

Abstract
This Review provides an overview of the emerging concepts of catalysts, membranes, and membrane electrode assemblies (MEAs) for water electrolyzers with anion-exchange membranes (AEMs), also known as zero-gap alkaline water electrolyzers. Much of the recent progress is due to improvements in materials chemistry, MEA designs, and optimized operation conditions. Research on anion-exchange polymers (AEPs) has focused on the cationic head/backbone/side-chain structures and key properties such as ionic conductivity and alkaline stability. Several approaches, such as cross-linking, microphase, and organic/inorganic composites, have been proposed to improve the anion-exchange performance and the chemical and mechanical stability of AEMs. Numerous AEMs now exceed values of 0.1 S/cm (at 60–80 °C), although the stability specifically at temperatures exceeding 60 °C needs further enhancement. The oxygen evolution reaction (OER) is still a limiting factor. An analysis of thin-layer OER data suggests that NiFe-type catalysts have the highest activity. There is debate on the active-site mechanism of the NiFe catalysts, and their long-term stability needs to be understood. Addition of Co to NiFe increases the conductivity of these catalysts. The same analysis for the hydrogen evolution reaction (HER) shows carbon-supported Pt to be dominating, although PtNi alloys and clusters of Ni(OH)2 on Pt show competitive activities. Recent advances in forming and embedding well-dispersed Ru nanoparticles on functionalized high-surface-area carbon supports show promising HER activities. However, the stability of these catalysts under actual AEMWE operating conditions needs to be proven. The field is advancing rapidly but could benefit through the adaptation of new in situ techniques, standardized evaluation protocols for AEMWE conditions, and innovative catalyst-structure designs. Nevertheless, single AEM water electrolyzer cells have been operated for several thousand hours at temperatures and current densities as high as 60 °C and 1 A/cm2, respectively.
1. Introduction
Hydrogen has played a key role throughout the industrial life of humankind, and the global demand for H2 has continuously increased, in fact tripling since 1975.1,2 Today’s global H2 production exceeds 70 million metric tons (MMTs)/year and is consumed by the oil and gas industry and by metal refineries or turned into value-added products such as NH3, feedstock chemicals such as CH3OH, or specialty chemicals.1 H2 can be considered a commodity of increasing need, with its importance already being reflected in its 117 billion US$ global market value.3Figure 1 shows the trend for the global demand for H2 divided into end-use sectors. The actual H2 demand per sector depends on the country, e.g., in the United States, the oil and gas sector consumes 80% of the produced H2. The future demand for H2 may experience an additional increase due to H2’s physical properties such as its high gravimetric standard heat of formation (being the highest among fuels)4 and its standard heat of formation value (H2’s high heating value is 142 MJ/kg5), which is up to 3 times higher than that for liquid hydrocarbon fuels.6 Unfortunately, the volumetric density of H2 of 8 MJ/L is 4 times lower than the 32 MJ/L value of gasoline,7 thus requiring a high storage volume or significant gas compression.2
Figure 1.

Historical trend of the global usage of H2 predominantly produced by utilizing a fossil fuel feedstock divided into industrial sectors. “Other pure” stands for applications needing high-purity H2, “DRI” stands for direct reduced iron steel production, and “Other mixed” stands for applications using H2 as a mixture gas, e.g., fuel or feedstock synthesis gas. Made from ref (1). Copyright 2019 U.S. Department of Energy.
Since the late 1950s, steam methane reforming (SMR) has been predominantly used to produce H2 followed by coal gasification and water electrolysis (WE), although the latter only contributes 2–4% to today’s global H2 production.8,9 The heating value of CH4 is high (the HHV is 55.5 MJ/kg5), making the production of H2 via SMR, which is in the range of 2 €/kg H2,9−11 economically attractive. Today, 96% of H2 is produced from fossil fuel-based feeds (48% from natural gas, 30% from heavy oils and naphtha, and 18% from coal).1,6 H2 production from fossil-based sources and moreover from CH4, which is an ∼30 times more potent greenhouse gas than CO2,12,13 is a net emitter of CO2 and other air pollutants. On the basis of calculations and a large set of reported data, Sun et al.14 concluded average CO2/H2 values of 9 kg/kg and 75.4 kg/MJ, translating into 720 MMT/year of CO2 emitted for 70 MMT/year H2 produced from SMR. The latter suggests that H2 produced from SMR alone contributes 1.7% to the 43.1 billion metric tons worldwide emissions from humans in 2019.15,16 Furthermore, the SMR process also produces H2 of low purity (95–98%), requiring upgrading steps such as pressure swing absorption for many applications (e.g., fuels, specialty chemicals, and the ceramic and electronics industries).17 A mechanical compression step, which can be costly, also needs to be added.18 WEs have an advantage of generating higher-purity and already partially compressed H2.
H2 as a fuel for the H2 economy is still a topic of interest.19,20 Much of the interest is driven by our increasing demand for energy based on cleaner sources.21 Correspondingly, H2 is considered for energy storage (ES) and as a fuel.11,21 The driving forces for the H2 economy are different across the globe depending on the resources, potential for energy generation, and political landscape of a country.
To understand the feasibility for a specific H2 production or storage route, technical gaps need to be identified and examined while keeping the cost in mind. The complete cycle needs to be considered including the end use of H2, which can be manifold and may be different depending on the locations of H2 production and consumption. H2 is suitable for short-, medium-, and long-term energy storage. H2 could, e.g., be reelectrified and injected back into the grid considering payback options known as power to power, which is used as a grid service to balance the grid when demand is high and production is low, or that known as price arbitrage, or to avoid the building of new grid connections.11,22−24 In addition, H2 can also be transformed to a liquid organic hydrogen carrier (LOHC), enabling safer transport for later use.25−29
In this Review, the feasibility of H2 produced from electrochemical water splitting coupled with renewable energies is of interest. The anion-exchange membrane water electrolysis (AEMWE) is one of three types of low-temperature (<100 °C) WEs. The other two are proton-exchange membrane water electrolysis (PEMWE) and traditional electrolysis, which uses highly caustic KOH as the electrolyte and a porous separator. Among the three low-temperature WEs, AEMWE is the least-mature technology, and prior to implementation, significant technological hurdles need to be overcome. Many of the hurdles lie in the AEMWE components’ chemistries, which will be discussed in this Review. References are made where needed to scientific knowledge established for well-studied PEMWEs and fuel cells (FCs). This Review differs from recent publications30−41 as it presents a comprehensive analysis of all components of an AEMWE up to the single-cell level and reviews AEMWE single-cell performances for cells that have shown at least 100 h of operation. Performance results and needs of materials development and engineering are given based on high-level analyses.
2. To and from H2 Produced via Water Electrolysis: Sources, Cost, Conversion, and Principles
Biomass
and water are renewable sources that potentially allow
clean H2 production.6,42 Vast amounts of biomass
are available, but the production of H2 from water is more
advanced; hence, H2 derived from biomass is viewed to be
implemented in the long term. H2 production from water splitting 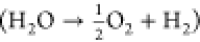 can be divided into low- and high-temperature
electrolysis, thermochemical water splitting, and photoelectrochemical
processes. Due to the absence of carbon-based reaction fuels, water
splitting offers the cleanest way of producing H2, provided
clean sources of electricity are used.
can be divided into low- and high-temperature
electrolysis, thermochemical water splitting, and photoelectrochemical
processes. Due to the absence of carbon-based reaction fuels, water
splitting offers the cleanest way of producing H2, provided
clean sources of electricity are used.
Fossil fuel-free energy sources, such as nuclear and renewables, can be low or CO2-free forms of energy. Nuclear offers several advantages as vast amounts are available and excess energy during low demands can be stored via, e.g., electrolysis. The outlet temperatures of nuclear reactors are in the range of 300–950 °C. Such a range can be attractive for higher-temperature electrolyzers, e.g., solid oxide electrolysis cells and thermochemical water splitting. However, these high-temperature electrolysis methods are not yet mature and suffer material-corrosion issues above 100 °C.6,43
Solar and wind provide intermittent forms of energy. Solar is currently the fastest growing energy source due to the many investments made globally. Solar energy supplied just above 2% of the global electricity usage in 2018, while wind energy provided ∼5%.44 The global capacity and usage of wind energy may well grow, as it is not costly and the technology is continuously advancing, even though on-shore wind farms require thousands of acres of land.44 Electrochemical WE is best suited in combination with wind energy, which calls for storage in the MW range.45 WE not only is able to provide large-scale storage but also offers medium- and longer-term storage unlike, e.g., flywheels, which are low cost but only allow short-term storage.46 In addition, WEs can accept high-current inputs per surface area, operate in dynamic modes, and can be ramped up quickly, which are all requirements for storage of intermittent energy sources.11,45,41 Batteries are not suitable as a storage option for wind energy because they only accept low currents per surface area and have high self-discharge rates.32,47,48 Thermal molten salts are another high-energy storage density option being developed.49 However, it is based on exchanging heat. The heat is stored in a molten salt (which is thermally insulated) and released when needed. In addition, a WE is better suited than a battery for operations in cold climates because WEs can be heated using internal electrical currents without compromising their lifetime.50,51 Unfortunately, the intermittent nature of renewable energy lowers the annual operating hours, thus increasing the cost of the technology.9
The coupling of wind and solar energy with WEs provides many advantages; nevertheless, clear challenges exist. Table 1 provides a summary of the H2 production characteristics from SMR and WEs. It is seen that a challenge of H2 produced by WEs is the cost, which can be captured in the sum of the operating (OPEX) and capital (CAPEX) investment costs.
Table 1. H2 Production Characteristics of Steam Methane Reforming (SMR) versus Electrochemical (<100 °C) Water Electrolyzers (WEs).
| characteristic | SMR | WE |
|---|---|---|
| feed | fossil fuel | H2O |
| estimated CO2 emissions per kg H2 (kg/kg H2) | 9a | 0.4–0.9b |
| global CO2 emissions (MMT/year) (2019) | 720 | 32–72b |
| % of global CO2 emissions 2019 (est) | 1.7 | 0.08–0.17b |
| H2 production cost (2019) (€/kgH2) | 2 | >3.8c |
| driving force for reaction | heat | electrical energy |
| catalysts | sulfur- and coke-tolerant (nickel, nanosized nickel, platinum, rhodium) | acidic: Pt (cathode), IrO2 (anode) |
| alkaline: nickel-based or Pt (cathode), often nickel-based (anode) | ||
| H2 purityd (%) | 95–98 | PEMWE: 99.9–99.999e |
| AEMWE: 99.4 |
Taken from ref (15).
The data are for traditional alkaline water electrolyzers coupled with wind energy and estimated from refs (52−55). Fewer carbon-footprint studies are available for water electrolysis coupled with wind than from SMR processes. However, all of these studies consistently show water electrolyzers coupled with wind to be one of the lowest CO2 emitters.
An estimate of 3.8 €/kg H2 is for coupling with wind, minimal operating hours of 7000 of 8760 per year, i.e., 80% capacity, a CAPEX of 800 €/kW, WE efficiency of 80%, and renewable electricity cost of 70 € M/Wh.10
H2 purity without purification processes as pressure or temperature swing adsorption.
Typically at 30 bar outlet pressure.
For low-temperature WEs the price of electricity is often taken as the OPEX value because electricity prices often dominate the cost of H2 production and reliable data for operating large-scale WEs are lacking.9 However, it is advisible to also include the cost of water (which needs to be of drinking water quality), specifically for operation at remote locations, as well as the WE maintenance costs.
The CAPEX cost is typically given as the investment cost per kW of electrical capacity and sometimes as the cost per nominal H2 production rate (in m3/h). In general, the definition of the CAPEX value in the form of cost per nominal capacity or cost per nominal H2 production rate is not complete.56 Neither of the two include relevant electrolyzer information such as the lifetime and the H2 production efficiency. A few approaches have been suggested to calculate the contribution of the CAPEX to the cost of H2 production in order to include the actual performance capabilities of different WEs.56 Villagra and Millet defined the CAPEX contribution to the total H2 cost (“CAPEX”) in €/kg of H2 produced as follows:56
| 1 |
In eq 1, n is the number of electrons (2 for H2 electrolysis), F is the Faraday constant (96 485 C/mole–), IC is the initial WE cost, LT and A are the lifetime and geometrical electrode area of the WE, respectively, MH2 is the molecular H2 weight, and jo is the operational current density. This definition gives a clearer indication of the WE characteristics that influence the H2 production cost as compared to the traditionally used CAPEX values. However, eq 1 does not include the efficiency of the WE, which could simply be introduced as a term in the dividend in eq 1.
From eq 1 it is seen that the CAPEX contribution to the cost of H2 produced decreases with an increase in the WE lifetime, electrode surface area, operating current density (j), and average efficiency of the H2 produced.56 Logically, a lower initial cost of the WE, which is influenced by materials and manufacturing costs, also reduces the cost of H2 production. To obtain the full cost of H2 production, the OPEX and CAPEX are combined. The joint OPEX and CAPEX costs define the technical targets the technology needs to achieve to be competitive for deployment. According to Table 1, WEs must produce H2 at a cost below 2 €/kgH2 to be cost competitive. The price for H2 production by WE coupled with wind energy could be below 3.8 €/kgH2 if the WE is used at an 80% annual capacity, has a CAPEX value of 800 €/kW, and has a cell efficiency of 80% at OPEX costs corresponding to renewable electricity costs of 70 €/MWh.9 Proost and others suggest that WEs could become more competitive to SMR as CAPEX prices of WEs are predicted to decrease with an increase in manufacturing (taking advantage of the economy of scale) and to a lesser extent also continue to decrease through additional research and development contributions.9,52,57 This seems reasonable considering that the CAPEX prices of PEMWEs decreased by 1 order of magnitude between 2000 and 2010 and continue to steadily decrease, as demonstrated in Table 2.18−59 These trends suggest that H2 production by WEs will become competitive if low-cost electricity is used.9,60
Table 2. Evolution of CAPEX Values for PEMWEs.
2.1. H2 Conversion to Chemical Raw Materials or LOHCs
A scheme demonstrating the coupling of wind with WE and the possible uses of the stored H2 is shown in Figure 2. Much of the wind resources are located in remote areas, and transport of H2 in pipelines is only feasible over limited distances in the 100–200-mile range.
Figure 2.

Schematic for the coupling of renewable (wind or solar) energy with water electrolysis. The figure shows options for short-, medium-, and long-term storage for the energy in the form of H2 and possible end uses including payback options. LOHC stands for liquid organic hydrogen carrier.
Several studies61,62 proposed to transform the H2 into a chemical raw material or LOHC (liquid organic hydrogen carrier), which are H2 carriers that can allow for safer transportation. The chemical raw materials could be CH3OH, dimethyl ether, gasoline, ammonia, and Fischer–Tropsch fuels. Examples of LOHC systems are N-alkylcarbazoles and derivatives.63 An early LOHC system was toluene/cyclohexane, but dehydrogenation in the liquid phase with easy condensation of the evaporated parts of the H2 carrier is also possible for higher-boiling aromatics and heteroaromatics.27,64,65 The LOHC is formed by a catalytic hydrogenation and a reversible dehydrogenation reaction. LOHC systems are liquids and can be used in the existing fuel infrastructure. LOHCs are also reloadable without the release of CO2. LOHCs offer higher volumetric energy densities than H2 and can be a room-temperature, long-term storage option. They could serve as a H2 supplier for arbitrary applications such as energy or specialty chemicals. CH3OH could be formed from CO2 concentrated from the atmosphere and H2 from electrolysis. CH3OH has a high acceptance level due to its similarity to existing fuels, although a 2014 techno-economic study showed that the cost of CH3OH via the route of using clean H2 is over the market price.66 Similarly, NH3 can be formed by electrolysis,67 which according to recent studies releases less CO2, when coupled with wind or solar energy, than the traditional Haber–Bosch process.68
2.2. Electrochemical Water Electrolyzers
Low-temperature water electrolyzers (WEs) can be divided into alkaline and acidic systems. They are further divided into finite and zero-gap electrolyzers (Figure 3). The schematics show the principles of a single WE cell, while an actual system consists of an assembly of many cells known as a stack. The anode and the cathode in a WE are spaced using a separator to avoid mixing of the H2 and O2 gases.
Figure 3.

Schematic of the three types of WEs as (a) traditional alkaline finite WE (AWE), (b) zero-gap PEMWE running under acidic conditions using an H+ conducting membrane, and (c) zero-gap AEMWE utilizing an OH– conducting membrane. The goal is to use noble metal-free catalysts for the cathode and anode of an AEMWE.
The terms finite and zero gap are related to the distance of the separator between the anode and the cathode, where the O2 evolution reaction (OER) and the H2 evolution reaction (HER) take place. Finite-gap alkaline WEs employ a porous separator and aqueous, e.g., 30 wt % (5 M) KOH, conducting solutions (Figure 3a).50,69 This is a proven technology and has been deployed in MW scales since the late 1950s.50,70 A well-known advantage of alkaline conditions, specifically pH > 13, is the stability of the non-platinum group metal (non-PGM)-based catalysts for the OER and HER, unlike for acidic media needing platinum group metal catalysts.42,44,50 Typically, high-surface-area Raney nickel electrodes are used in an infinite-gap alkaline electrolyzer.71,72 The use of a porous separator, such as Zircon and Perl UTP 500,73−75 calls for a large distance (>2 mm) between the anode and cathode to reduce H2 and O2 gas crossover, which unfortunately is accompanied by a high ohmic resistance due to the direct dependency of ionic resistance on electrolyte thickness. The latter limits the maximum current densities (jmax) that can be reached.46,56,76 Typically the jmax value for a finite-gap alkaline WE is 0.25 A/cm2, which is too low for integration with renewables, such as wind, that need ES technologies that are able to accept current densities in the several A/cm2 range as well as with fast dynamic responses.24,44 New WE designs are being developed that incorporate one electrode of minimized or even zero-gap distance to the separator.42,76−78 Examples explored are alkali-doped ion-solvating membranes in combination with, e.g., 24 wt % KOH electrolytes.79−81 Single-cell tests using a KOH-doped ion-solvating membrane and Raney nickel electrodes yielded a low cell voltage of 1.8 V at j values of 1.7 A/cm2.80
The zero-gap WE design reduces the internal resistance as thin polymer-based membranes of low H2 and O2 crossover are employed. Proton-exchange membranes (PEMs, also referred to as cation-exchange membranes) and anion-exchange membranes (AEMs) are used for acidic (Figure 3b) and alkaline (Figure 3c) zero-gap WEs, respectively. Consequently, zero-gap WEs are predicted to achieve higher j values than finite-gap electrolyzers. In the case of commercial PEMWEs, j values of up to 1–3 A/cm2 at lifetimes (LTs) of 15 000–20 000 h using membranes as thin as 50–200 μm PEMs have been demonstrated. PEMWEs are much more mature than AEMWEs. This is related to the fact that PEMs, which typically consist of a perfluorosulfonic acid that is known under the trademarks Nafion and Aquivion, have a significantly higher stability than anion-exchange membranes (AEMs), although the stability of Nafion is limited to 80 °C operations. In fact, PEMWEs using a Nafion separator are typically operated at 60 °C.18 Only in recent years have achievements been made to increase the stability of AEMs and single-cell AEMWEs run in the several A/cm2 range, although proof of extended durability and performance is still needed.73,77,82−84
Recent developments in the field of bipolar membranes (BPMs) have opened new opportunities.85 The BPM’s principal lies in linking the advantages of the PEM and AEM system where low-cost anode materials (alkaline media) and active and durable cathode catalysts (acidic media) are used. In the BPM system, a cation-exchange membrane (CEM) and an AEM are in direct contact to form a bipolar interface (Figure 4).85 A water dissociation or water recombination catalyst is added between the two membranes to enhance the performance.86−88 Activities of such bilayer catalysts have been shown to be close to those of alkaline HER catalysts.86
Figure 4.
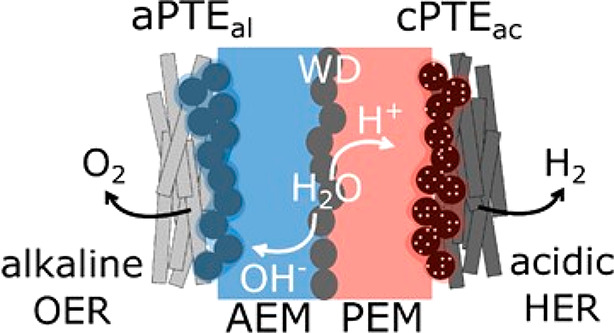
Schematic of a bipolar membrane (BPM) WE employing a solid AEM (blue) and PEM (red) with a water-dissociation (WD) catalyst layer located at the AEM|PEM interface. The OER and HER take place at the anode, indicated as aPTEal, and the cathode, indicated as cPTEac, respectively. Reprinted with permission from Open Access article.89 Copyright 2021 Royal Society of Chemistry under CC Attribution 3.0 Unported License https://creativecommons.org/licenses/by/3.0/.
Another difference between finite and zero-gap alkaline WEs is that zero-gap WEs operate on a pure water feed or dilute alkali electrolytes.90 The use of pure water theoretically eliminates issues related to the reaction of cations such as K+ with CO2 to form carbonates in OH– environments but requires an OH– conductive polymer, an anion-exchange ionomer (AEI), to be present in the catalyst layer.46,91,92 However, even at low KOH concentrations, or in pure water, the complete exclusion of CO2 is a challenge because CO2 is present in the air and can easily dissolve in water (0.75 g/L at 50 °C). Much of the research and development on zero-gap systems has focused on PEMWEs because the implementation of AEMWEs still strongly depends on the availability of AEMs, which show long-term stability at elevated temperatures, although low-power (e.g., 0.5–1 N·mH23/h) AEMWE systems are commercially available.39,93,94 The commercial system from Enapter (formerly Acta) offers high-purity (99.9%) H2 and 99.999% H2 with an optional dryer.94 An advantage of membranes, i.e., the zero-gap WEs, is to obtain a higher-purity H2 directly from the cell (section 6). WEs should last >50 000 h under high j values and ideally also under pressure of 50–80 bar and higher (≥60 °C) temperatures. Today’s commercial PEMWEs have shown long (at least 20 000 h) lifetimes at low temperatures and 30 bar.39,43
2.3. Thermodynamics for WEs
The water splitting reactions in acidic and alkaline media are overall comparable, although in alkaline media OH– is the conducting ion, while in acidic media H+ assumes this role. In the case of alkaline WEs, the reactions are as follows,
| 2 |
| 3 |
| 4 |
where SHE stands for standard hydrogen electrode and E° is the reversible potential. In alkaline conditions, the cathode needs two water molecules per H2 produced, and thus the water transport from the anode to the cathode is a crucial factor to be considered in the cell design, materials selection, and operation mode of the WE. The OH– needed at the anode is provided through the cathode reaction and needs to be transported through the catalyst layer and membrane to the catalyst sites in the anode layer.
The standard reversible potential (E°rev) for the water splitting reaction is −1.23 V, i.e., the reaction is endothermic and does not occur at a cell voltage (Ecell) below 1.23 V. For the water splitting reaction, the standard enthalpy and entropy are 285.84 and 163.6 kJ/molH2, respectively.43 This difference indicates a large entropy change of the reaction system when liquid H2O changes into the two gaseous products H2 and O2. Electrolysis at higher T values (>100 °C), or more precisely using a H2O steam reactant, reduces the energy requirements of the electrolysis as this entropy change is eliminated. Reference is also made to the thermoneutral voltage (E°tn), which for the water splitting reaction is 1.48 V, reflecting the transition point between endothermic and exothermic, i.e., the potential at which the reaction proceeds without heat input.
To understand the WE cell performance, the difference between Eanode and Ecathode, i.e., the cell potential (Ecell), is plotted versus j (Figure 5). Ecell depends on the reversible potential (Erev). However, an operating WE also experiences voltage losses as overpotentials (η) at the anode (ηan) and cathode (ηcat) and iR drops induced by the cell resistance (Rcell):
| 5 |
In eq 5, Rcell is a lump resistance term made of a number of resistances including contributions from the membrane, polar plates, interfaces, system circuits, and mass-transport losses. Mass-transport losses are losses that result from the nonstoichiometric supply of reactants to the active catalyst centers.95 The formation of oxygen and hydrogen bubbles is one possible effect. Product gas bubbles in contact with the electrodes reduce the electrode contact with the liquid water, which in turn decreases the active electrode areas. All of the voltage loss terms in eq 5 increase with an increase in the current (i), i.e., j as shown in Figure 5. For well-designed low-temperature WEs, iR drops across the catalyst layers and other components such as the gas diffusion layer (GDL) and bipolar plates (BPs) are negligible.96 For today’s AEMWEs, the membrane resistance dominates the voltage.97 This of course can change with the continued development of AEMs.
Figure 5.
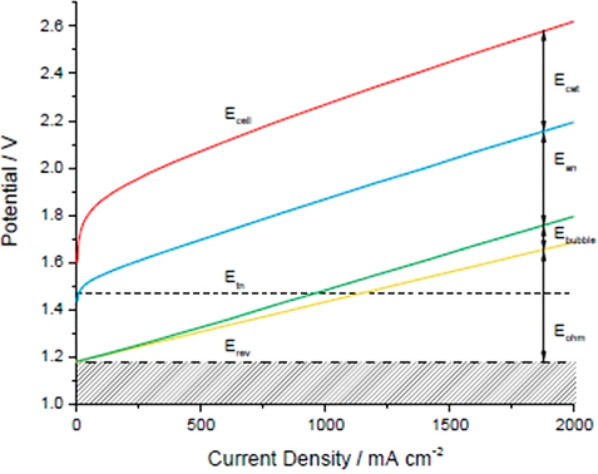
Typical potential (Ecell)–current density (j) curves using arbitrary values demonstrating the cumulative contributions of different voltage losses. The anode and cathode voltages (Ean and Ecat) can be reduced by using catalysts of higher activity and improved catalyst layers, while the ohmic voltage (Eohm) loss depends on both electrode conductivity and membrane ionic conductivity. Both,= potential losses due to gas bubble formation (Ebubble) and ohmic losses (Eohm) are reflected in the iRcell term shown in the simplified eq 5. Many factors influence the actual E losses thar are demonstrated in the figure. Reprinted with permission from (98). Copyright 2017 DTU Energy, Department of Energy and Energy Storage.
As seen from Figure 6, a higher temperature (T) lowers Ecell, which is beneficial for the electrochemical reactions, increases the counterion transport, and facilitates H2 and O2 separation as the gas solubility decreases with increasing T.99−101 A higher pressure (P) increases Ecell according to the Nernst equation, although the increase is not pronounced. In fact, the availability of compressed H2 directly from a WE is a benefit, reducing the cost of mechanical H2 compression provided that the compression remains in the 30–50 bar range.24,35,102 WE operation at higher pressures can require the reinforcement of thinner membranes (e.g., in the case of PEMWEs < 125 μm)103 to increase their mechanical strength and achieve higher WE efficiencies.
Figure 6.
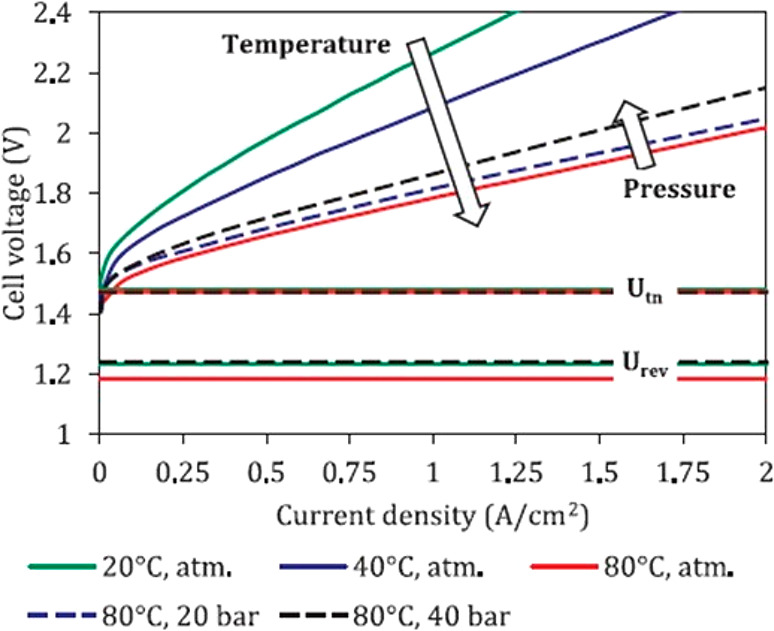
Cell voltage (Ecell)–curve as a function of the applied current density for a PEMWE. The influences of T and P are shown. The thermoneutral voltage of 1.48 V, labeled as Utn, and the reversible voltage of 1.23 V, labeled as Urev, are also shown. Reprinted with permission from ref (24). Copyright 2018 Elsevier.
2.4. Key Target Performance Characteristics
To make H2 production via PEMWEs economically competitive, the CAPEX cost needs to drop below 750 €/kW. This is at an electricity (OPEX) cost of <70 €/MWh.9 On the basis of extrapolations of experimental voltage versus current curves for single-cell PEMWEs, this goal has been suggested as feasible for WEs operated at ≥80 °C and current densities of 10 A/cm2, using a thin (25 μm) Nafion 212 membrane, and achieving a WE lifetime exceeding both 15 000 h and efficiencies of 75%.56 These data can be used as a guideline to approximate the target characteristics and potential cost savings when changing to more-abundant materials for the milder AEMWE conditions. This assumes that the economy of scaling to a fully integrated AEMWE system follows at least the same beneficial trend as observed for PEMWEs and relies on the development of AEMWE component materials matching lifetime and performance needs.18
Cost data for PEMWEs suggest that the stack makes up 60% of the CAPEX amount.11 The stack is made of the individual WE cell and the appropriate separators. The cell contains the heart of the WE: the membrane electrode assembly (MEA). The MEA is made of the anode and cathode catalyst layers, which are interfaced with the porous transport layers (PTLs) and sandwich the AEM. Typically in water electrolysis, metal-based PTLs are used at the anode; in alkaline conditions, a variety of Ni-based materials are used in single-cell tests. Because, in practice, the cathodic environment is less corrosive, more cost-efficient PTLs are in use at the cathode. These are mostly carbon-based (e.g., carbon fibers, carbon paper, or carbon cloth) and therefore often referred to as gas diffusion layers (GDLs); see, e.g., Figure 7.
Figure 7.

Schematic diagram of components for a single cell of an AEMWE. In this schematic, IrO2-based catalysts and a porous titanium transport layer (PTL) are used at the anode. (The titanium PTL is referred to as GDL in the schematic.) At the cathode (right-hand side), carbon-supported Pt (Pt/C) catalysts and a porous carbon GDL are used. H2O, OH–, and gas (H2 and O2) molecule flow are also indicated in the figure. Depending on the AEMWE, other catalyst compositions, e.g., Ni- and Fe-based anode catalysts, are often used. Reprinted with permission from ref (107). Copyright 2019 Elsevier.
The MEA, in turn, is sandwiched between two flow fields known as bipolar plates (BPs) that allow water, H2, and O2 to flow and conduct the current (Figure 7). For oxidative and acidic conditions, the BPs are typically made of costly titanium104−106 and dominate the cost, making up 51% of the stack costs, followed by the manufacturing costs of the MEA (10%) and the cost of the cathode (9%) and anode (8%) current collectors. The cost of the PEMWE anode catalyst and membrane are comparable at 6% and 5%, respectively, while research and development efforts resulted in cost reductions of the cathode to 1%. Changes in the BPs, e.g., by switching to stainless steel (even when noble metal coated for high potential corrosion protection),103 offer the potential for large reduction costs for AEMWEs compared to PEMWEs. In the case of other components such as the catalysts, the cost reduction by employing less-expensive materials alone is in the few to several % range. This indicates the need for AEM and catalyst improvements and calls for innovative material and component designs to assist in making AEMWEs viable.
3. HER and OER Catalysts
Enhancing the activity and stability of both the HER and OER electrocatalysts is crucial to make AEMWEs viable for large-scale deployment. OER catalyst improvements are urgently needed as the OER is a sluggish reaction, resulting in high overpotentials (ηs). A number of studies focused on gaining a detailed understanding of the HER and OER mechanisms in order to eventually create more-active catalysts. Reaction mechanistic understandings are important, but the creation of catalysts displaying high activities and stabilities in MEAs is crucial. Studies that focus on the AEMWE elecrocatalyst development often involve the preparation of catalyst powders. The catalyst powders can be subsequently transformed into catalyst layers (CLs) that can be up to several tenths of a micrometer thick. The activity of a catalyst measured in its powder form, i.e., prior to integration into a CL, and the activity of a catalyst in an actual CL of an MEA can be different (section 6). In some studies, thin catalyst films are also deposited onto solid and smooth electrode substrates such as gold foils. The latter can be valuable model catalysts, but for practical applications, porous current collectors enabling facile flow for the reactants and reaction products are needed. Therefore, the preparation of HER and OER electrocatalysts for AEMWEs often focuses on powder catalysts. However, the deposition of catalysts directly onto the porous and high-surface-area current collectors to be used in an MEA is also receiving attention, and recent literature has shown that such designs could open up AEMWE operation into high-current-density (>5 A/cm2) regimes.108
3.1. Metrics for Electrocatalysts
3.1.1. Mass and Intrinsic Activity
Both the HER and OER are heterogeneous reactions; thus, electron transfer from and to the reactants occurs across the electrode surface. Modifications of the electrocatalyst generally have the goal to lower the energy barrier of the reaction, which in electrocatalysis is observed as a lower overpotential (η) and an overall increase in the electrochemical activity. The two main approaches used to increase an electrocatalyst’s activity are (1) increasing the number of active surface sites and (2) increasing the catalyst’s intrinsic activity.109 An obvious strategy lies in increasing the electrochemical surface area (ECSA) of the electrocatalysts. Many approaches involve the preparation of catalysts of nanometer dimensions to reach maximal increases in the ratio of surface to bulk atoms. However, studies in actual AEMWE cells are needed to confirm if catalysts of nanosized dimensions (specifically if they are unsupported) retain their high-surface-area benefit. In an MEA, the electrocatalysts need to form an electronically conductive network without hindering the flow of reactants and products.
The exchange current density (jo) and the current measured at a specific η are indicators of the activity of a catalyst and are often presented as mass activity (current per catalyst mass) or intrinsic activity (current per ECSA). The mass activity is of practical relevance, but as already mentioned, the intrinsic activity is a measure of the actual catalytic activity. Unfortunately, an accurate measurement of the ECSA value of many electrocatalysts other than platinum, specifically when of high surface area, can be challenging,110,111 and it is further discussed in the activity testing procedure section presented in the Supporting Information. Therefore, grouping catalysts according to their intrinsic activities can be difficult. In addition, there are inconsistencies in the measurement of catalyst activities. Data are extracted for different electrolytes and are often reported as η at a specific current density per electrode area (cmgeom2). Such values are extremely difficult to compare because the loading of the catalysts on the electrode can be different and, of course, different catalysts can have widely different ESCA values. Another metric used in some studies is the turnover frequency (TOF), which is a function of the amount of H2 or O2 gas produced at a specific η resembling an equation as TOF in s–1: (the amount of gas produced at a specific η)/(F × ne × n), where F is the Faraday constant, ne is the number of electrons involved, and n is the number of catalyst atoms. However, there are a great deal of inconsistencies in calculating the TOF number specifically in the estimation of the amount of gas produced and the use of the number of catalyst atoms (n). For example, some authors use the total number of metal atoms of a catalyst, while others use the number of atoms on the catalyst surface; in some cases, the measured HER or OER current density is used as the amount of gas produced, while others measure the amount of gas produced. Therefore, the TOF values reported in the literature do not allow for an easy comparison of catalyst performances between different studies. If measured consistently, the TOF number could be a useful engineering metric. However, the consistent reporting of simply the current density (per mass and if possible per ECSA of the catalyst) at a specific η value (and preferably for the same electrolyte) rather than the TOF seems preferable for catalyst materials’ research purposes and presents fewer complications. Such an approach is consistent with a recent study by Anderson et al., who used a measurement protocol for OER catalysts, which reported current densities measured at a specific η value.112
The Tafel equation reflects kinetic information and yields the Tafel slope value (b) as follows: η = a + b × log(j). The Tafel slope yields reaction mechanistic information. To be valid, the Tafel slope needs to be determined at a η value exceeding RT/F, i.e., typically higher than at least 45–50 mV in order to neglect contributions from the back reaction.113 Smaller Tafel slopes are favorable as an increase in j, i.e., an increase in the HER and OER rates is accompanied by a smaller increase in both η and Ecell (eq 5). The η value is specific to a catalyst, indicating how the catalyst surface binds, interacts, and releases various reaction intermediates. The reaction kinetics are dependent on many experimental factors including the nature and morphology of the catalyst and the final electrode. Catalytic activities are influenced by the bulk and surface properties of a catalyst. It is well-known that catalyst activities can be tuned by means of alloying and introducing shape and ligand effects.114 Extrapolation of a Tafel plot to a η of 0, i.e., to the potential equaling the standard potential, yields jo. Tafel slope values need to be obtained from steady-state measurements (such as a constant-current or constant-potential experiment) because a Tafel slope depends on the surface coverage of adsorbed intermediate species. Many recent studies extracted mechanistic and Tafel slope information from slow-sweep linear voltammetry. Slow-sweep linear voltammetry does not provide steady-state conditions and hence can yield incorrect values. This has recently been emphasized by Anantharaj et al. and is demonstrated in Figure 8, which shows results for iR-corrected [ΔmV/Δdec] slopes extracted at different sweep rates for the example of a Co foil measured in 0.1 M KOH.115 It was demonstrated that the [ΔmV/Δdec] slopes depended on the sweep rate varying between 45 and 90 mV/dec, while the actual Tafel slope for this system extracted from constant-potential experiments yielded a value of 60 mV/dec.
Figure 8.

Demonstration of the erroneous impact of attempted Tafel slope measurements using slow-sweep voltage polarization, i.e., a nonsteady-state method. The data are for a Co foil measured using a 0.1 M KOH electrolyte. Reprinted with permission from ref (115). Copyright 2021 American Chemical Society.
Furthermore, the highest Tafel slope value measurable is 120 mV/dec (at 20 °C). Slopes exceeding 120 mV/dec are not Tafel slopes, i.e., their values cannot be interpreted as electrochemical reactions following Butler–Volmer behavior. Slopes higher than 120 mV/dec are observed and are a result of factors such as changes in the catalyst/electrode structure, which could be the formation of a resistive surface oxide and/or other changes in the catalyst structure.113
3.1.2. Metrics Including the Catalyst Stability
The development of catalysts often focuses on developing a material exhibiting a high electrocatalytic activity. However, the activity of a catalyst does not always correlate with the lifetime of a catalyst. Therefore, other metrics to assess catalysts can be useful, such as the recently suggested S-number.116 The S-number is the ratio between the amounts of evolved H2 or O2 gas versus the amount of dissolved catalyst metal.116,117 The amount of gas evolved is normalized using the ECSA value. The S-number appears to be a good indicator providing a comparative and balanced measure of catalytic activities and stability. However, care needs to be taken with the measurement of the S-number because the ECSA of a catalyst can change during the course of the measurement. Other similar metrics that could be useful reflect the catalyst utilization and lifetime in CL layers and MEAs for operating AEMWE conditions.
3.2. HER Catalysts
The kinetic pathway of the HER generally follows the Volmer–Heyrovskey or Volmer–Tafel mechanism.118 Both consist of water adsorption, followed by water dissociation (Volmer step, eq 6), and then either hydrogen dissociation via chemical desorption (Tafel step, eq 7) or electrochemical desorption (Heyrovsky step, eq 8) to form H2:118
| 6 |
| 7 |
| 8 |
In eqs 6–8, the * indicates a surface-bound species. Tafel slopes of −30, −40, or −120 mV/dec measured at 20 °C may be observed if the Heyrovsky, Tafel, or Volmer reaction, respectively, is the rate-determining step (rds).118−120 However, it is impossible to distinguish the actual reaction routes for the HER in the case of a −120 mV/dec Tafel slope.119 The energy barriers associated with each step play a role in determining the catalytic activity. It was suggested that the HER current density can be correlated with the calculated hydrogen-binding energy (HBE) on metal surfaces,121 and the HBE was shown to play a dominant role for the HER activity.121−124
The HER is one of the most studied electrochemical reactions, but compared to acidic conditions limited data is available in alkaline electrolytes. The HER activity decreases monotonically with increasing pH, supporting the theory of the higher HBE suppressing the catalytic activity.125 Furthermore, the HER takes place at more-negative potentials than the OER. Therefore, a higher number of stable materials are available for HER than for OER catalysts. These less-severe HER conditions also offer a wider range of electronically conductive and high-surface-area support materials for HER versus OER catalysts.
3.2.1. Platinum Group Metal-Based Catalysts
Among many systems studied, Pt and Pt-based catalysts show the highest intrinsic HER activities in alkaline and acidic electrolytes.126,127 Typical jo values for bulk and polycrystalline Pt measured in 0.1 M KOH are 0.62 ± 0.01 mA/cmPt2, and the HER kinetics for Pt are slowed by 2 orders of magnitude in alkaline versus acidic media due to an extra water-dissociation step.128 Similarly, the Tafel slope of Pt is favored (i.e., lower), namely, −30 mV/dec, in acidic solutions versus approximately −120 mV/dec for alkaline solutions.121,128 The following order was extracted from HER measurements carried out in 0.1 M KOH using smooth, single-metal bulk catalysts: Pt ≫ Pd > Ni > Fe ≈ Co > W > Cu > Au > Ag.121Figure 9a shows that the exchange current density, jo, and HBE values follow a Volcano-plot dependence in alkaline electrolytes, as is the case for acidic media. The HER activities, measured as jo, of these bulk metal electrodes show up to ∼4 orders of magnitude differences. A closer inspection of the Tafel slopes (Figure 9b) reported for this series shows high slopes from −90 to −216 mV/dec and only two catalysts, namely, W and Pt, show actual Tafel slopes, i.e., values less than −120 mV/dec. In the case of W, it is questionable if the HER was actually studied on the metal surface because, in aqueous solutions, the surface of tungsten will be covered with oxides, which are difficult to reduce to the metallic surface state in this electrolyte. The same could apply to the Ni, Fe, and Co catalysts studied because surface oxides form easily on these metals, and their complete reduction to the metallic surface state can be challenging. Additionally, hydride incorporation into metals such as Ni and Pd can further complicate HER activity measurements. In fact, a recent study using ambient-pressure X-ray photoelectron spectroscopy (XPS) suggests the formation of Pt–H components and their transformation and/or H intercalation in subsurface Pt layers to possibly take place on Pt in alkaline conditions.129
Figure 9.

HER results measured for bulk, single-metal electrodes in 0.1 M KOH. (a) jo versus calculated HBE (ΔH) values revealing a Volcano-plot relationship. (b) Tafel slope values as reported. The horizontal line at −120 mV/dec [shown in (b)] indicates the highest value a Tafel slope can display. (a, b) Reprinted with permission from ref (121). Copyright 2013 American Chemical Society.
Just as for acidic conditions, the surface orientation impacts the activity of a catalyst. The lower density and stepped surfaces of Pt are more active for the HER.130 Pt(110) exceeds the HER activities of Pt(100), and dense surfaces like Pt(111) show drastically lower activities.130 The use of single-crystal electrodes is not practical for AEMWE applications. However, the results show that tuning the catalyst’s morphology and working with nanoparticles can change the intrinsic activity in addition to increasing the surface-to-bulk atom ratio. The use and development of nanostructured and nanoengineered catalysts is important, but structural changes and agglomeration of small, specifically nanosized particles can take place during electrolysis, reducing the activity of a catalyst.131
The high cost of Pt is an issue for large scale applications. Correspondingly, Pt nanoparticles of <5 nm size, supported on carbon blacks such as Vulcan XC-72 and referred to as supported Pt/C catalysts, are often employed. These catalysts benefit from high ECSAs and correspondingly high mass activities. Sheng et al. carried out the careful extraction of jo values and activation energies (Eact) for the HER and H2 oxidation reaction (HOR) for bulk metal, polycrystalline Pt, and a commercial 46 wt % Pt/C catalyst in KOH electrolytes.128 The data shown in Table 3 suggest that the intrinsic exchange current density (jo,intr) and the Eact values are essentially the same for the bulk metal Pt and the 46 wt % Pt/C catalysts.
Table 3. Summary of Average HER/HOR Results for Polycrystalline Pt and Commercial Pt/C Catalysts128.
| electrolyte | jo,intra (at 21 ± 1.5 °C) (mA/cmPt2) | jo,massa (at 21 ± 1.5°C) (mA/mgPt) | Eact (kJ/mol) | Tafel slopea (at 21 ± 1.5°C) (mV/dec) | |
|---|---|---|---|---|---|
| Pt (pc)b | 0.1 M KOH | 0.62 ± 0.01 | n.a.c | 28.9 ± 4.3 | 109 |
| Pt/Cd | 0.1 M KOH | 0.57 ± 0.07 | 0.35 ± 0.05 | 29.5 ± 4 | n.r.c |
Measured at 21 ± 1.5 °C.
Polycrystalline bulk metal Pt.
n.a. and n.r. stand for not applicable and not reported, respectively.
Commercial 46 wt % Pt/C (Tanaka Kikinzoku International, Inc.). Measured ECSA = 62 m2/gPt.
It is important to validate studies of new catalysts by HER activity measurements of a commercially available Pt/C catalyst. Table S1 shows a summary of literature data for Pt/C catalysts as well as for other HER catalysts. Some of the HER (and also OER) activity data tables shown in the Supporting Information were built using data made available by Kibsgaard and Chorkendorff,132 but many additional catalysts and other relevant metrics (when available) such as the Tafel slopes, η range used for the Tafel slope measurements, and ECSA values were added in this Review. The reported HER characteristics for the Pt/C catalysts (most are commercial catalysts from a number of suppliers) differ substantially. The Tafel slope values show a large variation among the Pt/C catalysts, the majority of which are reported as negative slopes ranging between 36 and 55 mV/dec, while the slopes of two Pt/C catalysts are close to −120 mV/dec. A closer inspection of the η range used to extract the Tafel slopes (Table S1) reveals that the two Pt/C catalysts with the higher, i.e., close to −120 mV/dec, slope were measured at a valid η (>RT/F) range of >0.05 V. Furthermore, the majority of the HER activities shown in Table S1 were measured at 10 mA/cm2 geometrical electrode area (cmgeom2), which makes a direct comparison and validation of catalyst performances difficult because the catalyst loading on the electrode (mg/cmgeom2) can differ significantly. Mass and surface area normalized HER activities measured at the same η value are better for comparison; however, data for such measurements are rare. A plot of the mass activity of the Pt/C catalysts versus the corresponding η values, i.e., both measured at 10 mA/cmgeom2, is shown in Figure 10a and reveals an expected increase of jmass with η in an exponential manner. The latter is confirmed by plotting the same data as η versus the log10 of jmass (Figure 10b). Both plots demonstrate the scatter in the data, which can be at least partially ascribed to experimental variations as the majority of the studies use nonsteady-state polarization curves and different sweep rates for recording. The purpose of Figure 10b is to demonstrate the scatter in the results reported in different studies rather than suggesting the extraction of a Tafel slope, which would not be a valid approach using such data.
Figure 10.

Mass current density (jmass) for Pt/C catalysts reported in the literature versus the corresponding η value, both of which were measured at 10 mA/cmgeom2. Additional information about the Pt/C catalysts and the literature references are given in Table S1. (a) The data follow an exponential-type relationship, which is confirmed by (b), which shows essentially the same as (a) but as a plot of η versus the log 10 of jmass of the Pt/C catalysts.
The measurement of intrinsic activities is needed and can be obtained for Pt-based catalysts because the ECSA of Pt can be estimated using the charge resulting from adsorption and desorption of H (Hads/des).133,134 For the 15 Pt/C catalysts shown in Figure 10, ESCA values for three catalysts are reported. Only one group reported data that allow the estimation of the intrinsic HER activity at the same η (of −70 mV), suggesting intrinsic activities of 0.88 and 1.4 mA/cmPt2 for a commercial Pt/C and homemade Pt nanowire (NW) catalyst. The number of data points (measured at consistent conditions) is insufficient to draw conclusions and validate the activity values. However, the results emphasize the need for proper and consistent measurements and also for the establishment of a valid baseline using a Pt/C catalyst. Results reported for various HER catalysts are discussed in the following sections and will also be compared to the Pt/C activities shown in Table S1 and Figure 10.
3.2.1.1. Combinations of Pt and Ni
Combinations of Pt with Ni135 such as alloys and Ni deposits on Pt are recognized as being able to exceed the HER activity of Pt in alkaline media135−139 A synergistic effect between Pt and Ni exists, benefiting the HER. Xue et al. demonstrated this effect using a model catalyst formed by the growth of ultrathin Ni(OH)2 [and in subsequent work also thin NiFe(OH)2] clusters140 of 15–20% surface coverage onto Pt(111).135 The Ni(OH)2 clusters on Pt(111) demonstrated an 8-fold increase in the intrinsic activity compared to bare Pt(111), which was suggested to take place through a H-spillover mechanism from Pt to Ni(OH)2. The HER activity was further increased by adding cations such as Li+ to the electrolyte, which enhanced the formation of hydrogen intermediates. The same authors deposited such clusters on more practical Pt/C powder catalysts and also observed such a synergetic effect. Figure 11a shows a schematic demonstrating the H-spillover effect for the case of NiFe(OH)2 clusters on Pt. Furthermore, polarization curves (Figure 11b) and η values measured at 10 mA/cmgeom2 for Pt/C and Ni(OH)2 or NiFe(OH)2 clusters on a Pt/C powder catalyst (labeled as Ni@Pt/C or NiFe@Pt/C, respectively) are shown in Figure 11c. It is seen that the NiFe(OH)2 clusters formed on the Pt/C powder show the highest HER activity. The same is the case for NiFe(OH)2 clusters formed on bulk Pt(111) crystals (Figure 11d). Furthermore, NiCo(OH)2 clusters on Pt(111) show the lowest HER enhancement, i.e., lower than Ni(OH)2 and NiFe(OH)2 (Figure 11d).
Figure 11.
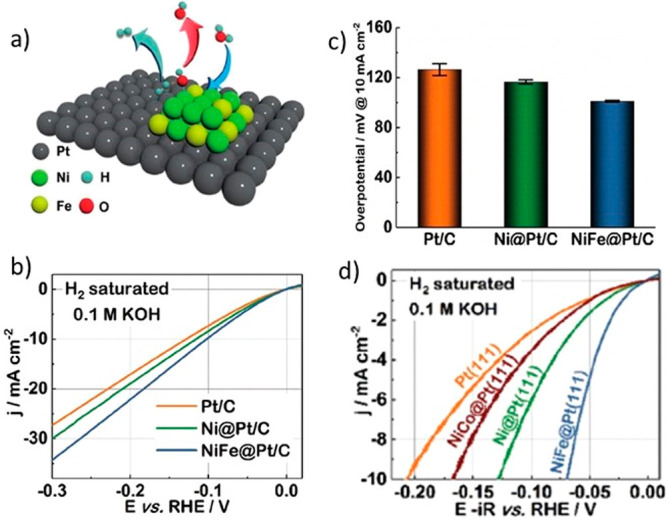
(a) H-spillover mechanism and enhancement of HER activities created by various Ni(OH)2-type clusters deposited on (b, c) Pt/C and (d) bulk Pt(111) crystals. A NiFe(OH)2 cluster on Pt is used to demonstrate the H-spillover mechanism in (a), while Ni(OH)2 and NiFe(OH)2 clusters are deposited on Pt/C powder catalysts for the polarization curves and η values shown in (b) and (c), respectively. (d) Polarization curves for NiCo(OH)2, Ni(OH)2, and NiFe(OH)2 clusters deposited onto bulk Pt(111). The abbreviations NiCo@, Ni@, and NiFe@ for the NiCo(OH)2, Ni(OH)2, and NiFe(OH)2 clusters, respectively, are used in the graphs. Reprinted with permission from ref (140). Copyright 2020 Wiley.
A number of studies report the synthesis of various forms of combined Pt and Ni-based catalyst powders with the goal to produce catalysts of higher HER activities by introducing the synergetic H-spillover effect. In most cases, the mass activity per mg Pt and the η values were measured at 0.01 A/cmgeom2 and are shown in Figure 12a. Data for the commercial Pt/C catalysts are also shown. Some authors also reported the intrinsic activities measured at η = 0.07 V and the ECSA values. These results are summarized in Figure 12b. Yin et al.138 formed Pt nanowires and also Pt nanoparticles on single-layer Ni(OH)2 sheets (the latter were formed by exfoliation of layered Ni(OH)2). At η = 0.07 V and in 1 M KOH, superior intrinsic activities (measured as j per Pt area) of up to approximately 8 and 3 times were reported for the two Pt catalysts formed on the single-layer Ni(OH)2 sheets compared to the commercial Pt/C and homemade Pt-only nanowires, respectively. The single Ni(OH)2 layers offer a high surface area for the dispersion of the Pt catalysts. However, a possible contribution of the high number of Ni(OH)2 surface sites to the HER was not considered in the j/cmPt2 measurements and cannot be completely ruled out on the basis of the reported measurements. The authors further reported that the combination of the Pt nanowire with the single-layer Ni(OH)2 structure also increases the catalyst’s stability, although the stability experiments were carried out over a short period of 4000 s.
Figure 12.
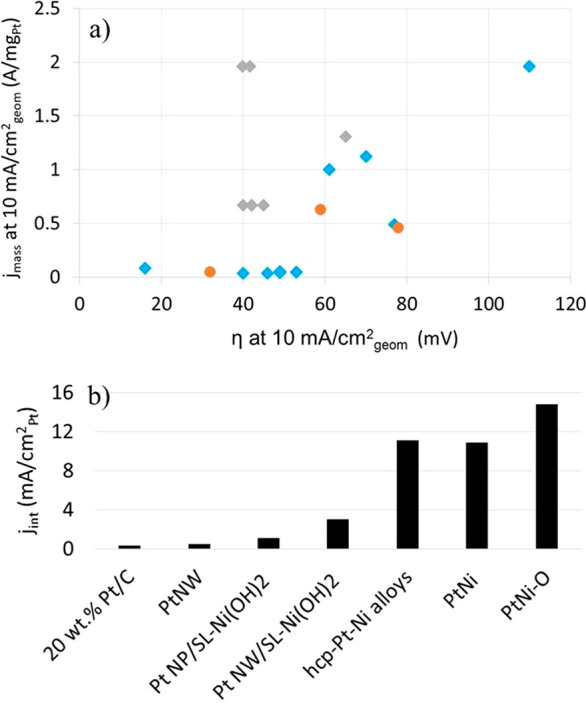
(a) Mass current activities per amount of Pt versus the corresponding η for various Pt–Ni catalysts, both measured at 10 mA/cmgeom2. (b) Plot of the intrinsic activity per ECSA of Pt (jint) measured at η = 70 mV for two Pt-based and a number of Pt–Ni-based catalysts. The data used for (a) and (b) are shown in Tables S2 and S3, respectively. The blue diamonds represent Pt/C, the gray diamonds represent PtxNiy alloys, and the orange circles represent the Pt nanosized catalysts wih Ni(OH) in (a).
Abbas et al. deposited Pt nanoparticles of 1.7–3.1 nm onto nickel urchin-like structures, referred to as xPt@Ni-SP.141 They reported that the HER mass activity per weight Pt in 1 M NaOH was up to 3.15 times higher for the xPt@Ni-SP catalysts compared to a 40 wt % Pt/C commercial catalyst. Differences in the intrinsic activities, measured as A/cmPt2, were smaller: the 0.75Pt@Ni-SP catalyst showed the highest increase of 1.3 times, while some xPt@Ni-SP catalysts exhibited a lower intrinsic activity over the Pt/C catalyst. The use of the nickel-based support might be beneficial to the catalysts’ long-term performance, as the authors reported a higher stability of the xPt@Ni-SP catalysts over the commercial carbon-supported Pt catalysts. Tafel slopes in the −30 mV/dec range were reported for all Ptx(x>0.5)@Ni-SP catalysts and the Pt/C catalysts, suggesting that the Volmer reaction was the rate-determining step. However, the reported Tafel slopes were extracted from nonsteady-state polarization curves. Chen et al. explored the deposition of Pt onto honeycomb-like NiO@Ni-film catalysts.137 The Ni films were actual Ni nanofoams that could also serve as current collectors in an MEA. The intrinsic HER activity per Pt surface area did not seem to vary remarkably among the catalysts. One catalyst, namely, Pt on the honeycomb-like NiO@Ni-nanofoam substrate, was reported to have a 15 times higher HER activity per mass of Pt compared to a commercial Pt/C catalyst. This increase may be at least partially due to a H-spillover effect. However, the direct deposition of the catalysts onto the current collector may also contribute to a higher mass activity by increasing the utilization of the catalyst (in this case the Pt onto honeycomb-like NiO@Ni-film) compared to a powder catalyst. Powder catalysts are typically transformed into electrodes using an ionomer and/or binder, which can block catalyst sites (see also section 6). Measurements of η at 10 mA/cmgeom2 showed an increase of ∼40% for both the Pt onto honeycomb-like NiO@Ni-film and the Pt/C powder catalysts over a period of 24 h. All of these results show that the combination of finely dispersed Pt on high-surface-area nickel present as, e.g., Ni(OH)2 layers can potentially offer HER catalysts of higher mass activity per Pt. It is therefore not surprising that other metal additions such as Fe and Co are being explored. It was already mentioned that the deposition of NiFe(OH)2 clusters on Pt(111) crystals further promotes the HER activity, suggesting that Fe assists Ni in the water-dissociation step.140 It is also claimed that Fe increases the conductivity and the oxidation state of Ni in its vicinity. Wang et al. decorated Co nanowires grown on a Ti mesh with Pt–Co alloys.142 Only one of the Pt–Co catalysts exceeded the HER mass activity of the commercial Pt/C catalyst, which seemed to be measured in A per geometrical electrode area, and only the less-active catalysts showed stable catalytic activities for 50 h.
However, the highest HER activities among these types of catalysts seem to be achieved by PtxNiy alloy particles, as suggested by the data also presented in Figure 12. Wang et al. prepared, by annealing, various Pt–Ni nanowire catalysts that were shown to consist of different alloy phases such as Pt3Ni4, Pt3Ni3, Pt3Ni2, Pt3Ni, and NiOx.136 They reported up to ∼12 times higher HER mass activity in 1 M KOH for their Pt–Ni nanowire catalysts than for a commercial Pt/C catalyst. The higher mass activity was assigned to the many interfaces of Pt3Ni and NiOx being created upon an optimized annealing process. The NiOx surface is proposed to accept the OH– produced in the H2O splitting reaction, while nearby Pt sites accept the Hads and produce the H2. The intrinsic HER activity was not measured, and the onset potential for the HER appears to be the same for all catalysts studied, including the commercial Pt/C catalyst.
3.2.1.2. Ru HER Catalysts
Ru is another PGM that is attracting attention as apotential HER catalyst for acidic as well as alkaline electrolytes. The ∼65 kcal/mol H-bonding energy of Ru is similar to that of Pt.141 While not as expensive as Pt, Ru is scarce. Therefore, Ru will only become a viable candidate for large-scale AEMWEs if Ru catalysts of high HER activities and long-term stability can be made using affordable materials and routes for the synthesis of both the catalyst and the support. Recent activities on the development of Ru-based HER catalysts for AEMWEs have focused on the formation and anchoring of Ru and also PtRu alloy nanosized particles on conductive carbon-based supports. High-surface-area carbon supports such as phosphorus carbon nanosheets and N-doped holey two-dimensional carbon sheets consisting of repeat units of, e.g., C2N structures were synthesized to allow the anchoring of the Ru-based particles.143 Density functional theory (DFT) calculations suggest that the H-binding energy is lowered for Ru particles embedded into these C2N and C2N2 structures and that both the Ru and the adjunct carbon atoms act as catalyst sites.141−144 The HER activities and catalyst loadings on the electrode seem to be given as total catalyst mass, i.e., including Ru and other components such as the supports in many of these studies. Furthermore, ECSA measurements are rare, which may be due to the fact that the reliable extraction of the ECSA values for Ru-based catalysts can be challenging. Double layer capacitance values and COads stripping measurements have been used to gain ECSA information, but Ru forms many different oxides at low potentials, each yielding a different Cdl value, and COads only adsorbs on metallic surfaces.110 Similarly, the method of Cu underpotential (Cuupd) deposition can be applied to catalyst sites in the metal state but not to oxides.110 Nevertheless, according to thin-layer catalyst measurements, some of the Ru-based catalysts show promise, as shown in Figure 13. Figure 13 is a plot of the mass activities versus the corresponding η values (both measured at 10 mA/cmgeom2) for Ru-based versus Pt/C powder catalysts. The results for the majority of HER activities for the Ru catalysts are underestimated due to the fact that the total catalyst weight is used for the mass activity calculation (the Ru loading for many of these catalysts does not seem to have been determined), while the activities for the Pt/C catalysts (black diamonds) and the supported Pt1Ru1.54 alloy (red cross) catalysts are per total noble metal weight. Figure 13 suggests that the Ru-based catalysts show mass activities as high as and exceeding that of Pt/C. In the case of the 2.5 nm Pt1Ru1.54 alloy catalyst formed on phosphorus carbon nanosheets, the activity per total noble metal loading seems to exceed that of the commercial Pt/C catalysts.145
Figure 13.
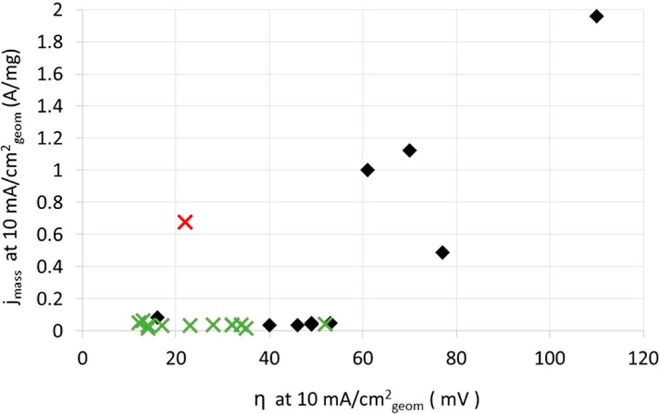
Mass activities (jmass) versus the corresponding η values of various supported catalysts, namely, Ru nanoparticles (green crosses), a 2.5 nm Pt1Ru1.54 alloy (red cross), and Pt/C (black diamonds). The jmass and η values are measured at 10 mA/cmgeom2 in 1 M KOH. The mass activities are measured in A/mg noble metal catalyst for the supported Pt1Ru1.54 alloy and the Pt/C catalysts, while in the case of the supported Ru catalysts, the mass activities are in mg per total catalyst, i.e., including the carbon support. Details about the catalysts, the actual values, and the corresponding references are given in Table S4.
The 2.5 nm Pt1Ru1.54 alloy catalyst reported to exceed the Pt/C catalyst and a homemade Pt catalyst supported on phosphorus carbon nanosheet were made by Li et al.145 The authors suggested that the observed enhancement of the Pt1Ru1.54 alloy catalysts was due to the electronic interactions between the nanosized Pt1Ru1.54 catalyst and the phosphorus carbon nanosheet, thus resulting in the enhancement of the H2O dissociation kinetics.
Mahmood et al. dispersed 1.6 nm averaged size Ru particles within holey, two-dimensional carbon nanosheets made of repeating C2N units.143Figure 14 demonstrated the formation and distribution as well as the embedment of the Ru nanoparticles within the layers of the high-surface-area nanosheets. The authors used Cuupd, COads stripping voltammetry, and Hads/des charges to estimate ECSA values and reported the number of active sites for the Ru/C2N to be ∼18% below those of the Pt/C2N and Pt/C catalysts. On the basis of the number of active sites estimated from these three methods, the TOF per active catalyst site (i.e., the intrinsic HER activity of the Ru/C2N) exceeded that of the commercial Pt/C catalysts by a factor of ∼1.7. It is assumed that the ECSA measurement for the Ru/C2N catalyst reflected the Ru sites in the metallic state, as discussed earlier. Only a small drop in the HER activity was reported after 10 000 potential cycles between 0.2 and −0.1 V versus the reversible hydrogen electrode in 1 M KOH. Details about the electrochemical experiments such as whether a high-surface-area Pt-free counter electrode was used were not given.
Figure 14.
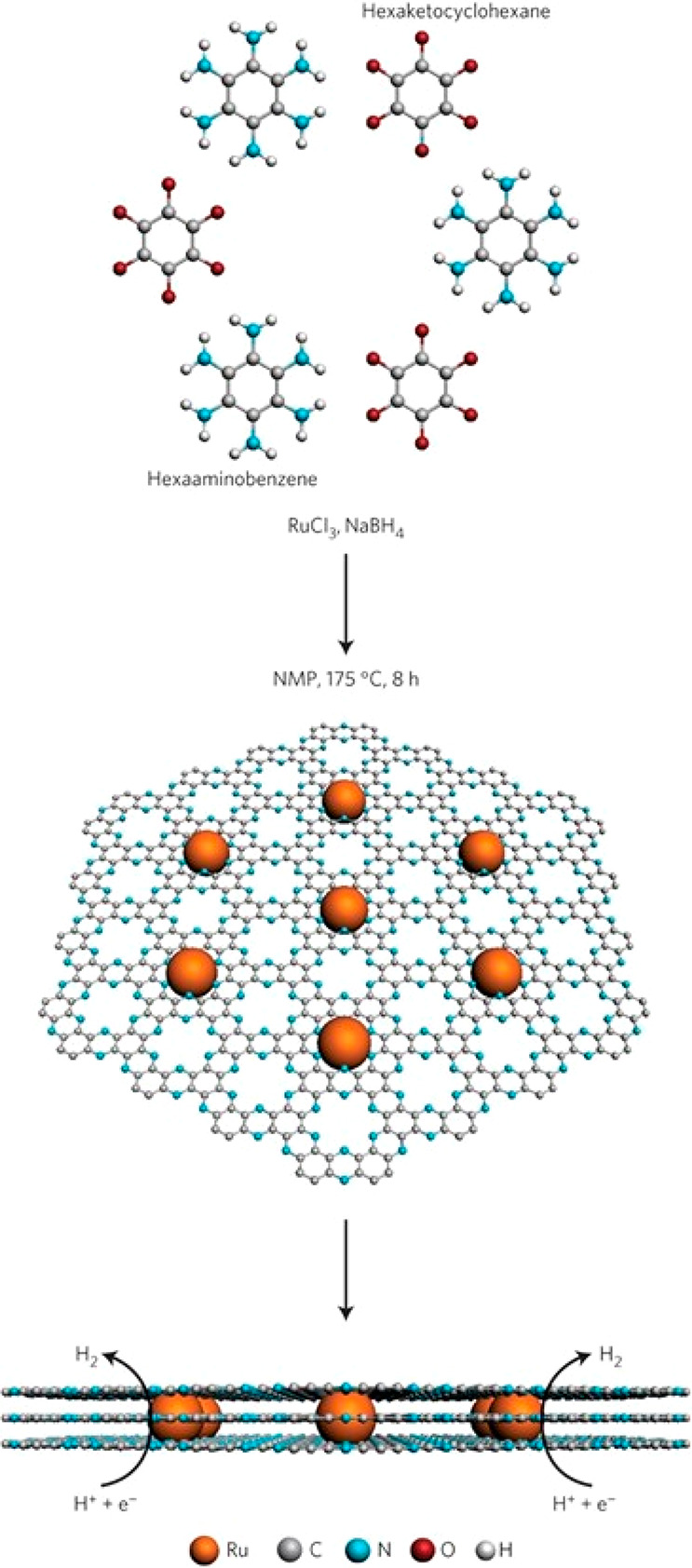
Schematic of the synthesis to form nanosized Ru catalysts embedded within holey, two-dimensional carbon nanosheets made of repeating C2N units. Reprinted with permission from ref (143). Copyright 2017 Springer Nature.
Other studies (the results of which are included in Figure 13 and Table S4) also focused on the dispersion of Ru on high-surface-area supports. Lu et al.144 formed Ru nanowires on N-doped carbon nanowires, Zheng et al.146 formed Ru particles of an average 2 nm size in C3N4 matrixes, while Xu et al.147 formed Ru particles of an average 1.5 nm size by pyrolysis at 350 °C using a carbon support of unspecified origin. These catalysts approached the mass activities of commercial Pt/C catalysts at comparable η values (Figure 13). As already noted, the authors appeared to give the loadings of the Ru catalysts as total catalyst loading, i.e., including the carbon support, while the mass activity for the Pt/C is per Pt metal.
Recent studies also involve the Ru–Ni system, which again often focuses on the dispersion of Ru (in the nanosized range) on Ni(OH)2-type matrixes, partially with the goal to take advantage of the two-dimensional high-surface-area structures that Ni-hydroxides can form. Ding et al.148 formed Ru–Ni nanoplates of ∼10–30 nm size, and Chen et al.149 formed RuNi as layered sheets (RuNi-LMH) on nickel nanofoams. Both groups reported lower η values measured at 10 mA/cmgeom2 compared to Pt/C catalysts. Similar to the RuNi system, RuCo catalysts are being explored. A nitrogen-doped carbon-supported Ru–Co alloy catalyst, formed by using the optimized annealing temperature of 600 °C, was reported to show a lower η of 34 versus 49 mV (measured at 10 mA/cmgeom2) versus a commercial Pt/C catalyst. The total catalyst loadings were ∼0.255 mg/cmgeom2, and the RuCo loading on the carbon seemed not to have been measured.150 The addition of Co to Ru was proposed to enhance the H* recombination step.150,151 Mao et al. formed Co-substituted Ru nanosheets and reported that the ∼30 nm Co atoms distributed among the Ru lattice had kinetics (measured as TOF) comparable to those of commercial Pt/C, Ru/C, and homemade RuCo alloy catalysts.152 Details about the calculation of the TOF numbers and the loading of the Pt/C catalyst do not seem to be presented.
In conclusion, activities reported for Ru-based HER catalysts, focusing on the dispersion and anchoring of the Ru catalysts, have shown promise in thin-layer electrode studies. However, a full understanding will require detailed analyses of these catalysts under AEMWE conditions as well as the determination of the Ru content of the catalysts. True Tafel slope measurements carried out under steady-state conditions within a valid η region are also needed. For water electrolysis in alkaline conditions, the stability of Ru is a concern, and it has been established that Ru catalysts have poor stability in alkaline conditions within the OER potential range. Therefore, thorough long-term stability measurements of these proposed Ru-based HER catalysts under conditions reflecting real AEMWE operations, i.e., involving intermittent periods and possible potential reversals, are important.
3.2.2. Ni-Based Catalysts without PGMs
3.2.2.1. Ni Metal HER Catalysts
Ni is an abundant metal and is used in traditional WE electrolyzers as an HER and OER catalyst, thus making it a candidate of high interest to replace Pt- or Ru-based catalysts for alkaline conditions.153−155 Ni metal shows good water adsorption, albeit the hydrogen-bonding energy to Ni metal is high and in general the rate-determining step is the H* recombination reaction.156,157 HER activities of Ni-only catalysts are lower than those for Pt/C catalysts; for example, at 10 mA/cm2, metallic Ni shows an ∼0.15 V higher η than that observed for Pt/C catalysts.157 Also, the HER activity of metallic Ni tends to decrease with the time of electrolysis, which is often attributed to hydride incorporation into the Ni lattice in the bulk and at the electrode surface.71,158 This is specifically strong for catalysts of small grain size, which correspondingly possess a high number of grain boundaries, where preferential H2 adsorption takes place.159,160 Corrosion is another factor eventually reducing the HER activity during electrolysis, as are changes in alkaline concentration induced by OH– adsorption.153 Ni metal surfaces can adsorb oxygen from the electrolyte and react to form NiO.161 NiO is transformed into NiOOH when cycled into positive potential regions, allowing the electrolyte to adsorb onto the surface, reacting with the NiO to form NiOOH penetrating further into the catalyst that results in lower HER activity.161
Alloying of Ni has been shown to change the HER activity, as for, e.g., binary NiMo and ternary NiCoMo alloys. An example of alloying Ni and Mo is shown in Figure 15, where the MoNi4 catalyst is reported to have a higher activity than Pt, although the loading of the MoNi4 on the current collector does not seem to be known.162 Other approaches are the interstitial doping of nickel with, e.g., nitrogen. Using this concept, Ni3N nanoparticles (np-Ni3N) were prepared and activities approaching those of commercial 20 wt % Pt/C and 20 wt % PtRu/C catalysts were reported.160 However, the active catalyst component loading of the np-Ni3N catalysts was significantly higher than those for the Pt/C and PtRu/C catalysts, namely, 0.16 mgNi3N/cmgeom2 versus 0.01 mgPt or PtRu/cmgeom2. The np-Ni3N catalyst is embedded in an N-doped graphitic support structure, which is proposed to alter the intrinsic catalytic properties of the Ni and possibly also stabilize the catalyst nanoparticles. Interstitial N-doping and embedment into the N-doped graphitic support of the Ni was carried out via a two-step reaction. The two reaction steps involved the decomposition reaction of preformed K2[Ni(CN)4] to form nanosheets made of Ni-cyano compounds at 450 °C followed by N-doping in a NH3 atmosphere also at 450 °C to form the 5–20 nm sized Ni3N nanoparticles. The N-doped graphitic lattice was formed simultaneously during this process.
Figure 15.

HER data extracted for Ni, Pt, and two Mo-based catalysts in 1 M KOH. (a, b) Slow-sweep polarization curves and Tafel slope values extracted from the polarization curves, respectively. (c) Comparison of Tafel slope values to other catalysts reported in the literature. (d) Results for a stabilization test carried out under potential cycling for the MoNi4 catalyst. Reprinted with permission from ref (162). Copyright 2017 Springer Nature.
CeO2 has been proposed as beneficial catalyst support to anchor metal catalyst sites. The system is of interest for both the HER and OER and may act as a bifunctional electrocatalyst.163−167 The metal-oxide interface of Ni and CeO2, with the latter being deposited on carbon nanotubes (CNTs), was reported to show a synergistic effect, and DFT calculations suggest that the interactions of Ni with CeO2 benefit the HBE, matching that of Pt/C.163 A lower η value for the Ni-CeO2/CNT catalyst compared to Ni/CNTs and CeO2–CNTs catalysts was reported, but the η value was higher than that measured for a commercial Pt/C catalyst of a 40% lower total metal loading. The addition of CeO2 was observed to enable the formation of Ni particles of 4 nm size, i.e., much smaller than the 50–100 nm Ni particles formed on CNTs free of CeO2. Therefore, some of the observed increases in the HER activity are likely due to an increase in the ECSA caused by the smaller Ni particle size of the Ni-CeO2/CNT versus Ni/CNT catalysts.
3.2.2.2. Non-PGM Ni Alloys and Mixtures: The Addition of Mo
Other attempts to increase and stabilize the activity of bare Ni catalysts involve the formation of binary and ternary alloys of Ni with different elements such as Co, Fe, Mo, Ce, Zn, and Cu to improve the catalytic activity, prevent hydride formation, and achieve a higher stability.168 Many of these studies originate from the development of HER catalysts for traditional alkaline WEs and also for artificial photosynthesis devices.169,170 Some reports suggest that Ni–Mo alloys have the highest HER activity among non-PGM catalysts30 and that the activity of the Ni–Mo alloys is further improved for ternary alloys.171−174 While Ni–Mo alloys show high HER activities among non-PGM catalysts, the actual measurements are not always conclusive due to issues measuring the ECSA and the intrinsic activities of these catalysts accurately.175 These measurements are complicated by the fact that catalysts of the Ni–Mo combination can have widely different particles sizes and porous structures. In addition, the Ni–Mo system exhibits a pseudocapacitance in the potential region, typically used to estimate the ECSA value.175 Therefore, increases in HER activities reported for Ni–Mo catalysts have been argued to be due to higher surface areas rather than actual beneficial intrinsic catalytic effects.176,177 Nevertheless, it has been shown using nonporous catalysts that Ni–Mo alloys can exhibit increased HER activity over Ni-only catalysts, as demonstrated in Figure 16. Figure 16 shows polarization curves for metallurgical Ni and Ni–Mo alloys of different compositions made by cutting the metallurgical rod into disc-shaped electrodes of the same size. The surfaces of the disc-shaped electrodes were carefully polished to produce smooth surfaces. However, Ni–Mo alloys do not yield the HER activities needed for large-scale AEMWE applications. The stability of Ni–Mo alloys also needs to be proven. Ni–Mo alloy particles of 50–200 nm size have shown stable currents of 0.02 A/cmgeom2 over 100 h in 2 M KOH using a catalyst loading of 1 mg/cmgeom2, but currents of 0.02 A/cmgeom2 are low and an analysis of dissolved metals was not performed.178
Figure 16.
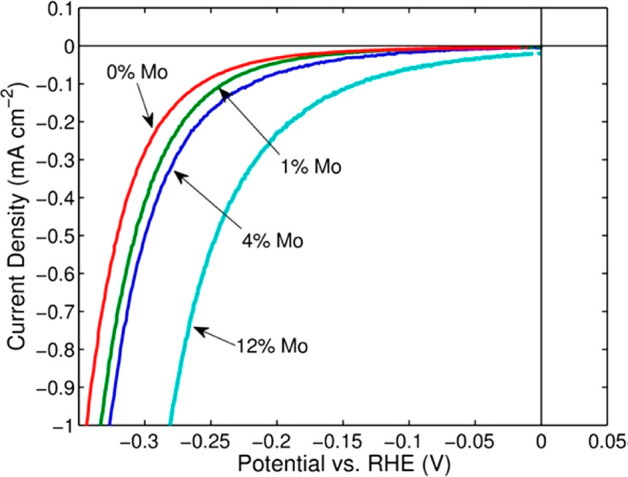
Polarization data of commercially available and metallurgically prepared Ni and Ni–Mo alloys with varying Mo content. Experiments were performed in 2 M KOH solutions. The Ni and Ni–Mo alloy samples were coin-sized samples prepared by cutting cylindrical rods and were carefully polished to create a smooth surface. Reprinted with permission from ref (178). Copyright 2013 American Chemical Society.
The addition of Mo to Ni, to form intermetallics or disorganized compounds, has been reported to improve the stability and the HER activity compared to Ni-only catalysts. In intermetallic compounds, the atomic fraction of Mo is much larger than that in disordered structures. Examples of intermetallics of Ni and Mo include Ni7Mo7, Ni3Mo, and Ni4Mo. In a recent study, polished samples of Ni7Mo7, Ni3Mo, and Ni4Mo were tested for the HER, and the metal dissolution was measured.179 Substantial dissolution of Mo occurred for Ni7Mo7, leading to an increase in the ECSA, while Ni3Mo and Ni4Mo did not show dissolution at potentials below 0 V versus the reversible hydrogen electrode. However, the stability range of Ni3Mo appears to be relatively narrow (between −0.25 and −1 V versus the reversible hydrogen electrode), and dissolution was reported to take place at open-circuit potentials.117 In comparison to intermetallics, in disordered structures the Mo content only needs to be a few atom % to be effective for the HER.175,179 For disordered structures, a Mo content of ∼10 atom % leads to the highest activity among Ni–Mo disordered catalysts.180
Many studies involve unsupported Ni–Mo catalysts of several hundred of nanometers in size.178 This is much larger than the sub-5-nm size that is typical for Pt/C catalyst,s and the larger size is at least partially responsible for the lower HER mass activities compared to Pt/C. To improve the electrical connectivity, Ni–Mo-based catalysts have been directly deposited onto Ni foam current collector substrates.181,182 A study of the formation of a ternary NixMoyFez alloy on Ni foam reported significantly lower η values than that for Ni only; however, the η values are still higher than that for Pt, and the catalyst loading is not known.174 Another two studies reported improved HER performances of their catalysts over commercial Pt/C powders.182,183 However, the loadings of the Ni–Mo catalysts were higher than the Pt/C catalyst loadings: approximately 55.8 and 44.3 mg/cmgeom2 of MoNi4/MoO2@Ni versus 2 mg/cmgeom2 Pt/C182 and 5.9 mg/cmgeom2 MoNi4/MoO3–x versus 1 mg/cmgeom2 Pt/C.183 Additional studies are needed to deposit lower amounts of Ni–Mo-based catalysts directly on the porous Ni current collectors, and as already mentioned, the long-term stability of Ni–Mo catalysts under AEMWE operating conditions needs to be proven.
In conclusion, it appears that the intrinsic activity of the combined Ni and Mo catalyst system can be higher than that for Ni-only catalysts, although the dissolution of Mo under intermittent electrolyzing conditions at high pH could be an issue. Many Ni–Mo catalysts are unsupported, representing particles in the tenth of a nanometer size range, and are unlikely to be comparable to the mass activity that can be achieved for the catalytically very active and nanosized catalysts such as Pt/C, PtNi/C, and various supported Ru catalysts discussed earlier.
3.2.3. Other HER Catalyst Groups
3.2.3.1. Mo Carbides
Another class of HER catalysts consists of Mo carbides, borides, and sulfides. Mo2C, MoC, or Mo catalysts embedded in nanocarbons,184 carbon nanosheets, CNTs, or boron–carbon–nitrogen are examples of Mo catalysts studied for the HER. Commercial Mo2C has shown an ∼5 times higher η than Pt/C.184 Encapsulation of Mo2C in nitrogen-doped porous CNT was reported to benefit the HER, and a η value (measured at 10 mA/cmgeom2) of 0.045 V for the embedded Mo2C compared to 0.033 V for the Pt/C catalyst was reported.185 A high loading of the embedded Mo2C catalyst of 0.728 mg/cmgeom2 was used. Other studies also reported that encapsulation benefited the HER activity by increasing the number of active sites with better water adsorption properties.186 Again, these studies tend to use high catalyst loadings to achieve the low η values reported, and in some cases Pt wires were used as counter electrodes. (The use of a Pt counter electrode can result in Pt dissolution and deposition of the dissolved Pt on the working electrode, i.e., onto the catalyst, which in turn can result in incorrect high HER activities.) Encapsulation has been reported to increase the catalyst’s stability by reducing both corrosion and agglomeration of the catalyst particles.185−187 Other results suggest that graphene-based structures offer electronic benefits, although the anchoring of the catalyst onto the graphene will need to be addressed.185−190 Mo-nitrides have similar HER activities as carbides,187,191 and the combination of Mo2C and Mo2N has been reported to lower the η value.187 In all cases, stability appears to be a recurring problem. Similarly, MoB also shows unstable performance in alkaline solutions.192
3.2.3.2. Transition Metal Phosphides and Sulfides
In acidic electrolytes, TM phosphides193 and sulfides194−196 have been reported to show notable HER activities. Therefore, Ni and cobalt phosphide catalysts such as Ni2P,197 Ni5P4, and Ni3P198 and CoP,199,200 Co2P, and CoP2, respectively, were also studied in alkaline media, but fast deactivation was observed.198 Poisoning of the catalyst surface or catalyst dissolution were suggested as possible mechanism for the observed deactivation.197,199 Phosphorus can perform in much the same way as N. These materials have high electrical conductivity and allow for higher catalyst loadings and high current densities, but the rapid catalyst deactivation is an issue.198,201
3.3. OER Catalysts
3.3.1. OER Reaction Mechanism and Stability Consideration
The OER involves four charge-transfer steps as follows, where the * indicates surface-adsorbed species:202
| 9 |
| 10 |
| 11 |
| 12 |
The OER free-energy diagram has an individual step height of 1.23 eV and a total change in free energy of 4.96 eV at standard conditions.202 As already discussed, the OER is a sluggish reaction. Changes in the binding energy of the reaction intermediates with the catalyst will change the overpotential. The minimal η for a catalyst surface that binds a reaction intermediate strongly is defined by the breaking of the reaction intermediate bond with the catalyst surface. Catalysts such as Mn, Co, Ir, and Ru oxides bind the reaction intermediates formed in the OER strongly and are predicted to show a minimum η of 0.37 V.203 NiO and TiO2 are examples of catalysts that show weak binding energies for OER intermediates and are defined by eqs 9 and 10.203
In the case of the acidic conditions of PEMWEs, only PGM OER catalysts have proven to show the stabilities needed for real applications. The alkaline pH of AEMWEs is viewed to offer a wide range of non-PGM OER catalysts, including transition metals such as Ni, Co, Fe, and Cu. However, this view is too simplistic, as long-term stability is an issue for many TM catalysts and also for catalyst supports. Therefore, the number of materials suitable as OER catalysts and supports is limited. Both the IrO2 and RuO2 OER catalysts, which are typically used in PEMWEs, show a lack of long-term stability in alkaline conditions. The RuO2 is the least stable of the two oxides, and the metallic counterparts of the oxides, i.e., Ir and Ru metal electrode catalysts, show poor stability.204 The stability issue is demonstrated in the Pourbaix diagrams shown in Figure 17 for Ni, Cu, Fe, and Cu, i.e., for four TMs of high interest as catalyst or catalyst components for AEMWE OER catalysts. The insets show the voltage and pH domains of relevance to AEMWEs considering different AEMWE feed modes, namely, water and dilute electrolytes. These domains are pH ∼7 for water, pH ∼9 for KHCO3, and a pH range of 9–12 for dilute NaOH or KOH. The colored areas suggest regions of corrosion for the corresponding element. An important point to note is that the stability of these TMs not only depends on the potential region but also on the pH and, hence, also on the nature of the solution feed to the AEMWE cell. The diagrams suggest that the stabilities of Co and Ni are jeopardized for pH 7 but are promising for a pH range exceeding 9. Indeed, NiO is known to have long-term stability at high pH and is used as an OER anode in traditional WEs. Copper is also suggested as a candidate for the pH range of ∼9–12. The diagram shown for Fe in Figure 17 ignores the formation of passivating iron oxides and shows that corrosion of iron may well occur under AEMWE operating conditions. It needs to be noted that Pourbaix diagrams are only guidelines based on thermodynamic information. They do not include reaction kinetics information, and experimental verification of the catalyst stability is needed. In addition, the stability of a catalyst is influenced by its chemical and physical structure, including its physical size. Nevertheless, Pourbaix diagrams provide insights and initial material stability guidelines.
Figure 17.

Pourbaix diagrams of cobalt, copper, iron, and nickel in aqueous electrolytes at ambient pressure and 25 °C. The inset shows the voltage–pH range that an anode catalyst may experience in an AEMWE. The diagrams were constructed from ref (204).
3.3.2. Challenges for OER Catalyst Supports
The harsh conditions of the OER also severely limit the number of stable support materials available for OER catalysts. The high surface area and electronically conductive carbon supports such as Vulcan XC-72, graphite, or potentially even CNTs, which are often used for the HER catalysts, are not suitable as a support for OER catalysts because carbon is easily oxidized and consumed during the OER. Therefore, non-carbon-supported OER catalysts are typical for AEMWEs, even though carbon-supported TM OER catalysts (including graphene, organic frameworks, and CNT supports) have been used, mainly at short experimental time scales. As already mentioned, the electronic conductivity of the support (or the bulk of a catalyst) also plays a role in the creation of effective OER catalysts. In fact, a conductive support can reduce electronic-conductivity limitations of certain catalysts, and, in the case of very thin (on the atomic-layer scale) catalysts, a support can alter the electronic properties of a catalyst and its lattice constants.203
3.3.3. OER Activities of IrO2 and RuO2
Research for OER catalysts for AEMWEs has become extensive over the past decade. Many different synthesis conditions are used, and the catalysts are not always fully characterized in terms of their size and structure. As for the HER catalysts, the majority of the OER catalysts studied for AEMWE applications are powder catalysts, and the activity of newly prepared OER catalysts are often measured in thin-layer electrode setups and/or directly in a single AEMWE cell; also, the establishment of a baseline catalyst is needed. OER activity comparisons to commercial Ir-oxide catalysts, which are still considered state-of-the-art catalysts in terms of initial activity, are reported in some studies. However, the reported OER activities for Ir-oxide catalysts vary significantly. This may be partially due to the different forms and properties Ir-oxide can have depending on the method used for their preparation. Some of the possible differences are demonstrated in Figure 18, which shows transmission electron microscopy images for four different Ir-oxides. Ir-oxide powder catalysts are often made by thermal composition of a precursor salt like hydrous IrCl3. It is known that the annealing temperature influences the ECSA, water content, and likely the electronic conductivity and crystallinity of the resulting oxide catalysts, and a higher temperature results in a more crystalline and compact oxide.205 Amorphous IrOx has a higher catalytic activity for the OER than that for the crystalline and rutile IrO2, which may be due to the open structure of the hydrous and amorphous IrOx versus the compact structure of IrO2. However, the stability of the former is lower.116 A hydrous and amorphous IrOx form can also be formed on the surface of an Ir metal, possibly assisting with the OER activity.206 Many commercial suppliers sell Ir-oxide powders as IrO2 or hydrous IrO2·H2O. Researchers should develop a practice of characterizing the as-received commercial Ir-oxides to understand the type of Ir-oxide structure being studied. At a minimum, the characterization of the as-received Ir-oxide including X-ray photon spectroscopy (XPS) of the Ir and O regions, an X-ray diffraction (XRD) pattern, and an examination of the redox chemistry of the as-received Ir-oxide by cyclic voltammetry (CV) needs to be carried out. In this Review, the labeling provided in the corresponding publications for the Ir-oxides will be used (which most commonly is IrO2), even though an actual proof of the structure is typically not provided.
Figure 18.

Transmission electron microscopy images for various Ir-based catalysts are as follows: (a–c) Ir particles, (d–f) nanosized Ir particles, (g–i) Ir black from Umicore, (j–l) amorphous IrOx from (a) TKK and (b) the rutile form of IrO2. Reprinted with permission from ref (207). Copyright 2019 Elsevier.
Consistent baseline data for the OER activity of Ir-oxide catalysts in alkaline conditions are rare. In a recent study, Anderson et al. suggested baseline control studies and also reviewed published data.112 The literature summary suggests mass activities of approximately 11 and 60 A/g in 0.1 M KOH and at a η of 0.35 V for two IrO2 powders from two different suppliers. More data is available for 1 M KOH electrolytes, and mass activity data for IrO2 powders vary between 9 and 275 A/g at η = 0.35 V, as summarized in Table S6. Stoerzinger et al. carried out a study on single-crystal IrO2 (and also RuO2) catalysts.205 They reported that the (100) surface was intrinsically more active than the thermodynamically more stable (110) surface for both oxides at pH 13 and correlated these OER activities with the density of uncoordinated metal sites of the crystal faces. The results are summarized in Table 4, which also includes intrinsic OER data for commercial IrO2 and RuO2 powder catalysts reported by Anderson et al.112 As seen in Table 4, the RuO2 single-crystal surfaces show significantly higher activities over the IrO2 equivalents. The activities of these single-crystal IrO2 and RuO2 catalysts are significantly lower than the intrinsic activities of the commercial IrO2 and RuO2 particles measured in 1 M KOH (Table 4). The single crystals of IrO2 and RuO2 used in the study were the rutile forms of the oxides and were formed as thin layers on (001)-oriented SrTiO3 and MgO substrates. All of these characteristics, i.e., possible differences in the crystalline catalyst structure, thin layers, and a potential substrate effect may be responsible for some of the intrinsic OER activity differences observed between the catalyst powders and the single-crystal films. The Tafel slope values for the four single-crystal electrodes are consistent with results by Lyons and Floquet,113 who reported that IrO2 and RuO2 catalysts exhibit low- and high-η Tafel slope regions. As shown in Table 4, at lower η values (<0.3 V), the Tafel slopes of all four single-crystal oxide surfaces were similar in the 60 mV/dec range, and an increase to 90 and 140 mV/dec for IrO2 and RuO2, respectively, was observed for the higher-η (>0.4 V) region. As already discussed in the HER section, a true Tafel slope cannot exceed a value of 120 mV/dec. The 140 mV/dec slopes for the RuO2 single crystal may be due to changes in the oxide surface structure taking place at high η values.
Table 4. Intrinsic OER Activities for IrO2 and RuO2 in KOH Electrolytes.
| catalyst | electrolyte | jinta (μA/cm2) | low-η Tafel slopea (mV/dec) | high-η Tafel slopea (mV/dec) | ref |
|---|---|---|---|---|---|
| IrO2 (100)b | 0.1 M KOH | 3 | 55 | 93 | (205) |
| IrO2 (110)b | 0.1 M KOH | 5 | 61 | 85 | (205) |
| IrO2 powder | 1 M KOH | 171c | n.r.d | 106 | (112) |
| RuO2 (100)b | 0.1 M KOH | 182 | 54 | 144 | (205) |
| RuO2 (110)b | 0.1 M KOH | 65 | 56 | 141 | (205) |
| RuO2 powder | 1 M KOH | 43c | n.r.d | 83 | (205) |
The intrinsic activities (jint) and Tafel slopes are measured at 23.5 °C in the case of the two powder catalysts. The temperature is presumably the same and/or within 3 °C for the single-crystal electrodes. Furthermore, double layer (Cdl) measurements were used to estimate the surface area of the commercial IrO2 and RuO2 powders. All jint and Tafel slopes values shown in Table 4 seem to have been measured from nonsteady-state measurements.
All of the IrO2 and RuO2 single crystals were thin films in the rutile form.
The jint values were measured at a η value of 0.35 V.
n.r. stands for not reported.
OER mass activity results reported for Ir-oxide powder catalysts show a large discrepancy, as demonstrated in Figure 19. Figure 19 shows the mass activities and the corresponding η values measured at 10 mA/cmgeom2. Figure 19 also illustrates the sluggishness of the OER, as the η value needed to achieve the same mass current density is substantially higher for the OER catalysts than for the HER catalysts (Figure 10). Furthermore, an exponential dependence of the mass activity on the η value is not evident. In fact, the majority of the catalysts suggest a zero (or very small) slope dependence on η. The exact reasons for this are unknown. It could be partially due to different properties, including differences in the ECSA values, of the Ir-oxide powder catalysts studied from the various suppliers, and it may be partially due to the fact that many of these results are obtained from slow-sweep polarization curves rather than from actual steady-state measurements. Among the data in the 0.35–0.4 V η range, one catalyst shows a substantially higher OER activity (red arrow and IrOx (2) in Figure 19a). This catalyst is reported as IrOx, possibly indicating an amorphous Ir-oxide form.208 The OER activity for this IrOx catalyst was extracted from steady-state Tafel slope measurements, thus adding validity to the data reported for these measurements of the catalyst. Figure 19a shows the results for one other IrOx catalyst [red arrow and IrOx (1)] for which the OER activity is seen to lie within the wide scatter of the other Ir-oxide catalysts.
Figure 19.
Comparison of mass activities (jmass) reported for commercial Ir-oxide powder catalysts versus the corresponding overpotential (η). Both jmass and the corresponding η values were measured at 10 mA/cmgeom2. (b) Enlarged version of (a) demonstrating the variability in the reported data for the lower η range. Details of the Ir-oxide mass loadings on the electrodes and references are given in Table S6. The majority of the Ir-oxides were reported to be IrO2, with the exception of two oxides that are referred to as IrOx, as indicated in (a).
All of these results reveal the necessity of studies establishing baselines for selected catalysts carried out as proper steady-state measurements and in reference to their preparation method and detailed physical and chemical characterization. The lack of true comparative studies including both mass and intrinsic activities for a large number of well-characterized Ir-oxide powder catalysts complicates the identification of the best-performing OER catalysts. Due to the challenges of accurately determining the ECSA values for many of these catalysts, mass activities are often reported and, as for HER catalysts, the mass activity of a catalyst is a characteristic of high practical relevance; however, it cannot be directly translated into an intrinsic activity.
3.3.4. Ni-Based OER Catalysts
High-surface-area nickel and nickel alloy catalysts have been of key interest for traditional alkaline water electrolyzers that runs on high-concentration (in the 30 wt % range) KOH electrolytes, and the catalysts are directly formed on nickel mesh current collectors. Reports for the synthesis methods of unsupported and high-surface-area alloy powder OER catalysts are scarce. In fact, it is challenging to synthesize unsupported alloy powder catalysts with the high surface areas needed for AEMWE OER catalysts. It is noteworthy that Ni alloys have shown promising intrinsic OER activities that can be high specifically when freshly prepared.209−211 Similar to the case of HER catalysts, the most promising Ni-alloy OER catalysts seem to be NixFey and NixCozFey, although Ni-based alloys containing, e.g., Al and Mo have also been suggested.212,213 Unfortunately, Al and Mo suffer stability issues at high potentials in high pH conditions, a fact that is often neglected. In the cases of the alloy and metal catalysts, it can be assumed that an oxide and/or an (oxy)hydroxide form is involved in the OER, as at least the surface of these catalysts will be transformed into oxidized forms upon exposure to the high potentials and high pH typical for OER conditions.204,214
Not surprisingly, Ni-based catalysts in the nanosize range have been synthesized, and some of the highest OER mass activities (measured in thin-layer electrode cells) have been reported for these high-surface-area catalysts. For example, at η = 0.35 V and in 1 M KOH, a mass activity of 1795 A/gcatalyst was reported for a 5 nm Ni-shell catalyst formed on an ∼50 nm Fe core particle.215 In addition to the high surface area, the combination of a thin Ni shell on a Fe core appears to increase the intrinsic catalytic activity of the Ni. Studies will need to be carried out to understand if the high activity of such a nanosized catalyst will also be observed in an MEA and if nanoparticles and core–shell catalysts possess long-term stability under real AEMWE operating conditions. However, recent reports for single AEMWE cells run on 1 M KOH electrolytes used a commercial NiFe2O4 OER catalyst. Details of the catalyst are not given, but the supplier, US Research Nanomaterials, focuses on nanosized catalysts. Long-term operation of up to several thousand hours at reasonably high j values of 1 A/cm2 is shown, suggesting that at least the latter type of Fe-containing catalysts could be suitable for AEMWE applications despite the fact that the Fe-to-Ni ratio of this catalyst is very high (Table 6).
Table 6. State-of-the-Art AEMWE Cells and Experimental Parametersa.
| catalyst
loading (mg/cm2) |
operating
conditions |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| study | anode | cathode | AEI loading (wt %) anode/cathode | membrane, thickness (μm) | IECmembrane (mmol/g or meq/g) | electrolyte | j (A/cm2) | Ecell (V) | time (h) | degradation rate (μV/h) | ref |
| 1 | IrO2, 3 | Pt black, 3 | F-PAE, 2.5/2.5 | ATM-PP | 1.7/(2.7 for F-PAE) | H2O | 0.2 | 1.9 | 2000 | 350 | (457) |
| 2 | FexNiyOOH-20F, 4.8 | Pt/C, 0.94 | PAP-TP-85 | PAP-TP-85, 30 | 2.4 | H2O | 0.2 | 1.63 | 150 | 560 | (424) |
| 3 | Ir black, 2.7 | Pt/C, 2.7 | Sustainion XA-9, 5 | Sustainion 37–50, 50 | ∼1.1 | H2O | 0.5 | 1.9 | 170 | 705 | (428) |
| 4 | IrO2, 2.6 | Pt black (HiSPEC 1000 TM), 2.4 | A-Radel, 22/27 | A-201 Tokuyama, 28 | 2.0 | H2O | 0.2 | 1.8 | 535 | 747 | (83) |
| 5 | Ni2Fe3, 3 | PtRu/C, 2 | TMA-53, 20/20 | HTMA-DAPP, 26 | 2.6/(2.6 for TMA-53) | H2O | 0.2 | 1.75 | 170 | 1470 | (414) |
| 6 | IrO2, 0.6 | Pt/C, 0.3 | BPN | quaternized polyphenylene | n/ab | H2O | 0.2 | 2.1 | 100 | 1800 | (415) |
| 7 | IrO2, 1.2 | Pt/C, 0.3 | PFOTFPh-TMA-C8, 25 wt % | PFOTFPh-TMA-C8, 23 | 2.7 | H2O | 200 | 1.65 | 120 | 2250 | (458) |
| 8 | IrO2, 2.3–2.8 | Pt black, 2.3–2.8 | PiperION (TP-85), 10 | PiperION (TP-85), 50 | 2.02–2.37 | H2O | 0.5 | 1.85 | 175 | 2571 | (465) |
| 9 | Ni–Fe–Ox, 5 | Ni–Fe–Co, 5 | Sustainion XB-7 | PBI-based AEM (KOH doped) | n/ab | 1 M KOH | 1 | 2.09 | 100 | –100 | (461) |
| 10a | NiFe2O4, 1.8 | modified Raney nickel, 14.5 | Nafionc | Sustainion grade Td, 50–80 | ∼1.1 | 1 M KOH | 1 | 1.83 | 1920 | 0.7 ± 0.2 | (453) |
| 10b | NiFe2O4, 1.8 | modified Raney nickel, 2.7 | Nafionc | Sustainion X37-50, 50–80 | ∼1.1 | 1 M KOH | 1 | 1.85 | 12180, 10100 | 0.7 ± 0.2 | (453) |
| 11 | Raney Ni–Fe | Raney Ni–Fe | no AEI | PFTP-13 AEM, ∼30 | 2.82 | 1 M KOH | 0.5 | ∼1.5 | 1000 | ±close to 0 | (463) |
| 12 | NiFeOOH, electrodeposited | Pt/C, 0.4 | Fumion, 30 (cathode only) | FAA-3, 50 | 1.4–1.6 | 1 M KOH | 0.5 | ∼1.6 | 100 | ±close to 0 | (108) |
| 13 | NiFeOx, 2 | NiFeCo, 3 | Nafionc | Sustainion 37–50, 50 | ∼1.1 | 1 M KOH | 1 | 1.9 | 1920 | 5 | (462) |
| 14 | IrO2, 1.2 | Pt/C, 0.3 | PFOTFPh-TMA-C8, 25 wt % | PFOTFPh-TMA-C8, 53 | 2.7 | 1 M KOH | 200 | 1.6 | 135 | 370 | (458) |
| 15 | IrO2, 2 | Pt/C, 0.5 | PFTP, 10/PFBP, 25 | PFTP-13 AEM, ∼30 | 2.82 | 1 M KOH | 0.5 | 1.6 | 1100 | 455 | (463) |
| 16 | NiAl, 47.9 (plasma sprayed) | NiAlMo, 42.7 (plasma sprayed) | no AEI | HMT-PMBI, 50 | 2.52 | 1 M KOH | 1 | 2.05 | 145 | 650 | (464) |
| 17 | Raney Ni–Fe, 20 | Raney Ni–Fe, 20 | no AEI | Sustainion 37–50, 50 | ∼1.1 | 1 M KOH | 0.5 | ∼1.93 | 250 | 1200 | (463) |
| 18 | NiCo2O4, 8 | Pt, 0.3 | qPPO, 90/10 (catalyst/ionomer) | Dowex Marathon A and LDPE blende | 2.45 | 1.95 M KOH | 0.3 | 1.8 | 100 | 200 | (466) |
| 19 | Ni foamf | Ni foamf | PSEBS-CM-DABCO, 100 | 0.76 | 1.95 M KOH | 0.3 | 2.26 | 160 | 20 | (467) | |
| 20 | NiCo2O4, 8 | Pt, 0.3 | qPPO, 90/10 (catalyst/polymer) | qPPO, TMA quaternized, 200 | 1.46 | 1.95 M KOH | 0.3 | 1.8 | 400 | 100 | (468) |
| 21 | NiCo2O4, 8 | Pt, 0.3 | PSEBS-CM-TMA/PTFE, ∼5 wt % or 95/5 | PSEBS-CM-TMA, 100 | 0.75 | 1.95 M KOH | 0.3 | 1.76 | 800 | 50 | (469) |
| 22 | CuCoOx (Acta 3030), 36 | Ni/(CeO2–La2O3)/C (Acta 4030), 7.4 | PTFEc, 10/10 | A-201 Tokuyama, 28 | 1.8 | 1 wt % K2CO3 | 0.47 | 1.85 | 1000 | 200 | (84) |
| 23 | Ni0.7Co0.3Ox, 2 | Pt/C (40 wt %), 1 | AS-4, 5 | in-house prepared APEg | n/ab | 1 wt % KHCO3 | 0.1 | 2.03 | 550 | 200 | (470) |
| 24a | CuCoOx (Acta 3030), ∼30 | Ni/(CeO2–La2O3)/C (Acta 4030), ∼7.4 | alkaline ionomer (I2, Acta Spa), ∼9 | A-201 Tokuyama, 28 | 1.8 | 1 wt % K2CO3 | 0.5 | 1.98 | 200 | 500 | (235) |
| 24b | CuCoOx (Acta 3030), ∼30 | Ni/(CeO2–La2O3)/C (Acta 4030), ∼7.4 | alkaline ionomer (I2, Acta Spa), ∼9 | FAA-3-PP-75 | 1.4–1.6 | 1 wt % K2CO3 | 0.5 | 2.05 | 200 | 2380 | (235) |
All AEMWE cells were run for at least 100 h. Additional experimental conditions are given in Table S20.
n/a indicates that information was not available in publication or corresponding Supporting Information. F-PAE, partially fluorinated poly(arylene ether); PAP, poly(aryl piperidinium); ATM-PP, poly(phenylene) with benzylic methylammonium groups; BPN, quaternized biphenylene AEI; TMA, trimethylammonium-functionalized polystyrenes; HTMA-DAPP, hexamethyltrimethylammonium-functionalized Diels–Alder polyphenylene; qPPO, quaternized polyphenylene oxide (with trimethylamine, TMA); PFOTFPh-TMA-Cx, TMA-modified poly(fluorene-alt-tetrafluorophenylene) with x being the number of carbon atoms in the alkyl side chain; PFTP, poly(fluorenyl-co-terphenyl piperidinium-8); PFBP, poly(fluorenyl-co-biphenyl piperidinium-14); LDPE, low-density polyethylene; PSEBS, polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene, from technical data sheet.
Nafion and PTFE may act as binders rather than AEIs’ in all of these cases, i.e., studies 6–8, OH–-conducting electrolytes were fed to the AEMWE cell.
PTFE-reinforced Sustainion.
Dowex Marathon A is milled anion-selective particles (Dow, predominant particles between 10 and 30 μm size, IEC = 3.90 mequiv/g) blended with LDPE and a water-soluble additive (3.4 wt %). They are press-molded between poly(ethylene terephthalate) (0.3 mm thick) films.
Ni foams served as PTL and GDL; no catalyst was added onto the foams.
No further information was supplied by the authors on the membrane other than in-house prepared solid alkaline polymer electrolyte (APE).
Much of the recent OER catalyst development for AEMWEs has focused on the oxy(hydroxide) forms of nickel. The exact nature of these oxy(hydroxides) can vary, and hence, the abbreviation MOxHy is preferably used. It is known that nickel-based catalysts on their own have a low catalytic activity for the OER, while the incorporation of iron into the nickel lattice can substantially enhance the OER activity.169,216 The iron appears to play a key role for the observed enhancement and facilitates the formation of high-surface-area structures. Fe atoms easily replace Ni atoms in the oxide/(oxy)hydroxide lattice. In the as-synthesized catalysts, the Fe is present as 3+ and the Ni is present as 2+, thus creating a change in the overall charge, which is compensated by the intercalation of anions such as carbonate and also water molecules, creating layered and high-surface-area structures that can also facilitate ion transport.217 The layered double helix (LDH) structures are composed of Ni and Fe layers and can be formed as two-dimensional layered sheet structures. The sheets are often only a few nanometers thick. Figure 20 demonstrates the layered high-surface-area structures, experimental XRD patterns, and corresponding structures.
Figure 20.

(a) Scanning electron microscopy (SEM) images, (b) XRD patterns, and (c) unit cell structures for Ni(OH)2 and NiFeOxHy catalysts. (b) XRD patterns for different amounts of Fe in the NiFeOxHy catalysts. (c) Interlayer of H2O in the open LDH structure of the NiFeOxHy catalyst. Reprinted with permission from ref (169). Copyright 2014 American Chemical Society.
3.3.5. Iron Contribution to OER Catalysts
Many past studies have ignored the fact that, due to the similarity of Fe and Ni (with atomic numbers of 28 and 26, respectively), iron is easily incorporated into the nickel (as well as the Co) lattice. Corrigan reported in 1987 that even trace Fe impurities present in the KOH electrolytes can decrease the Tafel slope and also increase the OER activity of the Ni catalysts, as shown in Figure 21.218 Iron is typically also present in nickel precursor salts and KOH electrolytes unless high-purity chemicals are used. Chemical and electrochemical methods have also been proposed for the removal of iron from commercial KOH electrolytes.219,220 Fe is easily incorporated into the nickel lattice during the synthesis and/or upon potential cycling when NiOOH is formed unless iron-free chemicals are used. Thus, reports that ignore the contribution of iron cannot be used for reliable catalyst activity data interpretation.
Figure 21.

Effect of a 1 ppm Fe impurity in a 25 wt % KOH electrolyte on the cyclic voltammogram (CV) characteristics of a nickel oxide thin-film electrode. The steep increase in current at ∼0.52 V seen for the CV curve containing the Fe impurity (lower graph) is due to the Fe-impurity-facilitated OER. Reprinted with permission from ref (218). Copyright 1987 IOP Publishing.
In the case of single-metal (oxy)hydroxides, i.e., when Fe impurities are absent, FeOxHy on its own exhibits a higher OER activity than other single-metal (oxy)hydroxides, as follows: FeOxHy > CoOxHy > NiOxHy > MnOxHy.221 This relatively recent study compared (oxy)hydroxides formed by electrodeposition as thin films of similar and low mass in 1 M Fe-free KOH at η values of 0.45 V and used high-purity, i.e., low Fe content, precursor salts for the synthesis. Therefore, this trend may well reflect a more accurate order for the OER activity of single-metal (oxy)hydroxides than seen in previous studies.221Figure 22a shows the trends of the single-metal (oxy)hydroxide catalysts evaluated as TOF calculated per total metal sites of the thin catalyst films, which were assumed to be surface sites, while Figure 22b shows the CV characteristic of the catalyst films. The beneficial effect of Fe incorporation into Ni- and Co-(oxy)hydroxides is also demonstrated in Figure 22, which shows that Ni0.71Fe0.29OxHy has the highest TOF followed by Co0.59Fe0.41OxHy.
Figure 22.
OER activity trends for various thin-film catalysts made using Fe-free precursors and Fe-free 1 M KOH electrolyte solutions. (a) η plotted versus the corresponding TOF number for the catalysts. (b) CV characteristics. The mass of the thin films was used to calculate the TOF number. Reprinted with permission from ref (221). Copyright 2015 American Chemical Society.
Activity increases of 30–1000 times have been reported due to Fe incorporation in NiOxHy single-metal and multimetal catalysts.222 This large range can be assigned to the many different catalyst-synthesis procedures used, which in turn results in catalysts with many different chemical, structural, and physical properties. Dionigi and Strasser presented a review that also includes the many different synthesis routes for these oxides.223 Again, a thorough characterization of the as-prepared catalysts is often lacking and requires more attention. NizFez–1OxHy-type catalysts have shown high OER mass activities when tested in thin-electrode-layer setups, and recent single AEMWE cell studies are also promising. The long-term stability of iron still needs to be proven, although recent Pourbaix diagram calculations for an iron-doped Ni-NiOOH system suggest that the iron doping improves the pH stability range by ∼2.5 units for both the acidic and alkaline conditions.224
An optimum OER activity at 5–10 atom % Fe, which levels off at 30–50 atom % Fe, of total TM mass has been reported for NiOxHy films (Figure 23).225 Above the 30–50 atom % range, Fe forms Fe2O3 and transitions to the unstable FeOOH, which also reduces the catalytic effect of the incorporated Fe. Furthermore, the location of Fe in the structure is key to reaching high catalytic activities. It is suggested that Fe located at the surface and edges and incorporated into defects accounts for most of the activity increase as compared to Fe present in the bulk of the catalyst.226 This is also the case for Fe incorporation into Co catalysts.227 This explains why small Fe contents can lead to orders of magnitude higher activities. In addition to assisting in the formation of high-surface-area, open, two-dimensional layered structures, Fe can also affect the electronic conductivity of a catalyst. Fe added to the bulk of Ni catalysts appears to impact the redox behavior, which can be seen in a shift of the Ni2+/Ni3+ reaction to higher potentials.228
Figure 23.

Influence of the atom % of Fe incorporated into NiOxHy films. (a) CV characteristics of the films showing a continued shift of the redox peaks of the Ni2+/Ni3+ reaction to more-positive potentials, while the onset potential for the OER is shifted to lower values up to 15 atom % Fe, followed by an increase for the higher Fe atom % concentrations. (b) Linear increase of the Ni2+/Ni3+ redox reaction potentials with increasing atom % Fe, while the number of electrons transferred in the film shows a linear decrease. Reprinted with permission from ref (228). Copyright 2013 American Chemical Society.
Single-metal Co catalysts, which can possess good electronic conductivities, have shown decent but not high OER activities.229 However, similar to NiOxHy, Fe impurities from the electrolyte can incorporate into the CoxOxHy structure, which can lead to higher OER activities.230,231 Unlike the Ni(Fe)OxHy system, the incorporation of Fe into the CoOxHy structure from the electrolyte occurs at a much slower rate. Intentional incorporation of 40–60 atom % Fe into CoOxHy yielded a 100 times increase in catalytic activity compared to Fe-free CoOxHy.222,230 However, the incorporation of large amounts of Fe can dilute the high electronic conductivity of the cobalt host.222 Furthermore, a rapid OER activity decay can be observed in CoFeOxHy catalysts due to the formation of less-conductive Fe oxide phases. The latter is not stable under the alkaline OER conditions seen in Fe leaching from the catalyst.222 The high OER activity places CoFeOxHy just behind NiFeOxHy as the most promising bimetallic catalyst.
3.3.6. Ternary MnMmOxHy Catalysts
To further tune the intrinsic OER activity, ternary catalysts mostly including the same TM elements as discussed earlier are also explored. NiFeCoOxHy is one of the most promising ternary catalysts. The 3+ oxidation states of both Ni and Co are electronically conductive and need to be reached for the OER (neither the Ni nor Co 2+ oxide states are conductive). The addition of Co to NiFeOxHy increases the electrical conductivity (the electronically conductive Co3+ is formed at lower potentials than the Ni3+.232 Therefore, the addition of Co to Ni-based catalysts facilitates the transition to a more-conductive Ni phase.208,219,232,233 As an example, the electronic benefit of the Co in NiCoFeOxHy catalysts has also been reported for thin spin-cased and subsequently annealed films that showed 22 mV/dec lower Tafel slopes (58 versus 77 mV/dec for NiCoFeOxHy versus NiFeOxHy).232 However, the effect of the Co addition on the intrinsic OER activity was reported to be minimal: the most active NiCoFeOxHy film showed only an ∼1.5 times higher intrinsic activity (measured as TOF per surface atom mass) than the Co-free NiFeOxHy film. The increased electronic conductivity of such catalysts containing Co may have other benefits such as improving the performance of MEAs. Differences in the electronic conductivities of catalysts are often not accounted for and could well affect the performance of the catalyst when incorporated into CLs (section 6).
The positive impact of Fe on the OER activity, at least in the short term, of Ni- and Co-based catalysts has been established, but the question regarding its specific role in the catalyst structure remains unanswered. One popular theory is that Fe is the active site, and it has been suggested that Fe3+ acts as a Lewis acid promoting the formation of higher Ni4+ oxidation states.220 In any case, the LDH structures promote reactant access to active sites due to their open structure.
3.3.7. CoCu-Based Catalysts
CoCu-based catalysts are also of interest.234 The use of Co makes a catalyst expensive, but based on the Pourbaix diagrams, both Cu and Co may show the stability needed for OER catalysts for AEMWEs. In fact, single AEMWE cells employing a commercial CuCoOx (Acta 3030) anode catalyst were operated for >100 h (Table 6).84,235 Copper on its own is a poor OER catalyst, but its incorporation into CoOOH creates a catalyst of higher activity than both Cu(OH)2 and CoOOH; however, the mechanism of the enhanced OER activity is not well-understood. Early studies using Cu-incorporated Co3O4 catalysts (CuxCo3–xO4, 0 ≤ x < 1) involved single-AEMWE-cell tests and OER catalyst loadings in the 3 mg/cm2 MEA area range.90,236,237 The substitution of Co into the spinel Co3O4 lattice shifted both the Co3+/4+ redox reaction and the η for the OER to lower potentials. The authors showed promising AEMWE cell performances featuring a cell voltage of 1.8 V at 1 A/cm2, and the composition of Cu0.7Co2.3O4 was reported as the most active. A thermal decomposition method was used to prepare the catalysts, and the average catalyst particle size reported was in the 20–30 nm range, although larger particles may be present according to the transmission electron microscopy (TEM) images shown. Alternative synthesis routes may result in the optimization of the catalyst particle size and increases in the ECSA, which in turn could result in higher catalyst and AEMWE cell performances. More recent studies have explored the synthesis of high-surface-area, Cu-substituted Co3O4 catalysts,238−240 Karmakar and Srivastava synthesized Cu0.3Co2.7O4 nanochains.240 The smallest catalyst particles were in the 10–26 nm range. Jang et al. used a low-temperature and pH-adjusted coprecipitation method to form Cu0.5Co2.5O4 and Co3O4 catalysts.239 They reported Cu0.5Co2.5O4 particles of 3–4 nm size to be the smallest and most-active OER catalyst. In 1 M KOH, at 10 mA/cmgeom2 and for Cu0.5Co2.5O4 catalyst loadings of 0.5 mg/cmgeom2, a η value of 285 mV was reported. The Tafel slope of 79 mV/dec for this catalyst is also lower than the 98 mV/dec slope measured for the IrO2 catalyst, but it is higher than the slopes for NizFez–1OxHy catalysts. The half-cell performance measured for the Cu0.5Co2.5O4 catalyst on a Ni foam current collector yielded a cell voltage of 1.8 V at 1.3 A/cm2, and a decline of ∼15 mV was observed in the polarization curve recorded after 2000 h at 10 mA/cmgeom2. However, the catalyst loading on the Ni foam was high, namely, 10 mg/cm2, and the current density used for such a stability test is low. Therefore, the improvement over previous AMEWE cell tests seems minor. It is possible that the coprecipitation method used by Jang et al.239 also formed some larger particles, as in fact particle-size control without a stabilizer using the coprecipitation method is a challenge. Park et al. used the approach to form nanostructured CuCo2O4 catalysts directly on the Ni foam current collector.241 A cell voltage of 1.8 V was achieved at 1 A/cm2, showing a higher performance at a higher current density than that for a commercial IrO2 powder catalyst, although the loading of the CuCo2O4 versus the IrO2 catalyst on the Ni foam was significantly higher, namely, 23 versus 4 mg/cm2, respectively. The result for CuxCo3–xO4, 0 ≤ x < 1, makes this system interesting, but methods to form higher-ECSA catalysts need to be found.
3.3.8. Perovskites
Perovskites are another class of materials studied extensively as catalysts for the OER in alkaline media. The general formula of the perovskite structure is ABO3, where A and B are cations of different size. Perovskite catalysts are composed of rare and alkaline earth metals at site A and 3d TM at site B. The variation in the OER activity for a perovskite is correlated with the eg orbital filling, indicating an eg closer to unity to be more active.242 A decade ago, Ba0.5Sr0.5Co0.8Fe0.2O3−δ was reported with an intrinsically high OER activity,243 but it is unstable under the oxidative conditions that are relevant for AEMWEs. Further work showed that Ba2+ and Sr2+ leached, leaving behind a less-active Fe–Co surface.244 Recently, a double cubic perovskite, Pr0.5Ba0.5CoO3−δ, with the highest OER activity and an increased stability among perovskites was reported.242 According to computational studies, the position of the O p-band center relative to the Fermi level can explain the different OER activities observed for perovskite catalysts.243,127,245 Fermi levels closer and overlapping to the O p-band are linked to higher activities. Unfortunately, it is challenging to determine the exact binding energy of M–O as the surface is altered due to leaching and redeposition of metal cations during the OER.
3.3.9. Chalcogenide, Sulfide, and Phosphide Dopants
Research has also focused on chalcogenides, sulfides, and phosphides as dopants for TM catalysts because this class of materials has shown promising HER activities.246−248 Many studies report that TM sulfides and phosphides are better OER catalysts than the TM-only equivalent.246,249−251 Metal sulfides, phosphides, and nitrides are thermodynamically unstable under oxidizing conditions;204,252 hence, it is expected that these catalysts are oxidized to (oxy)hydroxides. However, detailed experimental support is lacking. Researchers have acknowledged the formation of the oxide and hydroxide phases at the surface, leaving the core, if anything, as sulfides, phosphides, and chalcogenides.253,254 The nature of the resulting structure may well have an enhanced catalytic activity due to the creation of defect sites or a higher surface area. In operando and postmortem analyses are needed to elude the mechanism.
3.3.10. Catalyst OER Activities in a Thin Layer Versus a Single AEMWE Cell
Measuring catalytic activities in a thin-layer electrode setup typically represent short-term measurements. Therefore, the subsequent evaluation of promising catalysts in single AEMWE cells or a half-cell setup are needed. The importance of this is also highlighted by the substantially higher Tafel slopes that have been measured in single-cell AEMWEs compared to slopes determined from thin-layer setups. Slopes higher than 120 mV/dec have been observed in AEMWEs, indicating that other factors than catalytic reactions determine this slope.255
Xu et al.208 made a series of single-, bi-, and multi-metal oxide OER catalysts. Catalysts showing higher thin-layer and single-cell AEMWE performances have been reported, but their study represented the measurement of both the OER activities for thin-layer electrodes and their subsequent evaluation in single-cell AEMWE performances for a large number of catalysts. Mixed Ni-, Co-, and Fe-oxide catalysts (namely, Co3O4, CoFeOx, NiFeOxHy, NiCoOx, NiCoOx:Fe, and NiCoFeOx) were made and also compared to a commercial IrOx (Proton OnSite) catalyst. Fe was likely incorporated into the Co3O4 and NiCoOx catalysts from the KOH electrolyte, and the authors used the formula “IrOx” for the commercial catalyst, possibly reflecting an amorphous Ir-oxide, as already discussed in section 3.3.3. It can be seen (Figure 24) that their catalysts referred to as NiCoOx:Fe, NiCoFeOx, and NiCoOx outperformed the commercial IrOx catalyst in the AEMWE cell, while the OER activity in a thin-catalyst-layer setup showed a different order. The mass activity (in A/mgcat) measured for the thin catalyst layers at a η value of 0.35 V showed the following order: NiFeOxHy (633 A/mg) > IrOx (273 A/mg) > CoFeOx (61 A/mg) > Co3O4 (30 A/mg) > NiCoFeOx (18 A/mg) ≈ NiCoOx:Fe (17.2 A/mg) > NiCoOx (8.8 A/mg). The order of the Tafel slopes measured in the thin-catalyst-layer setup was relatively similar to the order of mass activity as the lowest slopes in the 40–49 mV/dec range were observed for NiFeOxHy, CoFeOx, and Co3O4 as well as 47 mV/dec for IrOx, while NiCoFeOx, NiCoOx:Fe, and NiCoOx showed slopes of 54, 55, and 53 mV/dec, respectively. The authors suggested that some of the discrepancy between the thin-catalyst-layer setup data and the single-AEMWE-cell evaluation was due to the differences in electrical conductivity of the powder catalysts, as NiFeOxHy, Co3O4, and CoFeOx showed lower electronic conductivities than the other catalysts. Some of it may also reflect stability issues of the catalysts such as the NiFeOxHy catalyst, which seemed to show that one of the highest OER mass activities reported in a thin-catalyst-layer setup contained a high amount of Fe. It is possible that some of the high OER activity observed in the thin-layer and short-term experiment originated from a single Fe-oxide phase. Dissolution of Fe from a single Fe-oxide phase may well occur. The overall results clearly indicate that measurements in single AEMWE cells are needed to confirm the performance of a catalyst.
Figure 24.

(a) Tafel slopes measured in a thin-catalyst-layer setup, (b) conductivity of the as-prepared powders, and (c) performance of the OER catalysts in a single-cell AEMWE for a range of OER catalyst powders. The Tafel slopes in (a) were extracted from steady-state measurements in 1 M KOH electrolytes at 20 °C. The AEMWE performances were measured under a pure water feed at 50 °C. Reprinted with permission from ref (208). Copyright 2019 American Chemical Society.
3.3.11. Role of Lattice Oxygen for OER Catalysts
The high potentials driving the OER and the presence of lattice oxygen in most OER catalysts raise the question of what role lattice oxygen plays in the reaction and also in the stability of the catalysts. DFT calculations suggest that the release of lattice (bulk) oxygen in a system such as NiFeOxHy is feasible because the energetics for the OER at the surface and in the bulk are comparable.256 One study reports a linear increase of the OER current with increasing NiFeOxHy loadings (up to 0.1 mg/cm2) on a flat electrode surface.257 This was taken as support for activity from lattice oxygen, while others claim the opposite.258 Isotope-labeled water experiments coupled to a high-sensitivity detection method suggest that lattice oxygen contributes to the OER for many catalysts, including Au surface oxides,259 IrO2/Ti,260 Co3O4 spinel,261 some Ru-based catalysts,262,263 and perovskites including La0:5Sr0:5CoO3−γ, Pr0:5Ba0:5CoO3−γ, and SrCoO3−γ.264 Release of lattice oxygen was not observed for crystalline RuO2 structures265 nor for perovskites with low metal–oxygen bonds.264Figure 25 shows an example of isotope-labeling and Raman spectroscopy experiments for Ni and NiFe LDH catalysts suggesting the contribution of lattice oxygen in the case of Ni but not for the NiFe LDH catalyst.
Figure 25.

Isotope-exchange experiments and in situ Raman spectra of 18O-labeled (top) Ni and (middle) NiFe LDH, indicating the frequency shift and contribution of oxygen lattice for Ni, while the frequency remains constant for NiFe LDH. The figures at the bottom show the suggested scheme for O2 involvement (a) with and (b) without Fe. Reprinted with permission from ref (266). Copyright 2019 Wiley.
The literature results show that bulk oxidation is observed for specific compositions and catalyst structures. The lattice oxygen activities seem to vary depending on the nature of the catalyst, crystallinity, and operating conditions, as observed for the case of NiFeOxHy nanoparticles.214,266 Establishing an understanding of the factors determining the role of lattice oxygen will help in tuning the morphology, structure, and composition of the catalysts.
3.4. Stability of HER and OER Catalysts
As discussed throughout the catalyst section, HER and OER catalyst stability measurements relevant to AEMWE operating conditions are needed. Many different types of electrochemical methods such as chronoamperometric, chronopotentiometric, potential steps, and/or potential cycling have been applied to probe the stability of HER and OER catalysts. Therefore, consistent measurement procedures are needed, and suggested stability-evaluation protocols are shown in the Supporting Information.
Relevant factors that are potentially responsible for the deactivation of a catalyst, such as surface poisoning, morphology changes, and metal dissolution, can be missed by applying only electrochemical techniques. Ideally, the electrochemical catalyst stability studies are coupled in situ with analytical techniques, which are capable of quantifying dissolved metal components of the catalyst such as inductively coupled plasma mass spectrometry/optical emission spectroscopy (ICP-MS/OES).267 Electrochemical quartz crystal microbalance measurements (EQCM) can be useful to study mass changes of the electrode in situ.268 However, the EQCM response needs to be linear in order to avoid misinterpretations, as discussed by Moysiadou and Hu.269 Effort is being devoted to develop in situ techniques, such as in situ SEM/TEM or XRD, but these are far from being able to work under real operating conditions and are more suitable for the study of model catalysts. Therefore, the coupling of the electrochemical measurements with analytical techniques such as ICP-MS/OES is currently preferred.
The detection limits of ICP-MS are low [as low as 10 parts per trillion (ppt)], and hence, ICP-MS works best for solutions of low metal concentrations. ICP-MS is best-suited for acidic electrolytes,270 while the high cation concentration of alkaline electrolytes introduces calibration issues. The ICP-MS calibration becomes very challenging for cation concentrations exceeding 0.05 M,271 and ICP-OES, which is a less-sensitive instrument, is better suited for alkaline electrolytes. In the case of alkaline electrolytes, specific care needs to be taken to ensure complete metal dissolution in order to produce reliable ICP results.272 The additional step of acidifying the electrolyte is needed, hence presenting a challenge to in situ electrochemical–ICP measurements involving alkaline electrolytes. Some examples of catalyst-stability measurements, which included ICP-MS/OES measurements, were carried out for Co2P HER catalysts.273 Preferential dissolution of P over Co was shown to take place upon potential cycling, leaving a Co-rich surface of approximately twice the ESCA area after 2000 potential cycles. The dissolution of P resulted in an ECSA that doubled upon 2000 CV cycles.
The design of the electrochemical stability measurements reflecting conditions relevant to AEMWE operation is challenging. Many parameters such as the intrinsic activity, structure, composition, ECSA, conductivity of the catalyst, and feed solution of the AEMWE need to be considered for the design of the catalyst-stability experiments, as summarized in Figure 26 The formation of H2 and O2 gas bubbles is also a concern because they can block catalyst sites and consequently influence the stability measurements. In addition to physically blocking access to active sites, gas bubbles, if trapped in the structure of the catalyst and/or in a CL, can result in structural damage upon violent release.274 Gas bubble trapping on an electrode surface is specifically pronounced when a horizontal electrode, like a classic rotating ring disc electrode (RDE), is used and made worse upon rotating the electrode, as gas will be pulled into the center of the electrode by the rotational forces. A challenge is the time scale for the stability experiments, and catalysts capable of delivering high currents (>1 A/mg) for several thousands of hours are needed. It is evident that the design of accelerated stability tests (which reflect relevant AEMWE operating conditions and include information such as intermittent, startup, and shutdown operation and consider the development of potential hot spots and local pH fluctuations in a CL layer) is not trivial.
Figure 26.

Schematic diagram summarizing the important factors in designing an electrode for high-rate water splitting. Reprinted with permission from ref (274). Copyright 2021 American Chemical Society.
The electrochemical-stability measurements need to be complemented with a thorough characterization of the catalysts’ composition and structure (using XRD and XPS) at least before and after the measurement. Furthermore, electrochemical characterization of the electrode before and after the stability experiments is needed to show data such as CVs reflecting potential changes in the redox characteristics and ECSA value of the catalyst.
3.4.1. HER and OER Catalyst Stability
The stability of the OER catalysts is generally of greater concern than that for the HER catalysts. However, HER catalysts also undergo activity changes during electrolysis and even during intermittent periods. These activity changes can be caused by decomposition of the catalyst, hydride formation (section 3.1), agglomeration of nanosized catalyst particles, adsorption of the ionomer (section 6), deposition of dissolved metal cations on the catalysts, and H2 gas blockage of catalyst sites.
Reasons for catalyst-activity loss during the OER can be manifold and as simple as resulting from physical loss of the catalyst particles. However, chemical and structural alterations of the catalysts can also take place during the OER, such as dissolution of catalyst components and structural and compositional changes, which can enhance or decrease the OER activity. Generally, an increase in the anode potential within the OER region leads to an exponential increase in metal dissolution, which is taken as confirmation that the OER and catalyst dissolution are correlated.271 Some key points can also be taken from the numerous OER catalyst studies carried out for dominantly Ir- and Ru-based catalysts in acidic electrolytes.116 For example, the formation of an insulating oxide layer favors stability. Also, the metal-dissolution rate for crystalline structures is generally lower than that for amorphous structures due to a stronger metal-to-metal bonding energy. However, a stability test may suggest that a catalyst is very stable, but the catalyst’s OER activity may be inferior.271
Anodic dissolution is the most-probable cause of metal dissolution during the OER and is typically viewed as a continuous process, but catalysts can show instabilities during startup and shutdown periods, as demonstrated in a recent study by Speck et al.275 Metal dissolution of nine electrodes that were either 3d, 4d, or 5d transition metals was quantified (in acidic and alkaline electrolytes) using a flow cell and ICP-MS analysis. Either a low potential or a negative current was initially applied with the intent to reduce the metal surface. This was followed by a step to a potential where metal oxide formation was expected to occur, prior to reducing the metal surface again. The metal-dissolution rate was determined for the oxide formation and for the transient conditions, i.e., when the metal oxide surfaces were reduced. Results are shown in Figure 27. The study suggests that the rate of metal dissolution under oxide-formation conditions is proportional to the d-shell of the TM electrodes. For example, for Ir and Au metals that are within the same d-shell structure, metal dissolution is more pronounced for Au Ir. Transient dissolution was not observed for the 3d TMs, while transient metal dissolution occurred for 4d and 5d TMs at different rates. Overall, the results suggest that startup and shutdown should be avoided to decrease cathodic transient corrosion.
Figure 27.

Dissolution rates during transient measurements for nine different metal electrodes (as indicated in the graphs). The metals are grouped according to their electronic structure, i.e., either 3d, 4d, or 5d. Three metals per group were selected. The studies were carried out in 0.05 M NaOH. Reprinted with permission from ref (275). Copyright 2021 Wiley.
The study also suggested that the bonding energy of the metal–metal atoms and the affinity of the metal for oxygen are two determining descriptors of the catalyst’s stability during the OER. It was suggested that metals with higher cohesive energy (or metal–metal bonding energy) are less prone to dissolution, although this statement will need to be viewed in the context of the presence and nature of the electrolye. Additionally, a high oxygen-adsorption energy also favors the dissolution of metals with a higher affinity for oxygen because they are prone to incorporation of oxygen under oxidation conditions. Unlike the case of HER catalysts, the dissolution rate of OER catalysts in an open circuit has not been studied, but it would be useful for the selection of OER catalysts for AEMWEs.
Another effect of metal dissolution is the change in catalyst composition causing changes in catalytic activity. A study focusing on CoOx, CoFeOx, CoFeNiOx, and NiOx catalysts analyzed the total mass and composition of TM oxide catalysts before and after chronopotentiometric measurements in 1 M KOH for 6 h, as shown in Figure 28.269 All catalysts underwent noticeable compositional changes during the 6 h chronopotentiometric experiment (Figure 28). The first notable change was the incorporation of Fe if Fe was not already present in the catalyst structure [CoOx (a) and NiOx (c)]. Furthermore, the two catalysts without Co, namely, FeNiOx (c) and NiOx (d) showed minimal metal dissolution during the initial 6 h, while the Co-containing catalysts [CoOx, CoFeOx, and CoFeNiOx (a–c)] showed mass losses in the 10–20% range. These results suggest that Co, at least in the catalyst used by this research group, induces a stability issue. Using electrochemical impedance spectroscopy (EIS), the authors determined that the dissolution rate varied over the initial 6 h, and afterward, the catalysts were considered stable. This time-dependent behavior may be due to the dissolution of very reactive catalyst components including defects and undercoordinated surface sites, followed by the establishment of an apparent stable state such as a protective oxide layer.276
Figure 28.

Changes in the masses of various thin-film catalysts before and after constant-current experiments at 5 mA/cm2 for 6 h in 1 M KOH. Different loadings of the catalyst were used, as indicated in the figure (loadings 1, 2, and 3). The names of the catalysts and the catalyst compositions (measured before and after 6 h of chronopotentiometric experiments) are also shown in the graphs.269
Theoretical and experimental studies suggest that low-coordinated sites such as defects, edges, and steps are more likely corrosion sites.276 However, this descriptor is extremely difficult to quantify and replicate.
3.4.2. Differences between Model Electrochemical Stability Studies Versus an AEMWE
Many stability measurements utilize traditional electrochemical cells such as an H-cell or a flow cell, where the catalyst is often immersed in an aqueous electrolyte. However, a catalyst experiences significantly different conditions in a thin-layer setup (which is typically an aqueous model system) than in an MEA of an operating AEMWE. A comparison between an aqueous model system and an MEA is presented in Figure 29. One of the main differences is the electrolyte/electrode interface. In a model system, the electrode is completely immersed in the electrolyte, while in an AEMWE, the catalyst layer experiences a higher exposure to the gaseous reactant environment. In an MEA, the catalyst is also surrounded with other components such as an ionomer enabling OH– transport between anode and cathode (section 6). The electrode architecture in an aqueous model system can involve a powder catalyst but also compact thin films preferably deposited onto stable electrode surfaces of low HER and/or OER activity.
Figure 29.

Differences a catalyst can experience in a traditional electrochemical experiment labeled as an aqueous model system (AMS) and in an MEA of an AEMWE cell. The differences can be the electrode architecture, the electrolyte, reactant and product transport, and the operating conditions. Reprinted with permission from ref (277). Copyright 2021 Elsevier.
To close the gap between model studies and real operating systems, the use of a half-cell gas diffusion electrode (GDE) coupled to an ICP-MS/OES is favored.278 Researchers have studied Pt during the HER in a half-cell GDE and acidic conditions, and some findings apply to alkaline conditions. It was found that, contrary to what was observed for model systems, the dissolution of the Pt metal increased when the overall metal loading was decreased. Also, the metal dissolution in a half-cell GDE was lower because of the limited interactions with the electrolyte and the presence of the membrane. The ionomer also plays a role.278 For mass-transport-limitation reasons, one could also expect Ostwald ripening and local redeposition on existing particles to become more likely.279 Overall, one may speculate that the most general trends observed in a traditional electrochemical setup translate into a half-cell GDE system or an MEA. However, the intrinsic catalyst activities are expected to be different for evaluations carried out for a model versus a half-cell GDE system or an MEA due to the drastically different system architecture and operating conditions.
4. Anion-Exchange Membranes
So far, comparably low ionic conductivity and low durability of AEMs have been the major obstacles for the large-scale introduction of AEMWEs. However, recent advances, in increasing both the OH– conductivity and the alkaline stability of AEMs, have fueled AEMWE development.83,280−282 Much of the research over the past decade has focused on developing AEMs for alkaline anion-exchange membrane fuel cells (AEMFCs); hence, in this section, reference to AEM performance in AEMFCs is made when adequate. AEMs are made from anion-exchange polymers (AEPs) consisting of cationic headgroups attached to the polymeric backbones. Extensive research is being carried out on these AEPs and many reviews exist, but a performance comparison is difficult due to the lack of consistent evaluation conditions. Therefore, there is a need to advance the understanding of the general performance-relevant parameters in an AEMWE cell. Critical characteristics of AEMs, such as the ion-exchange capacity (IEC), OH– conductivity, chemical and mechanical stabilities, water uptake, and swelling of AEMs, depend on numerous factors including the AEP structure and operational parameters such as the electrolyte and humidification. A high ionic OH– conductivity exceeding 0.1 S/cm is preferred.32 High chemical and mechanical stabilities at j >3 A/cm2 and T > 60 °C, in the presence of O2 and in alkaline conditions, are needed. Many AEMs break down at temperatures exceeding 60 °C. The early research view was that cationic headgroups are accountable for the IEC, ionic conductivity, and chemical stability, while the polymer backbones are responsible for the mechanical and thermal stability.40 It is now recognized that both the mechanism and the rate of degradation are influenced by the complete structure of the AEP, thus calling for studies of the entire polymer rather than individual headgroups and backbones. Previous reviews identified many possible degradation pathways in alkaline media:40,283,284 (1) nucleophilic substitution (SN2) benzyl substitution, (2) SN2 methyl substitution, (3) β-elimination substitution, (4) ylide intermediated rearrangements, (5) SN2 Ar aryl ether cleavage in the polymer backbone, (6) ring opening (e.g., imidazolium (IM)), (7) SN2 methyl substitution IM, (8) heterocycle deprotonation IM, (9) SN2 and ring opening (e.g., in piperidinium, pyrrolidinium, and morpholinium), (10) ring opening (N-spirocyclic ammonium), (11) dehydrofluorination (polymer backbone), (12) nucleophilic addition and displacement (pyridinium), and (13) nucleophilic degradation (guanidinium). Most quaternary amines (QAs) and IM groups are prone to degradation under alkaline conditions via the Hofmann degradation, SN2, or ring-opening reaction, especially at elevated temperatures and high-pH conditions. Recent research has also shown that the degradation pathways depend on the AEP structure and the test conditions. For example, β-elimination has been shown to take place for AEMs tested at 80 °C for NaOH concentrations below 4 M, while the methyl substitution reaction is found to be predominant at 120 °C for NaOH concentrations of 8 M and at 100 °C and a low relative humidity (RH) of 5%.285
4.1. AEM Structures and Their Impact on Alkaline Stability
4.1.1. Cationic Headgroups
The cationic headgroups provide the exchange sites for OH– and are described by the IEC value. Many headgroups contain nitrogen, e.g., quaternary ammonium/tertiary diamines,286−288 (benz)imidazolium, guanidinium, and pyridinium.289−292 QAs are the most popular headgroups due to their promising ionic conductivity in an AEM, comparatively high stability, and ease of synthesis. Some nitrogen-free cationic headgroups have also shown promise with regards to ionic conductivity values and stability, e.g., sterically shielded phosphonium and sulphonium headgroups293−296 and ligand–metal complexes (Figure 30).297,298
Figure 30.
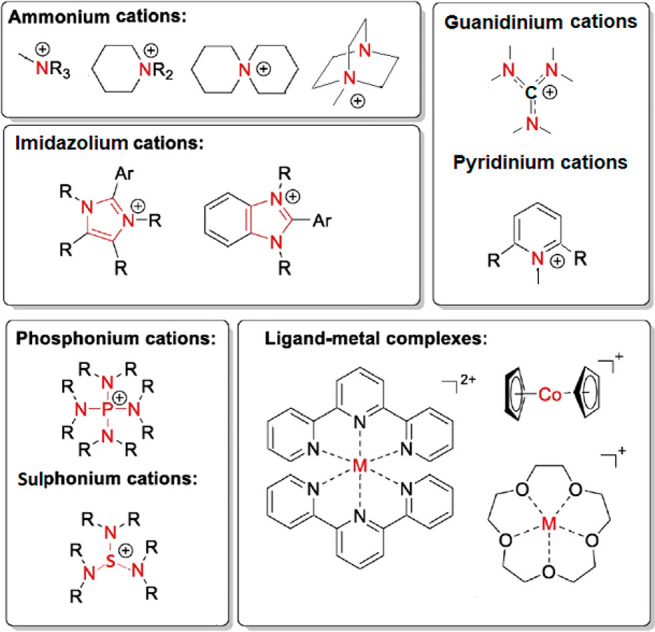
Examples of different types of common alkaline-stable cations for AEMs. Adapted with modification from ref (36).
To improve the stability of cation groups, many researchers designed molecules that are devoid of β-hydrogen or have a minimal number of β-hydrogen to suppress preferential Hofmann elimination.299 However, AEMs without β-hydrogens still show degradation due to other mechanisms, e.g., the SN2 mechanism, which occurs via direct nucleophilic attack of OH– anions on nitrogen atoms in the ammonium group, resulting in alcohol departure, or on the carbon atoms bonded with it, resulting in amine byproducts (Figure 31).300 Marino and Kreuer301 carried out an ex situ stability study on an extensive number of QA headgroups in their salt form using the same testing conditions, including controlling factors such as the temperature, solvent, and degree of solvation. The study reported that β-protons were less susceptible to nucleophilic attacks than previously suggested, whereas the presence of benzyl groups, nearby heteroatoms, or other electron-withdrawing species significantly promoted the degradation.301
Figure 31.
Hofmann elimination and nucleophilic degradation occurring via ammonium group displacement.
Further approaches to increase the alkaline stability of cation headgroups are (i) to introduce groups that enable a steric hindrance and, hence, shield the AEP from OH– attacks or (ii) to introduce groups with an electron-donor effect on or near the cations, countering the fact that electron-deficient cationic moieties appended to polymer backbones in AEMs are the most susceptible sites to OH– attacks.
A simplified stability trend for common cations used in AEMs has been proposed: pentasubstituted IM > C2-aryl benzimidazolium > simple IM; N-spirocyclic piperidinium > piperidinium > pyridinium; tetrakisaminophosphonium > triarylphosphonium.302 Besides the steric hindrance effect, particular requirements of stereochemistry can also enhance the alkaline AEM stabilities. The following are examples of the latter: Bauer and Strathmann303 studied a monoquaternized 1,4-diazabicyclo[2.2.2]octane (DABCO) cation tethered to a poly(ether sulfone) (PES) and found that the resulting AEP was highly resilient to OH– attacks. DABCO contains β-hydrogen, but the rigid cage structure in DABCO effectively hinders antiperiplanar conformation of the N atoms with β-hydrogen. Antiperiplanar conformations are a prerequisite for Hofmann elimination (Figure 32).
Figure 32.
Conformational analysis of DABCO (one of the syn-periplanar structures is highlighted in red).
The introduction of electron-donating groups in close vicinity of the cations is also investigated. The goal is to hinder OH– attacks by increasing the electron density of the cation. Bis-quaternary ammonium cross-linkers are more susceptible to degradation if two QA cations are close to each other, as two quaternized nitrogen centers strengthen the local environment for electron deficiency.304 The stability of alkyltrimethylammonium is higher than that for benzyltrimethylammonium (BTMA). The fact that imidazolium groups with electron-donating substituents improve the alkaline stability over conventional IM groups also supports the electron-donor strategy.305 Unsubstituted IMs generally exhibit poor chemical stability in strong alkaline conditions by ring-opening reactions such as SN2 reactions, heterocycle deprotonations, and substituent deprotonations.306 As mentioned, the stability could be increased by replacing the H in the β-position with electron-donating groups, such as methyl or butyl groups. Price et al.307 proposed that the effectiveness of increasing alkaline stability of imidazolium cations was higher for electronic stabilization of the C-2 position versus steric stabilization of the C-2 position. Some IM groups with large substituents can exceed the TMA (trimethylammonium) benchmark; however, the OH– conductivity of IM-based AEPs is lower than that for QA-based AEPs.308 Diesendruck and Dekel found that the alkaline stability of BTMA groups, 6-azonia-spiro-[5.5]-undecane (ASU), and large-steric-hindrance imidazolium groups are affected by the λ value (λ = number of water molecules per OH–) at room temperature.309 The relationship between the λ value and the current density has not yet been revealed experimentally.308
Introducing resonance-stabilized structures in or near cationic groups opens another potential antidegradation pathway. The positive charges are delocated over more than one N (or P) atom by using aromatic diamine or multiple N or P systems, leading to resonance stabilization, as seen for, e.g., the heterocyclic imidazolium system.35 Another example of a resonance-stabilized AEM structure is n-alkylaminophosphonium in poly(ethylene) backbones.294 Compared to QA, quaternary phosphonium (QP) cations have attracted less attention. It was found that QP cations containing three trimethylphenyl groups exhibited extremely high alkaline stability exceeding 64 times that of benzyltrimethylammonium.306 Their alkaline stability is improved by introducing certain bulky groups. This is due to the strong electron-donating ability of the substitution groups, which can conjugate with the phosphonium cation. However, it is difficult to obtain AEMs with high IECs and good mechanical properties, as the compatibility of quaternary phosphonium with polymer matrixes can be poor. Guanidinium-based cations are also viewed to form resonance-stabilized structures due to charge delocalization along several moieties. Unfortunately, guanidinium does not seem to effectively increase the stability due to its high susceptibility to nucleophilic OH– attacks.310
A recent approach to enhance alkaline stability is using cyclic cations as monoquaternized ammonium groups, but these cyclic ammonium groups still degrade mainly via nucleophilic substitution by a ring-opening mechanism in alkaline conditions. However, due to their ring strain, five- and six-ring cyclic ammonium groups have high alkaline stability, even higher than that of seven- or eight-membered rings.301,311−314 Some of these groups are low-cost and commercially available.
Metal-based cation groups with organic moieties have been researched and present promising stability at 80 °C in concentrated alkali solutions. Unlike the univalent cations, multivalent metal cations possess the ability to coordinate with more than one anion per cation center. Therefore, the incorporation of multivalent cations in AEMs facilitates the improvement of the IEC, resulting in higher ion conductivities.306 Examples of the latter are ruthenium, cobalt, and nickel, in bis(terpyridine) structure and permethyl cobaltocenium, but the corresponding materials can be costly and their synthesis tends to be complex. Tethering metal-based cation groups to a polymer backbone is difficult, and AEPs with metal-based cation groups do not yet show the ion conductivities and low water uptake needed for AEMWEs.
4.1.2. Backbones
Polymeric AEM backbones are base polymers free of cationic moieties. Examples are poly(arylene ether)-based backbones [e.g., polyphenylene oxide, poly(arylene ether sulfone), poly(arylene ether ketone), and poly(arylene ether phosphine oxide)], polyolefin-based backbones [e.g., polyethylene, polystyrene, polynorborene, and polytetrafluoroethylene], polyphenylene-based backbones, and backbones containing cationic moieties [e.g., poly(benzimidazole) and poly(phosphazene)].36 Some show very promising alkaline stability; for example, polyphenylene-based AEMs with ketone tethers linked to guanidinium cations were stable in 0.5 M KOH at 80 °C for thousands of hours.315
4.2. Ionic Conductivity and Other Physical Properties
4.2.1. Ionic-Conductivity Measurement Procedures
The ionic conductivity is a key parameter that is determined by a number of factors such as the IEC, water uptake, microphase separation, type and number of cationic groups, and spacing and clustering of the latter. Measurements of the ionic conductivity and other physical properties of AEMs need to be evaluated and standardized because current methodologies strongly vary between laboratories. The ionic conductivity depends on operation conditions such as the temperature and the viscosity of the electrolyte. Furthermore, the liquid-electrolyte compositions employed for the measurements often vary, such as using NaOH versus KOH solutions, and also in terms of the alkali concentrations. Furthermore, the measured ionic-conductivity value can be lower than the actual value of the OH– conductivity because OH– ions are quickly exchanged by CO32– and HCO3– ions if present in the system.316 This can be enabled by measurements under ambient conditions where CO2 is part of the air.
Consequently, the need for systematic measurements has been highlighted to ensure the determination of the actual OH– conductivity value rather than an apparently lower value resulting from carbonate infiltration into the material of interest.317 Therefore, prior to the ionic-conductivity measurements, the removal of air and CO32– and HCO3– ions in the N2 and H2O measuring atmospheres is suggested (Figure 33).
Figure 33.
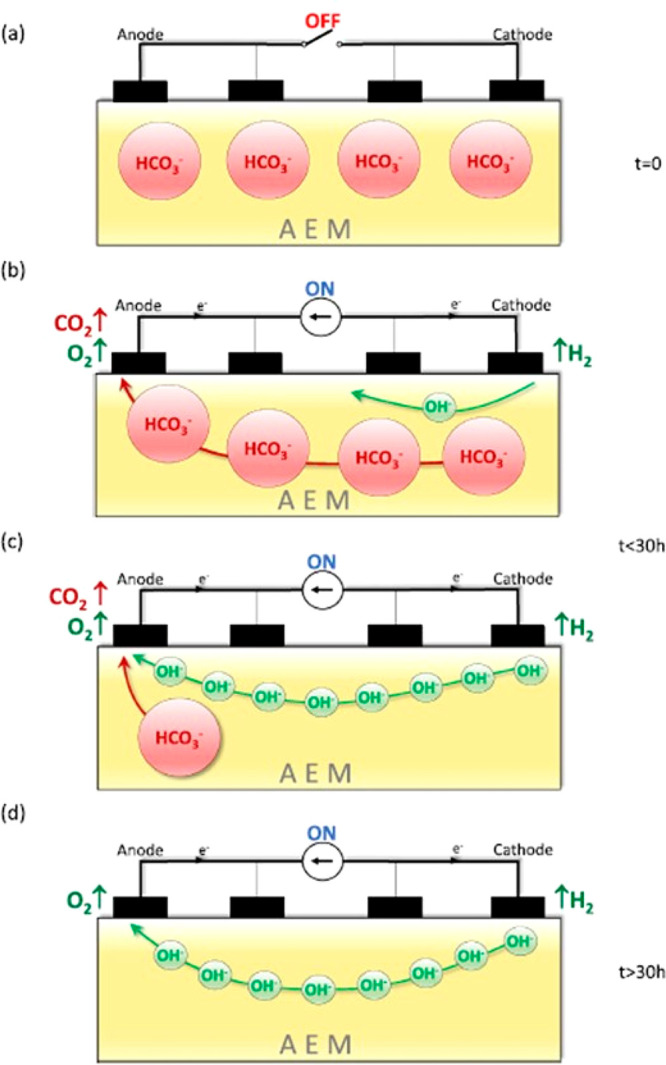
Schematic illustration of the processes taking place in the AEM while applying the direct current under the conditions of the conductivity measurement carried out under N2 and H2O atmospheres. Adapted from ref (316) with permission. The black rectangular boxes show the sensor electrodes and the anode and cathode for the two H2O splitting reactions. Closing the circuit turned the system on, allowing a low (typically 100 μA) current flow. The measurement setup shown in the figure yielded in-plane values.
In terms of the measurement procedure, the ionic conductivity is also often measured using four-point measurements determining in-plane values. However, only measurements across the membrane actually determine OH– conductivity between the cathode and anode and, thus, the value that is relevant for an operating water electrolyzer. Therefore, measurements of ionic conductivity should be performed in the through-plane direction.
4.2.2. Methods To Improve Critical AEM Properties
High ionic conductivities of AEMs can be achieved by increasing the IEC value, which is a measurement of fixed cation groups’ concentration. However, there is an optimum value, as high IECs result in high swelling ratios and large water uptake values, both of which are associated with the reduction of the membrane’s mechanical strength. This can be very pronounced, and a material with a very high IEC might be gel-like rather than a solid membrane, which is not desired for AEMWE applications. Therefore, an optimum value between ionic conductivity and mechanical membrane stability has to be achieved.
Strong swelling of the membrane material can lead to delamination of the AEM and catalyst layer. Gas and accompanied bubble evolution in operating AEMWEs was found to cause catalyst delamination upon extensive swelling, especially above 50 °C and for j values >0.5 A/cm2.318 It is very challenging to lower the swelling ratio of AEMs below the target values, which are dry/wet dimensional changes ≤1% in the machine direction and ≤4% in the transverse direction,32 without compromising the ionic-conductivity value. Most reported AEMs with ionic conductivities exceeding 0.05 S/cm at 20 °C show swelling ratios in the range of 20–40% and maximum tensile strengths of 16–34 MPa when fully hydrated.284 In addition to the ionic-conductivity value and dry/wet dimensions, other target values of AEMs for AEMWE applications are desired such as tensile strength >15 MPa, elongation at break >100%, area-specific resistance (ASR) ≤0.07 Ω cm2, and stability ≤0.07 Ω cm2 after 2000 h in an AEMWE.32
Material-design solutions at the nano- to microlevel are being developed to improve the ionic conductivity and reduce swelling. Currently, the commercial Tokuyama A201 is the AEM with the lowest reported swelling ratio of 6% TD (transverse direction) and 2% MD (machine direction). The ionic conductivity of this AEM is 0.042 S/cm at 90% RH at 20 °C.36 A parapolyphenylene-based AEM shows a remarkably low swelling ratio of 9.5%, a promising tensile strength of 35 MPa, and ionic conductivities of 0.049 S/cm at 30 °C and 0.137 S/cm at 80 °C.319
4.3. Performance-Enhancing Strategies
As discussed, in addition to alkaline stability, AEMs must simultaneously possess high OH– conductivity while maintaining mechanical integrity. The following paragraphs discuss methodologies that have shown some promise to create such membrane properties.
4.3.1. Cross-linking
Cross-linking creates chemical bonds between molecules contained in an ion-conducting polymer with the goal to reduce swelling; ideal cross-linking also maintains the high ionic conductivity of the AEP and AEM. Cross-linking is regarded as a straightforward way to improve thermal, mechanical, and physiochemical AEP properties. High mechanical stabilities are of extra high importance for thin (less than ∼50 μm) AEMs and for AEMWE operation at high differential pressure. Cross-linking can be physical or chemical. Physical cross-linking introduces ion–ion320 or van der Waals interactions321 between molecules. Chemical cross-linking refers to reagents covalently connected to the AEP. Such cross-linkable reagents can be small compounds, oligomers, or even end groups. Chemical cross-linking can be done as a one-step synthesis322−324 or as a post-cross-linking step. Cross-linking approaches have been explored as thiol–ene chemistry,325,326 Menshutkin reaction between halo-methylated polymer and commercially available diamines,327 ring-opening metathesis polymerization,328 olefin metathesis,329 and thermal cross-linking.330 Cross-linking has been shown to be beneficial, but swelling cannot be completely eliminated.328,293,331 The swelling and corresponding OH– attacks on the AEP backbones and the functional groups can be reduced, but the ionic conductivity and processabiliy will be reduced if the linking occurs via the ion-conducting end groups. Multication side-chain or end-group cross-link strategies have been proven as effective; for example, Chen et al.332 reported a series of multication cross-linked membranes with high OH– conductivity (0.155 S/cm at 80 °C) and good dimensional and alkaline stability. Lee et al.333 prepared a series of end-group cross-linked polysulfone (PSF) membranes by introducing a benzyne group at the end of the PSF polymer chain. The cross-linking improved the ionic conductivity (0.11 S/cm at 80 °C) and dimensional stability.
If not appropriately applied, cross-linking can result in poor AEP and AEM properties, for example, cross-linkers with long chains were shown to induce crystallinity into AEMs, compromising many physicochemical properties such as reducing the hydrophilicity.334 A study of poly(2,6-dimethyl-1,4-phenylene oxide) using hydrophilic cross-linkers that contained ethylene oxide (EO) showed that the presence of long EO cross-linkers increases the degree of crystallinity but reduces both the ionic conductivity and the alkaline stability.334 If not dosed correctly, cross-linking can be too strong, resulting in mechanically brittle AEMs and possibly poor alkaline stability. Furthermore, additional reaction steps complicate the processing of the membrane.40
Interaction of interpenetrating polymer networks (IPNs) allows for surpassing the mechanical strength of the original polymer with high IECs.335 Theoretically, IPN AEMs create networks made of a continuous ion-conductive phase, while non-ion-conductive networks maintain the mechanical stability. The networks interlace on the molecular scale without being covalently bonded. Examples are an IPN AEM based on poly(vinyl alcohol)/polyethylenimine and an IPN AEM cross-linked quaternized poly(epichlorohydrin)/polytetrafluoroethylene (PTFE).336−339 Reported IPN AEMs do not yet meet the mechanical strength requirements but show potential. For example, a cross-linked poly(vinyl alcohol)/cross-linked poly(vinyl benzyl-N-methyl piperidinium) IPN AEM yielded a high ionic conductivity of 0.258 S/cm at 80 °C, a moderate IEC value of 1.75 mmol/g, and an encouraging tensile strength of 9.3 MPa in the wet AEM state.340
4.3.2. Microphase Separation
The ion-conducting polymers can contain wetting (typically the ion-conducting part) and nonwetting parts. When such a polymer is in contact with a liquid such as water or an electrolyte, the polymer molecules can reorient in a manner such that the wetting parts of the polymer are in contact with the liquid, resulting in the formation of liquid clusters. If the molecules contain specific spatial properties, the wetting and nonwetting parts create two phases. This effect is called microphase separation. The formation of a hydrophilic/hydrophobic microphase separation structure is relevant for the preparation of high-performance AEMs; percolating-liquid ionically conducting domains, which are called ion channels, can be created.341
An important design criterion is to maximize the population of percolated ionic domains to enhance the ionic conductivity, although the ion-conductive domains need to be uniformly distributed across the AEM.342 Such a microphase-separation-structure control approach is promising to achieve both high ionic conductivity and high mechanical stability. The backbones, tethering chains, and molecular structures of the headgroups strongly influence the formation of the AEM microphase, altering the ionic conductivity and the water uptake.343,344 Correspondingly, the location, type, and concentration of cations and hydrophobic side chains need to be tuned in order to achieve optimum 3D phase-separation structures, and these are regularly investigated.345 Important strategies for optimum phase separations are the location of the wetting ion-conducting moiety in side chains or multiblock copolymers containing wetting and nonwetting alternating sections.40,346−349 In the aforementioned side-chain-type AEPs, the side-chain length, characterized by, e.g., the number of alkyl spacers, has been suggested to have a significant effect on the AEP’s performance, with five or six alkyl spacers being the optimum design.350 To reveal the morphology–properties relationship, atomic force microscopy (AFM) and transmission electron microscopy (TEM) have been employed. As an example, both the hydrophilicity and the flexibility of ionic side chains have been shown to play crucial roles in fabricating high-performance AEMs.317 This is shown in Figure 34, where the increased hydrophilicity and flexibility of ionic side chains (named TQAPPO) showed well-defined and well-distributed hydrophilic microphase separations.
Figure 34.

AFM tapping phase images revealing the architecture–morphology–properties relationship of AEMs (BQAPPO and TQAPPO). The x–y scales in the AFM images are 100 nm per square. The bright and dark domains in AFM images are designated as the hydrophobic and hydrophilic phases, respectively. Adapted with permission from ref (317). Copyright 2015 Springer Nature.
4.3.3. Organic/Inorganic Composite AEMs
Organic/inorganic composites are another strategy to improve AEM performance (Figure 35). Composite AEMs consist of two classes: mixed-matrix membranes embedding inorganic nanoparticles in organic AEPs and membranes made of an inert porous support filled with AEPs.351 Mixed-matrix membranes are gaining popularity due to a wide range of embedding materials such as metal ions, metal oxides, silica, functionalized nanoparticles, graphene oxide, and carbon nanotubes.352 The particles and the porous support membrane in composite AEMs are typically nonionic and curb water uptake, while the polycations provide high ionic loadings, facilitating ion conduction. Previous work encompassed composite AEMs that showed an increase in ionic conductivity as well as thermal, chemical, and mechanical stability while reducing the water uptake.352−355 However, the validation of the results in AEMWE cells is often missing.
Figure 35.

(a) Structure of a composite copoly(arylene ether sulfone)/nano-ZrO2 AEM designed to simultaneously achieve a high ionic conductivity, low water uptake, and improved thermal, mechanical, and chemical stabilities. Adapted with permission from ref (357). Copyright 2014 Royal Society of Chemistry. The blue dots are nano-ZrO2. (b) Porous-sandwich structure composite AEMs. Adapted with permission from ref (358). Copyright 2018 Elsevier. (c) Electric-field-oriented and magnetic-field-oriented composite AEMs. Adapted with permission from refs (359 and 360). Copyright 2014 and 2018 Royal Society of Chemistry, respectively.
The nanoparticles need to be uniformly dispersed in the organic phase, and they need to be alkaline-resistant.356 Correspondingly, particles such as silica and alumina are not recommended, while zirconia particles are promising. A poly(vinyl alcohol) (PVA)/PDDA/nano-ZrO2 composite AEM with 2.5 wt % ZrO2 showed properties such as a maximum tensile strength of 13.96 MPa and an elongation of 229%, while the 1.5 wt % nano-ZrO2 AEM yielded the highest ionic-conductivity value of 0.032 S/cm at 20 °C. Single-AEMWE-cell results using nano-ZrO2 incorporated into a commercial Sustainion membrane show promise and highlight the potential to increase H2 and O2 separation.303
Composite AEMs strengthened with porous, woven, or electrospun substrates have shown enhanced ionic conductivities and reduced swelling. The substrates are usually chemically inert, and mechanically stable AEPs such as high-density polyethylene, polypropylene, polystyrene, polyimide, or polyolefin are used. A 125-μm-thick noncomposite AEM showed a mechanical failure at 2000 h, while a thinner (60–90 μm) reinforced version did not fail over 4500 h in an AEMWE cell at 30 bar and 80 °C.35 Chen et al.358,360 designed a series of QA-functionalized LDH/poly(p-phenylene oxide) (PPO) composite membranes with a porous sandwich structure with a high ionic conductivity of 122 mS/cm at 80 °C. Another interesting approach is to design aligned composite membranes. Fan et al.359 and Chen et al.360 designed electric- and magnetic-field-oriented composite membrane series, respectively. The ionic conductivities of aligned composite membranes displayed improvements of 39% for electric-field-oriented composite membranes and 55% for magnetic-field-oriented composite membranes over the corresponding nonaligned composite membranes.
4.4. Promising AEM Examples and New Research Directions
In sections 4.1–4.3, critical AEP and AEM properties and methods of improving them were discussed, while in this section a summary highlighting promising AEM developments and research direction trends is presented. Before 2010, an ionic conductivity of 0.010 S/cm at 60–80 °C was the target for AEMs.361 Since then the target has increased 10-fold,284 and several AEMs have exceeded 0.2 S/cm.362−367 Current research on AEMs often aims on increasing the mechanical and chemical stabilities as well as breaking the operational temperature limits. Chen and Lee summarized the ex situ durability, OH– conductivity, and water uptake of different types of AEPs (shown in Figure 36).350N,N-Dimethylpiperidinium (DMP)-type AEMs displayed an outstanding alkaline stability (ex situ durability) and a relatively high conductivity. Some reinforced polynorbornene (PNB)-, 6-azonia-spiro-[5.5]-undecane (ASU)-, and aryl ether-free BTMA-type AEMs also show overall high performances.350
Figure 36.

Comparison between the water uptake, OH– conductivity (σ), and ex situ stability of typical BTMA-, DMP-, ASU-, side-chain-, imidazolium-, phosphonium/sulfonium-, cobaltocenium-, and ruthenium-type AEPs. The water uptake (Wu) corresponds to the σ value at the same temperature (most AEPs are recorded at 80 °C, but some for the side-chain-, imidazolium-, sulfonium-, and ruthenium-type AEPs are plotted at room temperature and 60 °C due to insufficient information). The alkaline stability was recorded based on the temporal stability of AEPs in 1 M NaOH or KOH at 80 °C with degradation <10%, and some of the stable AEPs were evaluated at harsher conditions. Adapted with permission from ref (350). Copyright 2021 Elsevier.
On the basis of intensive research efforts achieved over the past decade, future trends in AEP designs are emerging. AEPs with noncyclic QAs are still a focus, mainly because they are easy to obtain and have stable AEP backbones, and the properly designed side chains avoid both Hofmann elimination and counterion condensation. Heterocycloaliphatic QAs will likely attract more attention because they show high stability in alkaline solutions.301 The alkaline stability of heterocycloaliphatic QA cations critically depends on their position in the AEP structure, the ring size, the presence of an additional heteroatom, and ring-substitution patterns.368 Spirocyclic QA cations are a special type of aliphatic heterocyclic QA with unique structures. This class of AEPs exhibit extraordinary alkaline stability because the spirocyclic structure has a high transition-state energy against degradation reactions.369 Reported studies include examples such as incorporating QA salts into the polymer backbone, attaching them directly onto the aliphatic or aromatic polymer backbone, and introducing them as a cross-linker to form a network.312,313,370,371 In terms of investigating the relationship between the alkaline stability and the cation structure of AEPs, in situ AEMWE cell tests are needed. The importance of in situ AEMWE cell testing of AEMs has been highlighted by a study carried out by Meek et al.,372 who also defined a testing protocol. Some AEPs exhibited excellent ex situ durability.314 However, in situ and ex situ results are generally not in agreement. For example, it was found that BTMA-PPO with poor alkaline stability showed acceptable in situ durability at 0.1 A/cm2, while side-chain-type PPO exhibited a significant voltage loss.373
With regards to AEP backbones, ether-free backbones are a preferred choice for structure design, as polybenzimidazole (PBI)-, polyphenylene-, and polyolefin-type AEMs have been widely explored. PBI- and polyphenylene-type AEPs have high thermal and good chemical stabilities.374 The unique benzimidazole repeating units in the backbone provide a high density of electronegative pyridine nitrogens (—N=) and can form hydrogen bonds to conduct OH–. However, their processability and ionic conductivity are too low for AEMWEs. Current research focuses on improving the ionic conductivity by alkali doping and enhancing the solubility by introducing ether bonds into the main chains. To improve the performance of doped PBI, several approaches have been developed, such as tuning the porosity, building sandwiched-porous PBI, and fabricating AEP blend systems.375−378
In Table 5 the structures of state-of-the-art AEPs associated with good ex situ ionic conductivity and alkaline stability are summarized to guide future AEP design. The examples shown in Table 5 exceed both ion-conductivity values of 0.1 S/cm at 80 °C and an ex situ stability of 500 h.350 AEMs tested specifically in AEMWE single cells are summarized in Table S19. Furthermore, protocols for AEM evaluation have been discussed in the literature, as mentioned earlier.314
Table 5. Summary of Recent Research Progress for AEPs of Ionic Conductivity Exceeding 0.1 S/cm at 80 °C and Ex Situ Stability Longer than 500 h.




5. Membrane Electrode Assembly
Within the membrane electrode assembly (MEA), the membrane and catalysts are integrated into a functional unit. The membrane must provide mechanical stability during the compression of the MEA and the transport layers as well as enable ionic transport while inhibiting gas and electron crossover. The catalyst layer can be seen as the central interfacial layer in an MEA, where all transport pathways including chemical species, ions, and electrons need to come together at the reactive centers, which is the catalyst particle surface. In this section, we first discuss how to design optimum catalyst-layer structures. This is followed by a discussion on possible pathways for integration of the catalyst layer with the membrane and the manufacturing of MEAs.
5.1. Catalyst-Layer Design
Catalyst-layer design is about achieving optimum conditions for the transport pathways of the involved species. Here electrons need to be transported through electronically conducting pathways, which are the catalyst particles and the metals of the current collectors. Usually, the ions are transported via both the liquid and the solid electrolyte, and the reactants are liquid water and gases that are transported through the pores. These catalyst particles and the transport pathways are intertwined, which has to be considered for catalyst-layer design and which goes beyond the concept of catalyst activity.399 Catalyst-layer design seeks to find simple ways of manufacturing porous structures that have high-density reaction points within the so-called triple-phase (gas/solid/liquid) boundary (Figure 37).400 This is a three-dimensional area within the catalyst layer where the reactions (eqs 2 and 3) occur.400 Here the catalyst is the solid, the OH– conductor is the “liquid”, and H2 and O2 are involved as gaseous species. To achieve a high number of these sites, the integration of the catalyst into the MEA in terms of the catalyst-layer structure is critical. The anion-exchange ionomer (AEI), an AEP, is the sole OH– conductor in the case of a water-only feed, while OH– conductivity is facilitated by an alkali electrolyte feed. The AEI needs to be integrated with the catalysts to provide high OH– conductivity without blocking the catalyst sites and to allow for catalyst-layer porosity, facilitating the escape of the H2 and O2 products. Also the AEI acts as a binder to create a mechanically stable catalyst layer from the catalyst powder. In addition, the catalyst sites need to be electronically connected to the current collector for the electrochemical reactions to occur. High electronic conductivities of the catalysts and supports are needed, but the electronic conductivities on their own do not directly yield the highest-performing MEA, thus emphasizing the importance of the manufacturing methods and the catalyst integration into the MEA.401
Figure 37.
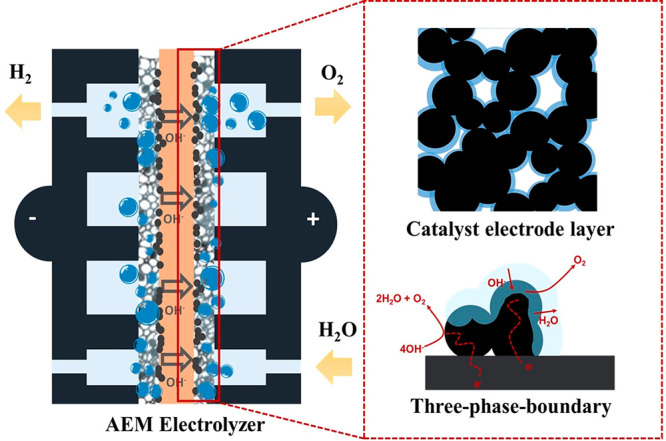
Simplified schematic of the triple-phase (gas, liquid, and solid) boundary for the OER showing the catalyst particles (black) that are in direct contact with the current collector (shown as a gray bar in the figure). The OH–-conducting AEI acting as an electrolyte and often also as a binder is shown in blue. In an actual MEA, the catalyst particles form up to several-micrometer-thick layers, and electronic conductions through the catalyst layer (from catalyst particle to adjunct catalyst particles) are needed.
It is crucial that the AEI is dispersed in a manner achieving maximal catalyst utilization and facilitating OH– transport from the cathode to the anode catalyst sites through a continuous and highly conductive pathway. The OH– conductivity of a catalyst layer can be 1 order of magnitude lower than that for the equivalent AEM and is influenced by the catalyst layer’s tortuosity. The latter can be seen as the mean deviation of traveling time from the shortest possible connection within a porous material.31 The optimal AEI loading typically ranges between 5 and 20 wt %, depends on numerous factors, and must be experimentally evaluated.402
An example of the influence of the AEI loading on various MEA properties and performance is shown in Figure 38. For these MEAs, tested in a single cell, the lowest voltage at a particular current density value is found for the 20 wt % AEI loading. The Nyquist plots (Figure 38b) further suggest that the high-frequency resistance (HFR) and the resistances for the anode and cathode charge-transfer reactions are the lowest for the 20 wt % AEI loading.107 The SEM images show differences in pore structures and the appearance of secondary pores for higher AEI loadings. The latter are suggested to lower the cell performance.
Figure 38.

(a) Polarization curves and (b) Nyquist plots for AEMWEs with different (10, 20, and 30 wt %) AEI loadings at 50 °C and (c) field-emission (FE)-SEM images of the MEAs fabricated using the different AEI loadings. KOH (1 M) at 1 mL/min was fed to the anode and cathode. Reprinted with permission from ref (107). Copyright 2019 Elsevier.
An understanding of how a specific AEI, which also acts as a binder for the catalyst particles, behaves in regards to factors such as its swelling and conductivity in feed electrolytes with different pH values will be crucial in understanding the AEI’s influence on transport in the pore phases.403
Mayerhöfer et al. recently studied the effect of 10 and 30 wt % AEI loadings in the anode catalyst layer on the AEMWE performance in water and 0.1 M KOH (as illustrated in Figure 39a and b).403 It was found that the employed AEI by itself, even at 30 wt %, was not able to supply the required OH– species and the basic environment for the PGM-free OER catalyst sites during pure water feed operations (Figure 39c). However, the performance increased by 20–45 times at a cell voltage of 1.8 V when a 0.1 M KOH feed was used.
Figure 39.
SEM images of 2 mg/cm2 CuCoOx anode catalyst layers with (a) 10 wt % and (b) 30 wt % ionomer loadings.404 (c) Polarization curves of the single AEMWE cells for pure water (dashed lines) and for 0.1 M KOH (solid lines) feed. (d) iR-corrected linear sweep voltammograms of the CuCoOx anode catalyst layers with varying ionomer contents at pH 12.7 (solid) and pH 7 (dashed) of a 0.05 M phosphate buffer solution in a scanning-flow-cell measurement. A Pt loading of 0.5 mg/cm2 was used at the cathode. (c, d) Reprinted with permission from ref (403). Copyright 2022 Elsevier.
In addition, a three-electrode scanning-flow-cell (SFC) experiment was conducted to investigate the anode catalyst layer. It was shown that a higher binder content can block the catalyst site and consequently lower the catalyst activity (Figure 39d) for the higher-pH feed. This effect was ascribed to the changes in the membrane- and contact resistances due to different swelling behaviors of the materials in the respective feed solutions. This shows that the role of catalyst-layer binders can differ significantly depending on the feed solutions.
The concept of not needing an AEI if an alkali electrolyte feed is applied has also been considered in a few studies.31,80 For this situation, the AEI may merely act as a catalyst particle binder and the liquid alkali electrolyte may be the vital OH– provider.
The AEI also influences the pH and hydrophobicity of the catalyst layer. The AEI needs to have the appropriate chemical properties, which include maintaining a pH favoring the catalyst’s chemical stability and hydrophobicity and the need to be mechanically stable, to prevent catalyst detachment. For example, the pKa of the conjugated acid of the QA cationic group or of some AEIs is lower than that for KOH, ∼10 versus 15.405 The choice of KOH (or other liquid alkali electrolytes) feeds also seems preferable for stability reasons in the case of, e.g., Ni-based catalysts. A high pH can also enhance the OER kinetics depending on the reaction order of the catalyst.318 Chemical similarity between the AEI and AEM allows for a low interfacial resistance and similar swelling, which helps to prevent delamination of the catalyst layer from the AEM.32 The functional groups and the backbone structure of the AEIs can be used to tune the hydrophobicity and chemical properties of the triple-phase boundary, which allows for pH adjustment and alters H2O availability at the catalyst sites. Chemical groups of the AEI can react and create negative side effects at both electrodes.406 In AEMFCs, aromatic AEI groups have been shown to adsorb on the Pt cathode catalysts, lowering the catalytic utilization (Figure 40).294,406−408 Experiments combined with DFT studies suggest that the adsorption of aromatic AEI fragments on Pt metal surfaces decreases as follows: p-terphenyl ≥ m-terphenyl > diphenyl ether > benzene ≥ o-terphenyl > 9,9-dimethyl fluorine, and the parallel adsorption of the adsorbed phenol ring on the Pt surface has a negative effect.409 The specific adsorption of QA cations and benzyl-group interactions with Pt can be lowered by utilizing large, rigid cations and nonrotatable phenyl groups,410 although the unsubstituted phenyl in polyaromatic backbones stays adsorbed on Pt well into positive potential regions.411 The adsorption energy depends on the catalyst. In the case of benzene, it may be lower for bimetallic surfaces such as Pt alloyed with Mo, Ni, or Ru.412,413
Figure 40.

(Top) Dependency of the performance for an AEMFC (y-axes) on the adsorption of phenyl from the ionomer for different cathode (H2 oxidation) catalysts. The lower figure shows DFT-calculated adsorption energies for different substituted benzenes on Pt as a function of the system size (C atoms per molecule). Adapted with permission from ref (409). Copyright 2019 American Chemical Society.
In addition, catalyst-site blocking side effects such as lowering the pH, possibly causing dissolution of TM catalysts, can also occur.414 This has been proposed for AEIs containing phenyl groups in the backbone structure, which can be oxidized to acidic phenolic compounds (Figure 41).
Figure 41.

Phenyl oxidation of (a) benzyltrimethylammonium hydroxide and (b) polyaromatic AEI at OER potentials. Adapted with permission from ref (415). Copyright 2019 American Chemical Society.
DFT calculations suggest that phenyl adsorbed on the electrode surface in parallel or lying positions is most susceptible to oxidation, and both positions are observed at potentials as high as 1.6 V (Figure 42). The adsorption energy depends on the surface as follows: PtO2 (110) > IrO2 (110) > PtO (110) > IrO( 110) > La0.85Sr0.15CoO3 (001) > La0.85Sr0.15CoO3 (111).
Figure 42.

(a) Polarization curves of an AEM water electrolyzer before and after the 100 h test at 2.1 V at 80 °C. (b) 1H NMR spectra of the anode AEI before and after the durability test. The inset in (b) is the expanded view of the oxidized phenol peak in the 1H NMR spectra; * denotes other expected oxidation sites. Adapted with permission from ref (415). Copyright 2019 American Chemical Society.
Studies also involve the use of PTFE as a binder in catalyst layers.85,416−419 PTFE is nonionomeric; hence, in the absence of an AEI, a liquid alkaline electrolyte needs to be fed to provide OH– conductivity. PTFE could play a role in tailoring the hydrophobicity/hydrophilicity of the catalyst layers to avoid flooding and gas blockage at critical locations. The PTFE loading needs to be sufficient to act as a binder but limited to avoid catalyst blockage and negative effects on the catalyst layer’s porosity.420 For FCs, many studies report on the optimal PTFE (and AEI) loading, including visualization experiments on water distribution within the MEA. However, an understanding of the effect of PTFE loading on the AEMWE performance is lacking, and studies of operando neutron scattering for water-distribution visualization could be useful.421 An attractive feature of PTFE is to allow electrode sintering at T values exceeding 300 °C, assisting in bonding the catalyst particles to the PTL and GDL, but care needs to be taken on how the catalyst-layer morphology is affected by high-temperature treatments.422
It can be summarized that the durability of an AEI in pure water-fed AEMWEs, especially at the anode under high operating potentials, is considered a limiting factor. Li et al.31 in a recent review distinguished between the durability-limiting factors for water-fed and concentrated KOH-fed AEMWEs, which among others included ionomer poisoning, ionomer detachment, and instability of the AEM. Alternative electrode-fabrication methods that exclude the use of an AEI in the catalyst layer are being reported more frequently, and such studies for AEMWE cells were recently reviewed by López-Fernández et al.423 The relevant AEMWE studies, which reported performance measurements for a minimum of 100 h, are listed in Table 6 and discussed further in section 7. These include reports for a unified electrode design where the catalyst layer was integrated within the GDL in a single component by means of growing the OER catayst directly on the substrate, such an Ni foam.85,424
5.2. MEA Design
To form an MEA, the catalyst can be deposited directly either on the membrane, referred to as the catalyst-coated membrane (CCM) technique, or on a substrate, referred to as the catalyst-coated substrate (CCS) technique. Typical preparation methods for coating the substrate, which for AEMWEs is typically a choice of either a GDL or PTL, include wet routes whereby the catalyst powder and ionomer are mixed with a suitable solvent to create a stable ink or slurry. The latter are applied by spraying or painting onto the GDL support. These techniques, adopted from the fuel-cell field, have been optimized for PEMWEs425 and, more recently, for AEMWEs. To reduce waste and the use of large amounts of solvents typically associated with the wet-route MEA-fabrication methods, alternative thin-film deposition methods are being investigated. Chemical vapor deposition, atomic layer deposition, ion beam sputtering deposition, or magnetron sputtering are examples of such thin-film deposition methods.
The CCS approach allows easier control to fabricate robust and stable catalyst layers by depositing the catalyst inks and slurries directly onto an appropriate substrate. Alternatively, the CCM approach holds the benefit of improved contact of the catalyst layer with the membrane interface, resulting in improved ionic conductivity, which is seen in a decrease of the interfacial contact resistance. The main concerns are that the stability of the ionomer could be compromised and superficial changes of the membrane such as swelling can be introduced during the catalyst-deposition process. Comparisons between CCM- and CCS-fabricated MEAs reported in the literature are not simple because many factors such as the membrane stability, ionomer and membrane compatibility, and deposition technique may differ significantly.
A recent review by Miller et al.30 illustrated this exactly, as they showed the average current density recorded at 1.8 V235,426 was similar, namely, ∼200 mA/cm2, for CCS- and CCM-fabricated MEAs. The single-cell AEMWE performance was found to largely depend on the operating temperature, catalyst type (PGM versus non-PGM), and electrolyte for the different fabricated MEAs. Other studies have reported optimal performances with a CCM-cathode and CCS-anode electrode configuration, as the cell stability for the CCM approach was poor due to delamination of anode catalyst particles.427
Another strategy for reducing the interfacial contact resistance between the membrane and the CCS-formed anode is the inclusion of a microporous layer (MPL) between the PTL and MEA, as illustrated in Figure 43. Improved electrical connection and liquid/gas transport were achieved for a NiMPL-PTL while operating with a water feed.428
Figure 43.

Schematics of different AEMWE cells including (A) only PTL and (B) the addition of a NiMPL-PTL on the anode and the cathode. (C) AEMWE-cell performances measured at 60 °C for water feed and for configured PTL/PTL (commercial Ni mesh) and NiMPL-PTL/NiMPL-PTL. (D) Electrochemical impedance spectroscopy measurements for the two cell configurations at 0.5 A/cm2 (from 50 kHz to 100 mHz). Reprinted with permission from ref (428). Copyright 2021 Elsevier.
Determining the preparation parameters influencing the MEA and catalyst layers is somewhat of a trial approach due to the many variables involved. Some of the knowledge acquired for PEMWEs and FCs can be extrapolated to AEMWEs. In addition, molecular dynamic modeling of the MEA components in conjunction with experimental verification could advance the field more rapidly. In general, hot-pressing the MEA is favorable to increase the contact between the catalyst layer and the AEM, although the AEM may dry out. The Tg values of the AEM and AEI play an important role in determining the hot-pressing temperature. Control is needed to avoid AEM compression specifically for AEMWEs fed with liquids as compared to the gaseous feed of a FC, which benefits from pressures anywhere between 2 and 200 kg/cm2 at 120–195 °C for 50–300 s.429
The MEA components need to be optimized in tandem to address factors such as water management to avoid both drying out and flooding. In the case of AEMFCs, it is now believed that maximum performance cannot be achieved due to water flooding.421 Many of these issues have been addressed for PEM-based FCs and WEs, but in the case of alkaline conditions, the imbalance of water produced and consumed at the anode and cathode is larger than that for acidic conditions. The source of OH– needed for the OER at the anode is in abundant supply when operated with a liquid alkali electrolyte as 1 M KOH, while the OER for the water-only feed depends on OH– being supplied through the water splitting reaction taking place at the cathode. In the electrochemical reactions of an AEMWE, 1 mol of H2O is produced at the anode and 2 mol are consumed at the cathode, while for a PEMWE, 1 mol is consumed at the anode and 0 mol are consumed at the cathode. Even though H2O is produced at the anode and consumed at the cathode, the water feed at the anode seems to become the preferred feeding mode for AEMWEs. This mode reduces the need for H2O and H2 separation, thus delivering a higher-purity H2 from the cell.430 However, the best cell performance and highest operating current densities have been reached by feeding electrolyte to both the cathode and anode, which also reduces the risk of anode dehydration and increases water transport to the cathode.83,107 Future strategies to tailor defined MEA and transport-layer structures directing the liquid and gas feed to specifically defined areas are important for the design of novel and effective electrode architectures. This could include tailoring the hierarchical porosities of the catalyst layers along the in-plane (electrode-to-electrode) direction and utilizing modifiers that repel H2O or maybe even capture H2O. The optimization of flow rates and KOH concentrations are both relevant to the design of an actual AEMWE and have so far received limited attention in the literature.430
5.3. Current Collectors, Bipolar Plates, and Flow-Field Design
The current collector, which can be referred to as either PTL (such as porous metal framework) or GDL (such as woven carbon fibers) in an electrolyzer, serves to convey the electric current between bipolar plates and the respective anode and cathode CLs while mechanically supporting the membrane. It provides the pathway for electrolyte and reaction products between respective compartments and CLs. The support is either a fiber, foam, or woven metal network, as illustrated in Figure 44, and is designed with a large specific surface area for increased contact between the CL and membrane. Ideally it should have relatively small pore sizes (1–100 μm), a high porosity (>60%), and a thickness between 0.3 and 1 mm.35
Figure 44.

SEM and optical microscopy images for different metal substrates, as follows: (a) 0.065 mm Ni wire mesh, (b) nonwoven stainless steel, (c) nonwoven Ni, (d) nonwoven C–Ni conductive composite, (e) GDE, and (f) stainless steel web. Reprinted with permission from ref (35). Copyright 2020 Royal Society of Chemistry.
At the interface between the electrodes, current collectors, and bipolar plates, contact resistances in the absence of passivation layers could lead to significant contributions to the cell resistance.78 Therefore, material selection and uniform contact between the former components are of high importance to ensure the long durability needed for WEs.
The thermodynamic stability of Ni foam280,431 and likewise of stainless steel (SS)432,433 felts, in combination with their ability to passivize at anodic potential in alkaline media, favor their use in AEMWE as anode substrates.30 However, common SS 316 is likely to leach Fe into the KOH electrolyte, specifically at elevated temperatures of 80 °C and over time.98 Therefore, SS 316 is likely not suitable as a long-term cell material, specifically at the anode. The carbon GDLs commonly used in FCs are restricted to use at the cathode in AEMWEs due to carbon corrosion in the presence of OH– ions, which tend to operate as nucleophilic intermediates and can accelerate degradation in the highly oxidative environment of AEMWE anodes.434
The bipolar plate’s role includes contacting cells and thus ensuring optimal reactant and product flow along a stack by means of manifolds incorporated in them. The flow-field design is closely connected to this aspect and affects the distribution of water as the reactant and the removal of produced gas and also needs to establish a firm electrical contact with the GDL and PTL. Different geometries exist, such as single and multiple serpentine, parallel column, and cascade pattern (as depicted in Figure 45), of which there is currently no optimal design.435 The optimal design is dependent on effective sealing of the cells for different pressures and operation at different cell sizes. Another consideration is the optimal supply of liquid water to the anode side of the cell and how this distribution effect can also serve as a temperature control of the cell or stack during operation. Most common for PEMWEs remains the use of the parallel channel design as it is proven to show a lower overpotential,436 although flow-distribution limitations at higher operating current densities are increasingly being investigated for WEs.437,438
Figure 45.

Flow-field designs commonly applied in PEMWEs: (A) single serpentine, (B) multiple serpentine, and (C) parallel column. Reprinted with permission from ref (435). Copyright 2019 Royal Society of Chemistry.
6. Operational Modes and Performance
There are three operational modes in a WE. For today’s AEMWEs, the different modes are achieved at the following approximate conditions: (i) kinetic control for j < 0.3 A/cm2, (ii) cell electric resistance for 0.3 A/cm2 < j < 1.5 A/cm2, and (iii) mass-transport effect for j > 1.5 A/cm2. An AEMWE will operate at high j values, i.e., in the mass-transport zone.
A benefit of running at higher j values is the increase in the OH– transport of the AEM due to the higher ratio of OH– versus CO32– ions.439 However, the operating j values should always be below the critical current density (jcrt), identified as the j value for which mass-transport losses and gas saturation limit the cell performance.440 Exceeding jcrt will lead to nonuniform gas distribution and nonuniform j values at the catalyst sites, resulting in the formation of undesired hot spots. This can result in the drying and, hence, degradation of the AEM. To limit gas-bubble formation at the surface of the electrode, limiting j is a good strategy. A few more suitable strategies can be considered such as releasing gas bubbles, including passive and active approaches like optimizing the catalyst layer, GDL, and PTL geometries and pore sizes;416,441,442 applying coatings;417 and the addition of surfactants.418 The electrolyte and reactant flow rates always need to allow fast transport, reducing the risk of creating mass-transport limitations and other failures.72,99,100 At higher j values, a higher flow rate is needed to match the consumption rate of the reactants.440
Electrolytes used in AEMWEs can be categorized into 3 groups: hydroxide solutions, carbonate solutions, and pure water plus the solid electrolyte. An advantage of the water-only feed is the absence of OH–, thus allowing the device to run at higher temperatures without a loss of the mechanical stability of the AEM.414 The ionic conductivity of the AEM often exceeds 0.08 S/cm, which in theory is sufficient to enable OH– transport from the cathode to the anode.35 However, using pure water in combination with the AEI as electrolyte presents a few hurdles. This includes the need to develop stable AEIs of high ionic conductivities for neutral and mildly alkaline pH. The overall cell resistance is also higher compared to the combination of thin AEMs and liquid alkali electrolytes.
Diluted KOH solutions, typically between 3 and 10 wt %, are preferred to ease the nucleophilic OH– attack on the AEM and AEI, but milder alkaline electrolytes are less effective in assisting in the OH– transport within the catalyst layer. The KOH conductivity drops ∼1 order of magnitude from 0.178 to 0.02 S/cm when changing from 5 to 0.5 wt % KOH.101 For low electrolyte concentrations, there is no buffer effect; hence, rapid and undesired pH changes in the AEM and the catalyst layers can take place, and small CO2 concentrations dissolved in the electrolyte increase the Ecell value. Effects of the H2O and different alkali electrolyte concentrations on the Ecell–j performance and also the high-frequency resistance (HFR) of single MEA cells are known.318Figure 46 shows the benefit of a higher KOH concentration in lowering the Ecell value. The lower Ecell values are partially due to lowering the resistance reflected in the HFR in Figure 46b but also due to additional benefits such as an increase in the effective ECSA of the catalysts using liquid alkali electrolyte feeds. Depending on the reaction order of the catalyst with respect to the pH, the catalyst kinetics can be enhanced and the catalyst stability can be altered with pH.419,443−445 In fact, the development of catalysts of zero reaction order may allow operation with H2O-only feed.318
Figure 46.

(a) Voltage (Ecell) and (b) high-frequency resistance (HFR) versus j curves. The catalysts are at the cathode, PtRu/C 0.36 mgPt/cm2, and at the anode, IrO2 0.75 mgIr/cm2. Dilute KOH or deionized (DI) water serves as the liquid electrolyte. Hexamethyltrimethylammonium-functionalized Diels–Alder polyphenylene (HTMA-DAPP) is used as the AEM and AIE. The AEM wet thickness is 50 μm. All the measurements were conducted at 60 °C and ambient pressure. Reprinted with permission from ref (446). Copyright 2021 IOP Publishing.
Figure 47 shows the breakdown for the individual overpotential components depending on the electrolyte concentration. Figure 47d shows the enhancement of the anode and cathode kinetics and the decrease in the HFR for higher KOH concentrations.
Figure 47.

Applied voltage breakdown for (a) water, (b) 0.01 M KOH, and (c) 1 M KOH. The dashed line shows the location corresponding to the largest j in (a). (d) Bar graph of the applied-voltage breakdowns at 0.56 A/cm2 (indicated by the dashed lines). The cell overpotential is broken down into the following: anode kinetic losses (blue), cathode kinetic losses (green), high-frequency resistance (HFR) loss (red), catalyst-layer (CL) ohmic loss (yellow), and ion-exchange loss (gray). The anode kinetic losses are further broken down into three parts: anode kinetic losses due to gas-bubble coverage (light blue), anode kinetic losses due to low pH (medium blue), and intrinsic kinetics loss (dark blue). Reprinted with permission from ref (446). Copyright 2021 IOP Publishing.
There is a threshold for which an increase in the catalyst loading leads to a decrease in conductivity due to the resistance to mass transport created by the catalyst-layer thickness.318 Mass-transport limitations are prominent at higher loadings and when operating at high j values. The short-term Ecell versus j curves in Figures 46 and 47 were obtained for relatively low catalyst loadings, specifically for the anode catalyst layer. Anode catalyst loadings of 2 mg/cm2 are more typical, and depending on the catalyst, loadings as high as 10 mg/cm2 have been employed.
The (Bi)carbonate solutions pH values range between neutral and pH 12, and they have been used as an alternative to NaOH or KOH feeds. The conductivity of a (bi)carbonate solution compared with KOH is lower; thus, more-concentrated electrolytes are often employed. For example, a comparison is made of the Ecell–j and Rcell–j of two identical cells, both fed with electrolytes of pH ∼12, but with one being 0.01 M KOH and the other being 0.72 M K2CO3; resistances of 0.3–0.4 Ω/cm2 and 0.1–0.2 Ω/cm2 were measured.447 Dilute (bi)carbonate electrolytes may reduce AEM and AEI stability issues, but the site blockage through carbonate deposition remains a problem and the long-term impact on catalyst utilization (and membrane blockage) needs to be determined.30 This is supported by a mathematical model, which suggests that the increased voltages result from the Nernstian voltage difference across the AEM (and also AEI) when OH– is replaced with CO3– rather than from a less-conductive K2CO3 electrolyte.448
The inclusion of a solid-state membrane in the AEMWE also has the benefit of allowing pressurization of the cathode and compression of the H2, up to 30 bar.449 This facilitates hydrogen storage as compared to the traditional alkaline electrolysis.59 Additionally, the separation of electrodes by means of the AEM ensures that H2 crossover from the cathode to the anode is limited and the risk of forming explosive gas mixtures (>4% of H2 in O2) is reduced.450,451 Factors such as operating current, membrane thickness, pressure, and temperature influence the gas crossover, which, if increased, affects the efficiency of the cell negatively and leads to H2 loss. It is known that for higher current densities and thicker membranes the gas crossover decreases.78 However, the H2 crossover increases linearly with an increase in the H2 partial pressure and could become an issue for pressured systems in the operating range of 30–60 bar.35 Studies reporting H2 crossover measurements include one by Ito et al.452 carried out at 8.5 bar using a Tokyama A201 as the AEM and Pt/C and CuCoOx cathode and anode catalysts, respectively. The Ito et al. study showed that the H2 crossover was 0.16 times that of a PEMWE system. Motealleh et al.453 performed a long-term study at atmospheric pressure and were able to decrease the H2 crossover by 56% by reinforcing a Sustainion membrane with 2% zirconia.
Limited by long-term stability, typically low-temperature PEMWEs and AEMWEs operate below 80 °C. A recent review by Lohmann-Richters et al.454 considered the challenges to increase the operating temperature for AEMWEs in order to increase the current density and the possibilities to capitalize on this unique advantage by means of thermal management. Thus far, it has been demonstrated that a performance with current densities of 3.75 A/cm2 at 1.75 V and 200 °C is obtainable by the implementation of mixed Ni, Fe, and/or Co and Raney-Ni–Mo as anode and cathode catalysts, respectively, albeit with porous zirconia as a diaphragm. It is concluded that further improvements with regards to components costs and possibly stability would be required to advance the latter technology.
7. Single-Cell AEMWEs Exceeding 100 h of Operation
Optimization of single AEMWE cells has resulted in significant stability and activity improvements in less than a decade. A comparison of the most-significant AEMWE single-cell performance and durability limiting factors for different operation modes (varying liquid electrolytes) using both PGM and PGM-free catalysts was provided by Li et al.31 To date, high current densities in the several A/cm2 range have been reported, e.g., 5.3 A/cm2 at 1.8 V in 1 M NaOH and 2.7 A/cm2 at 1.8 V in pure-water feed.414 These current densities and Ecell values are comparable to the single-cell results of PEMWEs. However, the biggest challenge remaining is achieving good durability at sufficiently high current density (most work currently aims for 1 A/cm2) to achieve acceptable Ecell values below 1.9 V for run times >100 h. Until recently, only a few studies have reported AEMWE single-cell measurements exceeding 100 h. These studies are listed in Table 6 and grouped in the order of the electrolyte, first for water and then for liquid alkali electrolytes, followed by the voltage-degradation rate (μV/h) in increasing order. Additional information is given in Table S20.
Due to the many different conditions such as catalyst, catalyst loading, AEI, AEM, and electrolyte, a direct comparison is not straightforward, but a number of observations can be made. On average, AEMWE cells operated on a water feed show a higher voltage degradation than liquid alkali electrolytes when operated at higher constant current (0.2 A/cm2 compared to minimum 0.5 and 1 A/cm2 for carbonate- and hydroxide-based electrolytes, respectively).424 From the eight durability studies found that operated on pure water, only two reported the use of PGM-free anodes (studies 2 and 5, Table 6), while all of the others include the use of baseline state-of-the-art (SoA) PEMWE catalysts, IrO2 and Pt/C. Both observations may be related to the fact that the pH in an AEMWE run on pure water is close to neutral or perhaps even drops below 7 in the anode catalyst layer. The pH drop is due to the consumption of OH– during the OER, possibly facilitating degradation of the anode catalysts such as NiFe.455,456 Furthermore, high IECs of the AEIs likely provide a higher-pH environment, benefiting the OER kinetics. However, high IECs also lead to a higher water uptake, which can result in the detachment of the catalysts and, hence, higher degradation rates (e.g., studies 4, 5, 7, and 8).
The goal of studies 1 and 6 was to understand the cell durability and performance by studying different cation-functionalized polyaromatic AEMs and AEIs.415,457 The AEM [made using poly(phenylene) as the backbone of the AEP with benzylic methylammonium groups (ATM-PP)] of study 1 shows only a gradual cell-performance loss. This suggests that the backbone degradation of AEM and AEI is delayed by slowing the cation degradation when using benzyltrimethylammonium (BTMA)-functionalized polyaromatics (study 1457). However, this AEM needs to be operated in the absence of caustic solutions and below 60 °C to limit backbone degradation.457 The tendency of phenyl groups in the AEP backbone to be oxidized under OER potentials has been found to detrimentally influence the performance by forming acidic phenols at the anode, as determined in study 5 for the quaternized biphenylene ionomer (BPN).414 The phenol-formation rate of unsubstituted phenyl groups was found to be much higher in comparison to ammonium-substituted phenyl groups.457 More recently, Soni et al. (study 7) demonstrated that an acceptable durability with high IEC (2.9 mequiv/g) can be achieved by introducing long alkyl side chains (Cx, x = 8) to the AEP backbone and effectively reducing the phenyl fraction that is susceptible to electrochemical oxidation.411,458 In addition, the poly(fluorene-alt-tetrafluorophenylene) (PFOTFPh) polymer backbone contains nonrotatable fluorene moieties, which are less likely to be absorbed on the Pt catalyst surface and therefore further serve to suppress phenyl absorption of the AEP backbone.410 The same PFOTFPh-TMA-C8 (only thicker membrane, 53 μm) prepared MEA in 1 M KOH (study 13) approached near 100% in faradaic efficiency due to a lower measured H2 permeability (8 times lower as compared with Nafion 211) and only 120 mV higher for current densities measured up to 2 A/cm2.
In study 5, 0.2 A/cm2 at 1.75 V in H2O was achieved.414 However, the large voltage degradation indicates limitations. It was found that, even in the presence of a high IEC ionomer (TMA-70, IEC = 3.3 mequiv/g), the non-PGM anode catalyst particles were washed out. Exchanging the AEI with a lower IEC TMA-53 (IEC = 2.6 mequiv/g) showed an increased binding strength for long-term operation. The combination of the same IEC (2.6 mequiv/g) AEIs and AEMs (HTMA-DAPP AEM and TMA-53 ionomer) and, hence, similar swelling proved to be more stable in cell tests.414
Xiao et al. (study 2) demonstrated the advantages obtainable with a non-PGM, self-supported, fluoride-incorporated nickel–iron oxy/hydroxide (FexNiyOOH-nF) catalyst directly grown onto a compressed Ni foam support.424 Increased catalyst utilization and an improved contact between the catalyst, PTL, and membrane ensured better long-term durability.
Razmjooei et al.428 (study 3) demonstrated that, by introducing a nickel-based microporous layer (MPL), an improved contact at the interface between MEA and PTL resulted in a reduced ohmic resistance and ultimately improved cell performance with Sustainion (IEC of ±1.1 mmol/g). Furthermore, an MPL designed with the appropriate pore size and distribution could serve to decrease mass-transport losses and enable operating AEMWEs at higher current densities (comparable to a PEMWE).459,460
Vincent et al.461 (study 9) demonstrated cell performances of 2.09 V at 1 A/cm2 (1.88 V at j = 0.6 A/cm2) in 1 M KOH at 60 °C. The maintained Ecell (first 100 h) compares well with that of study 12, which also used a Ni-based catalyst in the anode and cathode and 1 M KOH at 60 °C but with Sustainion as the AEI in the catalyst layers and an in-house prepared PBI AEM. Detailed information about the preparation or thickness of the AEM itself is not given, but the work illustrates the successful preparation of a low-cost Ni-based catalyst with apparent high stability and performance.
The most impressive performances regarding low voltage degradation have been achieved by PGM-catalyst-free cells implementing the use of a Sustainion membrane (studies 10a, 10b, and 13).453,462 According to data extracted from a peer-reviewed technical report published by Dioxide Materials, these membranes show by far the lowest degradation, indicated in the voltage increase of only 5 μV/h (study 13), although a 1 M KOH electrolyte was circulated to achieve an Ecell of 1.9 V at a j of 1 A/cm2.462 The Sustainion membrane is based on a polystyrene-based membrane with a quaternized imidazolium headgroup. This membrane is argued to achieve its high OH– conductivity through the K+ of the electrolyte in combination with its high water uptake; hence, it is suggested that it is not an actual AEM.32 The small degradation rate of the Sustainion membrane was confirmed by others (study 10).453 In fact, the latter group reported the longest-stability single-cell AEMWE performances to date of over 10 000 and 12 000 h for Sustainion XC37-50 and grade-T membranes (a PTFE- reinforced membrane), respectively, in 1 M KOH and at 1 A/cm2.453
The MEAs, using commercial Tokuyama and Sustainion membranes, employed either Nafion or PTFE in the catalyst layer, i.e., no actual AEIs, indicating that the OH– conductivity in the catalyst layers is provided by the alkali electrolyte feed. Chen et al.463 achieved a record density for AEMWEs of 7.68 A/cm2 at 2 V with 1 M KOH at 60 °C by using PGM catalysts (study 15). By using the same high IEC AEM (>2.8 mmol/g), namely, poly(fluorenyl-co-terphenyl piperidinium-13) (PFTP-13), and Ni–Fe composite catalysts (study 11), a current density of 1.62 A/cm2 at 2 V was measured in 1 M KOH. More importantly, the AEMWE study was based on running a dry cathode for applications where high-purity hydrogen is of importance. The authors found that an AEM with high IEC (∼2.8 mmol/g) and diffusivity (9–11 × 10 –8 cm2/s) was required for ensuring the high cell performance with only an anode feed. Furthermore, including a high-IEC ionomer (3.43 mmol/g) in the cathode electrode (25 wt % loading) had a subsequent high water uptake. This serves to secure water molecules diffusing through the AEM from the anode to the cathode electrode for electrochemical reactions. The durability of PGM-free electrodes, Ni–Fe/Ni foam, and PFTP AEM (study 15) demonstrated superb stability at an applied current 0.5 A/cm2 in 1 M KOH and can be regarded as one of the best-performing and durable AEMWE single-cell results so far along with commercial Sustainion (studies 10 and 13).
Another ultrahigh current density of 4 A/cm2 for 12 h measuring a stable cell voltage of ∼2.05 V was reported by Park et al.107 At 0.5 A/cm2 (study 12) the measured cell voltage showed not even a slight decrease over the 100 h measurement. The authors demonstrated an improved (2-fold) and durable performance of their in-house prepared three-dimensional unified electrode whereby the catalyst NiFeOOH was directly formed by electrodeposition onto the current collector as compared to conventional electrodes (0.5 mg/cm2 IrO2 anode). The atomic Fe/Ni ratio was optimized (3:3) to obtain an optimum balance between catalytic activity and electrical conductivity, which resulted in an improved cell performance from the reduced ohmic resistance. Beyond the enhanced catalyst utilization, the larger pore-size distribution of the unified electrode design also serves to improve the mass transport of the reactant and product.
Another longer-term study (study 16) employing electrodes without the addition of an AEI was presented by Wang et al.464 They managed to obtain a performance comparable to that of a PEM electrolyzer at 60 °C (1 A/cm2 at 1.8 V) with their atmospheric-plasma-sprayed (APS) NiAlMo layer anode, a 1 M KOH feed, and a hexamethyl-p-terphenyl poly(benzimidazolium) (HMT-PMBI) AEM. The performance was stable for 145 h.
Study 18 reports the use of a heterogeneous AEM, which consists of anion-exchange particles (Dowex Marathon particles, 10–30 μm, IEC of 3.9 mequiv/g) blended with a low-LDPE matrix and water-soluble additive for the purpose of increasing the conductivity of the membrane.466 The conductivity of the heterogeneous membrane was improved by 75% for an optimized water-soluble additive content of 3.4 wt %. However, this performance is limited to the use of a liquid alkali electrolyte to ensure efficient OH– transport through the membrane to provide contact between the anion-selective particles. A trimethylamine quaternized PPO was employed as the AEI in study 20.468 An identical MEA was tested in 1 M KOH at 70 °C, and the impact of membrane degradation (an IEC decrease of 5.7% after 100 h of operation) was observed, resulting in a more pronounced cell-voltage degradation of 400 μV/h. It was concluded that such a heterogeneous membrane’s performance highly depends on the liquid electrolyte’s conductivity, and to ensure an enhanced lifetime (>100 h) of the membrane in 1 M KOH electrolyte, a cell temperature of 50 °C could be considered limiting.
In a follow-up study by Hnát et al.467 (study 19), the stronger base DABCO was investigated as the quaternization agent for the chosen backbone polymer, a poly(styrene–ethylene–butylene–styrene). Increases in the operation temperature and electrolyte concentration ensured higher electrolysis efficiency but increased the degradations of the AEM and AEI. Therefore, the strategy to decrease the weak spots in the AEP structure was to lower the IEC and use 10 wt % KOH as the electrolyte. The heterogeneous membrane proved promising, albeit with a higher Ecell of 2.25 V, which was a consequence of the bare Ni foam used as a current collector and catalyst for the long-term measurements. It was concluded by the research group of Žitka et al.469 that the polymer matrix PPO quaternized with either DABCO or TMA eventually (after 400 h) suffered from degradation of the backbone hydrolysis mechanism and the performance deteriorated at operation temperatures of 60 °C in 10 wt % KOH. They found more promise in combining their TMA-quaternized PSEBS in both membrane and AEI (study 21), which operated for 800 h and showed a low voltage degradation.
Study 22 used a catalyst-coated-substrate preparation method using PTFE instead of an actual OH–-conducting AEI at the anode.84 The cell was stable for 1000 h at 0.47 A/cm2 on a 1 wt % K2CO3 feed.
Chi et al.470 (study 23) focused on evaluating the durability of nickel/cobalt oxide as non-PGM and compared the cell performance with commercial Acta 3030 as the OER catalysts. Their prepared Ni0.7Co0.3Ox showed good stability and outperformed (0.2 versus 0.1 A/cm2 at 2 V) the cell with a commercial anode catalyst. Detailed information about the membrane besides that it was prepared in-house were not presented.
Study 24 by Vincent et al.235 also reported the use of commercial Acta 3030 and 3040 as OER and HER catalysts, respectively. Very high catalyst loadings (∼30 mg/cm2) were used, and it is the first study to report the use of Acta supplied ionomer (I2) for longer-term studies, compared to Tokoyama (24a) and (24b) FAA-3-PP-75 membranes in 1 wt % K2CO3 at 60 °C.
8. Establishing Protocols for Single-Cell AEMWE Evaluation
Electrochemical characterization of suitable AEMWE materials in full cells typically entails performance and durability measurements. Due to varying operating conditions, care needs to be taken when comparing the AEMWE cell performances. The comparison of AEMWE test results remains difficult due to the absence of uniform testing protocols.101 Typical reports of high-performing AEMWE cells include short-term tests recording slow-sweep polarization (j–Ecell) curves. The recording of j–Ecell curves is most often preceded by a break-in procedure that serves to activate the catalyst layer and conditions the AEM. This procedure may vary depending on the AEM and catalyst used. Such a break-in typically entails applying a constant Ecell in the range of 1.6 and 2.2 V,35,98,471 which is incrementally (in 0.2 V steps) increased for varying holding times to reach pseudostable current densities at the applied Ecell. In other cases, slow-sweep j–Ecell curves are repeatedly recorded until pseudostable curves are obtained.
Short-term tests can show promising results, but the durability of the cell and the components require a longer evaluation (>1000 h) and even intermittent tests to allow for a dynamic durability assessment. So far, the long-term tests vary between operating at a constant j of 0.5 or 1 A/cm2 and a few intermittent-mode studies entailing cycling between a high and low (e.g., between 1 and 1.8 V) Ecell limit.84,472 Frensch et al.473 discussed the limits of past AEMWE durability studies typically conducted under continuous feed at constant j or by cycling within narrow Ecell ranges. They suggested a dynamic evaluation of the stability for ∼1000 h. A voltage bias of 1.95 V was applied by interrupting the steady operation at varying time intervals (such as every 2–10 h and every 100 h). The effect on the cell performance was evaluated from electrochemical impedance spectroscopy (EIS) data recorded before and after each rest period. The frequency of the rest times was found to significantly affect the cell stability, and recoverable and irreversible losses associated with either nonpermanent gas-bubble formation or AEM degradation (likely due to drying during more frequent rest times) were distinguished.
There remains the need to properly define protocols for full-cell stability tests while keeping the end application in mind (e.g., intermittent operation when coupled with renewable energy storage). The inclusion of characterization techniques such as high-frequency resistance (HFR) or full EIS in support of j–Ecell curves allows for a breakdown of the associated (kinetic, ohmic, and mass-transport region) losses and identifies the limitations of the AEM, interfaces, catalyst layers, and other MEA characteristics. Slow-sweep voltammetry yields information on the catalytic activity specifically when studied in half-cell MEAs. Floating electrode and gas diffusion electrode (GDE) half-cell setups have been developed. The floating electrode best suits electrochemical reactions with gas as the reactant but does not allow one to change parameters such as temperature, pressure, and feed flow rate.474 The GDE half-cell setup seems more suitable for comparing catalyst activity in an AEM or BPM system, and parameters such as feed flow rate, T, and P, as well as different components such as PTL, GDL, AEI, and catalyst loading, can be tuned to mimic the real system.475−477 On the basis of this information, a testing protocol for the evaluation of single-cell AEMWEs is proposed in the Supporting Information.
9. Developments on AEMWE Stack Designs
Operational modes of single AEMWE cells will need to be tuned to optimize the performance of AEMWE stacks. An initial AEMWE stack study by Bouzek and co-workers430 was aimed at verifying suitable MEA design parameters in order to ensure that high-purity product gases were produced with their developed gas-separator system. A 3-cell stack consisting of 5 × 5 cm2 nickel foam electrodes (no catalyst was added) and the commercial heterogeneous anion-selective membrane Ralex reinforced with PP mesh (Mega, Czech Republic) was studied. To decrease the oxygen contamination due to H2 crossover, the electrolyte feed arrangement was adjusted to feed electrolyte to the anode compartment only. This allowed for water molecules to diffuse from the anode to the cathode due to the membrane’s hydrophilic nature while facilitating the pressurization of the H2 produced at the cathode without the need for separation from the liquid phase. In addition, a sufficiently high pH at the anode was maintained withstanding dilution from produced water, thus preventing the dissolution of the Ni electrode, which can take place upon positive electrode polarization at pH < 9.478 The authors developed a mathematical model allowing for stack-performance validation, thus aiding in the scale-up of future zero-gap stack developments and the understanding of the impact of limiting factors.
Apart from the AEMWE single-cell studies, studies of commercial-size stack operation of active areas exceeding 60 cm2 in the water electrolysis literature are few.479 The first study thus far to apply their own developed electrocatalysts to an AEMWE stack system was recently reported by Park et al.480 They demonstrated the activity and durability of their own developed NiCoO and NiCo alloys as the HER and CuCoO as the OER electrocatalysts at a commercial scale. The catalysts were incorporated in a 5-cell AEMWE stack system (active area of 64 cm2). Interestingly, their 5-cell stack performed better than a similar material’s single-cell measurement, achieving 740 mA/cm2 at 1.65 V per cell as compared to the measured 540 mA/cm2 at 1.85 V cell voltage for the single AEMWE cell. It is important to understand the influence of the electrolyte behavior as the stacks increase in number and size and the impact of design that allows for optimum laminar and turbulent flow of electrolyte, which directly affects the cell performance.481 Attention on fluid mechanical analysis to better understand and further improve performance remains an important topic as the AEMWE technology develops. Furthermore, cell degradation for the 5-cell stack was measured at a voltage degradation per stack of 2 mV/h with an initial cell efficiency calculated at 69% and a H2 purity of 99.995% measured by gas chromatography.
10. Summary and Outlook
Much progress in AEMWE single-cell performance has been made, and PGM-free anodes are being incorporated. AEMWE cells frequently approach j values exceeding 1 A/cm2 that are needed for large-scale applications, and one long-term study has shown a high durability in combination with a low cell voltage (<2 V) for 10 000 h. Concrete conclusions regarding the most promising compositions of an MEA cannot be made at this point due to the many different variables and inconsistent evaluation protocols used. However, higher performances for cells run on dilute alkali electrolytes rather than H2O and equipped with PGM-free anodes are observed. The dilute alkaline-fed cells (1 M KOH) in combination with PGM-free anodes have shown improvements in the stability (voltage-degradation rates of ≤5 μV/h were observed) for minimal measurement times of 1000 h.453−463 The stability of MEA components, specifically the catalysts and AEMs, are still limiting the widespread implementation of AEMWEs. To increase the stability, it is key to understand the degradation mechanism employing a diverse range of characterization techniques. The development of in situ characterization techniques to identify the mechanism leading to polymer degradation, including in situ studies at higher temperatures, would be helpful. This includes the need to understand the interactions between the catalysts with the AEM and AEIs. The in situ identification of chemical and physical changes specifically for the sluggish anode catalysts when operating at high j values and Ecell values would also be of value.415
Changes in the Rcell and, hence, also the Ecell value over time need to be understood. Many factors can increase Rcell including blockage by H2 and O2, carbonate deposition, and corrosion of the current collectors. The overall Rcell value can be measured in situ using HFR,83 but a resistance increase can originate from many sources, making it difficult, but necessary, to decouple the various sources. EIS could provide insight into the ohmic resistance of the AEM as well as monitoring in situ changes taking place in, e.g., catalyst-layer characteristics such as the ionic conductivity and catalytic activities.
The alkaline stability (specifically at >60 °C) is still a concern for AEMs. Systematic investigation into different microphase-separated structures at the molecular level, prediction of species transport, and water solvation within microphase-separated materials by multiscale molecular simulation will assist in moving the field of AEM design forward.284 The peralkylammoniums will likely continue to be among the most studied and stable classes of cations, while all-carbon backbones, aryl ether-free, and PBI structure will likely continue as a research focus. The materials’ processability and cost will need to be considered. More extensive and standardized device testing is needed.36
The most active and stable catalysts for the HER include various types of Pt–Ni and Pt/C nanosized systems. Well-dispersed nanosized PtRu and Ru catalysts also show promise, although a full characterization of the latter, including the estimation of the Ru content and stability studies including intermittent conditions, are needed because Ru and RuO2 are known to show poor stability in alkaline media at more-positive potentials. It appears that much of the progress for the latter has been made by utilizing high-surface-area carbon supports featuring N-groups and layered structures to allow the dispersion (on the <3 nm scale) and the embedment of the ruthenium catalysts. Such approaches, i.e., the use of modified carbon supports other than the typically used Vulcan XC-72, may be of benefit to other catalysts to achieve a smaller particle size (in the case of, e.g., Mo-based catalysts) and to possibly avoid agglomeration of the nanosized catalyst particles during electrolysis. PGM-free catalyst options such as the combination of Ni and Mo have shown some promise. The addition of Mo to Ni has been shown to increase the HER activity over Ni-only catalysts. However, the intrinsic activities for these Ni–Mo catalysts are still below the activity of Pt/C catalysts, and the stability of Mo needs to be increased. It appears that the particle sizes of many of the PGM-free HER catalysts are larger than those for the state-of-the-art Pt/C catalysts, which in combination with lower intrinsic activities reduces the mass activity of the resulting catalysts.
The most promising OER catalysts in terms of high mass activity and a low OER onset potential include Ni–Fe-based catalysts. These catalysts typically show nanoscale features and are present as supported particles, core–shell type structures, or high-surface-area layered structures of hydrous oxides such as NiFeOxHy and NiCoFeOxHy. Many Ni–Fe–Co catalysts have slightly higher activities than Ni–Fe catalysts, which may be related to an increase in conductivity introduced by Co. Ni–Fe–Mo-based catalysts have also been reported; however, the stability of Mo reflecting real operating conditions needs to be addressed. CoCu catalysts have attracted interest for early AEMWE single-cell studies, and the addition of Cu into the Co-oxide spinel lattice was shown to improve the intrinsic OER activity. However, mass OER activities and the performance in single AEMWE cells are not as high as for Ir-oxides and Ni–Fe-based catalysts. This could be at least partially due to a larger particle size and size distribution of the CoCu catalysts. Ir-oxides are the state-of-the-art OER catalyst in acidic electrolytes, but the stability of Ir-oxides is poorer in alkaline versus acidic media. Many different forms of Ir-oxide exist, and significant differences in the OER activity and stability are observed. The intrinsic OER activity of Ir-based catalysts follows the order Ir-metal > amorphous IrOx > rutile IrO2, while the stability order is reversed, i.e., Ir-metal < amorphous IrOx < rutile IrO2. More care needs to be taken when selecting baseline catalysts, such as, e.g., an Ir-oxide for the evaluation of newly developed catalysts. This includes the need for careful physical and electrochemical characterization of the baseline catalyst before and after electrocatalytic-activity measurements. Ni–Fe- and Ni–Co–Fe-based catalysts have shown higher activities than Ir-oxides in both thin-layer cells and single-cell AEMWE tests. The stabilities of these Ni–Fe- and Ni–Co–Fe-based catalysts need to be proven, but promising results have been revealed in single-cell AEMWE tests. Nevertheless, careful studies of the performance and stability of specifically the high-surface-area and hydrous NiFeOxHy and NiCoFeOxHy catalysts are needed that also reflect real AEMWE conditions. Studies shedding light on the stability and potential degradation mechanism of the Ni–Fe-based catalysts under AEMWE conditions will further advance this technology.
The questions of increasing the anode catalyst activity and overcoming catalyst dissolution remain. Metal dissolution of PGM catalysts has been extensively studied in a standard 3-electrode setup or using in situ detection techniques, but only a few reports for metal dissolution of PGM-free catalysts exist. To the best of our knowledge, no studies exist discussing metal dissolution in an AEMWE cell. Higher j values are typically applied in an MEA (as compared to thin-layer catalyst evaluation), and a catalyst experiences different conditions (such as being flooded) in a thin-layer setup versus an MEA setup.
It is difficult to compare the activities of different catalysts due to the different experimental conditions used. These include catalyst loading, catalyst surface area, measurement methods, temperature, purity and concentration of the electrolyte, and purity of the salts used for the catalyst synthesis, specifically considering Fe impurities. Comparing activities based on j values normalized for the geometric area or the ECSA can be unreliable if the sample is, for example, highly porous or if the normalization is carried out using different methods to measure the ECSA. However, the extraction of ECSA-related data for the catalysts being studied is needed. Unfortunately, reliable methods to determine ECSA values for oxide and/or oxide-covered catalysts do not exist. However, trends can be established using a combination of well-described electrochemical methods such as Cdl, Cuupd, and charge values extracted from redox reactions. Catalyst stability needs to be examined more rigorously using analytical methods such as ICP-MS/OES analyses to quantify the catalyst dissolution. Uniform protocols should be used to evaluate the catalyst activity as well as the stability of the catalysts. The catalyst-stability studies also need to include measurements that consider real AEMWE conditions such as fluctuations that a catalyst can experience in the CL of an MEA as well as startup and shutdown conditions. In all cases, i.e., for activity and stability measurements, the catalysts need to be well-characterized before and after the measurements using electrochemical methods as well as physical-characterization methods. The use of steady-state measurements to extract mass and intrinsic HER and OER information and to construct a valid Tafel plot is of high importance. Currently, large discrepancies for both the HER and OER catalysts reported in the literature exist, which are likely at least partially due to using nonsteady-state methods as polarization curves.
The actual Pt amounts on the cathode in an AEMWE approach small values such as 0.5 mg Pt/C/cm2,482 and Pt–Ni alloys and potentially also Ru-based catalysts may further lower the cathode Pt loadings. Anode catalyst loadings in the 2 mg/cm2 and higher range are still typically used due to the sluggish OER kinetics.482 The use of non-PGM catalysts and possibly high-surface-area and multimetal Ni-based catalysts seems the most promising route for OER catalysts for AEMWE applications. To achieve this, as well as to achieve reductions of the OER catalyst loadings, innovative designs of the anode catalyst layer in addition to the use of highly active and stable catalysts could help. Developing novel catalyst-layer and catalyst designs that allow for efficient water and counterion transport while using highly active catalysts in small amounts seems to be an effective strategy to enhance the performance of AEMWEs.102,483 This might include designs similar to the thin OER catalyst-layer structure developed by 3M, which has shown promise for high performance and long-term stability for low loading of PGM anode catalysts in PEMWEs.102 Other CL designs explored for AEMWEs are the direct formation of the HER and OER catalysts on the porous current collector, instead of transforming catalyst powder catalysts into CLs, that subsequently need to be applied to either the porous current collector substrate or the membrane, known as CCS and CCM methods, respectively. Direct formation of catalysts on the porous and high-surface-area current collectors potentially offers a higher catalyst utilization and improved catalyst/electrode interactions. Such designs are in line with a proposal to consider a PGM-free catalyst with less sensitivity to local pH fluctuations and less interaction with the AEI. Recent studies have demonstrated promising durability advances with self-supported PGM-free electrodes and omitting the inclusion of an AEI but with a supporting electrolyte.453,463 Also missing are PTL and GDL studies where the effect of pore size and distribution of different support designs on mass- and charge-transport limitations are compared and optimized for AEMWEs by modeling and are validated experimentally. Besides the optimum electrode design, the AEM and AEIs still require adequate ion-exchange capacity and water diffusivity for high-performance AEMWEs.463,483 The earlier-mentioned considerations must be tailored for the chosen cell operating mode to ensure high performance while keeping in mind the relevant durability-limiting factors.31
Acknowledgments
We acknowledge the support from the National Research Council of Canada’s (NRC-CNRC) Materials for Clean Fuels Challenge Program for the joint project between the NRC-CNRC and HI ERN. We also thank Dr. Phil De Luna (NRC-CNRC) for proofreading the manuscript.
Biographies
Naiying Du is a Research Council Officer at the National Research Council of Canada (NRC). Before joining NRC, she completed her Ph.D. in Polymer Chemistry and Physics at Graduate University of the Chinese Academy of Sciences. Her research interests focus on an emerging area design, synthesis, and characterization of advanced polymer materials for energy efficiency and sustainability; microporous polymers for CO2 capture and storage; and functional polymers for printable organic electronics and sensors.
Claudie Roy is now a Science & Technology Advisor at Natural Resources Canada (NRCAN) working for the Office of Energy Research and Development. Prior to joining NRCAN she was a researcher at the National Research Council of Canada focusing on energy storage and conversion applications such as industrial hydrogen production and CO2 conversion. Before joining the NRC, she did her Ph.D. at the Technical University of Denmark, where she gained experience in the field of materials design and synthesis, characterization, and electrocatalysis for energy applications.
Retha Peach is a team leader at the Helmholtz Institute Erlangen-Nürnberg for Renewable Energy with a focus on analyzing and optimizing the interface engineering aspects of water electrolysis technologies. Before joining HI ERN, she completed her Ph.D. at the North West University of South Africa (NWU). Her experience includes the fabrication of blended membranes and membrane electrode assemblies that are suitable for alkaline and basic operated water electrolysis in low and intermediate temperature ranges.
Matthew Turnbull is a researcher at the National Research Council of Canada focusing on materials characterization and surface analysis, with a focus on X-ray methods and electron microscopy. He earned his Ph.D. in physical and analytical chemistry at the University of Western Ontario, working on the design and characterization of thin-film, nanoparticle-based solar cell devices.
Simon Thiele is the leader of the Electrocatalytic Interface Engineering research department at the Helmholtz Institute Erlangen-Nürnberg for Renewable Energy. Also since 2018, he is a professor at the Department of Chemical and Biological Engineering at the technical faculty of the Friedrich-Alexander-University Erlangen-Nürnberg. His research focuses on novel concepts for electrochemical conversion devices such as fuel cells and electrolysers.
Christina Bock is a senior researcher at the National Research Council of Canada (NRC) leading the Energy Storage Materials and Formulations team. She has extensive experience on materials development, characterization, and integration in small devices for energy conversion and energy storage materials. She has published extensively on platinum- and nickel-based catalysts. She is the recipient of several awards and served as Treasurer and President of the Electrochemical Society, Inc. She is in the process of leaving NRC but will remain with the organization as a consultant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrev.1c00854.
Tables for HER activity measurements, tables for OER activity measurements, protocols to evaluate HER and OER catalysts, protocol to evaluate HER and OER catalyst stability, ECSA measurements for HER/OER activity reporting, summary of AEMs evaluated in single AEMWE cells, additional data for state-of-the-art single-cell AEMWE tests, and protocol for single-cell AEMWE testing abbreviations and symbols (PDF)
Author Contributions
C.B. designed the review, wrote the first two sections and a large fraction of section 3, and contributed to and edited all of the other sections and the Supporting Information. N.D. wrote section 4. C.R. contributed to sections 3 and 7 and the testing protocols and organized the references. R.P. contributed to and led the organization of sections 5–9 and the testing protocols. S.T. edited all sections of the review. M.T. contributed to section 3. All of the authors designed the figures and reviewed the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Connelly E.; Elgowainy A.; Ruth M.. Current Hydrogen Market Size: Domestic and Global; DOE Hydrogen and Fuel Cells Program: 2019; pp 1–5. [Google Scholar]
- The Future of Hydrogen; International Energy Agency: 2019; p 18. [Google Scholar]
- Sample Report: Hydrogen Generation Market Analyses; Grand View Research (CVR), 2020; pp 6–7.
- Probstein R. F.; Hicks R. E.. Synthetic Fuels; McGraw-Hill: New York, 1982; p 20. [Google Scholar]
- The engineering toolbox. https://www.engineeringtoolbox.com/ (accessed Oct 2020).
- Pinsky R.; Sabharwall P.; Hartvigsen J.; O’Brien J. Comparative Review of Hydrogen Production Technologies for Nuclear Hybrid Energy Systems. Prog. Nucl. Energy 2020, 123, 103317. 10.1016/j.pnucene.2020.103317. [DOI] [Google Scholar]
- Office of Energy Efficiency and Renewable Energy. Hydrogen Production: Natural Gas Reforming. https://www.energy.gov./eere/fuelcells/hydrogen-production-natural-gas-reforming (accessed Apr 2021).
- Schoots K.; Ferioli F.; Kramer G. J.; van der Zwaan B. C. C. Learning Curves for Hydrogen Production Technology: An Assessment of Observed Cost Reductions. Int. J. Hydrog. Energy 2008, 33, 2630–2645. 10.1016/j.ijhydene.2008.03.011. [DOI] [Google Scholar]
- Proost J. State-of-the Art CAPEX Data for Water Electrolysers, and Their Impact on Renewable Hydrogen Price Settings. Int. J. Hydrog. Energy 2019, 44, 4406–4413. 10.1016/j.ijhydene.2018.07.164. [DOI] [Google Scholar]
- Bourgeois R.Alkaline Electrolysis Final Technical Report; Report no. DOE/FC/14223-1; General Electric Global Research Center: Niskayuna, NY, 2006; pp 4–9.
- Bertuccioli L.; Chan A.; Hart D.; Lehner F.; Madden B.; Standen E.. Development of water electrolysis in the European Union; Fuel Cells Hydrogen Joint Undertaking: Lausanne, 2014; pp 7–25. [Google Scholar]
- Yvon-Durocher G.; Allen A. P.; Bastviken D.; Conrad R.; Gudasz C.; St-Pierre A.; Thanh-Duc N.; Del Giorgio P. A. Methane Fluxes Show Consistent Temperature Dependence across Microbial to Ecosystem Scales. Nature 2014, 507, 488–491. 10.1038/nature13164. [DOI] [PubMed] [Google Scholar]
- Princeton University. A more potent greenhouse gas than carbon dioxide, methane emissions will leap as Earth warms. www.sciencedaily.com/releases/2014/03/140327111724.htm (accessed Oct 2020).
- Sun P.; Young B.; Elgowainy A.; Lu Z.; Wang M.; Morelli B.; Hawkins T. Criteria Air Pollutants and Greenhouse Gas Emissions from Hydrogen Production in U.S. Steam Methane Reforming Facilities. Environ. Sci. Technol. 2019, 53, 7103–7113. 10.1021/acs.est.8b06197. [DOI] [PubMed] [Google Scholar]
- International Energy Agency . Global CO2 emissions in 2019. https://www.iea.org/articles/global-co2-emissions-in-2019 (accessed Dec 2020).
- Harvey C. N. G.CO2 Emissions Will Break Another Record in 2019. Scientific American; https://www.scientificamerican.com/article/co2-emissions-will-break-another-record-in-2019/ (accessed Feb 2022).
- Bacquart T.; Arrhenius K.; Persijn S.; Rojo A.; Auprêtre F.; Gozlan B.; Moore N.; Morris A.; Fischer A.; Murugan A.; Bartlett S.; et al. Hydrogen Fuel Quality from Two Main Production Processes: Steam Methane Reforming and Proton Exchange Membrane Water Electrolysis. J. Power Sources 2019, 444, 227170. 10.1016/j.jpowsour.2019.227170. [DOI] [Google Scholar]
- Hamdan M.PEM Electrolyzer Incorporating an Advanced Low-Cost Membrane; DOE/GO/18065-22; U.S. Department of Energy: 2013.
- Andrews J.; Shabani B. Re-Envisioning the Role of Hydrogen in a Sustainable Energy Economy. Int. J. Hydrog. Energy 2012, 37, 1184–1203. 10.1016/j.ijhydene.2011.09.137. [DOI] [Google Scholar]
- Staffell I.; Scamman D.; Velazquez Abad A.; Balcombe P.; Dodds P. E.; Ekins P.; Shah N.; Ward K. R. The Role of Hydrogen and Fuel Cells in the Global Energy System. Environ. Sci. Technol. 2019, 12, 463–491. 10.1039/C8EE01157E. [DOI] [Google Scholar]
- Apostolou D.; Enevoldsen P.; Xydis G. Supporting Green Urban Mobility - The Case of a Small-Scale Autonomous Hydrogen Refuelling Station. Int. J. Hydrog. Energy 2019, 44, 9675–9689. 10.1016/j.ijhydene.2018.11.197. [DOI] [Google Scholar]
- Götz M.; Lefebvre J.; Mörs F.; McDaniel Koch A.; Graf F.; Bajohr S.; Reimert R.; Kolb T. Renewable Power-to-Gas: A Technological and Economic Review. Renew. Energy 2016, 85, 1371–1390. 10.1016/j.renene.2015.07.066. [DOI] [Google Scholar]
- Lee B.; Heo J.; Kim S.; Sung C.; Moon C.; Moon S.; Lim H. Economic Feasibility Studies of High Pressure PEM Water Electrolysis for Distributed H2 Refueling Stations. Energy Convers. Manag. 2018, 162, 139–144. 10.1016/j.enconman.2018.02.041. [DOI] [Google Scholar]
- Buttler A.; Spliethoff H. Current Status of Water Electrolysis for Energy Storage, Grid Balancing and Sector Coupling via Power-to-Gas and Power-to-Liquids: A Review. Renew. Sust. Energy Rev. 2018, 82, 2440–2454. 10.1016/j.rser.2017.09.003. [DOI] [Google Scholar]
- Teichmann D.; Arlt W.; Wasserscheid P.; Freymann R. A Future Energy Supply Based on Liquid Organic Hydrogen Carriers (LOHC). Environ. Sci. Technol. 2011, 4, 2767–2773. 10.1039/c1ee01454d. [DOI] [Google Scholar]
- Dong Y.; Yang M.; Yang Z.; Ke H.; Cheng H. Catalytic Hydrogenation and Dehydrogenation of N-Ethylindole as a New Heteroaromatic Liquid Organic Hydrogen Carrier. Int. J. Hydrog. Energy 2015, 40, 10918–10922. 10.1016/j.ijhydene.2015.05.196. [DOI] [Google Scholar]
- Geburtig D.; Preuster P.; Bösmann A.; Müller K.; Wasserscheid P. Chemical Utilization of Hydrogen from Fluctuating Energy Sources - Catalytic Transfer Hydrogenation from Charged Liquid Organic Hydrogen Carrier Systems. Int. J. Hydrog. Energy 2016, 41, 1010–1017. 10.1016/j.ijhydene.2015.10.013. [DOI] [Google Scholar]
- Preuster P.; Papp C.; Wasserscheid P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-Free Hydrogen Economy. Acc. Chem. Res. 2017, 50, 74–85. 10.1021/acs.accounts.6b00474. [DOI] [PubMed] [Google Scholar]
- Niermann M.; Drünert S.; Kaltschmitt M.; Bonhoff K. Liquid Organic Hydrogen Carriers (LOHCs)-Techno-Economic Analysis of LOHCs in a Defined Process Chain. Environ. Sci. Technol. 2019, 12, 290–307. 10.1039/C8EE02700E. [DOI] [Google Scholar]
- Miller H. A.; Bouzek K.; Hnat J.; Loos S.; Bernäcker C. I.; Weißgärber T.; Röntzsch L.; Meier-Haack J. Green Hydrogen from Anion Exchange Membrane Water Electrolysis: A Review of Recent Developments in Critical Materials and Operating Conditions. Sustain. Energy Fuels. 2020, 4, 2114–2133. 10.1039/C9SE01240K. [DOI] [Google Scholar]
- Li D.; Motz A. R.; Bae C.; Fujimoto C.; Yang G.; Zhang F.-Y.; Ayers K. E.; Kim Y. S. Durability of Anion Exchange Membrane Water Electrolyzers. Energy Environ. Sci. 2021, 14, 3393–3419. 10.1039/D0EE04086J. [DOI] [Google Scholar]
- Henkensmeier D.; Najibah M.; Harms C.; Zitka J.; Hnát J.; Bouzek K. Overview: State-of-the Art Commercial Membranes for Anion Exchange Membrane Water Electrolysis. J. Electrochem. Energy Convers. Storage 2021, 18, 024001. 10.1115/1.4047963. [DOI] [Google Scholar]
- Lindquist G. A.; Xu Q.; Oener S. Z.; Boettcher S. W. Membrane Electrolyzers for Impure-Water Splitting. Joule 2020, 4, 2549–2561. 10.1016/j.joule.2020.09.020. [DOI] [Google Scholar]
- Brauns J.; Turek T. Alkaline Water Electrolysis Powered by Renewable Energy: A Review. Processes 2020, 8, 248–271. 10.3390/pr8020248. [DOI] [Google Scholar]
- Mamlouk M.; Manolova M.. Chapter 6: Alkaline Anionic Exchange Membrane Water Electrolysers. In Electrochemical Methods for Hydrogen Production; RSC Energy and Environment Series; Royal Society of Chemistry: Cambridge, 2020; pp 80–252; 10.1039/9781788016049-00180. [DOI] [Google Scholar]
- You W.; Noonan K. J. T.; Coates G. W. Alkaline-Stable Anion Exchange Membranes: A Review of Synthetic Approaches. Prog. Polym. Sci. 2020, 100, 101177–101189. 10.1016/j.progpolymsci.2019.101177. [DOI] [Google Scholar]
- Ayers K.; Danilovic N.; Ouimet R.; Carmo M.; Pivovar B.; Bornstein M. Perspectives on Low-Temperature Electrolysis and Potential for Renewable Hydrogen at Scale. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 219–239. 10.1146/annurev-chembioeng-060718-030241. [DOI] [PubMed] [Google Scholar]
- Abbasi R.; Setzler B. P.; Lin S.; Wang J.; Zhao Y.; Xu H.; Pivovar B.; Tian B.; Chen X.; Wu G.; Yan Y. A Roadmap to Low-Cost Hydrogen with Hydroxide Exchange Membrane Electrolyzers. Adv. Mater. 2019, 31, 1805876. 10.1002/adma.201805876. [DOI] [PubMed] [Google Scholar]
- Vincent I.; Bessarabov D. Low Cost Hydrogen Production by Anion Exchange Membrane Electrolysis: A Review. Renew. Sust. Energy Rev. 2018, 81, 1690–1704. 10.1016/j.rser.2017.05.258. [DOI] [Google Scholar]
- Hagesteijn K. F. L.; Jiang S.; Ladewig B. P. A Review of the Synthesis and Characterization of Anion Exchange Membranes. J. Mater. Sci. 2018, 53, 11131–11150. 10.1007/s10853-018-2409-y. [DOI] [Google Scholar]
- Pletcher D.; Li X. Prospects for Alkaline Zero Gap Water Electrolysers for Hydrogen Production. Int. J. Hydrog. Energy 2011, 36, 15089–15104. 10.1016/j.ijhydene.2011.08.080. [DOI] [Google Scholar]
- Nikolaidis P.; Poullikkas A. A Comparative Overview of Hydrogen Production Processes. Renew. Sust. Energy Rev. 2017, 67, 597–611. 10.1016/j.rser.2016.09.044. [DOI] [Google Scholar]
- Grigoriev S. A.; Fateev V. N.; Bessarabov D. G.; Millet P. Current Status, Research Trends, and Challenges in Water Electrolysis Science and Technology. Int. J. Hydrog. Energy 2020, 45, 26036–26058. 10.1016/j.ijhydene.2020.03.109. [DOI] [Google Scholar]
- Center for climate and energy solutions. Renewable Energy; https://www.c2es.org/content/renewable-energy/ (accessed Nov 2020).
- Carmo M.; Fritz D. L.; Mergel J.; Stolten D. A Comprehensive Review on PEM Water Electrolysis. Int. J. Hydrog. Energy 2013, 38, 4901–4934. 10.1016/j.ijhydene.2013.01.151. [DOI] [Google Scholar]
- Sabihuddin S.; Kiprakis A. E.; Mueller M. A Numerical and Graphical Review of Energy Storage Technologies. Energies 2015, 8, 172–216. 10.3390/en8010172. [DOI] [Google Scholar]
- Zhang Y.; Campana P. E.; Lundblad A.; Yan J. Comparative Study of Hydrogen Storage and Battery Storage in Grid Connected Photovoltaic System: Storage Sizing and Rule-Based Operation. Appl. Energy 2017, 201, 397–411. 10.1016/j.apenergy.2017.03.123. [DOI] [Google Scholar]
- Pletcher D.; Li X.; Wang S. A Comparison of Cathodes for Zero Gap Alkaline Water Electrolysers for Hydrogen Production. Int. J. Hydrog. Energy 2012, 37, 7429–7435. 10.1016/j.ijhydene.2012.02.013. [DOI] [Google Scholar]
- Zablocki A.Fact sheet: Energy storage (2019). Environmental and Energy Study Institute; https://www.eesi.org/papers/view/energy-storage-2019 (accessed Dec 2020).
- Schmidt O.; Gambhir A.; Staffell I.; Hawkes A.; Nelson J.; Few S. Future Cost and Performance of Water Electrolysis: An Expert Elicitation Study. Int. J. Hydrog. Energy 2017, 42, 30470–30492. 10.1016/j.ijhydene.2017.10.045. [DOI] [Google Scholar]
- Eichman J.; Harrison K.; Peters M.; Eichman J.; Harrison K.; Peters M.. Novel Electrolyzer Applications: Providing More Than Just Hydrogen; Report no. NREL/TP-5400-61758; National Renewable Energy Lab (NREL): Golden, CO, 2014; pp 1–35. [Google Scholar]
- Yadav D.; Banerjee R. Net Energy and Carbon Footprint Analysis of Solar Hydrogen Production from the High-Temperature Electrolysis Process. Appl. Energy 2020, 262, 114503. 10.1016/j.apenergy.2020.114503. [DOI] [Google Scholar]
- Cetinkaya E.; Dincer I.; Naterer G. F. Life Cycle Assessment of Various Hydrogen Production Methods. Int. J. Hydrog. Energy 2012, 37, 2071–2080. 10.1016/j.ijhydene.2011.10.064. [DOI] [Google Scholar]
- Koroneos C.; Dompros A.; Roumbas G.; Moussiopoulos N. Life Cycle Assessment of Hydrogen Fuel Production Processes. Int. J. Hydrog. Energy 2004, 29, 1443–1450. 10.1016/j.ijhydene.2004.01.016. [DOI] [Google Scholar]
- Bhandari R.; Trudewind C. A.; Zapp P. Life Cycle Assessment of Hydrogen Production via Electrolysis - A Review. J. Clean. Prod. 2014, 85, 151–163. 10.1016/j.jclepro.2013.07.048. [DOI] [Google Scholar]
- Villagra A.; Millet P. An Analysis of PEM Water Electrolysis Cells Operating at Elevated Current Densities. Int. J. Hydrog. Energy 2019, 44, 9708–9717. 10.1016/j.ijhydene.2018.11.179. [DOI] [Google Scholar]
- Saba S. M.; Müller M.; Robinius M.; Stolten D. The Investment Costs of Electrolysis - A Comparison of Cost Studies from the Past 30 Years. Int. J. Hydrog. Energy 2018, 43, 1209–1223. 10.1016/j.ijhydene.2017.11.115. [DOI] [Google Scholar]
- Ayers K. E.; Capuano C.; Anderson E. B. Recent Advances in Cell Cost and Efficiency for PEM-Based Water Electrolysis. ECS Trans. 2012, 41, 15–22. 10.1149/1.3684798. [DOI] [Google Scholar]
- Nuttall L. J.; Fickett A. P.; Titterington W. A.. Hydrogen Generation By Solid Polymer Electrolyte Water Electrolysis. Hydrogen Energy; Springer: Boston, MA, 1975; pp 441–455. [Google Scholar]
- Danilovic N.; Ayers K. E.; Capuano C.; Renner J. N.; Wiles L.; Pertoso M. (Plenary) Challenges in Going from Laboratory to Megawatt Scale PEM Electrolysis. ECS Trans. 2016, 75, 395–402. 10.1149/07514.0395ecst. [DOI] [Google Scholar]
- Yang M.; Cheng G.; Xie D.; Zhu T.; Dong Y.; Ke H.; Cheng H. Study of Hydrogenation and Dehydrogenation of 1-Methylindole for Reversible Onboard Hydrogen Storage Application. Int. J. Hydrog. Energy 2018, 43, 8868–8876. 10.1016/j.ijhydene.2018.03.134. [DOI] [Google Scholar]
- Uhrig F.; Kadar J.; Müller K. Reliability of Liquid Organic Hydrogen Carrier-Based Energy Storage in a Mobility Application. Energy Sci.Eng. 2020, 8, 2044–2053. 10.1002/ese3.646. [DOI] [Google Scholar]
- Emel’Yanenko V. N.; Varfolomeev M. A.; Verevkin S. P.; Stark K.; Müller K.; Müller M.; Bösmann A.; Wasserscheid P.; Arlt W. Hydrogen Storage: Thermochemical Studies of N-Alkylcarbazoles and Their Derivatives as a Potential Liquid Organic Hydrogen Carriers. J. Phys. Chem. C 2015, 119, 26381–26389. 10.1021/acs.jpcc.5b10392. [DOI] [Google Scholar]
- Taube M.; Rippin D. W.; Cresswell D. L.; Knecht W. A System of h y d r o g e n - p o w e r e d Vehicles with Liquid Organic Hydrides. Int. J. Hydrog. Energy 1983, 8, 213–225. 10.1016/0360-3199(83)90067-8. [DOI] [Google Scholar]
- Scherer G. W. H.; Newson E.; Wokaun A. Economic Analysis of the Seasonal Storage of Electricity with Liquid Organic Hydrides. Int. J. Hydrog. Energy 1999, 24, 1157–1169. 10.1016/S0360-3199(98)00177-3. [DOI] [Google Scholar]
- Tremel A.; Wasserscheid P.; Baldauf M.; Hammer T. Techno-Economic Analysis for the Synthesis of Liquid and Gaseous Fuels Based on Hydrogen Production via Electrolysis. Int. J. Hydrog. Energy 2015, 40, 11457–11464. 10.1016/j.ijhydene.2015.01.097. [DOI] [Google Scholar]
- Andersen S. Z.; Čolić V.; Yang S.; Schwalbe J. A.; Nielander A. C.; McEnaney J. M.; Enemark-Rasmussen K.; Baker J. G.; Singh A. R.; Rohr B. A.; et al. A Rigorous Electrochemical Ammonia Synthesis Protocol with Quantitative Isotope Measurements. Nature 2019, 570, 504–508. 10.1038/s41586-019-1260-x. [DOI] [PubMed] [Google Scholar]
- Grundt T.; Christiansen K. Hydrogen by Water Electrolysis as Basis for Small Scale Ammonia Production. A Comparison with Hydrocarbon Based Technologies. Int. J. Hydrog. Energy 1982, 7, 247–257. 10.1016/0360-3199(82)90088-X. [DOI] [Google Scholar]
- Ursua A.; Sanchis P.; Gandia L. M. Hydrogen Production from Water Electrolysis : Current Status and Future Trends. Proc. IEEE Inst Electr Electron Eng. 2012, 100, 410–426. 10.1109/JPROC.2011.2156750. [DOI] [Google Scholar]
- Kreuter W.; Hofmann H. Electrolysis: The Important Energy Transformer in a World of Sustainable Energy. Int. J. Hydrog. Energy 1998, 23, 661–666. 10.1016/S0360-3199(97)00109-2. [DOI] [Google Scholar]
- Hall D. S.; Bock C.; MacDougall B. R. The Electrochemistry of Metallic Nickel: Oxides, Hydroxides, Hydrides and Alkaline Hydrogen Evolution. J. Electrochem. Soc. 2013, 160, F235–F243. 10.1149/2.026303jes. [DOI] [Google Scholar]
- Schalenbach M.; Kasian O.; Mayrhofer K. J. J. An Alkaline Water Electrolyzer with Nickel Electrodes Enables Efficient High Current Density Operation. Int. J. Hydrog. Energy 2018, 43, 11932–11938. 10.1016/j.ijhydene.2018.04.219. [DOI] [Google Scholar]
- Varcoe J. R.; Atanassov P.; Dekel D. R.; Herring A. M.; Hickner M. A.; Kohl P. A.; Kucernak A. R.; Mustain W. E.; Nijmeijer K.; Scott K.; Xu T.; Zhuang L. Anion-Exchange Membranes in Electrochemical Energy Systems. Environ. Sci. Technol. 2014, 7, 3135–3191. 10.1039/C4EE01303D. [DOI] [Google Scholar]
- In Lee H.; Dung D. T.; Kim J.; Pak J. H.; Kim S. k.; Cho H. S.; Cho W. C.; Kim C. H. The Synthesis of a Zirfon-Type Porous Separator with Reduced Gas Crossover for Alkaline Electrolyzer. Int. J. Energy Res. 2020, 44, 1875–1885. 10.1002/er.5038. [DOI] [Google Scholar]
- Renaud R.; LeRoy R. L. Separator Materials for Use in Alkaline Water Electrolysers. Int. J. Hydrog. Energy 1982, 7, 155–166. 10.1016/0360-3199(82)90142-2. [DOI] [Google Scholar]
- Phillips R.; Edwards A.; Rome B.; Jones D. R.; Dunnill C. W. Minimising the Ohmic Resistance of an Alkaline Electrolysis Cell through Effective Cell Design. Int. J. Hydrog. Energy 2017, 42, 23986–23994. 10.1016/j.ijhydene.2017.07.184. [DOI] [Google Scholar]
- Marini S.; Salvi P.; Nelli P.; Pesenti R.; Villa M.; Berrettoni M.; Zangari G.; Kiros Y. Advanced Alkaline Water Electrolysis. Electrochim. Acta 2012, 82, 384–391. 10.1016/j.electacta.2012.05.011. [DOI] [Google Scholar]
- Schalenbach M.; Tjarks G.; Carmo M.; Lueke W.; Mueller M.; Stolten D. Acidic or Alkaline? Towards a New Perspective on the Efficiency of Water Electrolysis. J. Electrochem. Soc. 2016, 163, F3197–F3208. 10.1149/2.0271611jes. [DOI] [Google Scholar]
- Diaz L. A.; Hnát J.; Heredia N.; Bruno M. M.; Viva F. A.; Paidar M.; Corti H. R.; Bouzek K.; Abuin G. C. Alkali Doped Poly (2,5-Benzimidazole) Membrane for Alkaline Water Electrolysis: Characterization and Performance. J. Power Sources 2016, 312, 128–136. 10.1016/j.jpowsour.2016.02.032. [DOI] [Google Scholar]
- Kraglund M. R.; Carmo M.; Schiller G.; Ansar S. A.; Aili D.; Christensen E.; Jensen J. O. Ion-Solvating Membranes as a New Approach towards High Rate Alkaline Electrolyzers. Environ. Sci. Technol. 2019, 12, 3313–3318. 10.1039/C9EE00832B. [DOI] [Google Scholar]
- Kraglund M. R.; Aili D.; Jankova K.; Christensen E.; Li Q.; Jensen J. O. Zero-Gap Alkaline Water Electrolysis Using Ion-Solvating Polymer Electrolyte Membranes at Reduced KOH Concentrations. J. Electrochem. Soc. 2016, 163, F3125–F3131. 10.1149/2.0161611jes. [DOI] [Google Scholar]
- Ayers K. E.; Anderson E. B.; Capuano C. B.; Niedzwiecki M.; Hickner M. A.; Wang C.-Y.; Leng Y.; Zhao W. Characterization of Anion Exchange Membrane Technology for Low Cost Electrolysis. ECS Trans. 2013, 45, 121–130. 10.1149/04523.0121ecst. [DOI] [Google Scholar]
- Leng Y.; Chen G.; Mendoza A. J.; Tighe T. B.; Hickner M. A.; Wang C. Y. Solid-State Water Electrolysis with an Alkaline Membrane. J. Am. Chem. Soc. 2012, 134, 9054–9057. 10.1021/ja302439z. [DOI] [PubMed] [Google Scholar]
- Pavel C. C.; Cecconi F.; Emiliani C.; Santiccioli S.; Scaffidi A.; Catanorchi S.; Comotti M. Highly Efficient Platinum Group Metal Free Based Membrane-Electrode Assembly for Anion Exchange Membrane Water Electrolysis. Angew. Chem., Int. Ed. 2014, 53, 1378–1381. 10.1002/anie.201308099. [DOI] [PubMed] [Google Scholar]
- Giesbrecht P. K.; Müller A. M.; Read C. G.; Holdcroft S.; Lewis N. S.; Freund M. S. Vapor-Fed Electrolysis of Water Using Earth-Abundant Catalysts in Nafion or in Bipolar Nafion/Poly(Benzimidazolium) Membranes. Sustain. Energy Fuels. 2019, 3, 3611–3626. 10.1039/C9SE00672A. [DOI] [Google Scholar]
- Oener S. Z.; Foster M. J.; Boettcher S. W. Accelerating Water Dissociation in Bipolar Membranes and for Electrocatalysis. Science 2020, 369, 1099–1103. 10.1126/science.aaz1487. [DOI] [PubMed] [Google Scholar]
- Mayerhöfer B.; McLaughlin D.; Böhm T.; Hegelheimer M.; Seeberger D.; Thiele S. Bipolar Membrane Electrode Assemblies for Water Electrolysis. ACS Appl. Energy Materials 2020, 3, 9635–9644. 10.1021/acsaem.0c01127. [DOI] [Google Scholar]
- Chen Y.; Wrubel J. A.; Klein W. E.; Kabir S.; Smith W. A.; Neyerlin K. C.; Deutsch T. G. High-Performance Bipolar Membrane Development for Improved Water Dissociation. ACS Appl. Polym.Mater. 2020, 2, 4559–4569. 10.1021/acsapm.0c00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerhöfer B.; Ehelebe K.; Speck F. D.; Bierling M.; Bender J.; Kerres J. A.; Mayrhofer K. J. J.; Cherevko S.; Peach R.; Thiele S. On the Effect of Anion Exchange Ionomer Binders in Bipolar Electrode Membrane Interface Water Electrolysis. J. Mater. Chem. A 2021, 9, 14285–14295. 10.1039/D1TA00747E. [DOI] [Google Scholar]
- Wu X.; Scott K. CuxCo3-xO4 (0 ≤ x < 1) Nanoparticles for Oxygen Evolution in High Performance Alkaline Exchange Membrane Water Electrolysers. J. Mater. Chem. 2011, 21, 12344–12351. 10.1039/c1jm11312g. [DOI] [Google Scholar]
- Cao Y. C.; Wu X.; Scott K. A Quaternary Ammonium Grafted Poly Vinyl Benzyl Chloride Membrane for Alkaline Anion Exchange Membrane Water Electrolysers with No-Noble-Metal Catalysts. Int. J. Hydrog. Energy 2012, 37, 9524–9528. 10.1016/j.ijhydene.2012.03.116. [DOI] [Google Scholar]
- Parrondo J.; Arges C. G.; Niedzwiecki M.; Anderson E. B.; Ayers K. E.; Ramani V. Degradation of Anion Exchange Membranes Used for Hydrogen Production by Ultrapure Water Electrolysis. RSC Adv. 2014, 4, 9875–9879. 10.1039/c3ra46630b. [DOI] [Google Scholar]
- Acta S.p.A. Acta Powered by Nature. https://www.hydrogenfuelnews.com/acta-new-alkaline-electrolyzer/855465/ (accessed Jun 2021).
- Enapter. The AEM Electrolyser: Making Green Hydrogen Easy. https://www.enapter.com/aem-electrolyser (accessed Feb 2022).
- Phillips R.; Gannon J. F. W.; Dunnill W. C.. Alkaline Electrolysers. In Electrochemical Methods for Hydrogen Production; Scott K., Ed.; Royal Society of Chemistry: 2019; Vol. 25, pp 28–58. [Google Scholar]
- Kang Z.; Mo J.; Yang G.; Retterer S. T.; Cullen D. A.; Toops T. J.; Green J. B.; Mench M. M.; Zhang F. Y. Investigation of Thin/Well-Tunable Liquid/Gas Diffusion Layers Exhibiting Superior Multifunctional Performance in Low-Temperature Electrolytic Water Splitting. Environ. Sci. Technol. 2017, 10, 166–175. 10.1039/C6EE02368A. [DOI] [Google Scholar]
- An L.; Zhao T. S.; Chai Z. H.; Tan P.; Zeng L. Mathematical Modeling of an Anion-Exchange Membrane Water Electrolyzer for Hydrogen Production. Int. J. Hydrog. Energy 2014, 39, 19869–19876. 10.1016/j.ijhydene.2014.10.025. [DOI] [Google Scholar]
- Kraglund M. R.Alkaline Membrane Water Electrolysis with Non-Noble Catalysts. Ph.D. Thesis, DTU Energy, Department of Energy Conversion and Storage, 2017; p 28. [Google Scholar]
- Fang Y. H.; Liu Z. P. Tafel Kinetics of Electrocatalytic Reactions: From Experiment to First-Principles. ACS Catal. 2014, 4, 4364–4376. 10.1021/cs501312v. [DOI] [Google Scholar]
- Tse E. C. M.; Gewirth A. A. Effect of Temperature and Pressure on the Kinetics of the Oxygen Reduction Reaction. J. Phys. Chem. A 2015, 119, 1246–1255. 10.1021/acs.jpca.5b00572. [DOI] [PubMed] [Google Scholar]
- Miller H. A.; Bouzek K.; Hnat J.; Loos S.; Bernäcker C. I.; Weißgärber T.; Röntzsch L.; Meier-Haack J. Green Hydrogen from Anion Exchange Membrane Water Electrolysis: A Review of Recent Developments in Critical Materials and Operating Conditions. Sustain. Energy Fuels. 2020, 4, 2114–2133. 10.1039/C9SE01240K. [DOI] [Google Scholar]
- Debe M. K.; Hendricks S. M.; Vernstrom G. D.; Meyers M.; Brostrom M.; Stephens M.; Chan Q.; Willey J.; Hamden M.; Mittelsteadt C. K.; Capuano C. B.; Ayers K. E.; Anderson E. B. Initial Performance and Durability of Ultra-Low Loaded NSTF Electrodes for PEM Electrolyzers. J. Electrochem. Soc. 2012, 159, K165–K176. 10.1149/2.065206jes. [DOI] [Google Scholar]
- Ayers K. E.; Anderson E. B.; Capuano C.; Carter B.; Dalton L.; Hanlon G.; Manco J.; Niedzwiecki M. Research Advances towards Low Cost, High Efficiency PEM Electrolysis. ECS Trans. 2010, 33, 3–15. 10.1149/1.3484496. [DOI] [Google Scholar]
- Lopata J.; Kang Z.; Young J.; Bender G.; Weidner J. W.; Shimpalee S. Effects of the Transport/Catalyst Layer Interface and Catalyst Loading on Mass and Charge Transport Phenomena in Polymer Electrolyte Membrane Water Electrolysis Devices. J. Electrochem. Soc. 2020, 167, 064507. 10.1149/1945-7111/ab7f87. [DOI] [Google Scholar]
- Liu C.; Carmo M.; Bender G.; Everwand A.; Lickert T.; Young J. L.; Smolinka T.; Stolten D.; Lehnert W. Performance Enhancement of PEM Electrolyzers through Iridium-Coated Titanium Porous Transport Layers. Electrochem. Commun. 2018, 97, 96–99. 10.1016/j.elecom.2018.10.021. [DOI] [Google Scholar]
- Mergel J.; Fritz D. L.; Carmo M.. Stack Technology for PEM Electrolysis. In Hydrogen Science and Engineering : Materials, Processes, Systems and Technology; Wiley-VCH Verlag: 2016; pp 331–357. [Google Scholar]
- Park J. E.; Kang S. Y.; Oh S. H.; Kim J. K.; Lim M. S.; Ahn C. Y.; Cho Y. H.; Sung Y. E. High-Performance Anion-Exchange Membrane Water Electrolysis. Electrochim. Acta 2019, 295, 99–106. 10.1016/j.electacta.2018.10.143. [DOI] [Google Scholar]
- Park J. E.; Park S. B.; Kim M. J.; Shin H.; Kang S. Y.; Cho Y. H.; Sung Y. E. Three-Dimensional Unified Electrode Design Using a NiFeOOH Catalyst for Superior Performance and Durable Anion-Exchange Membrane Water Electrolyzers. ACS Catal. 2022, 12, 135–145. 10.1021/acscatal.1c04117. [DOI] [Google Scholar]
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Nørskov J. K.; Jaramillo T. F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, 6321. 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Bock C.; MacDougall B. Novel Method for the Estimation of the Electroactive Pt Area. J. Electrochem. Soc. 2003, 150, E377–E383. 10.1149/1.1586924. [DOI] [Google Scholar]
- Hall D. S.; Bock C.; MacDougall B. R. An Oxalate Method for Measuring the Surface Area of Nickel Electrodes. J. Electrochem. Soc. 2014, 161, H787–H795. 10.1149/2.0711412jes. [DOI] [Google Scholar]
- Anderson G. C.; Pivovar B. S.; Alia S. M. Establishing Performance Baselines for the Oxygen Evolution Reaction in Alkaline Electrolytes. J. Electrochem. Soc. 2020, 167, 044503–044515. 10.1149/1945-7111/ab7090. [DOI] [Google Scholar]
- Lyons M. E. G.; Floquet S. Mechanism of Oxygen Reactions at Porous Oxide Electrodes. Part 2 - Oxygen Evolution at RuO2, IrO2 and IrxRu1-xO2 Electrodes in Aqueous Acid and Alkaline Solution. Phys. Chem. Chem. Phys. 2011, 13, 5314–5335. 10.1039/c0cp02875d. [DOI] [PubMed] [Google Scholar]
- Lischka M.; Mosch C.; Groß A. Tuning Catalytic Properties of Bimetallic Surfaces: Oxygen Adsorption on Pseudomorphic Pt/Ru Overlayers. Electrochim. Acta 2007, 52, 2219–2228. 10.1016/j.electacta.2006.03.113. [DOI] [Google Scholar]
- Anantharaj S.; Noda S.; Driess M.; Menezes P. W. The Pitfalls of Using Potentiodynamic Polarization Curves for Tafel Analysis in Electrocatalytic Water Splitting. ACS Energy Lett. 2021, 6, 1607–1611. 10.1021/acsenergylett.1c00608. [DOI] [Google Scholar]
- Geiger S.; Kasian O.; Ledendecker M.; Pizzutilo E.; Mingers A. M.; Fu W. T.; Diaz-Morales O.; Li Z.; Oellers T.; Fruchter L.; Ludwig A.; Mayrhofer K. J. J.; Koper M. T. M.; Cherevko S. The Stability Number as a Metric for Electrocatalyst Stability Benchmarking. Nat. Catal. 2018, 1, 508–515. 10.1038/s41929-018-0085-6. [DOI] [Google Scholar]
- Wang Z.; Zheng Y. R.; Montoya J.; Hochfilzer D.; Cao A.; Kibsgaard J.; Chorkendorff I.; Nørskov J. K. Origins of the Instability of Nonprecious Hydrogen Evolution Reaction Catalysts at Open-Circuit Potential. ACS Energy Lett. 2021, 6, 2268–2274. 10.1021/acsenergylett.1c00876. [DOI] [Google Scholar]
- Lei C.; Wang Y.; Hou Y.; Liu P.; Yang J.; Zhang T.; Zhuang X.; Chen M.; Yang B.; Lei L.; Yuan C.; Qiu M.; Feng X. Efficient Alkaline Hydrogen Evolution on Atomically Dispersed Ni-Nx Species Anchored Porous Carbon with Embedded Ni Nanoparticles by Accelerating Water Dissociation Kinetics. Environ. Sci. Technol. 2019, 12, 149–156. 10.1039/C8EE01841C. [DOI] [Google Scholar]
- Kubisztal J.; Budniok A.; Lasia A. Study of the Hydrogen Evolution Reaction on Nickel-Based Composite Coatings Containing Molybdenum Powder. Int. J. Hydrog. Energy 2007, 32, 1211–1218. 10.1016/j.ijhydene.2006.11.020. [DOI] [Google Scholar]
- Shinagawa T.; Garcia-Esparza A. T.; Takanabe K. Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion. Sci. Rep. 2015, 5, 13801. 10.1038/srep13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W.; Myint M.; Chen J. G.; Yan Y. Correlating the Hydrogen Evolution Reaction Activity in Alkaline Electrolytes with the Hydrogen Binding Energy on Monometallic Surfaces. Environ. Sci. Technol. 2013, 6, 1509–1512. 10.1039/c3ee00045a. [DOI] [Google Scholar]
- Durst J.; Siebel A.; Simon C.; Hasché F.; Herranz J.; Gasteiger H. A. New Insights into the Electrochemical Hydrogen Oxidation and Evolution Reaction Mechanism. Environ. Sci. Technol. 2014, 7, 2255–2260. 10.1039/C4EE00440J. [DOI] [Google Scholar]
- Sheng W.; Zhuang Z.; Gao M.; Zheng J.; Chen J. G.; Yan Y. Correlating Hydrogen Oxidation and Evolution Activity on Platinum at Different PH with Measured Hydrogen Binding Energy. Nat. Commun. 2015, 6, 5848. 10.1038/ncomms6848. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang G.; Li G.; Huang B.; Pan J.; Liu Q.; Han J.; Xiao L.; Lu J.; Zhuang L. Pt-Ru Catalyzed Hydrogen Oxidation in Alkaline Media: Oxophilic Effect or Electronic Effect? Environ. Sci. Technol. 2015, 8, 177–181. 10.1039/C4EE02564D. [DOI] [Google Scholar]
- Zheng J.; Sheng W.; Zhuang Z.; Xu B.; Yan Y. Universal Dependence of Hydrogen Oxidation and Evolution Reaction Activity of Platinum-Group Metals on PH and Hydrogen Binding Energy. Sci. Adv. 2016, 2, e1501602. 10.1126/sciadv.1501602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasatti S. Work Function, Electronegativity, and Electrochemical Behaviour of Metals. J. Electroanal. Chem. Interface Electrochem. 1972, 39, 163–184. 10.1016/S0022-0728(72)80485-6. [DOI] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Logadottir A.; Kitchin J. R.; Chen J. G.; Pandelov S.; Stimming U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23–J26. 10.1149/1.1856988. [DOI] [Google Scholar]
- Sheng W.; Gasteiger H. A.; Shao-Horn Y. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs Alkaline Electrolytes. J. Electrochem. Soc. 2010, 157, B1529. 10.1149/1.3483106. [DOI] [Google Scholar]
- Stoerzinger K. A.; Favaro M.; Ross P. N.; Yano J.; Liu Z.; Hussain Z.; Crumlin E. J. Probing the Surface of Platinum during the Hydrogen Evolution Reaction in Alkaline Electrolyte. J. Phys. Chem. B 2018, 122, 864–870. 10.1021/acs.jpcb.7b06953. [DOI] [PubMed] [Google Scholar]
- Markovića N. M.; Sarraf S. T.; Gasteiger H. A.; Ross P. N. Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Alkaline Solution. J. Chem. Soc., Faraday Trans. 1996, 92, 3719–3725. 10.1039/FT9969203719. [DOI] [Google Scholar]
- Cui C.; Ahmadi M.; Behafarid F.; Gan L.; Neumann M.; Heggen M.; Cuenya B. R.; Strasser P. Shape-Selected Bimetallic Nanoparticle Electrocatalysts: Evolution of Their Atomic-Scale Structure, Chemical Composition, and Electrochemical Reactivity under Various Chemical Environments. Faraday Discuss. 2013, 162, 91–112. 10.1039/c3fd20159g. [DOI] [PubMed] [Google Scholar]
- Kibsgaard J.; Chorkendorff I. Considerations for the Scaling-up of Water Splitting Catalysts. Nat. Energy. 2019, 4, 430–433. 10.1038/s41560-019-0407-1. [DOI] [Google Scholar]
- Yang L. X.; Bock C.; Macdougall B.; Park J. The Role of the WOx Ad-Component to Pt and PtRu Catalysts in the Electrochemical CH3OH Oxidation Reaction. J. Appl. Electrochem. 2004, 34, 427–438. 10.1023/B:JACH.0000016628.81571.f4. [DOI] [Google Scholar]
- Conway B. E.; Angerstein-Kozlowska H.; Sharp W. B. A.; Criddle E. E. Ultrapurification of Water for Electrochemical and Surface Chemical Work by Catalytic Pyrodistillation. Anal. Chem. 1973, 45, 1331–1336. 10.1021/ac60330a025. [DOI] [Google Scholar]
- Subbaraman R.; Tripkovic D.; Strmcnik D.; Chang K. C.; Uchimura M.; Paulikas a P.; Stamenkovic V.; Markovic N. M. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces. Science 2011, 334, 1256–1260. 10.1126/science.1211934. [DOI] [PubMed] [Google Scholar]
- Wang P.; Jiang K.; Wang G.; Yao J.; Huang X. Phase and Interface Engineering of Platinum-Nickel Nanowires for Efficient Electrochemical Hydrogen Evolution. Angew. Chem., Int. Ed. 2016, 55, 12859–12863. 10.1002/anie.201606290. [DOI] [PubMed] [Google Scholar]
- Chen Z. J.; Cao G. X.; Gan L. Y.; Dai H.; Xu N.; Zang M. J.; Dai H.-B.; Wu H.; Wang P. Highly Dispersed Platinum on Honeycomb-like NiO@Ni Film as a Synergistic Electrocatalyst for the Hydrogen Evolution Reaction. ACS Catal. 2018, 8, 8866–8872. 10.1021/acscatal.8b02212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Zhao S.; Zhao K.; Muqsit A.; Tang H.; Chang L.; Zhao H.; Gao Y.; Tang Z. Ultrathin Platinum Nanowires Grown on Single-Layered Nickel Hydroxide with High Hydrogen Evolution Activity. Nat. Commun. 2015, 6, 6430. 10.1038/ncomms7430. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Chen Q.; Zhang J.; Li H.; Jiang Y.; Shen S.; Fu G.; Lu B. A.; Xie Z.; Zheng L. Platinum-Nickel Alloy Excavated Nano-Multipods with Hexagonal Close-Packed Structure and Superior Activity towards Hydrogen Evolution Reaction. Nat. Commun. 2017, 8, 15131. 10.1038/ncomms15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S.; Haid R. W.; Kluge R. M.; Ding X.; Garlyyev B.; Fichtner J.; Watzele S.; Hou S.; Bandarenka A. S. Enhancing the Hydrogen Evolution Reaction Activity of Platinum Electrodes in Alkaline Media Using Nickel-Iron Clusters. Angew. Chem., Int. Ed. 2020, 59, 10934–10938. 10.1002/anie.202000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas S. A.; Kim S. H.; Iqbal M. I.; Muhammad S.; Yoon W. S.; Jung K. D. Synergistic Effect of Nano-Pt and Ni Spine for HER in Alkaline Solution: Hydrogen Spillover from Nano-Pt to Ni Spine. Sci. Rep. 2018, 8, 2986. 10.1038/s41598-018-21396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Ren X.; Luo Y.; Wang L.; Cui G.; Xie F.; Wang H.; Xie Y.; Sun X. An Ultrafine Platinum-Cobalt Alloy Decorated Cobalt Nanowire Array with Superb Activity toward Alkaline Hydrogen Evolution. Nanoscale 2018, 10, 12302–12307. 10.1039/C8NR02071J. [DOI] [PubMed] [Google Scholar]
- Mahmood J.; Li F.; Jung S. M.; Okyay M. S.; Ahmad I.; Kim S. J.; Park N.; Jeong H. Y.; Baek J. B. An Efficient and PH-Universal Ruthenium-Based Catalyst for the Hydrogen Evolution Reaction. Nat. Nanotechnol. 2017, 12, 441–446. 10.1038/nnano.2016.304. [DOI] [PubMed] [Google Scholar]
- Lu B.; Guo L.; Wu F.; Peng Y.; Lu J. E.; Smart T. J.; Wang N.; Finfrock Y. Z.; Morris D.; Zhang P.; Li N.; Gao P.; Ping Y.; Chen S. Ruthenium Atomically Dispersed in Carbon Outperforms Platinum toward Hydrogen Evolution in Alkaline Media. Nat. Commun. 2019, 10, 631. 10.1038/s41467-019-08419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Pei W.; He J.; Liu K.; Qi W.; Gao X.; Zhou S.; Xie H.; Yin K.; Gao Y.; He J.; Zhao J.; Hu J.; Chan T. S.; Li Z.; Zhang G.; Liu M. Hybrids of PtRu Nanoclusters and Black Phosphorus Nanosheets for Highly Efficient Alkaline Hydrogen Evolution Reaction. ACS Catal. 2019, 9, 10870–10875. 10.1021/acscatal.9b03506. [DOI] [Google Scholar]
- Zheng Y.; Jiao Y.; Zhu Y.; Li L. H.; Han Y.; Chen Y.; Jaroniec M.; Qiao S. Z. High Electrocatalytic Hydrogen Evolution Activity of an Anomalous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 16174–16181. 10.1021/jacs.6b11291. [DOI] [PubMed] [Google Scholar]
- Xu C.; Ming M.; Wang Q.; Yang C.; Fan G.; Wang Y.; Gao D.; Bi J.; Zhang Y. Facile Synthesis of Effective Ru Nanoparticles on Carbon by Adsorption-Low Temperature Pyrolysis Strategy for Hydrogen Evolution. J. Mater. Chem. A 2018, 6, 14380–14386. 10.1039/C8TA03572E. [DOI] [Google Scholar]
- Ding J.; Shao Q.; Feng Y.; Huang X. Ruthenium-Nickel Sandwiched Nanoplates for Efficient Water Splitting Electrocatalysis. Nano Energy 2018, 47, 1–7. 10.1016/j.nanoen.2018.02.017. [DOI] [Google Scholar]
- Chen X.; Wan J.; Wang J.; Zhang Q.; Gu L.; Zheng L.; Wang N.; Yu R. Atomically Dispersed Ruthenium on Nickel Hydroxide Ultrathin Nanoribbons for Highly Efficient Hydrogen Evolution Reaction in Alkaline Media. Adv. Mater. 2021, 33, 2104764. 10.1002/adma.202104764. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Zhu Y.; Chen Y.; Lu Y.; Lin Q.; Zhang L.; Tao S.; Zhang X.; Wang H. RuCo Alloy Bimodal Nanoparticles Embedded in N-Doped Carbon: A Superior PH-Universal Electrocatalyst Outperforms Benchmark Pt for the Hydrogen Evolution Reaction. J. Mater. Chem. A 2020, 8, 12810–12820. 10.1039/D0TA04491A. [DOI] [Google Scholar]
- Rao Y.; Wang S.; Zhang R.; Jiang S.; Chen S.; Yu Y.; Bao S.; Xu M.; Yue Q.; Xin H.; Kang Y. Nanoporous V-Doped Ni5P4Microsphere: A Highly Efficient Electrocatalyst for Hydrogen Evolution Reaction at All PH. ACS Appl. Mater. 2020, 12, 37092–37099. 10.1021/acsami.0c08202. [DOI] [PubMed] [Google Scholar]
- Mao J.; He C. T.; Pei J.; Chen W.; He D.; He Y.; Zhuang Z.; Chen C.; Peng Q.; Wang D.; Li Y. Accelerating Water Dissociation Kinetics by Isolating Cobalt Atoms into Ruthenium Lattice. Nat. Commun. 2018, 9, 4958. 10.1038/s41467-018-07288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colli A. N.; Girault H. H.; Battistel A. Non-Precious Electrodes for Practical Alkaline Water Electrolysis. Materials 2019, 12, 1336. 10.3390/ma12081336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoney G. G. The Tension of Metallic Films Deposited by Electrolysis. Proc. R. Soc. London, Ser. A 1909, 82, 172–175. [Google Scholar]
- Quaino P.; Juarez F.; Santos E.; Schmickler W. Volcano Plots in Hydrogen Electrocatalysis-Uses and Abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. 10.3762/bjnano.5.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstajić N.; Popović M.; Grgur B.; Vojnović M.; Šepa D. On the Kinetics of the Hydrogen Evolution Reaction on Nickel in Alkaline Solution - Part I. Mechanism. J. Electroanal. Chem. 2001, 512, 16–26. 10.1016/S0022-0728(01)00590-3. [DOI] [Google Scholar]
- Lasia A.; Rami A. Kinetics of Hydrogen Evolution on Nickel Electrodes. J. Electroanal. Chem. 1990, 294, 123–141. 10.1016/0022-0728(90)87140-F. [DOI] [Google Scholar]
- Men Y.; Li P.; Zhou J.; Chen S.; Luo W. Trends in Alkaline Hydrogen Evolution Activity on Cobalt Phosphide Electrocatalysts Doped with Transition Metals. Cell Rep. 2020, 1, 100136. 10.1016/j.xcrp.2020.100136. [DOI] [Google Scholar]
- Delvaux A.; Lumbeeck G.; Idrissi H.; Proost J. Effect of Microstructure and Internal Stress on Hydrogen Absorption into Ni Thin Film Electrodes during Alkaline Water Electrolysis. Electrochim. Acta 2020, 340, 135970. 10.1016/j.electacta.2020.135970. [DOI] [Google Scholar]
- Wang T.; Wang M.; Yang H.; Xu M.; Zuo C.; Feng K.; Xie M.; Deng J.; Zhong J.; Zhou W.; Cheng T.; Li Y. Weakening Hydrogen Adsorption on Nickel via Interstitial Nitrogen Doping Promotes Bifunctional Hydrogen Electrocatalysis in Alkaline Solution. Environ. Sci. Technol. 2019, 12, 3522–3529. 10.1039/C9EE01743G. [DOI] [Google Scholar]
- Takada N.; Uchiyama T.; Uchimoto Y.; Yamagishi H.; Ohta T.; Orikasa Y. Soft X-Ray Absorption Spectroscopic Study on Nickel Electrode for Alkaline Water Electrolysis. Mem. SR Cent. Ritsumeikan Univ. 2019, 21, 3–5. 10.34521/srmemoirs.21.0_3. [DOI] [Google Scholar]
- Zhang J.; Wang T.; Liu P.; Liao Z.; Liu S.; Zhuang X.; Chen M.; Zschech E.; Feng X. Efficient Hydrogen Production on MoNi4 Electrocatalysts with Fast Water Dissociation Kinetics. Nat. Commun. 2017, 8, 15437. 10.1038/ncomms15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Z.; Liu W.; Yin L. C.; Fang R.; Li M.; Altman E. I.; Fan Q.; Li F.; Cheng H. M.; Wang H. Metal/Oxide Interface Nanostructures Generated by Surface Segregation for Electrocatalysis. Nano Lett. 2015, 15, 7704–7710. 10.1021/acs.nanolett.5b03709. [DOI] [PubMed] [Google Scholar]
- Cossar E.; Oyarce Barnett A.; Seland F.; Baranova E. A. The Performance of Nickel and Nickel-Iron Catalysts Evaluated as Anodes in Anion Exchange Membrane Water Electrolysis. Catalysts 2019, 9, 814. 10.3390/catal9100814. [DOI] [Google Scholar]
- Song X. Z.; Zhu W. Y.; Wang X. F.; Tan Z. Recent Advances of CeO2-Based Electrocatalysts for Oxygen and Hydrogen Evolution as Well as Nitrogen Reduction. ChemElectroChem. 2021, 8, 996–1020. 10.1002/celc.202001614. [DOI] [Google Scholar]
- Tian L.; Liu H.; Zhang B.; Liu Y.; Lv S.; Pang L.; Li J. Ni and CeO2 Nanoparticles Anchored on Cicada-Wing-like Nitrogen-Doped Porous Carbon as Bifunctional Catalysts for Water Splitting. ACS Appl. Nano Mater. 2022, 5, 1252–1262. 10.1021/acsanm.1c03850. [DOI] [Google Scholar]
- Zhang Y.; Liao W.; Zhang G. A General Strategy for Constructing Transition Metal Oxide/CeO2 Heterostructure with Oxygen Vacancies toward Hydrogen Evolution Reaction and Oxygen Evolution Reaction. J. Power Sources 2021, 512, 230514. 10.1016/j.jpowsour.2021.230514. [DOI] [Google Scholar]
- Barati Darband G.; Aliofkhazraei M.; Rouhaghdam A. S. Facile Electrodeposition of Ternary Ni-Fe-Co Alloy Nanostructure as a Binder Free, Cost-Effective and Durable Electrocatalyst for High-Performance Overall Water Splitting. J. Colloid Interface Sci. 2019, 547, 407–420. 10.1016/j.jcis.2019.03.098. [DOI] [PubMed] [Google Scholar]
- Trotochaud L.; Young S. L.; Ranney J. K.; Boettcher S. W. Nickel-Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. JACS 2014, 136, 6744–6753. 10.1021/ja502379c. [DOI] [PubMed] [Google Scholar]
- McCrory C. C. L.; Jung S.; Ferrer I. M.; Chatman S. M.; Peters J. C.; Jaramillo T. F. Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. 10.1021/ja510442p. [DOI] [PubMed] [Google Scholar]
- Gong M.; Dai H. A Mini Review of NiFe-Based Materials as Highly Active Oxygen Evolution Reaction Electrocatalysts. Nano Res. 2015, 8, 23–39. 10.1007/s12274-014-0591-z. [DOI] [Google Scholar]
- Solmaz R.; Kardaş G. Electrochemical Deposition and Characterization of NiFe Coatings as Electrocatalytic Materials for Alkaline Water Electrolysis. Electrochim. Acta 2009, 54, 3726–3734. 10.1016/j.electacta.2009.01.064. [DOI] [Google Scholar]
- Los P.; Lasia A. Electrocatalytic Properties of Amorphous Nickel Boride Electrodes for Hydrogen Evolution Reaction in Alkaline Solution. J. Electroanal. Chem. 1992, 333, 115–125. 10.1016/0022-0728(92)80385-H. [DOI] [Google Scholar]
- Badrnezhad R.; Nasri F.; Pourfarzad H.; Jafari S. K. Effect of Iron on Ni-Mo-Fe Composite as a Low-Cost Bifunctional Electrocatalyst for Overall Water Splitting. Int. J. Hydrogen Energy 2021, 46, 3821–3832. 10.1016/j.ijhydene.2020.10.174. [DOI] [Google Scholar]
- Csernica P. M.; McKone J. R.; Mulzer C. R.; Dichtel W. R.; Abruña H. D.; DiSalvo F. J. Electrochemical Hydrogen Evolution at Ordered Mo7Ni7. ACS Catal. 2017, 7, 3375–3383. 10.1021/acscatal.7b00344. [DOI] [Google Scholar]
- Conway B. E.; Bai L. H2 Evolution Kinetics at High Activity Ni-Mo-Cd Electrocoated Cathodes and Its Relation to Potential Dependence of Sorption of H. Int. J. Hydrogen Energy 1986, 11, 533–540. 10.1016/0360-3199(86)90020-0. [DOI] [Google Scholar]
- Jakšic J. M.; Vojnović M. V.; Krstajić N. V. Kinetic Analysis of Hydrogen Evolution at Ni-Mo Alloy Electrodes. Electrochim. Acta 2000, 45, 4151–4158. 10.1016/S0013-4686(00)00549-1. [DOI] [Google Scholar]
- Mckone J. R.; Sadtler B. F.; Werlang C. A.; Lewis N. S.; Gray H. B. Ni-Mo Nanopowders for Efficient Electrochemical Hydrogen Evolution. ACS Catal. 2013, 3, 166–169. 10.1021/cs300691m. [DOI] [Google Scholar]
- Rößner L.; Schwarz H.; Veremchuk I.; Zerdoumi R.; Seyller Th.; Armbrüster M. Durability of Mo-Ni Intermetallic Compounds in the Hydrogen Evolution Reaction. ChemRxiv 2020, 1–11. 10.26434/chemrxiv.12639032.v1. [DOI] [PubMed] [Google Scholar]
- Faid A. Y.; Oyarce Barnett A.; Seland F.; Sunde S. Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis. Catalysts 2018, 8, 614. 10.3390/catal8120614. [DOI] [Google Scholar]
- Zhang J.; Wang T.; Liu P.; Liao Z.; Liu S.; Zhuang X.; Chen M.; Zschech E.; Feng X. Efficient Hydrogen Production on MoNi4 Electrocatalysts with Fast Water Dissociation Kinetics. Nat. Commun. 2017, 8, 15437. 10.1038/ncomms15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-Y.; Zhang Y.; Zhang X.; Tang T.; Luo H.; Niu S.; Dai Z.-H.; Wan L.-J.; Hu J.-S. Self-Templated Fabrication of MoNi4/MoO3-x Nanorod Arrays with Dual Active Components for Highly Efficient Hydrogen Evolution. Adv. Mater. 2017, 29, 1703311. 10.1002/adma.201703311. [DOI] [PubMed] [Google Scholar]
- Wu H. B.; Xia B. Y.; Yu L.; Yu X. Y.; Lou X. W. Porous Molybdenum Carbide Nano-Octahedrons Synthesized via Confined Carburization in Metal-Organic Frameworks for Efficient Hydrogen Production. Nat. Commun. 2015, 6, 6512. 10.1038/ncomms7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Yu G.; Li G. D.; Sun Y.; Asefa T.; Chen W.; Zou X. Coupling Mo2C with Nitrogen-Rich Nanocarbon Leads to Efficient Hydrogen-Evolution Electrocatalytic Sites. Angew. Chem., Int. Ed. 2015, 54, 10752–10757. 10.1002/anie.201504376. [DOI] [PubMed] [Google Scholar]
- Lu C.; Tranca D.; Zhang J.; Rodríguez Hernández F.; Su Y.; Zhuang X.; Zhang F.; Seifert G.; Feng X. Molybdenum Carbide-Embedded Nitrogen-Doped Porous Carbon Nanosheets as Electrocatalysts for Water Splitting in Alkaline Media. ACS Nano 2017, 11, 3933–3942. 10.1021/acsnano.7b00365. [DOI] [PubMed] [Google Scholar]
- Anjum M. A. R.; Lee M. H.; Lee J. S. BCN Network-Encapsulated Multiple Phases of Molybdenum Carbide for Efficient Hydrogen Evolution Reactions in Acidic and Alkaline Media. J. Mater. Chem. A 2017, 5, 13122–13129. 10.1039/C7TA03407E. [DOI] [Google Scholar]
- Tang C.; Hu Q.; Li F.; He C.; Chai X.; Zhu C.; Liu J.; Zhang Q.; Zhu B.; Fan L. Coupled Molybdenum Carbide and Nitride on Carbon Nanosheets: An Efficient and Durable Hydrogen Evolution Electrocatalyst in Both Acid and Alkaline Media. Electrochim. Acta 2018, 280, 323–331. 10.1016/j.electacta.2018.05.129. [DOI] [Google Scholar]
- Wang L.; Li Y.; Yin X.; Wang Y.; Lu L.; Song A.; Xia M.; Li Z.; Qin X.; Shao G. Comparison of Three Nickel-Based Carbon Composite Catalysts for Hydrogen Evolution Reaction in Alkaline Solution. Int. J. Hydrog. Energy 2017, 42, 22655–22662. 10.1016/j.ijhydene.2017.06.215. [DOI] [Google Scholar]
- Elias L.; Hegde A. C. Electrolytic Synthesis of Ni-W-MWCNT Composite Coating for Alkaline Hydrogen Evolution Reaction. J. Mater. Eng. Perform. 2018, 27, 1033–1039. 10.1007/s11665-018-3134-z. [DOI] [Google Scholar]
- Huang Y.; Hu L.; Liu R.; Hu Y.; Xiong T.; Qiu W.; Balogun M. S.; Pan A.; Tong Y. Nitrogen Treatment Generates Tunable Nanohybridization of Ni5P4 Nanosheets with Nickel Hydr(Oxy)Oxides for Efficient Hydrogen Production in Alkaline, Seawater and Acidic Media. Appl. Catal., B 2019, 251, 181–194. 10.1016/j.apcatb.2019.03.037. [DOI] [Google Scholar]
- Lv Z.; Tahir M.; Lang X.; Yuan G.; Pan L.; Zhang X.; Zou J. J. Well-Dispersed Molybdenum Nitrides on a Nitrogen-Doped Carbon Matrix for Highly Efficient Hydrogen Evolution in Alkaline Media. J. Mater. Chem. A 2017, 5, 20932–20937. 10.1039/C7TA06981B. [DOI] [Google Scholar]
- Vrubel H.; Hu X. Molybdenum Boride and Carbide Catalyze Hydrogen Evolution in Both Acidic and Basic Solutions. Angew. Chem., Int. Ed. 2012, 51, 12703–12706. 10.1002/anie.201207111. [DOI] [PubMed] [Google Scholar]
- Kibsgaard J.; Tsai C.; Chan K.; Benck J. D.; Nørskov J. K.; Abild-Pedersen F.; Jaramillo T. F. Designing an Improved Transition Metal Phosphide Catalyst for Hydrogen Evolution Using Experimental and Theoretical Trends. Environ. Sci. Technol. 2015, 8, 3022–3029. 10.1039/C5EE02179K. [DOI] [Google Scholar]
- Kibsgaard J.; Jaramillo T. F.; Besenbacher F. Building an Appropriate Active-Site Motif into a Hydrogen-Evolution Catalyst with Thiomolybdate [Mo3S13]2- Clusters. Nat. Chem. 2014, 6, 248–253. 10.1038/nchem.1853. [DOI] [PubMed] [Google Scholar]
- Jaramillo T. F.; Jørgensen K. P.; Bonde J.; Nielsen J. H.; Horch S.; Chorkendorff I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. 10.1126/science.1141483. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Moses P. G.; Bonde J.; Jørgensen K. P.; Nielsen J. H.; Horch S.; Chorkendorff I.; Nørskov J. K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. 10.1021/ja0504690. [DOI] [PubMed] [Google Scholar]
- Popczun E. J.; McKone J. R.; Read C. G.; Biacchi A. J.; Wiltrout A. M.; Lewis N. S.; Schaak R. E. Nanostructured Nickel Phosphide as an Electrocatalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. 10.1021/ja403440e. [DOI] [PubMed] [Google Scholar]
- Laursen A. B.; Wexler R. B.; Whitaker M. J.; Izett E. J.; Calvinho K. U. D.; Hwang S.; Rucker R.; Wang H.; Li J.; Garfunkel E.; Greenblatt M.; Rappe A. M.; Dismukes G. C. Climbing the Volcano of Electrocatalytic Activity While Avoiding Catalyst Corrosion: Ni3P, a Hydrogen Evolution Electrocatalyst Stable in Both Acid and Alkali. ACS Catal. 2018, 8, 4408–4419. 10.1021/acscatal.7b04466. [DOI] [Google Scholar]
- Jiang N.; You B.; Sheng M.; Sun Y. Electrodeposited Cobalt-Phosphorous-Derived Films as Competent Bifunctional Catalysts for Overall Water Splitting. Angew. Chem., Int. Ed. 2015, 54, 6251–6254. 10.1002/anie.201501616. [DOI] [PubMed] [Google Scholar]
- Tian J.; Liu Q.; Asiri A. M.; Sun X. Self-Supported Nanoporous Cobalt Phosphide Nanowire Arrays : An. J. Am. Chem. Soc. 2014, 136, 7587–7590. 10.1021/ja503372r. [DOI] [PubMed] [Google Scholar]
- Menezes P. W.; Indra A.; Das C.; Walter C.; Göbel C.; Gutkin V.; Schmeißer D.; Driess M. Uncovering the Nature of Active Species of Nickel Phosphide Catalysts in High-Performance Electrochemical Overall Water Splitting. ACS Catal. 2017, 7, 103–109. 10.1021/acscatal.6b02666. [DOI] [Google Scholar]
- Rossmeisl J.; Qu Z. W.; Zhu H.; Kroes G. J.; Nørskov J. K. Electrolysis of Water on Oxide Surfaces. J. Electroanal. Chem. 2007, 607, 83–89. 10.1016/j.jelechem.2006.11.008. [DOI] [Google Scholar]
- Frydendal R.; Busch M.; Halck N. B.; Paoli E. A.; Krtil P.; Chorkendorff I.; Rossmeisl J. Enhancing Activity for the Oxygen Evolution Reaction: The Beneficial Interaction of Gold with Manganese and Cobalt Oxides. ChemCatChem. 2015, 7, 149–154. 10.1002/cctc.201402756. [DOI] [Google Scholar]
- Pourbaix M.Atlas of Chemical and Electrochemical Equilibria in the Presence of a Gaseous Phase; National Association of Corrosion Engineers: Houston, TX, 1974. [Google Scholar]
- Stoerzinger K. A.; Qiao L.; Biegalski M. D.; Shao-Horn Y. Orientation-Dependent Oxygen Evolution Activities of Rutile IrO2 and RuO2. J. Phys. Chem. Lett. 2014, 5, 1636–1641. 10.1021/jz500610u. [DOI] [PubMed] [Google Scholar]
- Bock C.; Birss V. I. Anion and Water Involvement in Hydrous Ir Oxide Redox Reactions in Acidic Solutions. J. Electroanal. Chem. 1999, 475, 20–27. 10.1016/S0022-0728(99)00330-7. [DOI] [Google Scholar]
- Tan X.; Shen J.; Semagina N.; Secanell M. Decoupling Structure-Sensitive Deactivation Mechanisms of Ir/IrOx Electrocatalysts toward Oxygen Evolution Reaction. J. Catal. 2019, 371, 57–70. 10.1016/j.jcat.2019.01.018. [DOI] [Google Scholar]
- Xu D.; Stevens M. B.; Cosby M. R.; Oener S. Z.; Smith A. M.; Enman L. J.; Ayers K. E.; Capuano C. B.; Renner J. N.; Danilovic N.; Li Y.; Wang H.; Zhang Q.; Boettcher S. W. Earth-Abundant Oxygen Electrocatalysts for Alkaline Anion-Exchange-Membrane Water Electrolysis: Effects of Catalyst Conductivity and Comparison with Performance in Three-Electrode Cells. ACS Catal. 2019, 9, 7–15. 10.1021/acscatal.8b04001. [DOI] [Google Scholar]
- Plata-Torres M.; Torres-Huerta A. M.; Domínguez-Crespo M. A.; Arce-Estrada E. M.; Ramírez-Rodríguez C. Electrochemical Performance of Crystalline Ni-Co-Mo-Fe Electrodes Obtained by Mechanical Alloying on the Oxygen Evolution Reaction. Int. J. Hydrog. Energy 2007, 32, 4142–4152. 10.1016/j.ijhydene.2007.05.023. [DOI] [Google Scholar]
- Kim K. H.; Zheng J. Y.; Shin W.; Kang Y. S. Preparation of Dendritic NiFe Films by Electrodeposition for Oxygen Evolution. RSC Adv. 2012, 2, 4759–4767. 10.1039/c2ra20241g. [DOI] [Google Scholar]
- Potvin E.; Brossard L. Electrocatalytic Activity of Ni-Fe Anodes for Alkaline Water Electrolysis. Mater. Chem. Phys. 1992, 31, 311–318. 10.1016/0254-0584(92)90192-B. [DOI] [Google Scholar]
- Gerken J. B.; Shaner S. E.; Massé R. C.; Porubsky N. J.; Stahl S. S. A Survey of Diverse Earth Abundant Oxygen Evolution Electrocatalysts Showing Enhanced Activity from Ni-Fe Oxides Containing a Third Metal. Environ. Sci. Technol. 2014, 7, 2376–2382. 10.1039/C4EE00436A. [DOI] [Google Scholar]
- Chen J. Y. C.; Miller J. T.; Gerken J. B.; Stahl S. S. Inverse Spinel NiFeAlO4 as a Highly Active Oxygen Evolution Electrocatalyst: Promotion of Activity by a Redox-Inert Metal Ion. Environ. Sci. Technol. 2014, 7, 1382–1386. 10.1039/c3ee43811b. [DOI] [Google Scholar]
- Roy C.; Sebok B.; Scott S. B.; Fiordaliso E. M.; Sørensen J. E.; Bodin A.; Trimarco D. B.; Damsgaard C. D.; Vesborg P. C. K.; Hansen O.; Stephens I. E. L.; Kibsgaard J.; Chorkendorff I. Impact of Nanoparticle Size and Lattice Oxygen on Water Oxidation on NiFeOxHy. Nat. Catal. 2018, 1, 820–829. 10.1038/s41929-018-0162-x. [DOI] [Google Scholar]
- Candelaria S. L.; Bedford N. M.; Woehl T. J.; Rentz N. S.; Showalter A. R.; Pylypenko S.; Bunker B. A.; Lee S.; Reinhart B.; Ren Y.; Ertem S. P.; Coughlin E. B.; Sather N. A.; Horan J. L.; Herring A. M.; Greenlee L. F. Multi-Component Fe-Ni Hydroxide Nanocatalyst for Oxygen Evolution and Methanol Oxidation Reactions under Alkaline Conditions. ACS Catal. 2017, 7, 365–379. 10.1021/acscatal.6b02552. [DOI] [Google Scholar]
- Smith A. M.; Trotochaud L.; Burke M. S.; Boettcher S. W. Contributions to Activity Enhancement via Fe Incorporation in Ni-(Oxy)Hydroxide/Borate Catalysts for near-Neutral pH Oxygen Evolution. ChemCommun. 2015, 51, 5261–5263. 10.1039/C4CC08670H. [DOI] [PubMed] [Google Scholar]
- Hall D. S.; Lockwood D. J.; Bock C.; MacDougall B. R. Nickel Hydroxides and Related Materials: A Review of Their Structures, Synthesis and Properties. Proc. Math. Phys. Eng. Sci. 2015, 471, 20140792. 10.1098/rspa.2014.0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan D. A. The Catalysis of the Oxygen Evolution Reaction by Iron Impurities in Thin Film Nickel Oxide Electrodes. J. Electrochem. Soc. 1987, 134, 377–384. 10.1149/1.2100463. [DOI] [Google Scholar]
- Burke M. S.; Zou S.; Enman L. J.; Kellon J. E.; Gabor C. A.; Pledger E.; Boettcher S. W. Revised Oxygen Evolution Reaction Activity Trends for First-Row Transition-Metal (Oxy)Hydroxides in Alkaline Media. J. Phys. Chem. Lett. 2015, 6, 3737–3742. 10.1021/acs.jpclett.5b01650. [DOI] [PubMed] [Google Scholar]
- Spanos I.; Tesch M. F.; Yu M.; Tüysüz H.; Zhang J.; Feng X.; Müllen K.; Schlögl R.; Mechler A. K. Facile Protocol for Alkaline Electrolyte Purification and Its Influence on a Ni-Co Oxide Catalyst for the Oxygen Evolution Reaction. ACS Catal. 2019, 9, 8165–8170. 10.1021/acscatal.9b01940. [DOI] [Google Scholar]
- Burke M. S.; Zou S.; Enman L. J.; Kellon J. E.; Gabor C. A.; Pledger E.; Boettcher S. W. Revised Oxygen Evolution Reaction Activity Trends for First-Row Transition-Metal (Oxy)Hydroxides in Alkaline Media. J. Phys. Chem. Lett. 2015, 6, 3737–3742. 10.1021/acs.jpclett.5b01650. [DOI] [PubMed] [Google Scholar]
- Burke M. S.; Kast M. G.; Trotochaud L.; Smith A. M.; Boettcher S. W. Cobalt-Iron (Oxy)Hydroxide Oxygen Evolution Electrocatalysts: The Role of Structure and Composition on Activity, Stability, and Mechanism. J. Am. Chem. Soc. 2015, 137, 3638–3648. 10.1021/jacs.5b00281. [DOI] [PubMed] [Google Scholar]
- Dionigi F.; Strasser P. NiFe-Based (Oxy)Hydroxide Catalysts for Oxygen Evolution Reaction in Non-Acidic Electrolytes. Adv. Energy Mater. 2016, 6, 1600621. 10.1002/aenm.201600621. [DOI] [Google Scholar]
- Zhou Y.; López N. The Role of Fe Species on NiOOH in Oxygen Evolution Reactions. ACS Catal. 2020, 10, 6254–6261. 10.1021/acscatal.0c00304. [DOI] [Google Scholar]
- Lee S.; Bai L.; Hu X. Deciphering Iron-Dependent Activity in Oxygen Evolution Catalyzed by Nickel-Iron Layered Double Hydroxide. Angew. Chem., Int. Ed. 2020, 59, 8072–8077. 10.1002/anie.201915803. [DOI] [PubMed] [Google Scholar]
- Stevens M. B.; Trang C. D. M.; Enman L. J.; Deng J.; Boettcher S. W. Reactive Fe-Sites in Ni/Fe (Oxy)Hydroxide Are Responsible for Exceptional Oxygen Electrocatalysis Activity. J. Am. Chem. Soc. 2017, 139, 11361–11364. 10.1021/jacs.7b07117. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Curto A.; Rodríguez-Fernández J.; Wang Z.; Parikh A.; Fester J.; Dong M.; Vojvodic A.; Lauritsen J. v. The Effect of Fe Dopant Location in Co(Fe)OOHx Nanoparticles for the Oxygen Evolution Reaction. ACS Nano 2021, 15, 18226–18236. 10.1021/acsnano.1c07219. [DOI] [PubMed] [Google Scholar]
- Louie M. W.; Bell A. T. An Investigation of Thin-Film Ni-Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen. J. Am. Chem. Soc. 2013, 135, 12329–12337. 10.1021/ja405351s. [DOI] [PubMed] [Google Scholar]
- Blakemore J. D.; Gray H. B.; Winkler J. R.; Müller A. M. Co3O4 Nanoparticle Water-Oxidation Catalysts Made by Pulsed-Laser Ablation in Liquids. ACS Catal. 2013, 3, 2497–2500. 10.1021/cs400639b. [DOI] [Google Scholar]
- Ye S. H.; Shi Z. X.; Feng J. X.; Tong Y. X.; Li G. R. Activating CoOOH Porous Nanosheet Arrays by Partial Iron Substitution for Efficient Oxygen Evolution Reaction. Angew. Chem., Int. Ed. 2018, 57, 2672–2676. 10.1002/anie.201712549. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Nellist M. R.; Enman L. J.; Xiang J.; Boettcher S. W. Modes of Fe Incorporation in Co-Fe (Oxy)Hydroxide Oxygen Evolution Electrocatalysts. ChemSusChem 2019, 12, 2015–2021. 10.1002/cssc.201801975. [DOI] [PubMed] [Google Scholar]
- Stevens M. B.; Enman L. J.; Korkus E. H.; Zaffran J.; Trang C. D. M.; Asbury J.; Kast M. G.; Toroker M. C.; Boettcher S. W. Ternary Ni-Co-Fe Oxyhydroxide Oxygen Evolution Catalysts: Intrinsic Activity Trends, Electrical Conductivity, and Electronic Band Structure. Nano Res. 2019, 12, 2288–2295. 10.1007/s12274-019-2391-y. [DOI] [Google Scholar]
- Long X.; Xiao S.; Wang Z.; Zheng X.; Yang S. Co Intake Mediated Formation of Ultrathin Nanosheets of Transition Metal LDH - An Advanced Electrocatalyst for Oxygen Evolution Reaction. ChemCommun. 2015, 51, 1120–1123. 10.1039/C4CC08856E. [DOI] [PubMed] [Google Scholar]
- Chauhan M.; Reddy K. P.; Gopinath C. S.; Deka S. Copper Cobalt Sulfide Nanosheets Realizing a Promising Electrocatalytic Oxygen Evolution Reaction. ACS Catal. 2017, 7, 5871–5879. 10.1021/acscatal.7b01831. [DOI] [Google Scholar]
- Vincent I.; Kruger A.; Bessarabov D. Development of Efficient Membrane Electrode Assembly for Low Cost Hydrogen Production by Anion Exchange Membrane Electrolysis. Int. J. Hydrog. Energy 2017, 42, 10752–10761. 10.1016/j.ijhydene.2017.03.069. [DOI] [Google Scholar]
- Cao Y. C.; Wu X.; Scott K. A Quaternary Ammonium Grafted Poly Vinyl Benzyl Chloride Membrane for Alkaline Anion Exchange Membrane Water Electrolysers with No-Noble-Metal Catalysts. Int. J. Hydrog. Energy 2012, 37, 9524–9528. 10.1016/j.ijhydene.2012.03.116. [DOI] [Google Scholar]
- Wu X.; Scott K. A Polymethacrylate-Based Quaternary Ammonium OH - Ionomer Binder for Non-Precious Metal Alkaline Anion Exchange Membrane Water Electrolysers. J. Power Sources 2012, 214, 124–129. 10.1016/j.jpowsour.2012.03.069. [DOI] [Google Scholar]
- Wei Y.; Ren X.; Ma H.; Sun X.; Zhang Y.; Kuang X.; Yan T.; Ju H.; Wu D.; Wei Q. CoC2O4·2H2O Derived Co3O4 Nanorods Array: A High-Efficiency 1D Electrocatalyst for Alkaline Oxygen Evolution Reaction. ChemCommun. 2018, 54, 1533–1536. 10.1039/C7CC08423D. [DOI] [PubMed] [Google Scholar]
- Jang M. J.; Yang J.; Lee J.; Park Y. S.; Jeong J.; Park S. M.; Jeong J. Y.; Yin Y.; Seo M. H.; Choi S. M.; Lee K. H. Superior Performance and Stability of Anion Exchange Membrane Water Electrolysis: PH-Controlled Copper Cobalt Oxide Nanoparticles for the Oxygen Evolution Reaction. J. Mater. Chem. A 2020, 8, 4290–4299. 10.1039/C9TA13137J. [DOI] [Google Scholar]
- Karmakar A.; Srivastava S. K. Interconnected Copper Cobaltite Nanochains as Efficient Electrocatalysts for Water Oxidation in Alkaline Medium. ACS Appl. Mater. 2017, 9, 22378–22387. 10.1021/acsami.7b03029. [DOI] [PubMed] [Google Scholar]
- Park Y. S.; Jang M. J.; Jeong J.; Park S. M.; Wang X.; Seo M. H.; Choi S. M.; Yang J. Hierarchical Chestnut-Burr like Structure of Copper Cobalt Oxide Electrocatalyst Directly Grown on Ni Foam for Anion Exchange Membrane Water Electrolysis. ACS Sustain. Chem. Eng. 2020, 8, 2344–2349. 10.1021/acssuschemeng.9b06767. [DOI] [Google Scholar]
- Grimaud A.; May K. J.; Carlton C. E.; Lee Y. L.; Risch M.; Hong W. T.; Zhou J.; Shao-Horn Y. Double Perovskites as a Family of Highly Active Catalysts for Oxygen Evolution in Alkaline Solution. Nat. Commun. 2013, 4, 2439. 10.1038/ncomms3439. [DOI] [PubMed] [Google Scholar]
- Suntivich J.; May K. J.; Gasteiger H. A.; Goodenough J. B.; Shao-horn Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. 10.1126/science.1212858. [DOI] [PubMed] [Google Scholar]
- May K. J.; Carlton C. E.; Stoerzinger K. A.; Risch M.; Suntivich J.; Lee Y. L.; Grimaud A.; Shao-Horn Y. Influence of Oxygen Evolution during Water Oxidation on the Surface of Perovskite Oxide Catalysts. J. Phys. Chem. Lett. 2012, 3, 3264–3270. 10.1021/jz301414z. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Menezes P. W.; Panda C.; Loos S.; Bunschei-Bruns F.; Walter C.; Schwarze M.; Deng X.; Dau H.; Driess M. A Structurally Versatile Nickel Phosphite Acting as a Robust Bifunctional Electrocatalyst for Overall Water Splitting. Environ. Sci. Technol. 2018, 11, 1287–1298. 10.1039/C7EE03619A. [DOI] [Google Scholar]
- Guo Y.; Park T.; Yi J. W.; Henzie J.; Kim J.; Wang Z.; Jiang B.; Bando Y.; Sugahara Y.; Tang J.; Yamauchi Y. Nanoarchitectonics for Transition-Metal-Sulfide-Based Electrocatalysts for Water Splitting. Adv. Mater. 2019, 31, 1807134. 10.1002/adma.201807134. [DOI] [PubMed] [Google Scholar]
- Anantharaj S.; Ede S. R.; Sakthikumar K.; Karthick K.; Mishra S.; Kundu S. Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe, Co, and Ni: a review. ACS Catal. 2016, 6, 8069–8097. 10.1021/acscatal.6b02479. [DOI] [Google Scholar]
- Liu K.; Zhang C.; Sun Y.; Zhang G.; Shen X.; Zou F.; Zhang H.; Wu Z.; Wegener E. C.; Taubert C. J.; Miller J. T.; Peng Z.; Zhu Y. High-Performance Transition Metal Phosphide Alloy Catalyst for Oxygen Evolution Reaction. ACS Nano 2018, 12, 158–167. 10.1021/acsnano.7b04646. [DOI] [PubMed] [Google Scholar]
- Xuan C.; Wang J.; Xia W.; Peng Z.; Wu Z.; Lei W.; Xia K.; Xin H. L.; Wang D. Porous Structured Ni-Fe-P Nanocubes Derived from a Prussian Blue Analogue as an Electrocatalyst for Efficient Overall Water Splitting. ACS Appl. Mater. 2017, 9, 26134–26142. 10.1021/acsami.7b08560. [DOI] [PubMed] [Google Scholar]
- Dutta A.; Pradhan N. Developments of Metal Phosphides as Efficient OER Precatalysts. J. Phys. Chem. Lett. 2017, 8, 144–152. 10.1021/acs.jpclett.6b02249. [DOI] [PubMed] [Google Scholar]
- Jin S. Are Metal Chalcogenides, Nitrides, and Phosphides Oxygen Evolution Catalysts or Bifunctional Catalysts?. ACS Energy Lett. 2017, 2, 1937–1938. 10.1021/acsenergylett.7b00679. [DOI] [Google Scholar]
- Dutta A.; Samantara A. K.; Dutta S. K.; Jena B. K.; Pradhan N. Surface-Oxidized Dicobalt Phosphide Nanoneedles as a Nonprecious, Durable, and Efficient OER Catalyst. ACS Energy Lett. 2016, 1, 169–174. 10.1021/acsenergylett.6b00144. [DOI] [Google Scholar]
- Mabayoje O.; Shoola A.; Wygant B. R.; Mullins C. B. The Role of Anions in Metal Chalcogenide Oxygen Evolution Catalysis: Electrodeposited Thin Films of Nickel Sulfide as “Pre-Catalysts. ACS Energy Lett. 2016, 1, 195–201. 10.1021/acsenergylett.6b00084. [DOI] [Google Scholar]
- Gupta G.; Scott K.; Mamlouk M. Performance of Polyethylene Based Radiation Grafted Anion Exchange Membrane with Polystyrene-b-Poly (Ethylene/Butylene)-b-Polystyrene Based Ionomer Using NiCO2O4 Catalyst for Water Electrolysis. J. Power Sources 2018, 375, 387–396. 10.1016/j.jpowsour.2017.07.026. [DOI] [Google Scholar]
- Doyle A. D.; Bajdich M.; Vojvodic A. Theoretical Insights to Bulk Activity Towards Oxygen Evolution in Oxyhydroxides. Catal. Lett. 2017, 147, 1533–1539. 10.1007/s10562-017-2010-z. [DOI] [Google Scholar]
- Batchellor A. S.; Boettcher S. W. Pulse-Electrodeposited Ni-Fe (Oxy)hydroxide Oxygen Evolution Electrocatalysts with High Geometric and Intrinsic Activities at Large Mass Loadings. ACS Catal. 2015, 5, 6680–6689. 10.1021/acscatal.5b01551. [DOI] [Google Scholar]
- Morales-Guio C. G.; Mayer M. T.; Yella A.; Tilley S. D.; Grätzel M.; Hu X. An Optically Transparent Iron Nickel Oxide Catalyst for Solar Water Splitting. J. Am. Chem. Soc. 2015, 137, 9927–9936. 10.1021/jacs.5b05544. [DOI] [PubMed] [Google Scholar]
- Diaz-Morales O.; Calle-Vallejo F.; De Munck C.; Koper M. T. M. Electrochemical Water Splitting by Gold: Evidence for an Oxide Decomposition Mechanism. Chem. Sci. 2013, 4, 2334–2343. 10.1039/c3sc50301a. [DOI] [Google Scholar]
- Fierro S.; Nagel T.; Baltruschat H.; Comninellis C. Investigation of the Oxygen Evolution Reaction on Ti/IrO2 Electrodes Using Isotope Labelling and on-Line Mass Spectrometry. Electrochem. Commun. 2007, 9, 1969–1974. 10.1016/j.elecom.2007.05.008. [DOI] [Google Scholar]
- Amin H. M. A.; Baltruschat H. How Many Surface Atoms in Co3O4Take Part in Oxygen Evolution? Isotope Labeling Together with Differential Electrochemical Mass Spectrometry. Phys. Chem. Chem. Phys. 2017, 19, 25527–25536. 10.1039/C7CP03914J. [DOI] [PubMed] [Google Scholar]
- Macounova K.; Makarova M.; Krtil P. Oxygen Evolution on Nanocrystalline RuO2 and Ru0.9Ni0.1O2-δ Electrodes - DEMS Approach to Reaction Mechanism Determination. Electrochem. Commun. 2009, 11, 1865–1868. 10.1016/j.elecom.2009.08.004. [DOI] [Google Scholar]
- Wohlfahrt-Mehrens M.; Heitbaum J. Oxygen Evolution on Ru and RuO2 Electrodes Studied Using Isotope Labelling and On-Line Mass Spectrometry. J. Electroanal. Chem. 1987, 237, 251–260. 10.1016/0022-0728(87)85237-3. [DOI] [Google Scholar]
- Grimaud A.; Diaz-Morales O.; Han B.; Hong W. T.; Lee Y. L.; Giordano L.; Stoerzinger K. A.; Koper M. T. M.; Shao-Horn Y. Activating Lattice Oxygen Redox Reactions in Metal Oxides to Catalyse Oxygen Evolution. Nat. Chem. 2017, 9, 457–465. 10.1038/nchem.2695. [DOI] [PubMed] [Google Scholar]
- Stoerzinger K. A.; Diaz-Morales O.; Kolb M.; Rao R. R.; Frydendal R.; Qiao L.; Wang X. R.; Halck N. B.; Rossmeisl J.; Hansen H. A.; et al. Orientation-Dependent Oxygen Evolution on RuO2 without Lattice Exchange. ACS Energy Lett. 2017, 2, 876–881. 10.1021/acsenergylett.7b00135. [DOI] [Google Scholar]
- Lee S.; Banjac K.; Lingenfelder M.; Hu X. Oxygen Isotope Labeling Experiments Reveal Different Reaction Sites for the Oxygen Evolution Reaction on Nickel and Nickel Iron Oxides. Angew. Chem., Int. Ed. 2019, 58, 10295–10299. 10.1002/anie.201903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasian O.; Geiger S.; Mayrhofer K. J. J.; Cherevko S. Electrochemical On-Line ICP-MS in Electrocatalysis Research. Chem. Rec. 2019, 19, 2130–2142. 10.1002/tcr.201800162. [DOI] [PubMed] [Google Scholar]
- Frydendal R.; Paoli E. A.; Knudsen B. P.; Wickman B.; Malacrida P.; Stephens I. E. L.; Chorkendorff I. Benchmarking the Stability of Oxygen Evolution Reaction Catalysts: The Importance of Monitoring Mass Losses. ChemElectroChem. 2014, 1, 2075–2081. 10.1002/celc.201402262. [DOI] [Google Scholar]
- Moysiadou A.; Hu X. Stability Profiles of Transition Metal Oxides in the Oxygen Evolution Reaction in Alkaline Medium. J. Mater. Chem. A 2019, 7, 25865–25877. 10.1039/C9TA10308B. [DOI] [Google Scholar]
- Cherevko S.; Zeradjanin A. R.; Topalov A. A.; Kulyk N.; Katsounaros I.; Mayrhofer K. J. J. Dissolution of Noble Metals during Oxygen Evolution in Acidic Media. ChemCatChem. 2014, 6, 2219–2223. 10.1002/cctc.201402194. [DOI] [Google Scholar]
- Zeradjanin A. R.; Masa J.; Spanos I.; Schlögl R. Activity and Stability of Oxides During Oxygen Evolution Reaction---From Mechanistic Controversies Toward Relevant Electrocatalytic Descriptors. Front. Energy Res. 2021, 8, 613092. 10.3389/fenrg.2020.613092. [DOI] [Google Scholar]
- Schalenbach M.; Kasian O.; Ledendecker M.; Speck F. D.; Mingers A. M.; Mayrhofer K. J. J.; Cherevko S. The Electrochemical Dissolution of Noble Metals in Alkaline Media. Electrocatalysis 2018, 9, 153–161. 10.1007/s12678-017-0438-y. [DOI] [Google Scholar]
- Zhang Y.; Gao L.; Hensen E. J. M.; Hofmann J. P. Evaluating the Stability of Co2P Electrocatalysts in the Hydrogen Evolution Reaction for Both Acidic and Alkaline Electrolytes. ACS Energy Lett. 2018, 3, 1360–1365. 10.1021/acsenergylett.8b00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou T.; Wang S.; Li Y. Perspective on High-Rate Alkaline Water Splitting. ACS Mater. 2021, 3, 224–234. 10.1021/acsmaterialslett.0c00536. [DOI] [Google Scholar]
- Speck F. D.; Zagalskaya A.; Alexandrov V.; Cherevko S. Periodicity in the Electrochemical Dissolution of Transition Metals. Angew. Chem. 2021, 60, 13343–133449. 10.1002/anie.202100337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy C.; Rao R. R.; Stoerzinger K. A.; Hwang J.; Rossmeisl J.; Chorkendorff I.; Shao-Horn Y.; Stephens I. E. L. Trends in Activity and Dissolution on RuO2 under Oxygen Evolution Conditions: Particles versus Well-Defined Extended Surfaces. ACS Energy Lett. 2018, 3, 2045–2051. 10.1021/acsenergylett.8b01178. [DOI] [Google Scholar]
- Ehelebe K.; Escalera-López D.; Cherevko S. Limitations of Aqueous Model Systems in the Stability Assessment of Electrocatalysts for Oxygen Reactions in Fuel Cell and Electrolyzers. Curr. Opin. Electrochem. 2021, 29, 100832. 10.1016/j.coelec.2021.100832. [DOI] [Google Scholar]
- Ehelebe K.; Knöppel J.; Bierling M.; Mayerhöfer B.; Böhm T.; Kulyk N.; Thiele S.; Mayrhofer K. J. J.; Cherevko S. Platinum Dissolution in Realistic Fuel Cell Catalyst Layers. Angew. Chem., Int. Ed. 2021, 60, 8882–8888. 10.1002/anie.202014711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafforgue C.; Chatenet M.; Dubau L.; Dekel D. R. Accelerated Stress Test of Pt/C Nanoparticles in an Interface with an Anion-Exchange Membrane - An Identical-Location Transmission Electron Microscopy Study. ACS Catal. 2018, 8, 1278–1286. 10.1021/acscatal.7b04055. [DOI] [Google Scholar]
- Xiao L.; Zhang S.; Pan J.; Yang C.; He M.; Zhuang L.; Lu J. First Implementation of Alkaline Polymer Electrolyte Water Electrolysis Working Only with Pure Water. Environ. Sci. Technol. 2012, 5, 7869–7871. 10.1039/c2ee22146b. [DOI] [Google Scholar]
- Kreuer K. D. Ion Conducting Membranes for Fuel Cells and Other Electrochemical Devices. Chem. Mater. 2014, 26, 361–380. 10.1021/cm402742u. [DOI] [Google Scholar]
- Aili D.; Wright A. G.; Kraglund M. R.; Jankova K.; Holdcroft S.; Jensen J. O. Towards a Stable Ion-Solvating Polymer Electrolyte for Advanced Alkaline Water Electrolysis. J. Mater. Chem. A 2017, 5, 5055–5066. 10.1039/C6TA10680C. [DOI] [Google Scholar]
- Mustain W. E.; Chatenet M.; Page M.; Kim Y. S. Durability Challenges of Anion Exchange Membrane Fuel Cells. Environ. Sci. Technol. 2020, 13, 2805–2838. 10.1039/D0EE01133A. [DOI] [Google Scholar]
- Arges C. G.; Zhang L. Anion Exchange Membranes’ Evolution toward High Hydroxide Ion Conductivity and Alkaline Resiliency. ACS Appl. Energy Materials 2018, 1, 2991–3012. 10.1021/acsaem.8b00387. [DOI] [Google Scholar]
- Park E. J.; Maurya S.; Hibbs M. R.; Fujimoto C. H.; Kreuer K. D.; Kim Y. S. Alkaline Stability of Quaternized Diels-Alder Polyphenylenes. Macromolecules 2019, 52, 5419–5428. 10.1021/acs.macromol.9b00853. [DOI] [Google Scholar]
- Ponce-González J.; Whelligan D. K.; Wang L.; Bance-Soualhi R.; Wang Y.; Peng Y.; Peng H.; Apperley D. C.; Sarode H. N.; Pandey T. P.; Divekar A. G.; Seifert S.; Herring A. M.; Zhuang L.; Varcoe J. R. High Performance Aliphatic-Heterocyclic Benzyl-Quaternary Ammonium Radiation-Grafted Anion-Exchange Membranes. Environ. Sci. Technol. 2016, 9, 3724–3735. 10.1039/C6EE01958G. [DOI] [Google Scholar]
- Dai P.; Mo Z. H.; Xu R. W.; Zhang S.; Lin X.; Lin W. F.; Wu Y. X. Development of a Cross-Linked Quaternized Poly(Styrene-b-Isobutylene-b-Styrene)/Graphene Oxide Composite Anion Exchange Membrane for Direct Alkaline Methanol Fuel Cell Application. RSC Adv. 2016, 6, 52122–52130. 10.1039/C6RA08037E. [DOI] [Google Scholar]
- Wang J.; He G.; Wu X.; Yan X.; Zhang Y.; Wang Y.; Du L. Crosslinked Poly (Ether Ether Ketone) Hydroxide Exchange Membranes with Improved Conductivity. J. Membr. Sci. 2014, 459, 86–95. 10.1016/j.memsci.2014.01.068. [DOI] [Google Scholar]
- Lin X.; Liang X.; Poynton S. D.; Varcoe J. R.; Ong A. L.; Ran J.; Li Y.; Li Q.; Xu T. Novel Alkaline Anion Exchange Membranes Containing Pendant Benzimidazolium Groups for Alkaline Fuel Cells. J. Membr. Sci. 2013, 443, 193–200. 10.1016/j.memsci.2013.04.059. [DOI] [Google Scholar]
- Wang J.; Gu S.; Kaspar R. B.; Zhang B.; Yan Y. Stabilizing the Imidazolium Cation in Hydroxide-Exchange Membranes for Fuel Cells. ChemSusChem 2013, 6, 2079–2082. 10.1002/cssc.201300285. [DOI] [PubMed] [Google Scholar]
- Guo D.; Lai A. N.; Lin C. X.; Zhang Q. G.; Zhu A. M.; Liu Q. L. Imidazolium-Functionalized Poly(Arylene Ether Sulfone) Anion-Exchange Membranes Densely Grafted with Flexible Side Chains for Fuel Cells. ACS Appl. Mater. 2016, 8, 25279–25288. 10.1021/acsami.6b07711. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang Y.; Cao T.; Yang Y.; Xiong Y.; Xu S.; Xu Z. Highly Conductive Alkaline Anion Exchange Membrane Containing Imidazolium-Functionalized Octaphenyl Polyhedral Oligomeric Silsesquioxane Filler. J. Membr. Sci. 2017, 541, 474–482. 10.1016/j.memsci.2017.07.037. [DOI] [Google Scholar]
- Gu S.; Cai R.; Yan Y. Self-Crosslinking for Dimensionally Stable and Solvent-Resistant Quaternary Phosphonium Based Hydroxide Exchange Membranes. ChemCommun. 2011, 47, 2856–2858. 10.1039/c0cc04335d. [DOI] [PubMed] [Google Scholar]
- Noonan K. J. T.; Hugar K. M.; Kostalik H. A.; Lobkovsky E. B.; Abruña H. D.; Coates G. W. Phosphonium-Functionalized Polyethylene: A New Class of Base-Stable Alkaline Anion Exchange Membranes. J. Am. Chem. Soc. 2012, 134, 18161–18164. 10.1021/ja307466s. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Gu S.; Wang J.; Liu Y.; Herring A. M.; Yan Y. Tertiary Sulfonium as a Cationic Functional Group for Hydroxide Exchange Membranes. RSC Adv. 2012, 2, 12683–12685. 10.1039/c2ra21402d. [DOI] [Google Scholar]
- Hossain M. A.; Jang H.; Sutradhar S. C.; Ha J.; Yoo J.; Lee C.; Lee S.; Kim W. Novel Hydroxide Conducting Sulfonium-Based Anion Exchange Membrane for Alkaline Fuel Cell Applications. Int. J. Hydrog. 2016, 41, 10458–10465. 10.1016/j.ijhydene.2016.01.051. [DOI] [Google Scholar]
- Zha Y.; Disabb-Miller M. L.; Johnson Z. D.; Hickner M. A.; Tew G. N. Metal-Cation-Based Anion Exchange Membranes. J. Am. Chem. Soc. 2012, 134, 4493–4496. 10.1021/ja211365r. [DOI] [PubMed] [Google Scholar]
- Kwasny M. T.; Tew G. N. Expanding Metal Cation Options in Polymeric Anion Exchange Membranes. J. Mater. Chem. A 2017, 5, 1400–1405. 10.1039/C6TA07990C. [DOI] [Google Scholar]
- Koronka D.; Miyatake K. Anion Exchange Membranes Containing No β-Hydrogen Atoms on Ammonium Groups: Synthesis, Properties, and Alkaline Stability. RSC Adv. 2021, 11, 1030–1038. 10.1039/D0RA09308D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki S.; Douglin J. C.; Kostuch A.; Dekel D. R.; Kruczała K. Are Radicals Formed during Anion-Exchange Membrane Fuel Cell Operation?. J. Phys. Chem. Lett. 2020, 11, 7630–7636. 10.1021/acs.jpclett.0c02349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M. G.; Kreuer K. D. Alkaline Stability of Quaternary Ammonium Cations for Alkaline Fuel Cell Membranes and Ionic Liquids. ChemSusChem 2015, 8, 513–523. 10.1002/cssc.201403022. [DOI] [PubMed] [Google Scholar]
- Hugar K. M.; You W.; Coates G. W. Protocol for the Quantitative Assessment of Organic Cation Stability for Polymer Electrolytes. ACS Energy Lett. 2019, 4, 1681–1686. 10.1021/acsenergylett.9b00908. [DOI] [Google Scholar]
- Bauer B.; Strathmann H.; Effenberger F. Anion-Exchange Membranes with Improved Alkaline Stability. Desalination 1990, 79, 125–144. 10.1016/0011-9164(90)85002-R. [DOI] [Google Scholar]
- Komkova E. N.; Stamatialis D. F.; Strathmann H.; Wessling M. Anion-Exchange Membranes Containing Diamines: Preparation and Stability in Alkaline Solution. J. Membr. Sci. 2004, 244, 25–34. 10.1016/j.memsci.2004.06.026. [DOI] [Google Scholar]
- Hugar K. M.; Kostalik H. A.; Coates G. W. Imidazolium Cations with Exceptional Alkaline Stability: A Systematic Study of Structure-Stability Relationships. J. Am. Chem. Soc. 2015, 137, 8730–8737. 10.1021/jacs.5b02879. [DOI] [PubMed] [Google Scholar]
- Tao Z.; Wang C.; Zhao X.; Li J.; Guiver M. D. Progress in High-Performance Anion Exchange Membranes Based on the Design of Stable Cations for Alkaline Fuel Cells. Adv. Mater. Technol. 2021, 6, 2001220. 10.1002/admt.202001220. [DOI] [Google Scholar]
- Price S. C.; Williams K. S.; Beyer F. L. Relationships between Structure and Alkaline Stability of Imidazolium Cations for Fuel Cell Membrane Applications. ACS Macro Lett. 2014, 3, 160–165. 10.1021/mz4005452. [DOI] [PubMed] [Google Scholar]
- Chen N.; Lee Y. M. Anion Exchange Polyelectrolytes for Membranes and Ionomers. Prog. Polym. Sci. 2021, 113, 101345. 10.1016/j.progpolymsci.2020.101345. [DOI] [Google Scholar]
- Diesendruck C. E.; Dekel D. R. Water - A Key Parameter in the Stability of Anion Exchange Membrane Fuel Cells. Curr. Opin. Electrochem. 2018, 9, 173–178. 10.1016/j.coelec.2018.03.019. [DOI] [Google Scholar]
- Kim D. S.; Labouriau A.; Guiver M. D.; Kim Y. S. Guanidinium-Functionalized Anion Exchange Polymer Electrolytes via Activated Fluorophenyl-Amine Reaction. Chem. Mater. 2011, 23, 3795–3797. 10.1021/cm2016164. [DOI] [Google Scholar]
- Gu F.; Dong H.; Li Y.; Sun Z.; Yan F. Base Stable Pyrrolidinium Cations for Alkaline Anion Exchange Membrane Applications. Macromolecules 2014, 47, 6740–6747. 10.1021/ma5015148. [DOI] [Google Scholar]
- Olsson J. S.; Pham T. H.; Jannasch P. Poly(N,N-Diallylazacycloalkane)s for Anion-Exchange Membranes Functionalized with N-Spirocyclic Quaternary Ammonium Cations. Macromolecules 2017, 50, 2784–2793. 10.1021/acs.macromol.7b00168. [DOI] [Google Scholar]
- Pham T. H.; Olsson J. S.; Jannasch P. N-Spirocyclic Quaternary Ammonium Ionenes for Anion-Exchange Membranes. J. Am. Chem. Soc. 2017, 139, 2888–2891. 10.1021/jacs.6b12944. [DOI] [PubMed] [Google Scholar]
- Meek K. M.; Nykaza J. R.; Elabd Y. A. Alkaline Chemical Stability and Ion Transport in Polymerized Ionic Liquids with Various Backbones and Cations. Macromolecules 2016, 49, 3382–3394. 10.1021/acs.macromol.6b00434. [DOI] [Google Scholar]
- Kim Y. S.Advanced Materials for Fully-Integrated MEAs in AEMFCs. In 2018 U.S. DOE Hydrogen and Fuel Cells Program and Vehicle Technologies Office Annual Merit Review and Peer Evaluation Meeting; U.S. Department of Energy: 2017; pp 1–29.
- Ziv N.; Dekel D. R. A Practical Method for Measuring the True Hydroxide Conductivity of Anion Exchange Membranes. Electrochem. Commun. 2018, 88, 109–113. 10.1016/j.elecom.2018.01.021. [DOI] [Google Scholar]
- He Y.; Pan J.; Wu L.; Zhu Y.; Ge X.; Ran J.; Yang Z. J.; Xu T. A Novel Methodology to Synthesize Highly Conductive Anion Exchange Membranes. Sci. Rep. 2015, 5, 13417. 10.1038/srep13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G.; Scott K.; Mamlouk M. Performance of Polyethylene Based Radiation Grafted Anion Exchange Membrane with Polystyrene-b-Poly (Ethylene/Butylene)-b-Polystyrene Based Ionomer Using NiCO2O4 Catalyst for Water Electrolysis. J. Power Sources 2018, 375, 387–396. 10.1016/j.jpowsour.2017.07.026. [DOI] [Google Scholar]
- Peng H.; Li Q.; Hu M.; Xiao L.; Lu J.; Zhuang L. Alkaline Polymer Electrolyte Fuel Cells Stably Working at 80 °C. J. Power Sources 2018, 390, 165–167. 10.1016/j.jpowsour.2018.04.047. [DOI] [Google Scholar]
- Kwon S.; Lee B.; Kim T. H. High Performance Blend Membranes Based on Densely Sulfonated Poly(Fluorenyl Ether Sulfone) Block Copolymer and Imidazolium-Functionalized Poly(Ether Sulfone). Int. J. Hydrog. Energy 2017, 42, 20176–20186. 10.1016/j.ijhydene.2017.06.040. [DOI] [Google Scholar]
- Lee B.; Lim H.; Chae J. E.; Kim H. J.; Kim T. H. Physically-Crosslinked Anion Exchange Membranes by Blending Ionic Additive into Alkyl-Substituted Quaternized PPO. J. Membr. Sci. 2019, 574, 33–43. 10.1016/j.memsci.2018.12.053. [DOI] [Google Scholar]
- Pan J.; Li Y.; Zhuang L.; Lu J. Self-Crosslinked Alkaline Polymer Electrolyte Exceptionally Stable at 90 °C. ChemCommun. 2010, 46, 8597–8599. 10.1039/c0cc03618h. [DOI] [PubMed] [Google Scholar]
- Bai T. T.; Cong M. Y.; Jia Y. X.; Ma K. K.; Wang M. Preparation of self-crosslinking anion exchange membrane with acid block performance from side-chain type polysulfone. J. Membr. Sci. 2020, 599, 117831. 10.1016/j.memsci.2020.117831. [DOI] [Google Scholar]
- Hossain M. M.; Wu L.; Liang X.; Yang Z.; Hou J.; Xu T. Anion Exchange Membrane Crosslinked in the Easiest Way Stands out for Fuel Cells. J. Power Sources 2018, 390, 234–241. 10.1016/j.jpowsour.2018.04.064. [DOI] [Google Scholar]
- Tibbits A. C.; Mumper L. E.; Kloxin C. J.; Yan Y. S. A Single-Step Monomeric Photo-Polymerization and Crosslinking via Thiol-Ene Reaction for Hydroxide Exchange Membrane Fabrication. J. Electrochem. Soc. 2015, 162, F1206–F1211. 10.1149/2.0321510jes. [DOI] [Google Scholar]
- Zhang X.; Cao Y.; Zhang M.; Huang Y.; Wang Y.; Liu L.; Li N. Enhancement of the Mechanical Properties of Anion Exchange Membranes with Bulky Imidazolium by “Thiol-Ene” Crosslinking. J. Membr. Sci. 2020, 596, 117700. 10.1016/j.memsci.2019.117700. [DOI] [Google Scholar]
- Park J. S.; Park S. H.; Yim S. D.; Yoon Y. G.; Lee W. Y.; Kim C. S. Performance of Solid Alkaline Fuel Cells Employing Anion-Exchange Membranes. J. Power Sources 2008, 178, 620–626. 10.1016/j.jpowsour.2007.08.043. [DOI] [Google Scholar]
- Robertson N. J.; Kostalik H. A. IV; Clark T. J.; Mutolo P. F.; Abruña H. D.; Coates G. W. Tunable High Performance Cross-Linked Alkaline Anion Exchange Membranes for Fuel Cell Applications. J. Am. Chem. Soc. 2010, 132, 3400–3404. 10.1021/ja908638d. [DOI] [PubMed] [Google Scholar]
- Wang L.; Hickner M. A. Low-Temperature Crosslinking of Anion Exchange Membranes. Polym. Chem. 2014, 5, 2928–2935. 10.1039/C3PY01490H. [DOI] [Google Scholar]
- Wu L.; Pan Q.; Varcoe J. R.; Zhou D.; Ran J.; Yang Z.; Xu T. Thermal Crosslinking of an Alkaline Anion Exchange Membrane Bearing Unsaturated Side Chains. J. Membr. Sci. 2015, 490, 1–8. 10.1016/j.memsci.2015.04.046. [DOI] [Google Scholar]
- Dai P.; Mo Z. H.; Xu R. W.; Zhang S.; Wu Y. X. Cross-Linked Quaternized Poly(Styrene-b-(Ethylene-Co-Butylene)-b-Styrene) for Anion Exchange Membrane: Synthesis, Characterization and Properties. ACS Appl. Mater. 2016, 8, 20329–20341. 10.1021/acsami.6b04590. [DOI] [PubMed] [Google Scholar]
- Chen N.; Lu C.; Li Y.; Long C.; Li Z.; Zhu H. Tunable Multi-Cations-Crosslinked Poly(Arylene Piperidinium)-Based Alkaline Membranes with High Ion Conductivity and Durability. J. Membr. Sci. 2019, 588, 117120–117129. 10.1016/j.memsci.2019.05.044. [DOI] [Google Scholar]
- Lee K. H.; Cho D. H.; Kim Y. M.; Moon S. J.; Seong J. G.; Shin D. W.; Sohn J. Y.; Kim J. F.; Lee Y. M. Highly Conductive and Durable Poly(Arylene Ether Sulfone) Anion Exchange Membrane with End-Group Cross-Linking. Environ. Sci. Technol. 2017, 10, 275–285. 10.1039/C6EE03079C. [DOI] [Google Scholar]
- Sung S.; Mayadevi T. S.; Min K.; Lee J.; Chae J. E.; Kim T. H. Crosslinked PPO-Based Anion Exchange Membranes: The Effect of Crystallinity versus Hydrophilicity by Oxygen-Containing Crosslinker Chain Length. J. Membr. Sci. 2021, 619, 118774. 10.1016/j.memsci.2020.118774. [DOI] [Google Scholar]
- Singh A. K.; Zhou L.; Shinde A.; Suram S. K.; Montoya J. H.; Winston D.; Gregoire J. M.; Persson K. A. Electrochemical Stability of Metastable Materials. Chem. Mater. 2017, 29, 10159–10167. 10.1021/acs.chemmater.7b03980. [DOI] [Google Scholar]
- Lin J.; Yan X.; He G.; Chen W.; Zhen D.; Li T.; Ma L.; Wu X. Thermoplastic Interpenetrating Polymer Networks Based on Polybenzimidazole and Poly (1, 2-Dimethy-3-Allylimidazolium) for Anion Exchange Membranes. Electrochim. Acta 2017, 257, 9–19. 10.1016/j.electacta.2017.09.126. [DOI] [Google Scholar]
- Wang Y.; Wan H.; Wang D.; Wang J.; Wang L.; Feng R. Preparation and Characterization of A Semi-Interpenetrating Network Alkaline Anion Exchange Membrane. Fibers and Polym. 2018, 19, 11–21. 10.1007/s12221-018-7768-5. [DOI] [Google Scholar]
- Xue J.; Liu L.; Liao J.; Shen Y.; Li N. Semi-Interpenetrating Polymer Networks by Azide-Alkyne Cycloaddition as Novel Anion Exchange Membranes. J. Mater. Chem. A 2018, 6, 11317–11326. 10.1039/C8TA02177E. [DOI] [Google Scholar]
- Zeng L.; Zhao T. S.; Wei L.; Zeng Y. K.; Zhang Z. H. Polyvinylpyrrolidone-Based Semi-Interpenetrating Polymer Networks as Highly Selective and Chemically Stable Membranes for All Vanadium Redox Flow Batteries. J. Power Sources 2016, 327, 374–383. 10.1016/j.jpowsour.2016.07.081. [DOI] [Google Scholar]
- Zeng L.; He Q.; Liao Y.; Kuang S.; Wang J.; Ding W.; Liao Q.; Wei Z. Anion Exchange Membrane Based on Interpenetrating Polymer Network with Ultrahigh Ion Conductivity and Excellent Stability for Alkaline Fuel Cell. Research 2020, 2020, 1–11. 10.34133/2020/4794706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arges C. G.; Kambe Y.; Dolejsi M.; Wu G. P.; Segal-Pertz T.; Ren J.; Cao C.; Craig G. S. W.; Nealey P. F. Interconnected Ionic Domains Enhance Conductivity in Microphase Separated Block Copolymer Electrolytes. J. Mater. Chem. A 2017, 5, 5619–5629. 10.1039/C6TA10838E. [DOI] [Google Scholar]
- Wang L.; Magliocca E.; Cunningham E. L.; Mustain W. E.; Poynton S. D.; Escudero-Cid R.; Nasef M. M.; Ponce-González J.; Bance-Souahli R.; Slade R. C. T.; Whelligan D. K.; Varcoe J. R. An Optimised Synthesis of High Performance Radiation-Grafted Anion-Exchange Membranes. Green Chem. 2017, 19, 831–843. 10.1039/C6GC02526A. [DOI] [Google Scholar]
- Heitner-Wirguin C. Recent Advances in Perfluorinated Ionomer Membranes: Structure, Properties and Applications Carla. J. Membr. Sci. 1996, 120, 1–33. 10.1016/0376-7388(96)00155-X. [DOI] [Google Scholar]
- Yamaguchi T.; Miyata F.; Nakao S. I. Pore-Filling Type Polymer Electrolyte Membranes for a Direct Methanol Fuel Cell. J. Membr. Sci. 2003, 214, 283–292. 10.1016/S0376-7388(02)00579-3. [DOI] [Google Scholar]
- Chen X. C.; Wong D. T.; Yakovlev S.; Beers K. M.; Downing K. H.; Balsara N. P. Effect of Morphology of Nanoscale Hydrated Channels on Proton Conductivity in Block Copolymer Electrolyte Membranes. Nano Lett. 2014, 14, 4058–4064. 10.1021/nl501537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Leng Y.; Hickner M. A.; Wang C. Y. Highly Stable, Anion Conductive, Comb-Shaped Copolymers for Alkaline Fuel Cells. J. Am. Chem. Soc. 2013, 135, 10124–10133. 10.1021/ja403671u. [DOI] [PubMed] [Google Scholar]
- Lin B.; Xu F.; Su Y.; Zhu Z.; Ren Y.; Ding J.; Yuan N. Facile Preparation of Anion-Exchange Membrane Based on Polystyrene-b-Polybutadiene-b-Polystyrene for the Application of Alkaline Fuel Cells. Ind. Eng. Chem. Res. 2019, 58, 22299–22305. 10.1021/acs.iecr.9b05314. [DOI] [Google Scholar]
- Liu L.; Li D.; Xing Y.; Li N. Mid-Block Quaternized Polystyrene-b-Polybutadiene-b-Polystyrene Triblock Copolymers as Anion Exchange Membranes. J. Membr. Sci. 2018, 564, 428–435. 10.1016/j.memsci.2018.07.055. [DOI] [Google Scholar]
- Zhu L.; Pan J.; Wang Y.; Han J.; Zhuang L.; Hickner M. A. Multication Side Chain Anion Exchange Membranes. Macromolecules 2016, 49, 815–824. 10.1021/acs.macromol.5b02671. [DOI] [Google Scholar]
- Chen N.; Lee Y. M. Anion exchange polyelectrolytes for membranes and ionomers. Prog. Polym. Sci. 2021, 113, 101345–101379. 10.1016/j.progpolymsci.2020.101345. [DOI] [Google Scholar]
- Ran J.; Wu L.; He Y.; Yang Z.; Wang Y.; Jiang C.; Ge L.; Bakangura E.; Xu T. Ion Exchange Membranes: New Developments and Applications. J. Membr. Sci. 2017, 522, 267–291. 10.1016/j.memsci.2016.09.033. [DOI] [Google Scholar]
- Kickelbick G. Concepts for the Incorporation of Inorganic Building Blocks into Organic Polymers on a Nanoscale. Prog. Polym. Sci. 2003, 28, 83–114. 10.1016/S0079-6700(02)00019-9. [DOI] [Google Scholar]
- Wu Y.; Wu C.; Xu T.; Fu Y. Novel Anion-Exchange Organic-Inorganic Hybrid Membranes Prepared through Sol-Gel Reaction of Multi-Alkoxy Precursors. J. Membr. Sci. 2009, 329, 236–245. 10.1016/j.memsci.2008.12.056. [DOI] [Google Scholar]
- Bakangura E.; Wu L.; Ge L.; Yang Z.; Xu T. Mixed Matrix Proton Exchange Membranes for Fuel Cells: State of the Art and Perspectives. Prog. Polym. Sci. 2016, 57, 103–152. 10.1016/j.progpolymsci.2015.11.004. [DOI] [Google Scholar]
- Samsudin A. M.; Hacker V. Preparation and Characterization of PVA/PDDA/Nano-Zirconia Composite Anion Exchange Membranes for Fuel Cells. Polymers 2019, 11, 1399. 10.3390/polym11091399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberty-Robert C.; Vallé K.; Pereira F.; Sanchez C. Design and Properties of Functional Hybrid Organic-Inorganic Membranes for Fuel Cells. Chem. Soc. Rev. 2011, 40, 961–1005. 10.1039/c0cs00144a. [DOI] [PubMed] [Google Scholar]
- Li X.; Tao J.; Nie G.; Wang L.; Li L.; Liao S. Cross-Linked Multiblock Copoly(Arylene Ether Sulfone) Ionomer/Nano-ZrO2 Composite Anion Exchange Membranes for Alkaline Fuel Cells. RSC Adv. 2014, 4, 41398–41410. 10.1039/C4RA06519K. [DOI] [Google Scholar]
- Chen N.; Long C.; Li Y.; Wang D.; Zhu H. High-Performance Layered Double Hydroxide/Poly(2,6-Dimethyl-1,4-Phenylene Oxide) Membrane with Porous Sandwich Structure for Anion Exchange Membrane Fuel Cell Applications. J. Membr. Sci. 2018, 552, 51–60. 10.1016/j.memsci.2018.01.045. [DOI] [Google Scholar]
- Fan J.; Zhu H.; Li R.; Chen N.; Han K. Layered Double Hydroxide-Polyphosphazene-Based Ionomer Hybrid Membranes with Electric Field-Aligned Domains for Hydroxide Transport. J. Mater. Chem. A 2014, 2, 8376–8385. 10.1039/c4ta00686k. [DOI] [Google Scholar]
- Chen N.; Wang D.; Long C.; Li Y.; Lu C.; Wang F.; Zhu H. Magnetic Field-Oriented Ferroferric Oxide/Poly(2,6-Dimethyl-1,4-Phenylene Oxide) Hybrid Membranes for Anion Exchange Membrane Applications. Nanoscale 2018, 10, 18680–18689. 10.1039/C8NR06048G. [DOI] [PubMed] [Google Scholar]
- Varcoe J. R.; Slade R. C. T. Prospects for Alkaline Anion-Exchange Membranes in Low Temperature Fuel Cells. Fuel Cells 2005, 5, 187–200. 10.1002/fuce.200400045. [DOI] [Google Scholar]
- Wang L.; Bellini M.; Miller H. A.; Varcoe J. R. A High Conductivity Ultrathin Anion-Exchange Membrane with 500+ h Alkali Stability for Use in Alkaline Membrane Fuel Cells That Can Achieve 2 W cm–2 at 80 °C. J. Mater. Chem. A 2018, 6, 15404–15412. 10.1039/C8TA04783A. [DOI] [Google Scholar]
- Mandal M.; Huang G.; Kohl P. A. Highly Conductive Anion-Exchange Membranes Based on Cross-Linked Poly(Norbornene): Vinyl Addition Polymerization. ACS Appl. Energy Materials 2019, 2, 2447–2457. 10.1021/acsaem.8b02051. [DOI] [Google Scholar]
- Chen W.; Mandal M.; Huang G.; Wu X.; He G.; Kohl P. A. Highly Conducting Anion-Exchange Membranes Based on Cross-Linked Poly(Norbornene): Ring Opening Metathesis Polymerization. ACS Appl. Energy Materials 2019, 2, 2458–2468. 10.1021/acsaem.8b02052. [DOI] [Google Scholar]
- Wang L.; Peng X.; Mustain W. E.; Varcoe J. R. Radiation-Grafted Anion-Exchange Membranes: The Switch from Low- to High-Density Polyethylene Leads to Remarkably Enhanced Fuel Cell Performance. Environ. Sci. Technol. 2019, 12, 1575–1579. 10.1039/C9EE00331B. [DOI] [Google Scholar]
- Zhu L.; Yu X.; Peng X.; Zimudzi T. J.; Saikia N.; Kwasny M. T.; Song S.; Kushner D. I.; Fu Z.; Tew G. N.; Mustain W. E.; Yandrasits M. A.; Hickner M. A. Poly(Olefin)-Based Anion Exchange Membranes Prepared Using Ziegler-Natta Polymerization. Macromolecules 2019, 52, 4030–4041. 10.1021/acs.macromol.8b02756. [DOI] [Google Scholar]
- He R.; Wen P.; Zhang H. N.; Guan S.; Xie G.; Li L. Z.; Lee M. H.; Li X. D. In-Situ Photocrosslinked Hydroxide Conductive Membranes Based on Photosensitive Poly(Arylene Ether Sulfone) Block Copolymers for Anion Exchange Membrane Fuel Cells. J. Membr. Sci. 2018, 556, 73–84. 10.1016/j.memsci.2018.03.088. [DOI] [Google Scholar]
- Dang H. S.; Jannasch P. A Comparative Study of Anion-Exchange Membranes Tethered with Different Hetero-Cycloaliphatic Quaternary Ammonium Hydroxides. J. Mater. Chem. A 2017, 5, 21965–21978. 10.1039/C7TA06029G. [DOI] [Google Scholar]
- Gu L.; Dong H.; Sun Z.; Li Y.; Yan F. Spirocyclic Quaternary Ammonium Cations for Alkaline Anion Exchange Membrane Applications: An Experimental and Theoretical Study. RSC Adv. 2016, 6, 94387–94398. 10.1039/C6RA22313C. [DOI] [Google Scholar]
- Pham T. H.; Olsson J. S.; Jannasch P. Poly(Arylene Alkylene)s with Pendant N-Spirocyclic Quaternary Ammonium Cations for Anion Exchange Membranes. J. Mater. Chem. A 2018, 6, 16537–16547. 10.1039/C8TA04699A. [DOI] [Google Scholar]
- Pham T. H.; Jannasch P. Aromatic Polymers Incorporating Bis-N-Spirocyclic Quaternary Ammonium Moieties for Anion-Exchange Membranes. ACS Macro Lett. 2015, 4, 1370–1375. 10.1021/acsmacrolett.5b00690. [DOI] [PubMed] [Google Scholar]
- Meek K. M.; Antunes C. M.; Strasser D.; Owczarczyk Z. R.; Neyerlin A.; Pivovar B. S. High-Throughput Anion Exchange Membrane Characterization at NREL. ECS Trans. 2019, 92, 723–731. 10.1149/09208.0723ecst. [DOI] [Google Scholar]
- Liu L.; Chu X.; Liao J.; Huang Y.; Li Y.; Ge Z.; Hickner M. A.; Li N. Tuning the Properties of Poly(2,6-Dimethyl-1,4-Phenylene Oxide) Anion Exchange Membranes and Their Performance in H2/O2 Fuel Cells. Environ. Sci. Technol. 2018, 11, 435–446. 10.1039/C7EE02468A. [DOI] [Google Scholar]
- Wu Q. X.; Pan Z. F.; An L. Recent Advances in Alkali-Doped Polybenzimidazole Membranes for Fuel Cell Applications. Renew. Sust. Energy Rev. 2018, 89, 168–183. 10.1016/j.rser.2018.03.024. [DOI] [Google Scholar]
- Xing B.; Savadogo O. Hydrogen/Oxygen Polymer Electrolyte Membrane Fuel Cells (PEMFCs) Based on Alkaline-Doped Polybenzimidazole (PBI). Electrochem. Commun. 2000, 2, 697–702. 10.1016/S1388-2481(00)00107-7. [DOI] [Google Scholar]
- Zarrin H.; Jiang G.; Lam G. Y. Y.; Fowler M.; Chen Z. High Performance Porous Polybenzimidazole Membrane for Alkaline Fuel Cells. Int. J. Hydrog. Energy 2014, 39, 18405–18415. 10.1016/j.ijhydene.2014.08.134. [DOI] [Google Scholar]
- Zeng L.; Zhao T. S.; An L.; Zhao G.; Yan X. H. A High-Performance Sandwiched-Porous Polybenzimidazole Membrane with Enhanced Alkaline Retention for Anion Exchange Membrane Fuel Cells. Environ. Sci. Technol. 2015, 8, 2768–2774. 10.1039/C5EE02047F. [DOI] [Google Scholar]
- Diaz L. A.; Coppola R. E.; Abuin G. C.; Escudero-Cid R.; Herranz D.; Ocón P. Alkali-Doped Polyvinyl Alcohol - Polybenzimidazole Membranes for Alkaline Water Electrolysis. J. Membr. Sci. 2017, 535, 45–55. 10.1016/j.memsci.2017.04.021. [DOI] [Google Scholar]
- Wang J.; Zhao Y.; Setzler B. P.; Rojas-Carbonell S.; Ben Yehuda C.; Amel A.; Page M.; Wang L.; Hu K.; Shi L.; Gottesfeld S.; Xu B.; Yan Y. Poly(Aryl Piperidinium) Membranes and Ionomers for Hydroxide Exchange Membrane Fuel Cells. Nat. Energy. 2019, 4, 392–398. 10.1038/s41560-019-0372-8. [DOI] [Google Scholar]
- Zhang J.; Zhang K.; Liang X.; Yu W.; Ge X.; Shehzad M. A.; Ge Z.; Yang Z.; Wu L.; Xu T. Self-Aggregating Cationic-Chains Enable Alkaline Stable Ion-Conducting Channels for Anion-Exchange Membrane Fuel Cells. J. Mater. Chem. A 2021, 9, 327–337. 10.1039/D0TA11011F. [DOI] [Google Scholar]
- Pham T. H.; Olsson J. S.; Jannasch P. Effects of the: N -Alicyclic Cation and Backbone Structures on the Performance of Poly(Terphenyl)-Based Hydroxide Exchange Membranes J. Mater. Chem. A 2019, 7, 15895–15906. 10.1039/C9TA05531B. [DOI] [Google Scholar]
- Zhu H.; Li Y.; Chen N.; Lu C.; Long C.; Li Z.; Liu Q. Controllable Physical-Crosslinking Poly(Arylene 6-Azaspiro[5.5] Undecanium) for Long-Lifetime Anion Exchange Membrane Applications. J. Membr. Sci. 2019, 590, 117307–117316. 10.1016/j.memsci.2019.117307. [DOI] [Google Scholar]
- Chen N.; Lu C.; Li Y.; Long C.; Zhu H. Robust Poly(Aryl Piperidinium)/N-Spirocyclic Poly(2,6-Dimethyl-1,4-Phenyl) for Hydroxide-Exchange Membranes. J. Membr. Sci. 2019, 572, 246–254. 10.1016/j.memsci.2018.10.067. [DOI] [Google Scholar]
- Chen N.; Long C.; Li Y.; Lu C.; Zhu H. Ultrastable and High Ion-Conducting Polyelectrolyte Based on Six-Membered N-Spirocyclic Ammonium for Hydroxide Exchange Membrane Fuel Cell Applications. ACS Appl. Mater. Interfaces 2018, 10, 15720–15732. 10.1021/acsami.8b02884. [DOI] [PubMed] [Google Scholar]
- Fan J.; Wright A. G.; Britton B.; Weissbach T.; Skalski T. J. G.; Ward J.; Peckham T. J.; Holdcroft S. Cationic Polyelectrolytes, Stable in 10 M KOHaq at 100 °C. ACS Macro Lett. 2017, 6, 1089–1093. 10.1021/acsmacrolett.7b00679. [DOI] [PubMed] [Google Scholar]
- You W.; Padgett E.; MacMillan S. N.; Muller D. A.; Coates G. W. Highly Conductive and Chemically Stable Alkaline Anion Exchange Membranes via ROMP of Trans-Cyclooctene Derivatives. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (20), 9729–9734. 10.1073/pnas.1900988116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Wang Y.; France-Lanord A.; Wang Y.; Wu Y. C. M.; Lin S.; Li Y.; Grossman J. C.; Swager T. M. Ionic Highways from Covalent Assembly in Highly Conducting and Stable Anion Exchange Membrane Fuel Cells. J. Am. Chem. Soc. 2019, 141, 18152–18159. 10.1021/jacs.9b08749. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen W.; Li T.; Yan X.; Zhang Y.; Zhang F.; Wu X.; Pang B.; Li J.; He G. Ultra-Thin Quaternized Polybenzimidazole Anion Exchange Membranes with throughout OH-conducive Highway Networks for High-Performance Fuel Cells. J. Mater. Chem. A 2021, 9, 7522–7530. 10.1039/D0TA11717J. [DOI] [Google Scholar]
- Zhang Z.; Wu L.; Varcoe J.; Li C.; Ong A. L.; Poynton S.; Xu T. Aromatic Polyelectrolytes via Polyacylation of Pre-Quaternized Monomers for Alkaline Fuel Cells. J. Mater. Chem. A 2013, 1, 2595–2601. 10.1039/c2ta01178f. [DOI] [Google Scholar]
- Lee W. H.; Mohanty A. D.; Bae C. Fluorene-Based Hydroxide Ion Conducting Polymers for Chemically Stable Anion Exchange Membrane Fuel Cells. ACS Macro Lett. 2015, 4, 453–457. 10.1021/acsmacrolett.5b00145. [DOI] [PubMed] [Google Scholar]
- Lee W. H.; Kim Y. S.; Bae C. Robust Hydroxide Ion Conducting Poly(Biphenyl Alkylene)s for Alkaline Fuel Cell Membranes. ACS Macro Lett. 2015, 4, 814–818. 10.1021/acsmacrolett.5b00375. [DOI] [PubMed] [Google Scholar]
- Lee W. H.; Park E. J.; Han J.; Shin D. W.; Kim Y. S.; Bae C. Poly(Terphenylene) Anion Exchange Membranes: The Effect of Backbone Structure on Morphology and Membrane Property. ACS Macro Lett. 2017, 6, 566–570. 10.1021/acsmacrolett.7b00148. [DOI] [PubMed] [Google Scholar]
- Mahmoud A. M. A.; Elsaghier A. M. M.; Otsuji K.; Miyatake K. High Hydroxide Ion Conductivity with Enhanced Alkaline Stability of Partially Fluorinated and Quaternized Aromatic Copolymers as Anion Exchange Membranes. Macromolecules 2017, 50, 4256–4266. 10.1021/acs.macromol.7b00401. [DOI] [Google Scholar]
- Ahmed Mahmoud A. M.; Miyatake K. Optimization of the Pendant Chain Length in Partially Fluorinated Aromatic Anion Exchange Membranes for Alkaline Fuel Cells. J. Mater. Chem. A 2018, 6, 14400–14409. 10.1039/C8TA04310H. [DOI] [Google Scholar]
- Mohanty A. D.; Ryu C. Y.; Kim Y. S.; Bae C. Stable Elastomeric Anion Exchange Membranes Based on Quaternary Ammonium-Tethered Polystyrene-b-Poly(Ethylene-Co-Butylene)-b-Polystyrene Triblock Copolymers. Macromolecules 2015, 48, 7085–7095. 10.1021/acs.macromol.5b01382. [DOI] [Google Scholar]
- Gao X.; Yu H.; Qin B.; Jia J.; Hao J.; Xie F.; Shao Z. Enhanced Water Transport in AEMs Based on Poly(Styrene-Ethylene-Butylene-Styrene) Triblock Copolymer for High Fuel Cell Performance. Polym. Chem. 2019, 10, 1894–1903. 10.1039/C8PY01618F. [DOI] [Google Scholar]
- Wang L.; Brink J. J.; Liu Y.; Herring A. M.; Ponce-González J.; Whelligan D. K.; Varcoe J. R. Non-Fluorinated Pre-Irradiation-Grafted (Peroxidated) LDPE-Based Anion-Exchange Membranes with High Performance and Stability. Energy Environ. Sci. 2017, 10 (10), 2154–2167. 10.1039/C7EE02053H. [DOI] [Google Scholar]
- Cha S. M.; Park J. E.; Kim S.; Han S. H.; Sang H. S.; Shin S.-H.; Yang S. H.; Kim T.-H.; Yu D. M.; So S.; et al. Poly(carbazole)-based anion-conducting materials with high performance and durability for energy conversion devices. Energy Environ. Sci. 2020, 13, 3633–3645. 10.1039/D0EE01842B. [DOI] [Google Scholar]
- Etzold B. J. M.; Krewer U.; Thiele S.; Dreizler A.; Klemm E.; Turek T. Understanding the Activity Transport Nexus in Water and CO2 Electrolysis: State of the Art, Challenges and Perspectives. Chem. Eng. J. 2021, 424, 130501. 10.1016/j.cej.2021.130501. [DOI] [Google Scholar]
- Mo J.; Kang Z.; Retterer S. T.; Cullen D. A.; Toops T. J.; Green J. B.; Mench M. M.; Zhang F. Y. Discovery of True Electrochemical Reactions for Ultrahigh Catalyst Mass Activity in Water Splitting. Sci. Adv. 2016, 2, e1600690 10.1126/sciadv.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Stevens M. B.; Cosby M. R.; Oener S. Z.; Smith A. M.; Enman L. J.; Ayers K. E.; Capuano C. B.; Renner J. N.; Danilovic N.; Li Y.; Wang H.; Zhang Q.; Boettcher S. W. Earth-Abundant Oxygen Electrocatalysts for Alkaline Anion-Exchange-Membrane Water Electrolysis: Effects of Catalyst Conductivity and Comparison with Performance in Three-Electrode Cells. ACS Catal. 2019, 9, 7–15. 10.1021/acscatal.8b04001. [DOI] [Google Scholar]
- Mo J.; Kang Z.; Yang G.; Li Y.; Retterer S. T.; Cullen D. A.; Toops T. J.; Bender G.; Pivovar B. S.; Green J. B.; Zhang F. Y. In Situ Investigation on Ultrafast Oxygen Evolution Reactions of Water Splitting in Proton Exchange Membrane Electrolyzer Cells. J. Mater. Chem. A 2017, 5, 18469–18475. 10.1039/C7TA05681H. [DOI] [Google Scholar]
- Mayerhöfer B.; Speck F. D.; Hegelheimer M.; Bierling M.; Abbas D.; McLaughlin D.; Cherevko S.; Thiele S.; Peach R. Electrochemical- and Mechanical Stability of Catalyst Layers in Anion Exchange Membrane Water Electrolysis. Int. J. Hydrog. Energy 2022, 47, 4304–4314. 10.1016/j.ijhydene.2021.11.083. [DOI] [Google Scholar]
- Hegelheimer M.Optimization of Noble Metal Free Catalyst Layers and Porous Transport Electrode Structures in Anion Exchange Membrane Water Electrolysis. Master’s Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Department Chemistry and Bioingengineering, 2020; p46. [Google Scholar]
- Hall H. K. Jr. Correlation of the Base Strengths of Amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. 10.1021/ja01577a030. [DOI] [Google Scholar]
- Bates M. K.; Jia Q.; Ramaswamy N.; Allen R. J.; Mukerjee S. Composite Ni/NiO-Cr2O3 Catalyst for Alkaline Hydrogen Evolution Reaction. J. Phys. Chem. C 2015, 119, 5467–5477. 10.1021/jp512311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Zhou L.; Shinde A.; Suram S. K.; Montoya J. H.; Winston D.; Gregoire J. M.; Persson K. A. Electrochemical Stability of Metastable Materials. Chem. Mater. 2017, 29, 10159–10167. 10.1021/acs.chemmater.7b03980. [DOI] [Google Scholar]
- Mohanty A. D.; Tignor S. E.; Krause J. A.; Choe Y. K.; Bae C. Systematic Alkaline Stability Study of Polymer Backbones for Anion Exchange Membrane Applications. Macromolecules 2016, 49, 3361–3372. 10.1021/acs.macromol.5b02550. [DOI] [Google Scholar]
- Matanovic I.; Maurya S.; Park E. J.; Jeon J. Y.; Bae C.; Kim Y. S. Adsorption of Polyaromatic Backbone Impacts the Performance of Anion Exchange Membrane Fuel Cells. Chem. Mater. 2019, 31, 4195–4204. 10.1021/acs.chemmater.9b01092. [DOI] [Google Scholar]
- Maurya S.; Noh S.; Matanovic I.; Park E. J.; Narvaez Villarrubia C.; Martinez U.; Han J.; Bae C.; Kim Y. S. Rational Design of Polyaromatic Ionomers for Alkaline Membrane Fuel Cells with > 1 W cm–2 Power Density. Environ. Sci. Technol. 2018, 11, 3283–3291. 10.1039/C8EE02192A. [DOI] [Google Scholar]
- Maurya S.; Fujimoto C. H.; Hibbs M. R.; Narvaez Villarrubia C.; Kim Y. S. Toward Improved Alkaline Membrane Fuel Cell Performance Using Quaternized Aryl-Ether Free Polyaromatics. Chem. Mater. 2018, 30, 2188–2192. 10.1021/acs.chemmater.8b00358. [DOI] [Google Scholar]
- Lehwald S.; Ibach H.; Demuth J. E. Vibration Spectroscopy of Benzene Adsorbed on Pt(111) and Ni(111). Surf. Sci. 1978, 78, 577–590. 10.1016/0039-6028(78)90234-0. [DOI] [Google Scholar]
- Park E. J.; Kim Y. S. Quaternized Aryl Ether-Free Polyaromatics for Alkaline Membrane Fuel Cells: Synthesis, Properties, and Performance-a Topical Review. J. Mater. Chem. A 2018, 6, 15456–15477. 10.1039/C8TA05428B. [DOI] [Google Scholar]
- Li D.; Park E. J.; Zhu W.; Shi Q.; Zhou Y.; Tian H.; Lin Y.; Serov A.; Zulevi B.; Baca E. D.; Fujimoto C.; Chung H. T.; Kim Y. S. Highly Quaternized Polystyrene Ionomers for High Performance Anion Exchange Membrane Water Electrolysers. Nat. Energy 2020, 5, 378–385. 10.1038/s41560-020-0577-x. [DOI] [Google Scholar]
- Li D.; Matanovic I.; Lee A. S.; Park E. J.; Fujimoto C.; Chung H. T.; Kim Y. S. Phenyl Oxidation Impacts the Durability of Alkaline Membrane Water Electrolyzer. ACS Appl. Mater. 2019, 11, 9696–9701. 10.1021/acsami.9b00711. [DOI] [PubMed] [Google Scholar]
- Duan J.-A.; Dong X.; Yin K.; Yang S.; Chu D. A Hierarchical Superaerophilic Cone: Robust Spontaneous and Directional Transport of Gas Bubbles. Appl. Phys. Lett. 2018, 113, 203704. 10.1063/1.5054623. [DOI] [Google Scholar]
- Tiwari P.; Tsekouras G.; Wagner K.; Swiegers G. F.; Wallace G. G. A New Class of Bubble-Free Water Electrolyzer That Is Intrinsically Highly Efficient. Int. J. Hydrog. Energy 2019, 44, 23568–23579. 10.1016/j.ijhydene.2019.07.100. [DOI] [Google Scholar]
- Weissenborn P. K.; Pugh R. J. Surface Tension of Aqueous Solutions of Electrolytes: Relationship with Ion Hydration, Oxygen Solubility, and Bubble Coalescence. J. Colloid Interface Sci. 1996, 184, 550–563. 10.1006/jcis.1996.0651. [DOI] [PubMed] [Google Scholar]
- Trzesniewski B. J.; Diaz-Morales O.; Vermaas D. A.; Longo A.; Bras W.; Koper M. T. M.; Smith W. A. In Situ Observation of Active Oxygen Species in Fe-Containing Ni-Based Oxygen Evolution Catalysts: The Effect of PH on Electrochemical Activity. J. Am. Chem. Soc. 2015, 137, 15112–15121. 10.1021/jacs.5b06814. [DOI] [PubMed] [Google Scholar]
- Cho M. K.; Park H. Y.; Lee H. J.; Kim H. J.; Lim A.; Henkensmeier D.; Yoo S. J.; Kim J. Y.; Lee S. Y.; Park H. S.; Jang J. H. Alkaline Anion Exchange Membrane Water Electrolysis: Effects of Electrolyte Feed Method and Electrode Binder Content. J. Power Sources 2018, 382, 22–29. 10.1016/j.jpowsour.2018.02.025. [DOI] [Google Scholar]
- Omasta T. J.; Park A. M.; Lamanna J. M.; Zhang Y.; Peng X.; Wang L.; Jacobson D. L.; Varcoe J. R.; Hussey D. S.; Pivovar B. S.; Mustain W. E. Beyond Catalysis and Membranes: Visualizing and Solving the Challenge of Electrode Water Accumulation and Flooding in AEMFCs. Environ. Sci. Technol. 2018, 11, 551–558. 10.1039/C8EE00122G. [DOI] [Google Scholar]
- Ito H.; Kawaguchi N.; Someya S.; Munakata T.; Miyazaki N.; Ishida M.; Nakano A. Experimental Investigation of Electrolytic Solution for Anion Exchange Membrane Water Electrolysis. Int. J. Hydrog. Energy 2018, 43, 17030–17039. 10.1016/j.ijhydene.2018.07.143. [DOI] [Google Scholar]
- López-Fernández E.; Sacedón C. G.; Gil-Rostra J.; Yubero F.; González-Elipe A. R.; de Lucas-Consuegra A. Recent Advances in Alkaline Exchange Membrane Water Electrolysis and Electrode Manufacturing. Molecules 2021, 26, 6326. 10.3390/molecules26216326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J.; Oliveira A. M.; Wang L.; Zhao Y.; Wang T.; Wang J.; Setzler B. P.; Yan Y. Water-Fed Hydroxide Exchange Membrane Electrolyzer Enabled by a Fluoride-Incorporated Nickel-Iron Oxyhydroxide Oxygen Evolution Electrode. ACS Catal. 2021, 11, 264–270. 10.1021/acscatal.0c04200. [DOI] [Google Scholar]
- Bladergroen B.; Su H.; Pasupathi S.; Linkov V.. Overview of Membrane Electrode Assembly Preparation Methods for Solid Polymer Electrolyte Electrolyzer. In Electrolysis; Kleperis J., Linkov V., Eds.; IntechOpen: 2012; pp 45–60. [Google Scholar]
- Mahrous A.-F. M.; Sakr I. M.; Balabel A.; Ibrahim K. Experimental Investigation of the Operating Parameters Affecting Hydrogen Production Process through Alkaline Water Electrolysis. Int. J. Therm. Environ. Eng. 2010, 2, 113–116. 10.5383/ijtee.02.02.009. [DOI] [Google Scholar]
- Ito H.; Miyazaki N.; Sugiyama S.; Ishida M.; Nakamura Y.; Iwasaki S.; Hasegawa Y.; Nakano A. Investigations on Electrode Configurations for Anion Exchange Membrane Electrolysis. J. Appl. Electrochem. 2018, 48, 305–316. 10.1007/s10800-018-1159-5. [DOI] [Google Scholar]
- Razmjooei F.; Morawietz T.; Taghizadeh E.; Hadjixenophontos E.; Mues L.; Gerle M.; Wood B. D.; Harms C.; Gago A. S.; Ansar S. A.; Friedrich K. A. Increasing the Performance of an Anion-Exchange Membrane Electrolyzer Operating in Pure Water with a Nickel-Based Microporous Layer. Joule 2021, 5, 1776–1799. 10.1016/j.joule.2021.05.006. [DOI] [Google Scholar]
- Zawodzinski T. A. Jr.; Springer T. E.; Uribe F.; Gottesfeld S. Characterization of Polymer Electrolytes for Fuel Cell Applications. Solid State Ion. 1993, 60, 199–211. 10.1016/0167-2738(93)90295-E. [DOI] [Google Scholar]
- Hnát J.; Kodým R.; Denk K.; Paidar M.; Žitka J.; Bouzek K. Design of a Zero-Gap Laboratory-Scale Polymer Electrolyte Membrane Alkaline Water Electrolysis Stack. Chem. Ing. Technol. Chem. Ing. Technol. 2019, 91, 821–832. 10.1002/cite.201800185. [DOI] [Google Scholar]
- Vincent I.; Kruger A.; Bessarabov D. Hydrogen Production by Water Electrolysis with an Ultrathin Anion-Exchange Membrane (AEM). Int. J. Electro. Chem. Sci. 2018, 13, 11347–11358. 10.20964/2018.12.84. [DOI] [Google Scholar]
- Liu Z.; Sajjad S. D.; Gao Y.; Yang H.; Kaczur J. J.; Masel R. I. The Effect of Membrane on an Alkaline Water Electrolyzer. Int. J. Hydrog. Energy 2017, 42, 29661–29665. 10.1016/j.ijhydene.2017.10.050. [DOI] [Google Scholar]
- Xu Q.; Oener S. Z.; Lindquist G.; Jiang H.; Li C.; Boettcher S. W. Integrated Reference Electrodes in Anion-Exchange-Membrane Electrolyzers: Impact of Stainless-Steel Gas-Diffusion Layers and Internal Mechanical Pressure. ACS Energy Lett. 2021, 6, 305–312. 10.1021/acsenergylett.0c02338. [DOI] [Google Scholar]
- Wu X.; Scott K.; Xie F.; Alford N. A Reversible Water Electrolyser with Porous PTFE Based OH– Conductive Membrane as Energy Storage Cells. J. Power Sources 2014, 246, 225–231. 10.1016/j.jpowsour.2013.07.081. [DOI] [Google Scholar]
- Bystron T.; Paidar M.; Klicpera T.; Schuster M.; Bouzek K.. Proton Exchange Membrane Water Electrolysers: Materials, Construction and Performance. In Electrochemical Methods for Hydrogen Production; Scott K., Ed.; Royal Society of Chemistry: 2019; pp 83–85. [Google Scholar]
- Ito H.; Maeda T.; Nakano A.; Hasegawa Y.; Yokoi N.; Hwang C. M.; Ishida M.; Kato A.; Yoshida T. Effect of Flow Regime of Circulating Water on a Proton Exchange Membrane Electrolyzer. Int. J. Hydrog. Energy 2010, 35, 9550–9560. 10.1016/j.ijhydene.2010.06.103. [DOI] [Google Scholar]
- Amores E.; Contreras A.; Rodriguez L.; Carrero M.. Flow Field Optimization for PEM Water Electrolysis Cell Using Computational Fluid Dynamic Simulations. In European Hydrogen Energy Conference; Fuel Cells and Hydrogen Joint Undertaking: Malaga, 2018; pp 342–343.
- Li H.; Nakajima H.; Inada A.; Ito K. Effect of Flow-Field Pattern and Flow Configuration on the Performance of a Polymer-Electrolyte-Membrane Water Electrolyzer at High Temperature. Int. J. Hydrog. Energy 2018, 43, 8600–8610. 10.1016/j.ijhydene.2018.02.171. [DOI] [Google Scholar]
- Siroma Z.; Watanabe S.; Yasuda K.; Fukuta K.; Yanagi H. Mathematical Modeling of the Concentration Profile of Carbonate Ions in an Anion Exchange Membrane Fuel Cell. J. Electrochem. Soc. 2011, 158, B682–B689. 10.1149/1.3576120. [DOI] [Google Scholar]
- Lee J. K.; Lee C.; Fahy K. F.; Zhao B.; LaManna J. M.; Baltic E.; Jacobson D. L.; Hussey D. S.; Bazylak A. Critical Current Density as a Performance Indicator for Gas-Evolving Electrochemical Devices Critical Current Density as a Performance Indicator for Gas-Evolving Electrochemical Devices. Cell Rep. Phys. Sci. 2020, 1, 100147. 10.1016/j.xcrp.2020.100147. [DOI] [Google Scholar]
- Yu C.; Cao M.; Dong Z.; Li K.; Yu C.; Wang J.; Jiang L. Aerophilic Electrode with Cone Shape for Continuous Generation and Efficient Collection of H2 Bubbles. Adv. Funct. Mater. 2016, 26, 6830–6835. 10.1002/adfm.201601960. [DOI] [Google Scholar]
- Ahn S. H.; Choi I.; Park H. Y.; Hwang S. J.; Yoo S. J.; Cho E.; Kim H. J.; Henkensmeier D.; Nam S. W.; Kim S. K.; Jang J. H. Effect of Morphology of Electrodeposited Ni Catalysts on the Behavior of Bubbles Generated during the Oxygen Evolution Reaction in Alkaline Water Electrolysis. ChemCommun. 2013, 49, 9323–9325. 10.1039/c3cc44891f. [DOI] [PubMed] [Google Scholar]
- Giordano L.; Han B.; Risch M.; Hong W. T.; Rao R. R.; Stoerzinger K. A.; Shao-Horn Y. PH Dependence of OER Activity of Oxides: Current and Future Perspectives. Catal. Today 2016, 262, 2–10. 10.1016/j.cattod.2015.10.006. [DOI] [Google Scholar]
- Shi Y.; Xie R.; Liu X.; Zhang N.; Aruta C.; Yang N. Tunable PH-Dependent Oxygen Evolution Activity of Strontium Cobaltite Thin Films for Electrochemical Water Splitting. Phys. Chem. Chem. Phys. 2019, 21, 16230–16239. 10.1039/C9CP02278C. [DOI] [PubMed] [Google Scholar]
- Yang C.; Batuk M.; Jacquet Q.; Rousse G.; Yin W.; Zhang L.; Hadermann J.; Abakumov A. M.; Cibin G.; Chadwick A.; Tarascon J. M.; Grimaud A. Revealing PH-Dependent Activities and Surface Instabilities for Ni-Based Electrocatalysts during the Oxygen Evolution Reaction. ACS Energy Lett. 2018, 3, 2884–2890. 10.1021/acsenergylett.8b01818. [DOI] [Google Scholar]
- Liu J.; Kang Z.; Li D.; Pak M.; Alia S. M.; Fujimoto C.; Bender G.; Kim Y. S.; Weber A. Z. Elucidating the Role of Hydroxide Electrolyte on Anion-Exchange-Membrane Water Electrolyzer Performance. J. Electrochem. Soc. 2021, 168, 054522. 10.1149/1945-7111/ac0019. [DOI] [Google Scholar]
- Wang L.; Weissbach T.; Reissner R.; Ansar A.; Gago A. S.; Holdcroft S.; Friedrich K. A. High Performance Anion Exchange Membrane Electrolysis Using Plasma-Sprayed, Non-Precious-Metal Electrodes. ACS Appl. Energy Materials 2019, 2, 7903–7912. 10.1021/acsaem.9b01392. [DOI] [Google Scholar]
- Stanislaw L. N.; Gerhardt M. R.; Weber A. Z. Modeling Electrolyte Composition Effects on Anion-Exchange-Membrane Water Electrolyzer Performance. ECS Trans. 2019, 92, 767–779. 10.1149/09208.0767ecst. [DOI] [Google Scholar]
- Nel Hydrogen Electrolysers: The World’s Most Efficient and Reliable Electrolysers; Nel Hydrogen: 2019; pp 1–9; http://img-admin.exponews.com.au.s3.amazonaws.com/exhibitors/e/nel-electrolysers-brochure-2018-pd-0600-0125-web_18041145.pdf (accessed Jan 2022).
- Janssen H.; Bringmann J. C.; Emonts B.; Schroeder V. Safety-Related Studies on Hydrogen Production in High-Pressure Electrolysers. Int. J. Hydrog. Energy 2004, 29, 759–770. 10.1016/j.ijhydene.2003.08.014. [DOI] [Google Scholar]
- Fateev V. N.; Grigoriev S. A.; Korobtsev S. V.; Porembskiy V. I.. Hydrogen Safety Aspects Related to High Pressure PEM Water Electrolysis. Proceedings of Second International Conference on Hydrogen Safety; International Association for Hydrogen Safety: 2007; pp 1–8.
- Ito H.; Kawaguchi N.; Someya S.; Munakata T. Pressurized Operation of Anion Exchange Membrane Water Electrolysis. Electrochim. Acta 2019, 297, 188–196. 10.1016/j.electacta.2018.11.077. [DOI] [Google Scholar]
- Motealleh B.; Liu Z.; Masel R. I.; Sculley J. P.; Richard Ni Z.; Meroueh L. Next-Generation Anion Exchange Membrane Water Electrolyzers Operating for Commercially Relevant Lifetimes. Int. J. Hydrog. Energy 2021, 46, 3379–3386. 10.1016/j.ijhydene.2020.10.244. [DOI] [Google Scholar]
- Lohmann-Richters F. P.; Renz S.; Lehnert W.; Müller M.; Carmo M. Review—Challenges and Opportunities for Increased Current Density in Alkaline Electrolysis by Increasing the Operating Temperature. J. Electrochem. Soc. 2021, 168, 114501. 10.1149/1945-7111/ac34cc. [DOI] [Google Scholar]
- Truong V. M.; Tolchard J. R.; Svendby J.; Manikandan M.; Miller H. A.; Sunde S.; Yang H.; Dekel D. R.; Oyarce Barnett A. Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells. Energies 2020, 13, 582. 10.3390/en13030582. [DOI] [Google Scholar]
- Dekel D. R.; Amar M.; Willdorf S.; Kosa M.; Dhara S.; Diesendruck C. E. Effect of Water on the Stability of Quaternary Ammonium Groups for Anion Exchange Membrane Fuel Cell Applications. Chem. Mater. 2017, 29, 4425–4431. 10.1021/acs.chemmater.7b00958. [DOI] [Google Scholar]
- Choe Y. K.; Fujimoto C.; Lee K. S.; Dalton L. T.; Ayers K.; Henson N. J.; Kim Y. S. Alkaline Stability of Benzyl Trimethyl Ammonium Functionalized Polyaromatics: A Computational and Experimental Study. Chem. Mater. 2014, 26, 5675–5682. 10.1021/cm502422h. [DOI] [Google Scholar]
- Soni R.; Miyanishi S.; Kuroki H.; Yamaguchi T. Pure Water Solid Alkaline Water Electrolyzer Using Fully Aromatic and High-Molecular-Weight Poly(Fluorene- Alt-Tetrafluorophenylene)-Trimethyl Ammonium Anion Exchange Membranes and Ionomers. ACS Appl. Energy Materials 2021, 4, 1053–1058. 10.1021/acsaem.0c01938. [DOI] [Google Scholar]
- Lettenmeier P.; Kolb S.; Sata N.; Fallisch A.; Zielke L.; Thiele S.; Gago A. S.; Friedrich K. A. Comprehensive Investigation of Novel Pore-Graded Gas Diffusion Layers for High-Performance and Cost-Effective Proton Exchange Membrane Electrolyzers. Environ. Sci. Technol. 2017, 10, 2521–2533. 10.1039/C7EE01240C. [DOI] [Google Scholar]
- Schuler T.; Ciccone J. M.; Krentscher B.; Marone F.; Peter C.; Schmidt T. J.; Büchi F. N. Hierarchically Structured Porous Transport Layers for Polymer Electrolyte Water Electrolysis. Adv. Energy Mater. 2020, 10, 1903216. 10.1002/aenm.201903216. [DOI] [Google Scholar]
- Vincent I.; Lee E. C.; Kim H. M. Highly Cost-Effective Platinum-Free Anion Exchange Membrane Electrolysis for Large Scale Energy Storage and Hydrogen Production. RSC Adv. 2020, 10, 37429–37438. 10.1039/D0RA07190K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczur J. J.; Yang H.; Liu Z.; Sajjad S. D.; Masel R. I. Carbon Dioxide and Water Electrolysis Using New Alkaline Stable Anion Membranes. Front. Chem. 2018, 6, 263. 10.3389/fchem.2018.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.; Paek S. Y.; Lee J. Y.; Park J. H.; Lee S. Y.; Lee Y. M. High-Performance Anion Exchange Membrane Water Electrolyzers with a Current Density of 7.68 A cm–2 and a Durability of 1000 h. Energy. Sci. Technol. 2021, 14, 6338–6348. 10.1039/D1EE02642A. [DOI] [Google Scholar]
- Wang L.; Weissbach T.; Reissner R.; Ansar A.; Gago A. S.; Holdcroft S.; Friedrich K. A. High Performance Anion Exchange Membrane Electrolysis Using Plasma-Sprayed, Non-Precious-Metal Electrodes. ACS Appl. Energy Materials 2019, 2, 7903–7912. 10.1021/acsaem.9b01392. [DOI] [Google Scholar]
- Lindquist G. A.; Oener S. Z.; Krivina R.; Motz A. R.; Keane A.; Capuano C.; Ayers K. E.; Boettcher S. W. Performance and Durability of Pure-Water-Fed Anion Exchange Membrane Electrolyzers Using Baseline Materials and Operation. ACS Appl. Mater. 2021, 13, 51917–51924. 10.1021/acsami.1c06053. [DOI] [PubMed] [Google Scholar]
- Hnát J.; Paidar M.; Schauer J.; Bouzek K. Polymer Anion-Selective Membrane for Electrolytic Water Splitting: The Impact of a Liquid Electrolyte Composition on the Process Parameters and Long-Term Stability. Int. J. Hydrog. Energy 2014, 39, 4779–4787. 10.1016/j.ijhydene.2014.01.086. [DOI] [Google Scholar]
- Hnát J.; Plevová M.; Žitka J.; Paidar M.; Bouzek K. Anion-Selective Materials with 1,4-Diazabicyclo[2.2.2]Octane Functional Groups for Advanced Alkaline Water Electrolysis. Electrochim. Acta 2017, 248, 547–555. 10.1016/j.electacta.2017.07.165. [DOI] [Google Scholar]
- Schauer J.; Hnát J.; Brožová L.; Žitka J.; Bouzek K. Anionic Catalyst Binders Based on Trimethylamine-Quaternized Poly(2,6-Dimethyl-1,4-Phenylene Oxide) for Alkaline Electrolyzers. J. Membr. Sci. 2015, 473, 267–273. 10.1016/j.memsci.2014.09.011. [DOI] [Google Scholar]
- Žitka J.; Peter J.; Galajdová B.; Pavlovec L.; Pientka Z.; Paidar M.; Hnát J.; Bouzek K. Anion Exchange Membranes and Binders Based on Polystyrene-Block-Poly (Ethylene-Ran-Butylene)-Block-Polystyrene Copolymer for Alkaline Water Electrolysis. Desal. Water Treat. 2019, 142, 90–97. 10.5004/dwt.2019.23411. [DOI] [Google Scholar]
- Chi J.; Yu H.; Li G.; Fu L.; Jia J.; Gao X.; Yi B.; Shao Z. Nickel/Cobalt Oxide as a Highly Efficient OER Electrocatalyst in an Alkaline Polymer Electrolyte Water Electrolyzer. RSC Adv. 2016, 6, 90397–90400. 10.1039/C6RA19615B. [DOI] [Google Scholar]
- Niaz A. K.; Akhtar A.; Park J. Y.; Lim H. T. Effects of the Operation Mode on the Degradation Behavior of Anion Exchange Membrane Water Electrolyzers. J. Power Sources 2021, 481, 229093. 10.1016/j.jpowsour.2020.229093. [DOI] [Google Scholar]
- Carbone A.; Zignani S. C.; Gatto I.; Trocino S.; Aricò A. S. Assessment of the FAA3–50 Polymer Electrolyte in Combination with a NiMn2O4 Anode Catalyst for Anion Exchange Membrane Water Electrolysis. Int. J. Hydrog. Energy 2020, 45, 9285–9292. 10.1016/j.ijhydene.2020.01.150. [DOI] [Google Scholar]
- Frensch S. H.; Fouda-Onana F.; Serre G.; Thoby D.; Araya S. S.; Kær S. K. Influence of the Operation Mode on PEM Water Electrolysis Degradation. Int. J. Hydrog. Energy 2019, 44, 29889–29898. 10.1016/j.ijhydene.2019.09.169. [DOI] [Google Scholar]
- Zalitis C. M.; Kramer D.; Kucernak A. R. Electrocatalytic Performance of Fuel Cell Reactions at Low Catalyst Loading and High Mass Transport. Phys. Chem. Chem. Phys. 2013, 15, 4329–4340. 10.1039/c3cp44431g. [DOI] [PubMed] [Google Scholar]
- Inaba M.; Jensen A. W.; Sievers G. W.; Escudero-Escribano M.; Zana A.; Arenz M. Benchmarking High Surface Area Electrocatalysts in a Gas Diffusion Electrode: Measurement of Oxygen Reduction Activities under Realistic Conditions. Environ. Sci. Technol. 2018, 11, 988–994. 10.1039/C8EE00019K. [DOI] [Google Scholar]
- Zhang D.; Zeng K. Evaluating the Behavior of Electrolytic Gas Bubbles and Their Effect on the Cell Voltage in Alkaline Water Electrolysis. Ind. Eng. Chem. Res. 2012, 51, 13825–13832. 10.1021/ie301029e. [DOI] [Google Scholar]
- Ehelebe K.; Seeberger D.; Paul M. T. Y.; Thiele S.; Mayrhofer K. J. J.; Cherevko S. Evaluating Electrocatalysts at Relevant Currents in a Half-Cell: The Impact of Pt Loading on Oxygen Reduction Reaction. J. Electrochem. Soc. 2019, 166, F1259–F1268. 10.1149/2.0911915jes. [DOI] [Google Scholar]
- Beverskog B.; Puigdomenech I. Revised Pourbaix diagrams for nickel at 25–300 °C. Corros. Sci. 1997, 39, 969–980. 10.1016/S0010-938X(97)00002-4. [DOI] [Google Scholar]
- Immerz C.; Paidar M.; Papakonstantinou G.; Bensmann B.; Bystron T.; Vidakovic-Koch T.; Bouzek K.; Sundmacher K.; Hanke-Rauschenbach R. Effect of the MEA design on the performance of PEMWE single cells with different sizes. J. Appl. Electrochem. 2018, 48, 701–711. 10.1007/s10800-018-1178-2. [DOI] [Google Scholar]
- Park Y. S.; Jeong J.; Noh Y.; Jang M. J.; Lee J.; Lee K. H.; Lim D. C.; Seo M. H.; Kim W. B.; Yang J.; Choi S. M. Commercial anion exchange membrane water electrolyzer stack through non-precious metal electrocatalysts. Appl. Catal., B 2021, 292, 120170. 10.1016/j.apcatb.2021.120170. [DOI] [Google Scholar]
- Li M.; Duan K.; Djilali N.; Sui P.-C. Flow sharing and turbulence phenomena in proton exchange membrane fuel cell stack headers. Int. J. Hydrogen Energy 2019, 44, 30306–30318. 10.1016/j.ijhydene.2019.09.140. [DOI] [Google Scholar]
- Bernt M.; Hartig-Weiß A.; Tovini M. F.; El-Sayed H. A.; Schramm C.; Schröter J.; Gebauer C.; Gasteiger H. A. Current Challenges in Catalyst Development for PEM Water Electrolyzers. Chem. Ing. Technol. 2020, 92, 31–39. 10.1002/cite.201900101. [DOI] [Google Scholar]
- Hao M.; Charbonneau V.; Fomena N. N.; Gaudet J.; Bruce D. R.; Garbarino S.; Harrington D. A.; Guay D. Hydrogen Bubble Templating of Fractal Ni Catalysts for Water Oxidation in Alkaline Media. ACS Appl. Energy Materials 2019, 2, 5734–5743. 10.1021/acsaem.9b00860. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.