of the band at 2174 cm–1 indicative of triple bond vibration with lower bond strength. On the basis of these measurements, the acetylenic units were considered as active sites for hydrogen adsorption. As shown in Figure 27d, the acetylenic units are initially activated by the absorption of light. This then induces the attraction of more electron density into the C≡C causing attenuation in bond strength and observed as a shift to lower wavenumbers in the EC-Raman spectra. The role of thiophene units in these polymeric systems is limited to band gap tuning and therefore cannot act as active centers for HER since it is less susceptible to photoactivation as demonstrated by EC-Raman spectroscopy.308
Figure 27.
(a) EC-Raman spectra of pDET/Cu under 647 nm laser excitation. (b) EC-Raman spectra of pDET under 594 nm laser excitation. (c) Potential-dependent currents for HER as a function of excitation wavelength. (d) Photoelectrocatalytic reaction scheme for photoinduced hydrogen evolution. Adapted with permission from ref (308). Copyright 2021 Royal Society of Chemistry.
Suppes et al.143 have also investigated the mechanism of HER by employing a solution-processed regioregular RR-P3HT polymer (prepared over a Ni catalyst) as a photocathode. Given the involvement of hydrogen (as hydride or proton) in the intermediate step309 and the susceptibility of P3HT polymer to protonation in the presence of strong acid,310 the behavior of RR-P3HT in 0.1 M H2SO4 was monitored by PL experiments. The PL intensity of the RR-P3HT quenches upon exposure to the aqueous acid solution (Figure 28a) to an extent comparable with that for a pyridine-containing protonated pi-conjugated system.311 This together with the high tendency of exciton diffusion through the film312 indicates that protons may be involved in the interfacial charge transfer reaction. As such, the mechanism was proposed to proceed via an intermediate that involves the protonation of P3HT at the polymer|electrolyte interface, followed by the reception of photogenerated electrons from the bulk to release hydrogen. The initial interfacial acid–base reaction at the P3HT surface has been suggested to be the driving force for the transfer of photogenerated electrons from the bulk to the surface.147 When the protonated p-type P3HT polymer is brought in contact with an electrolyte solution, the Fermi level of the semiconductor equilibrates with the redox potential of the electrolyte by transferring holes (majority carriers) from the electrode to the electrolyte. As such, the Fermi level shifts to a more negative potential, and the redox potential in the Helmholtz layer of the electrolyte decreases until equilibrium is reached. At equilibrium, the reduction in the density of holes near the surface induces the formation of a space charge layer (SCL) with an electric field that gives rise to band bending. The separation of the excitons generated within the SCL under illumination is driven by the band bending, such that holes move away from the interface with the electrolyte and electrons move toward it. The model describes the transfer of electrons from the bulk to the surface (Figure 28b) although these regions were not defined concerning the SCL. It is unclear whether photogenerated electrons from outside the SCL can reach surface sites. If recombination outside the SCL outcompetes transport to the SCL and surface, which is expected based on the short lifetimes of excitons in the bulk, this implies that the structure of the polymer film should be matched to the SCL width. We also note the possibility that charge accumulation at the interface with the electrolyte breaks down charge neutrality within the polymer. In this case, the effective space charge is represented by a single layer within the bulk of the semiconductor, and charge separation could occur far from the surface.313 The mechanism proposes active sites on P3HT, though it should be noted that metal-centered active sites from residual Ni from the synthetic route in P3HT cannot be discounted.314,315
Figure 28.

(a) Photoluminescence spectra of RR-P3HT films (a spectrum of the dry film in red and a spectrum of the film in contact with 0.1 M H2SO4 in black) at 550 nm excitation wavelength. (b) Proposed mechanism of RR-P3HT photocathodic activity in an aqueous solution. Adapted with permission from ref (143). Copyright 2013 Royal Society of Chemistry.
The following will discuss the key factors affecting the charge dynamics of polymer photoelectrodes that have been explored, namely junction formation, porosity and surface area, and crystallinity.
5.2. Architectural Modifications and the Charge Carrier Dynamics
5.2.1. Junctions
Considering inorganic semiconductors, a high dielectric constant reduces the exciton binding energy that holds the electron and hole together as a pair to levels comparable to the available thermal energy at room temperature. The excitons will thus spontaneously dissociate, and exciton dynamics typically are not relevant under standard operating conditions. In contrast, organic semiconductors have a low dielectric constant which increases the exciton binding energies to an extent greater than the thermal energy.316 The excitons persist at room temperature, and their dynamics will impact device function. Similar to BHJ photovoltaics, a blend of separate donor and acceptor polymers can be adopted in polymer-based organic photoelectrodes to generate free charges.317 Another approach is differential doping of a material to form a homojunction, such as in bulk homojunction PV318 and typical silicon PV.319 This strategy can also be applied to polymer-based organic photoelectrodes to promote the generation of free charges. Junctions (homojunctions and heterojunctions) have thus been actively pursued in the design of more efficient organic PEC cells.
In 2017, Ruan et al.104 investigated the influence of a nanojunction between bulk carbon nitride (G-CN) and a B-doped CN nanolayer on the photoanodic performance of the photoelectrode. They used electrochemical impedance spectroscopy (EIS) to establish that the formation of bilayer homojunction (s-BCN) dictates the separation of charge carriers in the photoanode. s-BCN photoanode produced a photocurrent density of 0.103 mA/cm2 at 1.23 V vs RHE in 0.1 M Na2SO4 under one sun irradiation. This outperformed the bare G-CN photoanode which showed a photocurrent density of 0.0106 mA/cm2 at 1.23 V vs RHE. The IPCE at 400 nm was nearly 10% for s-BCN compared to 1% for CN. The improved water oxidation by s-BCN was attributed to the bilayer heterojunction which improved charge transfer to the electrolyte. The EIS Nyquist plots in Figure 29a showed a decrease in the semicircle diameter for s-BCN compared to G-CN, which translates to a 3-fold increase in electron transfer conductivity.
Figure 29.

(a) Nyquist plots of G-CN and s-BCN obtained by applying a sine wave with amplitude of 5.0 mV over the frequency range from 10 kHz to 0.1 Hz. Inset: Periodic on/off photocurrent response of G-CN and s-BCN electrodes in 0.1m Na2SO4 with 0 V bias versus Ag/AgCl. (b) Potential dependence of the rate constant kt and kr for s-BCN and G-CN samples. Illumination: 365 nm UV light. Adapted with permission from ref (104). Copyright 2017 Wiley-VCH.
A more refined kinetics analysis was provided by intensity-modulated photocurrent spectroscopy (IMPS). The kinetics of charges are represented by the first-order rate constant of surface recombination (kr) and interfacial charge transfer (kt). This assumption may not hold for the multielectron, multiproton water oxidation reaction. However, similar analyses have been performed to elucidate the rate constants of the competing productive charge transfer and unproductive charge recombination processes for inorganic photocatalytic systems.320−323 As shown in Figure 29b, kr decreases with increasing potential, typical for an ideal semiconductor|electrolyte interface. This is attributed to the stronger band bending caused by the more positive applied bias, enhancing charge separation, and suppressing charge recombination at the surface. Additionally, at low potential, kr in G-CN is lower than in s-BCN due to the introduction of surface defects during B doping which acts as charge recombination centers. In effect, a high concentration of dopants may promote surface recombination and compromise PEC performance.324,325 For photoelectrodes based on CNx, doping past the optimal level mostly introduces trap states which serve as recombination centers irrespective of the redox reaction being monitored.101 At high potentials, dopant-induced surface recombination can be mitigated by the applied bias which directs electrons away from the surface and to the counter electrode.104 As seen in Figure 29b, kt for s-BCN at 0.2 V vs Ag/AgCl was approximately 10 times larger than that of G-CN. Also, at 0.4 V vs Ag/AgCl and under 365 nm UV light irradiation, the ratio of kt to kr was larger in s-BCN than in G-CN. The authors, therefore, concluded that more charges were available for oxygen evolution in s-BCN than in the bulk G-CN because of the nanojunction formation. This can be understood by considering the energetics of the system. The B-doping shifts the valence band of the CNx upward and generates a driving force for photoexcited holes to transfer from G-CN to the B-doped CNx layer. Electrons can flow in the opposite direction under a weak bias. The opposite movement of photogenerated charges induces charge separation which dramatically improves the photoanode performance.
Functionalization with heteroatoms was also attempted to improve the performance of a CNx photoelectrode. Fang et al.91 investigated this option by blending S-containing compounds into non-S precursors in the weight ratio of 1:1, 1:2, 1:5, and 1:10, in the synthesis of CNx films. The S acted as a directing agent326 for the growth of the polymeric CNx films on FTO as well as connections to assist charge migration. A photoanodic performance of 0.100 mA/cm2 was recorded at 1.23 V vs RHE under AM 1.5 illumination in a NaOH electrolyte solution. This compared favorably to the nondoped CNx (0.008 mA/cm2) and was accredited to the limited defects along with the interfaces and the reduced charge recombination associated with the heterojunction formation.327,328 The maximized photoanodic performance was achieved with films synthesized from a 1:2 weight ratio of S:non-S-containing compounds. This was consistent with the EIS Nyquist plot in which the polymeric CNx (PCNx) films made from the 1:2 weight ratio presented the smallest diameter compared with the other weight ratios, signifying efficient charge transport. Also, the optimized system exhibited efficient charge separation which was characterized by the dramatic decrease in emission peak at 470 nm compared to the high emission intensity observed for the other samples. This is an indication that radiative recombination was minimized as a result of exciton dissociation.329
To better understand the role of heterojunctions, Shalom and co-workers studied the charge recombination process between CNx and mesoporous TiO2 using time-resolved photoluminescence (trPL) and TAS.330 They reported the electron injection rate from the CNx excited states to the TiO2 conduction band by steady-state and trPL and the hole extraction kinetics using various liquid electrolytes and solid-state hole conductors. They observed that the emission spectrum of CNx on glass displayed a wide band centered at 510 nm. The emission was drastically quenched when CNx was deposited on TiO2 to create a heterojunction, indicative of CNx excited state quenching. The magnitude of the emission quenching of the CNx/TiO2 heterojunction film compared with that of CNx on glass was used to calculate an electron injection yield of about 90%. The emission of CNx/TiO2 also decayed faster than that of CNx alone, consistent with electron transfer. The recombination processes of CNx/TiO2 and CNx on glass were studied by TAS. The transient spectrum of CNx/TiO2 at 50 μs after excitation at 532 nm showed a photoinduced absorption band from 600 nm increasing toward the near-IR (NIR) spectral region. In contrast, the transient spectrum of CNx, the reference material, showed a negligible signal, consistent with the typical absorption onset of <450 nm for CNx, indicating that transient signals originated from the photoinduced electron transfer and not the deactivation of the CNx excited states. The ground state absorption of the CNx/TiO2 samples extended into the NIR range, suggesting that the interface plays a role in modifying the electronic structure of CNx. Also, the hole transfer process between the excited states in CNx and different hole acceptors was studied. Notably, P3HT coated on CNx/TiO2 films showed an enhanced TAS signal in the region from 750 to 925 nm.330 This signal was due to the charge transfer complementarity that occurred from the P3HT to TiO2 and CNx since P3HT coated on CNx/glass showed no signals. The spectrum for P3HT/CNx/TiO2 was attributed to the individual contributions from the oxidized CNx and P3HT formed by electron injection into the CB of TiO2.
Decorating the surfaces of organic photoelectrodes with cocatalysts presents a promising approach toward improving performance. For example, Fan et al.331 showed the incorporation of a layered double hydroxide (LDH) onto the structure of CNx. In their work, CNx was grown on an FTO substrate and the NiCo-LDH was electrochemically deposited on top. The resulting CNx/NiCo-LDH composite generated a photocurrent of 0.0118 mA/cm2 at 0.6 V vs SCE, representing an increase of almost 3 orders of magnitude compared to bare CNx. The EIS data demonstrated that the bare CNx film had the highest charge resistance. The charge transfer initially decreased when loading with NiCo-LHD up to 10 mC of charge passed. Beyond this optimal cocatalyst loading, the charge transfer resistance increased. This may be due to the formation of surface recombination sites at high amounts of additives, as we discussed above.
5.2.2. Porosity
The growth of porous organic materials with considerably large surface area for PEC OER and HER has also been investigated. If exciton dissociation (free charges generation) can occur at the semiconductor|electrolyte interface, then increasing the surface area could potentially result in maximizing the photocurrent, and the conventional donor:acceptor type heterojunction would not be needed to generate charges. However, this may not apply to all systems as other studies287,332,333 have shown that charge generation could proceed on faster time scales as opposed to exciton diffusion to the interface. Polymeric CNx electrodes with high porosity have recently been prepared for solar water splitting.87,97 Lv et al.87 reported a photoanode based on CNx synthesized from cyanamide, melamine, or dicyandiamide (DCDA; note that the authors used the incorrect name dicyanamide) by a two-step vapor deposition. The obtained CNx films displayed uniform morphologies with the DCDA-based film possessing the highest surface area, as a result of its smaller particle size. Under 1 sun illumination, the optimized CN-3g-dycyanamide films gave rise to a stable anodic photocurrent of 0.063 mA/cm2 at 1.23 V vs RHE (at pH 7) with an onset potential of 0.41 V vs RHE and a maximum IPCE of 6.6% at 350 nm. The stable anodic photocurrent delivered, implied that generation, separation, transport, and recombination of the charge carriers attained equilibrium as shown in the transient current curves of Figure 30a. Compared with melamine and cyanamide monomers, the superior PEC performance of DCDA-based CNx films was linked to enhanced light absorption and improved charge transfer at the photocatalyst|electrolyte interface as a result of a large surface area. The lower charge resistance at the photocatalyst|electrolyte interface of the optimized film (Figure 30b) is in line with its higher photoactivity.
Figure 30.

(a) Transient photocurrent density and (b) EIS spectra measured at 1.23 V versus RHE under light illumination of the CN films prepared using different amounts of dicyanamide. Adapted with permission from ref (87). Copyright 2017 Elsevier.
A relatively simple pathway for growing highly porous and large-scale CNx films with controllable chemical and photophysical properties has recently been reported by Peng et al.96 Employing the doctor-blade technique, CNx films were grown on a FTO substrate using a supramolecular paste containing typical CNx precursors and barbituric acid. The resulting uniform and transparent films with a large surface area were presented as CN(x), x = 0, 0.05, 0.1, and 0.15 concerning the mass of barbituric acid used as a source of carbon doping. The advantage of high porosity and increased surface area was demonstrated by the increase in photocurrent from 0.0033 mA/cm2 to 0.0075 mA/cm2 at 1.23 V vs RHE (in 0.1 M KOH) between the less-porous (g-CN(0)) and more porous (g-CN(0.1)) films (Figure 31b). It was anticipated that increasing the C-doping would correspond to higher electronic conduction and effective charge transfer (Figure 31a). Past the optimal doping of x = 0.1, the performance decreases as a result of enhanced recombination induced by the excess carbon sites. Heterojunctions could also be prepared by the doctor-blade technique employed. The g-CN(0)/g-CN(0.1) heterojunction is thought to facilitate charge separation despite the type-I heterojunction (Figure 31c): both electrons and holes would transfer to g-CN(0.1) if only considering the energetics. This is characterized by a strong fluorescence quenching indicative of reduced recombination.
Figure 31.

(a) Nyquist plot of the CN films at 1.23 V vs RHE in dark condition. (b) Photocurrent of the CN films at 1.23 V vs RHE in 0.1 M KOH aqueous solution under one sun. (c) Energy diagram of the g-CN(0)/g-CN(0.1)/FTO junction. Adapted with permission from ref (96). Copyright 2018 Wiley-VCH.
5.2.3. Crystallinity
A final design strategy that has attracted the interest of researchers with tangible prospects in facilitating charge transfer in organic photoelectrodes is crystallinity. Ruan et al.94 reported the effect of crystallinity on the PEC performance of CNx films. Their findings established that long-lived charge carriers reside in more poorly crystalline samples, due to deeper trap states. To explore the impact of trap states, bulk CNx, porous CNx, and compact CNx films were fabricated, representing moderate crystallinity, low crystallinity, and high crystallinity, respectively. Crystallinity was assessed from the full width at half-maximum (fwhm) values of the 110 XRD peak at 27.5° (1.0°, 1.1°, and 0.7° for bulk, porous, and compact, respectively) where lower fwhm values represent higher crystallinity.94,334,335 Open circuit voltage decay (OCVD) measurements were used to better understand the role of trap states. The average charge lifetime was determined by fitting a biexponential function to the decay curves (Figure 32). Photovoltage decay in the compact CNx film (Figure 32b) was much faster than bulk (Figure 32a) and porous (Figure 32c) samples. The average electron lifetimes were found to be 0.9 s for compact, 5.0 s for bulk, and 12.8 s for the porous sample (Figure 32d). The short average lifetime indicated rapid charge recombination in the absence of electron donor, while the longer electron lifetimes were mainly attributable to the severe electron trap effect. The long-lived electrons in the bulk and porous samples are mostly trapped at deep levels and located at low energy levels, hence, incapable of participating in redox reactions.287 In effect, the migration of electrons in the photoelectrode was hindered by deep trap states, and reducing the deep trap state density promotes charge transfer efficiency and boosts photocurrent density. The lower trap state density in compact CNx films enabled more charges to reach the interface and participate in redox reactions. This is also supported by the large photovoltages recorded.
Figure 32.

Open circuit voltage decay (OCVD) plots of (a) bulk CNx, (b) compact CNx, and (c) porous CNx with 150 W xenon lamp illumination from the electrolyte–electrode (EE) side. (d) Calculated average charge lifetimes in the g-C3N4 films. (Generated photovoltage ΔV is the difference in voltage between dark and illumination conditions). Adapted with permission from ref (94). Copyright 2019 Royal Society of Chemistry.
Recently, an engineered CNx photocathode with a considerable amount of trap states as a result of N-defects and C–OH terminal groups has been presented by Ruan et al.122 The material showed improved properties compared to pristine CNx with respect to conductivity and the lifetime of shallow-trapped charges. Quantitatively, conductivity increased by 2 orders of magnitude, and the lifetime of the shallow-trapped charges increased by 3 orders of magnitude in the optimized def-g-C3N4 compared with ref-g-C3N4. The average electron lifetime increased from 0.9 to 5.5 s as trap states were introduced, in line with a reduction in charge carrier mobility associated with charge trapping in defect-rich samples. In the absence of trap states, the decay kinetics of CNx after about 50 ns of photoexcitation may be described by a power-law function. Nonetheless, this description may not hold for decay kinetics that precedes 50 ns due to the influence of emission. As shown in Figure 33, the negative TAS signal observed for def-g-C3N4 is significant up to tens of microseconds whereas the electron lifetime in the shallow emissive state of ref-g-C3N4 is less than 50 ns.
Figure 33.

TAS kinetics of ref-g-C3N4, def-g-C3N4-1, def-g-C3N4-2, and def-g-C3N4-5 samples under N2 atmosphere after 355 nm excitation (200 Hz, 850 μJ/cm2/pulse), monitored with a 660 nm probe. Adapted from ref (122). Copyright 2020 American Chemical Society.
Similarly, Shalom and co-workers reported a simple method to grow a densely packed CNx film by crystallization of CNx monomers on a FTO substrate, followed by thermal condensation.95 The photoanode prepared from melamine delivered a photocurrent of 0.116 mA/cm2 at 1.23 V vs RHE and up to a 1 V shift of the onset potential under one sun in 0.1 M KOH (pH = 13) aqueous solution with IPCE of 8.5% at 400 nm. However, the photocurrent decreases to 0.071 mA/cm2 in a neutral electrolyte (NaH2PO4, pH = 7) and further reduces to 0.064 mA/cm2 in an acidic electrolyte (H2SO4, pH = 0.2) at an onset potential below 0.3 V vs RHE. To elucidate the charge separation mechanism, electron lifetime, and hole extraction kinetics, TAS measurements of the CNx film in various electrolytes at 1.23 V vs RHE were taken (Figure 34a). As could be expected from the increased photocurrents, the electron lifetime increases from 0.73 ms in an acidic electrolyte to 1.09 ms in a basic electrolyte. This suggests that the photoanode response displays progressive enhancement with increasing pH, similar to the behavior reported for hematite photoanodes.336,337 Generally, the surface charge density of semiconductors increases as pH decreases, and this affects the energetics of the semiconductor. As charges accumulate at the surface of the semiconductor, the Fermi level of the semiconductor equilibrates with these surface states instead of the redox couple of the solution, which reduces the extent of band bending338 and hence leads to poor charge separation. This may account for why photoanodic performance was maximized in the presence of the basic electrolyte and declined in the acidic electrolyte. As observed in Figure 34b, the presence of triethanolamine (TEOA) further increased the electron lifetime from about 1.09 to 2.26 ms because of a faster hole extraction, which suppressed electron–hole recombination. As a result, the photocurrent of the CNx photoelectrode doubled to 0.245 mA/cm2 following the addition of 10% TEOA into the 0.1 M KOH.
Figure 34.

(a) TAS spectra (delay time unspecified) of a CNx film soaked in different aqueous solutions. (b) TAS decay of a CNx film soaked in 0.1 M KOH, and 0.1 M KOH containing a 10% TEOA aqueous solution monitored at 850 nm. Adapted with permission from ref (95). Copyright 2018 Wiley-VCH.
An insight into carbon-based conjugated systems in relation to their catalytic activity is that the rate of exposure of the active sites on the catalyst is paramount, and it is linked to the dimensional state of the molecule. Higher-dimensional state systems would have limited exposure of their active sites/species as opposed to systems of lower-dimensional states.338 The easy manipulation of organic systems from one-dimensional state to the other via the introduction of new linker groups339 suggests the relevance of this to the catalysis community. The activity of substrate-bound polymer photoelectrodes is usually influenced by the interaction between the substrate and the photoelectrode. A strong combination between polymer photoelectrodes and substrates, which is achieved through irreversible chemical interactions, opposes resistance and promotes charge transportation at higher levels than those which are loosely bound via weak van der Waals forces.120 Efforts should therefore be directed at techniques that strongly bind photoelectrodes to their substrates to enhance PEC performance.
One question that remains unanswered despite the relevant information provided in this section is how material design strategies affect the behavior of charge carriers and their impact on device performance. In other words, the requisite information needed to fill the gap in our current understanding of the complexity of the charge carrier dynamics in relation to device performance is still lacking. The limited operando studies of organic-based photoelectrodes are immediately thought of as the main contributing factor to our present lack of understanding. Spectroscopic and electrochemical measurements that concurrently monitor key processes of charge separation, charge transfer, and recombination alongside the photocurrent density of organic-based photoelectrodes have been rarely reported. There are not enough studies that use techniques such as trPL and TAS to afford us information on these key processes and provide guidance for material and system optimization. Interestingly, of the material design strategies of junction formation, porosity, and crystallinity, only the charge carrier dynamics of junctions were characterized by both trPL and TAS measurements. Neither of these techniques was used to describe the charge carrier dynamics of porous systems. Only TAS was utilized to establish that in crystalline CNx photoanode charge separation is enhanced, whereas electron–hole recombination declines as pH increases, leading to an increase in photoanodic performance.95 Most of the measurements performed on polymer photoelectrodes to describe the behavior of charges were found to employ electrochemical-based techniques. However, analyses of these measurements to give information on charge recombination kinetics other than charge transfer kinetics are yet to be reported. For instance, EIS measurement was utilized to establish that charge transfer at the photocatalyst|electrolyte interface of a porous photoelectrode material proceeds at a faster rate compared with its nonporous counterpart.87 In contrast, the recombination kinetics of these charges was not reported. Expanded analyses of EIS responses and other modulated electrooptical techniques applied to organic photoelectrodes will greatly improve our understanding.104,320,323,340 Additionally, coupling two or more spectroscopic techniques to explore and monitor the excited state structural and carrier populations, alongside the different photochemical/photophysical processes within these polymer photoelectrodes will provide rich information that will improve our understanding of their inner workings. For instance, performing integrated time-resolved Raman and TAS measurements on polymer photoelectrodes will afford us information on the excited state structural dynamics341 and charge carrier dynamics, respectively, which are key considerations for the architecture of next-generation efficient photoelectrodes.
6. Theoretical Modeling on Organic/Polymer-Based Semiconductors
The organic semiconductor materials used in polymer-based photoelectrodes are significant members of the family of functional materials, with great performance which sometimes supersedes that of their inorganic counterparts. However, the best performances of these organic materials are usually subject to addressing certain challenges at the atomic, molecular, and morphological length scales. This presents a daunting but exciting task to the computational community, including chemists, physicists, materials, and data scientists. In view of this, various methods and models have been developed with wide applications in the potential quantification and rationalization of the electronic properties of organic materials.
Generally, the interplay between the chemistry of the molecular core and the intermolecular factors of organic semiconductor materials dictate their performance and key electronic properties.342 These properties are manipulative and, hence, serve as an inspiration to both experimentalists and theorists. The common pitfalls encountered by organic and polymer semiconductors are their instability and lower charge mobility, which are influenced by either chemical or physical factors. Thus, the advanced understanding of the chemical and physical factors that determine the properties and performance of organic and polymer-based semiconductors is crucial toward improving their stability.342
For many π-conjugated polymer-based semiconductors, the magnitude of their charge transport properties is a function of the strength of the electronic coupling between adjacent cores, which has been shown to be consistent with the exchange component of the total interaction energy.343,344 As such, the fundamental challenges that have been identified in this arena are (1) the lack of universal theory of the charge transport and (2) the role of material’s morphology on the charge transport.345−347 Models of inorganic crystalline and amorphous solids have been used to discuss the charge carrier dynamics in organic semiconductors in the past. However, several limitations arise in the use of these models, particularly due to the differences in interaction between nuclear and electronic degrees of freedom that exist in both organic and inorganic solids.345 In response, newly developed methods for modeling charge transport in organic and polymer-based semiconductor materials turn to consolidate conventional quantum chemical methods and traditional methods used in soft-matter modeling.345 These methods are phenomenological theories, which are based on a simplified model of transport. Then a correlation between device response (e.g., current, mobility, voltage, or electric field) and parameters that can be used in the context of describing experimental results is deduced. The applicability of soft-matter modeling methods to organic semiconductor emanates from the fact that crystalline organic solids, held together by van der Waals forces are relatively soft and this softness impacts the charge transport. This modeling strategy is mostly used for organic electronics such as liquid crystals (LCs)348 and hence will not be critically looked at in this review.
The Gaussian disorder model (GDM) and its recent improvements349 and the multiple trapping model350 have emerged as the widely used models for describing the charge transport in polymeric semiconductors. The GDM describes the hopping transport across a disordered single or multiple component organic solid where charge transporting elements, which can either be the molecules participating in transport or segments of a main chain polymer that are separated by topological defects, are identified as sites whose energies of their hole or electron transporting states are subject to a Gaussian distribution of energies.351 This model has been developed from numerical simulations based on Monte Carlo (MC) simulations to an analytical approach.352 GDM based on numerical simulations has revealed that there is the likelihood of charge carrier hopping within the Gaussian DOS of a diluted system to accomplish a random walk subsequent to a relaxation to a dynamic equilibrium energy below the center of DOS (Figure 35). From the analytical approach in the GDM framework, this hopping is initiated from the energy levels above the transport energy (Et) and proceeds downward to spatially neighboring sites with lower energies under quasi-equilibrium conditions as displayed in Figure 35. However, a hop to a site below Et is followed by an exponentially faster upward hopping mediated by thermal activation in the vicinity of Et with a sequential hop to deeper energy levels below Et.353
Figure 35.

Relaxation of an injected charge carrier as a function of time in Gaussian DOS (left). Adapted with permission from ref (351). Copyright 1993 Wiley-VCH. Hopping transport of injected charge carrier via the states at thermal energy in Gaussian DOS (middle) and space (right). Adapted with permission from ref (354). Copyright 2021 IOP Publishing, Ltd.
The multiple trapping model includes a mobility edge (ME) energy with an exponential density of shallow traps extending to lower energies.350 The ME model adopts the principle that there is a defined energy (the ME) in the density of states (DOS) that segregates mobile states from localized states. This suggests that immobile trapped carriers are temporarily made mobile if they reach mobile states by the thermal excitation. It is essential to note that when thermal activation predominates, carriers do not necessarily hop to the mobility edge but strictly to the energy at which the vibration of the wave function decreases toward the Boltzmann factor.350 The ME model is applicable to materials that are polycrystalline, where the mobile states are extended band states of the crystallites and the trapped states are located in the disordered regions between the crystallites. Thus, hopping directly between trapped states is a competing transport mechanism.355 The multiple trapping model further describes hopping of photoexcited carriers in amorphous materials such as a-As2Se3356 and a-Si:H,357 as one which occurs directly between localized, band-tail states giving rise to a new path for energy deactivation at low temperatures. At times beyond the segregation time (time expended for the separation of mobile and localized states), e.g., 1 μs in a-As2Se3 and a-Si:H, the mobility is characterized by thermal excitation leading to multiple trapping. However, at lower times, charges move directly to lower energy states.355 Typically, the rate at which carriers hop away from deep or shallow states is smaller for deeper initial energies based on the exponential dependence of the rate on energy difference (effectively an activation energy) and on wave function overlap on energy. This invokes energy-induced state divisions which could be either fast or slow. Shallower states have fast detrapping rates, and carriers will depart as fast as they arrive and accumulate in deeper states where detrapping rates are slow and hence, carrier population is high. The energy which separates these fast and slow states is time- and temperature-dependent and is referred to as demarcation energy.355 The demarcation energy is the energy that defines the instance where the hopping-away rate is equivalent to the inverse of observation time. The mobility values obtained from the multiple trapping model of organic-based semiconductors respond quickly to small variations of trap characteristics when carrier concentrations are small. Again, the mobility shows temperature dependence (Arrhenius relation of mobility), and this has been demonstrated by both experimental and theoretical findings for the diketopyrrolopyrrole-naphthalene copolymer (PDPP-TNT)358 as displayed in Figure 36.
Figure 36.
Simulated and experimental average mobilities of the diketopyrrolopyrrole-naphthalene copolymer (PDPP-TNT) for various temperatures. Adapted with permission from359. Copyright 2020 American Institute of Physics.
Polymer-based semiconductors are usually characterized by a π-conjugated backbone. Typically, charge transport in polymer-based semiconductors is affected by conformational disorder in the polymer backbone356 and chemical defects.357 The pronounced conformational disorder in polymers, especially those containing aromatic rings causes deviations from planarity in the polymer backbone, and this leads to a decrease in charge mobility along the polymer chains. The above phenomenon has been investigated by Choi et al.,356 using PANI- and PPy-doped with perchlorate as conducting polymers. They reported that distortions in the aromatic ring orientations of PPy and PANI are driven by temperature and film thickness, respectively. Within 220–240 K, the PPy doped with a perchlorate counterion exhibited structural changes which induced ring orientation and subsequent modification of charge mobility. Thicker films of PANI on gold displayed an out-of-plane tilt of the aromatic backbone with different conductance. Also, defects in molecules may act as trap states358 which largely affects charge mobility. The dependence of charge mobility on morphological features of polymer semiconductors have also been reported.360 Morphological studies performed on regioregular P3HT has revealed that low molecular weight (MW) films have a higher degree of crystallinity than high MW films. In contrast, high MW, less-ordered films favor high charge mobility because the films are longer than the domains, thereby minimizing the effects of grain boundaries by bridging neighboring grains. The charge transfer between polymer chains and the persistence length are dependent on the molecular packing.361 Hence, accounting for the physical nature of the interactions behind the molecular packing and their subsequent connection to the charge transport properties is essential for modeling and designing crystalline organic semiconductors.362 The molecular packing can be tuned by various intermolecular interactions including hydrogen bonding, halogen bonding, and π–π stacking.344 The influence of intermolecular interactions on molecular packing and the consequent effect on charge transport has been demonstrated and reported for perylene diimide dyes (PDIs).362 The intermolecular electrostatic O-π and O–H interactions in PDIs cumulatively impact the solid-state/molecular packing and the dimensionality and directionality of the charge percolation network. Charge percolation is a transport process in anisotropic polymer materials where there are different charge transport paths.363 Two-dimensional π-stacking motifs in PDIs is beneficial to high electron transport unlike one-dimensional π-stacking motifs which have obstructed electron transport due to dynamic disorders in intermolecular couplings and energies.
These different properties of polymer semiconductors have been studied using quantum mechanical and molecular simulation methods364 to gain insights into the optical and transport properties of π-conjugated polymers,365 thereby serving as a guide for the design of next generation systems. Theoretical simulations have aided in the computation of effective mass tensor for relevant bands which are useful in the estimation of the exciton binding energy associated with the lowest electronic LUMO–HOMO transition in π-conjugated polymeric semiconductors.365 The associated binding energy of an effective mass of 0.09me is about 0.2 eV and an exciton radius of about 20 Å. These details are integral to the understanding of the optoelectronic properties of the polymer. Recently, DFT calculations have been used to derive a transport model for polycrystalline polymer-based semiconductor by combining the contributions of the electronic structure of the crystalline domain with a model of interaction at grain boundaries (interface between two crystallites).366 The study focused on crystalline poly(3-alkylthiophene) (P3AT), an organic polymer with high charge mobility, to construct a reasonable density of states model combined with transport mechanisms for easy calculation and comparison of its conduction. The model adopted three assumptions viz., (1) transport is confined in a single plane of the lamella; hence, a two-dimensional density of states is used for the ordered lamella, (2) an exponential band tail of localized states represents the disordered regions between the ordered grains, and (3) transport in the ordered region/lamella is characterized by constant mobility. The main findings revealed that structural ordering in polymers has two significant effects. These include a higher mobility transport path accessible to holes due to the presence of lamella in the polymer structure. Second, the volume of amorphous material is decreased, causing a reduction in associated localized-state density. Hence, even if transport is dominated by hopping in the disordered material, the conductivity will increase because the Fermi energy moves further up the DOS for a given gate voltage, and the exponential increase in hopping conductivity increasingly offsets the reduced volume of material.
Other models such as atomistic molecular dynamic (MD) simulations and static lattice (SL) calculations have jointly been used to investigate the structure of poly(3-butyltiophene) (P3BT) as a function of temperature and pressure.367 In P3BT, the thiophene rings are connected at the two carbon atoms in the 1-position relative to the sulfur atom, with alkyl substituents sitting at one of the two 3-positions, and this conforms to the general lattice structure of P3ATs.368 It has been established through atomistic simulation that the butyl side chain in P3BT can assume cis and gauche conformations which are thermally accessible. This may induce torsional distortions that trigger the deformation of the π-conjugated polymer backbone by energy transmission from thermally induced sources stored in the alkyl side chain and can be observed as a dilation of the forbidden gap.368 Similarly, atomistic MD and SL simulations conducted on P3AT have revealed that at ca. 6–7 GPa, planarization of the polymer main chains is initiated. This process is usually accompanied by a spontaneous tilting in the alkyl side chain. However, it is archetypal of the polymer to exhibit a reduction in this “tilting” at pressures beyond 10 GPa.369 Thus, it is evidential that temperature and pressure largely affect the structure of π-conjugated systems, and therefore, the scientific community can capitalize on this to compound the existing database on the structure activity relationship (SAR) of organic polymers. The static considerations adopted in modeling are gradually being replaced and/or complemented by dynamic considerations where the time-dependent nuclear motions inform the overall charge dynamics of the material.370 For instance, the intramolecular transport of charge carriers in poly(phenylenevinylene) (PVP) and P3HT was calculated by tight-binding approximation combined with static disorder along the chain.371 The polymers were modeled by a chain of sites that are consistent with the monomer repeat-units, and therefore, charge migration on this chain is described by the Hamiltonian.370 Next, the results revealed that the distinct effects of static and dynamic disorder on particle motion trigger two stages of particle propagation. In that, at short times the particles move ballistically (i.e., exhibits maximum velocity over a short period of time), while at lengthy times their propagation is diffusive with higher dispersions for higher degree of static disorder. Basically, static disorder creates a friction for the particle propagation, whereas the dynamic disorder may drive or impede the intersite transfer depending on the degree of static disorder.371
Despite the inability of the current developed modeling methods to quantitatively predict the charge transport characteristics of the polymeric semiconductor materials used in making the photoelectrodes, the major progress that has been made in this field so far cannot be overlooked. This has paved the way for researchers and experimentalists in the community to study an array of plausible materials and their properties prior to their use as the main semiconductor for the design of photoelectrodes. Increasing the diversity of the polymers investigated experimentally and computationally is needed to help generate a more holistic understanding of the relationship between physical, chemical, and electronic properties of conjugated polymers.
7. Conclusion and Outlook
Solar fuel synthesis stands to make a significant impact on a sustainable future. Especially, the PEC approach for producing H2 from water and high value-added chemicals such as methanol and ethanol by utilization of CO2 waste has the potential to develop a zero carbon-emission economy. The limitations of inorganic materials for engineering photoanodes and photocathodes were discussed in the review, and the advantages such as low cost, earth abundance, ease of band position tuning, and sustainability of the polymer photoelectrodes were detailed. As the polymer-based materials have been extensively studied for particle suspension-based photocatalysis, our focus in this review is to bring attention to emerging polymer-based photoelectrodes for PEC reactions, which generates reduction and oxidation products separately, thus avoiding the back reaction and mitigating the risk of explosion in situ.
The emerging polymers including CNx, COF, MOF, P3HT, etc. have shown noticeable photocurrent density for either the reduction or oxidation reaction under one sun irradiation. A few strategies have been reviewed to improve the photocurrent density, i.e., the intimate contact between the polymer and the substrate, appropriate defects engineering, microstructure, doping, and junction structure. Furthermore, the key role of the molecular/nanoparticle cocatalyst has been highlighted in conjunction with the preparation methods, including both activity improvement and stability enhancement. Among these, engineering BHJ has been considered as one of the key strategies to enhance the photocurrent density.147
Engineering the polymer BHJ, especially using P3HT:PCBM (donor:acceptor) and sandwiching it between electron and hole acceptor layers, has been considered an efficient way to increase the photocurrent density of the photocathode for the reduction half-reaction. For instance, a benchmark photocathodic current density of 8 mA/cm2 at 0 V vs RHE was achieved using the P3HT:PCBM BHJ architecture positioned between the CuI hole selective layer and the Pt-decorated TiO2 electron acceptor layer.148 A similar approach was used to engineer the photoanode as well using benzodithiophene-based polymer PBDTTTPD as the electron donor and naphthalenediimide-based polymer PNDITCVT as the electron acceptor, which achieved a high water oxidation photocurrent density of 2 mA/cm2 at 1.23 V vs RHE, in which mesoporous ZnO was used as the electron transport layer and Co3O4 as the water oxidation cocatalyst.118 Furthermore, a BHJ between p-type polymer PBDB-T and n-type polymer ITIC showed the record oxidation photocurrent density of 15 mA/cm2 at 1.23 V vs RHE, in the presence of oxidation cocatalyst NiFe-LDHs.116
It must be noted that with the exception of the above system, the general performance of BHJ photoanodes lags behind that of BHJ photocathodes.317 This is owed to the fact that polymers are usually n-type semiconductors, thus having favorable energetic alignment to match the electrochemical potential needed for efficient HER other than OER. Also, the high kinetic overpotential involved in OER372 further disfavors polymer-based semiconductors to meet the electrochemical potential requirement for efficient OER. The HOMO level position of polymer-based semiconductors is not positive enough to generate the needed thermodynamic driving force for water oxidation compared with the best performing metal oxide inorganic semiconductors. Typically, the composition of the VB maximum of metal oxide semiconductors is dominated by O 2p orbitals.81 The orbitals that dominate the HOMO of polymer-based semiconductors, analogous to the VB of metal oxide semiconductors, usually vary depending on the structure of the polymer. For instance, based on density functional theory (DFT) calculation, it has been established that the HOMO of heptazine which is the building block of CNx is localized on those N atoms in the heptazine ring that are directly bonded to two carbon atoms (N2C).373 This suggests that the HOMO of CNx comprises N 2p orbitals which are positioned at a more negative electronic potential than the O 2p orbitals.374 Thus, the O 2p orbitals in metal oxide semiconductors are positioned at a more positive electronic potential which readily favors water oxidation, as opposed to polymer-based semiconductors. Nonetheless, lateral bay extension of rylene diimides can lower the HOMO (shift to more positive potentials)375 sufficiently to thermodynamically allow water oxidation, as exemplified by the recently reported chlorinated rylene diimide-based acceptors.376 These acceptors were used in BHJ photoanodes with thiophenedicarboximide-benzodithiophene (TPD-BDT) donors and notably sustained PEC performance over 3 h under sacrificial electron donor conditions. Although all these aforementioned benchmarks demonstrate the significance of junctions and synergy between junctions and cocatalysts for PEC fuel synthesis, the challenge for the optimization of the general performance of polymer-based photoelectrodes, especially photoanodes, cannot be overemphasized.
Although many organic or polymer semiconductors have been widely used in organic solar cells and light-emitting diodes, few were used in PEC for solar fuel production. One major reason is the poor contact between the polymer semiconductors and the aqueous electrolyte377 at the interface or bad hydrophilicity of the polymer surface, which makes the reactant adsorption and charge injection into the reactant molecule challenging. The deposition of a layer of hydrophilic functional group would help to sort out this issue; however, it might screen the light absorption. In some cases, organic molecular dyes have been used as light-harvesting materials, which is further coated with hole and electron separation layers of inorganic metal oxides. In the view of energy levels, only limited polymer semiconductors have either suitable LUMO levels to drive the reduction reaction at the water interface or appropriate HOMO levels to produce high photocurrent.378 The next major reason is the short lifetime of the excitons in the polymer semiconductors, leading to a poor charge carrier separation and diffusion in the polymers.379 To increase the degree of polymer crystallinity improves the charge carrier separation, such as by controlled annealing in different atmosphere.71 Moreover, there are many steps involved during the PEC process, such as adsorption of water and protons, charge injection, bond breaking, intermediate formation, and desorption of the products, hence the mechanistic investigation is crucial but these are hardly investigated for organic polymer semiconductors as we discussed in Section 5. Another important limiting factor is the poor stability of the polymer semiconductors in aqueous solution under the strong light irradiation condition.
Polymer-based photoelectrodes have advantages in terms of cost, earth-abundance, ease of synthesis, etc. However, the PEC performance of polymer-based photoelectrodes, except the P3HT and PBDB-T-based ones listed above, is mainly 10 times smaller than their inorganic counterparts. There are many reasons demonstrated for not being competitive to the inorganic semiconductor-based photoelectrodes at present, including poor hydrophilicity and stability, moderate efficiency, a limited number of polymer semiconductors, and poor understanding of charge separation.58,380 However, demonstrations such as those by Comas Rojas et al.148 and Bourgeteau et al.145 have established that the photocurrent obtained by BHJ polymer-based photocathodes can be potentially comparable to that of their inorganic counterparts. Therefore, with the urgent need for an efficient and low-cost photoelectrode and taking into account the very recent fast development of polymer photoelectrodes, herein we would like to highlight several strategies to improve the PEC performance of the polymer-based photoelectrodes for solar fuel synthesis and elaborate on challenges remaining in the field that impede progress.
First tuning the bandgap by, for example, appropriate doping, modifying the polymer structure, and forming a junction is important to enhance light harvesting. Then the polymer photoelectrode and electrolyte interface has to be optimized by experimental and theoretical approaches, as it is crucial to determine the charge injection to the adsorbate for transforming small molecules into useful fuels and high-value chemicals. The thickness of the polymer film plays a crucial role as the charges in most of the polymers have a short diffusion length (between nanometer and micrometer). While a thick film can maximize light absorption, the desired thickness should balance the light absorption and charge diffusion, which can be achieved by optimizing the concentration/amount of precursors, and polymerization time during synthesis.
Next, the resistance of charge transfer has to be reduced. To achieve this, the contact between a substrate and the film must be improved, such as by depositing the film using appropriate pretreatment and deposition techniques. The PEC performance of the photoelectrodes also depends on the nature of the film (e.g., dense and degree of crystallinity) and the structure of the polymer, the film thus should be optimized by the film deposition conditions. With respect to the surface reaction, engineering the different facets to gain more active sites and better charge separation are also essential. To increase the contact area of the polymer–electrolyte interface can improve the adsorption of small molecules and enhance the surface reaction, which can be achieved by preparing the nanoarchitecture of the polymer film and/or porous surface morphology. More importantly, the stability of the polymer-based photoelectrode can be prolonged by using a transparent low-cost protective coating while it should not inhibit the photoabsorption of the polymer. The suitable electrocatalyst or cocatalyst thin layer can also provide better protection to the photoactive polymer film besides catalyzing the surface reaction.
This last point on stability can be seen as the “elephant in the room” for polymer-based photoelectrodes. While efforts have been made in the design of systems with robust interfacial layers and protective strategies to improve photocurrent stability, current systems are still short of necessary operational times. There is a clear need to have photocurrent stability beyond hundreds of minutes to make systems viable.381 Presently, lifetime testing of PEC cells beyond 48 h have rarely been reported.382 Generally, the instability of polymer-based PEC systems arises from poor stability of polymer-based semiconductor materials when in contact with an aqueous electrolyte. The inherent partial permeability of organic polymers to water383 exposes them to dissolution and subsequent delamination during practical application. Haro et al.146 revealed that the common organic hole transport layer PEDOT:PSS in a polymer photocathode dissolved in the presence of an aqueous electrolyte, causing delamination of the organic active layer. Notably, all OER systems highlighted in this report exhibit deactivation by manifesting gradual photocurrent decay over relatively shorter time scales unlike their inorganic counterparts.384 Of note, most of these systems were those without stability control mechanisms in the form of an additional protective layer made up of inorganic materials.58 For instance, the best photoanode reported herein is ITO/PBDBT/ITIC/GaIn@Ni/NiFe-LDHs, which displayed a photocurrent of 15.1 mA/cm2 at 1.23 V vs RHE. A stability test performed at 1.3 V vs RHE for 10 h revealed that 90% of the initial photocurrent density are retained on the passivated photoactive layer, whereas the same photoactive layer without passivation showed poor activity within a few minutes.116 This result highlights the benefits of a passivation layer, which appears to be necessary for protection of at least some of the polymer materials used.
It is without doubt that data on the lifetime of an electrode would support reliable technoeconomic analyses for PEC systems in the context of realistic lifetime projections. The PEC cell life span in addition to the solar-to-hydrogen (STH) efficiency has been shown to be an integral parameter in the life cycle energy balance of a PEC system.385 It is therefore necessary to develop standard lifetime testing of PEC cells to serve as a reference point in assessing the different organic and inorganic based prototypes that are being developed.50 While the utilization of protective coatings can be effective, it is essential to acknowledge that the current materials which are corrosion-sensitive may make large-scale production challenging since any irregularity in the protective layer would amount to increased production cost as a result of elevated failure rates and, hence, a compromise on the economic viability of the production technique.58 As has been established in Section 2 of this report, the fabrication process of the thin films dictates their microstructural properties which in turn influence their performance. It is required that the thin films exhibit an intimate contact with the conducting substrate for efficient charge transfer and better photoelectrode performance. If the contact between the photoelectrode and the conducting substrate is not strong, there is the likelihood of catalyst detachment during practical application, which is detrimental to photoelectrode response.86 Several strategies including a solvothermal method have been used to obtain intimate contact between the photoelectrode and the conducting substrate. The less durable physisorption approach386 of immobilizing molecular catalysts onto the photoabsorber has now been replaced with a chemical immobilization strategy which proceeds via strong anchor groups such as −COOH387 and PO3H2388 to ensure that the catalyst is covalently bound to the electrode surface.389 However, there are still reports of photocurrent instability assigned to catalyst detachment.390,391 It is unclear whether this is occasioned by interfacial behavior during practical application since information on photocurrent degradation in relation to the structure of the photoelectrode/catalyst is elusive in the field. Without knowledge of the photocurrent degradation mechanism, targeted improvements cannot be made. For example, it has been determined that the photocurrent degradation exhibited by solution-processed organohalide perovskite solar cells during practical application is an effect of the formation of light-activated metastable deep-level trap states.392 This limitation can immediately be addressed by operating the device at a lower temperature (0 °C) since photocurrent degradation normally exhibits a steep temperature-dependence.392 2D perovskites structures have also been used to greatly enhance the stability of typical 3D perovskites393 by inhibiting moisture-induced degradation at grain boundaries394 and improving crystallinity to reduce the number of active defects that can lead to degradation.395 Similar insights into the mechanism of photocurrent degradation of polymer photoelectrodes are needed to move the field closer to viable devices.
The application of polymer photoelectrodes as photoabsorbers for H2 generation by splitting water can be as efficient as the existing fossil fuel-based technologies for large-scale H2 production.396 However, the development of polymer-based photoelectrodes for large-scale H2 production and subsequent commercialization is still at the early stage. The most important factor to an immediate commercialization of this technology is the knowledge of the overall STH efficiency since the higher the STH efficiency, the better the energy return commensurable to their commercialization. Other important factors including the energy used to fabricate the PEC cell, the lifespan of the PEC cell relative to the energy payback time, and the cost of the energy output must be assessed. Today, the base-case conditions for full commercialization of PEC technology for H2 production include a 10-year lifetime for photoelectrodes with 10% STH and within a levelized cost range of 2–4 USD per kg of H2 as mentioned before.54 It must be noted that the new cost targets by 2031 are lower than the previous ones as brought forward by the US Department of Energy Hydrogen Shot, part of their Energy Earthshots program. For an inorganic semiconductor-based PEC cell with STH efficiency of 20%, it is estimated that the time span within which the cell can produce energy equivalent to its manufacturing energy (energy payback time) is 3 years.397 Even though polymer-based photoelectrodes can be carefully optimized to increase STH efficiency, it is unclear if the current optimization strategies may incur further cost and/or compromise the lifespan of the resulting PEC cells. Also, the commercial deployment of polymer-based photoelectrodes can only be an option in the foreseeable future if they can be appropriately tuned to deliver a stable performance over the extended lifetime of the resulting PEC device.50 The techno-economic analysis published very recently398 concluded that based on the levelized cost of hydrogen (LCOH2), PEC systems were not cost-competitive with existing alternatives on the market. However, it is important to note that the PEC system considered utilized an inorganic semiconductor photoactive layer (c-Si) which is usually cost-intensive. The PEC module cost for a Si-based system with 10% STH efficiency was estimated at 154 USD/m2. While the fabrication process cost was not detailed, the price of the c-Si light absorber was 31% (48 USD/m2) of the module cost.398 Another techno-economic analysis based on inorganic PEC to produce H2 determined that the PEC cell was ∼80% of the total project cost.399 The LCOH2 was at best ∼2 USD/kg, but the minimum cost of the PEC cell was 700 USD/m2. We are not aware of published techno-economic analyses of polymer-based PECs at this time but we can look at those of OPV to gain further insights. The cost of mass-produced OPV modules has been estimated to be in the vicinity of 10 USD/m2,400−402 much lower than the costs considered for the inorganic PECs. While a direct comparison cannot be made due to the different cell/module design (e.g., the additional need for membranes and cocatalysts for the PEC), it seems reasonable to expect that optimized organic PEC cells could cost <100 USD/m2 and lower the LCOH2 to the target of 1 USD/kg. More efforts should be channeled into the techno-economic analysis of PEC H2 production systems that utilizes polymer-based photoelectrode active layers and cost-effective cocatalysts.
Despite the appreciable amount of work over the past decades regarding the application of organic-based photoelectrodes for solar-driven reactions, only a few reports have presented comprehensive fundamental understanding, combining spectroscopic and electrochemical data that provide useful information on the behavior of the charge carriers generated in the photoelectrode and at the interface of photoelectrode and electrolyte. This is reflected in the rather limited number of papers we could identify that investigated the charge carrier dynamics in organic photoelectrodes. Section 5 is the comprehensive review of those we found. Notably, we did not identify any papers that describe the charge carrier dynamics of CO2 reduction by polymer PEC devices.
Regardless of the different mechanistic pathways assigned to the reduction reactions in organic and inorganic systems, it is incontrovertible that these reactions are highly selective to specific chemical sites/species within the system.403 For example, for metal-oxide inorganic photoelectrodes, HER proceeds via a homolytic or heterolytic path involving a metal hydride intermediate. In contrast, the HER mechanism on organic-based photoelectrodes tends not to be as straightforward as metal-oxide photoelectrodes, which have a similar general surface structure. The possible active sites of the organic material are broad, varying upon polymer structures and presenting some difficulty in assigning a general mechanism to HER. The operando EC-Raman spectroscopic studies with carbon-based conjugated polymer show that the active sites/species involved in HER may not necessarily be metal-centered catalytic sites but rather excited state antibonding orbitals with localized electrons.308 The previous studies404,405 on metal-centered inorganic electrocatalysts revealed that the physical location of reactive species could extend beyond the atomic surface of the metal center to some degree into the bulk material. This suggests that the number of reactive species and their intrinsic activity determines the overall catalytic activity of the electrocatalyst. We think that this concept may hold for organic systems, in particular for porous polymer photoelectrodes, albeit it is yet to be demonstrated and reported.
In line with an outlook on the development of cutting-edge techniques toward the engineering of highly efficient next-generation organic-based or polymer PEC devices, we propose 5 avenues to focus on. (1) There is a need for remarkable spectroscopic and electrochemical-based operando studies to fully describe the behavior of photogenerated charge carriers and their corresponding impact in a real device. This will deepen our understanding of the function of each component and provide effective strategies for photoelectrodes design. (2) Extend the study of charge carrier dynamics of organic-based PEC devices to other photoreactions, e.g., CO2 reduction or N2 reduction to fully understand the role of interfacial charge transfer and to take advantage of the tunability of organic materials. (3) Advancement of studies into charge carrier dynamics of PEC systems built from π-conjugated organic or polymer materials other than CNx and thiophene. As has been demonstrated with continued efforts in the understanding of inorganic photoelectrodes and organic PV systems, studying a breadth of materials will allow us to develop general design guidelines that link the effects of material design strategies with the performance of organic-based photoelectrodes. (4) Modeling is a powerful tool to screen photoelectrode and cocatalyst candidates, which not only save researchers time but also avoid wasting raw materials. (5) A promising technique toward the acceleration of polymer materials discovery is high-throughput experiments combined with artificial intelligence (AI).406−410 The increased rate at which materials can be tested and discovered holds tangible prospects in revolutionizing scientific discovery, with key impacts already being made in material science.411 The fast development of the discovery of polymer photocatalysts provides a large database. Utilizing such a large database, AI412 can also predict the stability issues such as due to the Ostwald ripening, particle migration, and coalescence of the photocatalyst or cocatalyst nanoparticles during the reaction, which will save the time for trial-and-error experimentation and enable lab-to-fab translation.
Finally, an important note for this society is that apart from the measurement of photocurrent density of polymer-based photoelectrodes, faradaic efficiency, IPCE, and STH should be reported in parallel in order to rationalize and fairly compare the performance of different photoelectrodes. On the basis of these, we believe that the performance of organic photocathodes is very promising and would gradually advance to levels, which are superior to, or at least comparable to their inorganic counterparts while by a very low-cost process.
Acknowledgments
M.T., Q.R., and J.T. acknowledge funding from UK EPSRC (EP/S018204/2), Leverhulme Trust (RPG-2017-122), and Royal Society Leverhulme Trust Senior Research Fellowship (SRF\R1\21000153). R.G. acknowledges the support of the NSERC and the Canada Foundation for Innovation (CFI) for operational and infrastructure support. Chaoqi Ai and Xinyu Ma are thanked for their valuable discussion on cocatalysts. J.T. and D.J. both thank the financial support from the Royal Society Newton Advanced Fellowship grant (NAF\R1\191163).
Biographies
Madasamy Thangamuthu is a senior postdoctoral fellow in the Department of Chemical Engineering at UCL. He attained his Ph.D. degree in electrochemistry in 2014 at Madurai Kamaraj University, Madurai, India. Then, he moved to the Swiss Federal Institute of Technology Lausanne (EPFL), Switzerland, for a postdoctoral research project through the Swiss government excellence scholarship to work on plasmonic heterogeneous photocatalysis including ammonia synthesis. In 2019, he joined Prof. Junwang Tang's Group at the Department of Chemical Engineering, UCL. His current research focuses on photocatalytic Z-scheme water splitting and decoupled water splitting.
Qiushi Ruan received his bachelor’s degree in 2014 at Shanghai Jiaotong University. He received his Ph.D. degree in 2020 at University College London under the supervision of Prof. Junwang Tang. Qiushi is currently working as a lecturer in Southeast University, China. His research focuses on polymer photoelectrochemical water splitting and photochemical metal–air batteries.
Peter Osei Ohemeng was born in Akim Oda, Eastern Region, Ghana. He received his Chemistry bachelor's degree in 2016 at the University of Ghana, Legon. He proceeded to obtain his master's degree in 2019 at the same university where he studied under the supervision of Dr. Enock Dankyi and Dr. Vitus Apalangya. Peter is currently a Ph.D. student under the supervision of Dr. Robert Godin at the Okanagan campus of UBC in Kelowna, British Columbia, Canada. His research focuses on the functionalization of carbon nitride as a means of ameliorating its photocatalytic performance toward the remediation of inorganic metal oxyanions in water.
Bing Luo is an Assistant Professor of the School of Chemical Engineering and Technology, Xi’an Jiaotong University. He received his B.S. degree in Optical Physics in 2013 and Ph.D. degree in Engineering Thermo-physics in 2019 at Xi’an Jiaotong University. After taking a visiting scholar at the University of Quebec, Canada, he started his career as a faculty member at Xi’an Jiaotong University in 2020. His research interests involve engineering of nanomaterials and surface plasmon polaritons and their applications in solar energy conversion, tip-enhanced Raman spectroscopy, and plasmonic photography.
Dengwei Jing obtained his Ph.D. degree from Xi’an Jiaotong University and is now a full Professor of the same university. His interest of study is now on energy conversion and utilization. He is the winner of the National Natural Science Foundation for Distinguished Young Scholars, the second prize winner of the National Natural Science Award of China, and the Newton Advanced Fellowship of Royal Society. He is the chief scientist of the China National Key R&D program and serves as an editorial board member of several well-known international journals. He has published more than 130 SCI-indexed papers and 2 English books and authored over 30 patents.
Robert Godin was born in Bathurst, New Brunswick, Canada. He has fostered an interest in photochemistry since the start of his higher education career. He first got introduced to photochemistry and spectroscopy techniques working with Prof. Tito Scaiano during his B.Sc. at the University of Ottawa. He continued to learn advanced optical techniques with Prof. Gonzalo Cosa during his Ph.D. at McGill University. Robert then completed a postdoctoral stint from 2015 to 2018 with Prof. James Durrant at Imperial College London and entered the field of solar energy conversion. In 2018, he established his independent group at the Okanagan campus of UBC in Kelowna, British Columbia, Canada. The group develops time-resolved spectroscopic tools to better understand carbon-based semiconductors for sustainable energy production, with a healthy dose of physical organic chemistry concepts.
Junwang Tang is the Member of Academia Europae, Fellow of RSC, and Professor of Chemistry and Material Engineering in the Department of Chemical Engineering at UCL. His research interests encompass photocatalytic small-molecule activation (e.g., CH4, N2, H2O, C6H6, and CO2) and microwave catalysis (e.g., catalytic plastic recycling), together with the investigation of the underlying charge dynamics and kinetics by state-of-the-art spectroscopies. In parallel, he also explores the design of the chemical reactors for the above-mentioned processes. Prof. Tang has also received many awards, the latest of which include the 2021 IChemE Andrew Medal, 2021 RSC Corday-Morgan Prize, and 2021 IChemE Innovative Product Award, in addition to the Royal Society Leverhulme Trust Senior Research Fellow 2021. He also sits on the Editorial Board of four international journals, e.g., the Editor of Applied Catalysis B, Associate Editor of Chin. J. Catal. and Asia-Pacific Journal of Chemical Engineering, and also sits on the Advisory Board of 7 other journals.
The authors declare no competing financial interest.
References
- Hall D. O.; Rao K.. Photosynthesis; Cambridge University Press, 1999. [Google Scholar]
- Ahmad T.; Zhang D. A Critical Review of Comparative Global Historical Energy Consumption and Future Demand: The Story Told so Far. Energy Rep. 2020, 6, 1973–1991. 10.1016/j.egyr.2020.07.020. [DOI] [Google Scholar]
- Saunders N.; Chapman S.. Fossil Fuel; Raintree, 2005. [Google Scholar]
- Lewis N. S. Research Opportunities to Advance Solar Energy Utilization. Science 2016, 351 (6271), aad1920. 10.1126/science.aad1920. [DOI] [PubMed] [Google Scholar]
- Global Energy Transformation: A Roadmap to 2050; IRENA, 2019.
- Hagfeldt A.; Boschloo G.; Sun L.; Kloo L.; Pettersson H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110 (11), 6595–6663. 10.1021/cr900356p. [DOI] [PubMed] [Google Scholar]
- Blanco H.; Faaij A. A Review at the Role of Storage in Energy Systems with a Focus on Power to Gas and Long-Term Storage. Renew. Sustain. Energy Rev. 2018, 81, 1049–1086. 10.1016/j.rser.2017.07.062. [DOI] [Google Scholar]
- Ball M.; Wietschel M.. The Hydrogen Economy: Opportunities and Challenges; Cambridge University Press, 2009. [Google Scholar]
- Lubitz W.; Tumas W. Hydrogen: An Overview. Chem. Rev. 2007, 107 (10), 3900–3903. 10.1021/cr050200z. [DOI] [PubMed] [Google Scholar]
- Nazir H.; Muthuswamy N.; Louis C.; Jose S.; Prakash J.; Buan M. E. M.; Flox C.; Chavan S.; Shi X.; Kauranen P.; Kallio T.; Maia G.; Tammeveski K.; Lymperopoulos N.; Carcadea E.; Veziroglu E.; Iranzo A.; Kannan A. M. Is the H2 Economy Realizable in the Foreseeable Future? Part III: H2 Usage Technologies, Applications, and Challenges and Opportunities. Int. J. Hydrog. Energy 2020, 45 (53), 28217–28239. 10.1016/j.ijhydene.2020.07.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Zhu Q.; Chen C.; Liu H.; Liu Z.; Zhao Z.; Zhang X.; Liu S.; Han B. Selective Electroreduction of Carbon Dioxide to Methanol on Copper Selenide Nanocatalysts. Nat. Commun. 2019, 10 (1), 677. 10.1038/s41467-019-08653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albo J.; Alvarez-Guerra M.; Castaño P.; Irabien A. Towards the Electrochemical Conversion of Carbon Dioxide into Methanol. Green Chem. 2015, 17 (4), 2304–2324. 10.1039/C4GC02453B. [DOI] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E. L.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119 (12), 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Guo S.-X.; Gandionco K. A.; Bond A. M.; Zhang J. Electrocatalytic Carbon Dioxide Reduction: From Fundamental Principles to Catalyst Design. Mater. Today Adv. 2020, 7, 100074. 10.1016/j.mtadv.2020.100074. [DOI] [Google Scholar]
- Liu A.; Gao M.; Ren X.; Meng F.; Yang Y.; Gao L.; Yang Q.; Ma T. Current Progress in Electrocatalytic Carbon Dioxide Reduction to Fuels on Heterogeneous Catalysts. J. Mater. Chem. A 2020, 8 (7), 3541–3562. 10.1039/C9TA11966C. [DOI] [Google Scholar]
- Zhu J.; Hu L.; Zhao P.; Lee L. Y. S.; Wong K.-Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120 (2), 851–918. 10.1021/acs.chemrev.9b00248. [DOI] [PubMed] [Google Scholar]
- Kalinci Y.; Hepbasli A.; Dincer I. Biomass-Based Hydrogen Production: A Review and Analysis. Int. J. Hydrog. Energy 2009, 34 (21), 8799–8817. 10.1016/j.ijhydene.2009.08.078. [DOI] [Google Scholar]
- Penconi M.; Rossi F.; Ortica F.; Elisei F.; Gentili P. L. Hydrogen Production from Water by Photolysis, Sonolysis and Sonophotolysis with Solid Solutions of Rare Earth, Gallium and Indium Oxides as Heterogeneous Catalysts. Sustainability 2015, 7, 9310. 10.3390/su7079310. [DOI] [Google Scholar]
- Schrauzer G. N.; Guth T. D. Photolysis of Water and Photoreduction of Nitrogen on Titanium Dioxide. J. Am. Chem. Soc. 1977, 99 (22), 7189–7193. 10.1021/ja00464a015. [DOI] [Google Scholar]
- Tee S. Y.; Win K. Y.; Teo W. S.; Koh L.-D.; Liu S.; Teng C. P.; Han M.-Y. Recent Progress in Energy-Driven Water Splitting. Adv. Sci. 2017, 4 (5), 1600337. 10.1002/advs.201600337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Domen K. Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies. Chem. Rev. 2020, 120 (2), 919–985. 10.1021/acs.chemrev.9b00201. [DOI] [PubMed] [Google Scholar]
- Walsh J. J.; Jiang C.; Tang J.; Cowan A. J. Photochemical CO2 reduction using structurally controlled g-C3N4. Phys. Chem. Chem. Phys. 2016, 18 (36), 24825–24829. 10.1039/C6CP04525A. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Suzuki H.; Xie J.; Tomita O.; Martin D. J.; Higashi M.; Kong D.; Abe R.; Tang J. Mimicking Natural Photosynthesis: Solar to Renewable H2 Fuel Synthesis by Z-Scheme Water Splitting Systems. Chem. Rev. 2018, 118 (10), 5201–5241. 10.1021/acs.chemrev.7b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X.; Song M.; Wang H.; Zhang X.; Sui N.; Zhang Q.; Colvin V. L.; Yu W. W. Latest Progress in Constructing Solid-State Z Scheme Photocatalysts for Water Splitting. Nanoscale 2019, 11 (23), 11071–11082. 10.1039/C9NR03218E. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Zhang L.; Yu J.; Wageh S.; Al-Ghamdi A. A.; Jaroniec M. Direct Z-Scheme Photocatalysts: Principles, Synthesis, and Applications. Mater. Today 2018, 21 (10), 1042–1063. 10.1016/j.mattod.2018.04.008. [DOI] [Google Scholar]
- Zhang W.; Mohamed A. R.; Ong W.-J. Z-Scheme Photocatalytic Systems for Carbon Dioxide Reduction: Where Are We Now?. Angew. Chem., Int. Ed. 2020, 59 (51), 22894–22915. 10.1002/anie.201914925. [DOI] [PubMed] [Google Scholar]
- Hisatomi T.; Domen K. Reaction Systems for Solar Hydrogen Production via Water Splitting with Particulate Semiconductor Photocatalysts. Nat. Catal. 2019, 2 (5), 387–399. 10.1038/s41929-019-0242-6. [DOI] [Google Scholar]
- Low J.; Jiang C.; Cheng B.; Wageh S.; Al-Ghamdi A. A.; Yu J. A Review of Direct Z-Scheme Photocatalysts. Small Methods 2017, 1 (5), 1700080. 10.1002/smtd.201700080. [DOI] [Google Scholar]
- Wang Y.; Shang X.; Shen J.; Zhang Z.; Wang D.; Lin J.; Wu J. C. S.; Fu X.; Wang X.; Li C. Direct and Indirect Z-Scheme Heterostructure-Coupled Photosystem Enabling Cooperation of CO2 Reduction and H2O Oxidation. Nat. Commun. 2020, 11 (1), 3043. 10.1038/s41467-020-16742-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng B.-J.; Putri L. K.; Kong X. Y.; Teh Y. W.; Pasbakhsh P.; Chai S.-P. Z-Scheme Photocatalytic Systems for Solar Water Splitting. Adv. Sci. 2020, 7 (7), 1903171. 10.1002/advs.201903171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalanoor B. S.; Seo H.; Kalanur S. S. Recent Developments in Photoelectrochemical Water-Splitting Using WO3/BiVO4 Heterojunction Photoanode: A Review. Mater. Sci. Energy Technol. 2018, 1 (1), 49–62. 10.1016/j.mset.2018.03.004. [DOI] [Google Scholar]
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q.; Santori E. A.; Lewis N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110 (11), 6446–6473. 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Ye K.-H.; Li H.; Huang D.; Xiao S.; Qiu W.; Li M.; Hu Y.; Mai W.; Ji H.; Yang S. Enhancing Photoelectrochemical Water Splitting by Combining Work Function Tuning and Heterojunction Engineering. Nat. Commun. 2019, 10 (1), 3687. 10.1038/s41467-019-11586-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara G.; Kumagai H.; Maeda K.; Kaeffer N.; Artero V.; Higashi M.; Abe R.; Ishitani O. Photoelectrochemical Reduction of CO2 Coupled to Water Oxidation Using a Photocathode with a Ru(II)-Re(I) Complex Photocatalyst and a CoOx/TaON Photoanode. J. Am. Chem. Soc. 2016, 138 (42), 14152–14158. 10.1021/jacs.6b09212. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Cheng J.; Xuan X.; Zhou J.; Cen K. CO2 Synergistic Reduction in a Photoanode-Driven Photoelectrochemical Cell with a Pt-Modified TiO2 Nanotube Photoanode and a Pt Reduced Graphene Oxide Electrocathode. ACS Sustain. Chem. Eng. 2016, 4 (12), 6344–6354. 10.1021/acssuschemeng.6b00909. [DOI] [Google Scholar]
- Zhao J.; Wang X.; Xu Z.; Loo J. S. C. Hybrid Catalysts for Photoelectrochemical Reduction of Carbon Dioxide: A Prospective Review on Semiconductor/Metal Complex Co-Catalyst Systems. J. Mater. Chem. A 2014, 2 (37), 15228–15233. 10.1039/C4TA02250E. [DOI] [Google Scholar]
- Wang Y.; He D.; Chen H.; Wang D. Catalysts in Electro-, Photo- and Photoelectrocatalytic CO2 Reduction Reactions. J. Photochem. Photobiol. C Photochem. Rev. 2019, 40, 117–149. 10.1016/j.jphotochemrev.2019.02.002. [DOI] [Google Scholar]
- Tuller H. L. Solar to Fuels Conversion Technologies: A Perspective. Mater. Renew. Sustain. Energy 2017, 6 (1), 3. 10.1007/s40243-017-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle K. E.; Warnan J.; Leung J. J.; Reuillard B.; Karmel I. S.; Reisner E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119 (4), 2752–2875. 10.1021/acs.chemrev.8b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Fan G.; Fu H.; Li Z.; Zou Z. Tandem Photoelectrochemical Cells for Solar Water Splitting. Adv. Phys. X 2018, 3 (1), 1487267. 10.1016/j.mset.2018.03.004. [DOI] [Google Scholar]
- Kuk S. K.; Ham Y.; Gopinath K.; Boonmongkolras P.; Lee Y.; Lee Y. W.; Kondaveeti S.; Ahn C.; Shin B.; Lee J.-K.; Jeon S.; Park C. B. Continuous 3D Titanium Nitride Nanoshell Structure for Solar-Driven Unbiased Biocatalytic CO2 Reduction. Adv. Energy Mater. 2019, 9 (25), 1900029. 10.1002/aenm.201900029. [DOI] [Google Scholar]
- Chen Y.; Feng X.; Liu Y.; Guan X.; Burda C.; Guo L. Metal Oxide-Based Tandem Cells for Self-Biased Photoelectrochemical Water Splitting. ACS Energy Lett. 2020, 5 (3), 844–866. 10.1021/acsenergylett.9b02620. [DOI] [Google Scholar]
- Sivula K. Metal Oxide Photoelectrodes for Solar Fuel Production, Surface Traps, and Catalysis. J. Phys. Chem. Lett. 2013, 4 (10), 1624–1633. 10.1021/jz4002983. [DOI] [PubMed] [Google Scholar]
- Nowotny J.; Bak T.; Nowotny M. K.; Sheppard L. R. Titanium Dioxide for Solar-Hydrogen I. Functional Properties. Int. J. Hydrog. Energy 2007, 32 (14), 2609–2629. 10.1016/j.ijhydene.2006.09.004. [DOI] [Google Scholar]
- Sivula K.; van de Krol R. Semiconducting Materials for Photoelectrochemical Energy Conversion. Nat. Rev. Mater. 2016, 1 (2), 15010. 10.1038/natrevmats.2015.10. [DOI] [Google Scholar]
- Moss B.; Babacan O.; Kafizas A.; Hankin A. A Review of Inorganic Photoelectrode Developments and Reactor Scale-Up Challenges for Solar Hydrogen Production. Adv. Energy Mater. 2021, 11 (13), 2003286. 10.1002/aenm.202003286. [DOI] [Google Scholar]
- Gopinath C. S.; Nalajala N. A Scalable and Thin Film Approach for Solar Hydrogen Generation: A Review on Enhanced Photocatalytic Water Splitting. J. Mater. Chem. A 2021, 9 (3), 1353–1371. 10.1039/D0TA09619A. [DOI] [Google Scholar]
- Ahmadi S.; Asim N.; Alghoul M. A.; Hammadi F. Y.; Saeedfar K.; Ludin N. A.; Zaidi S. H.; Sopian K. The Role of Physical Techniques on the Preparation of Photoanodes for Dye Sensitized Solar Cells. Int. J. Photoenergy 2014, 2014, 198734. 10.1155/2014/198734. [DOI] [Google Scholar]
- Banerjee T.; Podjaski F.; Kröger J.; Biswal B. P.; Lotsch B. V. Polymer Photocatalysts for Solar-to-Chemical Energy Conversion. Nat. Rev. Mater. 2021, 6 (2), 168–190. 10.1038/s41578-020-00254-z. [DOI] [Google Scholar]
- Pinaud B. A.; Benck J. D.; Seitz L. C.; Forman A. J.; Chen Z.; Deutsch T. G.; James B. D.; Baum K. N.; Baum G. N.; Ardo S.; Wang H.; Miller E.; Jaramillo T. F. Technical and Economic Feasibility of Centralized Facilities for Solar Hydrogen Production via Photocatalysis and Photoelectrochemistry. Energy Environ. Sci. 2013, 6 (7), 1983–2002. 10.1039/c3ee40831k. [DOI] [Google Scholar]
- Moniz S. J. A.; Shevlin S. A.; Martin D.; Guo Z. X.; Tang J. Visible-Light Driven Heterojunction Photocatalysts for Water Splitting- A Critical Review. Energy Environ. Sci. 2015, 8 (3), 731–759. 10.1039/C4EE03271C. [DOI] [Google Scholar]
- Bae D.; Seger B.; Vesborg P. C. K.; Hansen O.; Chorkendorff I. Strategies for Stable Water Splitting via Protected Photoelectrodes. Chem. Soc. Rev. 2017, 46 (7), 1933–1954. 10.1039/C6CS00918B. [DOI] [PubMed] [Google Scholar]
- Wu H.-L.; Li X.-B.; Tung C.-H.; Wu L.-Z. Recent Advances in Sensitized Photocathodes: From Molecular Dyes to Semiconducting Quantum Dots. Adv. Sci. 2018, 5 (4), 1700684. 10.1002/advs.201700684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Moniz S. J. A.; Wang A.; Zhang T.; Tang J. Photoelectrochemical Devices for Solar Water Splitting - Materials and Challenges. Chem. Soc. Rev. 2017, 46 (15), 4645–4660. 10.1039/C6CS00306K. [DOI] [PubMed] [Google Scholar]
- Günes S.; Neugebauer H.; Sariciftci N. S. Conjugated Polymer-Based Organic Solar Cells. Chem. Rev. 2007, 107 (4), 1324–1338. 10.1021/cr050149z. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Vogel A.; Sachs M.; Sprick R. S.; Wilbraham L.; Moniz S. J. A.; Godin R.; Zwijnenburg M. A.; Durrant J. R.; Cooper A. I.; Tang J. Current understanding and challenges of solar-driven hydrogen generation using polymeric photocatalysts. Nat. Energy 2019, 4 (9), 746–760. 10.1038/s41560-019-0456-5. [DOI] [Google Scholar]
- Rand B. P.; Genoe J.; Heremans P.; Poortmans J. Solar Cells Utilizing Small Molecular Weight Organic Semiconductors. Prog. Photovolt. Res. Appl. 2007, 15 (8), 659–676. 10.1002/pip.788. [DOI] [Google Scholar]
- Steier L.; Holliday S. A Bright Outlook on Organic Photoelectrochemical Cells for Water Splitting. J. Mater. Chem. A 2018, 6 (44), 21809–21826. 10.1039/C8TA07036A. [DOI] [Google Scholar]
- Sprick R. S.; Little M. A.; Cooper A. I. Organic Heterojunctions for Direct Solar Fuel Generation. Commun. Chem. 2020, 3 (1), 40. 10.1038/s42004-020-0288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Wang D.; Han H.; Li C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46 (8), 1900–1909. 10.1021/ar300227e. [DOI] [PubMed] [Google Scholar]
- Martin D. J.; Qiu K.; Shevlin S. A.; Handoko A. D.; Chen X.; Guo Z.; Tang J. Highly Efficient H2 Evolution from Water under visible light by Structure-Controlled Graphitic Carbon Nitride. Angew. Chem., Int. Ed. 2014, 53 (35), 9240–9245. 10.1002/anie.201403375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa H.; Ikeda S.; Aizawa M.; Yoshitake J.; Suzuki S. Polyacetylene Film: A New Electrode Material for Photoenergy Conversion. Synth. Met. 1981, 4 (1), 43–49. 10.1016/0379-6779(81)90056-4. [DOI] [Google Scholar]
- Kaneko M.; Nakamura H. Photoresponse of a Liquid Junction Polyaniline Film. J. Chem. Soc. Chem. Commun. 1985, 1985, 346–347. 10.1039/c39850000346. [DOI] [Google Scholar]
- Kenmochi T.; Tsuchida E.; Kaneko M.; Yamada A. Photoelectrochemical Response of Liquid Junction Poly(Thienylene). Electrochim. Acta 1985, 30 (10), 1405–1406. 10.1016/0013-4686(85)85022-2. [DOI] [Google Scholar]
- Yanagida S.; Kabumoto A.; Mizumoto K.; Pac C.; Yoshino K. Poly(p-Phenylene)-Catalysed Photoreduction of Water to Hydrogen. J. Chem. Soc. Chem. Commun. 1985, 1985, 474–475. 10.1039/c39850000474. [DOI] [Google Scholar]
- Wang S.; Zhang J.; Li B.; Sun H.; Wang S. Engineered Graphitic Carbon Nitride-Based Photocatalysts for Visible-Light-Driven Water Splitting: A Review. Energy Fuels 2021, 35 (8), 6504–6526. 10.1021/acs.energyfuels.1c00503. [DOI] [Google Scholar]
- Huang X.; Zhang Y.-B. Covalent Organic Frameworks for Sunlight-Driven Hydrogen Evolution. Chem. Lett. 2021, 50 (4), 676–686. 10.1246/cl.200834. [DOI] [PubMed] [Google Scholar]
- Meyer K.; Ranocchiari M.; van Bokhoven J. A. Metal Organic Frameworks for Photo-Catalytic Water Splitting. Energy Environ. Sci. 2015, 8 (7), 1923–1937. 10.1039/C5EE00161G. [DOI] [Google Scholar]
- Hao W.; Chen R.; Zhang Y.; Wang Y.; Zhao Y. Triazine-Based Conjugated Microporous Polymers for Efficient Hydrogen Production. ACS Omega 2021, 6 (37), 23782–23787. 10.1021/acsomega.1c02592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carminati S. A.; Rodríguez-Gutiérrez I.; de Morais A.; da Silva B. L.; Melo M. A.; Souza F. L.; Nogueira A. F. Challenges and Prospects about the Graphene Role in the Design of Photoelectrodes for Sunlight-Driven Water Splitting. RSC Adv. 2021, 11 (24), 14374–14398. 10.1039/D0RA10176A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Hippalgaonkar K.; Sprick R. S. Organic Materials as Photocatalysts for Water Splitting. J. Mater. Chem. A 2021, 9 (30), 16222–16232. 10.1039/D1TA03710B. [DOI] [Google Scholar]
- Chang X.; Wang T.; Yang P.; Zhang G.; Gong J. The Development of Cocatalysts for Photoelectrochemical CO2 Reduction. Adv. Mater. 2019, 31 (31), 1804710. 10.1002/adma.201804710. [DOI] [PubMed] [Google Scholar]
- Jian J.; Kumar R.; Sun J. Cu2O/ZnO p-n Junction Decorated with NiOx as a Protective Layer and Cocatalyst for Enhanced Photoelectrochemical Water Splitting. ACS Appl. Energy Mater. 2020, 3 (11), 10408–10414. 10.1021/acsaem.0c01198. [DOI] [Google Scholar]
- Li X.; Yu J.; Jaroniec M.; Chen X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119 (6), 3962–4179. 10.1021/acs.chemrev.8b00400. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y.; Kofuji Y.; Kanazawa S.; Sakamoto H.; Ichikawa S.; Tanaka S.; Hirai T. Platinum Nanoparticles Strongly Associated with Graphitic Carbon Nitride as Efficient Co-Catalysts for Photocatalytic Hydrogen Evolution under Visible Light. Chem. Commun. 2014, 50 (96), 15255–15258. 10.1039/C4CC06960A. [DOI] [PubMed] [Google Scholar]
- Reichert R.; Jusys Z.; Behm R. J. Au/TiO2 Photo(Electro)Catalysis: The Role of the Au Cocatalyst in Photoelectrochemical Water Splitting and Photocatalytic H2 Evolution. J. Phys. Chem. C 2015, 119 (44), 24750–24759. 10.1021/acs.jpcc.5b08428. [DOI] [Google Scholar]
- Osaki J.; Yoda M.; Takashima T.; Irie H. Selective Loading of Platinum or Silver Cocatalyst onto a Hydrogen-Evolution Photocatalyst in a Silver-Mediated All Solid-State Z-Scheme System for Enhanced Overall Water Splitting. RSC Adv. 2019, 9 (71), 41913–41917. 10.1039/C9RA09421K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Wang X.; Liu J.; Wang K.; Jin S.; Tan B. Palladium as a Superior Cocatalyst to Platinum for Hydrogen Evolution Using Covalent Triazine Frameworks as a Support. ACS Appl. Mater. Interfaces 2020, 12 (11), 12774–12782. 10.1021/acsami.9b21903. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Ida S.; Hyodo J.; Hagiwara H.; Ishihara T. Synthesis and Photocatalytic Activity of Rhodium-Doped Calcium Niobate Nanosheets for Hydrogen Production from a Water/Methanol System without Cocatalyst Loading. J. Am. Chem. Soc. 2011, 133 (45), 18034–18037. 10.1021/ja207103j. [DOI] [PubMed] [Google Scholar]
- Bedin K. C.; Muche D. N. F.; Melo Jr M. A.; Freitas A. L. M.; Gonçalves R. V.; Souza F. L. Role of Cocatalysts on Hematite Photoanodes in Photoelectrocatalytic Water Splitting: Challenges and Future Perspectives. ChemCatChem. 2020, 12 (12), 3156–3169. 10.1002/cctc.202000143. [DOI] [Google Scholar]
- Corby S.; Rao R. R.; Steier L.; Durrant J. R. The Kinetics of Metal Oxide Photoanodes from Charge Generation to Catalysis. Nat. Rev. Mater. 2021, 6, 1136–1155. 10.1038/s41578-021-00343-7. [DOI] [Google Scholar]
- Augustyński J.; Alexander B. D.; Solarska R. Metal Oxide Photoanodes for Water Splitting. Top. Curr. Chem. 2011, 303, 1–38. 10.1007/128_2011_135. [DOI] [PubMed] [Google Scholar]
- Song-Can W.; Feng-Qiu T.; Lian-Zhou W. Visible Light Responsive Metal Oxide Photoanodes for Photoelectrochemical Water Splitting: A Comprehensive Review on Rational Materials Design. Journal of Inorganic Materials. 2018, 33 (2), 173–197. 10.15541/jim20170352. [DOI] [Google Scholar]
- Sima M.; Vasile E.; Sima A.; Preda N.; Logofatu C. Graphitic Carbon Nitride Based Photoanodes Prepared by Spray Coating Method. Int. J. Hydrog. Energy 2019, 44 (45), 24430–24440. 10.1016/j.ijhydene.2019.07.243. [DOI] [Google Scholar]
- Mohamed N. A.; Safaei J.; Ismail A. F.; Mohamad Noh M. F.; Arzaee N. A.; Mansor N. N.; Ibrahim M. A.; Ludin N. A.; Sagu J. S.; Mat Teridi M. A. Fabrication of Exfoliated Graphitic Carbon Nitride, (g-C3N4) Thin Film by Methanolic Dispersion. J. Alloys Compd. 2020, 818, 152916. 10.1016/j.jallcom.2019.152916. [DOI] [Google Scholar]
- Bian J.; Li Q.; Huang C.; Li J.; Guo Y.; Zaw M.; Zhang R.-Q. Thermal Vapor Condensation of Uniform Graphitic Carbon Nitride Films with Remarkable Photocurrent Density for Photoelectrochemical Applications. Nano Energy 2015, 15, 353–361. 10.1016/j.nanoen.2015.04.012. [DOI] [Google Scholar]
- Lv X.; Cao M.; Shi W.; Wang M.; Shen Y. A New Strategy of Preparing Uniform Graphitic Carbon Nitride Films for Photoelectrochemical Application. Carbon 2017, 117, 343–350. 10.1016/j.carbon.2017.02.096. [DOI] [Google Scholar]
- Gu Q.; Gong X.; Jia Q.; Liu J.; Gao Z.; Wang X.; Long J.; Xue C. Compact Carbon Nitride Based Copolymer Films with Controllable Thickness for Photoelectrochemical Water Splitting. J. Mater. Chem. A 2017, 5 (36), 19062–19071. 10.1039/C7TA06081E. [DOI] [Google Scholar]
- Seo Y. J.; Das P. K.; Arunachalam M.; Ahn K.-S.; Ha J.-S.; Kang S. H. Drawing the Distinguished Graphite Carbon Nitride (g-C3N4) on SnO2 Nanoflake Film for Solar Water Oxidation. Int. J. Hydrog. Energy 2020, 45 (43), 22567–22575. 10.1016/j.ijhydene.2020.06.143. [DOI] [Google Scholar]
- Jia Q.; Zhang S.; Gao Z.; Yang P.; Gu Q. In Situ Growth of Triazine-Heptazine Based Carbon Nitride Film for Efficient (Photo)Electrochemical Performance. Catal. Sci. Technol. 2019, 9 (2), 425–435. 10.1039/C8CY02105H. [DOI] [Google Scholar]
- Fang Y.; Li X.; Wang X. Synthesis of Polymeric Carbon Nitride Films with Adhesive Interfaces for Solar Water Splitting Devices. ACS Catal. 2018, 8 (9), 8774–8780. 10.1021/acscatal.8b02549. [DOI] [Google Scholar]
- Wang F.; Bai S.; Tress W.; Hagfeldt A.; Gao F. Defects Engineering for High-Performance Perovskite Solar Cells. Npj Flex. Electron. 2018, 2 (1), 22. 10.1038/s41528-018-0035-z. [DOI] [Google Scholar]
- Celik E.; Ma Y.; Brezesinski T.; Elm M. T. Ordered Mesoporous Metal Oxides for Electrochemical Applications: Correlation between Structure, Electrical Properties and Device Performance. Phys. Chem. Chem. Phys. 2021, 23 (18), 10706–10735. 10.1039/D1CP00834J. [DOI] [PubMed] [Google Scholar]
- Ruan Q.; Bayazit M. K.; Kiran V.; Xie J.; Wang Y.; Tang J. Key Factors Affecting Photoelectrochemical Performance of G-C3N4 Polymer Films. Chem. Commun. 2019, 55 (50), 7191–7194. 10.1039/C9CC03084K. [DOI] [PubMed] [Google Scholar]
- Peng G.; Albero J.; Garcia H.; Shalom M. A Water-Splitting Carbon Nitride Photoelectrochemical Cell with Efficient Charge Separation and Remarkably Low Onset Potential. Angew. Chem., Int. Ed. 2018, 57 (48), 15807–15811. 10.1002/anie.201810225. [DOI] [PubMed] [Google Scholar]
- Peng G.; Xing L.; Barrio J.; Volokh M.; Shalom M. A General Synthesis of Porous Carbon Nitride Films with Tunable Surface Area and Photophysical Properties. Angew. Chem., Int. Ed. 2018, 57 (5), 1186–1192. 10.1002/anie.201711669. [DOI] [PubMed] [Google Scholar]
- Peng G.; Volokh M.; Tzadikov J.; Sun J.; Shalom M. Carbon Nitride/Reduced Graphene Oxide Film with Enhanced Electron Diffusion Length: An Efficient Photo-Electrochemical Cell for Hydrogen Generation. Adv. Energy Mater. 2018, 8 (23), 1800566. 10.1002/aenm.201800566. [DOI] [Google Scholar]
- Qin J.; Barrio J.; Peng G.; Tzadikov J.; Abisdris L.; Volokh M.; Shalom M. Direct Growth of Uniform Carbon Nitride Layers with Extended Optical Absorption towards Efficient Water-Splitting Photoanodes. Nat. Commun. 2020, 11 (1), 4701. 10.1038/s41467-020-18535-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.-X.; Liang H.-P.; Anito D. A.; Ding X.; Han B.-H. Emerging Applications of Porous Organic Polymers in Visible-Light Photocatalysis. J. Mater. Chem. A 2020, 8 (15), 7003–7034. 10.1039/D0TA00364F. [DOI] [Google Scholar]
- Xiong W.; Chen S.; Huang M.; Wang Z.; Lu Z.; Zhang R.-Q. Crystal-Face Tailored Graphitic Carbon Nitride Films for High-Performance Photoelectrochemical Cells. ChemSusChem 2018, 11 (15), 2497–2501. 10.1002/cssc.201801295. [DOI] [PubMed] [Google Scholar]
- Huang M.; Zhao Y.-L.; Xiong W.; Kershaw S. V.; Yu Y.; Li W.; Dudka T.; Zhang R.-Q. Collaborative Enhancement of Photon Harvesting and Charge Carrier Dynamics in Carbon Nitride Photoelectrode. Appl. Catal. B Environ. 2018, 237, 783–790. 10.1016/j.apcatb.2018.06.046. [DOI] [Google Scholar]
- Zhang Y.; Mori T.; Ye J.; Antonietti M. Phosphorus-Doped Carbon Nitride Solid: Enhanced Electrical Conductivity and Photocurrent Generation. J. Am. Chem. Soc. 2010, 132 (18), 6294–6295. 10.1021/ja101749y. [DOI] [PubMed] [Google Scholar]
- Xu J.; Cao S.; Brenner T.; Yang X.; Yu J.; Antonietti M.; Shalom M. Supramolecular Chemistry in Molten Sulfur: Preorganization Effects Leading to Marked Enhancement of Carbon Nitride Photoelectrochemistry. Adv. Funct. Mater. 2015, 25 (39), 6265–6271. 10.1002/adfm.201502843. [DOI] [Google Scholar]
- Ruan Q.; Luo W.; Xie J.; Wang Y.; Liu X.; Bai Z.; Carmalt C. J.; Tang J. A Nanojunction Polymer Photoelectrode for Efficient Charge Transport and Separation. Angew. Chem., Int. Ed. 2017, 56 (28), 8221–8225. 10.1002/anie.201703372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjule N.; Barrio J.; Xing L.; Volokh M.; Shalom M. Highly Efficient Polymeric Carbon Nitride Photoanode with Excellent Electron Diffusion Length and Hole Extraction Properties. Nano Lett. 2020, 20 (6), 4618–4624. 10.1021/acs.nanolett.0c01484. [DOI] [PubMed] [Google Scholar]
- Rieke P. C.; Armstrong N. R. Light-Assisted, Aqueous Redox Reactions at Chlorogallium Phthalocyanine Thin-Film Photoconductors: Dependence of the Photopotential on the Formal Potential of the Redox Couple and Evidence for Photoassisted Hydrogen Evolution. J. Am. Chem. Soc. 1984, 106 (1), 47–50. 10.1021/ja00313a010. [DOI] [Google Scholar]
- Kirner J. T.; Stracke J. J.; Gregg B. A.; Finke R. G. Visible-Light-Assisted Photoelectrochemical Water Oxidation by Thin Films of a Phosphonate-Functionalized Perylene Diimide Plus CoOx Cocatalyst. ACS Appl. Mater. Interfaces 2014, 6 (16), 13367–13377. 10.1021/am405598w. [DOI] [PubMed] [Google Scholar]
- Bornoz P.; Prévot M. S.; Yu X.; Guijarro N.; Sivula K. Direct Light-Driven Water Oxidation by a Ladder-Type Conjugated Polymer Photoanode. J. Am. Chem. Soc. 2015, 137 (49), 15338–15341. 10.1021/jacs.5b05724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Wagner P.; Tong L.; Wallace G. G.; Officer D. L.; Swiegers G. F. A Porphyrin-Doped Polymer Catalyzes Selective, Light-Assisted Water Oxidation in Seawater. Angew. Chem., Int. Ed. 2012, 51 (8), 1907–1910. 10.1002/anie.201107355. [DOI] [PubMed] [Google Scholar]
- Barona-Castano J.; Carmona-Vargas C.; Brocksom T.; de Oliveira K. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21 (3), 310. 10.3390/molecules21030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Fan H.; Bai F. Porphyrin-Based Photocatalysts for Hydrogen Production. MRS Bull. 2020, 45 (1), 49–56. 10.1557/mrs.2019.294. [DOI] [Google Scholar]
- Naruta Y.; Sasayama M.; Sasaki T. Oxygen Evolution by Oxidation of Water with Manganese Porphyrin Dimers. Angew. Chem., Int. Ed. Engl. 1994, 33 (18), 1839–1841. 10.1002/anie.199418391. [DOI] [Google Scholar]
- Sandanayaka A. S. D.; Murakami T.; Hasobe T. Preparation and Photophysical and Photoelectrochemical Properties of Supramolecular Porphyrin Nanorods Structurally Controlled by Encapsulated Fullerene Derivatives. J. Phys. Chem. C 2009, 113 (42), 18369–18378. 10.1021/jp9063577. [DOI] [Google Scholar]
- Abe T.; Nagai K.; Kabutomori S.; Kaneko M.; Tajiri A.; Norimatsu T. An Organic Photoelectrode Working in the Water Phase: Visible-Light-Induced Dioxygen Evolution by a Perylene Derivative/Cobalt Phthalocyanine Bilayer. Angew. Chem., Int. Ed. 2006, 45 (17), 2778–2781. 10.1002/anie.200504454. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yan D.; Shaffer D. W.; Ye X.; Layne B. H.; Concepcion J. J.; Liu M.; Nam C.-Y. Improved Stability and Performance of Visible Photoelectrochemical Water Splitting on Solution-Processed Organic Semiconductor Thin Films by Ultrathin Metal Oxide Passivation. Chem. Mater. 2018, 30 (2), 324–335. 10.1021/acs.chemmater.7b02889. [DOI] [Google Scholar]
- Yu J. M.; Lee J.; Kim Y. S.; Song J.; Oh J.; Lee S. M.; Jeong M.; Kim Y.; Kwak J. H.; Cho S.; Yang C.; Jang J.-W. High-Performance and Stable Photoelectrochemical Water Splitting Cell with Organic-Photoactive-Layer-Based Photoanode. Nat. Commun. 2020, 11 (1), 5509. 10.1038/s41467-020-19329-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Liu Y.; Cho H.-H.; Xia M.; Sekar A.; Primera Darwich B.; Wells R. A.; Yum J.-H.; Ren D.; Grätzel M.; Guijarro N.; Sivula K. A Hybrid Bulk-Heterojunction Photoanode for Direct Solar-to-Chemical Conversion. Energy Environ. Sci. 2021, 14 (5), 3141–3151. 10.1039/D1EE00152C. [DOI] [Google Scholar]
- Cho H.-H.; Yao L.; Yum J.-H.; Liu Y.; Boudoire F.; Wells R. A.; Guijarro N.; Sekar A.; Sivula K. A Semiconducting Polymer Bulk Heterojunction Photoanode for Solar Water Oxidation. Nat. Catal. 2021, 4 (5), 431–438. 10.1038/s41929-021-00617-x. [DOI] [Google Scholar]
- Gao Y.; Ding X.; Liu J.; Wang L.; Lu Z.; Li L.; Sun L. Visible Light Driven Water Splitting in a Molecular Device with Unprecedentedly High Photocurrent Density. J. Am. Chem. Soc. 2013, 135 (11), 4219–4222. 10.1021/ja400402d. [DOI] [PubMed] [Google Scholar]
- Xiong W.; Huang F.; Zhang R.-Q. Recent Developments in Carbon Nitride Based Films for Photoelectrochemical Water Splitting. Sustain. Energy Fuels 2020, 4 (2), 485–503. 10.1039/C9SE00785G. [DOI] [Google Scholar]
- Sagara N.; Kamimura S.; Tsubota T.; Ohno T. Photoelectrochemical CO2 Reduction by a P-Type Boron-Doped g-C3N4 Electrode under Visible Light. Appl. Catal. B Environ. 2016, 192, 193–198. 10.1016/j.apcatb.2016.03.055. [DOI] [Google Scholar]
- Ruan Q.; Miao T.; Wang H.; Tang J. Insight on Shallow Trap States-Introduced Photocathodic Performance in n-Type Polymer Photocatalysts. J. Am. Chem. Soc. 2020, 142 (6), 2795–2802. 10.1021/jacs.9b10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.-Y.; Lu X.-L.; Tang S.-F.; Yin X.-P.; Wei Z.-W.; Lu T.-B. Metal-Free 2D/2D Heterojunction of Graphitic Carbon Nitride/Graphdiyne for Improving the Hole Mobility of Graphitic Carbon Nitride. Adv. Energy Mater. 2018, 8 (16), 1702992. 10.1002/aenm.201702992. [DOI] [Google Scholar]
- Wang Z.; Zou G.; Feng C.; Ma Y.; Wang X.; Bi Y. Novel Composites of Graphitic Carbon Nitride and NiO Nanosheet Arrays as Effective Photocathodes with Enhanced Photocurrent Performances. RSC Adv. 2016, 6 (86), 83350–83355. 10.1039/C6RA18999G. [DOI] [Google Scholar]
- Ruan Q.The Investigation into Polymer Semiconductor Photoelectrodes for Light- Driven Water Splitting. Ph.D. Thesis, University College London, 2019. [Google Scholar]
- Dong Y.; Chen Y.; Jiang P.; Wang G.; Wu X.; Wu R. A Novel G-C3N4 Based Photocathode for Photoelectrochemical Hydrogen Evolution. RSC Adv. 2016, 6 (9), 7465–7473. 10.1039/C5RA23265A. [DOI] [Google Scholar]
- Wen P.; Sun Y.; Li H.; Liang Z.; Wu H.; Zhang J.; Zeng H.; Geyer S. M.; Jiang L. A Highly Active Three-Dimensional Z-Scheme ZnO/Au/g-C3N4 Photocathode for Efficient Photoelectrochemical Water Splitting. Appl. Catal. B Environ. 2020, 263, 118180. 10.1016/j.apcatb.2019.118180. [DOI] [Google Scholar]
- Wang Z.; Jin B.; Zou G.; Zhang K.; Hu X.; Park J. H. Rationally Designed Copper-Modified Polymeric Carbon Nitride as a Photocathode for Solar Water Splitting. ChemSusChem 2019, 12 (4), 866–872. 10.1002/cssc.201802495. [DOI] [PubMed] [Google Scholar]
- Basu M.; Zhang Z.-W.; Chen C.-J.; Lu T.-H.; Hu S.-F.; Liu R.-S. CoSe2 Embedded in C3N4: An Efficient Photocathode for Photoelectrochemical Water Splitting. ACS Appl. Mater. Interfaces 2016, 8 (40), 26690–26696. 10.1021/acsami.6b06520. [DOI] [PubMed] [Google Scholar]
- Pachfule P.; Acharjya A.; Roeser J.; Langenhahn T.; Schwarze M.; Schomäcker R.; Thomas A.; Schmidt J. Diacetylene Functionalized Covalent Organic Framework (COF) for Photocatalytic Hydrogen Generation. J. Am. Chem. Soc. 2018, 140 (4), 1423–1427. 10.1021/jacs.7b11255. [DOI] [PubMed] [Google Scholar]
- Stegbauer L.; Schwinghammer K.; Lotsch B. V. A Hydrazone-Based Covalent Organic Framework for Photocatalytic Hydrogen Production. Chem. Sci. 2014, 5 (7), 2789–2793. 10.1039/C4SC00016A. [DOI] [Google Scholar]
- Sick T.; Hufnagel A. G.; Kampmann J.; Kondofersky I.; Calik M.; Rotter J. M.; Evans A.; Döblinger M.; Herbert S.; Peters K.; Böhm D.; Knochel P.; Medina D. D.; Fattakhova-Rohlfing D.; Bein T. Oriented Films of Conjugated 2D Covalent Organic Frameworks as Photocathodes for Water Splitting. J. Am. Chem. Soc. 2018, 140 (6), 2085–2092. 10.1021/jacs.7b06081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter J. M.; Weinberger S.; Kampmann J.; Sick T.; Shalom M.; Bein T.; Medina D. D. Covalent Organic Framework Films through Electrophoretic Deposition—Creating Efficient Morphologies for Catalysis. Chem. Mater. 2019, 31 (24), 10008–10016. 10.1021/acs.chemmater.9b02286. [DOI] [Google Scholar]
- Wei S.; Zhang F.; Zhang W.; Qiang P.; Yu K.; Fu X.; Wu D.; Bi S.; Zhang F. Semiconducting 2D Triazine-Cored Covalent Organic Frameworks with Unsubstituted Olefin Linkages. J. Am. Chem. Soc. 2019, 141 (36), 14272–14279. 10.1021/jacs.9b06219. [DOI] [PubMed] [Google Scholar]
- Addicoat M. A.Photoelectrochemical Water Splitting by Triazine Based Covalent Organic Framework; Zenodo, 2020, 10.5281/zenodo.4081292. [DOI]
- Dai C.; He T.; Zhong L.; Liu X.; Zhen W.; Xue C.; Li S.; Jiang D.; Liu B. 2,4,6-Triphenyl-1,3,5-Triazine Based Covalent Organic Frameworks for Photoelectrochemical H2 Evolution. Adv. Mater. Interfaces 2021, 8 (7), 2002191. 10.1002/admi.202002191. [DOI] [Google Scholar]
- Xu S.; Sun H.; Addicoat M.; Biswal B. P.; He F.; Park S.; Paasch S.; Zhang T.; Sheng W.; Brunner E.; Hou Y.; Richter M.; Feng X. Thiophene-Bridged Donor-Acceptor Sp2-Carbon-Linked 2D Conjugated Polymers as Photocathodes for Water Reduction. Adv. Mater. 2021, 33 (1), 2006274. 10.1002/adma.202006274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Wang Z.; Wang L. Metal-Organic Framework-Based Materials for Solar Water Splitting. Small Sci. 2021, 1 (5), 2000074. 10.1002/smsc.202000074. [DOI] [Google Scholar]
- Beiler A. M.; McCarthy B. D.; Johnson B. A.; Ott S. Enhancing Photovoltages at P-Type Semiconductors through a Redox-Active Metal-Organic Framework Surface Coating. Nat. Commun. 2020, 11 (1), 5819. 10.1038/s41467-020-19483-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.; Chan B.; Proschogo N.; Solomon M. B.; Kepert C. J.; D’Alessandro D. M. A Cofacial Metal-Organic Framework Based Photocathode for Carbon Dioxide Reduction. Chem. Sci. 2021, 12 (10), 3608–3614. 10.1039/D0SC04691D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso J. C.; Stulp S.; de Brito J. F.; Flor J. B. S.; Frem R. C. G.; Zanoni M. V. B. MOFs Based on ZIF-8 Deposited on TiO2 Nanotubes Increase the Surface Adsorption of CO2 and Its Photoelectrocatalytic Reduction to Alcohols in Aqueous Media. Appl. Catal. B Environ. 2018, 225, 563–573. 10.1016/j.apcatb.2017.12.013. [DOI] [Google Scholar]
- Hou C.; Xu Q.; Wang Y.; Hu X. Synthesis of Pt@NH2-MIL-125(Ti) as a Photocathode Material for Photoelectrochemical Hydrogen Production. RSC Adv. 2013, 3 (43), 19820–19823. 10.1039/c3ra43188f. [DOI] [Google Scholar]
- Suppes G.; Ballard E.; Holdcroft S. Aqueous Photocathode Activity of Regioregular Poly(3-Hexylthiophene). Polym. Chem. 2013, 4 (20), 5345–5350. 10.1039/c3py00143a. [DOI] [Google Scholar]
- Bourgeteau T.; Tondelier D.; Geffroy B.; Brisse R.; Laberty-Robert C.; Campidelli S.; de Bettignies R.; Artero V.; Palacin S.; Jousselme B. A H2-Evolving Photocathode Based on Direct Sensitization of MoS3 with an Organic Photovoltaic Cell. Energy Environ. Sci. 2013, 6 (9), 2706–2713. 10.1039/c3ee41321g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeteau T.; Tondelier D.; Geffroy B.; Brisse R.; Cornut R.; Artero V.; Jousselme B. Enhancing the Performances of P3HT:PCBM-MoS3-Based H2-Evolving Photocathodes with Interfacial Layers. ACS Appl. Mater. Interfaces 2015, 7 (30), 16395–16403. 10.1021/acsami.5b03532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haro M.; Solis C.; Molina G.; Otero L.; Bisquert J.; Gimenez S.; Guerrero A. Toward Stable Solar Hydrogen Generation Using Organic Photoelectrochemical Cells. J. Phys. Chem. C 2015, 119 (12), 6488–6494. 10.1021/acs.jpcc.5b01420. [DOI] [Google Scholar]
- Fumagalli F.; Bellani S.; Schreier M.; Leonardi S.; Rojas H. C.; Ghadirzadeh A.; Tullii G.; Savoini A.; Marra G.; Meda L.; Grätzel M.; Lanzani G.; Mayer M. T.; Antognazza M. R.; Di Fonzo F. Hybrid Organic-Inorganic H2-Evolving Photocathodes: Understanding the Route towards High Performance Organic Photoelectrochemical Water Splitting. J. Mater. Chem. A 2016, 4 (6), 2178–2187. 10.1039/C5TA09330A. [DOI] [Google Scholar]
- Comas Rojas H.; Bellani S.; Fumagalli F.; Tullii G.; Leonardi S.; Mayer M. T.; Schreier M.; Grätzel M.; Lanzani G.; Di Fonzo F.; Antognazza M. R. Polymer-Based Photocathodes with a Solution-Processable Cuprous Iodide Anode Layer and a Polyethyleneimine Protective Coating. Energy Environ. Sci. 2016, 9 (12), 3710–3723. 10.1039/C6EE01655C. [DOI] [Google Scholar]
- Won D. H.; Chung J.; Park S. H.; Kim E.-H.; Woo S. I. Photoelectrochemical Production of Useful Fuels from Carbon Dioxide on a Polypyrrole-Coated p-ZnTe Photocathode under Visible Light Irradiation. J. Mater. Chem. A 2015, 3 (3), 1089–1095. 10.1039/C4TA05901H. [DOI] [Google Scholar]
- Hursán D.; Kormányos A.; Rajeshwar K.; Janáky C. Polyaniline Films Photoelectrochemically Reduce CO2 to Alcohols. Chem. Commun. 2016, 52 (57), 8858–8861. 10.1039/C6CC04050K. [DOI] [PubMed] [Google Scholar]
- Wahyuono R. A.; Seidler B.; Bold S.; Dellith A.; Dellith J.; Ahner J.; Wintergerst P.; Lowe G.; Hager M. D.; Wächtler M.; Streb C.; Schubert U. S.; Rau S.; Dietzek B. Photocathodes beyond NiO: Charge Transfer Dynamics in a π-Conjugated Polymer Functionalized with Ru Photosensitizers. Sci. Rep. 2021, 11 (1), 2787. 10.1038/s41598-021-82395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wan Y.; Ding Y.; Wu S.; Zhang Y.; Zhang X.; Zhang G.; Xiong Y.; Wu X.; Yang J.; Xu H. Conjugated Microporous Polymer Nanosheets for Overall Water Splitting Using Visible Light. Adv. Mater. 2017, 29 (38), 1702428. 10.1002/adma.201702428. [DOI] [PubMed] [Google Scholar]
- Jayanthi S.; Muthu D. V. S.; Jayaraman N.; Sampath S.; Sood A. K. Semiconducting Conjugated Microporous Polymer: An Electrode Material for Photoelectrochemical Water Splitting and Oxygen Reduction. ChemistrySelect 2017, 2 (16), 4522–4532. 10.1002/slct.201700505. [DOI] [Google Scholar]
- Zhang T.; Hou Y.; Dzhagan V.; Liao Z.; Chai G.; Löffler M.; Olianas D.; Milani A.; Xu S.; Tommasini M.; Zahn D. R. T.; Zheng Z.; Zschech E.; Jordan R.; Feng X. Copper-Surface-Mediated Synthesis of Acetylenic Carbon-Rich Nanofibers for Active Metal-Free Photocathodes. Nat. Commun. 2018, 9 (1), 1140. 10.1038/s41467-018-03444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Neumann C.; Zhang T.; Löffler M.; Wolf A.; Hou Y.; Turchanin A.; Zhang J.; Feng X. Poly(1,4-Diethynylbenzene) Gradient Homojunction with Enhanced Charge Carrier Separation for Photoelectrochemical Water Reduction. Adv. Mater. 2019, 31 (19), 1900961. 10.1002/adma.201900961. [DOI] [PubMed] [Google Scholar]
- Lattach Y.; Fortage J.; Deronzier A.; Moutet J.-C. Polypyrrole-Ru(2,2′-Bipyridine)32+/MoSx Structured Composite Film As a Photocathode for the Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2015, 7 (8), 4476–4480. 10.1021/acsami.5b00401. [DOI] [PubMed] [Google Scholar]
- Kamire R. J.; Majewski M. B.; Hoffeditz W. L.; Phelan B. T.; Farha O. K.; Hupp J. T.; Wasielewski M. R. Photodriven Hydrogen Evolution by Molecular Catalysts Using Al2O3-Protected Perylene-3,4-Dicarboximide on NiO Electrodes. Chem. Sci. 2017, 8 (1), 541–549. 10.1039/C6SC02477G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creissen C. E.; Warnan J.; Antón-García D.; Farré Y.; Odobel F.; Reisner E. Inverse Opal CuCrO2 Photocathodes for H2 Production Using Organic Dyes and a Molecular Ni Catalyst. ACS Catal. 2019, 9 (10), 9530–9538. 10.1021/acscatal.9b02984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata R.; Kumagai H.; Yamazaki Y.; Sahara G.; Ishitani O. Photoelectrochemical CO2 Reduction Using a Ru(II)-Re(I) Supramolecular Photocatalyst Connected to a Vinyl Polymer on a NiO Electrode. ACS Appl. Mater. Interfaces 2019, 11 (6), 5632–5641. 10.1021/acsami.8b05495. [DOI] [PubMed] [Google Scholar]
- Li T.-T.; Shan B.; Meyer T. J. Stable Molecular Photocathode for Solar-Driven CO2 Reduction in Aqueous Solutions. ACS Energy Lett. 2019, 4 (3), 629–636. 10.1021/acsenergylett.8b02512. [DOI] [Google Scholar]
- Wang D.; Hu J.; Sherman B. D.; Sheridan M. V.; Yan L.; Dares C. J.; Zhu Y.; Li F.; Huang Q.; You W.; Meyer T. J. A Molecular Tandem Cell for Efficient Solar Water Splitting. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (24), 13256–13260. 10.1073/pnas.2001753117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei V.; Reuillard B.; Reisner E. Bias-Free Solar Syngas Production by Integrating a Molecular Cobalt Catalyst with Perovskite-BiVO4 Tandems. Nat. Mater. 2020, 19 (2), 189–194. 10.1038/s41563-019-0501-6. [DOI] [PubMed] [Google Scholar]
- Jang Y. J.; Jeong I.; Lee J.; Lee J.; Ko M. J.; Lee J. S. Unbiased Sunlight-Driven Artificial Photosynthesis of Carbon Monoxide from CO2 Using a ZnTe-Based Photocathode and a Perovskite Solar Cell in Tandem. ACS Nano 2016, 10 (7), 6980–6987. 10.1021/acsnano.6b02965. [DOI] [PubMed] [Google Scholar]
- He J.; Janáky C. Recent Advances in Solar-Driven Carbon Dioxide Conversion: Expectations versus Reality. ACS Energy Lett. 2020, 5 (6), 1996–2014. 10.1021/acsenergylett.0c00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Prabhakar R. R.; Tan J.; Tilley S. D.; Moon J. Strategies for Enhancing the Photocurrent, Photovoltage, and Stability of Photoelectrodes for Photoelectrochemical Water Splitting. Chem. Soc. Rev. 2019, 48 (19), 4979–5015. 10.1039/C8CS00997J. [DOI] [PubMed] [Google Scholar]
- Cots A.; Bonete P.; Sebastián D.; Baglio V.; Aricò A. S.; Gómez R. Toward Tandem Solar Cells for Water Splitting Using Polymer Electrolytes. ACS Appl. Mater. Interfaces 2018, 10 (30), 25393–25400. 10.1021/acsami.8b06826. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Kaneko H.; Minegishi T.; Kubota J.; Domen K.; Lee J. S. Overall Photoelectrochemical Water Splitting Using Tandem Cell under Simulated Sunlight. ChemSusChem 2016, 9 (1), 61–66. 10.1002/cssc.201501401. [DOI] [PubMed] [Google Scholar]
- Bai Z.; Liu J.; Zhang Y.; Huang Z.; Gao Y.; Li X.; Du Y. Unassisted Solar Water Splitting Using a Cu2O/Ni(OH)2-ZnO/Au Tandem Photoelectrochemical Cell. J. Solid State Electrochem. 2020, 24 (2), 321–328. 10.1007/s10008-019-04437-9. [DOI] [Google Scholar]
- Lai Y.-H.; Lin K.-C.; Yen C.-Y.; Jiang B.-J. A Tandem Photoelectrochemical Water Splitting Cell Consisting of CuBi2O4 and BiVO4 Synthesized from a Single Bi4O5I2 Nanosheet Template. Faraday Discuss. 2019, 215 (0), 297–312. 10.1039/C8FD00183A. [DOI] [PubMed] [Google Scholar]
- Fan K.; Li F.; Wang L.; Daniel Q.; Gabrielsson E.; Sun L. Pt-Free Tandem Molecular Photoelectrochemical Cells for Water Splitting Driven by Visible Light. Phys. Chem. Chem. Phys. 2014, 16 (46), 25234–25240. 10.1039/C4CP04489D. [DOI] [PubMed] [Google Scholar]
- Li F.; Fan K.; Xu B.; Gabrielsson E.; Daniel Q.; Li L.; Sun L. Organic Dye-Sensitized Tandem Photoelectrochemical Cell for Light Driven Total Water Splitting. J. Am. Chem. Soc. 2015, 137 (28), 9153–9159. 10.1021/jacs.5b04856. [DOI] [PubMed] [Google Scholar]
- Duan L.; Fischer A.; Xu Y.; Sun L. Isolated Seven-Coordinate Ru(IV) Dimer Complex with [HOHOH]- Bridging Ligand as an Intermediate for Catalytic Water Oxidation. J. Am. Chem. Soc. 2009, 131 (30), 10397–10399. 10.1021/ja9034686. [DOI] [PubMed] [Google Scholar]
- Duan L.; Wang L.; Inge A. K.; Fischer A.; Zou X.; Sun L. Insights into Ru-Based Molecular Water Oxidation Catalysts: Electronic and Noncovalent-Interaction Effects on Their Catalytic Activities. Inorg. Chem. 2013, 52 (14), 7844–7852. 10.1021/ic302687d. [DOI] [PubMed] [Google Scholar]
- Duan L.; Araujo C. M.; Ahlquist M. S. G.; Sun L. Highly Efficient and Robust Molecular Ruthenium Catalysts for Water Oxidation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (39), 15584–15588. 10.1073/pnas.1118347109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L.; Bozoglian F.; Mandal S.; Stewart B.; Privalov T.; Llobet A.; Sun L. A Molecular Ruthenium Catalyst with Water-Oxidation Activity Comparable to That of Photosystem II. Nat. Chem. 2012, 4 (5), 418–423. 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- Wang L.; Duan L.; Wang Y.; Ahlquist M. S. G.; Sun L. Highly Efficient and Robust Molecular Water Oxidation Catalysts Based on Ruthenium Complexes. Chem. Commun. 2014, 50 (85), 12947–12950. 10.1039/C4CC05069J. [DOI] [PubMed] [Google Scholar]
- Duan L.; Xu Y.; Zhang P.; Wang M.; Sun L. Visible Light-Driven Water Oxidation by a Molecular Ruthenium Catalyst in Homogeneous System. Inorg. Chem. 2010, 49 (1), 209–215. 10.1021/ic9017486. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Sun L. Ru-Bda: Unique Molecular Water-Oxidation Catalysts with Distortion Induced Open Site and Negatively Charged Ligands. J. Am. Chem. Soc. 2019, 141 (14), 5565–5580. 10.1021/jacs.8b12862. [DOI] [PubMed] [Google Scholar]
- Wang D.; Marquard S. L.; Troian-Gautier L.; Sheridan M. V.; Sherman B. D.; Wang Y.; Eberhart M. S.; Farnum B. H.; Dares C. J.; Meyer T. J. Interfacial Deposition of Ru(II) Bipyridine-Dicarboxylate Complexes by Ligand Substitution for Applications in Water Oxidation Catalysis. J. Am. Chem. Soc. 2018, 140 (2), 719–726. 10.1021/jacs.7b10809. [DOI] [PubMed] [Google Scholar]
- Li L.; Duan L.; Xu Y.; Gorlov M.; Hagfeldt A.; Sun L. A Photoelectrochemical Device for Visible Light Driven Water Splitting by a Molecular Ruthenium Catalyst Assembled on Dye-Sensitized Nanostructured TiO2. Chem. Commun. 2010, 46 (39), 7307–7309. 10.1039/c0cc01828g. [DOI] [PubMed] [Google Scholar]
- Brimblecombe R.; Swiegers G. F.; Dismukes G. C.; Spiccia L. Sustained Water Oxidation Photocatalysis by a Bioinspired Manganese Cluster. Angew. Chem., Int. Ed. 2008, 47 (38), 7335–7338. 10.1002/anie.200801132. [DOI] [PubMed] [Google Scholar]
- Habisreutinger S. N.; Leijtens T.; Eperon G. E.; Stranks S. D.; Nicholas R. J.; Snaith H. J. Carbon Nanotube/Polymer Composites as a Highly Stable Hole Collection Layer in Perovskite Solar Cells. Nano Lett. 2014, 14 (10), 5561–5568. 10.1021/nl501982b. [DOI] [PubMed] [Google Scholar]
- Wee K.-R.; Brennaman M. K.; Alibabaei L.; Farnum B. H.; Sherman B.; Lapides A. M.; Meyer T. J. Stabilization of Ruthenium(II) Polypyridyl Chromophores on Nanoparticle Metal-Oxide Electrodes in Water by Hydrophobic PMMA Overlayers. J. Am. Chem. Soc. 2014, 136 (39), 13514–13517. 10.1021/ja506987a. [DOI] [PubMed] [Google Scholar]
- Ding X.; Gao Y.; Ye L.; Zhang L.; Sun L. Assembling Supramolecular Dye-Sensitized Photoelectrochemical Cells for Water Splitting. ChemSusChem 2015, 8 (23), 3992–3995. 10.1002/cssc.201500313. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Gao Y.; Ding X. A PMMA Overlayer Improving the Surface-Bound Stability of Photoanode for Water Splitting. Electrochim. Acta 2016, 207, 130–134. 10.1016/j.electacta.2016.04.125. [DOI] [Google Scholar]
- Ashford D. L.; Sherman B. D.; Binstead R. A.; Templeton J. L.; Meyer T. J. Electro-Assembly of a Chromophore-Catalyst Bilayer for Water Oxidation and Photocatalytic Water Splitting. Angew. Chem., Int. Ed. 2015, 54 (16), 4778–4781. 10.1002/anie.201410944. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Gu Z.-Y.; Arvapally R. K.; Chen Y.-P.; McDougald R. N.; Ivy J. F.; Yakovenko A. A.; Feng D.; Omary M. A.; Zhou H.-C. Rigidifying Fluorescent Linkers by Metal-Organic Framework Formation for Fluorescence Blue Shift and Quantum Yield Enhancement. J. Am. Chem. Soc. 2014, 136 (23), 8269–8276. 10.1021/ja5006866. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Keinan S.; Alibabaei L.; Luo H.; Ito A.; Meyer T. J. Controlled Electropolymerization of Ruthenium(II) Vinylbipyridyl Complexes in Mesoporous Nanoparticle Films of TiO2. Angew. Chem., Int. Ed. 2014, 53 (19), 4872–4876. 10.1002/anie.201402309. [DOI] [PubMed] [Google Scholar]
- Wang L.; Fan K.; Chen H.; Daniel Q.; Philippe B.; Rensmo H.; Sun L. Towards Efficient and Robust Anodes for Water Splitting: Immobilization of Ru Catalysts on Carbon Electrode and Hematite by in Situ Polymerization. Catal. Today 2017, 290, 73–77. 10.1016/j.cattod.2016.07.011. [DOI] [Google Scholar]
- Ashford D. L.; Lapides A. M.; Vannucci A. K.; Hanson K.; Torelli D. A.; Harrison D. P.; Templeton J. L.; Meyer T. J. Water Oxidation by an Electropolymerized Catalyst on Derivatized Mesoporous Metal Oxide Electrodes. J. Am. Chem. Soc. 2014, 136 (18), 6578–6581. 10.1021/ja502464s. [DOI] [PubMed] [Google Scholar]
- Lapides A. M.; Ashford D. L.; Hanson K.; Torelli D. A.; Templeton J. L.; Meyer T. J. Stabilization of a Ruthenium(II) Polypyridyl Dye on Nanocrystalline TiO2 by an Electropolymerized Overlayer. J. Am. Chem. Soc. 2013, 135 (41), 15450–15458. 10.1021/ja4055977. [DOI] [PubMed] [Google Scholar]
- Calvert J. M.; Schmehl R. H.; Sullivan B. P.; Facci J. S.; Meyer T. J.; Murray R. W. Synthetic and Mechanistic Investigations of the Reductive Electrochemical Polymerization of Vinyl-Containing Complexes of Iron(II), Ruthenium(II), and Osmium(II). Inorg. Chem. 1983, 22 (15), 2151–2162. 10.1021/ic00157a013. [DOI] [Google Scholar]
- Leem G.; Sherman B. D.; Burnett A. J.; Morseth Z. A.; Wee K.-R.; Papanikolas J. M.; Meyer T. J.; Schanze K. S. Light-Driven Water Oxidation Using Polyelectrolyte Layer-by-Layer Chromophore-Catalyst Assemblies. ACS Energy Lett. 2016, 1 (2), 339–343. 10.1021/acsenergylett.6b00171. [DOI] [Google Scholar]
- Lapides A. M.; Sherman B. D.; Brennaman M. K.; Dares C. J.; Skinner K. R.; Templeton J. L.; Meyer T. J. Synthesis, Characterization, and Water Oxidation by a Molecular Chromophore-Catalyst Assembly Prepared by Atomic Layer Deposition. The “Mummy” Strategy. Chem. Sci. 2015, 6 (11), 6398–6406. 10.1039/C5SC01752A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Sherman B. D.; Zhao Y.; He R.; Ghiviriga I.; Alibabaei L.; Meyer T. J.; Leem G.; Schanze K. S. Polymer Chromophore-Catalyst Assembly for Solar Fuel Generation. ACS Appl. Mater. Interfaces 2017, 9 (23), 19529–19534. 10.1021/acsami.7b05173. [DOI] [PubMed] [Google Scholar]
- Leem G.; Morseth Z. A.; Wee K.-R.; Jiang J.; Brennaman M. K.; Papanikolas J. M.; Schanze K. S. Polymer-Based Ruthenium(II) Polypyridyl Chromophores on TiO2 for Solar Energy Conversion. Chem. - Asian J. 2016, 11 (8), 1257–1267. 10.1002/asia.201501384. [DOI] [PubMed] [Google Scholar]
- Volpato G. A.; Marasi M.; Gobbato T.; Valentini F.; Sabuzi F.; Gagliardi V.; Bonetto A.; Marcomini A.; Berardi S.; Conte V.; Bonchio M.; Caramori S.; Galloni P.; Sartorel A. Photoanodes for Water Oxidation with Visible Light Based on a Pentacyclic Quinoid Organic Dye Enabling Proton-Coupled Electron Transfer. Chem. Commun. 2020, 56 (15), 2248–2251. 10.1039/C9CC09805D. [DOI] [PubMed] [Google Scholar]
- Jeon D.; Kim N.; Bae S.; Han Y.; Ryu J. WO3/Conducting Polymer Heterojunction Photoanodes for Efficient and Stable Photoelectrochemical Water Splitting. ACS Appl. Mater. Interfaces 2018, 10 (9), 8036–8044. 10.1021/acsami.7b19203. [DOI] [PubMed] [Google Scholar]
- Bonchio M.; Syrgiannis Z.; Burian M.; Marino N.; Pizzolato E.; Dirian K.; Rigodanza F.; Volpato G. A.; La Ganga G.; Demitri N.; Berardi S.; Amenitsch H.; Guldi D. M.; Caramori S.; Bignozzi C. A.; Sartorel A.; Prato M. Hierarchical Organization of Perylene Bisimides and Polyoxometalates for Photo-Assisted Water Oxidation. Nat. Chem. 2019, 11 (2), 146–153. 10.1038/s41557-018-0172-y. [DOI] [PubMed] [Google Scholar]
- Brimblecombe R.; Koo A.; Dismukes G. C.; Swiegers G. F.; Spiccia L. Solar Driven Water Oxidation by a Bioinspired Manganese Molecular Catalyst. J. Am. Chem. Soc. 2010, 132 (9), 2892–2894. 10.1021/ja910055a. [DOI] [PubMed] [Google Scholar]
- Harriman A. Photochemistry of Manganese Porphyrins. Part 8. Electrochemistry. J. Chem. Soc., Dalton Trans. 1984, 1984, 141–146. 10.1039/dt9840000141. [DOI] [Google Scholar]
- Li J.; Jiang Y.; Zhang Q.; Zhao X.; Li N.; Tong H.; Yang X.; Xia L. Immobilising a Cobalt Cubane Catalyst on a Dye-Sensitised TiO2 Photoanode via Electrochemical Polymerisation for Light-Driven Water Oxidation. RSC Adv. 2017, 7 (7), 4102–4107. 10.1039/C6RA24989B. [DOI] [Google Scholar]
- La Ganga G.; Puntoriero F.; Campagna S.; Bazzan I.; Berardi S.; Bonchio M.; Sartorel A.; Natali M.; Scandola F. Light-Driven Water Oxidation with a Molecular Tetra-Cobalt(Iii) Cubane Cluster. Faraday Discuss. 2012, 155 (0), 177–190. 10.1039/C1FD00093D. [DOI] [PubMed] [Google Scholar]
- McCool N. S.; Robinson D. M.; Sheats J. E.; Dismukes G. C. A Co4O4 “Cubane” Water Oxidation Catalyst Inspired by Photosynthesis. J. Am. Chem. Soc. 2011, 133 (30), 11446–11449. 10.1021/ja203877y. [DOI] [PubMed] [Google Scholar]
- Berardi S.; La Ganga G.; Natali M.; Bazzan I.; Puntoriero F.; Sartorel A.; Scandola F.; Campagna S.; Bonchio M. Photocatalytic Water Oxidation: Tuning Light-Induced Electron Transfer by Molecular Co4O4 Cores. J. Am. Chem. Soc. 2012, 134 (27), 11104–11107. 10.1021/ja303951z. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Yang X.; Li F.; Zhang Q.; Li S.; Tong H.; Jiang Y.; Xia L. Immobilization of a Molecular Cobalt Cubane Catalyst on Porous BiVO4via Electrochemical Polymerization for Efficient and Stable Photoelectrochemical Water Oxidation. Chem. Commun. 2019, 55 (10), 1414–1417. 10.1039/C8CC08802K. [DOI] [PubMed] [Google Scholar]
- Abe T.; Nagai K.; Ogiwara T.; Ogasawara S.; Kaneko M.; Tajiri A.; Norimatsu T. Wide Visible Light-Induced Dioxygen Evolution at an Organic Photoanode Coated with a Noble Metal Oxide Catalyst. J. Electroanal. Chem. 2006, 587 (1), 127–132. 10.1016/j.jelechem.2005.11.001. [DOI] [Google Scholar]
- Bledowski M.; Wang L.; Ramakrishnan A.; Khavryuchenko O. V.; Khavryuchenko V. D.; Ricci P. C.; Strunk J.; Cremer T.; Kolbeck C.; Beranek R. Visible-Light Photocurrent Response of TiO2-Polyheptazine Hybrids: Evidence for Interfacial Charge-Transfer Absorption. Phys. Chem. Chem. Phys. 2011, 13 (48), 21511–21519. 10.1039/c1cp22861g. [DOI] [PubMed] [Google Scholar]
- Wang L.; Bledowski M.; Ramakrishnan A.; König D.; Ludwig A.; Beranek R. Dynamics of Photogenerated Holes in TiO2-Polyheptazine Hybrid Photoanodes for Visible Light-Driven Water Splitting. J. Electrochem. Soc. 2012, 159 (7), H616–H622. 10.1149/2.010207jes. [DOI] [Google Scholar]
- Bledowski M.; Wang L.; Ramakrishnan A.; Bétard A.; Khavryuchenko O. V.; Beranek R. Visible-Light Photooxidation of Water to Oxygen at Hybrid TiO2-Polyheptazine Photoanodes with Photodeposited Co-Pi (CoOx) Cocatalyst. ChemPhysChem 2012, 13 (12), 3018–3024. 10.1002/cphc.201200071. [DOI] [PubMed] [Google Scholar]
- Wang L.; Mitoraj D.; Turner S.; Khavryuchenko O. V.; Jacob T.; Hocking R. K.; Beranek R. Ultrasmall CoO(OH)x Nanoparticles As a Highly Efficient “True” Cocatalyst in Porous Photoanodes for Water Splitting. ACS Catal. 2017, 7 (7), 4759–4767. 10.1021/acscatal.7b01466. [DOI] [Google Scholar]
- Vinoth S.; Ong W.-J.; Pandikumar A. Sulfur-Doped Graphitic Carbon Nitride Incorporated Bismuth Oxychloride/Cobalt Based Type-II Heterojunction as a Highly Stable Material for Photoelectrochemical Water Splitting. J. Colloid Interface Sci. 2021, 591, 85–95. 10.1016/j.jcis.2021.01.104. [DOI] [PubMed] [Google Scholar]
- Prakash J.; Prasad U.; Alexander R.; Bahadur J.; Dasgupta K.; Kannan A. N. M. Photoelectrochemical Solar Water Splitting: The Role of the Carbon Nanomaterials in Bismuth Vanadate Composite Photoanodes toward Efficient Charge Separation and Transport. Langmuir 2019, 35 (45), 14492–14504. 10.1021/acs.langmuir.9b02782. [DOI] [PubMed] [Google Scholar]
- Collomb M.-N.; Morales D. V.; Astudillo C. N.; Dautreppe B.; Fortage J. Hybrid Photoanodes for Water Oxidation Combining a Molecular Photosensitizer with a Metal Oxide Oxygen-Evolving Catalyst. Sustain. Energy Fuels 2020, 4 (1), 31–49. 10.1039/C9SE00597H. [DOI] [Google Scholar]
- Li S.; Jiang Y.; Jiang W.; Zhang Y.; Pan K.; Wang S.; Hu C.; Zhang L.-H.; Xia L. In-Situ Generation of g-C3N4 on BiVO4 Photoanode for Highly Efficient Photoelectrochemical Water Oxidation. Appl. Surf. Sci. 2020, 523, 146441. 10.1016/j.apsusc.2020.146441. [DOI] [Google Scholar]
- Park S.-Y.; Kim M.; Jung J.; Heo J.; Hong E. M.; Choi S. M.; Lee J.-Y.; Cho S.; Hong K.; Lim D. C. Stable Organic-Inorganic Hybrid Multilayered Photoelectrochemical Cells. J. Power Sources 2017, 341, 411–418. 10.1016/j.jpowsour.2016.12.017. [DOI] [Google Scholar]
- Vinoth S.; Pandikumar A. Ni Integrated S-GC3N4/BiOBr Based Type-II Heterojunction as a Durable Catalyst for Photoelectrochemical Water Splitting. Renew. Energy 2021, 173, 507–519. 10.1016/j.renene.2021.03.121. [DOI] [Google Scholar]
- Zhang Y.; Lv H.; Zhang Z.; Wang L.; Wu X.; Xu H. Stable Unbiased Photo-Electrochemical Overall Water Splitting Exceeding 3% Efficiency via Covalent Triazine Framework/Metal Oxide Hybrid Photoelectrodes. Adv. Mater. 2021, 33 (15), 2008264. 10.1002/adma.202008264. [DOI] [PubMed] [Google Scholar]
- Longchin P.; Mitoraj D.; Reyes O. M.; Adler C.; Wetchakun N.; Beranek R. Hybrid Photoanodes for Visible Light-Driven Water Oxidation: The Beneficial and Detrimental Effects of Nickel Oxide Cocatalyst. J. Phys. Energy 2020, 2 (4), 044001. 10.1088/2515-7655/abaec9. [DOI] [Google Scholar]
- Tran P. D.; Wong L. H.; Barber J.; Loo J. S. C. Recent Advances in Hybrid Photocatalysts for Solar Fuel Production. Energy Environ. Sci. 2012, 5 (3), 5902–5918. 10.1039/c2ee02849b. [DOI] [Google Scholar]
- Krawicz A.; Yang J.; Anzenberg E.; Yano J.; Sharp I. D.; Moore G. F. Photofunctional Construct That Interfaces Molecular Cobalt-Based Catalysts for H2 Production to a Visible-Light-Absorbing Semiconductor. J. Am. Chem. Soc. 2013, 135 (32), 11861–11868. 10.1021/ja404158r. [DOI] [PubMed] [Google Scholar]
- Krawicz A.; Cedeno D.; Moore G. F. Energetics and Efficiency Analysis of a Cobaloxime-Modified Semiconductor under Simulated Air Mass 1.5 Illumination. Phys. Chem. Chem. Phys. 2014, 16 (30), 15818–15824. 10.1039/C4CP00495G. [DOI] [PubMed] [Google Scholar]
- Cedeno D.; Krawicz A.; Doak P.; Yu M.; Neaton J. B.; Moore G. F. Using Molecular Design to Control the Performance of Hydrogen-Producing Polymer-Brush-Modified Photocathodes. J. Phys. Chem. Lett. 2014, 5 (18), 3222–3226. 10.1021/jz5016394. [DOI] [PubMed] [Google Scholar]
- Beiler A. M.; Khusnutdinova D.; Jacob S. I.; Moore G. F. Chemistry at the Interface: Polymer-Functionalized GaP Semiconductors for Solar Hydrogen Production. Ind. Eng. Chem. Res. 2016, 55 (18), 5306–5314. 10.1021/acs.iecr.6b00478. [DOI] [Google Scholar]
- Beiler A. M.; Khusnutdinova D.; Jacob S. I.; Moore G. F. Solar Hydrogen Production Using Molecular Catalysts Immobilized on Gallium Phosphide (111)A and (111)B Polymer-Modified Photocathodes. ACS Appl. Mater. Interfaces 2016, 8 (15), 10038–10047. 10.1021/acsami.6b01557. [DOI] [PubMed] [Google Scholar]
- Beiler A. M.; Khusnutdinova D.; Wadsworth B. L.; Moore G. F. Cobalt Porphyrin-Polypyridyl Surface Coatings for Photoelectrosynthetic Hydrogen Production. Inorg. Chem. 2017, 56 (20), 12178–12185. 10.1021/acs.inorgchem.7b01509. [DOI] [PubMed] [Google Scholar]
- Wadsworth B. L.; Beiler A. M.; Khusnutdinova D.; Reyes Cruz E. A.; Moore G. F. Interplay between Light Flux, Quantum Efficiency, and Turnover Frequency in Molecular-Modified Photoelectrosynthetic Assemblies. J. Am. Chem. Soc. 2019, 141 (40), 15932–15941. 10.1021/jacs.9b07295. [DOI] [PubMed] [Google Scholar]
- Nguyen N. P.; Wadsworth B. L.; Nishiori D.; Reyes Cruz E. A.; Moore G. F. Understanding and Controlling the Performance-Limiting Steps of Catalyst-Modified Semiconductors. J. Phys. Chem. Lett. 2021, 12 (1), 199–203. 10.1021/acs.jpclett.0c02386. [DOI] [PubMed] [Google Scholar]
- Wadsworth B. L.; Nguyen N. P.; Nishiori D.; Beiler A. M.; Moore G. F. Addressing the Origin of Photocurrents and Fuel Production Activities in Catalyst-Modified Semiconductor Electrodes. ACS Appl. Energy Mater. 2020, 3 (8), 7512–7519. 10.1021/acsaem.0c00919. [DOI] [Google Scholar]
- Andreiadis E. S.; Jacques P.-A.; Tran P. D.; Leyris A.; Chavarot-Kerlidou M.; Jousselme B.; Matheron M.; Pécaut J.; Palacin S.; Fontecave M.; Artero V. Molecular Engineering of a Cobalt-Based Electrocatalytic Nanomaterial for H2 Evolution under Fully Aqueous Conditions. Nat. Chem. 2013, 5 (1), 48–53. 10.1038/nchem.1481. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Chen H.; Tian H. Immobilization of a Cobalt Catalyst on Fullerene in Molecular Devices for Water Reduction. Chem. Commun. 2015, 51 (57), 11508–11511. 10.1039/C5CC03856A. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Borriello C.; Nenna G.; Rosentsveig R.; Di Luccio T. Dispersion of WS2 Nanotubes and Nanoparticles into Conducting Polymer Matrices for Application as LED Materials. Eur. Phys. J. B 2012, 85 (5), 160. 10.1140/epjb/e2012-20453-4. [DOI] [Google Scholar]
- Leem G.; Black H. T.; Shan B.; Bantang J. P. O.; Meyer T. J.; Reynolds J. R.; Schanze K. S. Photocathode Chromophore-Catalyst Assembly via Layer-By-Layer Deposition of a Low Band-Gap Isoindigo Conjugated Polyelectrolyte. ACS Appl. Energy Mater. 2018, 1 (1), 62–67. 10.1021/acsaem.7b00223. [DOI] [Google Scholar]
- Shao D.; Zheng L.; Feng D.; He J.; Zhang R.; Liu H.; Zhang X.; Lu Z.; Wang W.; Wang W.; Lu F.; Dong H.; Cheng Y.; Liu H.; Zheng R. TiO2-P3HT:PCBM Photoelectrochemical Tandem Cells for Solar-Driven Overall Water Splitting. J. Mater. Chem. A 2018, 6 (9), 4032–4039. 10.1039/C7TA09367E. [DOI] [Google Scholar]
- Guerrero A.; Haro M.; Bellani S.; Antognazza M. R.; Meda L.; Gimenez S.; Bisquert J. Organic Photoelectrochemical Cells with Quantitative Photocarrier Conversion. Energy Environ. Sci. 2014, 7 (11), 3666–3673. 10.1039/C4EE01775G. [DOI] [Google Scholar]
- Gustafson M. P.; Clark N.; Winther-Jensen B.; MacFarlane D. R. Organic Photovoltaic Structures as Photo-Active Electrodes. Electrochim. Acta 2014, 140, 309–313. 10.1016/j.electacta.2014.03.112. [DOI] [Google Scholar]
- Shao D.; Cheng Y.; He J.; Feng D.; Zheng L.; Zheng L.; Zhang X.; Xu J.; Wang W.; Wang W.; Lu F.; Dong H.; Li L.; Liu H.; Zheng R.; Liu H. A Spatially Separated Organic-Inorganic Hybrid Photoelectrochemical Cell for Unassisted Overall Water Splitting. ACS Catal. 2017, 7 (8), 5308–5315. 10.1021/acscatal.7b01002. [DOI] [Google Scholar]
- Rajasekar S.; Fortin P.; Tiwari V.; Srivastva U.; Sharma A.; Holdcroft S. Photocathodic Hydrogen Evolution from Catalysed Nanoparticle Films Prepared from Stable Aqueous Dispersions of P3HT and PCBM. Synth. Met. 2019, 247, 10–17. 10.1016/j.synthmet.2018.11.004. [DOI] [Google Scholar]
- Lai L.-H.; Gomulya W.; Berghuis M.; Protesescu L.; Detz R. J.; Reek J. N. H.; Kovalenko M. V.; Loi M. A. Organic-Inorganic Hybrid Solution-Processed H2-Evolving Photocathodes. ACS Appl. Mater. Interfaces 2015, 7 (34), 19083–19090. 10.1021/acsami.5b04550. [DOI] [PubMed] [Google Scholar]
- Shi W.; Yu W.; Li D.; Zhang D.; Fan W.; Shi J.; Li C. PTB7:PC61BM Bulk Heterojunction-Based Photocathodes for Efficient Hydrogen Production in Aqueous Solution. Chem. Mater. 2019, 31 (6), 1928–1935. 10.1021/acs.chemmater.8b04629. [DOI] [Google Scholar]
- Shi W.; Li D.; Fan W.; Ma J.; Li C.; Yu W.; Shi J.; Li C. Nonfullerene Bulk Heterojunction-Based Photocathodes for Highly Efficient Solar Hydrogen Production in Acidic and Neutral Solutions. Adv. Funct. Mater. 2020, 30 (46), 2003399. 10.1002/adfm.202003399. [DOI] [Google Scholar]
- Belarb E.; Blas-Ferrando V. M.; Haro M.; Maghraoui-Meherzi H.; Gimenez S. Electropolymerized Polyaniline: A Promising Hole Selective Contact in Organic Photoelectrochemical Cells. Chem. Eng. Sci. 2016, 154, 143–149. 10.1016/j.ces.2016.06.055. [DOI] [Google Scholar]
- Chae S. Y.; Lee M.; Je Kim M.; Cho J. H.; Kim B.; Joo O.-S. P-CuInS2/n-Polymer Semiconductor Heterojunction for Photoelectrochemical Hydrogen Evolution. ChemSusChem 2020, 13 (24), 6651–6659. 10.1002/cssc.202002123. [DOI] [PubMed] [Google Scholar]
- Bellani S.; Najafi L.; Martín-García B.; Ansaldo A.; Del Rio Castillo A. E.; Prato M.; Moreels I.; Bonaccorso F. Graphene-Based Hole-Selective Layers for High-Efficiency, Solution-Processed, Large-Area, Flexible, Hydrogen-Evolving Organic Photocathodes. J. Phys. Chem. C 2017, 121 (40), 21887–21903. 10.1021/acs.jpcc.7b05904. [DOI] [Google Scholar]
- Mezzetti A.; Fumagalli F.; Alfano A.; Iadicicco D.; Antognazza M. R.; di Fonzo F. Stable Hybrid Organic/Inorganic Photocathodes for Hydrogen Evolution with Amorphous WO3 Hole Selective Contacts. Faraday Discuss. 2017, 198 (0), 433–448. 10.1039/C6FD00216A. [DOI] [PubMed] [Google Scholar]
- Rojas H. C.; Bellani S.; Sarduy E. A.; Fumagalli F.; Mayer M. T.; Schreier M.; Grätzel M.; Di Fonzo F.; Antognazza M. R. All Solution-Processed, Hybrid Organic-Inorganic Photocathode for Hydrogen Evolution. ACS Omega 2017, 2 (7), 3424–3431. 10.1021/acsomega.7b00558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steier L.; Bellani S.; Rojas H. C.; Pan L.; Laitinen M.; Sajavaara T.; Di Fonzo F.; Grätzel M.; Antognazza M. R.; Mayer M. T. Stabilizing Organic Photocathodes by Low-Temperature Atomic Layer Deposition of TiO2. Sustain. Energy Fuels 2017, 1 (9), 1915–1920. 10.1039/C7SE00421D. [DOI] [Google Scholar]
- Yao L.; Guijarro N.; Boudoire F.; Liu Y.; Rahmanudin A.; Wells R. A.; Sekar A.; Cho H.-H.; Yum J.-H.; Le Formal F.; Sivula K. Establishing Stability in Organic Semiconductor Photocathodes for Solar Hydrogen Production. J. Am. Chem. Soc. 2020, 142 (17), 7795–7802. 10.1021/jacs.0c00126. [DOI] [PubMed] [Google Scholar]
- Ran J.; Zhang J.; Yu J.; Jaroniec M.; Qiao S. Z. Earth-Abundant Cocatalysts for Semiconductor-Based Photocatalytic Water Splitting. Chem. Soc. Rev. 2014, 43 (22), 7787–7812. 10.1039/C3CS60425J. [DOI] [PubMed] [Google Scholar]
- Zou X.; Zhang Y. Noble Metal-Free Hydrogen Evolution Catalysts for Water Splitting. Chem. Soc. Rev. 2015, 44 (15), 5148–5180. 10.1039/C4CS00448E. [DOI] [PubMed] [Google Scholar]
- Merki D.; Fierro S.; Vrubel H.; Hu X. Amorphous Molybdenum Sulfide Films as Catalysts for Electrochemical Hydrogen Production in Water. Chem. Sci. 2011, 2 (7), 1262–1267. 10.1039/C1SC00117E. [DOI] [Google Scholar]
- Yip H.-L.; Jen A. K.-Y. Recent Advances in Solution-Processed Interfacial Materials for Efficient and Stable Polymer Solar Cells. Energy Environ. Sci. 2012, 5 (3), 5994–6011. 10.1039/c2ee02806a. [DOI] [Google Scholar]
- Arai T.; Sato S.; Uemura K.; Morikawa T.; Kajino T.; Motohiro T. Photoelectrochemical Reduction of CO2 in Water under Visible-Light Irradiation by a p-Type InP Photocathode Modified with an Electropolymerized Ruthenium Complex. Chem. Commun. 2010, 46 (37), 6944–6946. 10.1039/c0cc02061c. [DOI] [PubMed] [Google Scholar]
- Sato S.; Arai T.; Morikawa T.; Uemura K.; Suzuki T. M.; Tanaka H.; Kajino T. Selective CO2 Conversion to Formate Conjugated with H2O Oxidation Utilizing Semiconductor/Complex Hybrid Photocatalysts. J. Am. Chem. Soc. 2011, 133 (39), 15240–15243. 10.1021/ja204881d. [DOI] [PubMed] [Google Scholar]
- Arai T.; Sato S.; Kajino T.; Morikawa T. Solar CO2 Reduction Using H2O by a Semiconductor/Metal-Complex Hybrid Photocatalyst: Enhanced Efficiency and Demonstration of a Wireless System Using SrTiO3 Photoanodes. Energy Environ. Sci. 2013, 6 (4), 1274–1282. 10.1039/c3ee24317f. [DOI] [Google Scholar]
- Nakada A.; Nakashima T.; Sekizawa K.; Maeda K.; Ishitani O. Visible-Light-Driven CO2 Reduction on a Hybrid Photocatalyst Consisting of a Ru(Ii) Binuclear Complex and a Ag-Loaded TaON in Aqueous Solutions. Chem. Sci. 2016, 7 (7), 4364–4371. 10.1039/C6SC00586A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata R.; Kumagai H.; Yamazaki Y.; Higashi M.; Abe R.; Ishitani O. Durable Photoelectrochemical CO2 Reduction with Water Oxidation Using a Visible-Light Driven Molecular Photocathode. J. Mater. Chem. A 2021, 9 (3), 1517–1529. 10.1039/D0TA07351B. [DOI] [Google Scholar]
- Tamaki Y.; Ishitani O. Supramolecular Photocatalysts for the Reduction of CO2. ACS Catal. 2017, 7 (5), 3394–3409. 10.1021/acscatal.7b00440. [DOI] [Google Scholar]
- Kuriki R.; Matsunaga H.; Nakashima T.; Wada K.; Yamakata A.; Ishitani O.; Maeda K. Nature-Inspired, Highly Durable CO2 Reduction System Consisting of a Binuclear Ruthenium(II) Complex and an Organic Semiconductor Using Visible Light. J. Am. Chem. Soc. 2016, 138 (15), 5159–5170. 10.1021/jacs.6b01997. [DOI] [PubMed] [Google Scholar]
- Kuriki R.; Yamamoto M.; Higuchi K.; Yamamoto Y.; Akatsuka M.; Lu D.; Yagi S.; Yoshida T.; Ishitani O.; Maeda K. Robust Binding between Carbon Nitride Nanosheets and a Binuclear Ruthenium(II) Complex Enabling Durable, Selective CO2 Reduction under Visible Light in Aqueous Solution. Angew. Chem., Int. Ed. 2017, 56 (17), 4867–4871. 10.1002/anie.201701627. [DOI] [PubMed] [Google Scholar]
- Blakemore J. D.; Gupta A.; Warren J. J.; Brunschwig B. S.; Gray H. B. Noncovalent Immobilization of Electrocatalysts on Carbon Electrodes for Fuel Production. J. Am. Chem. Soc. 2013, 135 (49), 18288–18291. 10.1021/ja4099609. [DOI] [PubMed] [Google Scholar]
- Schreier M.; Luo J.; Gao P.; Moehl T.; Mayer M. T.; Grätzel M. Covalent Immobilization of a Molecular Catalyst on Cu2O Photocathodes for CO2 Reduction. J. Am. Chem. Soc. 2016, 138 (6), 1938–1946. 10.1021/jacs.5b12157. [DOI] [PubMed] [Google Scholar]
- Orchanian N. M.; Hong L. E.; Skrainka J. A.; Esterhuizen J. A.; Popov D. A.; Marinescu S. C. Surface-Immobilized Conjugated Polymers Incorporating Rhenium Bipyridine Motifs for Electrocatalytic and Photocatalytic CO2 Reduction. ACS Appl. Energy Mater. 2019, 2 (1), 110–123. 10.1021/acsaem.8b01745. [DOI] [Google Scholar]
- Lee S.; Kim S.; Park C.; Moon G.; Son H.-J.; Baeg J.-O.; Kim W.; Choi W. Nafion-Assisted Noncovalent Assembly of Molecular Sensitizers and Catalysts for Sustained Photoreduction of CO2 to CO. ACS Sustain. Chem. Eng. 2020, 8 (9), 3709–3717. 10.1021/acssuschemeng.9b06797. [DOI] [Google Scholar]
- Yang W.; Kim J. H.; Hutter O. S.; Phillips L. J.; Tan J.; Park J.; Lee H.; Major J. D.; Lee J. S.; Moon J. Benchmark Performance of Low-Cost Sb2Se3 Photocathodes for Unassisted Solar Overall Water Splitting. Nat. Commun. 2020, 11 (1), 861. 10.1038/s41467-020-14704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kini G. P.; Jeon S. J.; Moon D. K. Design Principles and Synergistic Effects of Chlorination on a Conjugated Backbone for Efficient Organic Photovoltaics: A Critical Review. Adv. Mater. 2020, 32 (11), 1906175. 10.1002/adma.201906175. [DOI] [PubMed] [Google Scholar]
- Bronstein H.; Nielsen C. B.; Schroeder B. C.; McCulloch I. The Role of Chemical Design in the Performance of Organic Semiconductors. Nat. Rev. Chem. 2020, 4 (2), 66–77. 10.1038/s41570-019-0152-9. [DOI] [PubMed] [Google Scholar]
- Wan X.; Li C.; Zhang M.; Chen Y. Acceptor-Donor-Acceptor Type Molecules for High Performance Organic Photovoltaics - Chemistry and Mechanism. Chem. Soc. Rev. 2020, 49 (9), 2828–2842. 10.1039/D0CS00084A. [DOI] [PubMed] [Google Scholar]
- Kosco J.; Sachs M.; Godin R.; Kirkus M.; Francas L.; Bidwell M.; Qureshi M.; Anjum D.; Durrant J. R.; McCulloch I. The Effect of Residual Palladium Catalyst Contamination on the Photocatalytic Hydrogen Evolution Activity of Conjugated Polymers. Adv. Energy Mater. 2018, 8 (34), 1802181. 10.1002/aenm.201802181. [DOI] [Google Scholar]
- Mitchell E.; Law A.; Godin R. Experimental Determination of Charge Carrier Dynamics in Carbon Nitride Heterojunctions. Chem. Commun. 2021, 57 (13), 1550–1567. 10.1039/D0CC06841A. [DOI] [PubMed] [Google Scholar]
- Nielander A. C.; Shaner M. R.; Papadantonakis K. M.; Francis S. A.; Lewis N. S. A Taxonomy for Solar Fuels Generators. Energy Environ. Sci. 2015, 8 (1), 16–25. 10.1039/C4EE02251C. [DOI] [Google Scholar]
- McKone J. R.; Lewis N. S.; Gray H. B. Will Solar-Driven Water-Splitting Devices See the Light of Day?. Chem. Mater. 2014, 26 (1), 407–414. 10.1021/cm4021518. [DOI] [Google Scholar]
- Jacobsson T. J.; Fjällström V.; Edoff M.; Edvinsson T. Sustainable Solar Hydrogen Production: From Photoelectrochemical Cells to PV-Electrolyzers and Back Again. Energy Environ. Sci. 2014, 7 (7), 2056–2070. 10.1039/C4EE00754A. [DOI] [Google Scholar]
- Modestino M. A.; Haussener S. An Integrated Device View on Photo-Electrochemical Solar-Hydrogen Generation. Annu. Rev. Chem. Biomol. Eng. 2015, 6 (1), 13–34. 10.1146/annurev-chembioeng-061114-123357. [DOI] [PubMed] [Google Scholar]
- Licht S.; Wang B.; Mukerji S.; Soga T.; Umeno M.; Tributsch H. Efficient Solar Water Splitting, Exemplified by RuO2-Catalyzed AlGaAs/Si Photoelectrolysis. J. Phys. Chem. B 2000, 104 (38), 8920–8924. 10.1021/jp002083b. [DOI] [Google Scholar]
- Jacobsson T. J.; Fjällström V.; Sahlberg M.; Edoff M.; Edvinsson T. A Monolithic Device for Solar Water Splitting Based on Series Interconnected Thin Film Absorbers Reaching over 10% Solar-to-Hydrogen Efficiency. Energy Environ. Sci. 2013, 6 (12), 3676–3683. 10.1039/c3ee42519c. [DOI] [Google Scholar]
- Marinova N.; Valero S.; Delgado J. L. Organic and Perovskite Solar Cells: Working Principles, Materials and Interfaces. J. Colloid Interface Sci. 2017, 488, 373–389. 10.1016/j.jcis.2016.11.021. [DOI] [PubMed] [Google Scholar]
- Baxter J. B.; Richter C.; Schmuttenmaer C. A. Ultrafast Carrier Dynamics in Nanostructures for Solar Fuels. Annu. Rev. Phys. Chem. 2014, 65 (1), 423–447. 10.1146/annurev-physchem-040513-103742. [DOI] [PubMed] [Google Scholar]
- Peter L. M.; Upul Wijayantha K. G. Photoelectrochemical Water Splitting at Semiconductor Electrodes: Fundamental Problems and New Perspectives. ChemPhysChem 2014, 15 (10), 1983–1995. 10.1002/cphc.201402024. [DOI] [PubMed] [Google Scholar]
- Advances in Photoelectrochemical Water Splitting: Theory, Experiment and Systems Analysis; Tilley S. D.; Lany S.; van de Krol R., Eds.; Energy and Environment Series; The Royal Society of Chemistry, 2018. [Google Scholar]
- Osterloh F. E. Inorganic Nanostructures for Photoelectrochemical and Photocatalytic Water Splitting. Chem. Soc. Rev. 2013, 42 (6), 2294–2320. 10.1039/C2CS35266D. [DOI] [PubMed] [Google Scholar]
- Barroso M.; Mesa C. A.; Pendlebury S. R.; Cowan A. J.; Hisatomi T.; Sivula K.; Grätzel M.; Klug D. R.; Durrant J. R. Dynamics of Photogenerated Holes in Surface Modified α-Fe2O3 Photoanodes for Solar Water Splitting. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (39), 15640–15645. 10.1073/pnas.1118326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Le Formal F.; Kafizas A.; Pendlebury S. R.; Durrant J. R. Efficient Suppression of Back Electron/Hole Recombination in Cobalt Phosphate Surface-Modified Undoped Bismuth Vanadate Photoanodes. J. Mater. Chem. A 2015, 3 (41), 20649–20657. 10.1039/C5TA05826K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachäus C.; Abdi F. F.; Peter L. M.; van de Krol R. Photocurrent of BiVO4 Is Limited by Surface Recombination, Not Surface Catalysis. Chem. Sci. 2017, 8 (5), 3712–3719. 10.1039/C7SC00363C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellist M. R.; Laskowski F. A. L.; Qiu J.; Hajibabaei H.; Sivula K.; Hamann T. W.; Boettcher S. W. Potential-Sensing Electrochemical Atomic Force Microscopy for in Operando Analysis of Water-Splitting Catalysts and Interfaces. Nat. Energy 2018, 3 (1), 46–52. 10.1038/s41560-017-0048-1. [DOI] [Google Scholar]
- Qiu J.; Hajibabaei H.; Nellist M. R.; Laskowski F. A. L.; Hamann T. W.; Boettcher S. W. Direct in Situ Measurement of Charge Transfer Processes During Photoelectrochemical Water Oxidation on Catalyzed Hematite. ACS Cent. Sci. 2017, 3 (9), 1015–1025. 10.1021/acscentsci.7b00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin R.; Wang Y.; Zwijnenburg M. A.; Tang J.; Durrant J. R. Time-Resolved Spectroscopic Investigation of Charge Trapping in Carbon Nitrides Photocatalysts for Hydrogen Generation. J. Am. Chem. Soc. 2017, 139 (14), 5216–5224. 10.1021/jacs.7b01547. [DOI] [PubMed] [Google Scholar]
- O’Neill M.; Kelly S. M. Ordered Materials for Organic Electronics and Photonics. Adv. Mater. 2011, 23 (5), 566–584. 10.1002/adma.201002884. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Nawrocki R. A.; Voyles R. M.; Zhang H. H. Modeling of the Electrical Characteristics of an Organic Field Effect Transistor in Presence of the Bending Effects. Org. Electron. 2021, 88, 106000. 10.1016/j.orgel.2020.106000. [DOI] [Google Scholar]
- Khan Y.; Ahn Y.; Seo J. H.; Walker B. Ionic Moieties in Organic and Hybrid Semiconducting Devices: Influence on Energy Band Structures and Functions. J. Mater. Chem. C 2020, 8 (40), 13953–13971. 10.1039/D0TC03398G. [DOI] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; Nakano T.; Suzuki M.; Masuda T.; Inoue S.; Kimura T.; Nomura T.; Yonekura S.; Yu L.-J.; Sakamoto T.; Motomura T.; Chen J.-H.; Kato Y.; Noguchi T.; Tono K.; Joti Y.; Kameshima T.; Hatsui T.; Nango E.; Tanaka R.; Naitow H.; Matsuura Y.; Yamashita A.; Yamamoto M.; Nureki O.; Yabashi M.; Ishikawa T.; Iwata S.; Shen J.-R. Light-Induced Structural Changes and the Site of O = O Bond Formation in PSII Caught by XFEL. Nature 2017, 543 (7643), 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Kern J.; Chatterjee R.; Young I. D.; Fuller F. D.; Lassalle L.; Ibrahim M.; Gul S.; Fransson T.; Brewster A. S.; Alonso-Mori R.; Hussein R.; Zhang M.; Douthit L.; de Lichtenberg C.; Cheah M. H.; Shevela D.; Wersig J.; Seuffert I.; Sokaras D.; Pastor E.; Weninger C.; Kroll T.; Sierra R. G.; Aller P.; Butryn A.; Orville A. M.; Liang M.; Batyuk A.; Koglin J. E.; Carbajo S.; Boutet S.; Moriarty N. W.; Holton J. M.; Dobbek H.; Adams P. D.; Bergmann U.; Sauter N. K.; Zouni A.; Messinger J.; Yano J.; Yachandra V. K. Structures of the Intermediates of Kok’s Photosynthetic Water Oxidation Clock. Nature 2018, 563 (7731), 421–425. 10.1038/s41586-018-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Yamashita K.; Nakajima Y.; Ueno G.; Li H.; Yamane T.; Hirata K.; Umena Y.; Yonekura S.; Yu L.-J.; Murakami H.; Nomura T.; Kimura T.; Kubo M.; Baba S.; Kumasaka T.; Tono K.; Yabashi M.; Isobe H.; Yamaguchi K.; Yamamoto M.; Ago H.; Shen J.-R. An Oxyl/Oxo Mechanism for Oxygen-Oxygen Coupling in PSII Revealed by an x-Ray Free-Electron Laser. Science 2019, 366 (6463), 334–338. 10.1126/science.aax6998. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A. Missing Pieces in the Puzzle of Biological Water Oxidation. ACS Catal. 2018, 8 (10), 9477–9507. 10.1021/acscatal.8b01928. [DOI] [Google Scholar]
- Mesa C. A.; Francàs L.; Yang K. R.; Garrido-Barros P.; Pastor E.; Ma Y.; Kafizas A.; Rosser T. E.; Mayer M. T.; Reisner E.; Grätzel M.; Batista V. S.; Durrant J. R. Multihole Water Oxidation Catalysis on Haematite Photoanodes Revealed by Operando Spectroelectrochemistry and DFT. Nat. Chem. 2020, 12 (1), 82–89. 10.1038/s41557-019-0347-1. [DOI] [PubMed] [Google Scholar]
- Amin M.; Vogt L.; Szejgis W.; Vassiliev S.; Brudvig G. W.; Bruce D.; Gunner M. R. Proton-Coupled Electron Transfer During the S-State Transitions of the Oxygen-Evolving Complex of Photosystem II. J. Phys. Chem. B 2015, 119 (24), 7366–7377. 10.1021/jp510948e. [DOI] [PubMed] [Google Scholar]
- Liu E.; Li J.; Jiao L.; Doan H. T. T.; Liu Z.; Zhao Z.; Huang Y.; Abraham K. M.; Mukerjee S.; Jia Q. Unifying the Hydrogen Evolution and Oxidation Reactions Kinetics in Base by Identifying the Catalytic Roles of Hydroxyl-Water-Cation Adducts. J. Am. Chem. Soc. 2019, 141 (7), 3232–3239. 10.1021/jacs.8b13228. [DOI] [PubMed] [Google Scholar]
- Hu C.; Zhang L.; Gong J. Recent Progress Made in the Mechanism Comprehension and Design of Electrocatalysts for Alkaline Water Splitting. Energy Environ. Sci. 2019, 12 (9), 2620–2645. 10.1039/C9EE01202H. [DOI] [Google Scholar]
- Dubouis N.; Grimaud A. The Hydrogen Evolution Reaction: From Material to Interfacial Descriptors. Chem. Sci. 2019, 10 (40), 9165–9181. 10.1039/C9SC03831K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Hu R.; Bai H.; Chen H.; Lv F.; Liu L.; Wang S.; Tian H. Efficient Conjugated Polymer-Methyl Viologen Electron Transfer System for Controlled Photo-Driven Hydrogen Evolution. ACS Appl. Mater. Interfaces 2017, 9 (12), 10355–10359. 10.1021/acsami.7b00069. [DOI] [PubMed] [Google Scholar]
- Yu F.; Wang Z.; Zhang S.; Ye H.; Kong K.; Gong X.; Hua J.; Tian H. Molecular Engineering of Donor-Acceptor Conjugated Polymer/g-C3N4 Heterostructures for Significantly Enhanced Hydrogen Evolution Under Visible-Light Irradiation. Adv. Funct. Mater. 2018, 28 (47), 1804512. 10.1002/adfm.201804512. [DOI] [Google Scholar]
- Thoi V. S.; Sun Y.; Long J. R.; Chang C. J. Complexes of Earth-Abundant Metals for Catalytic Electrochemical Hydrogen Generation under Aqueous Conditions. Chem. Soc. Rev. 2013, 42 (6), 2388–2400. 10.1039/C2CS35272A. [DOI] [PubMed] [Google Scholar]
- Elgrishi N.; McCarthy B. D.; Rountree E. S.; Dempsey J. L. Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes. ACS Catal. 2016, 6 (6), 3644–3659. 10.1021/acscatal.6b00778. [DOI] [Google Scholar]
- Sun H.; Öner I. H.; Wang T.; Zhang T.; Selyshchev O.; Neumann C.; Fu Y.; Liao Z.; Xu S.; Hou Y.; Turchanin A.; Zahn D. R. T.; Zschech E.; Weidinger I. M.; Zhang J.; Feng X. Molecular Engineering of Conjugated Acetylenic Polymers for Efficient Cocatalyst-Free Photoelectrochemical Water Reduction. Angew. Chem., Int. Ed. 2019, 58 (30), 10368–10374. 10.1002/anie.201904978. [DOI] [PubMed] [Google Scholar]
- Ru C.; Wei Q.; Chen W.; Guan Q.; Zhang Q.; Ling Y.; Tao C.; Qin D.; Wu J.; Pan X. Tunable Conjugated Organoborane Oligomers for Visible-Light-Driven Hydrogen Evolution. ACS Energy Lett. 2020, 5 (2), 669–675. 10.1021/acsenergylett.0c00075. [DOI] [Google Scholar]
- Thorum M. S.; Hankett J. M.; Gewirth A. A. Poisoning the Oxygen Reduction Reaction on Carbon-Supported Fe and Cu Electrocatalysts: Evidence for Metal-Centered Activity. J. Phys. Chem. Lett. 2011, 2 (4), 295–298. 10.1021/jz1016284. [DOI] [Google Scholar]
- Liu W.; Zhang H.; Li C.; Wang X.; Liu J.; Zhang X. Non-Noble Metal Single-Atom Catalysts Prepared by Wet Chemical Method and Their Applications in Electrochemical Water Splitting. J. Energy Chem. 2020, 47, 333–345. 10.1016/j.jechem.2020.02.020. [DOI] [Google Scholar]
- Ly K. H.; Weidinger I. M. Understanding Active Sites in Molecular (Photo)Electrocatalysis through Complementary Vibrational Spectroelectrochemistry. Chem. Commun. 2021, 57 (19), 2328–2342. 10.1039/D0CC07376H. [DOI] [PubMed] [Google Scholar]
- Gimbert-Suriñach C.; Albero J.; Stoll T.; Fortage J.; Collomb M.-N.; Deronzier A.; Palomares E.; Llobet A. Efficient and Limiting Reactions in Aqueous Light-Induced Hydrogen Evolution Systems Using Molecular Catalysts and Quantum Dots. J. Am. Chem. Soc. 2014, 136 (21), 7655–7661. 10.1021/ja501489h. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Gunic E.; Miller L. L. Protonation of Oligothiophenes. Chem. Mater. 1995, 7 (2), 255–256. 10.1021/cm00050a004. [DOI] [Google Scholar]
- Adachi N.; Kaneko Y.; Sekiguchi K.; Sugiyama H.; Sugeno M. PH-Responsive Fluorescence Chemical Sensor Constituted by Conjugated Polymers Containing Pyridine Rings. Luminescence 2015, 30 (8), 1308–1312. 10.1002/bio.2898. [DOI] [PubMed] [Google Scholar]
- Broadwater S. J.; Hickey M. K.; McQuade D. T. A Comparison of Crystalline- and Graft Polymer-Based Chemosensors. J. Am. Chem. Soc. 2003, 125 (37), 11154–11155. 10.1021/ja035218a. [DOI] [PubMed] [Google Scholar]
- Böer K. W.Introduction to Space Charge Effects in Semiconductors; Springer Series in Solid-State Sciences; Springer: Berlin, 2009. [Google Scholar]
- Dumitru L. M.; Manoli K.; Magliulo M.; Ligonzo T.; Palazzo G.; Torsi L. A Hydrogel Capsule as Gate Dielectric in Flexible Organic Field-Effect Transistors. APL Mater. 2015, 3 (1), 014904. 10.1063/1.4901124. [DOI] [Google Scholar]
- Yong W.-W.; Lu H.; Li H.; Wang S.; Zhang M.-T. Photocatalytic Hydrogen Production with Conjugated Polymers as Photosensitizers. ACS Appl. Mater. Interfaces 2018, 10 (13), 10828–10834. 10.1021/acsami.7b18917. [DOI] [PubMed] [Google Scholar]
- Gregg B. A. Excitonic Solar Cells. J. Phys. Chem. B 2003, 107 (20), 4688–4698. 10.1021/jp022507x. [DOI] [Google Scholar]
- Yao L.; Rahmanudin A.; Guijarro N.; Sivula K. Organic Semiconductor Based Devices for Solar Water Splitting. Adv. Energy Mater. 2018, 8 (32), 1802585. 10.1002/aenm.201802585. [DOI] [Google Scholar]
- Choi M.-J.; García de Arquer F. P.; Proppe A. H.; Seifitokaldani A.; Choi J.; Kim J.; Baek S.-W.; Liu M.; Sun B.; Biondi M.; Scheffel B.; Walters G.; Nam D.-H.; Jo J. W.; Ouellette O.; Voznyy O.; Hoogland S.; Kelley S. O.; Jung Y. S.; Sargent Edward. H. Cascade Surface Modification of Colloidal Quantum Dot Inks Enables Efficient Bulk Homojunction Photovoltaics. Nat. Commun. 2020, 11 (1), 103. 10.1038/s41467-019-13437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu S.; Sontag D.. Crystalline Silicon Solar Cells: Homojunction Cells BT - Solar Cells and Modules; Shah A., Ed.; Springer International Publishing: Cham, 2020; pp 97–138. [Google Scholar]
- Li D.; Chen R.; Wang S.; Zhang X.; Zhang Y.; Liu J.; Yin H.; Fan F.; Shi J.; Li C. Unraveling the Kinetics of Photocatalytic Water Oxidation on WO3. J. Phys. Chem. Lett. 2020, 11 (2), 412–418. 10.1021/acs.jpclett.9b03340. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Wu S.; Li L.; Li L.; Li X. Regulating the Silicon/Hematite Microwire Photoanode by the Conformal Al2O3 Intermediate Layer for Water Splitting. ACS Appl. Mater. Interfaces 2019, 11 (6), 5978–5988. 10.1021/acsami.8b18681. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Wang D. Photocatalysis: Basic Principles, Diverse Forms of Implementations and Emerging Scientific Opportunities. Adv. Energy Mater. 2017, 7 (23), 1700841. 10.1002/aenm.201700841. [DOI] [Google Scholar]
- Zhang J.; García-Rodríguez R.; Cameron P.; Eslava S. Role of Cobalt-Iron (Oxy)Hydroxide (CoFeOx) as Oxygen Evolution Catalyst on Hematite Photoanodes. Energy Environ. Sci. 2018, 11 (10), 2972–2984. 10.1039/C8EE01346B. [DOI] [Google Scholar]
- Neumann B.; Bogdanoff P.; Tributsch H.; Sakthivel S.; Kisch H. Electrochemical Mass Spectroscopic and Surface Photovoltage Studies of Catalytic Water Photooxidation by Undoped and Carbon-Doped Titania. J. Phys. Chem. B 2005, 109 (35), 16579–16586. 10.1021/jp051339g. [DOI] [PubMed] [Google Scholar]
- Yu C.; Wu Z.; Liu R.; He H.; Fan W.; Xue S. The Effects of Gd3+ Doping on the Physical Structure and Photocatalytic Performance of Bi2MoO6 Nanoplate Crystals. J. Phys. Chem. Solids 2016, 93, 7–13. 10.1016/j.jpcs.2016.02.008. [DOI] [Google Scholar]
- Loera S.; Lima E.; Pfeiffer H.; Lara V. H. Synthesis of Aluminosilicates under High Pressure and Using Sulfur as Directing Agent. Cent. Eur. J. Chem. 2012, 10 (1), 105–112. [Google Scholar]
- Scarongella M.; Paraecattil A. A.; Buchaca-Domingo E.; Douglas J. D.; Beaupré S.; McCarthy-Ward T.; Heeney M.; Moser J.-E.; Leclerc M.; Fréchet J. M. J.; Stingelin N.; Banerji N. The Influence of Microstructure on Charge Separation Dynamics in Organic Bulk Heterojunction Materials for Solar Cell Applications. J. Mater. Chem. A 2014, 2 (17), 6218–6230. 10.1039/C3TA15112C. [DOI] [Google Scholar]
- Wang L.; Tahir M.; Chen H.; Sambur J. B. Probing Charge Carrier Transport and Recombination Pathways in Monolayer MoS2/WS2 Heterojunction Photoelectrodes. Nano Lett. 2019, 19 (12), 9084–9094. 10.1021/acs.nanolett.9b04209. [DOI] [PubMed] [Google Scholar]
- Perea-Causín R.; Brem S.; Malic E. Phonon-Assisted Exciton Dissociation in Transition Metal Dichalcogenides. Nanoscale 2021, 13 (3), 1884–1892. 10.1039/D0NR07131E. [DOI] [PubMed] [Google Scholar]
- Ohkita H.; Ito S. Transient Absorption Spectroscopy of Polymer-Based Thin-Film Solar Cells. Polymer 2011, 52 (20), 4397–4417. 10.1016/j.polymer.2011.06.061. [DOI] [Google Scholar]
- Fan X.; Wang T.; Gao B.; Xie X.; Zhang S.; Meng X.; Gong H.; Guo Y.; Huang X.; He J. Layered Double Hydroxides Decorated Graphic Carbon Nitride Film as Efficient Photoanodes for Photoelectrochemical Water Splitting. Catal. Today 2019, 335, 423–428. 10.1016/j.cattod.2019.01.034. [DOI] [Google Scholar]
- Vietmeyer F.; Frantsuzov P. A.; Janko B.; Kuno M. Carrier Recombination Dynamics in Individual CdSe Nanowires. Phys. Rev. B 2011, 83 (11), 115319. 10.1103/PhysRevB.83.115319. [DOI] [Google Scholar]
- Merschjann C.; Tschierlei S.; Tyborski T.; Kailasam K.; Orthmann S.; Hollmann D.; Schedel-Niedrig T.; Thomas A.; Lochbrunner S. Complementing Graphenes: 1D Interplanar Charge Transport in Polymeric Graphitic Carbon Nitrides. Adv. Mater. 2015, 27 (48), 7993–7999. 10.1002/adma.201503448. [DOI] [PubMed] [Google Scholar]
- Toyoda M.; Nanbu Y.; Nakazawa Y.; Hirano M.; Inagaki M. Effect of Crystallinity of Anatase on Photoactivity for Methyleneblue Decomposition in Water. Appl. Catal. B Environ. 2004, 49 (4), 227–232. 10.1016/j.apcatb.2003.12.012. [DOI] [Google Scholar]
- Inagaki M.; Imai T.; Yoshikawa T.; Tryba B. Photocatalytic Activity of Anatase Powders for Oxidation of Methylene Blue in Water and Diluted NO Gas. Appl. Catal. B Environ. 2004, 51 (4), 247–254. 10.1016/j.apcatb.2004.02.017. [DOI] [Google Scholar]
- Iandolo B.; Hellman A. The Role of Surface States in the Oxygen Evolution Reaction on Hematite. Angew. Chem., Int. Ed. 2014, 53 (49), 13404–13408. 10.1002/anie.201406800. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang H.; Ji H.; Ma W.; Chen C.; Zhao J. Pivotal Role and Regulation of Proton Transfer in Water Oxidation on Hematite Photoanodes. J. Am. Chem. Soc. 2016, 138 (8), 2705–2711. 10.1021/jacs.5b12069. [DOI] [PubMed] [Google Scholar]
- Du C.; Zhang M.; Jang J.-W.; Liu Y.; Liu G.-Y.; Wang D. Observation and Alteration of Surface States of Hematite Photoelectrodes. J. Phys. Chem. C 2014, 118 (30), 17054–17059. 10.1021/jp5006346. [DOI] [Google Scholar]
- Chandrasekharan Meenu P.; Datta S. P.; Singh S. A.; Dinda S.; Chakraborty C.; Roy S. Polyaniline Supported G-C3N4 Quantum Dots Surpass Benchmark Pt/C: Development of Morphologically Engineered g-C3N4 Catalysts towards “Metal-Free” Methanol Electro-Oxidation. J. Power Sources 2020, 461, 228150. 10.1016/j.jpowsour.2020.228150. [DOI] [Google Scholar]
- Klotz D.; Ellis D. S.; Dotan H.; Rothschild A. Empirical in Operando Analysis of the Charge Carrier Dynamics in Hematite Photoanodes by PEIS, IMPS and IMVS. Phys. Chem. Chem. Phys. 2016, 18 (34), 23438–23457. 10.1039/C6CP04683E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi H.; Tahara T. Tracking Ultrafast Structural Dynamics by Time-Domain Raman Spectroscopy. J. Am. Chem. Soc. 2021, 143 (26), 9699–9717. 10.1021/jacs.1c02545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryn’ova G.; Lin K.-H.; Corminboeuf C. Read between the Molecules: Computational Insights into Organic Semiconductors. J. Am. Chem. Soc. 2018, 140 (48), 16370–16386. 10.1021/jacs.8b07985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C.; Sears J. S.; Coropceanu V.; Brédas J.-L. Understanding the Density Functional Dependence of DFT-Calculated Electronic Couplings in Organic Semiconductors. J. Phys. Chem. Lett. 2013, 4 (6), 919–924. 10.1021/jz3021292. [DOI] [PubMed] [Google Scholar]
- Sutton C.; Risko C.; Brédas J.-L. Noncovalent Intermolecular Interactions in Organic Electronic Materials: Implications for the Molecular Packing vs Electronic Properties of Acenes. Chem. Mater. 2016, 28 (1), 3–16. 10.1021/acs.chemmater.5b03266. [DOI] [Google Scholar]
- Cheung D. L.; Troisi A. Modelling Charge Transport in Organic Semiconductors: From Quantum Dynamics to Soft Matter. Phys. Chem. Chem. Phys. 2008, 10 (39), 5941–5952. 10.1039/b807750a. [DOI] [PubMed] [Google Scholar]
- Wang L.; Nan G.; Yang X.; Peng Q.; Li Q.; Shuai Z. Computational Methods for Design of Organic Materials with High Charge Mobility. Chem. Soc. Rev. 2010, 39 (2), 423–434. 10.1039/B816406C. [DOI] [PubMed] [Google Scholar]
- Ortmann F.; Bechstedt F.; Hannewald K. Charge Transport in Organic Crystals: Theory and Modelling. Phys. Status Solidi B 2011, 248 (3), 511–525. 10.1002/pssb.201046278. [DOI] [Google Scholar]
- O’Neill M.; Kelly S. M. Liquid Crystals for Charge Transport, Luminescence, and Photonics. Adv. Mater. 2003, 15 (14), 1135–1146. 10.1002/adma.200300009. [DOI] [Google Scholar]
- Novikov S. V.; Dunlap D. H.; Kenkre V. M.; Parris P. E.; Vannikov A. V. Essential Role of Correlations in Governing Charge Transport in Disordered Organic Materials. Phys. Rev. Lett. 1998, 81 (20), 4472–4475. 10.1103/PhysRevLett.81.4472. [DOI] [Google Scholar]
- Salleo A.; Chen T. W.; Völkel A. R.; Wu Y.; Liu P.; Ong B. S.; Street R. A. Intrinsic Hole Mobility and Trapping in a Regioregular Poly(Thiophene). Phys. Rev. B - Condens. Matter Mater. Phys. 2004, 70 (11), 1–10. 10.1103/PhysRevB.70.115311. [DOI] [Google Scholar]
- Bässler H. Charge Transport in Disordered Organic Photoconductors a Monte Carlo Simulation Study. Phys. Status Solidi B 1993, 175 (1), 15–56. 10.1002/pssb.2221750102. [DOI] [Google Scholar]
- Monroe D. Hopping in Exponential Band Tails. Phys. Rev. Lett. 1985, 54 (2), 146–149. 10.1103/PhysRevLett.54.146. [DOI] [PubMed] [Google Scholar]
- Naito H.; Okuda M. Delayed Collection Field Experiment in Amorphous Arsenic Triselenide. J. Appl. Phys. 1993, 73 (3), 1246–1251. 10.1063/1.353265. [DOI] [Google Scholar]
- Lee J. Physical Modeling of Charge Transport in Conjugated Polymer Field-Effect Transistors. J. Phys. D: Appl. Phys. 2021, 54 (14), 143002. 10.1088/1361-6463/abd271. [DOI] [Google Scholar]
- Wilson B. A.; Kerwin T. P.; Harbison J. P. Optical Studies of Thermalization Mechanisms in A-Si:H. Phys. Rev. B 1985, 31 (12), 7953–7957. 10.1103/PhysRevB.31.7953. [DOI] [PubMed] [Google Scholar]
- Choi J.; Chipara M.; Xu B.; Yang C. S.; Doudin B.; Dowben P. A. Comparison of the π-Conjugated Ring Orientations in Polyaniline and Polypyrrole. Chem. Phys. Lett. 2001, 343 (3), 193–200. 10.1016/S0009-2614(01)00469-9. [DOI] [Google Scholar]
- Feng D.-Q.; Wisbey D.; Losovyj Ya. B.; Tai Y.; Zharnikov M.; Dowben P. A. Electronic Structure and Polymerization of a Self-Assembled Monolayer with Multiple Arene Rings. Phys. Rev. B 2006, 74 (16), 165425. 10.1103/PhysRevB.74.165425. [DOI] [Google Scholar]
- Jiang J.; Ling C.; Xu T.; Wang W.; Niu X.; Zafar A.; Yan Z.; Wang X.; You Y.; Sun L.; Lu J.; Wang J.; Ni Z. Defect Engineering for Modulating the Trap States in 2D Photoconductors. Adv. Mater. 2018, 30 (40), 1804332. 10.1002/adma.201804332. [DOI] [PubMed] [Google Scholar]
- Krutko O.; Dodabalapur A. Charge Transport and Dynamic Response of Organic and Polymer Transistors. J. Appl. Phys. 2020, 127 (10), 105501. 10.1063/1.5122767. [DOI] [Google Scholar]
- Kline R. J.; McGehee M. D. Morphology and Charge Transport in Conjugated Polymers. J. Macromol. Sci. Part C 2006, 46 (1), 27–45. 10.1080/15321790500471194. [DOI] [Google Scholar]
- Sirringhaus H.; Brown P. J.; Friend R. H.; Nielsen M. M.; Bechgaard K.; Langeveld-Voss B. M. W.; Spiering A. J. H.; Janssen R. A. J.; Meijer E. W.; Herwig P.; de Leeuw D. M. Two-Dimensional Charge Transport in Self-Organized, High-Mobility Conjugated Polymers. Nature 1999, 401 (6754), 685–688. 10.1038/44359. [DOI] [Google Scholar]
- Geng Y.; Li H.-B.; Wu S.-X.; Su Z.-M. The Interplay of Intermolecular Interactions, Packing Motifs and Electron Transport Properties in Perylene Diimide Related Materials: A Theoretical Perspective. J. Mater. Chem. 2012, 22 (39), 20840–20851. 10.1039/c2jm33369d. [DOI] [Google Scholar]
- Samukhin A. N.; Prigodin V. N.; Jastrabík L. Critical Percolation and Transport in Nearly One Dimension. Phys. Rev. Lett. 1997, 78 (2), 326–329. 10.1103/PhysRevLett.78.326. [DOI] [Google Scholar]
- Zade S. S.; Bendikov M. Twisting of Conjugated Oligomers and Polymers: Case Study of Oligo- and Polythiophene. Chem. - Eur. J. 2007, 13 (13), 3688–3700. 10.1002/chem.200600819. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Clark S. J.; Brand S.; Abram R. A. First-Principles Studies of the Structural and Electronic Properties of Poly-Para-Phenylene Vinylene. J. Phys.: Condens. Matter 2004, 16 (47), 8609–8620. 10.1088/0953-8984/16/47/013. [DOI] [Google Scholar]
- Street R. A.; Northrup J. E.; Salleo A. Transport in Polycrystalline Polymer Thin-Film Transistors. Phys. Rev. B 2005, 71 (16), 165202. 10.1103/PhysRevB.71.165202. [DOI] [Google Scholar]
- Xie H.; Corish J.; Morton-Blake D. A. Thermochromic Distortions in a Poly(3-Alkylthiophene): An Atomistic Simulation Investigation. Synth. Met. 2000, 113 (1), 65–72. 10.1016/S0379-6779(99)00304-5. [DOI] [Google Scholar]
- Corish J.; Feeley D. E.; Morton-Blake D. A.; Beniere F.; Marchetti M. Atomistic Investigation of Thermochromism in a Poly(3-Alkylthiophene). J. Phys. Chem. B 1997, 101 (48), 10075–10085. 10.1021/jp971734+. [DOI] [Google Scholar]
- O’Dwyer S.; Xie H.; Corish J.; Morton-Blake D. A. An Atomistic Simulation of the Effect of Pressure on Conductive Polymers. J. Phys.: Condens. Matter 2001, 13 (10), 2395–2410. 10.1088/0953-8984/13/10/329. [DOI] [Google Scholar]
- Grozema F. C.; van Duijnen P. Th.; Berlin Y. A.; Ratner M. A.; Siebbeles L. D. A. Intramolecular Charge Transport along Isolated Chains of Conjugated Polymers: Effect of Torsional Disorder and Polymerization Defects. J. Phys. Chem. B 2002, 106 (32), 7791–7795. 10.1021/jp021114v. [DOI] [Google Scholar]
- Siebbeles L. D. A.; Berlin Y. A. Quantum Motion of Particles along One-Dimensional Pathways with Static and Dynamic Energy Disorder. Chem. Phys. 1998, 238 (1), 97–107. 10.1016/S0301-0104(98)00311-5. [DOI] [Google Scholar]
- Gores H. J.; Schweiger H.-G.; Kim W.-K.. Optimization of Electrolyte Properties by Simplex Exemplified for Conductivity of Lithium Battery Electrolytes BT - Encyclopedia of Applied Electrochemistry; Kreysa G., Ota K.; Savinell R. F., Eds.; Springer: New York, NY, 2014; pp 1387–1392, 10.1007/978-1-4419-6996-5_443. [DOI] [Google Scholar]
- Liu N.; Li T.; Zhao Z.; Liu J.; Luo X.; Yuan X.; Luo K.; He J.; Yu D.; Zhao Y. From Triazine to Heptazine: Origin of Graphitic Carbon Nitride as a Photocatalyst. ACS Omega 2020, 5 (21), 12557–12567. 10.1021/acsomega.0c01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D.; Zheng Y.; Kobielusz M.; Wang Y.; Bai Z.; Macyk W.; Wang X.; Tang J. Recent Advances in Visible Light-Driven Water Oxidation and Reduction in Suspension Systems. Mater. Today 2018, 21 (8), 897–924. 10.1016/j.mattod.2018.04.009. [DOI] [Google Scholar]
- Jiang W.; Ye L.; Li X.; Xiao C.; Tan F.; Zhao W.; Hou J.; Wang Z. Bay-Linked Perylene Bisimides as Promising Non-Fullerene Acceptors for Organic Solar Cells. Chem. Commun. 2014, 50 (8), 1024–1026. 10.1039/C3CC47204C. [DOI] [PubMed] [Google Scholar]
- Sekar A.; Moreno-Naranjo J. M.; Liu Y.; Yum J.-H.; Darwich B. P.; Cho H.-H.; Guijarro N.; Yao L.; Sivula K. Bulk Heterojunction Organic Semiconductor Photoanodes: Tuning Energy Levels to Optimize Electron Injection. ACS Appl. Mater. Interfaces 2022, 14 (6), 8191–8198. 10.1021/acsami.1c21440. [DOI] [PubMed] [Google Scholar]
- Jayakumar J.; Chou H.-H. Recent Advances in Visible-Light-Driven Hydrogen Evolution from Water Using Polymer Photocatalysts. ChemCatChem. 2020, 12 (3), 689–704. 10.1002/cctc.201901725. [DOI] [Google Scholar]
- Kosco J.; Moruzzi F.; Willner B.; McCulloch I. Photocatalysts Based on Organic Semiconductors with Tunable Energy Levels for Solar Fuel Applications. Adv. Energy Mater. 2020, 10 (39), 2001935. 10.1002/aenm.202001935. [DOI] [Google Scholar]
- Forrest S. R. Excitons and the Lifetime of Organic Semiconductor Devices. Philos. Trans. A Math. Phys. Eng. Sci. 2015, 373 (2044), 20140320. 10.1098/rsta.2014.0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka K.; Winther-Jensen B.; Nishide H. Organic π-Conjugated Polymers as Photocathode Materials for Visible-Light-Enhanced Hydrogen and Hydrogen Peroxide Production from Water. Adv. Energy Mater. 2021, 11 (43), 2003724. 10.1002/aenm.202003724. [DOI] [Google Scholar]
- Chen Z.; Jaramillo T. F.; Deutsch T. G.; Kleiman-Shwarsctein A.; Forman A. J.; Gaillard N.; Garland R.; Takanabe K.; Heske C.; Sunkara M.; McFarland E. W.; Domen K.; Miller E. L.; Turner J. A.; Dinh H. N. Accelerating Materials Development for Photoelectrochemical Hydrogen Production: Standards for Methods, Definitions, and Reporting Protocols. J. Mater. Res. 2010, 25 (1), 3–16. 10.1557/JMR.2010.0020. [DOI] [Google Scholar]
- Ager J. W.; Shaner M. R.; Walczak K. A.; Sharp I. D.; Ardo S. Experimental Demonstrations of Spontaneous, Solar-Driven Photoelectrochemical Water Splitting. Energy Environ. Sci. 2015, 8 (10), 2811–2824. 10.1039/C5EE00457H. [DOI] [Google Scholar]
- Zhang K.; Yu Q.; Zhu L.; Liu S.; Chi Z.; Chen X.; Zhang Y.; Xu J. The Preparations and Water Vapor Barrier Properties of Polyimide Films Containing Amide Moieties. Polymers. 2017, 9, 677. 10.3390/polym9120677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K.; Li F.; Wang L.; Daniel Q.; Chen H.; Gabrielsson E.; Sun J.; Sun L. Immobilization of a Molecular Ruthenium Catalyst on Hematite Nanorod Arrays for Water Oxidation with Stable Photocurrent. ChemSusChem 2015, 8 (19), 3242–3247. 10.1002/cssc.201500730. [DOI] [PubMed] [Google Scholar]
- Sathre R.; Greenblatt J. B.; Walczak K.; Sharp I. D.; Stevens J. C.; Ager J. W.; Houle F. A. Opportunities to Improve the Net Energy Performance of Photoelectrochemical Water-Splitting Technology. Energy Environ. Sci. 2016, 9 (3), 803–819. 10.1039/C5EE03040D. [DOI] [Google Scholar]
- Clough A. J.; Yoo J. W.; Mecklenburg M. H.; Marinescu S. C. Two-Dimensional Metal-Organic Surfaces for Efficient Hydrogen Evolution from Water. J. Am. Chem. Soc. 2015, 137 (1), 118–121. 10.1021/ja5116937. [DOI] [PubMed] [Google Scholar]
- Weisz A. D.; Regazzoni A. E.; Blesa M. A. ATR-FTIR Study of the Stability Trends of Carboxylate Complexes Formed on the Surface of Titanium Dioxide Particles Immersed in Water. Solid State Ion. 2001, 143 (1), 125–130. 10.1016/S0167-2738(01)00840-2. [DOI] [Google Scholar]
- Queffélec C.; Petit M.; Janvier P.; Knight D. A.; Bujoli B. Surface Modification Using Phosphonic Acids and Esters. Chem. Rev. 2012, 112 (7), 3777–3807. 10.1021/cr2004212. [DOI] [PubMed] [Google Scholar]
- Niu F.; Wang D.; Li F.; Liu Y.; Shen S.; Meyer T. J. Hybrid Photoelectrochemical Water Splitting Systems: From Interface Design to System Assembly. Adv. Energy Mater. 2020, 10 (11), 1900399. 10.1002/aenm.201900399. [DOI] [Google Scholar]
- Kochergin Y. S.; Beladi-Mousavi S. M.; Khezri B.; Lyu P.; Bojdys M. J.; Pumera M. Organic Photoelectrode Engineering: Accelerating Photocurrent Generation via Donor-Acceptor Interactions and Surface-Assisted Synthetic Approach. J. Mater. Chem. A 2021, 9 (11), 7162–7171. 10.1039/D0TA11820F. [DOI] [Google Scholar]
- Wang M.; Yang Y.; Shen J.; Jiang J.; Sun L. Visible-Light-Absorbing Semiconductor/Molecular Catalyst Hybrid Photoelectrodes for H2 or O2 Evolution: Recent Advances and Challenges. Sustain. Energy Fuels 2017, 1 (8), 1641–1663. 10.1039/C7SE00222J. [DOI] [Google Scholar]
- Nie W.; Blancon J.-C.; Neukirch A. J.; Appavoo K.; Tsai H.; Chhowalla M.; Alam M. A.; Sfeir M. Y.; Katan C.; Even J.; Tretiak S.; Crochet J. J.; Gupta G.; Mohite A. D. Light-Activated Photocurrent Degradation and Self-Healing in Perovskite Solar Cells. Nat. Commun. 2016, 7 (1), 11574. 10.1038/ncomms11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Lin Q.; Chmiel F. P.; Sakai N.; Herz L. M.; Snaith H. J. Efficient Ambient-Air-Stable Solar Cells with 2D-3D Heterostructured Butylammonium-Caesium-Formamidinium Lead Halide Perovskites. Nat. Energy 2017, 2 (9), 17135. 10.1038/nenergy.2017.135. [DOI] [Google Scholar]
- Wang Q.; Chen B.; Liu Y.; Deng Y.; Bai Y.; Dong Q.; Huang J. Scaling Behavior of Moisture-Induced Grain Degradation in Polycrystalline Hybrid Perovskite Thin Films. Energy Environ. Sci. 2017, 10 (2), 516–522. 10.1039/C6EE02941H. [DOI] [Google Scholar]
- Azpiroz J. M.; Mosconi E.; Bisquert J.; De Angelis F. Defect Migration in Methylammonium Lead Iodide and Its Role in Perovskite Solar Cell Operation. Energy Environ. Sci. 2015, 8 (7), 2118–2127. 10.1039/C5EE01265A. [DOI] [Google Scholar]
- Hisatomi T.; Kubota J.; Domen K. Recent Advances in Semiconductors for Photocatalytic and Photoelectrochemical Water Splitting. Chem. Soc. Rev. 2014, 43 (22), 7520–7535. 10.1039/C3CS60378D. [DOI] [PubMed] [Google Scholar]
- Fan S.III-Nitride Nanowire Photoelectrodes: Design, Epitaxial Growth, and Solar-to-Fuels Production. Ph.D. Thesis, McGill University, 2016. [Google Scholar]
- Grimm A.; de Jong W. A.; Kramer G. J. Renewable Hydrogen Production: A Techno-Economic Comparison of Photoelectrochemical Cells and Photovoltaic-Electrolysis. Int. J. Hydrog. Energy 2020, 45 (43), 22545–22555. 10.1016/j.ijhydene.2020.06.092. [DOI] [Google Scholar]
- Schneidewind J. How Much Technological Progress Is Needed to Make Solar Hydrogen Cost-Competitive?. Adv. Energy Mater. 2022, 12, 2200342. 10.1002/aenm.202200342. [DOI] [Google Scholar]
- Mulligan C. J.; Wilson M.; Bryant G.; Vaughan B.; Zhou X.; Belcher W. J.; Dastoor P. C. A Projection of Commercial-Scale Organic Photovoltaic Module Costs. Sol. Energy Mater. Sol. Cells 2014, 120, 9–17. 10.1016/j.solmat.2013.07.041. [DOI] [Google Scholar]
- Machui F.; Hösel M.; Li N.; Spyropoulos G. D.; Ameri T.; Søndergaard R. R.; Jørgensen M.; Scheel A.; Gaiser D.; Kreul K.; Lenssen D.; Legros M.; Lemaitre N.; Vilkman M.; Välimäki M.; Nordman S.; Brabec C. J.; Krebs F. C. Cost Analysis of Roll-to-Roll Fabricated ITO Free Single and Tandem Organic Solar Modules Based on Data from Manufacture. Energy Environ. Sci. 2014, 7 (9), 2792–2802. 10.1039/C4EE01222D. [DOI] [Google Scholar]
- Chatzisideris M. D.; Laurent A.; Christoforidis G. C.; Krebs F. C. Cost-Competitiveness of Organic Photovoltaics for Electricity Self-Consumption at Residential Buildings: A Comparative Study of Denmark and Greece under Real Market Conditions. Appl. Energy 2017, 208, 471–479. 10.1016/j.apenergy.2017.10.003. [DOI] [Google Scholar]
- Prentice A. W.; Zwijnenburg M. A. Hydrogen Evolution by Polymer Photocatalysts; a Possible Photocatalytic Cycle. Sustain. Energy Fuels 2021, 5 (10), 2622–2632. 10.1039/D1SE00059D. [DOI] [Google Scholar]
- Francàs L.; Corby S.; Selim S.; Lee D.; Mesa C. A.; Godin R.; Pastor E.; Stephens I. E. L.; Choi K.-S.; Durrant J. R. Spectroelectrochemical Study of Water Oxidation on Nickel and Iron Oxyhydroxide Electrocatalysts. Nat. Commun. 2019, 10 (1), 5208. 10.1038/s41467-019-13061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy C.; Sebok B.; Scott S. B.; Fiordaliso E. M.; Sørensen J. E.; Bodin A.; Trimarco D. B.; Damsgaard C. D.; Vesborg P. C. K.; Hansen O.; Stephens I. E. L.; Kibsgaard J.; Chorkendorff I. Impact of Nanoparticle Size and Lattice Oxygen on Water Oxidation on NiFeOxHy. Nat. Catal. 2018, 1 (11), 820–829. 10.1038/s41929-018-0162-x. [DOI] [Google Scholar]
- P M. B.; L P. F. G.; D M. T.; F H.; M R. L.; E D. K.; R M.; E Y. L. P.; B R. M.; R D. J.; V L.; J N. G.; H S.; H Z. R.; S E. M.; H H. T.; J D. D.; A A.-G.; E H. J.; P B. C. Self-Driving Laboratory for Accelerated Discovery of Thin-Film Materials. Sci. Adv. 2021, 6 (20), eaaz8867 10.1126/sciadv.aaz8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M.; Tran K.; Min Y.; Wang C.; Wang Z.; Dinh C.-T.; De Luna P.; Yu Z.; Rasouli A. S.; Brodersen P.; Sun S.; Voznyy O.; Tan C.-S.; Askerka M.; Che F.; Liu M.; Seifitokaldani A.; Pang Y.; Lo S.-C.; Ip A.; Ulissi Z.; Sargent E. H. Accelerated Discovery of CO2 Electrocatalysts Using Active Machine Learning. Nature 2020, 581 (7807), 178–183. 10.1038/s41586-020-2242-8. [DOI] [PubMed] [Google Scholar]
- MacLeod B. P.; Parlane F. G. L.; Dettelbach K. E.; Elliott M. S.; Rupnow C. C.; Morrissey T. D.; Haley T. H.; Proskurin O.; Rooney M. B.; Taherimakhsousi N.; Dvorak D. J.; Chiu H. N.; Waizenegger C.; Ocean K.; Mokhtari M.; Berlinguette C. P. A Self-Driving Laboratory Advances the Pareto Front for Materials Properties. Nat. Commun. 2022, 13, 995. 10.1038/s41467-022-28580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave A.; Mitchell J.; Kandasamy K.; Wang H.; Burke S.; Paria B.; Póczos B.; Whitacre J.; Viswanathan V. Autonomous Discovery of Battery Electrolytes with Robotic Experimentation and Machine Learning. Cell Rep. Phys. Sci. 2020, 1 (12), 100264. 10.1016/j.xcrp.2020.100264. [DOI] [Google Scholar]
- Pendleton I. M.; Caucci M. K.; Tynes M.; Dharna A.; Nellikkal M. A. N.; Li Z.; Chan E. M.; Norquist A. J.; Schrier J. Can Machines “Learn” Halide Perovskite Crystal Formation without Accurate Physicochemical Features?. J. Phys. Chem. C 2020, 124 (25), 13982–13992. 10.1021/acs.jpcc.0c01726. [DOI] [Google Scholar]
- Gomes C. P.; Selman B.; Gregoire J. M. Artificial Intelligence for Materials Discovery. MRS Bull. 2019, 44 (7), 538–544. 10.1557/mrs.2019.158. [DOI] [Google Scholar]
- Hu S.; Li W. X. Sabatier Principle of Metal-Support Interaction for Design of Ultrastable Metal Nanocatalysts. Science 2021, 374 (6573), 1360–1365. 10.1126/science.abi9828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Addicoat M. A.Photoelectrochemical Water Splitting by Triazine Based Covalent Organic Framework; Zenodo, 2020, 10.5281/zenodo.4081292. [DOI]