

Abstract
Proximal tubular damage is an important prognostic determinant in various chronic kidney diseases (CKDs). Currently available diagnostic methods do not allow for early disease detection and are neither efficient. Indoxyl sulfate (IS) is an endogenous metabolite and protein‐bound uremic toxin that is eliminated via renal secretion, but accumulates in plasma during tubular dysfunction. Therefore, it may be suitable as a tubular function marker. To evaluate this, a fast bioanalytical method was developed and validated for IS in various species and a kidney cell line using LC–MS/MS. An isotope‐labeled IS potassium salt as an internal standard and acetonitrile (ACN) as a protein precipitant were used for sample pretreatment. The analyte was separated on a Polaris 3 C18‐A column by gradient elution using 0.1% formic acid in water and ACN, and detected by negative electrospray ionization in selected reaction monitoring mode. The within‐day (≤ 4.0%) and between‐day (≤ 4.3%) precisions and accuracies (97.7 to 107.3%) were within the acceptable range. The analyte showed sufficient stability at all conditions investigated. Finally, applying this assay, significantly higher plasma and lower urine concentrations of IS were observed in mice with diabetic nephropathy with tubular damage, which encourages validation toward its use as a biomarker.
Keywords: chronic kidney diseases, indoxyl sulfate, LC–MS/MS, renal tubular function, uremic toxins
1. INTRODUCTION
The kidney maintains body homeostasis by regulating fluid, electrolyte, and acid–base balances, and eliminates metabolic waste products through urine. Kidney diseases have become a global health issue, exceeding infectious diseases in causing hospital mortality. The prevalence of kidney dysfunction varies from 7 to 13%, depending on the country, indicating that more than 750 million people worldwide suffer from chronic kidney disease (CKD) (Delanaye et al., 2017). Every year, treating kidney diseases costs over $32 billion, posing a high financial burden on the public health budget (Manns et al., 2019). CKD has been divided into five stages based on (estimated) glomerular filtration rate [(e)GFR] along with albuminuria, which are used for disease diagnosis. However, CKD concerns more than only altered glomerular filtration. Tubular damage is an important component of many CKDs, such as diabetic nephropathy (DN) (Gilbert, 2017), with prognostic implications. Also in kidney transplant survival, tubular damage is an important prognostic determinant (Nankivell et al., 2018). Impaired tubular solute clearance may not decline in concordance with GFR. Tubular secretion is an active transporter‐mediated process, which can be influenced by various conditions. This includes the competition between retained solutes for urinary secretion, whereas glomerular filtration is a passive process and primarily depends on the size and charge selectivity of the basement membrane. Consequently, GFR offers a delayed reflection of pathological conditions in several diseases and drug toxicities. Increasing evidence suggests that proximal tubular injuries by many drugs, such as tenofovir and cisplatin, may be reflected by a change in tubular secretory clearance earlier than a decline in GFR (Townsend et al., 2003). Currently available diagnostic methods are not efficient in detecting tubular damage, which is more likely to occur at the early stage of CKD (Wang & Kestenbaum, 2018). Several tubular injury markers, such as kidney injury molecule‐1 (KIM‐1) and neutrophil gelatinase‐associated lipocalin (NGAL), are available for the diagnosis of acute injury. However, they may less likely be applicable for chronic tubular injury (Khawaja et al., 2019; Vaidya et al., 2010). Consequently, there is often a considerable delay in CKD diagnosis, indicating the need for reliable tubular function markers (Nanayakkara et al., 2012).
Indoxyl sulfate (IS, Figure 1a) is a protein‐bound small‐molecular weight organic anion solute, also known as 3‐indoxyl sulfate. It is derived from dietary l‐tryptophan, catalyzed into indole by the tryptophanase activity of gut microbiota and subsequently, after absorption and first‐pass metabolism in the liver, oxidized by cytochrome P450 (CYP)‐2E1 to form indoxyl and finally be converted to IS by sulfotransferase (SULT)‐1A1 (Ellis et al., 2016). In physiological conditions, more than 90% of IS binds reversibly to circulating albumin and, in the kidney, requires a carrier‐mediated secretory pathway for excretion. Renal tubular cells are equipped with organic anion transporter 1 (OAT1/SLC22A6) and OAT3 (SLC22A8), abundantly expressed at the basolateral membrane, that conduct intracellular uptake of IS (Enomoto et al., 2003). The metabolite is subsequently secreted to the tubular lumen by efflux transporters such as multidrug resistance proteins (MRP4) and breast cancer resistance protein (BCRP) at the apical membrane and, finally, excreted in the urine (Mutsaers et al., 2011). Once the tubular function is impaired, IS may not be secreted efficiently, leading to its reduced clearance and plasma accumulation. Evidence has shown that IS plasma concentration increases along with CKD progression, starting already at initial stages of kidney damage (Barreto et al., 2009; Lin et al., 2012). Lower IS clearance may predict CKD progression and all‐cause mortality (Wu et al., 2011), although contradictory findings have been reported as well (van den Brand et al., 2016).
FIGURE 1.
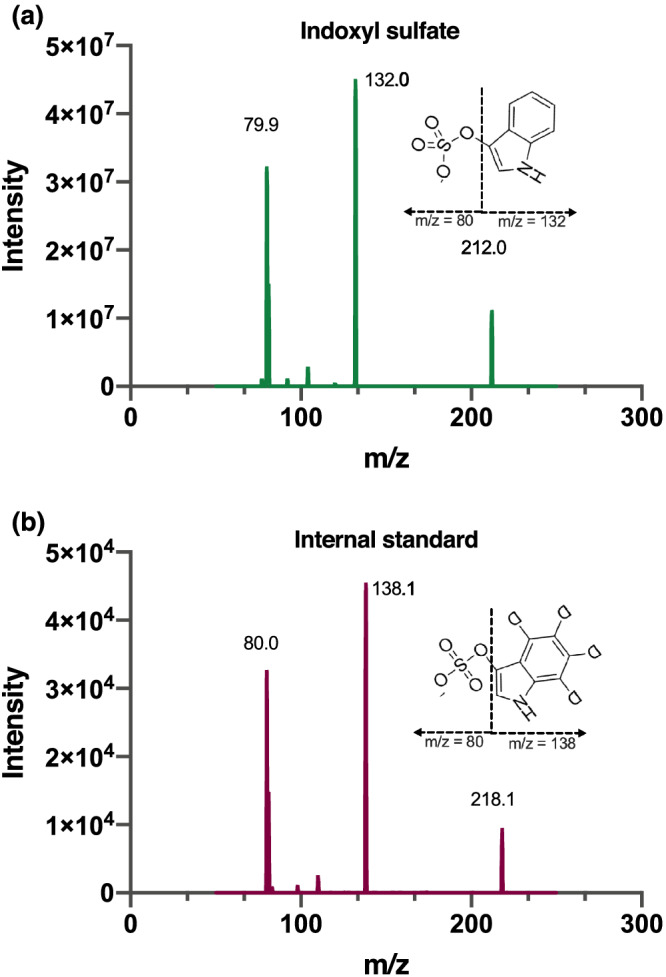
Product spectra of (a) indoxyl sulfate, m/z 212.0 @ –24 V and (b) internal standard (isotope labeled; 13C6), 218.0 @ –27 V
We hypothesize that IS may serve as a kidney tubular function marker. To test this, it is essential to have an efficient, fast, and reliable assay for IS. Various methods using HPLC or UPLC–MS/MS have been described (Lin et al., 2019; Poesen et al., 2015). However, the performance of a reliable biomarker should ideally be validated in multiple species before approaching clinical validation (Bonventre et al., 2010). Therefore, we developed a robust, sensitive, specific, accurate, and fast analytical method for IS measurement using LC–MS/MS and subsequently validated it in a variety of relevant matrices (plasma of human, goat, pig, rat, and mouse, and the lysate of human renal proximal tubular cells).
2. MATERIALS AND METHODS
2.1. Reagents
IS potassium salt and isotope‐labeled IS potassium salt (13C6, 99%) as internal standard were purchased from Sigma Aldrich (Zwijndrecht, The Netherlands) and Cambridge Isotope Laboratory (Tewksbury, Massachusetts, USA) (Merck, Schiphol‐Rijk, The Netherlands), respectively. Water (ULC–MS grade), acetonitrile (ACN) (HPLC‐S grade), and methanol (HPLC grade) were purchased from Biosolve (Valkenswaard, The Netherlands). Formic acid (analytical grade) was obtained from Merck (Darmstadt, Germany). Ultrapure water was produced by a Milli‐Q® Advantage A10 Water Purification Systems (Merck, The Netherlands). Bovine serum albumin (BSA) was obtained from Roche (Mannheim, Germany).
2.2. Sample collection
Pooled human and mouse ethylenediaminetetraacetic acid (EDTA) plasmas were supplied by Sera Laboratories (Haywards Health, West Sussex, UK). Individual lithium heparin goat, pig, and rat plasma were kindly provided by the Nephrology Department of University Medical Center, Utrecht, The Netherlands. Conditionally immortalized proximal tubular epithelial cells (ciPTEC) were used for cell lysate in the validation study. These cells were obtained from urine samples of healthy volunteers and overexpressing the organic anion transporter 1 (ciPTEC‐OAT1) as described by Nieskens et al. (Nieskens et al., 2016). The cells were collected and washed with cold HBSS and treated with 200 μL of 0.1 M NaOH (Sigma‐Aldrich) as lysis solution. Finally, the lysate was agitated for 10 min at room temperature.
2.3. Equipment
The LC–MS/MS system consisted of a DGU‐14A degasser, a CTO‐10Avp column oven, a Sil‐HTc autosampler, and two LC10‐ADvp‐ pumps (Shimadzu, Kyoto, Japan) and a Finnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer with electrospray ionization (Thermo Electron, Waltham, MA, USA). The Xcalibur software (version 1.4, Thermo Electron) was used to record and process the data.
2.4. LC‐MS/MS condition
Partial‐loop injections (10 μL) were made on a Polaris 3 C18‐A column (50 × 2 mm, dp = 3 μm, average pore diameter = 10 nm, Varian, Middelburg, The Netherlands) with a corresponding pre‐column (10 × 2 mm). The temperatures for the column and sample rack compartment were maintained, respectively, at 40°C and 4°C. A gradient (0.5 mL/min) using 0.1% (v/v) formic acid (FA) in water (solvent A) and ACN (solvent B) was programmed. The percentage of solvent B was increased linearly from 15 to 35% at 1.5 min after injection. This step was followed by flushing the column with 100% (v/v) ACN until 2.00 min, and finally, the column was reconditioned back to the starting conditions, 15% (v/v) ACN for 1 min resulting in a total run time of 3 min per sample. After injection, the whole elute was transferred into the electrospray probe at 1–2 min by switching the MS divert valve. In addition to equilibration at the end of the run (1 min), we performed an inter‐run equilibration during the injection procedure of the instrument (0.30 min). This was expected sufficient, because equilibration times for this type of column can be relatively short, even down to 2 column volumes (Stoll, 2019). A solvent mixture of 80% (v/v) of 0.1% FA and 20% (v/v) methanol at 0.5 mL/min was introduced to tune the electrospray in the negative ionization mode for optimization. The electrospray settings of the assay were a 5000 V spray voltage and a 320°C capillary temperature. The skimmer voltage was set off, and the nitrogen sheath, ion sweep, and auxiliary gases were set at 60, 0, and 15 arbitrary units, respectively. The selected reaction monitoring mode was used with argon as the collision gas at 1.4 mTorr and the tube lens off set 96 V for both IS and internal standard. IS was monitored at m/z 212.0 → 80.0 and 132.0 at −24 V and −21 V collision energies, and the internal standard at m/z 218.0 → 80.0 and 138.0 at −27 V and −21 V collision energies with a dwell time of 0.1 s for each transition. The mass resolution was set 0.7 for both separating quadrupoles.
2.5. Stock and working solutions
The stock solutions were prepared at 1000 μg/mL for IS and 100 μg/mL for the internal standard in ultrapure water. The working solution concentration of IS was made as 100 μg/mL in ultrapure water, and the internal standard was diluted 200‐fold in ACN to prepare the precipitating solution with a concentration of 0.5 μg/mL.
2.6. Sample pretreatment
The plasma of different species, as well as cell lysate, underwent the same protein precipitation procedure. About 20 μL of plasma or cell lysate or BSA, spiked with 10 μL of IS or water, was crushed in 80 μL of ACN containing 0.5 μg/mL of internal standard, which was followed by vortexing for 2 min and centrifuging for 2 min at 10,000g. After that, 64 μL of supernatant was collected in 1‐ml round‐bottom well of a polypropylene 96‐deep well plate and diluted with 200 μL of water. Finally, the plate was gently shaken before placing it in the autosampler for analysis.
2.7. Method validation
2.7.1. Calibration and quality control samples
The calibration and quality control (QC) samples were prepared to obtain standards at 100, 50, 25, 5, 1, 0.5, 0.1 μg/mL in 4.2% BSA in water and to obtain QCs at 75 (High), 5 (Medium), 0.25 (Low), and 0.1 (LLOQ) μg/ml. The lower limit of detection (LLOD) was estimated from the signal‐to‐noise ratio from a chromatogram of LLOQ spiked surrogate matrix. QC samples were prepared from a different stock solution, and each calibration sample was freshly prepared in duplicate prior to every measurement. Duplicate blank with no IS, and double blank with neither IS nor internal standard were also prepared, and the calibration curve was obtained by linear regression analysis.
2.8. Accuracy and precision
The accuracy and precision of QC samples were assessed by spiking the surrogate matrix (4.2% BSA) with all concentration levels of QC. The plasma of all animals and cell lysate were assessed by spiking with two concentrations of IS (50 and 10 μg/mL) together with the calibration curve. The 4.2% BSA and human plasma were assessed at three different days for both inter‐ and intra‐day accuracy and precision analysis, whereas the other matrices were assessed for intra‐day accuracy and precision. Equations 1 and 2 were used to calculate accuracy and precision, respectively. The endogenous concentration of IS in various matrices was contemplated in the accuracy calculation (see Equation 1).
| (1) |
| (2) |
The dilution integrity assay was performed by diluting the high level of QC by 10 and 20‐folds. All experiments were performed with six replicates for each sample.
2.9. Recovery and matrix effect
There are multiple ways to calculate the matrix effect. In the current study, the post‐extraction spike method was used by spiking the same concentration (QC high) of analyte in the extracted and unextracted matrices. Three different processes were used to assess the extraction recovery and matrix effect at high QC level using 6, 3, 4, 5, 6, and 6 replicates for the plasma of human, goat, pig, rat, mouse, and cell lysate, respectively. The responses in biological matrices were corrected for endogenous concentration. The following samples (A, B, and C) were used to calculate the recovery and matrix effect: (A) the blank plasma was spiked followed by the protein precipitation method described above, (B) the blank plasma was crushed with ACN to precipitate the protein, and the collected supernatant was subsequently spiked before following the previous extraction method, and (C) the original method of preparing QC samples without further treatment. The recovery and matrix effects were then estimated by calculating the absolute responses for A/B and B/C ratios, respectively. Further, relative matrix effects were calculated for individual human (n = 6), goat (n = 3), pig (n = 4), rat (n = 5), mouse (n = 6) and cell lysate (n = 6) samples.
2.10. Stability
The stability of IS was assessed in BSA and the plasma of human, pig, and mouse at high and low QC levels, whereas rat and cell lysate were assessed at medium QC level due to the limitation of matrices. The investigated conditions were 24 h at room temperature, three additional freeze–thaw cycles (1.5 h thawing at room temperature), and 2 months at – 30°C. Incurred sample reanalysis was performed on the plasma samples of mouse (n = 42). The samples were reanalyzed after being stored at – 30°C for 2 days. In addition, the stability under autosampler condition was assessed by reanalyzing QC samples after 5 days of storage at 4°C.
3. RESULTS
3.1. Method development and its validation
Validation of a bioanalytical method is paramount for reliable results in quantitative determination of analytes in various biological matrices. The bioanalytical method was validated according to EMA and FDA guidelines (Agency, 2011; Center for Drug Evaluation and Research & Center for Veterinary Medicine, 2020). A laboratory scheme based on these guidelines was used, and a complete validation process was performed except for the inter‐day precision study in goat, pig, rat, and mouse plasma, and cell lysate due to their limited availability. The validation parameters assessed in the current study were linearity, LLOQ, precision and accuracy, stability, recovery, and matrix effect. ESI‐MS/MS settings were optimized to obtain maximal sensitivity, for which a product spectrum is presented in Figure 1, enabling reproducible and quick quantification of IS. An appropriate regression model is vital to ensure accurate results. From the ratio of the peak areas of IS and internal standard, the concentrations were back‐calculated for six independent calibrations, performed on six different days, using the calibration curves of the respective runs. The deviation from the average of each level was observed as less than 1.9% (data not shown), allowing linear regression analysis. The average of the reproducible regression parameters of the linear regression functions (n = 6) were Y = 0.018 (± 0.010) + 0.26 (± 0.02) X with a coefficient of determination (R2) value of 0.994 ± 0.002 ( Table S1). Here, X is the IS concentration (μg/mL), and Y is the response relative to the internal standard. Figure 2 shows the chromatograms of blank and murine samples. LLOQ and LLOD were calculated at 0.1 and 0.03 μg/mL, respectively. The retention time was between 1.30 and 1.38 min, regardless of the matrix and the length of the sequence.
FIGURE 2.
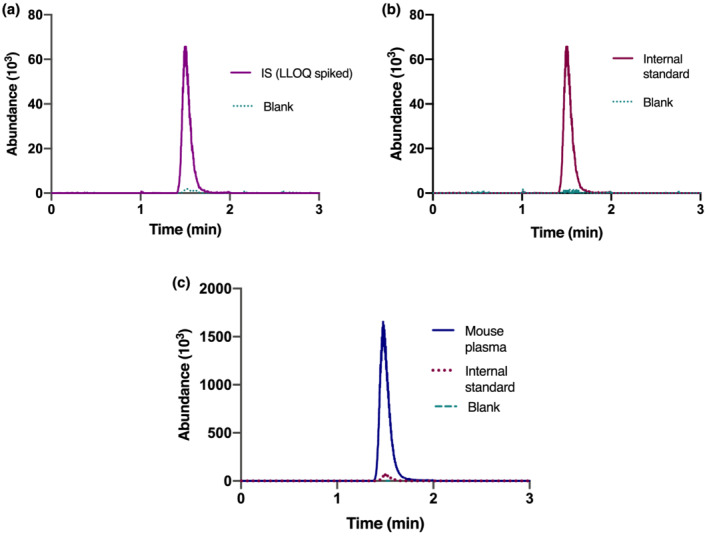
Representative chromatograms of (a) indoxyl sulfate and (b) internal standard for a blank and LLOQ (0.1 μg/mL) spiked surrogate matrix (BSA). Panel C represents the chromatograms for a mouse plasma sample
Accuracy describes the trueness of the concentrations measured in the samples containing a known amount of analyte, whereas precision describes reproducibility. The accuracy for QC samples and human plasma of this method was 97.7–104.8% (Table 1). Within‐day and between‐day precisions were ≤ 4.0 and ≤ 4.3%, respectively (Table 1). For the other matrices, accuracy was 100.8–107.3% and precision ≤2.1% (Table 1). Thus, it met the requirement of ≤15% variation (≤20% for LLOQ) (Agency, 2011; Center for Drug Evaluation and Research & Center for Veterinary Medicine, 2020). Deviation in accuracy and reproducibility in the 10‐ and 20‐fold diluted samples was <3%, which confirms the applicability and reliability of this method for samples with high analyte concentrations.
TABLE 1.
Accuracy and precision data for indoxyl sulfate in quality control samples (n = 18), human plasma (n = 18), animal plasma (n = 6) and cell sample (n = 6)
| Analytes | Nominal concentration (μg/ml) | Within‐day precision (%) | Between‐day precision (%) | Accuracy (%) |
|---|---|---|---|---|
| QC high | 75 | 1.2 | 1.9 | 97.7 |
| QC medium | 5 | 1 | 2.1 | 98.7 |
| QC low | 0.3 | 4 | 4.3 | 99.0 |
| LLOQ | 0.1 | 1.9 | 3.3 | 100.0 |
| Human plasma | 10 | 2.6 | 4 | 104.8 |
| 50 | 2 | 3.3 | 99.0 | |
| Goat plasma | 10 | 1 | 106.6 | |
| 50 | 1 | 102.5 | ||
| Pig plasma | 10 | 1.3 | 106.5 | |
| 50 | 1.9 | 101.5 | ||
| Rat plasma | 10 | 1.2 | 100.8 | |
| 50 | 1.3 | 106.6 | ||
| Mouse plasma | 10 | 1.3 | 100.9 | |
| 50 | 1.1 | 102.2 | ||
| Cell lysate | 10 | 2.1 | 107.3 |
Note: LLOQ, lower limit of quantification; QC, quality control.
The recoveries were calculated by comparing the peak area of unextracted with the extracted matrix. Extraction recoveries ranged from 90 to 99% (Table 2), suggesting that the losses in the extraction process were negligible. The matrix effect was calculated by comparing the response in the extracted authentic matrix and surrogate matrix. Ionization recoveries ranged from 87 to 113%, indicating a minor matrix effect. Relative matrix effects, representing the inter‐individual variability of the internal standard normalized matrix factor, were calculated for each animal and ranged between 100 and 110% (Table 2).
TABLE 2.
Data of recovery, matrix effect, and relative matrix effect in all plasma
| Matrices (plasma) | Recovery (%) | Matrix effect (%) | Relative matrix effect (%) | |
|---|---|---|---|---|
| Indoxyl sulfate | Internal standard | |||
| Humans (n = 6) | 90 ± 3 | 110 ± 2 | 100 ± 3 | 110% ± 2 |
| Goats (n = 3) | 99 ± 5 | 87 ± 4 | 87 ± 4 | 100 ± 4 |
| Pigs (n = 4) | 93 ± 2 | 112 ± 3 | 103 ± 2 | 109 ± 2 |
| Rats (n = 5) | 95 ± 3 | 111 ± 4 | 101 ± 2 | 109 ± 4 |
| Mice (n = 6) | 93 ± 2 | 113 ± 1 | 107 ± 3 | 106 ± 3 |
| Overall (n = 24) | 94 ± 4 | 108 ± 9 | 101 ± 6 | 107 ± 4 |
The stability of IS in various plasma and surrogate matrices at different concentrations and storage conditions is summarized in Table 3. All recoveries at 24 h room temperature and after three freeze–thaw cycles were between 96.3 and 102.1%. Recoveries after 2 months of storage at −30°C were between 91.4 and 102.4%. The autosampler stability of QC samples was observed as 95.9–109.3% relative recovery after 5 days storage at 4°C. This indicates that recovery of IS was acceptable at any storage condition. In addition, the chemical stability of IS was confirmed with both anticoagulants examined here.
TABLE 3.
Recovery of indoxyl sulfate at different storage conditions (n = 4)
| Analytes | Nominal concentration (μg/ml) | 24 h at room temperature (%) | 3 freeze–thaw cycles (%) | 2 months at −30°C (%) |
|---|---|---|---|---|
| Surrogate matrix | 2.5 | 100.6 ± 4 | 100.6 ± 2.2 | 95.2 ± 15.4 |
| 75 | 98.2 ± 1 | 96.3 ± 1.7 | 95.8 ± 1.4 | |
| Human plasma | 2.5 | 99.1 ± 2.3 | 97.9 ± 3.4 | 96.5 ± 0.6 |
| 75 | 99.8 ± 1.9 | 99.1 ± 0.8 | 97.5 ± 0.7 | |
| Pig plasma | 2.5 | 96.7 ± 6.7 | 96.6 ± 5.2 | 91.4 ± 1.9 |
| 75 | 100.1 ± 2 | 99.9 ± 1.3 | 96 ± 1.1 | |
| Mouse plasma | 2.5 | 99.6 ± 1.3 | 99.5 ± 2 | 102.4 ± 0.5 |
| 75 | 99.3 ± 0.6 | 100.5 ± 0.4 | 96.1 ± 1.1 | |
| Rat plasma | 5 | 100.1 ± 0.4 | 100.1 ± 2.3 | 102.4 ± 0.6 |
| Cell lysate | 5 | 101.2 ± 2.2 | 102.1 ± 1.2 | 97 ± 0.4 |
Incurred sample reanalysis was done by remeasuring mouse plasma samples after 1 week of storage at 4°C, to ensure the reproducibility of study samples and to verify the results from accuracy and precision analysis of IS spiked samples. The result of the incurred sample reanalysis is presented in Figure 3. Only 2 out of 42 samples exceeded the deviation of 20% (22 and 26%).
FIGURE 3.
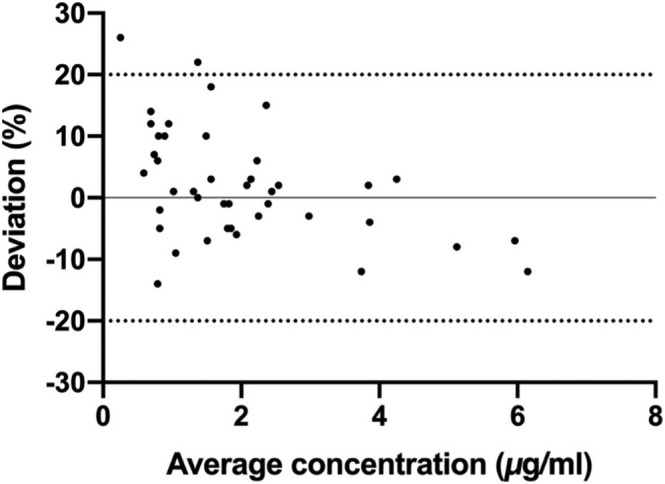
Bland–Altman plot represents a reanalysis of 42 samples of mouse plasma
3.2. Baseline levels of plasma IS in various species
The method was used to determine the baseline level of plasma IS in various healthy species obtained from multiple studies. Average plasma IS concentration in healthy humans, goats, pigs, rats, and mice were 0.5 (n = 6), 0.3 (n = 3), 0.2 (n = 6), 1.1 (n = 35), and 3.2 μg/mL (n = 18), respectively (Figure 4).
FIGURE 4.
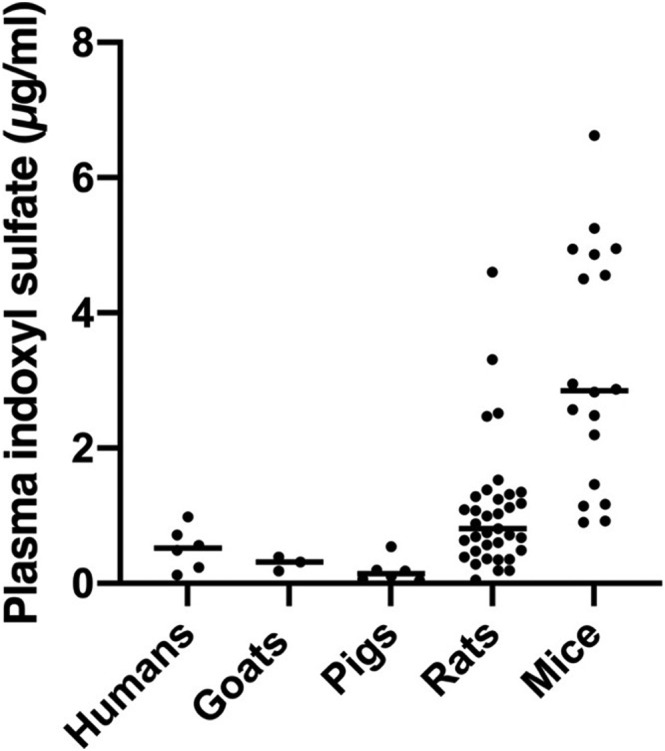
Baseline levels of plasma indoxyl sulfate (IS) in various healthy species. Each dot represents an individual animal
3.3. Altered IS concentration in plasma and urine of CKD mice
To validate the method developed in the context of CKD, we measured IS in a long‐term streptozotocin (STZ)‐induced mouse diabetic nephropathy (DN) model, which was characterized by severe tubular atrophy and interstitial fibrosis. The study design and animal handling procedure were reported previously (Falke et al., 2012) and concerned wild‐types and mice with a hemizygous deletion of Connective Tissue Growth Factor (CTGF/CCN2), an essential mediator of kidney fibrosis that is considerably upregulated in DN. Diabetes was induced in a subgroup of mice from both groups by a single intraperitoneal injection of 200 mg/kg streptozotocin (STZ).
Both at 6 and 8 months, IS plasma concentrations were over twofold higher in DN mice than in control mice (Figure 5a) and urine IS concentrations were approximately fourfold lower in DN mice than in control mice at both time points (Figure 5b).
FIGURE 5.
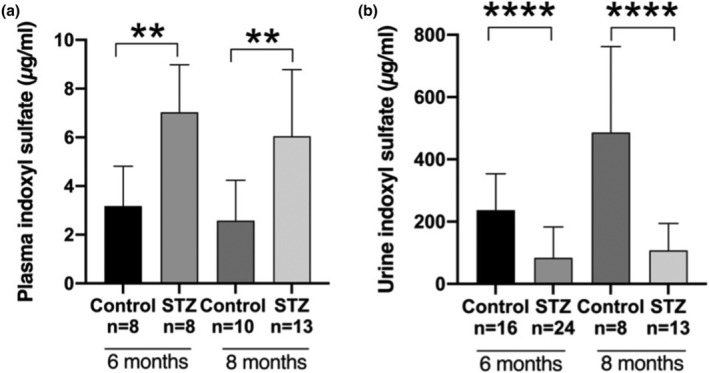
Indoxyl sulfate (IS) concentrations in control and diabetic nephropathy mice (STZ). At all time points, (a) elevated plasma IS concentrations and (b) decreased urinary IS concentrations were found in diabetic mice. Data are presented as mean ± SD of individual values obtained by using the current method. The non‐parametric Mann–Whitney U test was performed for statistical analysis
4. DISCUSSION
We developed a sensitive, efficient, reliable, and reproducible quantitative bioanalytical assay to detect IS using the LC–MS/MS technique and validated it in various species.
LC–MS/MS has long been used to measure IS in biological matrices due to its high level of sensitivity (Poesen et al., 2015; van Gelder et al., 2020). As compared to other studies, our assay shows superior analytical performance, demonstrating an improved process and extraction efficiency despite using a simple and fast protein precipitation process and a short chromatographic run time (3 versus ≥ 5 min for other methods (Olesova et al., 2020; Shu et al., 2016). The accuracy of our method ranged from 97.7 to 100% with an inter‐day variation up to 4.3% and intra‐day variation up to 4.0%, which is better than those of previously published methods that demonstrated an accuracy range of 95.0–105.3% and 90.3–99.1%, inter‐day variation of up to 7.3 and 13.2%, and intra‐day variation for up to 10.6 and 9.3%, respectively (Olesova et al., 2020; Shu et al., 2016). Notably, validation data of different species allowed us to explore species‐specific variation in the different parameters, which has not been reported earlier. Moreover, it provides a higher sensitivity by allowing a detection limit of 0.1 μg/mL, better reliability by enhanced validation parameters, and general applicability to both in vivo (in multiple species) and in vitro studies. The plasma protein precipitation was performed using a small volume of ACN, the most efficient organic precipitating agent for plasma, to restrict the dilution of samples (Mutsaers et al., 2013). At least six equally distributed concentration levels that comprise a physiologically relevant range are recommended for calibration samples. Based on the previously reported plasma concentration of IS in healthy and uremic humans, rats, and mice, seven concentrations within the range of 0.1–100 μg/mL were selected for the calibration samples used for validation of the assay. Since IS is an endogenous compound, a surrogate matrix approach was applied where 4.2% BSA in water was used as the matrix for the calibration samples.
A well‐known limitation of LC–MS‐based assays for quantification of analytes in complex matrices, for instance, plasma, is the matrix effect, involving ion suppression. The components of biological matrices, including phospholipids, salts, and proteins, are the primary sources of ion suppression, which may alter the ionization efficiency by several mechanisms such as preventing the analyte from accessing the charge, gaining charge, and remaining in the charged state, thereby resulting in incorrect data as well as weak accuracy and precision (Trufelli et al., 2011). Such effects can be compensated for by using an appropriate internal standard (Chiu et al., 2010). We have used an isotope‐labeled indoxyl sulfate potassium salt (13C6, 99% purity) as internal standard. Furthermore, a high organic flush at the end of each run was used to remove strongly retained matrix constituents to avoid ionization suppression in the long term.
We observed a distinct baseline level of plasma IS concentration among different species, but these levels are comparable to previously reported studies with human (Imazu et al., 2020; Poesen et al., 2015), goat (van Gelder et al., 2021), rat (Aoyama et al., 2002; Kikuchi et al., 2010), and mouse (Hung et al., 2016; Mishima et al., 2018). IS concentration in pig plasma has not been reported earlier.
As a tryptophan metabolite, this may be due to inter‐species variability in dietary protein intake and colonic metabolism processes. The gut microbial flora consumes dietary tryptophan to produce indole, which enters the portal system for further metabolism by the liver into IS. Genetic variations among different species in hepatic indole metabolism may also cause variation (Poesen et al., 2015).
In the mice with long‐term DN characterized by tubular damage, elevated concentrations of plasma IS and decreased concentrations of urinary IS suggest reduced renal clearance of IS. To investigate the hypothesized role of IS as a tubular function marker, further analysis is required, correlating IS concentrations and clearances with the extent of kidney tubular damage, tubular function parameters, and established tubular injury markers such as KIM‐1 and NGAL. However, we consider this beyond the scope of this paper that focuses on the validation of the assay as a first step. In addition, the biomarker value of IS as tubular function marker should be explored in various other kidney diseases and renal injury models.
The influence of plasma IS concentration on the tubular secretion of various commonly prescribed drugs in CKD, such as losartan, valsartan, and furosemide, was recently studied in vitro, and it was found that elevated plasma IS in CKD may competitively interfere with the transport of the drugs using the same transporter at the basolateral membrane of the proximal tubule cells (Masereeuw et al., 2000). As suggested by the validation data on in vitro kidney cell line, this method can be used to quantify intracellular IS concentration to determine the competitive transport via OAT1 in the presence of drugs and other toxins at various pathological conditions.
Our method also bears some limitations. The baseline concentration of indoxyl sulfate is close to the LLOQ, which warrants strategies to further improve the sensitivity of the assay. This could include an increase in injection volume, using a more sensitive detector and further optimization of the sample pretreatment procedure. Nevertheless, the sensitivity was sufficient for all our applications for which the assay was designed.
5. CONCLUSION
A simple, rapid, robust, and highly sensitive LC–MS/MS assay for quantification of IS in various species and a relevant kidney cell line has been developed and validated, which now allows for cross‐species analysis of IS as a translational biomarker for tubular function. The reliability of the assay was justified by good reproducibility, high accuracy, minimal matrix effect, and adequate recovery. Successful application of this method revealed a considerably higher plasma and lower urinary IS concentration in DN mice with tubular damage, suggesting that IS concentration may reflect kidney tubular injury. Further studies are needed to investigate whether this uremic toxin can be used as a tubular function biomarker in multiple species.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
FUNDING INFORMATION
This study has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie Grant agreement no. 813839.
AUTHORSHIP CONTRIBUTION STATEMENT
Sabbir Ahmed: Methodology, validation, formal analysis, investigation, writing—original draft, visualization. Rolf W. Sparidans: Conceptualization, methodology, resources, supervision. Jingyi Lu: Writing—review and editing, visualization. Silvia M. Mihaila: Writing—review and editing, supervision. Karin G.F. Gerritsen: Conceptualization, resources, writing—review and editing, supervision. Rosalinde Masereeuw: Conceptualization, resources, writing—review and editing, supervision, project administration.
INSTITUTIONAL REVIEW BOARD STATEMENT
The protocol code for the goat and pig trial is 2015‐II‐547‐014, and date of approval 1‐10‐2015. The rest of the plasma for method validation was commercially bought as mentioned in the method section. The information regarding diabetic nephropathy mice model was previously reported in the referred article. The experiments were performed with the approval of the Experimental Animal Ethics Committee of the University of Utrecht. In the current study, only the samples from wild‐type mice were investigated.
Supporting information
Table S1. Calibration data of Indoxyl sulfate (n = 6)
ACKNOWLEDGMENTS
The authors acknowledge the valuable support of Dr. Tri Nguyen, Prof. Dr. Roel Goldschmeding and Roel Broekhuizen.
Ahmed, S. , Sparidans, R. W. , Lu, J. , Mihaila, S. M. , Gerritsen, K. G. F. , & Masereeuw, R. (2022). A robust, accurate, sensitive LC–MS/MS method to measure indoxyl sulfate, validated for plasma and kidney cells. Biomedical Chromatography, 36(5), e5307. 10.1002/bmc.5307
Funding information European Union's Horizon 2020, Grant/Award Number: 813839
REFERENCES
- Aoyama, I. , Shimokata, K. , & Niwa, T. (2002). An oral adsorbent downregulates renal expression of genes that promote interstitial inflammation and fibrosis in diabetic rats. Nephron, 92, 635–651. 10.1159/000064108 [DOI] [PubMed] [Google Scholar]
- Barreto, F. C. , Barreto, D. V. , Liabeuf, S. , Meert, N. , Glorieux, G. , Temmar, M. , Choukroun, G. , Vanholder, R. , & Massy, Z. A. (2009). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clinical Journal of the American Society of Nephrology, 4, 1551–1558. 10.2215/CJN.03980609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre, J. V. , Vaidya, V. S. , Schmouder, R. , Feig, P. , & Dieterle, F. (2010). Next‐generation biomarkers for detecting kidney toxicity. Nature Biotechnology, 28, 436–440. 10.1038/nbt0510-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research , & Center for Veterinary Medicine . (2020). Bioanalytical Method Validation Guidance for Industry. Accessed August 19, 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry
- Chiu, M. L. , Lawi, W. , Snyder, S. T. , Wong, P. K. , Liao, J. C. , & Gau, V. (2010). Matrix effects—A challenge toward automation of molecular analysis. JALA: Journal of the Association for Laboratory Automation, 15, 233–242. 10.1016/j.jala.2010.02.001 [DOI] [Google Scholar]
- Delanaye, P. , Glassock, R. J. , & De Broe, M. E. (2017). Epidemiology of chronic kidney disease: Think (at least) twice! Clinical Kidney Journal, 10, 370–374. 10.1093/ckj/sfw154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, R. J. , Small, D. M. , Vesey, D. A. , Johnson, D. W. , Francis, R. , Vitetta, L. , Gobe, G. C. , & Morais, C. (2016). Indoxyl sulphate and kidney disease: Causes, consequences and interventions. Nephrology (Carlton), 21, 170–177. 10.1111/nep.12580 [DOI] [PubMed] [Google Scholar]
- Enomoto, A. , Takeda, M. , Taki, K. , Takayama, F. , Noshiro, R. , Niwa, T. , & Endou, H. (2003). Interactions of human organic anion as well as cation transporters with indoxyl sulfate. European Journal of Pharmacology, 466, 13–20. 10.1016/S0014-2999(03)01530-9 [DOI] [PubMed] [Google Scholar]
- European Medicines Agency . (2011). Guideline on bioanalytical method validation. Accessed 15 March. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf [DOI] [PubMed]
- Falke, L. L. , Dendooven, A. , Leeuwis, J. W. , Nguyen, T. Q. , van Geest, R. J. , van der Giezen, D. M. , Broekhuizen, R. , Lyons, K. , Stoop, R. , Kemperman, H. , Schlingemann, R. , Joles, J. A. , & Goldschmeding, R. (2012). Hemizygous deletion of CTGF/CCN2 does not suffice to prevent fibrosis of the severely injured kidney. Matrix Biology, 31, 421–431. 10.1016/j.matbio.2012.06.002 [DOI] [PubMed] [Google Scholar]
- Gilbert, R. E. (2017). Proximal Tubulopathy: Prime mover and key therapeutic target in diabetic kidney disease. Diabetes, 66, 791–800. 10.2337/db16-0796 [DOI] [PubMed] [Google Scholar]
- Hung, S. C. , Kuo, K. L. , Huang, H. L. , Lin, C. C. , Tsai, T. H. , Wang, C. H. , Chen, J. W. , Lin, S. J. , Huang, P. H. , & Tarng, D. C. (2016). Indoxyl sulfate suppresses endothelial progenitor cell‐mediated neovascularization. Kidney International, 89, 574–585. 10.1016/j.kint.2015.11.020 [DOI] [PubMed] [Google Scholar]
- Imazu, M. , Fukuda, H. , Kanzaki, H. , Amaki, M. , Hasegawa, T. , Takahama, H. , Hitsumoto, T. , Tsukamoto, O. , Morita, T. , Ito, S. , & Kitakaze, M. (2020). Plasma indoxyl sulfate levels predict cardiovascular events in patients with mild chronic heart failure. Scientific Reports, 10, 16528. 10.1038/s41598-020-73633-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja, S. , Jafri, L. , Siddiqui, I. , Hashmi, M. , & Ghani, F. (2019). The utility of neutrophil gelatinase‐associated Lipocalin (NGAL) as a marker of acute kidney injury (AKI) in critically ill patients. Biomarker Research, 7, 4. 10.1186/s40364-019-0155-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, K. , Itoh, Y. , Tateoka, R. , Ezawa, A. , Murakami, K. , & Niwa, T. (2010). Metabolomic search for uremic toxins as indicators of the effect of an oral sorbent AST‐120 by liquid chromatography/tandem mass spectrometry. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 878, 2997–3002. 10.1016/j.jchromb.2010.09.006 [DOI] [PubMed] [Google Scholar]
- Lin, C. J. , Liu, H. L. , Pan, C. F. , Chuang, C. K. , Jayakumar, T. , Wang, T. J. , Chen, H. H. , & Wu, C. J. (2012). Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Archives of Medical Research, 43, 451–456. 10.1016/j.arcmed.2012.08.002 [DOI] [PubMed] [Google Scholar]
- Lin, C. N. , Wu, I. W. , Huang, Y. F. , Peng, S. Y. , Huang, Y. C. , & Ning, H. C. (2019). Measuring serum total and free indoxyl sulfate and p‐cresyl sulfate in chronic kidney disease using UPLC‐MS/MS. Journal of Food and Drug Analysis, 27, 502–509. 10.1016/j.jfda.2018.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns, B. , Hemmelgarn, B. , Tonelli, M. , Au, F. , So, H. , Weaver, R. , Quinn, A. E. , & Klarenbach, S. (2019). The cost of Care for People with chronic kidney disease. Canadian Journal of Kidney Health and Disease, 6. 2054358119835521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masereeuw, R. , Van Pelt, A. P. , Van Os, S. H. G. , Willems, P. H. G. M. , Smits, P. , & Russel, F. G. M. (2000). Probenecid interferes with renal oxidative metabolism: A potential pitfall in its use as an inhibitor of drug transport. British Journal of Pharmacology, 131, 57–62. 10.1038/sj.bjp.0703541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima, E. , Fukuda, S. , Kanemitsu, Y. , Saigusa, D. , Mukawa, C. , Asaji, K. , Matsumoto, Y. , Tsukamoto, H. , Tachikawa, T. , Tsukimi, T. , Fukuda, N. N. , Ho, H. J. , Kikuchi, K. , Suzuki, C. , Nanto, F. , Suzuki, T. , Ito, S. , Soga, T. , Tomioka, Y. , & Abe, T. (2018). Canagliflozin reduces plasma uremic toxins and alters the intestinal microbiota composition in a chronic kidney disease mouse model. American Journal of Physiology. Renal Physiology, 315, F824–f33. 10.1152/ajprenal.00314.2017 [DOI] [PubMed] [Google Scholar]
- Mutsaers, H. A. , Engelke, U. F. , Wilmer, M. J. , Wetzels, J. F. , Wevers, R. A. , van den Heuvel, L. P. , Hoenderop, J. G. , & Masereeuw, R. (2013). Optimized metabolomic approach to identify uremic solutes in plasma of stage 3‐4 chronic kidney disease patients. PLoS ONE, 8, e71199. 10.1371/journal.pone.0071199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers, H. A. , van den Heuvel, L. P. , Ringens, L. H. , Dankers, A. C. , Russel, F. G. , Wetzels, J. F. , Hoenderop, J. G. , & Masereeuw, R. (2011). Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS ONE, 6, e18438. 10.1371/journal.pone.0018438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanayakkara, S. , Senevirathna, S. T. , Karunaratne, U. , Chandrajith, R. , Harada, K. H. , Hitomi, T. , Watanabe, T. , Abeysekera, T. , Aturaliya, T. N. , & Koizumi, A. (2012). Evidence of tubular damage in the very early stage of chronic kidney disease of uncertain etiology in the north Central Province of Sri Lanka: A cross‐sectional study. Environmental Health and Preventive Medicine, 17, 109–117. 10.1007/s12199-011-0224-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankivell, B. J. , Shingde, M. , Keung, K. L. , Fung, C. L. , Borrows, R. J. , O'Connell, P. J. , & Chapman, J. R. (2018). The causes, significance and consequences of inflammatory fibrosis in kidney transplantation: The Banff i‐IFTA lesion. American Journal of Transplantation, 18, 364–376. 10.1111/ajt.14609 [DOI] [PubMed] [Google Scholar]
- Nieskens, T. T. , Peters, J. G. , Schreurs, M. J. , Smits, N. , Woestenenk, R. , Jansen, K. , van der Made, T. K. , Röring, M. , Hilgendorf, C. , Wilmer, M. J. , & Masereeuw, R. (2016). A human renal proximal tubule cell line with stable organic anion transporter 1 and 3 expression predictive for antiviral‐induced toxicity. The AAPS Journal, 18, 465–475. 10.1208/s12248-016-9871-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesova, D. , Galba, J. , Piestansky, J. , Celusakova, H. , Repiska, G. , Babinska, K. , Ostatnikova, D. , Katina, S. , & Kovac, A. (2020). A novel UHPLC‐MS method targeting urinary Metabolomic markers for autism Spectrum disorder. Metabolites, 10. 10.3390/metabo10110443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poesen, R. , Mutsaers, H. A. , Windey, K. , van den Broek, P. H. , Verweij, V. , Augustijns, P. , Kuypers, D. , Jansen, J. , Evenepoel, P. , Verbeke, K. , Meijers, B. , & Masereeuw, R. (2015). The influence of dietary protein intake on mammalian tryptophan and phenolic metabolites. PLoS ONE, 10, e0140820. 10.1371/journal.pone.0140820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, C. , Chen, X. , Xia, T. , Zhang, F. , Gao, S. , & Chen, W. (2016). LC‐MS/MS method for simultaneous determination of serum p‐cresyl sulfate and indoxyl sulfate in patients undergoing peritoneal dialysis. Biomedical Chromatography, 30, 1782–1788. 10.1002/bmc.3753 [DOI] [PubMed] [Google Scholar]
- Stoll, D. R. (2019). Reversed‐Phase Liquid Chromatography and Water, Part II: Re‐equilibration of the Stationary Phase Following Gradient Elution. Accessed 25 November. https://www.chromatographyonline.com/view/reversed-phase-liquid-chromatography-and-water-part-ii-re-equilibration-stationary-phase-following-g
- Townsend, D. M. , Deng, M. , Zhang, L. , Lapus, M. G. , & Hanigan, M. H. (2003). Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. Journal of the American Society of Nephrology: JASN, 14, 1–10. 10.1097/01.ASN.0000042803.28024.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trufelli, H. , Palma, P. , Famiglini, G. , & Cappiello, A. (2011). An overview of matrix effects in liquid chromatography‐mass spectrometry. Mass Spectrometry Reviews, 30, 491–509. 10.1002/mas.20298 [DOI] [PubMed] [Google Scholar]
- Vaidya, V. S. , Ozer, J. S. , Dieterle, F. , Collings, F. B. , Ramirez, V. , Troth, S. , Muniappa, N. , Thudium, D. , Gerhold, D. , Holder, D. J. , Bobadilla, N. A. , Marrer, E. , Perentes, E. , Cordier, A. , Vonderscher, J. , Maurer, G. , Goering, P. L. , Sistare, F. D. , & Bonventre, J. V. (2010). Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nature Biotechnology, 28, 478–485. 10.1038/nbt.1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brand, J. A. , Mutsaers, H. A. , van Zuilen, A. D. , Blankestijn, P. J. , van den Broek, P. H. , Russel, F. G. , Masereeuw, R. , & Wetzels, J. F. (2016). Uremic solutes in chronic kidney disease and their role in progression. PLoS ONE, 11, e0168117. 10.1371/journal.pone.0168117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder, M. K. , de Vries, J. C. , Ahmed, S. , Monninkhof, A. S. , de Kort, G. A. P. , Vonken, E. P. A. , Hazenbrink, D. H. M. , Vaessen, K. R. D. , Nguyen, T. Q. , Verhaar, M. C. , Joles, J. A. , & Gerritsen, K. G. F. (2021). A uremic goat model created by subtotal renal artery embolization and gentamicin. Biology (Basel), 10, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder, M. K. , Middel, I. R. , Vernooij, R. W. M. , Bots, M. L. , Verhaar, M. C. , Masereeuw, R. , Grooteman, M. P. , Nubé, M. J. , van den Dorpel, M. A. , Blankestijn, P. J. , Rookmaaker, M. B. , & Gerritsen, K. G. F. (2020). Protein‐bound uremic toxins in hemodialysis patients relate to residual kidney function, are not influenced by convective transport, and do not relate to outcome. Toxins (Basel), 12. 10.3390/toxins12040234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , & Kestenbaum, B. (2018). Proximal tubular secretory clearance: A neglected partner of kidney function. Clinical Journal of the American Society of Nephrology, 13, 1291–1296. 10.2215/CJN.12001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, I. W. , Hsu, K. H. , Lee, C. C. , Sun, C. Y. , Hsu, H. J. , Tsai, C. J. , Tzen, C. Y. , Wang, Y. C. , Lin, C. Y. , & Wu, M. S. (2011). P‐Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrology, Dialysis, Transplantation, 26, 938–947. 10.1093/ndt/gfq580 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Calibration data of Indoxyl sulfate (n = 6)