

Abstract
Hydrogels are the most relevant biochemical scaffold due to their tunable properties, inherent biocompatibility, and similarity with tissue and cell environments. Over the past decade, hydrogels have developed from static materials to “smart” responsive materials adapting to various stimuli, such as pH, temperature, chemical, electrical, or light. Light stimulation is particularly interesting for many applications because of the capability of contact‐free remote manipulation of biomaterial properties and inherent spatial and temporal control. Moreover, light can be finely adjusted in its intrinsic properties, such as wavelength and intensity (i.e., the energy of an individual photon as well as the number of photons over time). Water is almost transparent for light in the photochemically relevant range (NIR–UV), thus hydrogels are well‐suited scaffolds for light‐responsive functionality. Hydrogels' chemical and physical variety combined with light responsiveness makes photoresponsive hydrogels ideal candidates for applications in several fields, ranging from biomaterials, medicine to soft robotics. Herein, the progress and new developments in the field of light‐responsive hydrogels are elaborated by first introducing the relevant photochemistries before discussing selected applications in detail.
Keywords: adaptive biomaterials, hydrogels, light‐responsive materials, smart materials
Originating from static scaffolds, hydrogels have entered the domain of stimuli‐responsive (smart) materials. Light as stimulus is of particular interest because it offers inherent spatiotemporal remote control over the stimulated response. Advances in the field of photoresponsive hydrogels are summarized with a focus on the molecular design and architecture, as well as the respective applications of these fascinating materials.

1. Introduction
Hydrogels are water‐swollen 3D networks of either physically or chemically crosslinked polymers.1 They are excellent soft materials with diverse applications in various fields due to their tunable chemistry structure and physical properties as well as excellent biocompatibility.2, 3, 4, 5 Hydrogel research has evolved from static materials to smart ones, whose structures and property show responsiveness to external stimuli, including temperature, pH, light, electric or magnetic fields, and the presence of enzymes or ion concentrations.6, 7, 8, 9, 10, 11 This unprecedented level of control over material properties has driven both medical and industrial fields into the realm of possibility: controlled drug delivery,12 chemical sensors,13 soft actuators or robotics,14, 15, 16 or scaffolds for dynamic 3D cell culture.17 Photoresponsiveness is attractive due to the inherent advantages of light as stimulus. Light is noninvasive and allows remote manipulation of materials without requiring additional reagents, thus, with limited byproducts. Modulation of irradiation parameters, such as light intensity and irradiation time, permits the precise control of irradiation dosage, thereby, the degree of photoreactions. Spatial control can be achieved in both 2D and 3D, and temporal control is possible by simply turning the respective light source on or off. Wavelength‐selective photochemical reactions can be used for orthogonal photomediation of hydrogel properties.18 Photoresponsive hydrogels, as externally tunable materials, strongly contribute to the field of soft smart materials in regard to the above‐mentioned applications.19 In this review, we elaborate on the design and the emerging applications of photoresponsive hydrogels. A comprehensive review about utilizing light for the formation of hydrogels is given by Mi and co‐workers.20 First, we discuss responsive modes and photochemical mechanisms of photoresponsive hydrogels. Following this, we highlight their applications for dynamic cell environments, smart biointerfaces, controlled drug delivery, photodriven actuators, and light‐healing materials. The review concludes with an outlook on the challenges and potential future approaches for photoresponsive hydrogels.
2. Light as Stimulus for Hydrogels
2.1. Macroscopic Hydrogel Photoresponses
The interaction of light with photoresponsive hydrogels can result in different responses, some of which are observable with the bare eye. Those include hydrogel formation or degradation, network contraction or expansion, and chemical modifications in the network, which then have an immediate consequence on materials properties (Figure 1 ). To distinguish between postmodifications and stimuli‐responsiveness, we herein define responsiveness as a controlled, in situ change of macroscopic materials properties because of induced molecular changes. Therefore, a light‐induced postmodification itself is not termed responsiveness, unless it leads to an immediate response of the system as a consequence of the chemical change (e.g., shrinking, repelling cells or proteins, etc.). Depending on the molecular mechanism it can be distinguished between reversible and irreversible photoresponsive processes.
Figure 1.
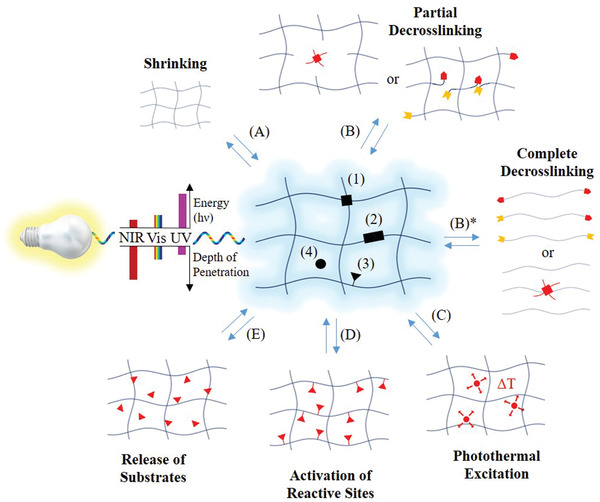
Molecular architecture and responses of a photoresponsive hydrogel (middle). The photoresponsive groups (black) can represent or be: 1) in the crosslinking points, 2) along the polymer or supramolecular backbone or 3) along the side chains, or 4) dissolved in the aqueous medium of hydrogels. Depending on location and type of photoresponsive moiety, photoresponses include shrinking (A) and partial de‐crosslinking (B), which is typically accompanied with an increase in water uptake and thus an increase in hydrogel volume. The opposite case, a photoinduced increase in crosslinking is typically accompanied by hydrogel shrinking. Complete de‐crosslinking leads to hydrogel degradation (i.e., liquification) (B)*. Further hydrogel responses are: C) photothermal excitation (i.e., a local increase in temperature), D) activation or deactivation of reactive sites, and E) release or capture of substrates.
Photoresponsive hydrogels can show volume changes via swelling or shrinkage due to water uptake or release upon light irradiation, respectively (Figure 1A). The swelling ratio is mainly determined by the network's hydrophilicity and crosslinking density, which can both be manipulated with light. An elegant approach is to influence the crosslinking of hydrogels via photoisomerization reactions. Thus, Harada and co‐workers developed hydrogels based on an α‐cyclodextrin (α‐CD)‐based [c2]daisy chain with an azobenzene derivative as the axis.21 Reversible photoisomerization of azobenzene units lead to a change in the average length of polymer chains in between two consecutive junctions and therefore to shrinking or swelling, respectively. A similar effect could be achieved by light‐induced cleavage of polymer backbones or crosslinking points (Figure 1B). The latter approach often lacks reversibility due to the irreversibility of most cleavage type reactions, which is why electrocyclic addition reactions, such as photodimerizations, have been explored to reversibly and repetitively modulate crosslinking in networks (Figure 1B, reverse reaction).
Incorporation of spiropyranes into hydrogels, which change their polarity upon photoisomerization, is also effective in remote changing the hydrogels' swelling properties. Both strategies produce reversible systems, given that no functional moiety is destroyed or removed upon heating, or reacted after isomerization. Photocontrolled changes in hydrogel volume are particularly useful for remote controlled actuators.22 Increasing or decreasing the stiffness of hydrogels can also be photoinduced by a variation of the degree of crosslinking, which is useful to design dynamic cell microenvironments.23, 24 In the border case of total loss of crosslinking, hydrogel liquification can be achieved.25 Such photoinduced gel–sol conversions (or vice versa) have been developed based on supramolecular hydrogels containing photoisomerizing compounds.26, 27 A typical example is the host–guest complex between azobenzene and cyclodextrine (CD), which can be used to form crosslinking points in hydrogel networks.28 Such hydrogels show reversible gel–sol transitions based on the cis–trans photoisomerization of azobenzene units, as only the trans azobenzene can serve as stable guest molecule. Another strategy to achieve reversible photoinduced gel–sol conversions is by introducing reversible covalent bonds (e.g., disulfides and trithiocarbonates) into the crosslinking points or the backbone of hydrogels, which spontaneously recombine after being cleaved by UV light.29, 30
Functional groups are critical to define the chemical, physical, and biological properties of hydrogels, such as wettability, catalysis, bioadhesion, etc. Hydrogels with photoresponsive functionalities enable the most precise spatiotemporal control over the expression of functional groups within hydrogel networks (Figure 1D,E), when compared to other stimuli such as temperature, pH, or ion concentrations. This can be achieved by introducing functional groups through photochemical addition reactions or activation of functional groups through the release of photocaging moieties or via photoisomerization processes. On the contrary, the removal of functionalities can be induced through photochemical cleavage reactions or deactivation of functionalities via photoaddition reactions. Two‐photon processes can help overcoming issues with light absorption and grant unreached spatial control in bulk. With such strategies, it is possible to not only pattern hydrogels with spatially defined functionalities but also to manipulate the display of functional groups in situ. These types of hydrogels remain of prime interest for mimicking the dynamic nature of the cellular microenvironment and controlled release techniques.
2.2. Molecular Mechanisms
Hydrogel photoresponsiveness usually arises from functional groups incorporated in the system and is not an inherent attribute of the polymers or supramolecules themselves, an exception being for example photothermally responsive, LCST‐based systems. An overview about relevant photoactive groups is given below. Type and placement of the photosensitive moieties must be chosen properly to achieve the desired response (see Section 2.1). The choice of a suitable light source is crucial to fit the respective needs of application. Generally, the emission spectrum of the light source and the absorption profile of the photoresponsive functionality should match in an uncontested region of the electromagnetic spectrum to increase the quantum yield and depth of light penetration. For hydrogels as bioscaffolds, potential harmful effects to organisms due to the irradiation must be considered. Out of the several types of photoresponsive molecules known, many have been employed to create photoresponsive hydrogels. The most relevant ones are introduced below, and representative examples are shown in Table 1 . The photoreaction types include cleavage, addition, exchange, and isomerization and are introduced in that order. Thereafter, photothermal agents are introduced.
Table 1.
Different photoreaction types and functional groups utilized for the design of photoresponsive hydrogels
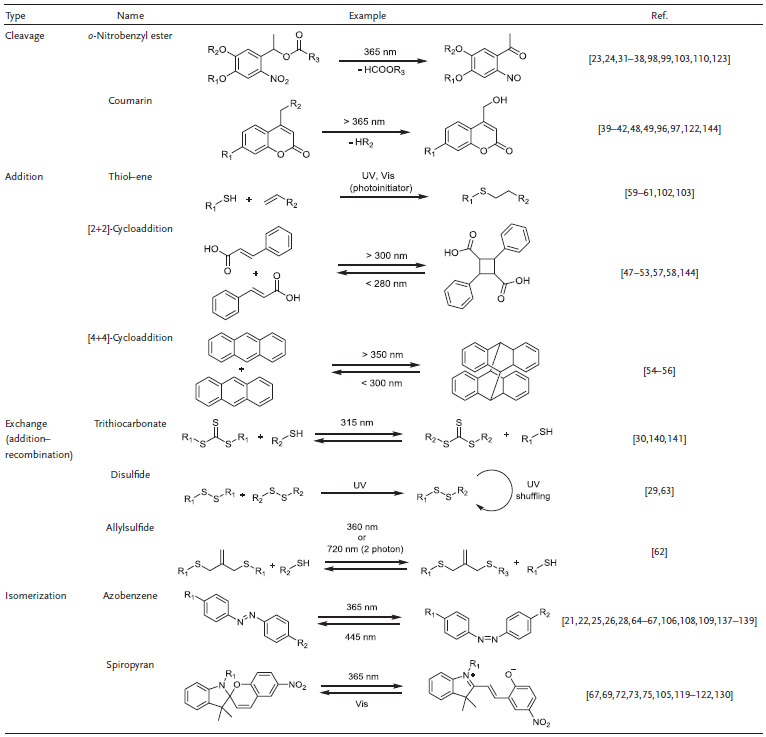
Derivatives of o‐nitrobenzyl compounds are by far the most widely applied photolabile group and therefore found their way into hydrogel chemistry.31, 32, 33 Because of their well‐known photolysis mechanism and adjustable chemical structure, they can serve as photocages34 or photolabile linkers.35 Their absorption profiles have been finely adjusted by modifying the aromatic structure.36 The parent structure, o‐nitrobenzyl ester, undergoes photolysis into carboxylic acid and o‐nitrosobenzyladehyde under irradiation with 260 nm light. Donor substitution, for example in 4,5‐dimethoxy o‐nitrobenzyl moieties, redshifts the wavelength required for cleavage to around 350 nm.37, 38 Coumarin derivatives are popular as alternatives to o‐nitrobenzyl derivatives due to their biocompatibility, fast cleavage rates, redshifted absorption, and two photon–induced reactions.39 Various modifications of coumarin derivatives have been developed to shift their absorption to a biologically relevant range. For example, 7‐diethylamino‐4‐thiocoumarinylmethyl derivative shows a high uncaging efficiency with blue light (470 nm, εϕ = 120 ± 25 m −1 cm−1).40 Zhu and co‐workers developed a new series of coumarin derivatives with an electron‐rich styryl moiety at the 3‐position with excellent absorption in the light region between 430 and 515 nm (blue‐green), large two‐photon absorption cross‐sections, and high uncaging quantum yields.41 Recently, Winter and co‐workers reported a family of boron‐dipyrromethene (BODIPY) photocages cleaved by green light (>500 nm).42 Other photolabile groups, such as triphenylmethane,43 p‐hydroxylphenacyl,44, 45 and 8‐bromo‐7‐hydroxylquinoline46 derivatives, are not of high significance for hydrogels yet.
Due to their orthogonality with many functional groups, electrocyclic reactions, such as [2+2], [4+4] or dipolar cycloadditions, are relevant reversible photoinduced addition reactions. Photodimerization reactions proceed by either a [2+2]‐ or a [4+4]‐cycloaddition mechanism with one π‐system being in an excited state, according to the Woodward–Hoffman rules. Both cycloadditions can be reversed upon exposure to light at suitable wavelength or via heating. Representative examples are [2+2]‐cycloaddition of cinnamates,47 coumarins,48, 49 maleimides,50, 51 stilbene,52, 53 and [4+4]‐cycloaddition of anthracenes.54, 55, 56 Many cycloaddition reactions can be activated with UV light. Due to the cleavage of C—C bonds, the reverse reaction requires light with higher energy. For example, stilbene derivatives undergo a [2+2]‐photodimerization when irradiated at about 340 nm, whereas the respective cyclobutane derivatives are cleaved by irradiation at 254 nm.57 The respective wavelengths can be redshifted into the visual light regime by changing the molecular structure of photoactive groups. Doi et al. recently reported the dimerization of trans‐carboxyl styrylpyrene under 455 nm visible light, which could be efficiently reversed by irradiation with 340 nm UV light.58 Thiol–ene “click” chemistry found widespread application in polymer chemistry and also made its way into the design of photoresponsive hydrogels.59, 60, 61 In addition, photoinduced exchange reactions, such as disulfide exchange chemistry and allyl sulfide/thiol exchanges, have been adopted in the design of light‐responsive hydrogels.62, 63 Notably, thiol‐ or disulfide reactions proceeding via a radical mechanism are fundamentally different from electrocyclic cycloadditions, which proceed via a concerted cyclic transition state. This is an important consideration when evaluating possible side reactions. Photoisomerization processes are the structural change of photoexcited molecules in between different isomers, such as cis–trans or constitutional isomers. These reactions are often reversible and produce no byproducts, which is desired for the design of photoresponsive hydrogels. Azobenzene is a well‐known example that undergoes photoinitiated (365 nm) transformation from trans‐configuration to cis‐configuration via a π–π* transition and can relax back to the more stable trans‐isomer by visible light irradiation (445 nm) or heating.64 This photoisomerization changes the molecular polarity and spatial demand, which influences the behavior in host–guest interaction. Azobenzene forms host–guest complexes with cyclodextrin whose stabilities depend on the cis or trans state of the azobenzene moiety.65 Thus, many designs for photoresponsive hydrogels based on azobenzene–CD host–guest interaction were invented, as reviewed lately.66 Spiropyran isomerizes from the hydrophobic close‐ring form to the hydrophilic open‐ring merocyanine zwitterion under the irradiation of 365 nm.67 If the spiropyrane isomerization is conducted in acidic environment, the merocyanine moiety is protonated to the respective cationic species, which influences the respective photoinduced reverse reaction.68, 69 The reverse isomerization can also be induced thermally. The sensitive wavelengths for both groups can be tuned by varying the substituents.70
The change from one isomer to the other is accompanied by a change in color, therefore pyrane‐based hydrogels possess inherent photochromic properties, which were lately shown to be tunable.71, 72 Their photochromic properties can be beneficial in terms of easy indication and monitoring of the photochemical reaction or for manufacturing sunglass‐like hydrogel contact lenses.73 As the changes in hydrophilicity upon spiropyrane isomerization affect the ability of hydrogels to swell, the ring opening or closing of spiropyranes can be used to reversibly toughen or change the volume of hydrogels.74, 75
Photothermal agents generate heat through irradiation. The local heating can be used to trigger reversible phase transitions of thermoresponsive polymers. This mechanism has been extensively used in the design of photoresponsive hydrogels. Photothermal agents can be inorganic nanomaterials and organic compounds. Noble metals (e.g., Au,76, 77 Ag,78 and Pt79) or oxide nanoparticles,80, 81 carbon nanomaterials,82 as well as black phosphorus83 are representatives of inorganic photothermal agents. Organic photothermal agents are also well developed and rely on cyanines,84 croconaines,85 diketopyrrolopyrroles,86 porphyrins,87 and conjugated polymers.88, 89, 90 Commonly used thermally responsive materials include poly(N‐isopropylacrylamide) (PNIPAAm), thermosensitive poly(ethylene glycol) (PEG) analogs, thermoresponsive elastin‐like peptides (ELPs), and thermosensitive liposomes.91, 92 This strategy is versatile to design hydrogels responsive to biocompatible NIR light; however, the damage of overheating to surrounded tissues need to be considered, at least in biological applications.93 In the same manner, the evaluation of bioorthogonality of other photochemical reactions remains of high importance, for example it was shown by Li et al. that tetrazole photoclick reactions are indeed not bioorthogonal and could be instead used for protein crosslinking.94
3. Applications of Photoresponsive Hydrogels
3.1. Dynamic Cell Microenvironment
In vitro cell biology aims to provide cell growing conditions as close to the natural role model as possible. Photoresponsive hydrogels are attractive scaffolds to mimic the dynamic nature of biomechanics in living tissues as they allow remote and contact‐free manipulation of the cell‐growing conditions. A brief review about engineering cell environments with light‐responsive hydrogels was recently given by Lin and co‐workers.95
Photocaged reactive groups are released, when their respective protecting group is cleaved via light irradiation. Incorporation of photocaged groups as structural components of hydrogels allows remote activation of reactive sites in situ. Shoichet and Luo paved the way in 2004 for photoreleased active sites in hydrogels by incorporating coumarin‐caged thiols into an agarose hydrogel.96 Upon two‐photon activation, free thiols are released and can undergo Michael reactions to pattern bioactive species. 3D channels of the adhesive peptide GRGDS can then confine cell migration into oligopeptide‐modified channels. This strategy was extended to pattern large proteins by combination of photocaging Michael reaction and orthogonal physical binding pairs.97 Later, in 2013, Lutolf and co‐workers developed a procedure that enabled in situ 3D cell manipulation via release of photocaged enzymes.98 Photocleavage of an o‐nitrobenzyl moiety from an enzymatic peptide substrate led to activation of this enzyme and enabled spatially defined enzymatic tethering of complex and physiologically relevant signaling proteins, extracellular matrix (ECM) proteins, and growth factors. Thus, guided 3D cell invasion within hydrogels was achieved (Figure 2 A). In addition to the control of tethering and release of biomolecules from hydrogels, photocleavage of 3‐(4,5‐dimethoxy‐2‐nitrophenyl)‐2‐butyl ester (DMNPB), photocaging the cyclic RGD peptide cyclo(Asp‐d‐Phe‐Lys‐Arg‐Gly) on the carboxylic side group of the aspartic acid residue, was shown to be effective in regulating cell adhesion, inflammation, and vascularization of hydrogels in vivo (Figure 2B).99 The former strategies required the design and incorporation of photocages to reactive or bioactive groups. In contrast, photopolymerizations provide an easy access toward photoreactive hydrogels when residual acrylate groups are present after gelation. Residual acrylates enable covalent immobilization of acrylate‐derivatized moieties via secondary light‐induced polymerization. Photopolymerizations of acrylates are fast, which minimizes the polymerization time and thus cell death, rendering acrylate polymerizations suitable for the photoinduced formation of hydrogels.100 This strategy has been extensively used for patterning of bioactive molecules ranging from short adhesion peptides to full‐length proteins, whose concentrations can be controlled by varying the precursor concentration and light dosage. By this approach, West and co‐workers guided cells in hydrogels both in 2D and 3D on the spatial microscale. Bioactive acrylate derivatives were immobilized in preformed PEG hydrogels via single‐photon and two‐photon photolithography.101 Local presentation of RGDS peptide guided cell migration to the RGDS‐containing regions. However, it is difficult to control and characterize the extent of acrylates remaining after the primary polymerization, i.e., the hydrogel formation. Photoaddition reactions have also been explored for the design of photoresponsive cell microenvironments. The thiol–ene reaction has attracted interest in biorelated applications due to its click‐like and biocompatible properties while not requiring Cu(I) catalyst, oxygen removal, or a photoinitiator. Anseth and co‐workers developed an enzymatically degradable hydrogel platform, where 3D photopatterning of biomolecules via thiol–ene reaction allows the direct observation of cellular behaviors in three dimensions in situ.102 The same group later developed a reversible patterning strategy based on the combination of two orthogonal photoreactions, namely, the photocoupling of thiol–ene and the photocleavage of an o‐nitrobenzyl moiety.103 Both the introduction and subsequent removal of RGDS peptide could be explicitly controlled in time and space by exposure to light. This strategy was even further extended to photopatterning of full length proteins via a biorthogonal reaction utilizing a photocaged oxime.104 Reversible photopatterning of biochemical ligands has also been achieved with a related reaction, the photoinitiated addition‐fragmentation chain transfer of an allyl sulfide group.62 It permits the iteratively repeatable immobilization of thiol‐containing peptides, but the exchanging process also inevitably decreases the stiffness of the hydrogel.
Figure 2.
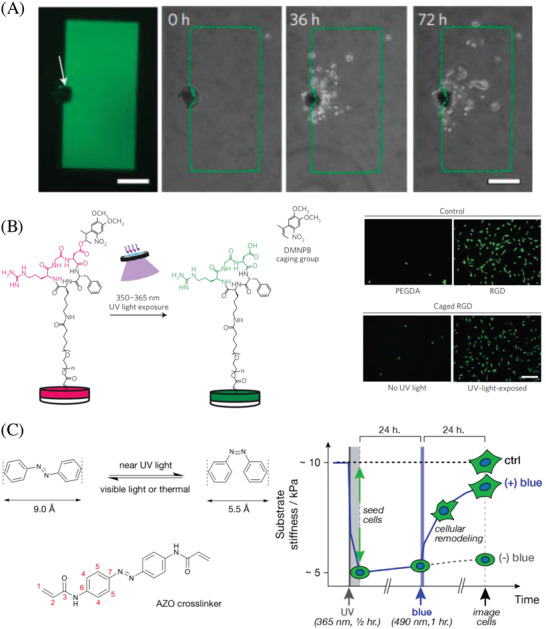
Light‐induced manipulation of cell behavior. A) Guided cell growth via enzymatic photopatterning. Cells invade only into the areas, where photocaged (inactive) enzymes have been remotely freed in situ (green rectangle). Reproduced with permission.98 Copyright 2019, Springer Nature. B) In vivo regulation of cell adhesion, inflammation and vascularization of biomaterials with UV light. Reproduced with permission.99 Copyright 2018, Springer Nature. C) On‐demand softening of a hydrogel due to the photoisomerization of an azobenzene‐containing dimethacrylate crosslinker. Reproduced with permission.106 Copyright 2012, American Chemical Society.
Cell manipulation via photoisomerization can be a result of either a chemical or a physical change in the hydrogel network upon isomerization. For constitution isomers, as in the case of spiropyranes, the chemical change upon isomerization is significant. Thus, dynamic control over human mesenchymal stem cell (hMSC) differentiation was achieved by Jiang and co‐workers by developing a photodynamic zwitterionic hydrogel based on the photoisomerization of spiropyran.105 Remarkably, the zwitterionic hydrogel could be continuously and reversibly shifted between a zwitterionic hydrophilic state and a hydrophobic state by shining either green or NIR light, respectively, which triggered or suspended the stem cell differentiation.
Stereoisomers (e.g., cis and trans azobenzene) do not significantly change chemical properties such as hydrophilicity upon isomerization. Nonetheless, they can be utilized to manipulate cells in hydrogels by tuning its stiffness. Exploiting the cis–trans photoisomerization of an azobenzene dimethacrylate crosslinker for in situ hydrogel softening, Wong and co‐workers were able to modulate the behavior of MSCs when exposed to blue light. As the light did not show to any influence on the MSCs, their behavior in the hydrogel can be regarded as pure mechano‐response (Figure 2C).106 Apart from photoisomerization, there are further strategies to photoinduce a change in hydrogels' physical properties to study the mechano‐responses of cells in situ. As discussed earlier, photoswitching the degree of crosslinking is useful for the remote control of the physical (mechanical) properties of hydrogels. Anseth and co‐workers developed photodegradable hydrogels based on PEG networks containing nitrobenzyl groups.24 The photolysis reaction allowed precise control of gel crosslinking density and stiffness in the presence of cells via UV, visible, or two‐photon irradiation. Temporal and spatial regulation of cell‐matrix mechanics interactions can be achieved with such photodegradable hydrogels. However, such photoablation is an irreversible process toward softening. To mimic the dynamic nature of ECM, it is necessary to design antagonistic or reversible photoresponsive hydrogels. The same group thus developed hydrogels with reversible mechanics based on hyaluronic acid modified with nitrobenzyl and methacrylate groups.23 Sequential photodegradation and photoinitiated crosslinking reactions made it possible to soften and then to stiffen the hydrogels. HMCs were used to demonstrate reversible cellular mechanosensing. Photoinitiated hydrogel softening (Young's modulus from ≈14 to 3.5 kPa) resulted in a decrease in the cell area and nuclear localization of Yes‐associated protein/transcriptional coactivator with PDZ binding motif (YAP/TAZ), which followed a reverse trend and increased upon subsequent stiffening (Young's modulus from ≈3.5 to 28 kPa). Suggs and co‐workers developed an alginate/calcium hydrogel capable of both dynamic stiffening and softening.107 The hydrogel stiffness depends on the calcium concentration and can be increased or decreased by controlled introduction of additional calcium or calcium chelators. NIR light–triggered release was achieved based on temperature‐sensitive liposomes encapsulated with gold nanorods to induce local heating upon irradiation. This system was effective to regulate fibroblast morphology based on dynamic phototuning of stiffness and demonstrated to be translatable to in vivo applications.
3.2. Controlled Drug Release
Controlled drug release is crucial for improving therapeutic efficacy and minimizing adverse effects due to the strong targeted features to regulate the distribution of drugs in vivo. Photoresponsive hydrogels can provide spatial and temporal control for drug release. The 3D networks of hydrogels can encapsulate various drugs, while the light‐induced gel‐to‐sol transition can be used to release the loaded drug.
Especially NIR‐responsive hydrogels provide great potentials for therapeutic treatment due to their deeper penetration in tissue and harmlessness (Figure 3 A). Gupta and co‐workers pioneered the design of photoresponsive nanogels based on the hydrophobic association of azobenzene‐modified dextran.108 Upon UV irradiation (365 nm), the cis–trans photoisomerization of azobenzene moieties results in weakening of hydrophobic interactions with concurrent expansion (swelling) of the inner hydrophobic cavities, which leads to faster rate of diffusion of the entrapped drug aspirin from the hydrophobic pockets of the gel network. A more recent example of an azobenzene host–guest interaction‐based release mechanism capable to photocontrol the release of a model drug was given by Anseth and co‐workers.109 Host–guest complexes between an azobenzene functionalized peptide as model drug and a four‐arm PEG‐based hydrogel functionalized with β‐cyclodextrin were shown to regulate the drug release under irradiation with 365 nm light. Noteworthy, the release rate even without light is high (≈75% after 2 h), showcasing the difficulty of designing a temporally well‐defined release mechanism in highly swelled hydrogels. To overcome the leakage problem of small‐sized drugs, Kim and co‐workers developed a photoresponsive hydrogel with mesoporous silica nanoparticles (MSN) by the molecular recognition between cyclodextrin‐modified MSN and six‐arm PEG with dodecyl terminal groups.110 MSN provides a rigid framework with nonporous reservoirs that can encapsulate a large amount of guest molecules, whose release can be blocked by “gatekeeper” cyclodextrin groups. The photolabile o‐nitrobenzyl linkers between cyclodextrin and MSN ensure not only the photodegradation of hydrogel networks but also the photoinduced removal of “gatekeeper” cyclodextrin groups, achieving the off–on release of encapsulated drugs.
Figure 3.
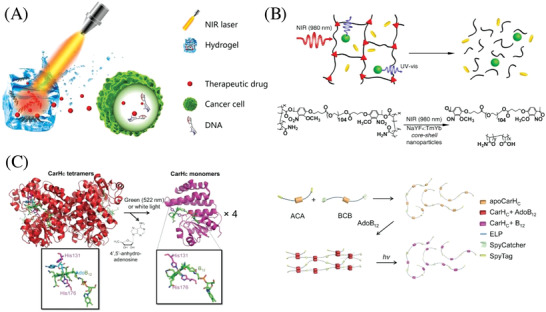
Controlled drug release based on photoresponsive hydrogels. A) Schematic illustration of a NIR‐triggered drug release from a hydrogel to induce apoptosis in cancer cells. Adapted with permission.113 Copyright 2012, American Chemical Society. B) Top: NIR‐triggered drug release based on the degradation of a photosensitive hydrogel encapsulating UCNPs. Bottom: Respective chemistry of NIR‐induced hydrogel degradation. Reproduced with permission.114 Copyright 2019, National Academy of Sciences. C) Left: Scheme of a photoresponsive protein hydrogel. The photodegradation is based on the light‐triggered disassembly of tetrameric CarHC accompanied by the degradation of AdoB12. Right: The hydrogel is synthesized by the polymerization of two telechelic proteins, ACA and BCB, through SpyTag and SpyCatcher chemistry and subsequent gelation with AdoB12. Reproduced with permission.112 Copyright 2019, National Academy of Sciences.
Supramolecular hydrogels are biomaterials, where molecular gelators are assembled into 3D fiber networks based on noncovalent interactions. Subtle structural changes in the constituent moieties greatly influence the hierarchical structures of obtained hydrogels. Thus, Hamachi and co‐workers have developed photoresponsive supramolecular hydrogels with glycolipid‐based hydrogelators bearing fumaric amide as photoswitching module.111 The cis–trans photoisomerization of fumaric amide effectively leads to assembly or disassembly of the self‐assembled supramolecular fibers to generate a hydrogel or the corresponding sol. Photoinduced gel‐to‐sol transition was used to accelerate the release of vitamin B12. The opposite process of photoinduced sol‐to‐gel conversion showed to be useful for restricting the rotary motion of a biomolecular motor protein (bead‐tethered F1‐ATPase) as well as the movement of bacteria (Escherichia coli) with spatiotemporal control.
Compared with synthetic polymer hydrogels, research in engineering photoresponsive protein hydrogels has just begun, even though protein hydrogels are attractive for biomedical applications owing to the ability to precisely control protein sequence, molecular weight, and tertiary structures. Sun and co‐workers created B12‐dependent photoresponsive protein hydrogels for controlled protein and stem cell release (Figure 3C).112 The CarHC domains tetramerize when binding to adenosylcobalamin (AdoB12) in the dark and can readily dissociate into monomers accompanied with a drastic protein conformational change caused by the cleavage of the C—Co bond on exposure to green (522 nm) or white light. The hydrogels are composed of entirely recombinant elastin‐like polypeptide‐fusion C‐terminal adenosylcobalamin binding domain (CarHC) protein using SpyTag‐SpyCatcher chemistry. The AdoB12‐dependent CarHC tetramerization was used for the formation of the hydrogel in the dark, which could undergo a rapid gel–sol transition caused by light‐induced CarHC disassembly. This system was shown to be effective to encapsulate and release both live cells and protein molecules upon white light exposure.
For in vivo applications, NIR light is more appealing than the rest of the photochemical wavelength spectrum, due to both the low health hazard and deeper light penetration. Cao and co‐workers developed an NIR‐light‐controlled drug delivery platform based on the composite hydrogels of low‐melting‐point agarose and PEGylated black phosphorous (BP) nanosheets.113 NIR‐induced (808 nm) heating due to the photothermal conversion of BP results in hydrolysis and subsequent melting of the hydrogel, which triggers the controlled release of encapsulated doxorubicin (DOX) (Figure 3A). In vivo experiments with tumor tissues demonstrated this system to be capable of accurately controlling the release of drugs to eradicate subcutaneous breast and melanoma cancers. The biodegradation of both BP and agarose allow this hydrogel to be easily excreted through urine after the treatment, which is promising for clinical translation. Upconversion luminescence converts NIR light into UV light, which can be used to design NIR light sensitive hydrogels. Thus, Zhao and co‐workers introduced upconversion nanoparticles into PEG hydrogels with photolabile linkers (Figure 3B).114 NIR light (980 nm) could then induce the gel–sol transition and trigger the release of biomacromolecules (proteins and enzymes) entrapped in the hydrogel without sacrificing their biocompatibility.
In addition to controlled drug release, photoresponsive hydrogels can be utilized to encapsulate and release live cells on‐demand. By taking advantage of the differences in reactivity of o‐nitrobenzyl derivatized linkers, Kasko and Griffin have developed a series of hydrogels with different photodegradation rate constants at 370 nm.115 Selective release of Human mesenchymal stem cells has been demonstrated without compromising cell viability, which is promising for controlled, sequential delivery of different cell populations. To drive drug release to a new level of site‐specific payload release, DeForest and co‐workers introduced a library of 17 peptide‐based crosslinkers, whose cleavage can be induced by three orthogonal stimuli (UV light, reductants, and matrix metalloproteinases). Hydrogels based on these multistimuli responsive crosslinkers and four‐arm poly(ethylene glycol) could be liquefied in the presence of any desired combination of those stimuli, based on whether the orthogonal cleaving functionalities in the crosslinkers were designed to be parallel, in series, or even both. This way highly selective and specific release conditions could be preprogrammed depending on the crosslinker design.116
3.3. Adaptive Surfaces
Surfaces are the interface between a material and its environment. They play a major role, whenever the shape of an interface or the interactions along an interface and not the bulk attributes are decisive for materials properties, which is the case for coatings, membranes or heterogenous catalysts. Despite their chemical nature, distinguishing features of surfaces are their physical properties, such as roughness, porosity or topology. Surface topologies responsive to light can be made from hydrogels. Broer and co‐workers have reported the dynamic surface of a pNIPAAm hydrogel with light‐absorbing CrO2.117 Upon flood irradiation, local heating leads to patterned swelling of hydrogels, thus forming concave structures. After switching off the light, pNIPAAm hydrogels become uniformly swollen generating a smooth surface. Hayward and co‐workers have reported the dynamic change of the wrinkled surface pattern allowing to displays or hide functionalized regions (Figure 4 A).118 A hybrid hydrogel of pNIPAAm loaded with iron oxide nanoparticles was used in this study. Under ambient light, the gel swelled in water, triggering an elastic creasing instability that sequesters functionalized regions at folded areas. Further flood exposure to UV light lead to shrinking of hydrogels, causing creases to unfold and “hidden” functionalities to display. This process is reversible, and it was also possible to independently actuate individually folded features through localized illumination. With photoresponsive hydrogels, it is convenient to control surface wettability and the diffusion and transport of molecules across the interface of materials. For example, ter Schiphorst et al. developed a photoresponsive cotton‐fabric modified with poly(NIPAAm‐co‐acrylic acid‐co‐spiropyran) hydrogel.119 Without light, the spiropyran (Sp) moiety isomerizes to the protonated, hydrophilic merocyanine, leading to water uptake from humid air and swelling of the hydrogel coating. Upon exposure to white light, the hydrophobic Sp isomer will restore, causing a shrinkage of fibers by 18% because of water release. After switching off the light, the fibers reswell to their original diameter, thus making this process cyclable.
Figure 4.
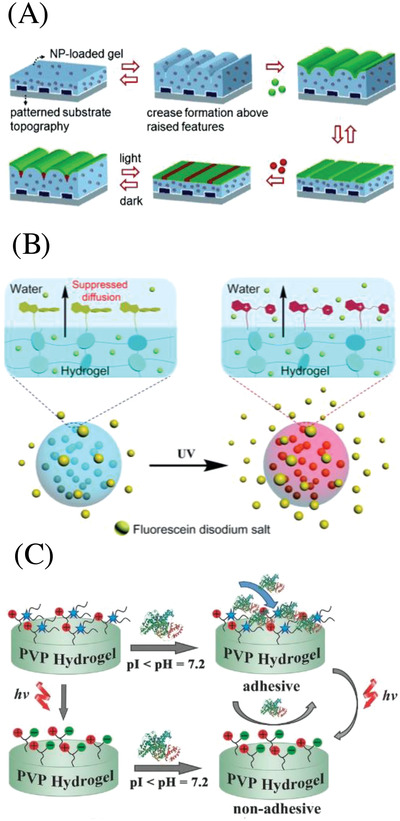
Adaptive surfaces based on photoresponsive hydrogels. A) Scheme showing the reversible switching of striped chemical patterns induced by exposure of PNIPAM hydrogels loaded with iron oxide nanoparticles to visible light. Reproduced with permission.118 Copyright 2019, Wiley‐VCH. B) Schematic illustration of light‐regulated molecular diffusion across a hydrogel layer based on the photoisomerization of spiropyran. Reproduced under the terms of the CC‐BY Creative Commons Attribution 3.0 Unported Licence ().120 Copyright 2019, Royal Society of Chemistry. C) Schematic illustration of regulated protein adhesion by phototriggered charge change on a hydrogel surface. Reproduced with permission.122 Copyright 2019, Wiley‐VCH.
In a different study, Chen et al. developed a strategy for covalent tethering of a photoresponsive spiropyrane layer (≈1.2 µm) to a hydrogel surface via quaternization reactions of tertiary amino groups with a iodide functionalized spiropyrane.120 The resulting hydrogel surface showed to be superhydrophobic (water contact angle of ≈142°–152°), which was attributed to both the hydrophobicity of Sp and the formed hierarchical micro/nanoscale surface roughness (Figure 4B). When the spiropyrane‐coated hydrogel was irradiated with UV light, the surface switched from superhydrophobic to hydrophilic, thus allowing photocontrolled diffusion of polar substrates in or out of the hydrogel, which was demonstrated for the inhibited release of fluorescein from a hydrogel in water. Another report making use of the change in hydrophilicity upon photoisomerization of spiropyrane is given by Liu and co‐workers, who showed regulated cell adhesion and surface‐mediated gene delivery based on a polyacrylamide hydrogel incorporating spiropyran moieties.121 UV light triggered the transition from the hydrophobic spiro form to the hydrophilic, zwitter ionic merocyanine form, leading to an increase in hydrophilicity by 13° as quantified by water contact angle (WCA) measurements, thus releasing mouse fibroblast L929 adherent cells. Visual light irradiation restored the surface to its original hydrophobic state, allowing repeated attachment and detachment of cells. Moreover, this reversible attach and release mechanism showed to be useful for light‐regulated release of gene‐modified cells after reverse gene transfection. Remote cells release from a hydrogels surface can also be induced by photoinduced surface charge changes, as previously shown by Zhu and co‐workers.122 Upon UV irradiation at 365 nm a coumarin moiety is cleaved from a caged lysine betaine zwitterion, consequently the surface transforms from an adhesive positive state to an antiadhesive zwitterionic state. With this technique, protein immobilization and directed cell adhesion could be accomplished with spatial and temporal control. Utilizing a similar approach, namely the photoinduced generation of zwitter ionic species on a hydrogel surface, Liu and Liu reported a hydrogel capable to not only kill bacteria on the surface due to a cationic charge, but also to consequently detach them from the surface via light to prevent further bacteria adhesion.123 Photocleavage of 4,5‐dimethoxy‐2‐nitrobenzyl group attached to a quaternary ammonium functionality with 365 nm light resulted in the change from a cationic to a zwitter ionic hydrogel surface. Due to this change, dead bacteria could be detached from the hydrogel surface (Figure 4C). As both the coumarin and the o‐nitrobenzyl based approaches for remote detaching of cells make use of irreversible photocleavage reactions, these systems are not cyclable in contrast to the spiropyrane photoisomerism‐based detach mechanism. The mechanical surface properties of hydrogels can also beexploited to manipulate the surface´s biological functionality including biophysical and bioadhesive properties. Thus, Yang et al. used a photodegradable hydrogel to control the magnitude and spatial organization of surface stiffness to direct cell fate.124 Light irradiation at 365 nm (10 W cm−2) was used to create stiffness patterns in situ, that lead to surface areas with either initial stiffness (Young's modulus ≈10 kPa) or softened areas (Young's modulus ≈2 kPa) with micrometer scale precision via masked irradiation. hMSCs seeded on hydrogels with higher concentrations of stiff regions display highly spread, elongated morphologies and higher Yes‐associated protein (YAP) activation, while randomized pattern induced round cell morphologies and low levels of YAP activation.
3.4. Soft Actuators
A distinguishing feature of living beings is the ability of active and controlled mobility. For humans it is our muscles, which work like actuators, that enable us to move so freely. The principle of an actuator is simple: an input of energy is converted into mechanical motion. Light‐triggered actuators are attractive from the point of remote manipulation of materials in applications like soft robotics, mechatronics or valves (microfluidics).125 Comprehensive reviews about soft smart actuators can be found elsewhere.126, 127, 128 Manufacturing soft actuators or molecular machines with fast and efficient light responsiveness is still a highly challenging field of research as full reversibility (e.g., contraction–expansion) is desired to generate functional materials.
Feringa and co‐workers showed how the light‐induced isomerization of a chiral helix‐like alkene could be used to manufacture a molecular motor (Figure 5 A).129 UV light (365 nm) causes isomerization of the stable (“Stable 1”) into the unstable isomer (“Unstable 1”), which then thermally (50 °C) releases the strain resulting from an unfavorable pseudoequatorial position of substituents via helix inversion leading to a full rotary cycle. The helix‐like alkene self‐assembles to nanofibers in aqueous solution, which can then be precipitated as hydrogel in Ca2+ solution to form strings with uniaxial nanofiber orientation (cm scale). Upon irradiation, the hydrogel strings bend toward the light with a flexion angle of 90° at a fast rate of ≈1.5° s−1. Relaxation into the original state was found to occur in less than 3 h, in accordance to the observed half‐life of the helix inversion of 2.7 h, which obviously cannot compete with biological actuators like muscles. Moreover, this process cannot be cycled, which was attributed to the instability of the supramolecular string at 50 °C. Takashima et al. designed a covalently crosslinked hydrogel with additional, reversibly light‐responsive crosslinking points based on host–guest interaction.22 Their hypothesis was, that partial de‐crosslinking will lead to an expansion, while the reformation of the additional crosslinking points causes a contraction of the hydrogel. The hydrogel was prepared from α‐CD‐modified acrylamide, azobenzene acrylamide and methylene bisacrylamide crosslinker. Due to the presence methylene bisacrylamide a covalently crosslinked network was formed, which was supported by additional reversible crosslinks due to the host–guest interaction between trans‐azobenzene and α‐CD moieties. UV light irradiation (365 nm) causes the azobenzene units to change to the cis‐state, disrupting the host–guest complexes and expanding the network. Shining visible light (430 nm) reversed this process and was observed to contract the hydrogel. Gels immersed in water were found to increase up to 124 wt% in weight upon UV irradiation and decrease back to 104 wt% upon visible light irradiation. Gel strings exposed to light sideways were found to bend away from the light source because of hydrogel expansion on the exposed side while visible light irradiation led to restoration of the original state. The system could be cycled for at least 5 times, but the light exposure time to reach flexion angle saturation were very long (≈55° after 60 min). Further work by Harada and co‐workers on α‐CD‐based systems led to greatly improved light‐responsive behavior (Figure 5B).21 In their novel network design, amino‐capped azobenzene moieties are covalently attached to α‐CD moieties, which dimerize to daisy chain type interlocked molecules ([c2]AzoCD2]. Those were then crosslinked via polycondensation reaction with succinimidyl ester capped four‐arm poly(ethylene glycol) to form a hydrogel network. UV irradiation (365 nm) lead to removal of the azobenzene moieties from the α‐CD cavities as consequence of the change from the trans‐ to the cis‐azobenzene isomer. As the four‐arm PEG crosslinked azobenzene moieties were interlocked through one α‐CD unit and covalently attached to another, they could not de‐crosslink (Figure 5B). Instead, a contraction was observed when the azobenzene moieties leave their α‐CD host, thus this system was complementary to their previous one, which expanded upon UV irradiation. Visible light could be used to regain the original state by inducing the isomerization back from cis‐azobenzene to trans‐azobenzene. Interestingly, this system enabled very fast (seconds) actuation responses to light in the dry state (xerogel), while the response time to both UV and visible light in the swollen state (hydrogel) was approximately 3 h. Recently, a poly(NIPAAm‐co‐SP‐co‐AA) hydrogel walker was synthesized based on the photoisomerization of spiropyranes.130 White light–induced isomerization from the zwitter‐ionic open‐ring form to the spiro form and thermal reisomerization enabled the hydrogel walker to perform a directed movement in one specific direction (Figure 5C). It must be noted, that the anisotropic movement is derived from the ratchet shape surface and not from the photochemistry itself. A similar principle was used earlier by ter Schiphorst and co‐workers to create a hydrogel‐based valve for application in microfluidic systems.131 Photothermal actuators are particularly interesting, where actuation with harmless irradiation (e.g., NIR) is required. Wang et al. reported the synthesis of composite hydrogels from thermoresponsive elastin‐like polypeptides with reduced‐graphene oxide sheets as photothermal agents. These materials exhibited rapid and tunable motions such as crawling with NIR light.132 Poly(NIPAAm) hydrogels with spatially defined doping with Nd2O3 and Yb2O3 particles were shown to be useful for wavelength sensitive actuation in the NIR region, a unique option arising from the sharp absorption bands of rare‐earth photothermal agents.81
Figure 5.
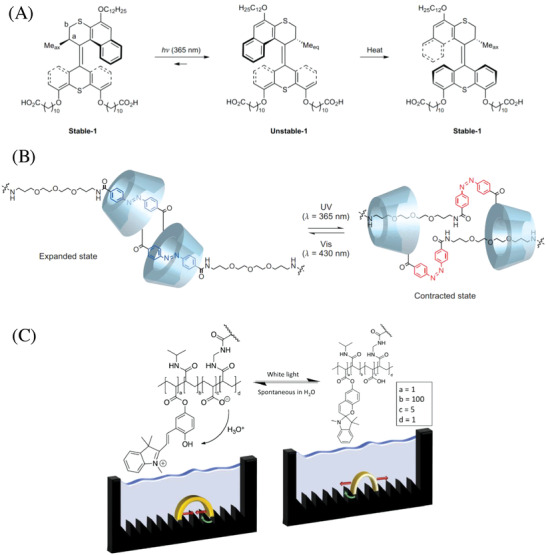
Photoresponsive hydrogel actuators. A) Reaction equation for the photoinduced isomerization of Stable 1 into Unstable 2 which thermally (50 °C) inverts back to Stable 1, thus completing a full molecular rotary cycle. Stable 1 could be shaped self‐assembled uniaxially oriented nanofibers on the cm scale, thus transferring molecular movement to the macroscopic domain (i.e., bending upon UV irradiation). Reproduced with permission.129 Copyright 2019, Springer Nature. B) Chemical structure and photoisomerization of an α‐CD‐based [c2]daisy chain with an azobenzene derivative as the axis. Reproduced with permission.21 Copyright 2016, Springer Nature. C) Anisotropic movement of a poly(NIPAAm‐co‐SP‐co‐AA) hydrogel walker on a ratchet shaped surface, based on the photoisomerization of a spiropyrane moiety. Reproduced with permission.130 Copyright 2019, Elsevier.
3.5. Healable Soft Materials
Material fatigue, environmental conditions, mechanical stress, or scratches lower the lifetime of materials, which then need to be replaced or worse, can lead to fatal accidents. Healing materials are promising candidates to overcome this issue and to prolong materials shelf life due to their inherent ability to repair themselves. For living beings, it is even more crucial to possess repair mechanisms, as even small damages can result in drastically reduced chances of reproduction or survival. Hydrogels as biomimetic materials are predestined to attempt getting closer toward the astonishing self‐repairing properties of living tissue, e.g., in flax133 or Delosperma cooperi succulent leaves.134 For a recent review with a broad scope of healing polymeric materials (gels, elastomers, and membranes) the reader is recommended an overview given by Pasparakis and Amaral.135 The focus in this chapter is on light‐induced self‐healing properties of hydrogels. Utilizing light has the advantage of being cheap and contact free, therefore reducing the risk of causing side effects to the repaired material. Another benefit of light‐driven repair mechanism is the precise spatial and temporal control, enabling repair on demand. It must be distinguished between light‐responsive self‐healing hydrogels and hydrogels in which the self‐healing mechanism is caused or controlled by light.136 The focus here is on the latter group. The chemical basis for self‐healing properties in hydrogels are generally dynamic interactions, like host–guest interaction or dynamic/reversible covalent bonds.
3.5.1. Photocontrolled Host–Guest Interaction
Condensated β‐cyclodextrin (β‐CD) oligomers hosting azobenzene acrylamide guest molecules were used as macro‐crosslinkers for copolymerization with acrylamide to form a hydrogel network.137 When the network structure is torn apart by physical damage, e.g., a knife cut, the hydrogel fragments could bind back together due to the dynamic nature of host–guest interactions between the azobenzene acrylamide units in the network backbone and the β‐CD crosslinker. By shining UV (365 nm) or visible (450 nm) light, the healing process could be selectively stopped or turned back on, respectively. The well‐known UV light–induced change from trans‐ to cis‐azobenzene rendered the host–guest interaction less favorable and therefore inhibited the healing process. Visible light induced the change back to the trans‐isomer, which readily formed host–guest complexes with β‐CD. The competition between free guest molecules, azobenzene and PEG chains (10 kDa) about a cyclodextrin host could be manipulated with UV (365 nm) to favor either PEG or trans‐azobenzene as guest molecule, resulting in a hydrogel or solution, respectively. Therefore, this hydrogel could be reshaped with the aid of UV light.138 Based on the same chemistry, Harada and co‐workers prepared a host‐polymer–guest‐polymer complex hydrogel. Again, it was possible to de‐crosslink the hydrogel with UV light (365 nm) resulting in liquification, which could be reversed with 430 nm light.25 Latter approach was further exploited to preformed CD and Azo gels with commonly used crosslinker N,N′‐methylenebis(acrylamide) (MBA) to allow light‐controlled attaching and detaching of hydrogel pieces (Figure 6 A).139
Figure 6.
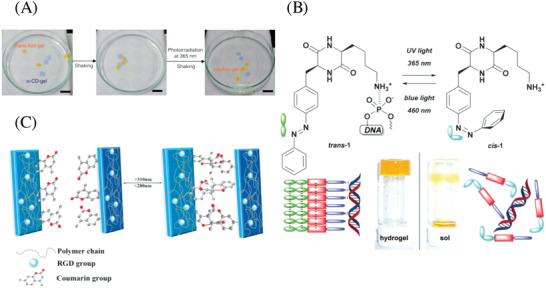
Photoinduced healing of hydrogels. A) Mixtures of azo‐functionalized hydrogels with α‐CD functionalized hydrogels attach or detach to each other depending on the light controlled cis or trans state of the azobenzene moiety. Reproduced with permission.139 Copyright 2013, Springer Nature. B) Reversible light‐induced gel–sol conversion based on the cis or trans state of an azobenzene unit in a low molecular weight gelator. The cis state does not allow supramolecular assembly, leading to liquification. Reproduced with permission.26 Copyright 2019, Royal Society of Chemistry. C) Photo‐crosslinking of damaged polyacrylamide hydrogels via [2+2]‐cycloaddition of coumarine functional groups in the polymers side chain. Reproduced with permission.144 Copyright 2019, Wiley‐VCH.
3.5.2. Phototriggered Exchange of Covalent Bonds
Another way of introducing self‐healing properties into molecules is based on dynamic covalent bonds, as present in disulfide bridges or trithiocarbonates. Anseth and co‐workers used a photoinitiator (lithium acylphosphinate, LAP) to cleave disulfide bridged hydrogels formed via oxidation of thiol‐capped four‐arm poly(ethylene glycol) macro‐crosslinkers.63 It was found that for photoinitiator concentrations below the concentration of crosslinks, the gel could be forced into different shapes following UV light (365 nm) irradiation. This is explained by the formation of C–S or P–S moieties and a free thiyl radical from the reaction of a disulfide with radicals derived from the photoinduced cleavage of LAP. The thiyl radicals rebuild disulfide bridges and therefore partly repair the network. As for two cleaved disulfide bridges only one new disulfide bridge can be formed, this approach is not a solution for long term stability. Nevertheless, it still offers interesting possibilities such as light‐induced covalent annealing of two hydrogel fragments. A very recent example making use of the dynamic nature of disulfides was given by Singha and co‐workers.29 The zwitter‐ion block‐copolymer PFMA‐b‐PDMAPMZ, consisting of poly(furfuryl methacrylate) (PFMA) and poly(N‐[3‐(dimethylamino)propyl]methacrylamide) (PDMAPMZ), was synthesized via chain‐extension RAFT (reversible addition‐fragmentation chain transfer) polymerization followed up by post functionalization with 1,3‐propane sultone. Dithiobismaleimidoethane (DTME) was then used to crosslink the polymer chains via Diels–Alder reaction of the maleimide moieties in the crosslinker (DTME) and the furfuryl moieties from PFMA in the polymer backbone. The resulting PFMA‐core zwitter ion‐shell micelles formed a physical hydrogel due to the ionic interaction of the shells. Notches made in vacuum dried hydrogels were healing when exposed to UV light. This was attributed to a synergistic combination of ionic interaction between polymer fragments and a UV‐triggered disulfide metathesis reaction (DSMR), i.e., disulfide bonds broken because of the irradiation and recombination to heal a notch in the surface.
Trithiocarbonates can be used as functional a group to enable the exchange of covalent bonds, as apparent by their use as RAFT agents.140 In 2011, the Matyjaszewski group first reported trithiocarbonates for photoinduced hydrogel healing.30 In a more recent example, Dong et al. demonstrated the use of a trithiocarbonate RAFT agent to form a hydrogel capable to heal macroscopic cuts under UV irradiation (365 nm).141 Polystyrene was synthesized using a symmetrical carbon acid‐capped trithiocarbonate RAFT agent, thereby introducing the RAFT group is in the middle of each polymer chain. The carboxylic acid chain ends were treated with 2‐bromoethanol to yield the respective ester and thus polystyrene with bromine end groups (Br–PS–Br). Poly(4‐vinylpyridine) was polymerized in a similar fashion. Crosslinking was achieved by quaternization of the pyridine moieties with the bromine end groups (NR3 +Br−). To study their self‐healing behavior, hydrogels were cut into pieces and irradiated for 60 min at 365 nm. Healing of the polymer pieces, observed with the naked eye, was attributed to the reshuffling reaction between the trithiocarbonate moieties present in the network. No assessment of the mechanical properties before and after healing was undertaken. Related functional groups, e.g., dithiocarbamates or dithiourethanes have already been exploited for light activated healing of polymer networks but not for hydrogels yet.142, 143
3.5.3. Photoinduced Supramolecular Disassembly and Reassembly
Pianowski et al. used an azobenzene‐containing cyclic dipeptide (PAP‐DKP‐Lys) as a low molecular weight gelator (LMWGs) to form light‐responsive supramolecular hydrogels (Figure 6B).26 Upon UV irradiation (365 nm) the supramolecular hydrogel turned into a solution as the azobenzene unit in PAP‐DKP‐Lys was forced into the cis‐isomer state, thereby distorting the π‐stacking that initially lead to self‐assembly and gelation. Blue light irradiation (460 nm, 10 W cm−2) could be used to “repair” the hydrogel by inducing a change back to the trans‐azobenzene unit in PAP‐DKP‐Lys, enabling efficient π‐stacking and therefore supramolecular self‐assembly. The mechanical properties of the hydrogel could be restored for more than 100 cycles as confirmed by rheological strain and frequency sweep measurements. The focus of this work was the light‐triggered release of dsDNA or doxorubicin, nonetheless it is herein suspected that the light‐induced liquefaction and regelation can be exploited to heal external damages.
3.5.4. Photo‐Crosslinking
Yu et al. presented a photohealing hydrogel based on the UV‐triggered [2+2]‐cycloaddition reaction between coumarines (Figure 6C).144 The hydrogel network was synthesized from acrylamide, 7‐(2‐methacryloyloxy‐ethoxy)‐4‐methylcoumarin and a poly(amidoamine) crosslinker. Damaged hydrogels could be repaired by shining UV light (365 nm) for up to 60 min due to photodimerization of coumarin moieties to a cyclobutane derivative along the main chains. Bisected hydrogels were shown to restore ≈89% of the original tensile strength upon 60 min irradiation (365 nm). The healing process could be partly reverted by irradiation of healed hydrogels with more energetic 254 nm UV light, which was attributed to the photocleavage of the cyclobutane moieties back to the initial coumarin moieties.
4. Conclusion and Outlook
During the last few years there has been growing interest in the development of photoresponsive hydrogels due to their unique dynamic physical and chemical properties, resembling those of soft matter found in living systems. Their diversity in terms of chemical composition and mechanical behavior combined with a responsiveness to light, a stimulus with various advantages such as being cheap, contact free, and spatiotemporally controllable, renders photoresponsive hydrogels ideal candidates for applications in a wide range of fields, ranging from chemistry and bioscience to materials and nanotechnologies. Photoresponsive hydrogels have been demonstrated as excellent scaffolds for soft actuators, healing materials, adaptive surfaces, dynamic cell microenvironments, and drug release. Although substantial effort has been dedicated to the design and investigation of light‐responsive hydrogels in different fields, most of the present systems are restricted to proof‐of‐concept studies. To expand their practical applications, there are several key challenges that need to be resolved. One of the major challenges is to develop photochemical reactions that are activated by low‐energy light (long wavelength) and trigger reactions with fast kinetics and high quantum efficiency. This also applies to photothermal processes. NIR‐responsive hydrogels with high energy efficiency are yet to develop even though these systems allow deep tissue penetration and precise control in 3D. On the way toward real applications, it is important to design hydrogel properties, including chemical compositions, physical properties, 3D structures, and porosity, which greatly affect the photoresponsive abilities. In addition, more efforts need to be paid to the study of the response kinetics to achieve fast, reliable, and reversible responses upon light stimulation. For most applications discussed, the use of hydrogels with reversible photoresponses is crucial or at least beneficial, a requirement which cannot be fulfilled by many photocleavage reactions. Photoisomerization reactions, photoinduced dynamic covalent chemistry, and electrocyclic reactions as well as wavelength orthogonal chemistries can contribute to overcome irreversibility. With suitable reversible systems at hand, even nonequilibrium systems can be realized, allowing additional intermediate states instead of just a phototriggered “on” or “off” state.145 The majority of photoresponsive hydrogels engineered to date connects a single response to a specific wavelength area. The development of hydrogels that can respond to several wavelengths in an intelligent way remains of prime importance to achieve selective control of multifunctional responsiveness or orthogonal photomodulation of two or more properties in the same system. Orthogonal photochemistry (“λ‐orthogonality”) has been exploited for hydrogel structuration already and seems promising for advancing wavelength‐selective light‐responsive hydrogels.146 Another challenge is the biocompatibility of light and the light‐responsive hydrogels when used as biomaterials. It is important to systematically evaluate the effects of light exposure, reactive intermediates, and photoproducts not only on cell or bacteria viability but also proteins, gene delivery, or metabolic activity both in vitro and in vivo. For photothermally responsive hydrogels, the toxicity to surrounding cells due to overheating also needs to be addressed. From a chemist's perspective, there are several interesting photoresponsive mechanisms and possibilities that have not yet found their way into photoresponsive hydrogel chemistry, such as photoswitchable ligands for metal coordination or more exotic functional groups such as diselenides or dithiourethanes. The field of dynamic covalent polymer chemistry can further influence and contribute to the design of light‐responsive hydrogels.147 Sophisticated illumination techniques such as 3D laser lithography demonstrate how to exploit the full potential of established responsive hydrogels by combining extreme spatial control with fine control over the light dosage.148 Photoresponsive hydrogels will continue to advance applications across different fields and further broaden our understanding of dynamic systems. Although many challenges need to be addresses, the potential for photoresponsive hydrogels is bright with vast developments and implementations awaiting.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
L.L. and J.M.S. contributed equally to this work. The research was supported by the ERC Starting Grant (ID: 337077‐DropCellArray).
Biographies
Lei Li received his B.S. degree in chemistry from Shandong University, China, in 2010, and then joined Peking University, China, as a Ph.D. student supervised by Prof. Zi‐chen Li. He worked on multicomponent polymerization and photoresponsive polymers, graduating in 2015. He then moved to Karlsruhe Institute of Technology, Germany, as postdoc to study biofunctional polymeric surfaces in the group of Pavel Levkin. In 2018, he became an associate researcher at Shandong University. His research focuses on the development of novel polymerization methods and the design of organosilicon materials.

Johannes M. Scheiger is a Ph.D. student in a joint project of Prof. Patrick Théato and Pavel Levkin. He received both his B.Sc. and M.Sc. degrees in chemistry from the Karlsruhe Institute of Technology (KIT), Germany, and completed his master thesis working on hydrogels with predefined lifetime in the group of Pavel Levkin. His research interests are focused on inorganic and polymer chemistry. He is currently working on the surface chemistry of polymers with high sulfur content.

Pavel A. Levkin is head of the Biofunctional Materials Systems research group at Karlsruhe Institute of Technology (KIT), Germany. He graduated from the Institute of Fine Chemical Technology, Moscow, and obtained his Ph.D. degree in organic chemistry from the University of Tübingen in Germany, which he followed with postdoctoral work at the University of California, Berkeley. He is the cofounder of ScreenFect GmbH and Aquarray GmbH. His research focuses on the development of functional and responsive materials, and surfaces for biomedical and biotechnological applications.

Li L., Scheiger J. M., Levkin P. A., Adv. Mater. 2019, 31, 1807333. 10.1002/adma.201807333
References
- 1. Varaprasad K., Raghavendra G. M., Jayaramudu T., Yallapu M. M., Sadiku R., Mater. Sci. Eng., C 2017, 79, 958. [DOI] [PubMed] [Google Scholar]
- 2. Drury J. L., Mooney D. J., Biomaterials 2003, 24, 4337. [DOI] [PubMed] [Google Scholar]
- 3. Hoffman A. S., Adv. Drug Delivery Rev. 2002, 54, 3. [Google Scholar]
- 4. Ullah F., Othman M. B. H., Javed F., Ahmad Z., Md Akil H., Mater. Sci. Eng., C 2015, 57, 414. [DOI] [PubMed] [Google Scholar]
- 5. Ruskowitz E. R., DeForest C. A., Nat. Rev. Mater. 2018, 3, 17087. [Google Scholar]
- 6. Ferreira N. N., Ferreira L. M. B., Cardoso V. M. O., Boni F. I., Souza A. L. R., Gremião M. P. D., Eur. Polym. J. 2018, 99, 117. [Google Scholar]
- 7. Liu Z., Faraj Y., Ju X.‐J., Wang W., Xie R., Chu L.‐Y., J. Polym. Sci., Part B: Polym. Phys. 2018, 56, 1306. [Google Scholar]
- 8. Duan J.‐j., Zhang L.‐n., Chin. J. Polym. Sci. 2017, 35, 1165. [Google Scholar]
- 9. Samal S. K., Dash M., Dubruel P., van Vlierberghe S., in Smart Polymers and Their Applications (Eds: Aguilar M. R., Román J. S.), Elsevier, Cambridge, UK: 2014, pp. 237–270. [Google Scholar]
- 10. Liu Z., Wei J., Faraj Y., Ju X.‐J., Xie R., Wang W., Chu L.‐Y., Can. J. Chem. Eng. 2018, 96, 2100. [Google Scholar]
- 11. Lim H. L., Hwang Y., Kar M., Varghese S., Biomater. Sci. 2014, 2, 603. [DOI] [PubMed] [Google Scholar]
- 12. Hoare T. R., Kohane D. S., Polymer 2008, 49, 1993. [Google Scholar]
- 13. Whitaker C. M., Derouin E. E., O'Connor M. B., Whitaker C. K., Whitaker J. A., Snyder J. J., Kaufmann N. R., Gilliard A. N., Reitmayer A. K., Macromol J.. Sci., Part A: Pure Appl. Chem. 2016, 54, 40. [Google Scholar]
- 14. Yang C., Liu Z., Chen C., Shi K., Zhang L., Ju X.‐J., Wang W., Xie R., Chu L.‐Y., ACS Appl. Mater. Interfaces 2017, 9, 15758. [DOI] [PubMed] [Google Scholar]
- 15. Ma Y., Ma S., Yang W., Yu B., Pei X., Zhou F., Liu W., Adv. Mater. Technol. 2018, 462, 1800467. [Google Scholar]
- 16. Han D., Farino C., Yang C., Scott T., Browe D., Choi W., Freeman J. W., Lee H., ACS Appl. Mater. Interfaces 2018, 10, 17512. [DOI] [PubMed] [Google Scholar]
- 17. Ma D., Zhou N., Zhang T., Hu K., Ma X.'e., Gu N., Colloids Surf. A 2017, 522, 97. [Google Scholar]
- 18. Truong V. X., Li F., Ercole F., Forsythe J. S., ACS Macro Lett. 2018, 7, 464. [DOI] [PubMed] [Google Scholar]
- 19. Tomatsu I., Peng K., Kros A., Adv. Drug Delivery Rev. 2011, 63, 1257. [DOI] [PubMed] [Google Scholar]
- 20. Yao H., Wang J., Mi S., Polymers 2018, 10, 11. [Google Scholar]
- 21. Iwaso K., Takashima Y., Harada A., Nat. Chem. 2016, 8, 625. [DOI] [PubMed] [Google Scholar]
- 22. Takashima Y., Hatanaka S., Otsubo M., Nakahata M., Kakuta T., Hashidzume A., Yamaguchi H., Harada A., Nat. Commun. 2012, 3, 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosales A. M., Vega S. L., DelRio F. W., Burdick J. A., Anseth K. S., Angew. Chem., Int. Ed. 2017, 56, 12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kloxin A. M., Kasko A. M., Salinas C. N., Anseth K. S., Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tamesue S., Takashima Y., Yamaguchi H., Shinkai S., Harada A., Angew. Chem., Int. Ed. 2010, 49, 7461. [DOI] [PubMed] [Google Scholar]
- 26. Pianowski Z. L., Karcher J., Schneider K., Chem. Commun. 2016, 52, 3143. [DOI] [PubMed] [Google Scholar]
- 27. Zhao Y.‐L., Stoddart J. F., Langmuir 2009, 25, 8442. [DOI] [PubMed] [Google Scholar]
- 28. Wu Y., Wu S., Tian X., Wang X., Wu W., Zou G., Zhang Q., Soft Matter 2011, 7, 716. [Google Scholar]
- 29. Banerjee S. L., Bhattacharya K., Samanta S., Singha N. K., ACS Appl. Mater. Interfaces 2018, 10, 27391. [DOI] [PubMed] [Google Scholar]
- 30. Amamoto Y., Kamada J., Otsuka H., Takahara A., Matyjaszewski K., Angew. Chem., Int. Ed. 2011, 50, 1660. [DOI] [PubMed] [Google Scholar]
- 31. Thomas S. W., Macromol. Chem. Phys. 2012, 213, 2443. [Google Scholar]
- 32. Zhao H., Sterner E. S., Coughlin E. B., Theato P., Macromolecules 2012, 45, 1723. [Google Scholar]
- 33. Claaßen C., Claaßen M. H., Gohl F., Tovar G. E. M., Borchers K., Southan A., Macromol. Biosci. 2018, 18, 1800104. [DOI] [PubMed] [Google Scholar]
- 34. Klán P., Šolomek T., Bochet C. G., Blanc A., Givens R., Rubina M., Popik V., Kostikov A., Wirz J., Chem. Rev. 2013, 113, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mikkelsen R. J. T., Grier K. E., Mortensen K. T., Nielsen T. E., Qvortrup K., ACS Comb. Sci. 2018, 20, 377. [DOI] [PubMed] [Google Scholar]
- 36. Hansen M. J., Velema W. A., Lerch M. M., Szymanski W., Feringa B. L., Chem. Soc. Rev. 2015, 44, 3358. [DOI] [PubMed] [Google Scholar]
- 37. Aujard I., Benbrahim C., Gouget M., Ruel O., Baudin J.‐B., Neveu P., Jullien L., Chem. – Eur. J. 2006, 12, 6865. [DOI] [PubMed] [Google Scholar]
- 38. Kim M. S., Diamond S. L., Bioorg. Med. Chem. Lett. 2006, 16, 4007. [DOI] [PubMed] [Google Scholar]
- 39. Trenor S. R., Shultz A. R., Love B. J., Long T. E., Chem. Rev. 2004, 104, 3059. [DOI] [PubMed] [Google Scholar]
- 40. Fournier L., Gauron C., Xu L., Aujard I., Le Saux T., Gagey‐Eilstein N., Maurin S., Dubruille S., Baudin J.‐B., Bensimon D., Volovitch M., Vriz S., Jullien L., ACS Chem. Biol. 2013, 8, 1528. [DOI] [PubMed] [Google Scholar]
- 41. Bao C., Fan G., Lin Q., Li B., Cheng S., Huang Q., Zhu L., Org. Lett. 2012, 14, 572. [DOI] [PubMed] [Google Scholar]
- 42. Goswami P. P., Syed A., Beck C. L., Albright T. R., Mahoney K. M., Unash R., Smith E. A., Winter A. H., J. Am. Chem. Soc. 2015, 137, 3783. [DOI] [PubMed] [Google Scholar]
- 43. Kurihara S., Ueno Y., Nonaka T., J. Appl. Polym. Sci. 1998, 67, 1931. [Google Scholar]
- 44. Conrad P. G., Givens R. S., Hellrung B., Rajesh C. S., Ramseier M., Wirz J., J. Am. Chem. Soc. 2000, 122, 9346. [Google Scholar]
- 45. Givens R. S., Rubina M., Wirz J., Photochem. Photobiol. Sci. 2012, 11, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhu Y., Pavlos C. M., Toscano J. P., Dore T. M., J. Am. Chem. Soc. 2006, 128, 4267. [DOI] [PubMed] [Google Scholar]
- 47. Zhang H., Li X., Lin Y., Gao F., Tang Z., Su P., Zhang W., Xu Y., Weng W., Boulatov R., Nat. Commun. 2017, 8, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Azagarsamy M. A., McKinnon D. D., Alge D. L., Anseth K. S., ACS Macro Lett. 2014, 3, 515. [DOI] [PubMed] [Google Scholar]
- 49. Ji W., Qin M., Feng C., Macromol. Chem. Phys. 2018, 219, 1700398. [Google Scholar]
- 50. Kang H., Liu H., Zhang X., Yan J., Zhu Z., Peng L., Yang H., Kim Y., Tan W., Langmuir 2011, 27, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shih H., Lin C.‐C., Biomacromolecules 2015, 16, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Castilla A. M., Draper E. R., Nolan M. C., Brasnett C., Seddon A., Mears L. L. E., Cowieson N., Adams D. J., Sci. Rep. 2017, 7, 8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Min K., Kim S., Kim S., Proc. Natl. Acad. Sci. USA 2017, 114, 6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wells L. A., Brook M. A., Sheardown H., Macromol. Biosci. 2011, 11, 988. [DOI] [PubMed] [Google Scholar]
- 55. Zheng Y., Micic M., Mello S. V., Mabrouki M., Andreopoulos F. M., Konka V., Pham S. M., Leblanc R. M., Macromolecules 2002, 35, 5228. [Google Scholar]
- 56. Truong V. X., Li F., Forsythe J. S., ACS Macro Lett. 2017, 6, 657. [DOI] [PubMed] [Google Scholar]
- 57. Doi T., Kashida H., Asanuma H., Org. Biomol. Chem. 2015, 13, 4430. [DOI] [PubMed] [Google Scholar]
- 58. Doi T., Kawai H., Murayama K., Kashida H., Asanuma H., Chem. – Eur. J. 2016, 22, 10533. [DOI] [PubMed] [Google Scholar]
- 59. Lowe A. B., Polym. Chem. 2014, 5, 4820. [Google Scholar]
- 60. Lowe A. B., Polym. Chem. 2010, 1, 17. [Google Scholar]
- 61. Kharkar P. M., Rehmann M. S., Skeens K. M., Maverakis E., Kloxin A. M., ACS Biomater. Sci. Eng. 2016, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gandavarapu N. R., Azagarsamy M. A., Anseth K. S., Adv. Mater. 2014, 26, 2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fairbanks B. D., Singh S. P., Bowman C. N., Anseth K. S., Macromolecules 2011, 44, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cembran A., Bernardi F., Garavelli M., Gagliardi L., Orlandi G., J. Am. Chem. Soc. 2004, 126, 3234. [DOI] [PubMed] [Google Scholar]
- 65. Pouliquen G., Amiel C., Tribet C., J. Phys. Chem. B 2007, 111, 5587. [DOI] [PubMed] [Google Scholar]
- 66. Zhang X., Ma X., Wang K., Lin S., Zhu S., Dai Y., Xia F., Macromol. Rapid Commun. 2018, 39, 1800142. [DOI] [PubMed] [Google Scholar]
- 67. Klajn R., Chem. Soc. Rev. 2014, 43, 148. [DOI] [PubMed] [Google Scholar]
- 68. Stumpel J. E., Ziółkowski B., Florea L., Diamond D., Broer D. J., Schenning A. P. H. J., ACS Appl. Mater. Interfaces 2014, 6, 7268. [DOI] [PubMed] [Google Scholar]
- 69. Ziółkowski B., Florea L., Theobald J., Benito‐Lopez F., Diamond D., Soft Matter 2013, 9, 8754. [Google Scholar]
- 70. Balmond E. I., Tautges B. K., Faulkner A. L., Or V. W., Hodur B. M., Shaw J. T., Louie A. Y., J. Org. Chem. 2016, 81, 8744. [DOI] [PubMed] [Google Scholar]
- 71. Feeney M. J., Thomas S. W., Macromolecules 2018, 51, 8027. [Google Scholar]
- 72. Vales T. P., Badon I. W. T., Kim H.‐J., Macromol. Res. 2018, 26, 950. [Google Scholar]
- 73. Yang X., Huang L., Zhou L., Xu H., Yi Z., Int. J. Polym. Sci. 2016, 2016, 1. [Google Scholar]
- 74. ter Schiphorst J., Melpignano G. G., Amirabadi H. E., Houben M. H. J. M., Bakker S., den Toonder J. M. J., Schenning A. P. H. J., Macromol. Rapid Commun. 2018, 39, 1700086. [DOI] [PubMed] [Google Scholar]
- 75. Sun Z., Liu S., Li K., Tan L., Cen L., Fu G., Soft Matter 2016, 12, 2192. [DOI] [PubMed] [Google Scholar]
- 76. Li L., Wang C., Huang Q., Xiao J., Zhang Q., Cheng Y., J. Mater. Chem. B 2018, 6, 2474. [DOI] [PubMed] [Google Scholar]
- 77. Zhang H., Guo S., Fu S., Zhao Y., Polymers 2017, 9, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reithofer M. R., Lakshmanan A., Ping A. T. K., Chin J. M., Hauser C. A. E., Biomaterials 2014, 35, 7535. [DOI] [PubMed] [Google Scholar]
- 79. Shi Q., Xia H., Li P., Wang Y.‐S., Wang L., Li S.‐X., Wang G., Lv C., Niu L.‐G., Sun H.‐B., Adv. Opt. Mater. 2017, 5, 1700442. [Google Scholar]
- 80. Yang Y., Tan Y., Wang X., An W., Xu S., Liao W., Wang Y., ACS Appl. Mater. Interfaces 2018, 10, 7688. [DOI] [PubMed] [Google Scholar]
- 81. Watanabe S., Era H., Kunitake M., Sci. Rep. 2018, 8, 13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dong X., Wei C., Liang J., Liu T., Kong D., Lv F., Colloids Surf., B 2017, 154, 253. [DOI] [PubMed] [Google Scholar]
- 83. Deng L., Xu Y., Sun C., Yun B., Sun Q., Zhao C., Li Z., Sci. Bull. 2018, 63, 917. [DOI] [PubMed] [Google Scholar]
- 84. Zhang J., Liu S., Hu X., Xie Z., Jing X., ACS Biomater. Sci. Eng. 2016, 2, 1942. [DOI] [PubMed] [Google Scholar]
- 85. Harmatys K. M., Battles P. M., Peck E. M., Spence G. T., Roland F. M., Smith B. D., Chem. Commun. 2017, 53, 9906. [DOI] [PubMed] [Google Scholar]
- 86. Grzybowski M., Gryko D. T., Adv. Opt. Mater. 2015, 3, 280. [Google Scholar]
- 87. Liang J., Dong X., Wei C., Ma G., Liu T., Kong D., Lv F., Carbohydr. Polym. 2017, 175, 440. [DOI] [PubMed] [Google Scholar]
- 88. Liu B. L., Conjugated Polymers for Biological and Biomedical Applications, Wiley‐VCH, Weinheim, Germany: 2018. [Google Scholar]
- 89. Sun T., Dou J.‐H., Liu S., Wang X., Zheng X., Wang Y., Pei J., Xie Z., ACS Appl. Mater. Interfaces 2018, 10, 7919. [DOI] [PubMed] [Google Scholar]
- 90. Xu L., Cheng L., Wang C., Peng R., Liu Z., Polym. Chem. 2014, 5, 1573. [Google Scholar]
- 91. Ward M. A., Georgiou T. K., Polymers 2011, 3, 1215. [Google Scholar]
- 92. Lutz J.‐F., Akdemir O., Hoth A., J. Am. Chem. Soc. 2006, 128, 13046. [DOI] [PubMed] [Google Scholar]
- 93. Shi K., Liu Z., Wei Y.‐Y., Wang W., Ju X.‐J., Xie R., Chu L.‐Y., ACS Appl. Mater. Interfaces 2015, 7, 27289. [DOI] [PubMed] [Google Scholar]
- 94. Li Z., Qian L., Li L., Bernhammer J. C., Huynh H. V., Lee J.‐S., Yao S. Q., Angew. Chem., Int. Ed. 2016, 55, 2002. [DOI] [PubMed] [Google Scholar]
- 95. Dong Y., Jin G., Hong Y., Zhu H., Lu T. J., Xu F., Bai D., Lin M., ACS Appl. Mater. Interfaces 2018, 10, 12374. [DOI] [PubMed] [Google Scholar]
- 96. Luo Y., Shoichet M. S., Nat. Mater. 2004, 3, 249. [DOI] [PubMed] [Google Scholar]
- 97. Wylie R. G., Ahsan S., Aizawa Y., Maxwell K. L., Morshead C. M., Shoichet M. S., Nat. Mater. 2011, 10, 799. [DOI] [PubMed] [Google Scholar]
- 98. Mosiewicz K. A., Kolb L., van der Vlies A. J., Martino M. M., Lienemann P. S., Hubbell J. A., Ehrbar M., Lutolf M. P., Nat. Mater. 2013, 12, 1072. [DOI] [PubMed] [Google Scholar]
- 99. Lee T. T., García J. R., Paez J. I., Singh A., Phelps E. A., Weis S., Shafiq Z., Shekaran A., Del Campo A., García A. J., Nat. Mater. 2015, 14, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Caliari S. R., Burdick J. A., Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hahn M. S., Miller J. S., West J. L., Adv. Mater. 2006, 18, 2679. [Google Scholar]
- 102. DeForest C. A., Polizzotti B. D., Anseth K. S., Nat. Mater. 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. DeForest C. A., Anseth K. S., Nat. Chem. 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. DeForest C. A., Tirrell D. A., Nat. Mater. 2015, 14, 523. [DOI] [PubMed] [Google Scholar]
- 105. Bai T., Sinclair A., Sun F., Jain P., Hung H.‐C., Zhang P., Ella‐Menye J.‐R., Liu W., Jiang S., Chem. Sci. 2016, 7, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lee I.‐N., Dobre O., Richards D., Ballestrem C., Curran J. M., Hunt J. A., Richardson S. M., Swift J., Wong L. S., ACS Appl. Mater. Interfaces 2018, 10, 7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stowers R. S., Allen S. C., Suggs L. J., Proc. Natl. Acad. Sci. USA 2015, 112, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Patnaik S., Sharma A. K., Garg B. S., Gandhi R. P., Gupta K. C., Int. J. Pharm. 2007, 342, 184. [DOI] [PubMed] [Google Scholar]
- 109. Nehls E. M., Rosales A. M., Anseth K. S., J. Mater. Chem. B 2016, 4, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Park C., Lee K., Kim C., Angew. Chem., Int. Ed. 2009, 48, 1275. [DOI] [PubMed] [Google Scholar]
- 111. Matsumoto S., Yamaguchi S., Ueno S., Komatsu H., Ikeda M., Ishizuka K., Iko Y., Tabata K. V., Aoki H., Ito S., Noji H., Hamachi I., Chem. – Eur. J. 2008, 14, 3977. [DOI] [PubMed] [Google Scholar]
- 112. Wang R., Yang Z., Luo J., Hsing I.‐M., Sun F., Proc. Natl. Acad. Sci. USA 2017, 114, 5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Qiu M., Wang D., Liang W., Liu L., Zhang Y., Chen X., Sang D. K., Xing C., Li Z., Dong B., Xing F., Fan D., Bao S., Zhang H., Cao Y., Proc. Natl. Acad. Sci. USA 2018, 115, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yan B., Boyer J.‐C., Habault D., Branda N. R., Zhao Y., J. Am. Chem. Soc. 2012, 134, 16558. [DOI] [PubMed] [Google Scholar]
- 115. Griffin D. R., Kasko A. M., J. Am. Chem. Soc. 2012, 134, 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Badeau B. A., Comerford M. P., Arakawa C. K., Shadish J. A., DeForest C. A., Nat. Chem. 2018, 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu D., Bastiaansen C. W. M., den Toonder J. M. J., Broer D. J., Langmuir 2013, 29, 5622. [DOI] [PubMed] [Google Scholar]
- 118. Yoon J., Bian P., Kim J., McCarthy T. J., Hayward R. C., Angew. Chem., Int. Ed. 2012, 51, 7146. [DOI] [PubMed] [Google Scholar]
- 119. ter Schiphorst J., van den Broek M., de Koning T., Murphy J. N., Schenning A. P. H. J., Esteves A. C. C., J. Mater. Chem. A 2016, 4, 8676. [Google Scholar]
- 120. Chen L., Yao X., Gu Z., Zheng K., Zhao C., Lei W., Rong Q., Lin L., Wang J., Jiang L., Liu M., Chem. Sci. 2017, 8, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang N., Li Y., Zhang Y., Liao Y., Liu W., Langmuir 2014, 30, 11823. [DOI] [PubMed] [Google Scholar]
- 122. Ming Z., Ruan X., Bao C., Lin Q., Yang Y., Zhu L., Adv. Funct. Mater. 2017, 27, 1606258. [Google Scholar]
- 123. Liu Q., Liu L., Langmuir 2019, 35, 1450. [DOI] [PubMed] [Google Scholar]
- 124. Yang C., DelRio F. W., Ma H., Killaars A. R., Basta L. P., Kyburz K. A., Anseth K. S., Proc. Natl. Acad. Sci. USA 2016, 113, E4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. ter Schiphorst J., Saez J., Diamond D., Benito‐Lopez F., Schenning A. P. H. J., Lab Chip 2018, 18, 699. [DOI] [PubMed] [Google Scholar]
- 126. Carrico J. D., Tyler T., Leang K. K., Int. J. Smart Nano Mater. 2017, 8, 144. [Google Scholar]
- 127. Banerjee H., Suhail M., Ren H., Biomimetics 2018, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ionov L., Mater. Today 2014, 17, 494. [Google Scholar]
- 129. Chen J., Leung F. K.‐C., Stuart M. C. A., Kajitani T., Fukushima T., van der Giessen E., Feringa B. L., Nat. Chem. 2018, 10, 132. [DOI] [PubMed] [Google Scholar]
- 130. Francis W., Dunne A., Delaney C., Florea L., Diamond D., Sens. Actuators, B 2017, 250, 608. [Google Scholar]
- 131. ter Schiphorst J., Coleman S., Stumpel J. E., Ben Azouz A., Diamond D., Schenning A. P. H. J., Chem. Mater. 2015, 27, 5925. [Google Scholar]
- 132. Wang E., Desai M. S., Lee S.‐W., Nano Lett. 2013, 13, 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Paul‐Victor C., Dalle Vacche S., Sordo F., Fink S., Speck T., Michaud V., Speck O., PLoS One 2017, 12, e0185958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Speck O., Schlechtendahl M., Borm F., Kampowski T., Speck T., Beilstein J. Nanotechnol. 2018, 9, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Amaral A. J. R., Pasparakis G., Polym. Chem. 2017, 8, 6464. [Google Scholar]
- 136. Chen H., Ma X., Wu S., Tian H., Angew. Chem., Int. Ed. 2014, 53, 14149. [DOI] [PubMed] [Google Scholar]
- 137. Yang Q., Wang P., Zhao C., Wang W., Yang J., Liu Q., Macromol. Rapid Commun. 2017, 38. [DOI] [PubMed] [Google Scholar]
- 138. Liao X., Chen G., Liu X., Chen W., Chen F., Jiang M., Angew. Chem., Int. Ed. 2010, 49, 4409. [DOI] [PubMed] [Google Scholar]
- 139. Yamaguchi H., Kobayashi Y., Kobayashi R., Takashima Y., Hashidzume A., Harada A., Nat. Commun. 2012, 3, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Nicolaÿ R., Kamada J., van Wassen A., Matyjaszewski K., Macromolecules 2010, 43, 4355. [Google Scholar]
- 141. Dong P., Cui K., Xu F., Jiang T., Ma Z., Polym. Int. 2018, 67, 868. [Google Scholar]
- 142. Gordon M. B., French J. M., Wagner N. J., Kloxin C. J., Adv. Mater. 2015, 27, 8007. [DOI] [PubMed] [Google Scholar]
- 143. Amamoto Y., Otsuka H., Takahara A., Matyjaszewski K., Adv. Mater. 2012, 24, 3975. [DOI] [PubMed] [Google Scholar]
- 144. Yu L., Xu K., Ge L., Wan W., Darabi A., Xing M., Zhong W., Macromol. Biosci. 2016, 16, 1381. [DOI] [PubMed] [Google Scholar]
- 145. Coleman S., ter Schiphorst J., Ben Azouz A., Bakker S., Schenning A. P. H. J., Diamond D., Sens. Actuators, B 2017, 245, 81. [Google Scholar]
- 146. Batchelor R. R., Blasco E., Wuest K. N. R., Lu H., Wegener M., Barner‐Kowollik C., Stenzel M. H., Chem. Commun. 2018, 54, 2436. [DOI] [PubMed] [Google Scholar]
- 147. Zou W., Dong J., Luo Y., Zhao Q., Xie T., Adv. Mater. 2017, 29, 1606100. [DOI] [PubMed] [Google Scholar]
- 148. Hippler M., Blasco E., Qu J., Tanaka M., Barner‐Kowollik C., Wegener M., Bastmeyer M., Nat. Commun. 2019, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]