

Abstract
A cluster of six genes, tRNATrp-secE-nusG-rplK-rplA-pkwR, was cloned and sequenced from a Corynebacterium glutamicum cosmid library and shown to be contiguous in the C. glutamicum genome. These genes encode a tryptophanyl tRNA, the protein translocase component SecE, the antiterminator protein NusG, and the ribosomal proteins L11 and L1 in addition to PkwR, a putative regulatory protein of the LacI-GalR family. S1 nuclease mapping analysis revealed that nusG and rplK are expressed as separate transcriptional units and rplK and rplA are cotranscribed as a single mRNA. A 19-nucleotide inverted repeat that appears to correspond to a transcriptional terminator was located in the 3′ region downstream from nusG. Northern analysis with different probes confirmed the S1 mapping results and showed that the secE-rplA four-gene region gives rise to four transcripts. secE was transcribed as a 0.5-kb monocistronic mRNA, nusG formed two transcripts of 1.4 and 1.0 kb from different initiation sites, and the two ribosomal protein genes rplK and rplA were cotranscribed as a single mRNA of 1.6 kb. A consensus L1 protein binding sequence was identified in the leader region of the rplK-rplA transcript, suggesting that expression of the rplK-rplA cluster was regulated by autogenous regulation exerted by the L1 protein at the translation level. The promoters of the nusG and rplK-rplA genes were subcloned in a novel corynebacterial promoter-probe vector and shown to confer strong expression of the reporter gene.
Ribosomal proteins of both gram-positive and gram-negative bacteria are involved in the translational control of the expression of genes for the initiation of physiological and morphological differentiation (29), although the molecular mechanisms involved are poorly known. One of these mechanisms, relA control, involved in adaptation of the cells to amino acid starvation, is mediated by the hyperphosphorylated guanosine tetraphosphate and pentaphosphate [(p)ppGpp]. These compounds are formed from GTP and the pyrophosphoryl group of ATP in a reaction mediated by the RelA factor that is associated with ribosomal proteins. The RelA protein becomes active when uncharged tRNA accumulates due to the lack of the corresponding amino acids, and ribosomes are unable to work (11).
In Escherichia coli, a functional ribosomal protein, L11, encoded by the rplK gene, is required for the activation of the RelA factor (10). Similarly, a functional rplK gene product is required for (p)ppGpp biosynthesis in Bacillus subtilis (41) and Streptomyces coelicolor (30, 32).
Corynebacterium glutamicum and Brevibacterium lactofermentum, renamed Corynebacterium lactofermentum (2), are widely used for industrial production of amino acids (22, 37). A large number of genes involved in primary metabolism have been cloned from corynebacteria (9, 24) and have been used to improve the production of amino acids (25).
Amino acid accumulation in corynebacteria follows a decrease in rRNA synthesis and growth (E. González-Lavado, C. Barreiro, and J. F. Martin, unpublished data). Initial evidence indicates that the growth rate of corynebacteria is inversely correlated with the cellular (p)ppGpp concentration (36). The roles of ribosomal proteins and the rel mechanism in the switch from the growth phase to the amino acid production phase in corynebacteria are of great interest.
A relA (also similar to spoT) gene of C. glutamicum encoding a bifunctional enzyme with (p)ppGpp synthetase and (p)ppGpp-degrading activity was cloned (45). However, the role of the L11 ribosomal protein in the synthesis of (p)ppGpp and in the switch from the growth phase to the amino acid accumulation stage in corynebacteria remains unknown. Ribosomal protein engineering is receiving increasing attention as a tool to modify growth-related control mechanisms (31). It was, therefore, of great interest to clone the gene encoding L11 and other ribosomal proteins to elucidate its role in the mechanism of rel control in C. glutamicum. We report the cloning, organization, and transcriptional analysis of a six-gene region of corynebacteria that contains the genes for the L11 and L1 ribosomal proteins.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All bacterial strains and plasmids used in this work are listed in Table 1. E. coli was grown in Luria Bertani broth (38) at 37°C. C. glutamicum ATCC 13032 and B. lactofermentum R31, renamed C. lactofermentum (2), a high-efficiency host strain for plasmid transformation (25, 40), were grown in trypticase-soy broth (TSB) at 30°C. E. coli transformants were selected in the presence of ampicillin (100 μg/ml), and C. glutamicum and C. lactofermentum transformants were selected in media with kanamycin (30 μg/ml).
TABLE 1.
Strains and plasmids used in this work
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| E. coli DH5α | F−recA1 endA1 gyrA96 thi-1 hsdR17 (rk−mk+) sup44 relA1 λ− (ϕ80dlacZΔM15) Δ(lacZYA-argF) U169 | 13 |
| E. coli WK6 mutS | F′ lacIqZΔM15 proAB Δ(lac-roAB) galE strA mutS215::Tn10 | 18 |
| C. glutamicum ATCC 13032 | Wild type; Nalr | ATCCa |
| B. lactofermentum R31 | MeLysr Aecr Nalr | 39 |
| Plasmids | ||
| pBluescript SK(+) or KS(+) | E. coli vector; AprlacZ ori f1 | 1 |
| M13K07 | M13-derived phage; Kmr; used as helper phage for ssDNAb isolation | 44, 27 |
| pUL880M | E. coli-Corynebacterium vector; Apr Hygr; neo gene without promoter used for promoter analysis | 21 |
| pULCE0 | pUL880M with new cloning sites | This work |
ATCC, American Type Culture Collection.
ssDNA, single-stranded DNA.
DNA isolation and manipulation.
E. coli plasmid DNA was obtained by alkaline lysis as described by Birnboim and Doly (3). Total C. glutamicum DNA was prepared as described by Martín and Gil (25), and C. lactofermentum and C. glutamicum plasmidic DNA were prepared by the method of Kieser (17, 25).
DNA manipulations were performed as described by Sambrook et al. (38). DNA fragments were isolated from agarose gels using the Geneclean II kit (Bio 101).
E. coli cells were transformed as described by Hanahan (12), whereas corynebacteria were transformed by electroporation (8, 25).
Southern hybridizations.
DNA fragments were vacuum blotted from 0.8% agarose gels to nylon membranes (42). Labeling of the DNA probes was done with digoxigenin (Boehringer Mannheim, Mannheim, Germany) according to the manufacturer's instructions.
DNA sequencing.
The nucleotide sequence of the cloned region was determined in both strands by the dideoxynucleotide chain termination method using an automatic DNA sequencer (ALF; Pharmacia). The nucleotide and deduced amino acid sequences were compared with those in the EMBL and GenBank databases by using the Clustal W program (43).
RNA extraction.
Total RNA from corynebacteria was extracted by a method based on that of Eikmanns et al. (9) except that the cell pellet, obtained after centrifugation, was frozen with liquid nitrogen and kept at −70°C before RNA extraction. The RNA concentration was determined spectrophotometrically by determining the absorbance at 260 nm.
S1 nuclease protection assays.
Low-resolution S1 mapping was performed as described by Sambrook et al. (38) using 200 μg of total C. glutamicum RNA and 100 to 150 ng of probe DNA. Treatment with S1 nuclease was made for 30 min (longer times were found to be less suitable). tRNA from Saccharomyces cerevisiae (type X-SA; Sigma) was used as a negative control for the hybridizations and the complete probe, or only the homologous part of the probe, was used as a positive control.
Northern hybridization.
Denaturing RNA electrophoresis was performed in 0.9% agarose gels in MOPS (morpholine propane sulfonic acid) buffer (20 mM MOPS, 5 mM sodium acetate, 1 mM EDTA [pH 7.0]) with 17% (vol/vol) formaldehyde. RNA (30 μg) was dissolved in denaturing buffer (50% formamide, 20% formaldehyde, 20% MOPS [5×]) with 10% DYE (38) and 1% ethidium bromide. DNA probes were labeled with 32P by nick translation (Promega).
Hybridizations were performed at 42°C with 50% formamide; membrane washings were carried out as described by Sambrook et al. (38) except that the second washing was done at 42°C. The labeled bands were detected by autoradiography with exposition times of 72 and 96 h.
Promoter-probe plasmids.
A new promoter-probe plasmid, pULCE0, was constructed from the vector pUL880M (23, 26). pUL880M was linearized with BglII. The protruding BglII ends were partially filled with dGTP and dATP using the Klenow fragment of the DNA polymerase. A polylinker containing the restriction sites BstXI and ClaI (sites that were absent in the rest of the vector) was synthesized and linked to the linearized pUL880M vector. The appropriate orientation of the polylinker was confirmed by PCR using the following primers: Oligo 1 (5′-TCGCCAGAGCTCTGGATCGATA-3′), identical to that used to construct the polylinker, and Oligo 2 (5′-GGGAGCGGCGATACCGTAAA-3′), internal to the kanamycin resistance gene of pUL880M. An amplification band of 804 bp was obtained that corresponded to the insertion of the polylinker in the correct orientation (Fig. 1). Finally, the region of the polylinker was sequenced in both strands (Fig. 1), and the functionality of the polylinker was confirmed by hydrolysis with restriction endonucleases. This vector (unlike pUL880M) allowed directed cloning of promoter sequences with several different cohesive ends.
FIG. 1.

Construction of the promoter-probe vector pULCE0. This vector was constructed by inserting a polylinker (shaded nucleotide sequence) with unique restriction sites (absent from other places in the vector) in the BglII site upstream from the promoterless kanamycin resistance marker. One of the BglII sites was removed (X) by appropriate selection of oligonucleotides (arrowhead). Reverse-type letters indicate BglII ends partially filled to avoid vector religation. KmR, kanamycin-resistance gene; HygR, hygromycin resistance gene; ApR, ampicillin resistance gene. ori E. coli is the colE1 plasmid replication origin, and ori B. lactofermentum corresponds to the replication origin of plasmid pBL1 of B. lactofermentum (synonymous with C. lactofermentum). Ttrp, transcriptional terminator of the C. lactofermentum tryptophan operon (to avoid readthrough expression).
Subcloning of the promoters.
The promoter region upstream of rplK, controlling expression of rplK-rplA, was amplified by PCR as a 225-bp fragment containing its own translation start codon and was subcloned in pULCE0. This promoter region was named PrplKA. The construction with the promoter PrplKA was introduced by transformation into C. lactofermentum R31 (a high-efficiency host strain for initial transformation), reisolated, and then transformed into C. glutamicum. Direct transformation of C. glutamicum with the ligation mixture was inefficient, but transformation with plasmid isolated from C. lactofermentum R31 was quite efficient.
Similarly, the promoter region upstream of nusG (named PnusG) was subcloned as a 567-bp fragment with its own translation start codon in the promoter-probe vector pULCE0 and was introduced first into C. lactofermentum and then into C. glutamicum.
Ten transformants containing the plasmid with the PrplKA promoter (named pULCErplKA) and another 10 containing the plasmid with the PnusG promoter (named pULCEnusG) were selected and used to test the promoter strength by growing them in plates of TSB medium with increasing concentrations of kanamycin (from 100 to 1,200 μg/ml).
Nucleotide sequence accession number.
The nucleotide sequence of the 4.42-kb fragment containing the tRNATrp-secE-nusG-rplK-rplA-pkwR cluster was deposited in the EMBL database under accession number AJ300822.
RESULTS
Cloning the DNA region encoding L11 and L1 ribosomal proteins.
Since the rplA and rplK genes (encoding the L1 and L11 proteins, respectively) are linked to the secE and nusG genes in some gram-positive bacteria, a search for this gene cluster in the cosmid library of C. glutamicum was made using two probes, A and B. Probe A corresponded to an 800-bp BglII-SalI fragment containing the secE gene of Streptomyces lividans (J. Blanco and J. F. Martín, unpublished data), and probe B was a 1,500-bp KpnI fragment containing the S. lividans nusG gene.
The results of Southern hybridization with total DNA of C. glutamicum digested with SalI showed a single 6.6-kb SalI band that hybridized with both probes. Similarly, a 5.5-kb BamHI fragment hybridized with both probes, suggesting that the secE and nusG genes are linked and probably maintain the same organization as in other gram-positive bacteria.
The C. glutamicum cosmid library was then hybridized with an 800-bp KpnI fragment containing the secE of S. lividans, and 10 positive cosmids were selected and reconfirmed by a second hybridization with the secE and nusG probes (see above). Cosmid pCG1 was selected and used for further studies. pCG1 DNA was digested with BamHI, and the hybridizing 5.5-kb band was subcloned in pBluescript SK(+) to form the new plasmid pB1.
The cloned DNA fragment comprises six genes, including the rplA-rplK cluster.
Cosmid pCG1 was mapped with restriction endonucleases. A PstI-BamHI region of 4.42 kb was entirely sequenced in both strands, showing a G+C content of 53.48%. Computer analysis of the sequence taking into account the codon usage of corynebacteria (23) revealed five open reading frames (ORFs) in the sequenced DNA fragment (Fig. 2A).
FIG. 2.

(a) Restriction map and locations of the six genes found in the sequenced 4.42-kb DNA fragment (see the text). The wavy arrows correspond to the transcripts of the genes. The shaded bars show the S1 protection probes, and the solid bars represent the probes used in Northern analysis (see Fig. 4 and 5). (B) Stem-and-loop structure formed in the intergenic region between nusG and rplK-rplA corresponding to an imperfect inverted repeat of 19 nt. (C) Alignment of the L1-binding sequence in the 5′ untranslated region of the rplK-rplA transcript with those of S. coelicolor (35) and S. griseus (20).
Upstream of the first ORF, we found a gene encoding a tRNATrp (73 bp). Comparison of the nucleotide sequences and the deduced amino acid sequences of the five ORFs with the sequence of proteins in the Swiss-Prot data bank indicates that the first, ORF1, encodes a protein of 111 amino acids that shows homology with the membrane protein SecE of different gram-positive and gram-negative bacteria (36.1% identity with SecE of Mycobacterium tuberculosis). ORF1 was separated from the tRNATrp gene by an intergenic region of 230 bp. A sequence of 60 bp present in the upstream region of ORF1 is identical to that of a promoter region that was randomly cloned as a transcription initiation region (34). Downstream from ORF1 and in the same orientation we found ORF2, which encodes a protein of 318 amino acids and a deduced molecular mass of 34.7 kDa that shows very high homology with the antiterminator protein NusG (48.9% identity with NusG from M. tuberculosis). Downstream from nusG, ORF3 and ORF4 encode proteins of 145 and 236 amino acids that show extensive homology with the ribosomal proteins L11 and L1, respectively (85.4% identity with the L11 protein of M. tuberculosis and 71.5% identity with the L1 protein of M. tuberculosis). These two genes have been named rplK and rplA, respectively.
ORF5 is located at the end of the sequenced fragment and in the opposite orientation to ORF4. It corresponds to a truncated gene (918 bp) encoding a protein fragment (lacking the N-terminal end) with significant homology (63.3% identity at the nucleotide level) with the pkwR gene of Thermomonospora curvata (14). PkwR is a putative regulatory protein that is a member of the LacI-GalR family of regulatory proteins.
These results clearly indicate that the region cloned in cosmid pCG1 contains the desired genes, rplK and rplA, for the ribosomal proteins L11 and L1. In some gram-positive bacteria, the genes rplJ and rplL, encoding other ribosomal proteins, map downstream of rplA. To study the possibility that a DNA rearrangement might have occurred during cosmid packaging, the entire region of cosmid pCG1 and the total DNA of C. glutamicum were digested in parallel and hybridized with a 1,820-bp HindIII-BamHI probe (Fig. 3) that contains both the complete rplA gene and the sequenced ORF5.
FIG. 3.

Southern hybridization showing that ORF5 (pkwR) is adjacent to rplA both in the cosmid pCG1 (lanes 1, 3, 5, 7, 9, and 11) and in the chromosome (lanes 2, 4, 6, 8, 10, and 12). Cosmid DNA and total DNA were digested with BamHI plus SpeI (lanes 1 and 2), BamHI plus SacI (lanes 3 and 4), BamHI plus EcoRI (lanes 5 and 6), BamHI plus BstXI (lanes 7 and 8), BamHI plus ApaI (lanes 9 and 10), and BamHI (lanes 11 and 12).
The results (Fig. 3) showed unequivocally that ORF5 (pkwR) is adjacent to rplA both in the chromosome and in the cosmid. In M. tuberculosis, the region downstream of rplA does not contain rplJ and rplL (5).
Transcriptional analysis of the secE-rplA region.
Knowledge of the expression of the ribosomal protein genes is extremely important to understand the downshift in growth rate and protein synthesis that leads to amino acid accumulation. Low-resolution S1 mapping with the “heterologous tail” probe procedure (33) was used to locate the transcription initiation regions that were studied in detail. The modified heterologous tail probe method allows us to distinguish in a simple form RNA-DNA hybrids from reannealed DNA-DNA formed by base pairing of the two strands of the probe.
Protection analysis of the nusG-rplK intergenic region was done with probe PvuII (1,721 bp) and, in the case of the rplK-rplA intergenic region, with the 855-bp PvuII fragment as the probe (Fig. 4).
FIG. 4.

Low-resolution S1 mapping experiments. The probes used in each protection experiment are shown below the cloned DNA fragment. Note that each probe contains a homologous region (solid) and a heterologous region (shaded) belonging to lacZ and lacI. (A) Protected bands in the nusG-rplK region. Lanes: 1, probe A; 2, homologous part of probe A; 3, negative control with probe A and tRNA; 4, S1 protection assay (protected bands are indicated by arrows); 5, molecular size marker. (B) Lanes: 1, probe B; 2, homologous part of probe B; 3, negative control with probe and tRNA; 4, S1 protection assay (60-min digestion with nuclease S1); 5, S1 protection assay (30-min digestion with nuclease S1) (the protected band is indicated by the arrow); 6, size marker. Sizes in nucleotides are indicated at the right. Commercial tRNA from S. cerevisiae was used as a negative control instead of C. glutamicum RNA.
Three positive protection bands for the nusG-rplK region were found (Fig. 4A); the largest one clearly corresponds to the reannealed probe with the tail (1.72 kb); the second, with a size of 950 bp, corresponds to the protected band of nusG mRNA; and the third corresponds to the protected mRNA of rplK (300 nucleotides [nt]). The lack of a protection band corresponding to the complete ApaI-SpeI fragment (without a tail) indicated that there was not a bicistronic transcript for both the nusG and rplK genes. The intergenic region between nusG and rplK-rplA contains a long (19-nt) inverted repeat forming a very stable stem-and-loop structure and a short poly(U) tail (Fig. 2B) that may work as a transcriptional terminator. The presence of this terminator structure is in agreement with the formation of separate transcripts from nusG and rplK-rplA. The 300-nt protected band corresponding to rplK indicates that the transcription start point of rplK is located about 180 nt upstream of the ATG translation initiation codon. In summary, all of the evidence indicates that there is a promoter region between nusG and rplK and that the two genes are expressed as separate transcripts.
Similarly, low-resolution S1 mapping of the rplK-rplA region showed two positive hybridizing bands (Fig. 4B) that corresponded exactly to the reannealed probe (with its tail) and to an RNA protection band of 725 nt that coincides with the expected size of a bicistronic transcript of both the rplK and rplA genes, i.e., both rplK and rplA are transcribed as a single mRNA (see Discussion).
Northern analysis of the secE-rplA region shows that it is transcribed as four separate mRNAs.
Northern analysis of the entire region confirmed the results obtained by low-resolution S1 mapping and provided evidence that the secE gene is expressed as a separate unit, unlike what occurs in E. coli. The results of Northern analysis with a secE probe (Fig. 5A) showed that secE forms a transcript of 0.5 kb that is clearly smaller than any of the other transcripts formed from the genes in the region. This mRNA size is in good agreement with the size of the secE gene (333 nt).
FIG. 5.

Northern analysis of transcripts of the cloned region with probes corresponding to secE, nusG, rplK-rplA, rplA, or rplK. The probes used are solid in Fig. 2A and were labeled by nick translation. Note that rplK and rplA are expressed as a single transcript of 1.6 kb.
Interestingly, hybridizations with the nusG probe revealed two transcripts of 1.4 and 1.0 kb (Fig. 5B), neither of which coincides with other transcripts in the region. This result indicated that there are two overlapping mRNAs for the nusG gene, probably differing in the transcription start point, because low-resolution S1 mapping indicates that there is a single 3′ end.
Northern hybridizations of the RNA with an rplK-rplA probe showed a single transcript of 1.6 kb (Fig. 5C). When the hybridization was repeated with individual probes internal to either rplA or rplK, the same 1.6-kb band appeared, thus confirming that both rplK and rplA are transcribed as a single bicistronic mRNA, in agreement with the conclusions of the S1 nuclease protection studies.
Upstream of the rplK-rplA cluster in the leader region of rplK mRNA, we have found a putative L1 binding sequence that shows strong conservation (Fig. 2C) with the reported L1 binding sequence in the rplK-rplA transcript of Streptomyces griseus (20) and S. coelicolor (35).
Promoter studies.
The promoter region upstream of secE was isolated previously (34) and was not studied further. The two other promoter regions upstream of nusG and rplK-rplA were analyzed by subcloning them in a new promoter-probe vector, pULCE0 (Fig. 1), to quantify their transcription initiation abilities. The subcloned promoter fragments contained the −35 and −10 regions and their ribosome binding sites.
All transformants expressing the kanamycin resistance marker from the PrplKA promoter were able to grow in the presence of up to 975 μg of kanamycin/ml, whereas the control untransformed strain was sensitive to 20 μg/ml. Similarly, the level of resistance to kanamycin of the transformants with the PnusG promoter was 800 μg/ml.
Figure 6 compares the efficiencies of PrplKA and PnusG with those of other endogenous promoters of the C. lactofermentum plasmid pBL1 (4). The efficiency of PrplKA was clearly higher than those of the other known corynebacterial promoters with the same marker and vector, indicating that there is a strong expression of the rplK-rplA operon in C. glutamicum.
FIG. 6.
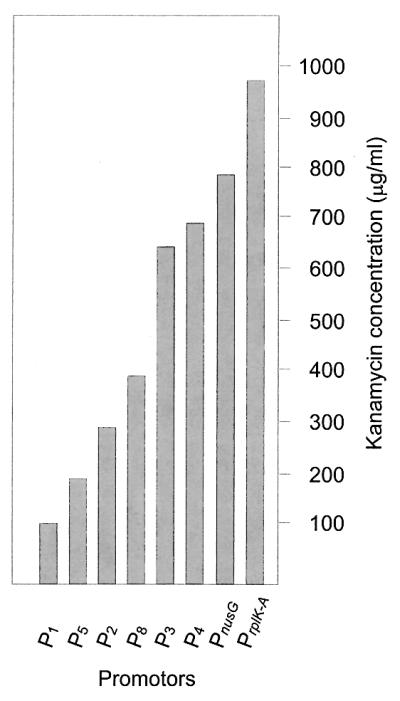
Levels of resistance to kanamycin (in solid TSB plates) conferred by expressing the kanamycin resistance gene of pULCE0 from the rplK-rplA promoter and from the nusG promoter in comparison with expression from the promoters obtained from pBL1 of C. lactofermentum (4).
DISCUSSION
The ribosomal protein L11 is involved in control of the expression of genes regulated by the rel mechanism (16, 30, 45). The L11 protein is required for ribosome-dependent accumulation of (p)ppGpp (32). We have cloned this gene (rplK) and found that it is associated with rplA (encoding the ribosomal protein L1) in a cluster with the secE, nusG, and pkwR genes, in addition to a tRNATrp gene located in the distal region (with respect to rplK-rplA) of the cluster.
There are notable differences in the conservation of the genes in this region in different bacteria. The rplK gene has been cloned from E. coli (6) and other gram-negative bacteria, and more recently from gram-positive microorganisms, including B. subtilis (19), M. tuberculosis (5), Streptomyces virginiae (15), and S. coelicolor (35). In all these organisms, as in C. glutamicum, the gene encoding L11 is clustered with the secE, nusG, and rplA genes.
A gene homologous to the pkwR gene of the actinomycete T. curvata (14) was located downstream from rplA and in opposite orientation to it in C. glutamicum. In M. tuberculosis (5), the region downstream of rplA contains five small ORFs in opposite orientation to that of rplA (4), and none of these ORFs correspond to the pkwR gene, whereas in B. subtilis (19), S. virginiae (15), S. coelicolor (http://www.sanger.ac.uk/Projects/S_coelicolor/), and other bacteria, such as E. coli and Thermotoga maritima (28), rplA is followed by the genes rplJ and rplL, encoding the ribosomal proteins L10 and L7/L12.
These results suggest that there are two types of organization of the rplK-rplA genes in actinomycetes: (i) the arrangement of rplK-rplA-pkwR that occurs in C. glutamicum and (ii) that known in species of Streptomyces (16, 20). The pkwR gene encodes a putative regulatory protein, a member of the LacI-GalR family of regulatory proteins, but its exact role is unknown (14). It is likely that the larger cluster of ribosomal proteins in Streptomyces species may have been formed by recruiting genes from other gram-positive or gram-negative bacteria.
Our results with transcriptional analysis indicate that rplK and rplA are transcribed as a single bicistronic mRNA, whereas secE and nusG form separate transcripts. Similar results have been reported in E. coli (6), S. virginiae (15), and S. griseus (20). The rplK-rplA promoter region of C. glutamicum is preceded by a putative transcriptional terminator (a long stem-and-loop structure), further supporting the conclusion that nusG forms a transcript separate from that of rplA-rplK.
The simultaneous transcription of rplK-rplA suggests that the formation of the ribosomal proteins L11 and L1 is coordinated as expected. In E. coli there is, in addition to transcriptional regulation, an elegant mechanism of autoregulation by the L1 protein at the translational level (7). The protein L1 binds to a box in the mRNA upstream of the rplK translation initiation site and prevents translation of the mRNA by the ribosomes. In this way, an excess of the ribosomal protein L1 prevents the wasteful synthesis of additional L1 and L11 proteins. Similarly, in the leader region of rplK-rplA we have found a putative L1 binding sequence that is similar to the L1 binding sequence of S. griseus (20) and S. coelicolor (35), suggesting that the rplK-rplA expression is also autoregulated in C. glutamicum.
Promoter analysis confirmed that nusG and rplK-rplA are preceded by separate promoters. In addition, a promoter cloned by random search of transcription-initiating activity by Pátek et al. (34) fully coincided with a fragment of the nucleotide sequence upstream of secE. The promoter region upstream of rplK-rplA is very efficient for transcription initiation, since it confers resistance to up to 975 μg of kanamycin/ml when coupled to the Tn5 kanamycin resistance gene, higher than previously reported promoters from corynebacteria available to us (24, 25). The nusG promoter is slightly less efficient in transcription initiation. The regulation of these promoters in response to nutrient starvation, osmotic stress, and the rel control is the subject of further research. These strong promoters may be used for efficient gene expression in corynebacteria.
ACKNOWLEDGMENTS
This research was supported by a grant from the European Union (BIO4-CT96-0145). Carlos Barreiro received a fellowship from the Ministry of Science and Technology (Spain), and Eva González- Lavado was granted a fellowship by the Basque Government (Vitoria, Spain). We thank H. Sahm for providing the initial cosmid library, A. Rodríguez-García for his scientific support, and M. Mediavilla, B. Martín, J. Merino, R. Barrientos, and M. Corrales for excellent technical assistance.
ADDENDUM IN PROOF
A related article on a defined deletion within the rplK gene will appear in Microbiology (L. Wehmeier, O. Brockmann-Gretza, A. Pisabarro, A. Tauch, A. Pühler, J. F. Martín, and J. Kalinowski, Microbiology, in press).
REFERENCES
- 1.Alting-Mees M A, Short J M. pBluescript II: gene mapping vectors. Nucleic Acids Res. 1989;17:9494. doi: 10.1093/nar/17.22.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amador E, Castro J M, Correia A, Martín J F. Structure and organization of the rrnD operon of Brevibacterium lactofermentum: analysis of the 16s rRNA gene. Microbiology. 1999;145:915–924. doi: 10.1099/13500872-145-4-915. [DOI] [PubMed] [Google Scholar]
- 3.Birnboim H C, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadenas R F, Martín J F, Gil J A. Construction and characterization of promoter-probe vectors for corynebacteria using the kanamycin-resistance reporter gene. Gene. 1991;98:117–121. doi: 10.1016/0378-1119(91)90113-p. [DOI] [PubMed] [Google Scholar]
- 5.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry III C E, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrel B G, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 6.Downing W L, Sullivan S L, Gottesman M E, Dennis P P. Sequence and transcriptional pattern of the essential Escherichia coli secE-nusG operon. J Bacteriol. 1990;172:1621–1627. doi: 10.1128/jb.172.3.1621-1627.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draper D E. How do proteins recognize specific RNA sites? New clues from autogenously regulated ribosomal proteins. Trends Biochem Sci. 1989;14:335–338. doi: 10.1016/0968-0004(89)90167-9. [DOI] [PubMed] [Google Scholar]
- 8.Dunican L K, Shivnan E. High frequency transformation of whole cells of amino acid producing coryneform bacteria using high voltage electroporation. Biotechnology. 1989;7:1067–1070. [Google Scholar]
- 9.Eikmanns B J, Thum-Schmitz N, Eggelin L, Lüdtke K, Sahm H. Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology. 1994;140:1817–1828. doi: 10.1099/13500872-140-8-1817. [DOI] [PubMed] [Google Scholar]
- 10.Friesen J F, Fiil N P, Parker J M, Haseltine W A. A new relaxed mutant of Escherichia coli with an altered 50S ribosomal subunit. Proc Natl Acad Sci USA. 1974;71:3465–3469. doi: 10.1073/pnas.71.9.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman E, Jakubowski H. Uncharged tRNA, protein synthesis, and the bacterial stringent response. Mol Microbiol. 1990;4:2035–2040. doi: 10.1111/j.1365-2958.1990.tb00563.x. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D. Studies on transformation of Escherichia coli. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D. Techniques for transformation of Escherichia coli, In: Glover D M, editor. DNA cloning. A practical approach. Oxford, United Kingdom: Irl. Press; 1985. pp. 109–135. [Google Scholar]
- 14.Janda L, Tichy P, Spizek J, Petricek M. A deduced Thermomonospora curvata protein containing serine/threonine protein kinase and WD-repeat domains. J Bacteriol. 1996;178:1487–1489. doi: 10.1128/jb.178.5.1487-1489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama M, Sakai Y, Okamoto S, Ihara F, Nihira T, Yamada Y. Gene organization in the ada-rplL region of Streptomyces virginiae. Gene. 1996;171:135–136. doi: 10.1016/0378-1119(96)00067-4. [DOI] [PubMed] [Google Scholar]
- 16.Kawamoto S, Zhang D, Ochi K. Sequence analysis of the ribosomal L11 protein gene (rplK=relC) in Streptomyces lavendulae using a deletion allele. J Antibiot. 1998;51:954–957. doi: 10.7164/antibiotics.51.954. [DOI] [PubMed] [Google Scholar]
- 17.Kieser T. Factors affecting the isolation of cccDNA from Streptomyces lividans and Escherichia coli. Plasmid. 1984;12:19–36. doi: 10.1016/0147-619x(84)90063-5. [DOI] [PubMed] [Google Scholar]
- 18.Kramer W, Drutsa V, Jansen H-W, Kramer B, Pflugfelder M, Fritz H K. The gapped duplex method DNA approach to oligonucleotide-directed mutation construction. Nucleic Acids Res. 1984;12:9441–9456. doi: 10.1093/nar/12.24.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 20.Küster C, Piepersberg W, Distler J. Cloning and transcriptional analysis of the rplKA-orf31-rplJL gene cluster of Streptomyces griseus. Mol Gen Genet. 1998;257:219–229. doi: 10.1007/s004380050642. [DOI] [PubMed] [Google Scholar]
- 21.Malumbres M. Clonación y caracterización molecular de los genes biosintéticos de treonina (thrC) y lisina de Brevibacterium lactofermentum. Ph.D. thesis. León, Spain: University of León; 1993. [Google Scholar]
- 22.Malumbres M, Martín J F. Molecular control mechanisms of lysine and threonine biosynthesis in amino acid-producing corynebacteria: redirecting carbon flow. FEMS Microbiol Lett. 1996;143:103–114. doi: 10.1111/j.1574-6968.1996.tb08468.x. [DOI] [PubMed] [Google Scholar]
- 23.Malumbres M, Gil J A, Martín J F. Codon preference in corynebacteria. Gene. 1993;134:15–24. doi: 10.1016/0378-1119(93)90169-4. [DOI] [PubMed] [Google Scholar]
- 24.Martín J F. Molecular genetics of amino acid-producing corynebacteria. In: Baumberg S, Hunter I, Rhodes M, editors. Microbial products: new approaches. Cambridge, United Kingdom: Cambridge University Press; 1989. pp. 25–29. [Google Scholar]
- 25.Martín J F, Gil J A. Corynebacteria. In: Demain A L, Davies J E, Atlas R M, Cohen G, Hershberger C L, Hu W-S, Sherman D H, Willson R C, Wu J H D, editors. Manual of industrial microbiology and biotechnology. 2nd ed. Washington, D.C.: ASM Press; 1999. pp. 379–391. [Google Scholar]
- 26.Martín J F, Cadenas R F, Malumbres M, Mateos L M, Guerrero C, Gil J A. Construction and utilization of promoter-probe and expression vectors in corynebacteria. Characterization of corynebacterial promoters. In: Heslot H, Davies J, Florent J, Bobichon L, Durand G, Penasse L, editors. Genetics of Industrial Microorganisms. Strasbourg, France: Société Française de Microbiologie; 1990. pp. 283–292. [Google Scholar]
- 27.Mead D A, Kemper B. Chimeric single-stranded DNA phage-plasmid cloning vectors. Bio/Technology. 1988;10:85–102. doi: 10.1016/b978-0-409-90042-2.50010-6. [DOI] [PubMed] [Google Scholar]
- 28.Nelson K E, Clayton R A, Gill S R, Gwinn M L, Dodson R J, Haft D H, Hickey E K, Peterson J D, Nelson W C, Ketchum K A, McDonald L, Utterback T R, Malek J A, Linher K D, Garrett M M, Stewart A M, Cotton M D, Pratt M S, Phillips C A, Richardson D, Heidelberg J, Sutton G G, Fleischmann R D, Eisen J A, Fraser C M, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 29.Ochi K. Occurrence of the stringent response in Streptomyces sp. and its significance for the initiation of morphological and physiological differentiation. J Gen Microbiol. 1986;132:2621–2631. doi: 10.1099/00221287-132-9-2621. [DOI] [PubMed] [Google Scholar]
- 30.Ochi K. Streptomyces relC mutants with an altered ribosomal protein ST-L11 and genetic analysis of a Streptomyces griseus relC mutant. J Bacteriol. 1990;172:4008–4016. doi: 10.1128/jb.172.7.4008-4016.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ochi K. Searching for novel ribosomal functions. Tanpakushitsu Kakusan Koso. 1999;44:1967–1974. [PubMed] [Google Scholar]
- 32.Ochi K, Zhang D, Kawamoto S, Hesketh A. Molecular and functional analysis of the ribosomal L11 and S12 protein genes (rplK and rpsL) of Streptomyces coelicolor A3(2) Mol Gen Genet. 1997;256:488–498. doi: 10.1007/pl00008614. [DOI] [PubMed] [Google Scholar]
- 33.Paradkar A S, Kwamena A A, Jensen S E. A pathway-specific transcriptional activator regulates late steps of clavulanic acid biosynthesis in Streptomyces clavuligerus. Mol Microbiol. 1998;27:831–843. doi: 10.1046/j.1365-2958.1998.00731.x. [DOI] [PubMed] [Google Scholar]
- 34.Pátek M, Eikmanns B J, Pátek J, Sahm H. Promoters from Corynebacterium glutamicum: cloning, molecular analysis and search for consensus motif. Microbiology. 1996;142:1297–1309. doi: 10.1099/13500872-142-5-1297. [DOI] [PubMed] [Google Scholar]
- 35.Puttikhunt C, Nihira T, Yamada Y. Cloning, nucleotide sequence and transcriptional analysis of the nusG gene of Streptomyces coelicolor A3(2), which encodes a putative transcriptional antiterminator. Mol Gen Genet. 1995;247:118–122. doi: 10.1007/BF00425829. [DOI] [PubMed] [Google Scholar]
- 36.Ruklisha M, Viesturs U, Labane L. Growth control and ppGpp synthesis in Brevibacterium flavum cells at various medium mixing rates and aeration intensities. Acta Biotechnol. 1995;15:41–48. [Google Scholar]
- 37.Sahm H, Eggeling L, Eikmanns B J, Krämer R. Metabolic design in amino acid producing bacterium Corynebacterium glutamicum. FEMS Microbiol Lett. 1995;16:243–252. [Google Scholar]
- 38.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 39.Santamaría R I, Gil J A, Martín J F. High-frequency transformation of Brevibacterium lactofermentum protoplasts by plasmid DNA. J Bacteriol. 1985;162:463–467. doi: 10.1128/jb.162.1.463-467.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santamaría R I, Martín J F, Gil J A. Identification of a promoter sequence in the plasmid pUL340 of Brevibacterium lactofermentum and construction of new cloning vectors for corynebacteria containing two selectable markers. Gene. 1987;56:199–208. doi: 10.1016/0378-1119(87)90137-5. [DOI] [PubMed] [Google Scholar]
- 41.Smith I, Paress P, Cabane K, Dubnau E. Genetics and physiology of the rel system of Bacillus subtilis. Mol Gen Genet. 1980;178:271–279. doi: 10.1007/BF00270472. [DOI] [PubMed] [Google Scholar]
- 42.Southern E M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 43.Thompson J D, Higgins D G, Gibson T J, Clustal W. Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vieira J, Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 45.Wehmeier L, Schäfer A, Burkovski A, Krämer R, Mechold U, Malke H, Pühler A, Kalinowski J. The role of the Corynebacterium glutamicum rel gene in (p)ppGpp metabolism. Microbiology. 1998;144:1853–1862. doi: 10.1099/00221287-144-7-1853. [DOI] [PubMed] [Google Scholar]