

Abstract
Mitochondrial dysfunction and oxidative stress are strongly implicated in Parkinson’s disease (PD) pathogenesis and there is evidence that mitochondrially-generated superoxide can activate NADPH oxidase 2 (NOX2). Although NOX2 has been examined in the context of PD, most attention has focused on glial NOX2, and the role of neuronal NOX2 in PD remains to be defined. Additionally, pharmacological NOX2 inhibitors have typically lacked specificity. Here we devised and validated a proximity ligation assay for NOX2 activity and demonstrated that in human PD and two animal models thereof, both neuronal and microglial NOX2 are highly active in substantia nigra under chronic conditions. However, in acute and sub-acute PD models, we observed neuronal, but not microglial NOX2 activation, suggesting that neuronal NOX2 may play a primary role in the early stages of the disease. Aberrant NOX2 activity is responsible for the formation of oxidative stress-related post-translational modifications of α-synuclein, and impaired mitochondrial protein import in vitro in primary ventral midbrain neuronal cultures and in vivo in nigrostriatal neurons in rats. In a rat model, administration of a brain-penetrant, highly specific NOX2 inhibitor prevented NOX2 activation in nigrostriatal neurons and its downstream effects in vivo, such as activation of leucine-rich repeat kinase 2 (LRRK2). We conclude that NOX2 is an important enzyme that contributes to progressive oxidative damage which in turn can lead to α-synuclein accumulation, mitochondrial protein import impairment, and LRRK2 activation. In this context, NOX2 inhibitors hold potential as a disease-modifying therapy in PD.
Keywords: Oxidative stress, NADPH oxidase 2 activity, Parkinson’s disease pathogenesis
Graphical Abstract
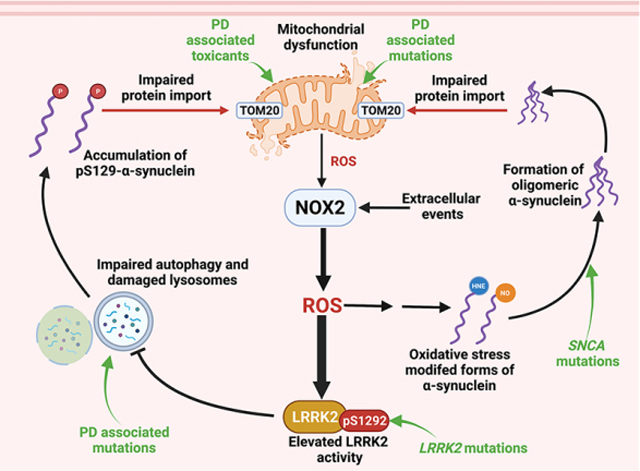
1. Introduction
The pathogenesis of Parkinson’s disease (PD) is multifactorial and the exact mechanisms accounting for nigrostriatal cell loss are still incompletely understood. Oxidative stress has long been implicated in PD (Jenner et al., 1992) and there is consensus on the exquisite vulnerability of dopamine (DA) neurons to oxidative stressors (Sanders and Timothy, 2013; Horowitz et al., 2011; Blesa et al., 2015). Increasing evidence points to the contribution of oxidative mechanisms in α-synuclein misfolding/aggregation (Giasson et al., 2000; Di Maio et al., 2016) and stimulation of LRRK2 activity (Di Maio et al., 2018; Keeney et al., 2021), all of which are key contributors to the progression of idiopathic PD (iPD). However, the therapeutic potential of antioxidant approaches for disease modification in PD has never been adequately tested. Agents that have been used in clinical trials, such as α-tocopherol or coenzyme Q10, have poor brain penetrance and there has been no good measure of target engagement. Moreover, it may be more efficacious to directly inhibit cellular pro-oxidant systems rather than quenching their final products, reactive oxygen species.
Although mitochondrial metabolism is considered an important source of ROS that relates to high levels of aerobic respiration in the brain, there are other major sources of ROS that may contribute to redox disturbances in neurodegeneration. NADPH oxidases (NOXs) are electron transferring membrane protein complexes that function as major enzymatic generators of superoxide anion and hydrogen peroxide (H2O2) (Bedard and Krause, 2007). Despite the ubiquitous expression of the NOX isoforms 1, 2 and 4 in the central nervous system (Ibi et al., 2006; Kim et al., 2005; Vallet et al., 2005; Tammariello et al., 2000), NOX2 is the predominant isoform in brain (Cheng et al., 2001) and has been considered a key player in brain aging (Fan et al., 2019). While a potential role of NOX2 has been examined in iPD (Belarbi et al., 2017) and in PD models (Wu et al., 2003; Zhang et al., 2018; Rodriguez-Pallares et al., 2007; Purisai et al., 2007; Hernandes et al., 2013), previous studies have focused on the relevance of NOX2 in PD-related microglial activation. The role of neuronal NOX2 in PD pathogenesis remains to be explored.
The subcellular localization for most of the NOX isoforms have been identified and varies by cell type. The compartmentalization required for activation of subcellular redox signaling pathways confers specialized cellular functions to the different NOX isoforms. NOX2 is predominantly associated with plasma membrane and endosomes (Ushio-Fukai, 2009). Endosomal trafficking internalizes and re-localizes the NOX2 complex in intracellular organelles, including the endoplasmic reticulum (ER) through inter-organelle membrane contact sites (MCSs) (Henne, 2017). Thus, NOX2 can be associated with the ER and functionally interact with ER-associated organelles, including mitochondria. In certain cellular systems, NOX2 is intimately related with mitochondrial function in a redox regulatory pathway recently termed “ROS-induced ROS production” (RIRP) (Daiber, 2010; Dikalov, 2011). In this scheme, mitochondrially-generated stimulates NOX2 activity and vice versa.
NOX2 (gp91phox) is constitutively associated with membrane-bound p22phox, comprising cytochrome b558. Functional NOX2 requires the association with the cytosolic regulatory subunits p67phox, p40phox to form the NOX2phox complex. A recent study shows that activation of Rac proteins, and their subsequent binding to the NOX2phox complex represents a key event of NOX2 activation (Hoang et al., 2021). However, the binding of the exclusive NOX2 “adaptor”, p47phox to cytochrome b558 is required for the recruitment p40phox and p67phox onto cytochrome b558 to form the active NOX2phox complex (Brown and Griendling, 2009). Thus, the activation of NOX2 mediated by the Rac proteins is strictly related to a p47phox-dependent assembly of the NOX2phox complex. As proof of the essential role of association of p47phox with the NOX2phox complex, p47phox “assembly inhibitors” are the first widely useful, isoform-specific inhibitors of NOX2 (Rey et al., 2001; Altenhofer et al., 2015; Li et al., 2019).
Here we report the development of a proximity ligation (PL) assay that measures the association of p47phox and NOX2 as a surrogate for NOX2 activity. Using this new assay, we examine the neuronal and microglial NOX2 activation state in human PD and in two PD-relevant models in rat and the downstream effects of NOX2 activity, including the formation of post-translationally modified α-synuclein, impaired mitochondrial protein import, and kinase activation of LRRK2. Furthermore, utilizing the bridged tetrahydroquinoline, CPP11G, a small molecule with selective NOX2 assembly inhibitory function (Cifuentes-Pagano et al., 2012; Cifuentes-Pagano et al., 2013), we explore the therapeutic potential of specific NOX2 inhibition in the rotenone model of PD.
2. Materials and methods
2.1. Study design
This study was designed to assess the role of NOX2 in iPD. To do so, we developed a new proximity ligation assay to assess the in-situ interaction of p47phox and NOX2 as a surrogate of NOX2 activity. This assay was validated using CRISPR/Cas9 gene-edited NOX2−/− HEK-293 cells and two highly specific NOX2 inhibitors. The validated assay was used to assess the NOX2 activation state in iPD postmortem brain tissue and in two rat models of PD. Additional studies examined the role of NOX2 activity as well as downstream consequences of NOX2 activity in vivo. All in vitro experiments were replicated at least three times, and key validation studies identifying CRISPR/Cas9 gene-edited cell lines by means of proximity ligation assays were analyzed by blinded assessors. In vivo experiments using rotenone treatment, with or without concomitant CPP11G treatment, were performed in a single cohort of rats (n = 6 per treatment group), and outcomes were analyzed by blinded assessors. Rats were randomized to treatment group. There was no exclusion of outliers.
2.2. Reagents
All reagents were purchased from Sigma-Aldrich, unless otherwise specified. Selective NOX2 inhibitors Nox2ds-tat and CPP11G were provided by Dr. Patrick J. Pagano.
Antibodies were sourced and used as follows:
2.3. Proximity ligation assays
Proximity ligation assay (PLA) (DUO92004–100RXN; DUO92002–100RXN; DUO92007– 100RXN, Sigma-Aldrich) was performed as previously described (Di Maio et al., 2016; Di Maio et al., 2018; Keeney et al., 2021) in 4% PFA (paraformaldehyde)–fixed tissue or cells. Samples were incubated with specific primary antibodies to the proteins to be detected. Secondary antibodies conjugated with oligonucleotides (provided in the Sigma kit) were added to the reaction and incubated. Ligation solution, consisting of two oligonucleotides and ligase, was added. In this assay, the oligonucleotides hybridize to the two proximity ligation probes and join to a closed loop if they are in proximity. Amplification solution, consisting of nucleotides and fluorescently labeled oligonucleotides, was added together with polymerase. The oligonucleotide arm of one of the proximity ligation probes acts as a primer for “rolling-circle amplification” using the ligated circle as a template, and this generates a concatemeric product. Fluorescently labeled oligonucleotides hybridize to the rolling circle amplification product. The proximity ligation signal was visible as a distinct fluorescent spot and was analyzed by confocal microscopy. Control experiments were run in parallel in (i) knockout cells of one of the proteins of interest in presence of an inhibitor of interaction of interest and (iii) primary antibodies deletion. For more details, refer to (Keeney et al., 2021). All information (catalog numbers and dilutions) regarding the antibodies used for the PLAs can be found in Table 1.
Table 1.
Detailed list of the antibodies used in this study.
| Host | Antibody | Catalog # | Vendor | Dilution | Use |
|---|---|---|---|---|---|
|
| |||||
| Mouse | NOX2/gp91phox | ab80897 | Abcam | 1:1000 (ICC) 1:500 (IHC) |
PLA NOX2 activity |
| Rabbit | NCF1[FPR13131–25] (p47phox) | ab181090 | Abcam | 1:1000 (ICC) 1:500 (IHC) |
PLA NOX2 activity |
| Rabbit | NOX1 | ab131088 | Abcam | 1:1000 (ICC) | ICC |
| Rabbit | NOX4 | ab133303 | Abcam | 1:1000 (ICC) | ICC |
| Mouse | α-synuclein | 610,787 | BD Transduction | 1:1000 (ICC) 1:500 (IHC) |
PLA 4-HNE-α-syn PLA α-syn-TOM20 |
| Rabbit | 4-hydroxynonenal | ab46545 | Abcam | 1:1000 (ICC) 1:500 (IHC) |
PLA 4-HNE-α-syn 4-HNE IHC |
| Rabbit | TOM20 | ab186735 | Abcam | 1:500 (IHC) | PLA α-syn-TOM20 |
| Mouse | α-synuclein (211) | ab80627 | Abcam | Conjugated to Probes | PLA oligomeric α-synuclein |
| Rabbit | LRRK2 (pS1292) | ab203181 | Abcam | 1:1000 (ICC) 1:500 (IHC) |
PLA pS1292–LRRK2 (LRRK2 activity) |
| Mouse | LRRK2 (N241A) | 75–253 | UC Davis | 1:1000 (ICC) 1:500 (IHC) |
PLA pS1292–LRRK2 (LRRK2 activity) |
| Mouse | Nitrated α-synuclein | MA5–16142 | Invitrogen | 1:500 (ICC) | NO-α-syn ICC |
| Sheep | Tyrosine Hydroxylase | AB1542 | Millipore | 1:2000 (IHC) | DA neurons marker |
| Rabbit | Iba1 | 019–19,741 | Wako | 1:500 (IHC) | Microglial marker |
| Mouse | α-synuclein (211) | Sc-12,767 | Santa Cruz | 1:500 (WB) (Cells lysates) | Western blot α-synuclein |
| Rabbit | α-synuclein (pS129) | ab51253 | Abcam | 1:1000 (WB) (Cells lysates) | Western blot pS129 α-syn |
| Rabbit | β-actin | ab8227 | Abcam | 1:10,000 (WB) (Cells lysates) | Protein loading control |
| Mouse | β-actin | ab8226 | Abcam | 1:10,000 (WB) (Cells lysates) | Protein loading control |
2.4. NOX2-p47phox co-Immunoprecipitation
Co-immunoprecipitation assay for NOX2-p47phox interaction was performed with Pierce™ Classic Magnetic IP/Co-IP Kit (Thermo Fisher; Catalog number: 88804). The assay was performed in HEK-293 cells lysates according with the procedure (for details, please refer to https://www.thermofisher.com/order/catalog/product/88804?SID=srch-srp-88804).
2.5. Dihydroethidium (DHE) staining
3 mM stock solution of DHE (D11347, Thermo Fisher) was made in DMSO and stored at −20 °C. Prior to use, the 3 mM DHE solution was thawed and protected from light, as DHE is sensitive to light. The 3 mM solution was diluted in cell culture media to reach a final concentration of 3 μM DHE. Old media was aspirated off each coverslip and replaced with the fresh media containing 3 μM DHE. Cells were incubated for 20 min at 37 °C protected from light. The cells were then gently washed with 1× PBS at room temperature for 5 min then fixed with 4% PFA for 20 min. Following fixation, cells were washed 3 × 10 min with 1× PBS. Depending on experiment, cells were either (i) further processed for staining or incubated with DAPI, washed with 1× PBS, and then mounted on glass slides for confocal microscopy imaging. Imaging parameters were set to avoid signal saturation when NOX2 was activated - and those parameters were kept constant across experimental conditions. By doing so, it was very difficult to detect DHE signal in the absence of NOX2 activity. In fact, by turning up the sensitivity (and thereby saturating the DHE signal), it was possible to detect the signal from rotenone alone.
2.6. Fluorescence measurements
Quantitative fluorescence measurements were made with an Olympus upright 3-laser scanning confocal microscope, taking care to ensure that images contained no saturated pixels. For quantitative comparisons, all imaging parameters (e.g., laser power, exposure, pinhole) were held constant across specimens. Depending on the specific experiment, readouts included fluorescence intensity in predefined regions of interest, such as tyrosine hydroxylase-positive dopaminergic neurons or Iba1-positive microglia.
2.7. Cell cultures
Primary ventral midbrain cultures.
Brains extracted from E-17 rat embryos were immersed in cold Neurobasal Medium supplemented with 200 I.U./ml Pennicillin, 200 l.U./ml Streptoycin (Cellgro). Following anatomical landmarks, the SN, without the VTA, was dissected from a coronal midbrain slice. The cells obtained by enzymatic dissociation followed by mechanical trituration, were plated at a density of 5 · 104 cells/cm2 in Neurobasal medium supplemented with 10% FBS and 200 I.U./ml Pennicillin, 200 l.U./ml Streptoycin (Cellgro). Experiments were performed at DIV-10. 24 h before the experiments, the cells were exposed to fresh Neurobasal medium, and all treatments were performed in the same culture medium.
HEK-293 cells were cultured in DMEM/F-12 (cat: 11320082, Thermo Scientific) supplemented with 10% Fetal Bovine Serum (FBS) and Pen-Strep. Cells were maintained at 37 °C and 5% CO2.
2.8. Animals
All experiments utilizing animals were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Retired male breeder Lewis rats were treated with rotenone as described (Greenamyre et al., 2010; Rocha et al., 2020). In brief, rats received a daily intraperitoneal (i.p.) injection of 2.8 mg/kg of rotenone. The acute (1 dose) and sub-acute (5 daily doses) cohorts were euthanized 24 h after the last injection. The endpoint cohort received 9–14 daily injections and were euthanized when they reached their behavioral endpoint.
For CPP11G treatment, rats were dosed with 15 mg/kg (Dr. Patrick J. Pagano) by oral gavage concomitantly with rotenone or vehicle (miglyol).
2.9. AAV2-mediated gene transfer
Vector inoculations were carried out in adult male Lewis rats retired breeders as previously described (Zharikov et al., 2015; Van Laar et al., 2020). 2 μL viral vector suspension was infused dorsal to the substantia nigra (− 5.8 mm anterior/posterior, ±2.2 mm right/left, − 7.5 mm ventral to bregma) using a Hamilton syringe with a 30-gauge needle (45° bevel) at a rate of 0.2 μL/min. AAV2-h-[SNCA] (right side) was infused at a concentration of 2.0 × 1012 GC/ml, giving a total dose 4.0 × 109 GC.
Rats underwent a post-surgery recovery (21 days) After recovery, the animals were euthanized 3, 6 and 12 weeks after the injection.
2.10. Human tissue
Paraffin-embedded midbrain sections were obtained from the University of Pittsburgh Brain Bank. All banked specimens underwent standardized premortem neurological and postmortem neuropathological assessment. Diagnoses were confirmed and staging performed by the study neuropathologist (JKK) by examination of H&E, alpha-synuclein, tau, silver and ubiquitin stains of key sections needed for Braak staging (Alafuzoff et al., 2009). The study design was reviewed and approved by the University of Pittsburgh Committee for Oversight of Research Involving the Dead. Midbrain sections from 6 PD/PDD patients and 7 control subjects, matched for age and postmortem intervals, were used for the analysis; 10–15 cells were imaged per brain section, 3 sections for each patient.
| Male/Female | Age | Brain weight | Postmortem interval | |
|---|---|---|---|---|
| Control | 4/3 | 67 ± 5 | 1248 ± 33 | 7.0 ± 0.8 |
| PD | 5/1 | 73 ± 4 | 1297 ± 29 | 9.8 ± 1.9 |
To eliminate endogenous fluorescence, human tissue was pre-treated with Sudan Black, an autofluorescence eliminating reagent according to the manufacturer’s instructions (Chemicon, Temecula, CA).
2.11. Detection of CPP11G and rotenone concentrations in rat brains
1.5 pmol CPP11H internal standard was added to 50 μl of rat brains homogenates and 300 μl acetonitrile was added for extraction. After sonication and centrifugation, the supernatants were analyzed for CPP11G and rotenone by high performance liquid chromatography-electrospray ionization-tandem mass spectrometry. After chromatographic resolution, CPP11G and rotenone were detected using a 5000 triple quadrupole mass spectrometer (Applied Biosystems, San Jose, CA) equipped with an electrospray ionization source (ESI) in positive mode. The following parameters were used for the analysis of CPP11G: declustering potential (DP) 80 V, entrance potential (EP) 10 V, collision cell exit potential (CXP) 15 V, and a desolvation temperature of 700 °C. Multiple reaction monitoring (MRM) transitions were used with the best collision energy: MRM 390.1/334.1, 390.1/278 and 390.1/207.9 with 20 CE eV, 35 eV, and 45 eV respectively. For rotenone analysis: DP 80 V, EP 5 V, 32 CE eV, CXP 13 V, MRM 395.3/367.2, 395.3/213.2 and 395.3/192.2. Quantitation of CPP11G in rat brain was performed using CPP11G calibration curves in presence of CPP11H as internal standard (MRM 416.1/97). Levels of CPP11G in rat brain were reported as pmol/mg protein. Quantitation of rotenone in rat brain was performed using an external calibration curve and levels of rotenone were reported as pmol/mg protein.
2.12. CRISPR/Cas9 genome editing of HEK-293 cells to produce NOXs knockouts cell lines
NOX1, NOX2 and NOX4 knockouts cell lines were generated by Synthego Corporation using CRISPR/Cas9 genome editing technology. We received the gene-edited cell clones that were grown and expanded for polymerase chain reaction (PCR), DNA sequence analyses and immunocytochemistry for the proteins of interest (Supplemental Fig. 5).
2.13. Statistical analyses
Each result presented here was derived from three to six independent experiments. For simple comparisons of two experimental conditions, two-tailed, unpaired t-tests were used. When AAV2 vector was injected into one hemisphere of the rat brain and the other hemisphere was used as a control, two-tailed paired t-tests were used. For comparisons of multiple experimental conditions, one-way or two-way ANOVA was used, and if significant overall, post hoc Bonferroni corrections for multiple pairwise comparisons were made. P values less than 0.05 were considered significant.
3. Results
3.1. NOX2 activity assay validation
We developed and validated a novel proximity ligation (PL) assay with excellent anatomical and cellular resolution that can provide information regarding NOX2 activation state, which is based on the in-situ detection of the p47phox–NOX2 interaction. Mitochondrial ROS are known to activate NOX2 (Dikalov, 2011; Hoang et al., 2021; Brown and Griendling, 2009), and in this context, we found an increased PL signal for NOX2 activity in primary rat ventral midbrain neuronal (pVMB) cultures exposed for 24 h to a sublethal concentration (50 nM) of the mitochondrial complex I inhibitor, rotenone (Fig. 1A, B; p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction). pVMB cultures were simultaneously assayed in parallel for superoxide production using dihydroethidium (DHE) and after rotenone treatment were found to have an increased signal consistent with increased NOX2 activity (Fig. 1A, C p. < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction). Co-treatment with the NOX2 peptidic assembly inhibitor, Nox2ds-tat (10 μM), was effective in preventing both PL and DHE signals (Fig. 1A–C; p < 0.0001 compared to rotenone; ANOVA with Bonferroni’s correction). The scrambled control peptide variant of Nox2ds-tat, Scr. Nox2ds-tat (10 μM), was without effect and did not prevent the rotenone-induced NOX2 activation or the associated superoxide production. Similarly, as shown in Fig. 1 D–F, rotenone treatment in wild type HEK- 293 cells, elicited a significant increase of NOX2 activity and superoxide production detected as p47phox-NOX2 PL signal and DHE fluorescence, respectively (p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction). Rotenone-related NOX2 activity and superoxide production were prevented in CRISPR/Cas9 gene-edited NOX2−/− HEK-293 cells (p < 0.0001 compared to rotenone treated wild type cells; ANOVA with Bonferroni’s correction) and in wild type HEK-293 cells co-treated with Nox2ds-tat (p < 0.0001 compared to rotenone; ANOVA with Bonferroni’s correction). Scr.Nox2ds-tat failed to prevent rotenone-induced NOX2 activation and the associated superoxide production in wild type HEK-293 cells. To assess the sensitivity of p47phox–NOX2 PL signal, we exposed wild type HEK-293 cells for 24 h to 50 nM rotenone and we performed simultaneous dose-response analyses of Nox2ds-tat or CPP11G (Li et al., 2019; Cifuentes-Pagano et al., 2013) by analyzing p47phox–NOX2 PL and superoxide products (DHE) (Fig. 1G, H). Both inhibitors dose-dependently blocked rotenone-induced NOX2 activity (PL signal) and, in parallel, reduced the generation of superoxide (DHE signal). A more conventional NOX2 assay, that lacks anatomical or subcellular resolution, depends on co-immunoprecipitation of p47phox with NOX2 to show activation of the enzyme. Using this assay (Supplemental Fig. 1), we confirmed that rotenone treatment activates NOX2, and Nox2ds-tat blocks this effect, as indicated by our PL assay.
Fig. 1.
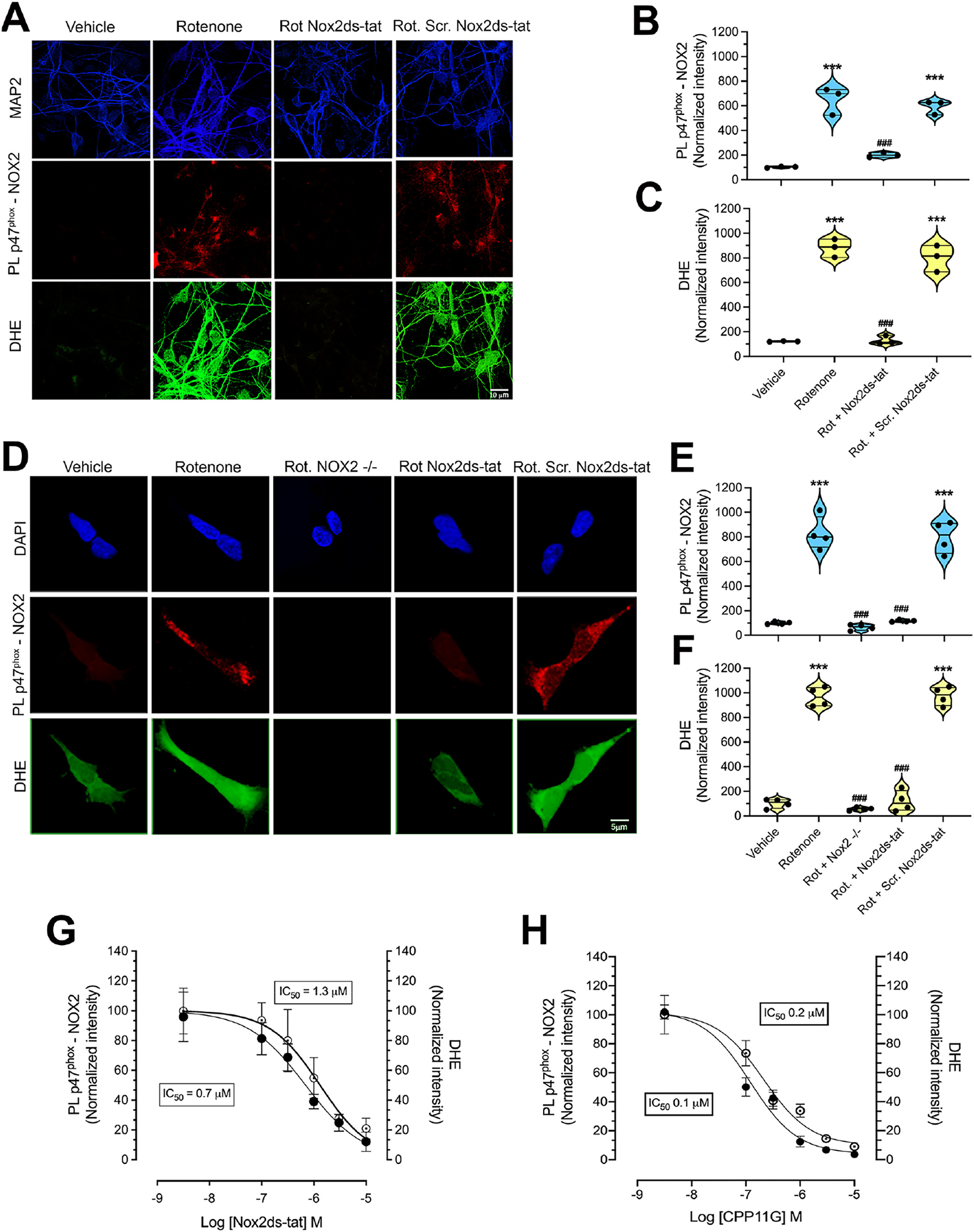
Validation of the p47phox-NOX2 proximity ligation assay as an index of NOX2 activity in situ.
(A) p47phox-NOX2 PL signal (red) and DHE-related fluorescent signal (green) in pVMB cultures. Cells treated for 24 h with a sublethal concentration (50 nM) of the complex I inhibitor, rotenone, showed increased PL signal for p47phox-NOX2 interaction and increased cytosolic superoxide production detected as DHE fluorescence. This cellular response to rotenone exposure was prevented in co-treatment with Nox2ds-tat, but not by the scrambled variant, Scr. Nox2ds-tat. (B, C) Quantification of the p47phox-NOX2 PL signal and DHE-related fluorescence intensity in pVMB cultures. Symbols represent the normalized means of the intensities (with vehicle treatment being set at 100) analyzed for each independent experiment (100–150 neurons/treatment group per experiment). Statistical analysis was performed as one-way ANOVA with Bonferroni’s correction (n = 3 independent experiments). (D) Similarly, a significant increase of PL signal for p47phox-NOX2 interaction and DHE fluorescence was observed in wild-type HEK-293 cells exposed to rotenone for 24 h. Both signals for p47phox-NOX2 interaction and DHE were prevented in CRISPR/Cas9 gene-edited NOX2−/− HEK-293 cells and by co-treatment with Nox2ds-tat in wild-type HEK-293. Scr. Nox2ds-tat co-treatment failed to prevent the rotenone induced increase in p47phox-NOX2 interaction and DHE fluorescence. (E, F) Quantification of the p47phox-NOX2 PL signal and DHE-related fluorescence intensity in HEK-293 cells. Symbols represent the normalized means of the intensities (with vehicle treatment being set at 100) analyzed for each independent experiment (100–150 cells/treatment group per experiment). Statistical analysis was performed as one-way ANOVA with Bonferroni’s correction (n = 4 independent experiments) In plots B, C, E and F, ***denotes p < 0.0001 significance compared vehicle; ###denotes p < 0.0001 significance compared to rotenone. (G, H) Dose response of PL p47phox-NOX2 and DHE signals to NOX2 inhibitors. HEK-293 cells were treated with rotenone (50 nM) for 24 h to activate NOX2 in the absence or presence of increasing concentrations of inhibitors (30 nM to 10 μM for both compounds). Quantification of the PL and DHE signals shows that, in a dose-dependent manner, the highly specific NOX2 inhibitors, Nox2ds-tat and CPP11G reduced the p47phox-NOX2 PL signal (black symbols) (IC50: 0.7 μM for Nox2ds-tat and 0.1 μM for CPP11G) paralleled by attenuation of DHE fluorescence (open symbols) (IC50: 1.3 μM for Nox2ds-tat and 0.2 μM for CPP11G; n = 3 independent experiments).
3.2. Direct evidence of NOX2 activity in brain tissue from iPD subjects
To determine whether NOX2 activation in substantia nigra pars compacta (SNpc) constitutes a relevant event in iPD, we performed PL p47phox–NOX2 assay in a blinded fashion in postmortem substantia nigra tissue sections from individuals who had died with PD and control subjects. Compared to control subjects, both nigrostriatal dopamine neurons and microglia from PD cases had a strong PL signal, indicating NOX2 activation (Fig. 2A, B; p < 0.0001 for DA neurons and p < 0.0001 for microglia compared to control patients; unpaired two-tailed t-test). To our knowledge, this is the first demonstration on a cell-by-cell basis that NOX2 activity is elevated in both nigrostriatal dopamine neurons and in microglia in individuals with PD.
Fig. 2.
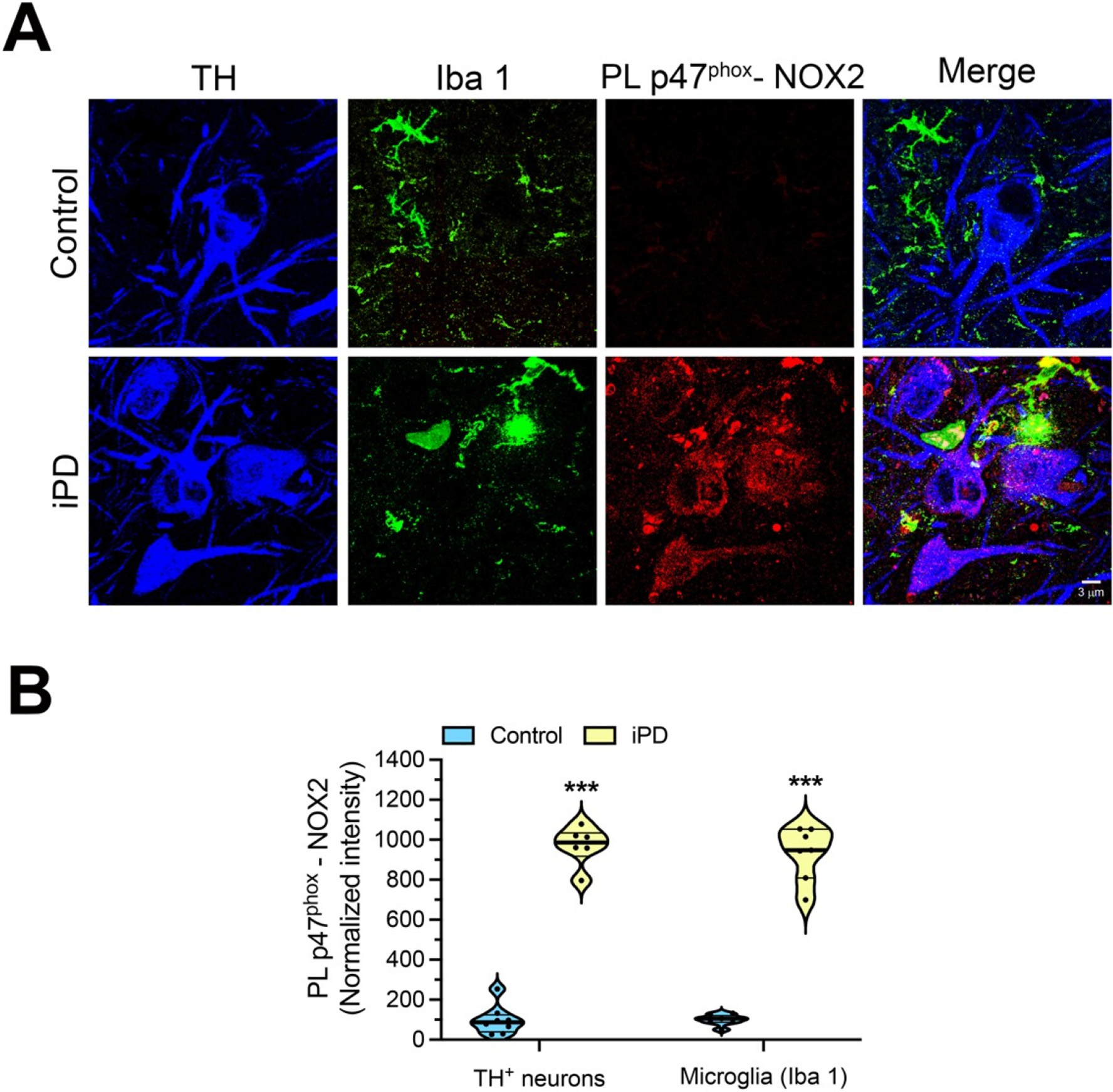
NOX2 activity in human iPD postmortem brain tissue.
(A) Representative images of p47phox-NOX2 PL signal (red) in midbrain sections from a healthy, age- matched control human brain (top row) and a brain from an individual with iPD (bottom row). Compared to control brains, the PD brains show a strong p47phox-NOX2 PL signal in tyrosine hydroxylase (TH) positive neurons (blue) and in microglia (Iba1; green). (B) Quantification of p47phox-NOX2 PL signal. Symbols represent the normalized mean of the intensities (with control being set to 100) analyzed for each patient (n = 7 control brains and 6 PD brains, 10–15 cells imaged per brain section, 3 sections for each subject). Statistical comparisons by unpaired two-tailed t-test for neurons and microglia. ***denotes p < 0.0001 significance compared to controls.
3.3. Neuronal NOX2 activation is an early phenomenon in PD animal models
Despite evidence that shows NOX2 is expressed in neurons (Kim et al., 2005), the topic remains controversial. To address this issue, we performed confocal immunofluorescent analysis in rat nigrostriatal neurons. As shown in Supplemental Fig. 2A (top row), SNpc dopamine neurons express both NOX2 and p47phox under basal conditions. Systemic inhibition of mitochondrial complex I with rotenone reproduces many features of PD in rats (Rocha et al., 2020). Utilizing a sub-acute rotenone model in rats (5 days treatment), we observed a modest, but significant increase of NOX2 expression (p < 0.05; unpaired t-test), but not p47phox (Supplemental Fig. 2 B, C). Performing p47phox-NOX2 correlation analysis (Pearson index; Supplemental Fig. 2C), we found a significant increase of the correlation index (colocalization) of p47phox with NOX2 in rotenone treated rats (p < 0.0001; unpaired t-test), consistent with the fact that NOX2 activity is related to p47phox-NOX2 association.
Previous studies reported evidence of microglial NOX2 upregulation in PD patients and in animal models of PD (Wu et al., 2003; Zhang et al., 2018; Rodriguez-Pallares et al., 2007; Purisai et al., 2007; Hernandes et al., 2013). However, the role of neuronal NOX2 remains relatively unexplored. Using the p47phox–NOX2 PL assay, we investigated the relative timing of NOX2 activation in microglia compared to dopamine neurons in (i) acute (1 day treatment), sub-acute (5 days treatment) and (iii) chronic (11–14 days treatment) rotenone models in rats (Fig. 3 A, B). Acute administration (1 day) of rotenone elicited NOX2 activation in rat dopamine neurons (p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction), but not in microglia. Under sub-acute conditions (rotenone treatment for 5 days), we still observed NOX2 activation in dopamine neurons (p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction), but this was now accompanied by an increase in microglial NOX2 activity (p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction). With chronic rotenone treatment, there was equivalent NOX2 activation in dopamine neurons and nearby microglia (p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction).
Fig. 3.
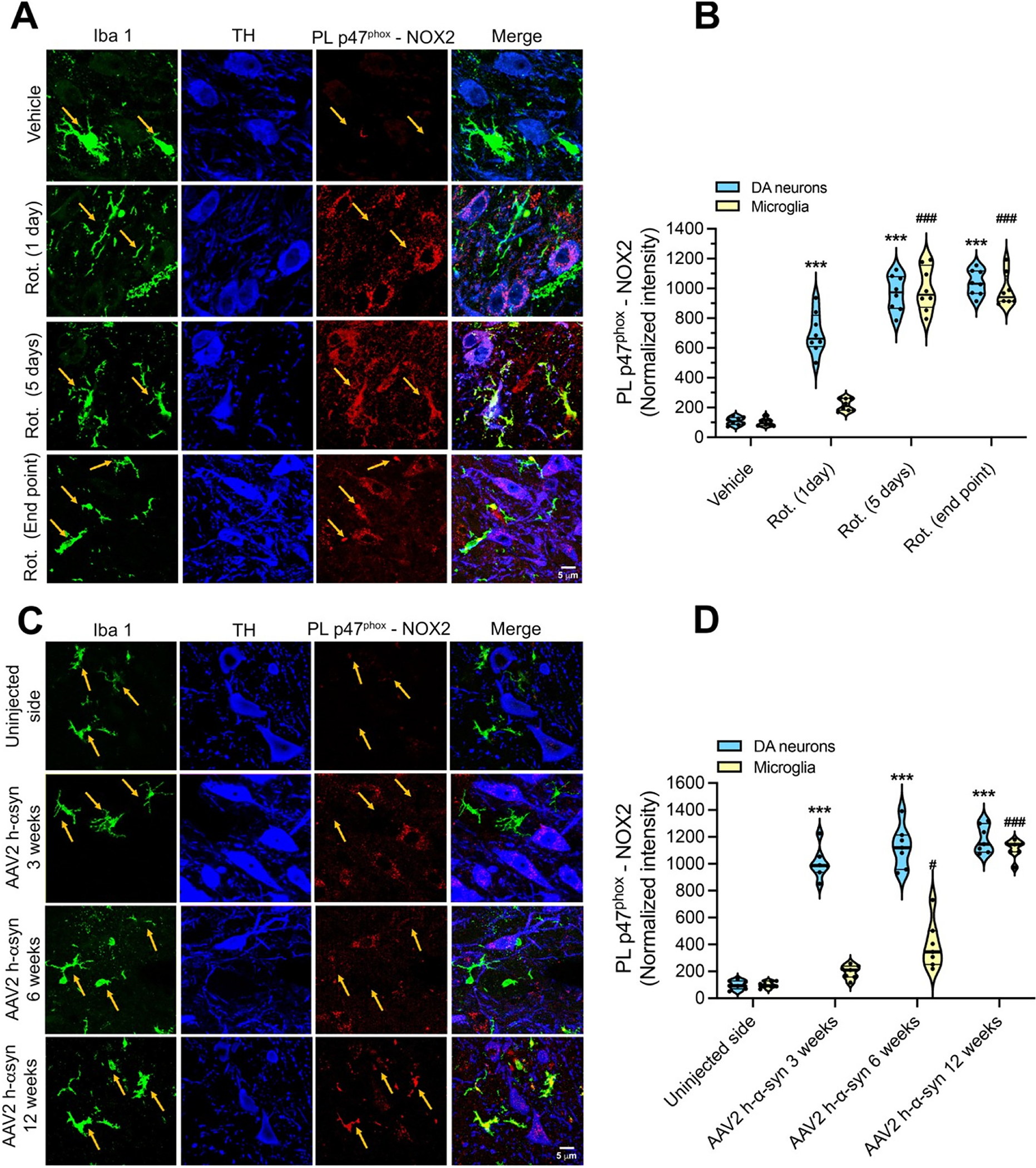
Dopamine neurons show earlier NOX2 activation than microglia in two rat models of PD.
(A) Representative images showing p47phox-NOX2 PL signal (red) in DA neurons (TH; blue) and microglia (Iba-1; green) in substantia nigra sections from rats treated with vehicle (first row), rotenone for 1 day (second row), 5 days (third row) or endpoint rotenone treatment (10–14 days; fourth row). Rats treated for 1 day showed activation of NOX2 in nigrostriatal DA neurons but not microglia. Five days of rotenone treatment elicited an increase in p47phox-NOX2 PL signal in both DA neurons and in microglia. A similar NOX2 activity state was detected in endpoint treated rats. (B) Quantification of microglial and neuronal NOX2 activity by PL p47phox-NOX2. Symbols represent the normalized mean intensity (with vehicle being set to 100) analyzed in single animals (n = 6 animals/treatment group: 4 sections/animal and 30–40 cells.
/Section were measured.). Statistical comparison was performed by one-way ANOVA with Bonferroni’s correction. ***denotes p < 0.0001 significance compared to vehicle for DA neurons; ###p < 0.0001 significance compared to vehicle for microglia. (C) Shown are p47phox-NOX2 PL signal in DA neurons (TH; blue) and microglia (Iba1; green) in the substantia nigra from rats that received a unilateral nigral injection of AAV2 h-α-syn. Three weeks after AAV2 h-α-syn injection (second row), a significant increase in p47phox-NOX2 PL signal, compared to the contralateral un-injected nigra (first row), was observed in DA neurons but not in microglia. Microglial NOX2 activity started to appear after 6 weeks from AAV2 h-α- syn injection (third row), with a maximal activation observed in the AAV2 h-α-syn injected nigra after 12 weeks. (D) Quantification of p47phox-NOX2 PL signal in nigrostriatal dopamine neurons and microglia from control (uninjected) and AAV2 h-α-syn injected rat brain hemispheres. Symbols represent the normalized mean intensity (with the uninjected hemisphere being set to 100) from each hemisphere in each animal (n = 6 animals/time-point) Statistical comparison was performed by one-way ANOVA with Bonferroni’s correction. ***denotes p < 0.0001 significance compared to control for DA neurons. #denotes p < 0.05 and ###denotes p < 0.0001 significance compared to control for microglia.
Using another more chronic model of PD, AAV2-mediated human α-synuclein over-expression in SNpc (Van Laar et al., 2020), we assessed neuronal and microglial NOX2 activation 3 weeks, 6 weeks, and 12 weeks post-AAV2 h-α-syn injection. GFP control does not show evidence of neurodegeneration or oxidative stress (Zharikov et al., 2015). At each of these time points, the PL assay revealed robust NOX2 activity in nigral dopamine neurons (Fig. 3C, D; p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction). A significant increase of microglial NOX2 activity was observed only after 12 weeks post AAV2 h-α-syn injection (Fig. 3C, D; p < 0.0001 compared to non-injected side; ANOVA with Bonferroni’s correction). Thus, as in the rotenone model, neuronal NOX2 activation precedes microglial NOX2 activation – in this case, by several weeks. This may indicate that neuronal NOX2 activation can be an early event in PD pathogenesis and that the resultant neuronal damage leads to microglial activation.
3.4. Rotenone activation of NOX2 depends on mitochondrial ROS
A growing body of evidence suggests that NOX2 can be activated by mitochondrial-derived ROS in a process known as RIRP. To assess this possibility, we explored if this phenomenon occurs in pVMB cultures by performing our NOX2 PL assay at various time points after exposure to rotenone (from 2 to 24 h). As shown in Fig. 4, we detected increased NOX2 activity after 2 h of 50 nM rotenone exposure with a maximal response at 4 h (Fig. 4A, B; p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction) and a sustained elevation for at least 24 h. Co-treatment with the mitochondrial superoxide scavenger, MitoTEMPO (25 nM), prevented rotenone induced NOX2 activity at all timepoints (Fig. 4A, B; p < 0.0001 compared to rotenone; ANOVA with Bonferroni’s correction). Consistent with these results, treatment of HEK-293 cells with rotenone (50 nM) for 24 h elicited NOX2 activation and superoxide production (detected as DHE fluorescence), both of which were prevented by co-treatment with MitoTEMPO (Supplemental Fig. 3; p < 0.0001 compared to rotenone; ANOVA with Bonferroni’s correction). Together, these data obtained in 2 distinct cell types, indicate that mitochondrial superoxide production can lead to NOX2 activation, thereby confirming a functional link between mitochondrial function and NOX2 activity. Importantly, these findings also rule out a direct effect of rotenone on NOX2 activity.
Fig. 4.
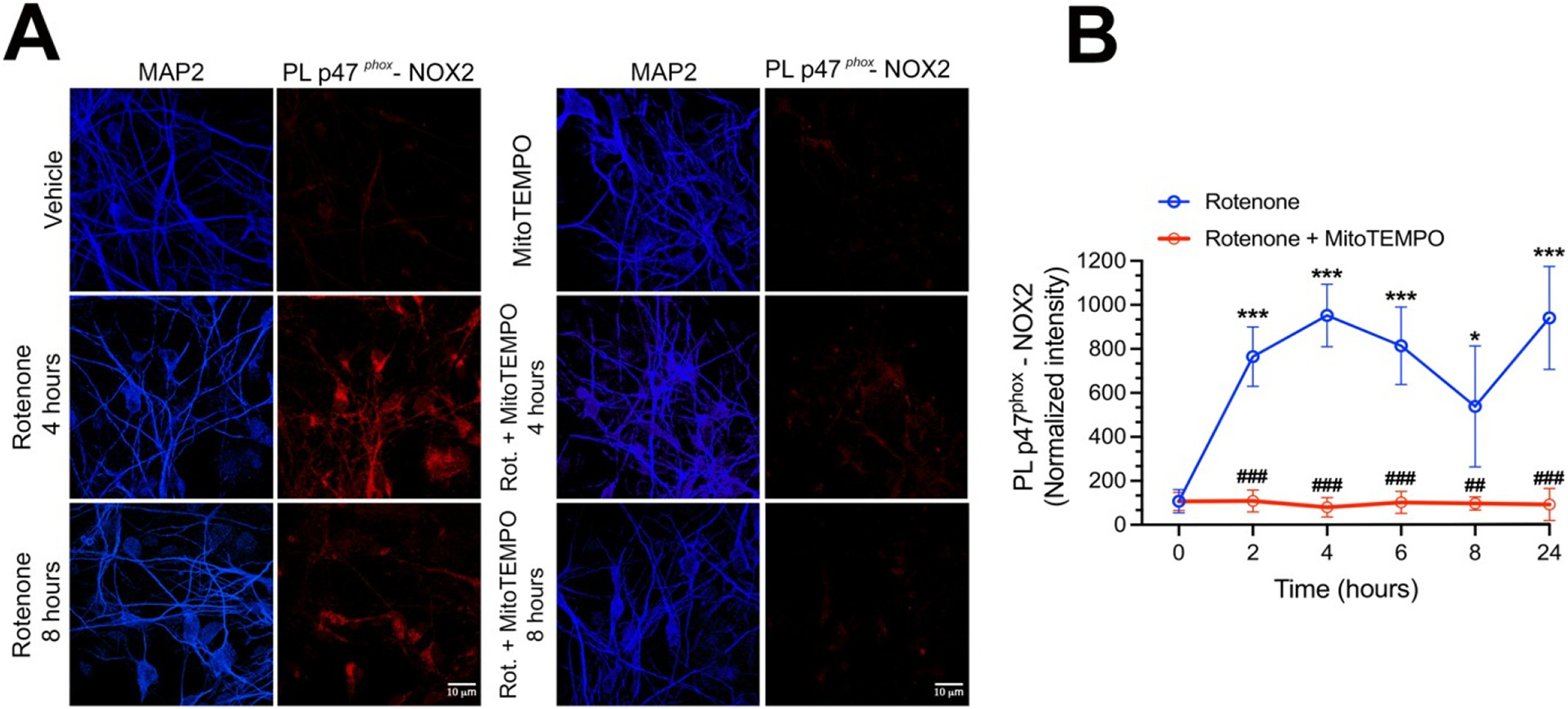
Rotenone induced mitochondrial superoxide elicits NOX2 activation.
(A) Representative images of p47phox-NOX2 PL signal (red) in pVMB neurons exposed 4 or 8 h to rotenone (left column), or rotenone + MitoTEMPO (right column). (B) Quantification and time course of PL signal for NOX2 activity induced by rotenone (blue line) or rotenone + MitoTEMPO (red line) exposure. As shown, rotenone elicited NOX2 activation (detected as p47phox-NOX2 PL signal) within 2 h and activation persisted for at least 24 h of rotenone exposure. Co-treatment with the mitochondrial superoxide scavenger, MitoTEMPO (25 nM) prevented NOX2 activity at all time points, indicating that mitochondrial ROS are responsible for NOX2 activation. Symbols represent the normalized average of cellular fluorescence intensities (with vehicle set to 100) analyzed in 3 independent experiments: (100–150 neurons/treatment group). Statistical comparison by one-way ANOVA with Bonferroni’s correction.
*denotes p < 0.05 and ***denotes p < 0.0001 significance compared to vehicle; ###denotes p < 0.0001 significance compared to rotenone.
3.5. Effects of NOX2 activity on α-synuclein
Post-translational modifications (PTM) of α-synuclein are believed to be important in the pathogenesis of PD and are associated with SNpc dopamine neuron degeneration (Alafuzoff et al., 2009). On the other hand, how the formation of such neurotoxic PTMs of α-synuclein occurs is not clear. Oxidative stress-related PTMs forms of α-synuclein, including 4-hydroxy-2-nonenal-α-synuclein (4-HNE-α-syn) and nitrated α-synuclein (NO-α-syn) have been shown to promote α-synuclein oligomerization (Oueslati et al., 2010; Xiang et al., 2013). To elucidate the role of NOX2 in α-synuclein modification, we used pVMB neuronal cultures. Rotenone treatment for 24 h elicited a significant accumulation of 4-HNE- α-syn adducts (detected as PL 4-HNE-α-syn) and, co-treatment with Nox2ds-tat, was effective in preventing this cellular response to rotenone. As control, co-treatment with Scr.Nox2ds-tat did not prevent rotenone-induced accumulation of 4-HNE-α-syn (Fig. 5 A, B; p < 0.0001 compared to vehicle; ANOVA with Bonferroni’s correction).
Fig. 5.
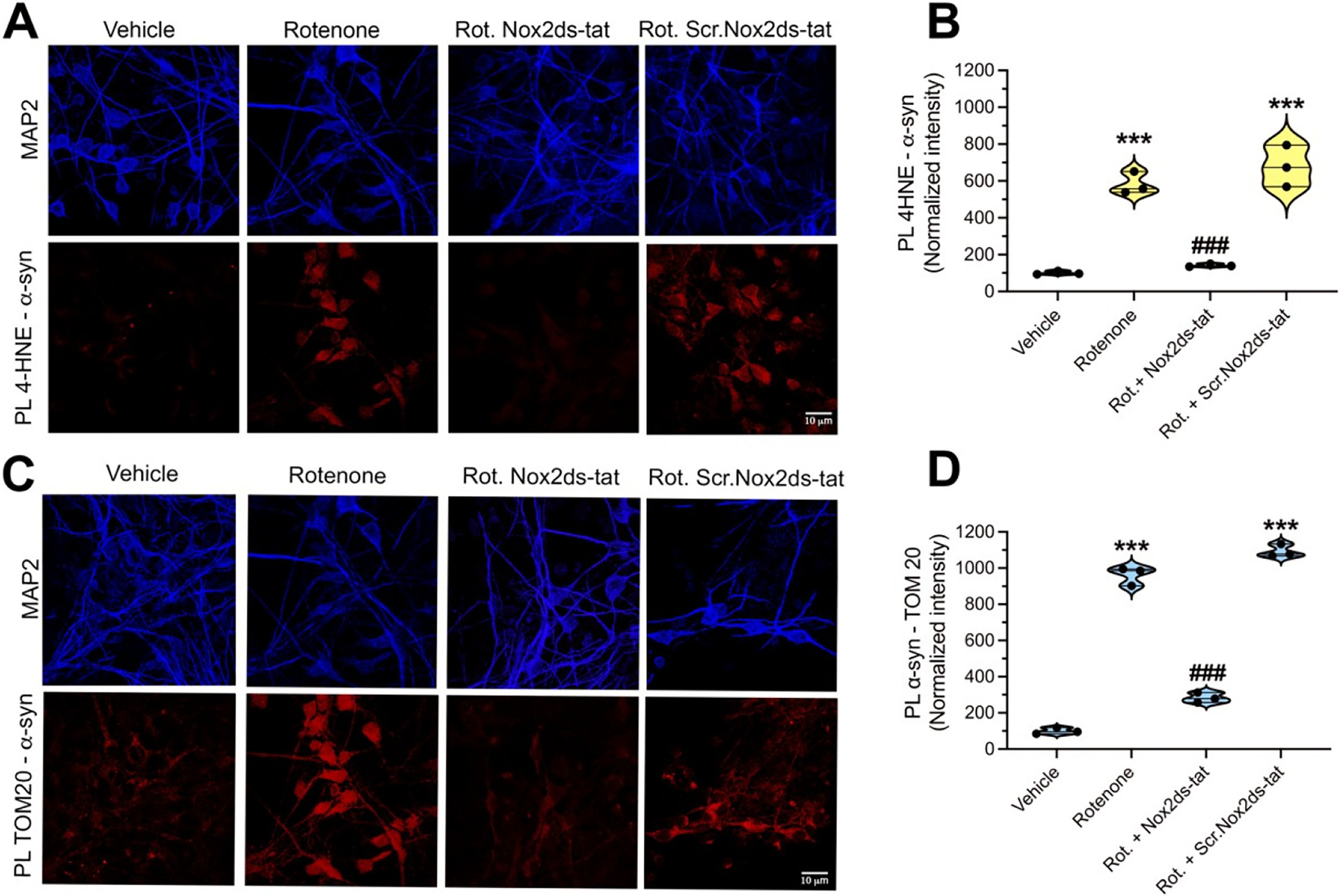
NOX2-dependent accumulation of PTM forms of α-synuclein.
(A) Confocal analysis of 4-HNE-α-syn adducts, detected as 4-HNE–α-syn PL signal, in pVMB neurons. Cells exposed to rotenone for 24 h showed significant increase of 4-HNE–α-syn, which was prevented by co-treatment with Nox2ds-tat but not Scr.Nox2ds-tat, indicating a role for NOX2 in the formation of this highly aggregable form of α-syn. (B) Quantification of PL 4-HNE–α-syn signal. Symbols represent the normalized means of the intensities (with vehicle set to 100) analyzed for each independent experiment (100–150 neurons/treatment group per experiment). Statistical analysis was performed as one-way ANOVA with Bonferroni’s correction (n = 3 independent experiments). ***denotes p < 0.0001 significance compared vehicle; ###denotes p < 0.0001 significance compared to rotenone. (C) Rotenone induced interaction of α-syn with TOM20 is prevented by NOX2 inhibition, consistent with the involvement of NOX2 in the cellular accumulation of posttranslational forms of α-syn able to interfere with TOM20- mediated protein import. (D) Quantification of the PL TOM20–α-syn signal. Symbols represent the normalized means of the intensities (with vehicle set to 100) analyzed for each independent experiment (100–150 neurons/treatment group per experiment). Statistical analysis was performed by one-way ANOVA with Bonferroni’s correction (n = 3 independent experiments). ***denotes p < 0.0001 significance compared to vehicle; ###denotes p < 0.0001 significance compared to rotenone.
We previously reported that certain modified forms of α-synuclein, including pSer129-α-synuclein and oligomeric α-synuclein, bind to TOM20 of the mitochondrial protein import machinery and cause mitochondrial protein import impairment and mitochondrial dysfunction (Di Maio et al., 2016). In the current study, we assessed the downstream consequences of NOX2-induced α-synuclein PTMs. Proximity ligation assays (α-synuclein–TOM20) in pVMB cultures exposed to rotenone for 24 h revealed a strong α-synuclein–TOM20 interaction. Co-treatment with Nox2ds-tat prevented rotenone-induced α-synuclein–TOM20 interaction; however, this interaction was still observed in pVMB co-treated with Scr.Nox2ds-tat (Fig. 5 C, D; p < 0.0001; ANOVA with Bonferroni’s correction).
We further explored the downstream effects of NOX2 activation on α-synuclein modifications using HEK-293 cells. In this system, we found that treatment of cells with rotenone for 24 h increased levels of total α-synuclein (Supplemental Fig. 4 A, B) and pSer129α-Syn (Supplemental Fig. 4C, D). Notably, the effects of rotenone were prevented in NOX2−/− HEK- 293 cells (P < 0.0001 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction) and by Nox2ds-tat co-treatment in wild-type cells (P < 0.0006 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction). Using HEK-293 cells, we further investigated the role of NOX2 activity in the formation of PTM forms of α-synuclein, including 4-HNE-α-syn, NO-α-syn and oligomeric α-syn. PL of 4-HNE-α-syn and immunofluorescence analysis of NO-α-syn revealed a robust increase of cellular levels of these oxidative stress-modified forms of α-synuclein in rotenone-treated HEK-293 cells. Formation of both rotenone induced 4-HNE-α-syn adducts and NO-α-Syn was prevented in NOX2−/− cells or wild-type cells co-treated with Nox2ds-tat (Supplemental Fig. 4 E, F for HNE-α-Syn; P < 0.0001 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction. Supplemental Fig. 4 G, H for NO-α-Syn; P < 0.0001 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction). We also assayed the rotenone-induced formation of oligomeric α-synuclein by proximity ligation assay (AS-PLA); as previously described (Roberts et al., 2019). This assay recognizes oligomeric species of α-synuclein (dimeric to higher molecular weight species), not monomeric or fibrillar α-synuclein (Roberts et al., 2019).Treatment with rotenone for 48 h elicited the formation of α- synuclein oligomers (Supplemental Fig. 4 I & J) and this was prevented in NOX2−/− HEK-293 cells (p < 0.0001 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction) and by Nox2ds-tat co-treatment in wild-type cells (p < 0.0001 compared to wild-type-treated cells; ANOVA with Bonferroni’s correction). Together these results indicate that oxidative modifications of α-synuclein may constitute a link between NOX2 activation and downstream mitochondrial dysfunction.
Neurons and microglia co-express other NOX isoforms, with most abundant being NOX1 and NOX4. We utilized CRISPR/Cas9 gene-edited NOX1−/− and NOX4−/− HEK-293 cells (Supplemental Fig. 5 A, B) to investigate the role of these NOX isoforms in rotenone-mediated formation of 4-HNE-α-syn. Rotenone induced formation of 4-HNE-α-syn was prevented only in NOX2−/− cells, not in NOX1−/− or NOX4−/− cells. These data demonstrate for the first time, NOX2 activity is critical for the formation and accumulation of an oxidative stress-related PTM forms of α-synuclein (Supplemental Fig. 5 B, C).
3.6. A brain-penetrant inhibitor prevents nigrostriatal NOX2 activation and its downstream effects
To provide evidence of CPP11G blood brain barrier (BBB) permeability and persistence in brain tissue, we performed mass spectrometry analysis to measure the presence of CPP11G in rat brains 24 h after a single oral administration of CPP11G (15 mg/kg). At this prolonged timepoint, CPP11G was readily detectable in whole brain lysates, indicating BBB penetrance and persistence of the NOX2 inhibitor (Supplemental Fig. 6A). Similarly, mass spectrometry analysis after a single intraperitoneal dose of rotenone (2.8 mg/kg), revealed its residual presence in brain 24 h after injection (Supplemental Fig. 6B). Mass spectrometry also showed that when rotenone was co-administered with CPP11G, it did not alter the brain concentration of the NOX2 inhibitor. Moreover, co-administration of CPP11G did not change the brain concentration of rotenone (Supplemental Fig. 6 A, B), suggesting that the two compounds do not interfere with the metabolism of each other.
In this study, we also examined whether the NOX2 inhibitor, CPP11G, was able to block rotenone induced NOX2 activation in SNpc dopamine neurons in vivo, as well as its downstream sequelae. Twenty-four hours after a single rotenone administration (2.8 mg/kg; i.p.), nigrostriatal neurons showed a marked elevation of NOX2 activity, detected as PL p47phox–NOX2 (Supplemental Fig. 6C, D). When CPP11G was co-administered with rotenone, it prevented the rotenone induced NOX2 activation in dopamine neurons when assessed 24 h after treatment.
The rotenone model of PD reproduces many features of the human disease, including oxidative stress, LRRK2 activation, and reduced mitochondrial protein import (Sanders and Timothy, 2013; Horowitz et al., 2011; Di Maio et al., 2016; Di Maio et al., 2018). To determine whether a NOX2 inhibitor could prevent rotenone induced, PD-relevant downstream effects in vivo, we treated rats for 5 days with rotenone (2.8 mg/kg/day, i.p.) with or without CPP11G (15 mg/kg/day, p.o.). Daily systemic administration of rotenone causes a progressive weight loss in rats, likely due to its deleterious effects on mitochondria. Here, 5 days of rotenone treatment caused a 10% weight loss compared to vehicle control animals. Co-treatment with CPP11G prevented rotenone-induced loss of weight, suggesting a critical role of NOX2 in the systemic toxicity of rotenone exposure (Supplemental Fig. 7).
Five days of rotenone treatment led to a robust increase in p47phox–NOX2 PL signal in nigrostriatal dopamine neurons, which was associated with an increase in lipid peroxidation, detected as 4-HNE levels (Fig. 6 A–D). Co-treatment with CPP11G effectively blocked the rotenone induced activation of NOX2 (p < 0.0001, two-way ANOVA with Bonferroni’s correction) and formation of 4-HNE (P < 0.0001; two-way ANOVA with Bonferroni’s correction). We previously reported that LRRK2 kinase activity can be stimulated by oxidative mechanisms, which may involve NOX2 activity (Di Maio et al., 2018). Here, we found that rats treated with rotenone for 5 days also showed robust LRRK2 kinase activity in dopamine neurons, and this was blocked by co-treatment with CPP11G (Fig. 7A, B), indicating that LRRK2 kinase activity is a downstream effect of NOX2 activation in an epidemiologically relevant model of PD.
Fig. 6.
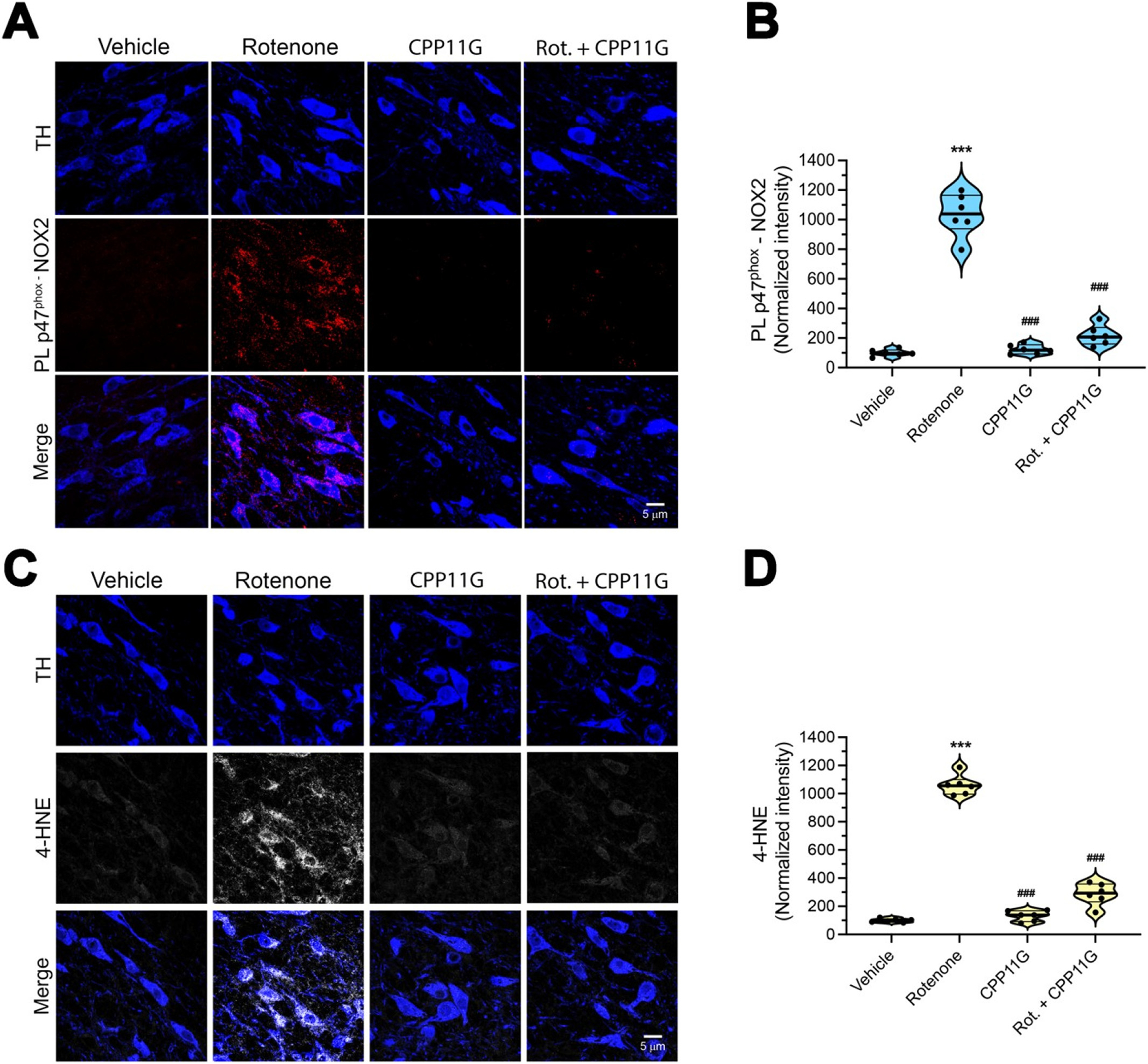
In vivo nigrostriatal NOX2 activation and oxidative damage in the rotenone model of PD are prevented by a brain penetrant NOX2 inhibitor.
(A) Representative images of p47phox-NOX2 PL signal (red) after 5 days of rotenone treatment ± CPP11G. Nigrostriatal DA neurons (TH, blue) showed a sustained NOX2 activity (red) that was prevented in animals that received co-treatment with CPP11G. (B) Quantification of p47phox-NOX2 proximity ligation signal. Each symbol represents the normalized intensity of an individual animal (with vehicle set to 100) from which 4 sections /animal and 30–40 cells /section were measured. Comparison by ANOVA with post hoc Bonferroni’s correction. (C) Fluorescence images of 4-hydroxynonenal (4-HNE) immunohistochemistry (gray) as a marker of lipid peroxidation. Five days of rotenone treatment caused a significant increase of 4- HNE in nigrostriatal neurons. Co-treatment with CPP11G prevented lipid peroxidation. (D) Quantification of 4-HNE fluorescence signal. Each symbol (n = 6) represents an individual animal as above. Comparison by ANOVA with Bonferroni’s correction. ***denotes p < 0.0001 significance compared to vehicle; ###denotes p < 0.0001 significance compared to rotenone.
Fig. 7.
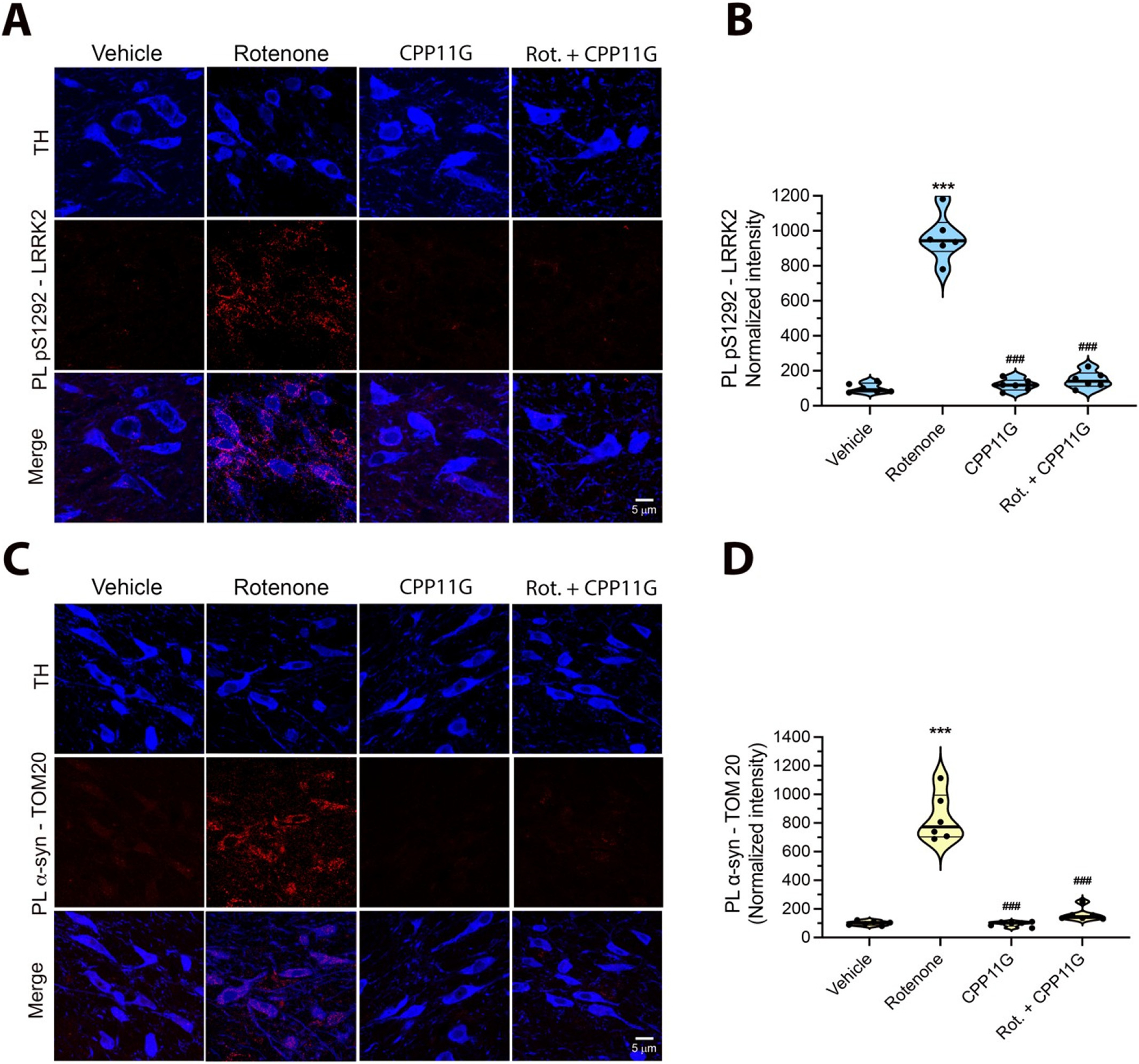
The brain penetrant NOX2 inhibitor, CPP11G, prevents rotenone-induced LRRK2 activation and α-synuclein-TOM20 interaction in rat nigrostriatal dopamine neurons.
(A) pSer1292-LRRK2 PL signal (red) in rats treated with vehicle (1st column), rotenone alone (2nd column), CPP11G alone (3rd column), or rotenone + CPP11G (4th column) for 5 days. Rotenone elicited a significant increase in pSer1292-LRRK2 PL signal, indicating LRRK2 kinase activation in nigrostriatal dopamine neurons (TH; blue). When co-administrated with rotenone, CPP11G blocked rotenone induced LRRK2 kinase activity. (B) Quantification of pSer1292-LRRK2 PL signal in nigrostriatal DA neurons. Each symbol represents the normalized intensity of an individual animal (with vehicle set to 100) from which 4 sections.
/Animal and 30–40 cells /section were measured. Statistical comparison by ANOVA with Bonferroni’s correction. (C) PL signal between α-synuclein and TOM20 (red). Rotenone treatment induced a strong PL signal for α-synuclein-TOM20 interaction. Co-administration of CPP11G prevented rotenone-mediated α- synuclein-TOM20 PL signal. (D) Quantification of α-synuclein-TOM20 PL signal in nigrostriatal dopamine neurons. Symbols represent individual animals as in B. Comparison by ANOVA with Bonferroni’s correction. ***denotes p < 0.0001 significance compared to vehicle; ###denotes p < 0.0001 significance compared to rotenone.
Similar to our previous results (Di Maio et al., 2016) in rats that were treated with rotenone to endpoint (10–14 days), we found that when rats were treated with rotenone for 5 days, there was a strong α-syn – TOM20 PL signal indicative of an aberrant interaction of α-syn with mitochondrial protein import machinery (P < 0.0001, two-way ANOVA with Bonferroni’s correction). Co-treatment with CPP11G prevented the interaction of α-synuclein with TOM20 (Fig. 7C, D), suggesting that α-synuclein-mediated mitochondrial protein import impairment is driven in part by NOX2 activity in vivo.
4. Discussion
Results of the current study validate a new in situ assay for NOX2 activity and reveal, for the first time, a sustained NOX2 activity in SNpc dopamine neurons and in microglia in iPD post-mortem human brain tissue and animal models of PD. Microglial activation (neuroinflammation) is a pathological feature of PD (Gerhard et al., 2006), and increased NOX2 expression in such post-mortem specimens raises the hypothesis that the contribution of NOX2 activity to PD pathogenesis may be primarily exerted through microglial oxidative stress-related neuroinflammatory responses (Rodriguez-Pallares et al., 2007). Moreover, in animal models of PD, there is upregulation of NOX2 (gp91phox) and phosphorylation of p47phox in SNpc microglial cells (Wu et al., 2003; Gerhard et al., 2006). However, neurons express NOX2 as well (Kim et al., 2005; Vallet et al., 2005; Tammariello et al., 2000; Cheng et al., 2001) and there remains a relative lack of knowledge on the possible role of neuronal NOX2 activity in PD pathogenesis. Until now, it has been difficult to assess the activity of NOX2 in situ, in fixed tissue, or with a cellular level of resolution. Despite the positive modulatory effects on NOX2 activity exerted by certain co-factors, including Rac proteins, the recruitment of p47phox to the NOX2 catalytic membrane complex constitutes the key step in NOX2 activation. In this context, we developed and validated a proximity ligation assay to detect the in-situ interaction of p47phox with NOX2, which allows assessment of NOX2 activation state with a cellular level of resolution (e.g., in dopamine neurons vs. surrounding microglia). Regarding the specificity of the PL assay, it correlates with (i) conventional IP (pulldown) methods and ROS production (DHE signal) and its downstream consequences, and (iii) it is supported by high resolution confocal imaging of the immunolocalization/translocation of NOX2 and p47phox upon activation. Moreover, it is important to note that the NOX2 PL signal is blocked dose-dependently by highly specific NOX2 assembly inhibitors that act by preventing the interaction between NOX2 and p47phox.
Applying this assay to human brain tissue sections from SNpc, we found that NOX2 was activated in both dopamine neurons and surrounding microglia in PD cases compared to controls. To delineate the relative temporal profiles of NOX2 activation in neurons versus microglia in SNpc, we performed in vivo experiments in two PD animal models, and found that neuronal NOX2 is activated acutely, followed in a delayed fashion by an increase in microglial NOX2 activation. In the α-syn overexpression model this delay amounted to several weeks. Thus, at least in these two in vivo models of PD, neuronal NOX2 activation appears to be primary, and this suggests that neuronal NOX2 plays a key role in the initiation of oxidative signaling that is, in turn, responsible for a cascade of pathogenic events, such as posttranslational modification of α-synuclein, mitochondrial impairment and LRRK2 activation, which contribute to PD onset and progression.
To address mechanistic questions, we employed in vitro cell culture systems and genetic and pharmacological approaches. Nox2ds-tat is a peptidic inhibitor that mimics a sequence in the cytosolic B-loop of NOX2 (Csanyi et al., 2011). As such, it binds the cytosolic regulatory protein, p47phox, which in turn prevents its translocation/binding to, and activation of, the NOX2 enzyme complex. The use of this highly selective inhibitor allowed us to dissect PD-relevant downstream events associated with NOX2 activation by applying the in vitro rotenone model in pVMB neurons and confirming key results in HEK-293 cells. Based on their neural crest origin, HEK-293 cells express several neuron-specific genes and share some cellular features with neurons (Shaw et al., 2002). We used HEK- 293 cells for gene-editing approaches, which allowed us to validate key results in CRISPR/Cas9 gene-edited NOX2−/−, NOX1−/− and NOX4 −/− cell lines.
Previous work in different (non-neuronal) experimental systems, has found crosstalk between mitochondria and NOX isoforms (Case et al., 2013; Desouki et al., 2005). As described here, we found that the mitochondrial complex I inhibitor, rotenone, generated a p47phox–NOX PL signal, that was associated with increased ROS production, and enhanced association of p47phox with NOX2, as assessed by co- immunoprecipitation and high-resolution confocal immunofluorescence imaging and colocalization analysis. As a potent complex I inhibitor, rotenone treatment is known to generate . In this context, rotenone induced activation of NOX2 was blocked by treatment with MitoTEMPO, a mitochondrially targeted scavenger. Overall, these results indicate that in neurons mitochondrially-derived ROS can activate NOX2, and in doing so, greatly amplify cellular ROS production via RIRP. Our results also indicate that rotenone does not activate NOX2 directly, as suggested by others (Zhou et al., 2012).
Accumulation of toxic forms of α-synuclein in dopaminergic neurons is believed to be central to nigrostriatal neurodegeneration in PD. We have previously reported that rotenone induced mitochondrial dysfunction leads to PTM forms of α-synuclein (e.g., pSer129-α-syn) and oligomeric-α-synuclein (Di Maio et al., 2016). The current study provides a compelling link between neuronal NOX2 activity and α-synuclein dysregulation: we provide evidence that NOX2 activation is an essential step in the process by which rotenone causes the formation of toxic species of α-synuclein. Indeed, in pVMB cultures co-treated with Nox2ds-tat and in NOX2−/− HEK-293 cells, rotenone had no effect on α-synuclein. This appears to be a specific effect of NOX2, since rotenone-induced accumulation of 4-HNE-α-syn still occurred in NOX1−/− and NOX4 −/− cells. Oxidative stress-related PTM forms of α-synuclein, including 4-HNE modified and nitrated forms, have a higher propensity to form oligomers compared to unmodified, monomeric α-synuclein (Qin et al., 2007; He et al., 2019). In this context, 4-HNE-α-syn is particularly and selectively toxic to dopamine neurons (Xiang et al., 2013; Bae et al., 2013). Our finding that NOX2 inhibition or knock out also prevents the formation of oligomeric α-synuclein, provides further insight into the key roles of 4-HNE-α-syn and NO-α-syn in the accumulation of toxic forms of α-synuclein.
In a previous study, we showed that certain PTM forms of α-synuclein, including oligomeric species, interact with mitochondrial protein import machinery causing mitochondrial impairment. In the present study, we show that mitochondrial ROS activate NOX2, and NOX2 inhibition prevents the accumulation of toxic forms of α-synuclein. Thus, there appears to be bidirectional signaling between mitochondria and NOX2 such that (i) mitochondrial ROS activates NOX2 and amplifies cellular ROS production via RIRP; NOX2-generated ROS leads to the formation of toxic species of α-synuclein, which (iii) impairs mitochondrial protein import and (iv) results in continued mitochondrial ROS production (Graphical abstract). In this scheme of oxidative stress and α-synuclein toxicity, inhibition of ROS production at its source, NOX2, may be much more efficient than attempting to scavenge ROS.
To examine the potential in vivo effects of nigrostriatal NOX2 activation, we used a sub-acute (5 day) rotenone treatment protocol which does not lead nigrostriatal neurodegeneration, but which reliably reproduces early pathological features of PD (Rocha et al., 2020). We previously reported that wildtype LRRK2 is activated in iPD, and this was reproduced in rotenone-treated rats (Di Maio et al., 2018). In the current study, we found that rotenone induced LRRK2 activation in nigrostriatal dopamine neurons was prevented by co-treatment with CPP11G, a highly specific small molecule NOX2 assembly inhibitor, further supporting previous results suggesting that LRRK2 is a redox sensitive kinase (Di Maio et al., 2018).
Given our findings that (i) LRRK2 is activated in nigral neurons in iPD (Di Maio et al., 2018), NOX2 activity is required for wildtype LRRK2 activation in vivo, and (iii) NOX2 is activated in nigral neurons in iPD, it appears likely this mechanism is operative in the human disease. Endolysosomal deficits and autophagic dysfunction contribute to the progression of PD (Rocha et al., 2020; Rocha et al., 2022). Interestingly, aberrant LRRK2 kinase activity has been shown to lead to autophagic dysfunction and endolysosomal deficits with the consequence of accumulation of α-synuclein (Di Maio et al., 2018; Rocha et al., 2020; Rocha et al., 2022). Treatment with a LRRK2 kinase inhibitor prevented these deficits in the rotenone rat model (50), suggesting that elevated kinase activity contributes to this process (Graphical Abstract). Thus, NOX2 may contribute to both the formation and the compromised disposal of toxic species of α-synuclein, directly via the formation of oxidative stress modified forms of α-synuclein and indirectly via LRRK2 kinase activity. A downstream consequence of the impaired degradation of oligomeric α-synuclein and pSer129-α-Syn is that they can bind to TOM20 and impair mitochondrial function (Di Maio et al., 2016; Di Maio et al., 2018). Consistent with our in vitro results, we found that in vivo inhibition of NOX2 also prevented this form of α-synuclein-induced mitochondrial toxicity (Graphical Abstract).
Systemic administration of rotenone to rats is invariably associated with progressive loss of body weight and even treatments that have successfully protected against nigrostriatal degeneration have not prevented this weight loss (unpublished results). An unexpected effect of systemic administration of CPP11G was prevention of rotenone-induced weight loss. This may suggest that the weight loss is a consequence of systemic NOX2 activation by rotenone. Given that PD is a “systemic” disease that affects the gastrointestinal system and the autonomic and peripheral nervous systems with α-synuclein and oxidative pathology, NOX2 inhibitors such as CPP11G may hold promise beyond central nervous system neuroprotection.
In conclusion, our results suggest that neuronal NOX2 plays a pivotal role in the pathogenesis of PD. It is activated by mitochondrial ROS and serves to amplify that signal, producing sufficient to produce lipid peroxidation, post-translational modification and oligomerization of α-synuclein, and increased LRRK2 kinase activity in neurons. Further, by both directly facilitating formation of pathogenic species of α-synuclein and indirectly inhibiting their autophagic degradation (via LRRK2 activation), NOX2 activity also amplifies the downstream mitochondrial toxicity of α-synuclein (Graphical abstract). Considering this apparent feed forward cycle, NOX2 presents an attractive target for the simultaneous therapeutic amelioration of ongoing α-synuclein toxicity and aberrant LRRK2 activity. Our in vivo studies, using an epidemiologically relevant model of PD (Greenamyre et al., 2010; Tanner et al., 2011) provide strong preclinical support for this strategy and for a selective and potent class of novel inhibitors that target NOX2 in disease.
Supplementary Material
Acknowledgements
The Graphical abstract was created using Biorender (https://biorender.com) (Publication License # BH23TK90PS).
Funding
This work was supported by research grants from the NIH (R01NS100744–01, R21NS112787, NS095387, and AG005133) and the Ri.MED Foundation, Italy. Support was also provided by the American Parkinson’s disease Association Center for Advanced Research at the University of Pittsburgh and the friends and family of Sean Logan. The brain bank has received support from the University of Pittsburgh Brain Institute.
Footnotes
Declaration of Competing Interest
The authors declare that they have no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105754.
Data and materials availability
All data associated with this study can be found in the paper and the Supplemental Materials.
References
- Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, Bogdanovic N, Bugiani O, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovács GG, Meyronet D, Monoranu C, Parchi P, Parkkinen L, Patsouris E, Roggendorf W, Rozemuller A, Stadelmann-Nessler AC, Streichenberger N, Thal DR, Kretzschmar H, 2009. Staging/typing of Lewy body related a-synuclein pathology: A study of the BrainNet Europe consortium. Acta Neuropathol. 117, 635–652. [DOI] [PubMed] [Google Scholar]
- Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH, 2015. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 23 (5), 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae EJ, Ho DH, Park E, Jung JW, Cho K, Hong JH, et al. , 2013. Lipid peroxidation product 4- hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid. Redox Signal. 18 (7), 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH, 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87 (1), 245–313. [DOI] [PubMed] [Google Scholar]
- Belarbi K, Cuvelier E, Destee A, Gressier B, Chartier-Harlin MC, 2017. NADPH oxidases in Parkinson’s disease: a systematic review. Mol. Neurodegener. 12 (1), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR, 2015. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 9, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DI, Griendling KK, 2009. Nox proteins in signal transduction. Free Radic. Biol. Med. 47 (9), 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Li S, Basu U, Tian J, Zimmerman MC, 2013. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 305 (1), H19–H28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD, 2001. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 269 (1–2), 131–140. [DOI] [PubMed] [Google Scholar]
- Cifuentes-Pagano E, Csanyi G, Pagano PJ, 2012. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell. Mol. Life Sci. 69 (14), 2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Pagano E, Saha J, Csanyi G, Ghouleh IA, Sahoo S, Rodriguez A, et al. , 2013. Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors. Medchemcomm. 4 (7), 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini DJ, Egana L, Lopes LR, et al. , 2011. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med. 51 (6), 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, 2010. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797 (6–7), 897–906. [DOI] [PubMed] [Google Scholar]
- Desouki MM, Kulawiec M, Bansal S, Das GM, Singh KK, 2005. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. 4 (12), 1367–1373. [DOI] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, et al. , 2016. Alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8 (342), 342ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, et al. , 2018. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 10 (451). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S, 2011. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51 (7), 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LM, Geng L, Cahill-Smith S, Liu F, Douglas G, McKenzie CA, et al. , 2019. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Invest. 129 (8), 3374–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. , 2006. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 21 (2), 404–412. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. , 2000. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 290 (5493), 985–989. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Cannon JR, Drolet R, Mastroberardino PG, 2010. Lessons from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 31 (4), 141–142 (author reply 2–3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yu Z, Chen S, 2019. Alpha-Synuclein nitration and its implications in Parkinson’s disease. ACS Chem. Neurosci. 10 (2), 777–782. [DOI] [PubMed] [Google Scholar]
- Henne WM, 2017. Discovery and roles of ER-Endolysosomal contact sites in disease. Adv. Exp. Med. Biol. 997, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandes MS, Cafe-Mendes CC, Britto LR, 2013. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxidative Med. Cell. Longev. 2013, 157857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang HM, Johnson HE, Heo J, 2021. Rac-dependent feedforward autoactivation of NOX2 leads to oxidative burst. J. Biol. Chem. 297 (2), 100982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz MP, Milanese C, Di Maio R, Hu X, Montero LM, Sanders LH, et al. , 2011. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid. Redox Signal. 15 (4), 855–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi M, Katsuyama M, Fan C, Iwata K, Nishinaka T, Yokoyama T, et al. , 2006. NOX1/NADPH oxidase negatively regulates nerve growth factor-induced neurite outgrowth. Free Radic. Biol. Med. 40 (10), 1785–1795. [DOI] [PubMed] [Google Scholar]
- Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD, 1992. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s disease research group. Ann. Neurol. 32 (Suppl), S82–S87. [DOI] [PubMed] [Google Scholar]
- Keeney MT, Hoffman EK, Greenamyre TJ, Di Maio R, 2021. Measurement of LRRK2 kinase activity by proximity ligation assay. Bio Protoc. 11 (17), e4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Shin KS, Chung YB, Jung KW, Cha CI, Shin DH, 2005. Immunohistochemical study of p47Phox and gp91Phox distributions in rat brain. Brain Res. 1040 (1–2), 178–186. [DOI] [PubMed] [Google Scholar]
- Li Y, Cifuentes-Pagano E, DeVallance ER, de Jesus DS, Sahoo S, Meijles DN, et al. , 2019. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol. 22, 101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, Fournier M, Lashuel HA, 2010. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145. [DOI] [PubMed] [Google Scholar]
- Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 25 (2), 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL, 2007. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J. Biol. Chem. 282 (8), 5862–5870. [DOI] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ, 2001. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ. Res. 89 (5), 408–414. [DOI] [PubMed] [Google Scholar]
- Roberts RF, Bengoa-Vergniory N, Alegre-Abarrategui J, 2019. Alpha-Synuclein proximity ligation assay (AS-PLA) in brain sections to probe for alpha-Synuclein oligomers. Methods Mol. Biol. 1948, 69–76. [DOI] [PubMed] [Google Scholar]
- Rocha EM, De Miranda BR, Castro S, Drolet R, Hatcher NG, Yao L, et al. , 2020. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol. Dis. 134, 104626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Keeney MT, Di Maio R, De Miranda BR, Greenamyre JT, 2022. Mar. LRRK2 and idiopathic Parkinson’s disease. Trends Neurosci. 45 (3), 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Pallares J, Parga JA, Munoz A, Rey P, Guerra MJ, Labandeira-Garcia JL, 2007. Mechanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 103 (1), 145–156. [DOI] [PubMed] [Google Scholar]
- Sanders LH, Timothy Greenamyre J., 2013. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 62, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL, 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16 (8), 869–871. [DOI] [PubMed] [Google Scholar]
- Tammariello SP, Quinn MT, Estus S, 2000. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J. Neurosci. 20 (1), RC53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. , 2011. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 119 (6), 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M, 2009. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 11 (6), 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, et al. , 2005. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 132 (2), 233–238. [DOI] [PubMed] [Google Scholar]
- Van Laar VS, Chen J, Zharikov AD, Bai Q, Di Maio R, Dukes AA, et al. , 2020. Alpha-Synuclein amplifies cytoplasmic peroxide flux and oxidative stress provoked by mitochondrial inhibitors in CNS dopaminergic neurons in vivo. Redox Biol. 37, 101695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, et al. , 2003. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 100 (10), 6145–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, Schaffer TE, et al. , 2013. Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 54, 71–83. [DOI] [PubMed] [Google Scholar]
- Zhang W, Gao JH, Yan ZF, Huang XY, Guo P, Sun L, et al. , 2018. Minimally toxic dose of lipopolysaccharide and alpha-Synuclein oligomer elicit synergistic dopaminergic neurodegeneration: role and mechanism of microglial NOX2 activation. Mol. Neurobiol. 55 (1), 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, et al. , 2015. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 125 (7), 2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhang F, Chen SH, Zhang D, Wilson B, Hong JS, et al. , 2012. Rotenone activates phagocyte NADPH oxidase by binding to its membrane subunit gp91phox. Free Radic. Biol. Med. 52 (2), 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study can be found in the paper and the Supplemental Materials.