

Abstract
Postpartum depression (PPD) is a common major depressive episode surrounding childbirth, with estimated rates ranging from 5.5% to 23.5% of all live births across Europe and the USA based on the presence of key symptoms. PPD has been associated with significant impairments in both maternal functioning and mother‐infant attachment, and these impairments can have lasting effects on the emotional and cognitive development of children. Although the precise pathophysiology of PPD is unknown, preclinical findings suggest that large fluctuations in neurosteroid hormone levels can induce physiological plasticity in the expression of functional GABAA receptors during pregnancy and the postpartum period, and that deficits in this plasticity may underpin a biological mechanism that contributes to the manifestation of depressive symptoms. Here, we review the controlled clinical trials to date that have assessed the efficacy of pharmacological treatments for PPD, including oestradiol, selective serotonin reuptake inhibitors, brexanolone (an iv formulation of allopregnanolone) and an investigational neuroactive steroid and GABAA positive allosteric modulator, zuranolone. Coupled with the GABAergic deficits implicated in major depressive disorder, these findings highlight not only the potential role of GABAA receptor plasticity in the pathophysiology of PPD, but also the novel therapeutic approach of using positive allosteric modulators targeting GABAergic transmission to treat women affected by PPD.
Keywords: GABA positive allosteric modulator, neuroactive steroid, postpartum depression
Although the precise pathophysiology of postpartum depression (PPD) is unknown, preclinical findings suggest that large fluctuations in neurosteroid hormone levels can induce physiological plasticity in the expression of functional GABAA receptors during pregnancy and the postpartum period, and that deficits in this plasticity may underpin a biological mechanism that contributes to the manifestation of depressive symptoms. These findings highlight the potential role of GABAA receptor plasticity in the pathophysiology of PPD, as well as the novel therapeutic approach of using positive allosteric modulators targeting GABAergic transmission to treat women affected by PPD.
![]()
1. INTRODUCTION
Postpartum depression (PPD) is a major depressive episode (MDE) associated with pregnancy and childbirth. In the 5th version of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐5), one of the specifiers of an MDE includes “peripartum onset” in recognition of the onset of symptoms that can occur during pregnancy or in the first 4 weeks following delivery. 1 Expert opinions vary greatly regarding the timing of symptom onset that defines PPD, ranging from the early postpartum period to 12 months after delivery. 2 , 3 , 4 , 5 In accordance with DSM‐5 criteria, the diagnosis of depression requires the presence of 5 or more of the following symptoms: depressed mood, diminished interest or pleasure in all (or almost all) activities, significant weight loss or gain, insomnia, psychomotor agitation or slowing, fatigue or loss of energy, feelings of worthlessness or excessive inappropriate guilt, diminished ability to think or concentrate, or recurrent thoughts of death or suicidal ideation. 1 Additionally, common PPD symptoms present as persistent sadness, anxiety, irritability, feelings of guilt, loss of interest, fatigue, trouble bonding with the baby, persistent doubts about the ability to care for the baby, or suicidal thoughts. 6 The impact of PPD can also extend to the entire family, including children, because several studies have implicated PPD as negative factor affecting the cognitive and emotional outcomes of infants. 7 , 8 Impairments in the quality of caregiving, mother‐infant interaction and attachment have also been identified as contributing factors to the burden of disease. 9 , 10
PPD may be underdiagnosed because of a lack of disease awareness, barriers for access to care and stigma 11 , 12 ; therefore, many studies report rates of PPD based on the presence of key symptoms. In the USA, 9.7%‐23.5% of women with a recent live birth experience symptoms of PPD. 13 Across eight European countries, 5.5%‐13.5% of mothers are reported to experience PPD symptoms each year. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Analyses that require a formal diagnosis of depression have reported PPD rates up to approximately 13%. 12 , 14 , 22 , 23 , 24 , 25 , 26 , 27 The predicted costs of untreated perinatal mood and anxiety disorders, including PPD, were estimated to be $14 billion in the USA in 2017 for affected mothers and their children. 28 A similar UK study showed that perinatal mental illness could cost society over £8 billion per each 1‐year birth cohort. 29 Up until recently, there have been limited pharmacological treatment options for PPD; the first PPD‐specific treatment, brexanolone (ZULRESSO®; Sage Therapeutics, Inc, Cambridge, MA, USA) was approved by the US Food and Drug Administration (FDA) in 2019. 30 Brexanolone is an iv formulation of allopregnanolone that is administered as a continuous infusion for 60 hours. 31 Traditional oral antidepressant treatments have been frequently used to treat these patients despite no specific indication for PPD; however, the literature has shown varied efficacy of such treatments, indicating that there remains a high unmet need among this population. 32 , 33 , 34 , 35
2. NEUROSTEROIDS AND PATHOPHYSIOLOGY OF PPD
Neurosteroids are produced from cholesterol in the brain and alter neuronal excitability through membrane receptors without direct effects on gene expression. 36 Early observations of the rapid central nervous system action of neurosteroids in the animal literature date back to 1941. 37 Based on their structural properties, neurosteroids produce a range of effects on excitatory or inhibitory neurotransmission. 38 In one of the earlier reports, Majewska et al 39 demonstrated that 3α‐hydroxy‐5α‐dihydroprogesterone and 3α,5α‐tetrahydrodeoxycorticosterone are potent modulators of the GABAA receptor. Animal studies have identified key enzymes for neurosteroid biosynthesis localised in several regions of the brain, including the cerebral cortex, hippocampus, dentate gyrus, thalamus, cerebellum, olfactory bulb, striatum and amygdala. 40 , 41 Neurosteroids are therefore assumed to act locally within specific corticolimbic circuitries, 41 and recent investigations have identified a site of action on synaptic and extrasynaptic GABAA receptors. 42 GABAA receptors are ligand‐gated anion channels typically comprised of α and β subunits (2 of each), along with a single γ subunit in synaptic receptors or a single δ subunit in extrasynaptic receptors (Figure 1). 42 Allopregnanolone, also known as 3α‐hydroxy‐5α ‐pregnan‐20‐one (ALLO), is a major pregnancy hormone and is also a highly potent positive allosteric modulator of both synaptic and extrasynaptic GABAA receptors (Figure 1). 43 , 44 , 45
FIGURE 1.
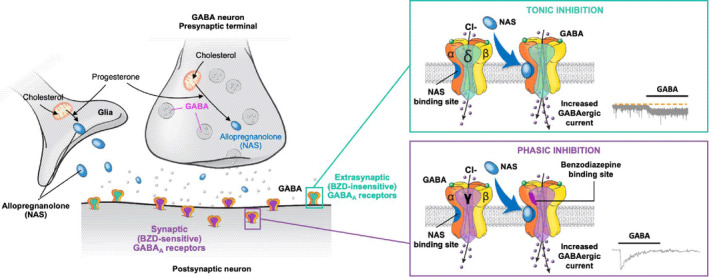
Mechanism of action of the positive allosteric modulator allopregnanolone on synaptic and extrasynaptic GABAA receptors. Allopregnanolone binds both synaptic and extrasynaptic GABAA receptors, acting as a positive allosteric modulator to increase the frequency and duration of channel opening, thus increasing hyperpolarizing GABAergic current. 46 , 93 BZD, benzodiazepine; GABA, γ‐aminobutyric acid; GABAA, γ‐aminobutyric acid type A; NAS, neuroactive steroid
Dysregulation of neurosteroidogenesis has been implicated in several neuropsychiatric disorders, including both PPD and unipolar depression, as reviewed in detail by Porcu and et al. 46 Consistent with initial preclinical findings, 47 cerebrospinal fluid levels of ALLO in depressed patients were found to be lower than in healthy controls at baseline and subsequently increased and normalised following treatment with the selective serotonin reuptake inhibitors (SSRIs) fluoxetine or fluvoxamine, 48 suggesting that SSRIs may mediate antidepressant/anxiolytic actions in part via promoting neurosteroid synthesis. Alterations in genetics/epigenetics, inflammation, neuroplasticity and the hypothalamic‐pituitary‐adrenal axis have been observed in both forms of depression. 49 , 50 , 51 Because these systems interact with each other and do not operate in isolation, it is important to recognise the potential role of biological triggers that contribute to the PPD mechanism of disease. Although the diagnostic criteria of PPD are the same as those for an MDE in the setting of major depressive disorder (MDD), the timing of symptom onset is often in the 4‐6 weeks following childbirth. 2 , 3 , 52 This may indicate a biological trigger for PPD, leading to downstream pathophysiological elements common to both MDD and PPD. For example, women with PPD have been shown to be more sensitive to hormonal fluctuations compared to women without PPD. 53 Furthermore, a seminal epidemiology study that assessed almost all women who gave birth in Sweden over an 11‐year period (n = 707 701) showed that a history of depression increases the risk for PPD by more than 20‐fold. 54
There is a considerable increase in hormone levels that peaks towards the end of pregnancy; this includes ALLO, which begins at < 20 nanomolar concentrations in early pregnancy and subsequently reaches 75‐110 nanomolar concentrations for most women. 43 The precipitous drop of these hormone levels around parturition (including to < 2 nanomolar concentrations for ALLO at 4‐6 weeks postpartum) has therefore been a focus of research as a potential biological trigger for PPD. 55 , 56 , 57 , 58 Although this drop in hormone levels may represent a trigger, it fails to explain why the majority of women do not experience PPD even though all experience this hormonal fluctuation and decline in ALLO. Further preclinical research has elucidated that there is down‐regulation of GABAA δ and γ2 subunits that parallel increasing ALLO levels during pregnancy, which is followed by functional up‐regulation back to baseline levels in the postpartum period. 59 These findings suggest that this physiological compensation may be impaired in PPD, and that the emergence of severe depressive symptoms in the postpartum period may be a result of insufficient up‐regulation of GABAA receptors. Maguire and Mody 59 also investigated the GABAA receptor plasticity with a focus on δ subunit knockout mice and found that both homozygotes and heterozygotes (Gabrd‐/‐ and Gabrd+/‐) displayed abnormal maternal behaviours leading to reduced survival of the offspring, which were ameliorated by the administration of δ subunit selective agonist THIP (4,5,6,7‐tetrahydroisoxazolo(5,4‐c)pyridin‐3‐ol) in Gabrd+/‐ mice. The GABAergic deficit hypothesis of depression stems from widely replicated observations of reduced GABA levels in plasma, cerebrospinal fluid and cortical brain tissue, as well as postmortem findings showing reduced expression of GABAergic markers in depressed patients, 60 thus placing GABAergic neural circuit deficits at the centre of the aetiology of depressive disorders. Notably, monoaminergic antidepressant use is linked with increased cortical GABA concentrations in both patients with depression and healthy volunteers, further supporting this view. 61 , 62 Collectively, the available evidence suggests that PPD may be associated with a trigger of rapidly declining neurosteroid levels at parturition on a background of impaired up‐regulation of the GABAA receptors in susceptible women, and the manifestation of depressive symptoms is based on the GABAergic deficits that are common to depressed patients. It follows that, because certain neurosteroids such as ALLO are highly potent modulators of GABAA receptors, their pharmacologic effects can be expected to improve symptoms of depression in patients with PPD based on these GABAergic deficits. 44 , 60 , 63
In addition, preclinical evidence suggests that ALLO has been shown to exert anti‐inflammatory actions through a number of possible mechanisms, including its effects on macrophages and microglia. 64 , 65 Other research has shown that GABAergic signalling reduces the production of proinflammatory cytokines through actions on antigen‐presenting cells. 66 Further research is needed to characterise the association between the anti‐inflammatory effects of ALLO and its mechanism of action in PPD.
3. RANDOMISED CONTROLLED TRIALS IN PATIENTS WITH PPD
Despite the availability of numerous antidepressant agents for MDD, none were approved specifically for PPD until the approval of brexanolone in 2019 (as summarised below and in Table 1). 31 Overall, there have been few rigorous clinical trials published that assess the efficacy and safety of pharmacologic agents for depression in patients with PPD. The findings from a systematic literature review conducted in 2017 and presented at a scientific congress, 35 which was subsequently updated in 2021 with newly included studies presented in this review, identified only 7 randomised, double‐blind, placebo‐ or active‐controlled trials that assessed pharmacologic therapies for the treatment of PPD aside from brexanolone or zuranolone (Table 1). 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 These trials primarily assessed the impact of oestradiol and SSRIs on patients with PPD.
TABLE 1.
Summary of double‐blind placebo‐ or active‐controlled RCTs of pharmacologic therapies for the treatment of PPD
|
Author (year); Country (N) |
Intervention(s) and comparator(s) | Primary endpoint; time period | Definition/criteria for PPD (timing of symptom onset) | Key efficacy outcomes |
|---|---|---|---|---|
| SSRIs and estradiol | ||||
| Hantsoo (2014) 70 ; US (N = 36) | Sertraline (n = 17) vs. placebo (n = 19) | HAMD‐19/CGI responsea and remissionb rates; 6 weeks | SCID‐confirmed MDD; HAMD‐19 ≥ 18 and < 32, CGI moderate severity (within 3 months of delivery) |
Week 6 Response: 59% vs. 26%; P = .05 Remission: 53% vs. 21%; P = .05 Mean HAMD‐19 score: 8.8 vs. 13.0 |
| O’Hara (2019) 69 ; US (N = 162) | Sertraline (n = 56) vs. placebo (n = 53) vs. IPT (n = 53) | Change in HAMD‐17 score over time; 12 weeksc | HAMD‐17 ≥ 12, SCID‐confirmed DSM‐IV diagnosis of MDD (within 6 months of delivery) |
No significant group by time interaction for HAMD‐17 scores (P = .454) or BDI scores (P = .063) A significant group by time interaction was noted for IDAS‐GD scores (P = .030) |
| Yonkers (2008) 68 ; US (N = 70) | Paroxetine (n = 35) vs. placebo (n = 35) | NRd; 8 weeks | HAMD‐17 ≥ 16 at initial visit, met DSM‐IV diagnostic criteria for MDD (within 3 months of delivery) |
HAMD‐17, week 8 Main effect vs. placebo: −1.62; P = .22 Remissione: 37% vs. 14% (OR = 3.5; 95% CI = 1.1‐11.5; P = .04) CGI‐S, week 8 Main effect vs. placebo: −0.48; P = .047 |
| Gregoire (1996) 67 ; UK (N = 61) | Estradiol (n = 34) vs. placebo (n = 27) | NRf | EPDS ≥ 14; MDE according to RDC and SADS interview (within 12 weeks of delivery) |
EPDS, month 1 Mean (SD): 13.3 (5.7) vs. 16.5 (5.3) Treatment effect vs. placebo: 4.38 (95% CI = 1.89‐6.87); P < .001 SADS‐C Treatment effect vs. placebo: 6.30 (95% CI = 1.70‐10.69); P < .02 |
| Wisner (2015) 71 ; US (N = 85) | Estradiol (n = 26) vs. sertraline (n = 30) vs. placebo (n = 29) | SIGH‐ADS29 responseg and remissionh; 8 weeks | 2 weeks of symptoms post‐birth (within 4 weeks of delivery) | Rates of response (42.3% vs. 63.3% vs. 58.6%; P = .26) and remission (26.9% vs. 30% vs. 31%; P = .94) did not differ across the 3 groups at week 8 |
| Wisner (2006) 72 , 73 , 74 , 75 ; US (N = 109) | Sertraline (n = 55) vs. nortriptyline (n = 54) | HAMD‐17 responsei and remissionj rates; 8 weeks | HAMD‐17 ≥ 18, SCID‐confirmed MDD (within 4 weeks of delivery) |
Week 8 Responded: 56% vs. 69%; P = .19 Remitted: 46% vs. 48%; P = .79 Nonresponse: 24% vs. 26% |
| Kashani (2017) 76 ; Iran (N = 68) |
Fluoxetine (n = 32) vs. saffron (n = 32) |
HAMD‐17 responsei and remissionk rates; 6 weeks | HAMD‐17 ≥ 10 and ≤ 18; DSM‐IV MDD (4‐12 weeks after delivery) |
No significant time by treatment interaction for HAMD‐17 score Remission: 21.9% vs. 18.8% (P = 1.00) Response: 50% vs. 40.6% (P = .61) |
| GABAA positive allosteric modulators | ||||
| Kanes (2017) 77 ; US (N = 21) | BRX90 (n = 10) vs. placebo (n = 11) | HAMD‐17 change from baseline; 60 hours | SCID‐I MDE and HAMD‐17 ≥ 26 (no earlier than third trimester and within 4 weeks of birth) |
Change from baseline in HAMD‐17 scores 60 hours: −20.97 vs. −8.75 (mean difference −12.2; 95% CI = −20.77 to −3.67; P = .0075) |
| Meltzer‐Brody (2018) 78 , 81 ; US (N = 138 [Study 1]; 108 [Study 2]) |
Study 1 BRX90 (n = 45) vs. BRX60 (n = 47) vs. placebo (n = 46) Study 2 BRX90 (n = 54) vs. placebo (n = 54) |
HAMD‐17 change from baseline; 60 hours | SCID‐I MDE and HAMD‐17 ≥ 26 (Study 1) or HAMD‐17 20‐25 (Study 2) (no earlier than third trimester and within 4 weeks of birth) |
LS mean change from baseline in HAMD‐17 scores, Study 1 60 hours: −17.7 vs. −19.5 vs. −14.0 Mean difference BRX90 vs. placebo: −3.7 (95% CI = −6.9 to −0.5; P = .0252) Mean difference BRX60 vs. placebo: −5.5 (95% CI = −8.8 to −2.2; P = .0013 LS mean change from baseline in HAMD‐17 scores, Study 2 60 hours: −14.6 vs. 12.1 Mean difference BRX90 vs. placebo: −2.5 (95% CI = −4.5 to −0.5); P = .0160 |
| Deligiannidis (2021) 85 ; US (N = 153) | Zuranolone (n = 77) vs. placebo (n = 76) | HAMD‐17 change from baseline; 15 days | SCID‐I MDE and HAMD‐17 ≥ 26 (no earlier than third trimester and within 4 weeks of birth) |
LS mean change from baseline in HAMD‐17 scores Day 15: −17.8 vs. −13.6; P = .003 |
BRX60, brexanolone 60 μg/kg/h; BRX90, brexanolone 90 μg/kg/h; CGI(‐S), Clinical Global Impressions Scale (‐Severity); CI, confidence interval; DSM, Diagnostic and Statistical Manual of Mental Disorders; EPDS, Edinburgh Postnatal Depression Scale; HAMD‐17, 17‐item Hamilton Depression Rating Scale; HAMD‐19, 19‐item Hamilton Depression Rating Scale; IDAS‐GD, Inventory of Depression and Anxiety Symptoms, General Depression scale; IPT, interpersonal therapy; LS, least squares; MDD, major depressive disorder; MDE, major depressive episode; NR, not reported; OR, odds ratio; PPD, postpartum depression; RCT, randomized controlled trial; RDC, research diagnostic criteria; SADS(‐C), Schedule for Affective Disorders and Schizophrenia(‐Current); SCID(‐I), Structured Clinical Interview for DSM‐IV (Axis I Disorders); SD, standard deviation; SIGH‐ADS29, Structured Interview Guide for the Hamilton Depression Rating Scale—Atypical Depression Symptoms; SSRI, selective serotonin reuptake inhibitor.
Response defined as HAMD‐19 ≤ 10, ≥ 50% decrease in HAMD‐19 score from baseline, and score of “much improved” or “very much improved” on the improvement scale of the CGI.
Remission defined as HAMD‐19 ≤ 7.
Primary analysis followed assessed time interactions based on follow‐up at 4, 8, and 12 weeks.
HAMD‐17, IDS‐SR, and CGI‐S scores assessed but primary endpoint unclear.
Remission defined as HAMD‐17 ≤ 8.
EPDS, SADS‐C scores assessed but primary endpoint and time point unclear (full study duration 6 months).
Response defined as ≥ 50% decrease in SIGH‐ADS29 score from baseline.
Remission defined as SIGH‐ADS29 ≤ 8.
Response defined as ≥ 50% decrease in HAMD‐17 score from baseline.
Remission defined as HAMD‐17 < 7.
Remission defined as HAMD‐17 ≤ 7.
In general, most studies were conducted across a small number of centres (eg, ≤ 3) and had fewer than 100 participants. In some cases, investigators suggested that they had difficulties meeting their planned numbers for enrollment, which occurred primarily because of strict inclusion criteria, inefficient recruitment methods and time constraints. 67 , 68 One study used multiple protocol amendments over a 2.5‐year period to expand its inclusion criteria, eventually allowing breastfeeding women to participate and relaxing the depressive symptom scale threshold for study entry (from 17‐item Hamilton Depression Rating Scale [HAMD‐17] ≥ 15 to ≥ 12). 69 Some studies also reported imbalances in baseline characteristics between active treatment and placebo groups; in 2 studies, these differences occurred in depressive symptom scales that were assessed as study outcomes. 68 , 70 Primary endpoints were not clearly stated in all studies but, in general, time points for the primary analyses ranged from 6 to 12 weeks, suggesting that the study investigators expected the onset of maximum treatment effect to occur over this period.
The findings from placebo‐controlled randomised controlled trials (RCTs) for the treatment of PPD were mixed. For example, a study that assessed high‐dose (200 μg) oestradiol in patients with severe PPD (mean Edinburgh Postnatal Depression Scale [EPDS] score > 21; PPD defined as MDE according to the Schedule for Affective Disorders and Schizophrenia‐Current [SADS‐C] interview and symptoms occurring within 12 weeks of delivery) demonstrated that those who received oestrogen improved rapidly within the first month, with a significantly greater improvement in mean EPDS score compared to placebo (overall treatment effect: 4.38 points; 95% confidence interval [Cl] = 1.89‐6.87). 67 Similar findings were observed with the SADS‐C scale 67 ; however, no other common depression symptom scales were measured in this trial. By contrast, another study that randomised patients with PPD (who showed 2 weeks of symptoms starting within 4 weeks of delivery; no symptom scale specified) to oestradiol, the SSRI sertraline or placebo, was stopped early because serum concentrations of oestradiol were not significantly different between any treatment groups at weeks 4 or 8. 71 The analysis reported no significant differences between treatment groups in either of the primary outcomes of rates of response (reduction of baseline Structured Interview Guide for the Hamilton Depression Rating Scale ‐ Atypical Depression Symptoms [SIGH‐ADS29] score by ≥ 50%) or remission (SIGH‐ADS29 score ≤ 8), although the study investigators noted that this was expected because the study was underpowered as a result of its early stoppage. 71
Additional studies that assessed SSRIs, including both sertraline and paroxetine, reported varying results in patients with PPD, often with between‐group differences only being reported for depressive symptoms scales that were not the primary outcome. For example, an 8‐week study of patients with PPD (defined as patients with symptom onset within 3 months of delivery who met DSM‐IV criteria for MDD, with HAMD‐17 ≥ 16) treated with paroxetine or placebo showed that there was only a significant between‐group improvement from baseline for the Clinical Global Impression ‐ Severity (CGI‐S) score, but not HAMD‐17 score. 68 This study also reported no significant difference between groups in the proportion of patients who achieved response criteria (CGI ‐ Improvement [CGI‐I] score of 1 or 2), although more patients achieved remission (HAMD‐17 ≤ 8) in the paroxetine group compared to placebo (37% vs 14%; P = .04). 68 It should be noted, however, that there was considerable dropout in this study, with 35 patients randomised to each treatment group and only 17 (paroxetine) and 14 (placebo) remaining in the study at end of the 8‐week treatment period. 68 Withdrawals because of lack of efficacy were reported for 6 and 7 patients, respectively. 68 In another study of 36 women with PPD (with Structured Clinical Interview for DSM‐IV [SCID]‐confirmed MDD, 19‐item HAMD [HAMD‐19] score ≥18 and <32, a CGI score of moderate severity, and onset within 3 months of delivery) randomised to either sertraline or placebo, patients in the sertraline‐treated group had significantly lower HAMD‐19 scores at baseline compared to the placebo group (20.6 vs 23.2; P = .03). 70 Despite this imbalance, the study measured response criteria according to the achievement of a HAMD‐19 score of ≤ 10 after 6 weeks of treatment, and it was concluded that there was a significantly greater proportion of women achieving response who were randomised to sertraline compared to placebo (59% vs 26%; P = .05). 70 Notably, the study reported no statistically significant differences in mean HAMD‐19 scores between groups at the end of week 6. 70 Another study that compared patients with PPD (with SCID‐confirmed MDD and HAMD‐17 score ≥12, and with symptoms within 6 months of delivery) who were treated with sertraline or placebo in a double‐blind manner (along with a third group that received interpersonal therapy unblinded) over 12 weeks showed that there were no significant differences in PPD symptoms between groups over time according to the HAMD‐17 (primary outcome) and Beck Depression Inventory scales. 69 The study did report statistically significant time to improvement for the Inventory of Depression and Anxiety Symptoms ‐ General Depression scale for the sertraline group compared to both interpersonal therapy and placebo, although there were no significant differences between any groups for response (≥ 50% reduction in HAMD‐17) or recovery (HAMD‐17 ≤ 7, typically defined as remission) criteria. 69
Three RCTs compared antidepressant agents head‐to‐head in patients with PPD; in addition to the study described above that compared oestradiol and sertraline along with a placebo arm (with no between‐group differences reported although the study was stopped early), 71 a separate study compared sertraline with nortriptyline in patients with SCID‐confirmed MDD and a HAMD‐17 score ≥ 18 within 4 weeks of delivery, 72 , 73 , 74 , 75 and another compared fluoxetine with saffron in patients with DSM‐IV MDD who had HAMD‐17 scores ≥10 and ≤18 and symptom onset between 4 and 12 weeks after delivery. 76 In both studies, there were no between‐group differences in the proportion of patients who achieved response or remission criteria at treatment end (weeks 8 and 6, respectively). 72 , 76
4. STUDIES OF NEUROACTIVE STERIODS THAT ARE GABAA POSITIVE ALLOSTERIC MODULATORS
The first RCT of brexanolone, an iv formulation of allopregnanolone, in PPD was conducted in 21 women with severe symptoms (total HAMD‐17 score ≥ 26) in 4 centres in the USA. 77 Major eligibility criteria included women who were up to 6 months postpartum and between the ages of 18 and 45 years with no significant medical findings, and an onset of depressive symptoms either during the last trimester of pregnancy or within 4 weeks after delivery. 77 Brexanolone or placebo was administered as a continuous infusion for 60 hours, and the primary endpoint was change from baseline in the HAMD‐17 total score at this time point. 77 Patients were followed‐up in a blinded manner for 30 days. 77 After 60 hours, the mean difference between the groups in change scores from baseline was −12.2 (95% CI = −20.77 to −3.67) (P = .0075) favouring the brexanolone group. 77 In addition, HAMD‐17 score improvements favoured the brexanolone group at 36, 48, 60 and 72 hours, and days 7 and 30. 77 Remission (HAMD‐17 total score ≤ 7) was observed in 7 of 10 patients in the brexanolone group compared to 1 of 11 patients in the placebo group at 60 hours (P = .0364), whereas response rates (50% or greater reduction from baseline in total HAMD‐17 score) did not reach statistical significance between groups (7 in 10 patients in the brexanolone group compared to 4 of 11 patients in the placebo group; P = .1450). 77 There were no serious adverse events (AEs) or discontinuations in either group, and the most frequent AEs in the brexanolone group were dizziness (2 patients in the brexanolone and 3 patients in the placebo group) and somnolence (2 patients in the brexanolone and none in the placebo group). 77 The results of this first RCT of a neuroactive steroid with a novel mechanism of action as a GABAA positive allosteric modulator, which demonstrated fast onset of improvement and relatively high remission rates, supported further development of brexanolone for the treatment of PPD.
Subsequent brexanolone studies in patients with PPD were conducted under an umbrella protocol that included two multicentre, randomised, double‐blind, placebo‐controlled phase 3 trials. 78 These clinical trials included common key eligibility criteria, including age range (18‐45 years), being up to 6 months postpartum and a requirement for being in good physical health. 78 Study 1 included women with a HAMD‐17 severity score ≥ 26, and randomised patients 1:1:1 to a single 60‐hour continuous infusion of either brexanolone 90 μg kg‐1 h‐1 (BRX90), brexanolone 60 μg kg‐1 h‐1 (BRX60) or placebo. 78 The primary endpoint was the change from baseline in the total HAMD‐17 score at 60 hours, and the follow‐up period lasted 30 days during which time participants remained blind to treatment. 78 In total, 138 women were randomised in study 1 to BRX90 (n = 45), BRX60 (n = 47) or placebo (n = 46). After 60 hours, the mean difference in change from baseline in HAMD‐17 scored between the placebo group and both brexanolone infusion groups was statistically significant (−5.5 [95% CI = −8.8 to −2.2], P = .0013 for BRX60; −3.7 [95% CI = −6.9 to −0.5], P = .0252 for BRX90). 78 In the BRX60 group, compared to the placebo group, HAMD‐17 total scores were significantly lower starting at 24 hours and all later timepoints, including 36, 48, 60 and 72 hours and 7 and 30 days (all P < .05). 78 In the BRX90 group, statistically significant changes in total HAMD‐17 scores were observed at 60 hours and 30 days (both P < .05). 78 Remission rates (HAMD‐17 ≤ 7) at 60 hours were 51.4% in the BRX60 group compared to 16.3% in the placebo group (P = .001) and 30.8% in the BRX90 group (P = .083 compared to placebo). 78 , 79 The response rate at 60 hours was 86.5% in the BRX60 group compared to 55.8% in the placebo group (P = .005) and 74.4% in the BRX90 group (P = .049 compared to placebo). 78 , 79 Brexanolone infusion was generally well tolerated; the most common treatment‐emergent AEs in the brexanolone groups were headache, dizziness and somnolence. 78 One patient in the BRX60 group experienced 2 serious AEs of suicidal ideation and an intentional overdose attempt during the follow‐up period, which was not considered to be treatment‐related. 78
For study 2, the HAMD‐17 severity score of included patients were within a range from 20 to 25, and patients were randomised 1:1 either to BRX90 (n = 54) or placebo (n = 54). 78 Similar to study 1, a 60‐hour infusion of brexanolone or placebo was administered, and patients remained blinded for 30 days with the primary endpoint of change from baseline in the total HAMD‐17 score at 60 hours. 78 The mean difference in HAMD‐17 change from baseline scores favoured the BRX90 group (−2.5 [95% CI = −4.5 to −0.5], P = .0160) and total HAMD‐17 scores were significantly lower in the BRX90 group compared to the placebo group at 48, 60 and 72 hours and 7 days (all P < .05). 78 After 60 hours, remission was achieved in 61.2% in the BRX90 group compared to 38.5% in the placebo group (P = .003) and response criteria were met for 75.5% in the BRX90 group compared to 59.6% in the placebo group (P =.017). 78 , 80 The most common treatment‐emergent AEs in the BRX90 group were headache, dizziness and somnolence. One patient in the BRX90 group experienced 2 serious AEs of altered state of consciousness and syncope that were considered to be treatment‐related. Based on these 3 RCTs, brexanolone infusion received the first approval from the FDA in March 2019 for the treatment of adults with PPD with a Risk Evaluation and Mitigation Strategy (REMS). 30 Under the REMS, healthcare facilities and pharmacies are required to be certified and patients are required to enroll before administration of brexanolone infusion with the goal of mitigating the potential risk of serious harm resulting from excessive sedation and sudden loss of consciousness during the infusion. 31
A post‐hoc analysis of an integrated trial dataset from the above 3 brexanolone RCTs indicated that brexanolone provided clinical meaningful patient‐level improvement and between‐group differences relative to placebo in the PPD population. 81 Furthermore, an indirect treatment comparison showed that brexanolone demonstrated a more rapid improvement and larger differences in change from baseline for the treatment of PPD relative to SSRIs using matching‐adjusted indirect comparisons that accounted for the differences in trial designs between inpatient and outpatient settings. 82
Zuranolone, also known as SAGE‐217/BIIB125, is a synthetic investigational neuroactive steroid GABAA receptor positive allosteric modulator. Similar to brexanolone, zuranolone has affinity for both synaptic and extrasynaptic GABAA receptors, although it has been designed with a pharmacokinetic provision of oral bioavailability and once‐daily dosing. 83 The efficacy of zuranolone treatment with respect to improving depressive symptoms was demonstrated in patients with MDD. 84 The first randomised, double‐blind, placebo‐controlled clinical trial of zuranolone in PPD included 153 women age 18‐44 years who were up to 6 months postpartum and who had an onset of symptoms no earlier than the last trimester and up to 4 weeks after delivery. 85 Key entry criteria included a total HAMD‐17 score ≥ 26 and medical stability. 85 Patients were randomised 1:1 to receive either 30 mg of zuranolone or matching placebo capsules for 14 days and were followed for another 4 weeks remaining blind to treatment. 85 Patients treated with zuranolone demonstrated a significantly greater decrease from baseline in the primary endpoint of day 15 HAMD‐17 total score (least squares mean −17.8 vs −13.6 points; P = .003). 85 At the last assessment point (day 45), a statistically significant separation between zuranolone and placebo was still observed (P = .003). 85 In addition, day 15 response and remission rates were significantly greater in the zuranolone group (72% and 45%, respectively) compared to the placebo group (48% and 23%, respectively; both P < .05). 85 These positive findings have encouraged further development of zuranolone for the treatment of PPD. At present, another phase 3 RCT is ongoing in the USA and Europe that is testing the efficacy and safety of 50 mg of zuranolone compared to placebo for the treatment of women with PPD. 86
Other analogues of ALLO have been tested for a range of neuropsychiatric disorders, although, to date, none have been assessed in patients with PPD. One example is ganaxolone, a 3β‐methylated synthetic analog of ALLO, which was shown to significantly decrease mean total Montgomery‐Åsberg Depression Rating Scale score over 8 weeks in a pilot study of 10 postmenopausal women with persistent depression. 87 Ganaxolone has also demonstrated mixed efficacy in different populations of patients with epilepsy. 88 Furthermore, in children with Fragile X syndrome, ganaxolone treatment did not result in a statistically significant improvement in CGI‐I score and most scales of anxiety and aberrant behaviour compared to placebo (although it did lead to a significant improvement in the depression subscale of the Anxiety, Depression and Mood Scale). 89 ALLO has also been tested in a phase 1b/2a clinical trial in 24 patients with early Alzheimer's disease; an exploratory efficacy analysis demonstrated that there were no differences between the ALLO and placebo groups at 12 weeks with respect to cognition scores and hippocampal volume. 90 Additional clinical trials of ALLO in patients with early or mild Alzheimer's disease are currently planned or underway. 91 , 92
5. CONCLUSIONS
PPD is a common mood disorder that is associated with substantial implications for mothers and their families, including children. Although there is more work to be done to characterise the pathophysiology of PPD and how it is delineated from MDD, the available preclinical data suggest that, in the context of large neurosteroid level fluctuations, deficits in GABAA receptor plasticity during pregnancy and the postpartum period may confer biological vulnerability to depression. Controlled clinical trials with monoaminergic antidepressants have shown delayed improvements and variable outcomes. Brexanolone infusion, an iv formulation of ALLO, received the first regulatory approval for the treatment of PPD after demonstrating significant improvements in depressive symptoms within 60 hours. Collectively, these findings support further evaluation of the possible impact that neurosteroids may have as positive allosteric modulators of GABAA receptors for the treatment of PPD.
AUTHOR CONTRIBUTIONS
Handan Gunduz‐Bruce: Conceptualisation; Data curation; Methodology; Writing – original draft; Writing – review & editing. Koji Takahashi: Conceptualisation; Data curation; Writing – original draft; Writing – review & editing. Ming‐Yi Huang: Conceptualisation; Data curation; Writing – original draft; Writing – review & editing.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jne.13019.
ACKNOWLEDGEMENTS
Handan Gunduz‐Bruce, Koji Takahashi and Ming‐Yi Huang are employees of Sage Therapeutics, Inc and own stock/stock options. We thank Marcia Reinhart of Tantalus Medical Communications (Victoria, Canada) for providing medical writing support, which was funded by Sage Therapeutics, Inc and Biogen in accordance with Good Publication Practice (GPP3) guidelines.
Gunduz‐Bruce H, Takahashi K, Huang M‐Y. Development of neuroactive steroids for the treatment of postpartum depression. J Neuroendocrinol. 2022;34:e13019. 10.1111/jne.13019
REFERENCES
- 1. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Association; 2013. [Google Scholar]
- 2. Stowe ZN, Hostetter AL, Newport DJ. The onset of postpartum depression: Implications for clinical screening in obstetrical and primary care. Am J Obstet Gynecol. 2005;192(2):522‐526. [DOI] [PubMed] [Google Scholar]
- 3. Kettunen P, Koistinen E, Hintikka J. Is postpartum depression a homogenous disorder: time of onset, severity, symptoms and hopelessness in relation to the course of depression. BMC Pregnancy Childbirth. 2014;14:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robertson E, Celasun N, Stewart DE. Risk factors for postpartum depression. In: Stewart DE, Robertson E, Dennis C‐L, Grace SL, Wallington T, eds. Postpartum depression: Literature review of risk factors and interventions. 2003. [Google Scholar]
- 5. American College of Obstetricians and Gynecologists . Screening for perinatal depression. ACOG Committee Opinion No. 757. Obstet Gynecol. 2018;132(5):e208‐212. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services NIoH, National Institute of Mental Health,. Perinatal Depression. https://www.nimh.nih.gov/health/publications/perinatal‐depression/index.shtml. Published 2021. Accessed March 25, 2021
- 7. Moore Simas TA, Huang M‐Y, Patton C, et al. The humanistic burden of postpartum depression: a systematic literature review. Curr Med Res Opin. 2019;35(3):383‐393. [DOI] [PubMed] [Google Scholar]
- 8. Murray L, Cooper PJ. Postpartum depression and child development. Psychol Med. 1997;27(2):253‐260. [DOI] [PubMed] [Google Scholar]
- 9. Parsons CE, Young KS, Rochat TJ, Kringelbach ML, Stein A. Postnatal depression and its effects on child development: a review of evidence from low‐ and middle‐income countries. Br Med Bull. 2012;101:57‐79. [DOI] [PubMed] [Google Scholar]
- 10. Murray L. The development of children of postnatally depressed mothers: Evidence from the Cambridge longitudinal study. Psychoanal Psychother. 2009;23(3):185‐199. [Google Scholar]
- 11. Pinto‐Foltz MD, Logsdon MC. Stigma towards mental illness: a concept analysis using postpartum depression as an exemplar. Issues Ment Health Nurs. 2008;29(1):21‐36. [DOI] [PubMed] [Google Scholar]
- 12. Sudhanthar S, Sheikh ZE, Thakur K. Postpartum depression screening: are we doing a competent job? BMJ Open Qual. 2019;8(4):e000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauman BL, Ko JY, Cox S, et al. Vital signs: postpartum depressive symptoms and provider discussions about perinatal depression ‐ United States, 2018. MMWR Morb Mortal Wkly Rep. 2020;69(19):575‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ban L, Gibson JE, West J, Fiaschi L, Oates MR, Tata LJ. Impact of socioeconomic deprivation on maternal perinatal mental illnesses presenting to UK general practice. Br J Gen Pract. 2012;62(603):e671‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banti S, Mauri M, Oppo A, et al. From the third month of pregnancy to 1 year postpartum. Prevalence, incidence, recurrence, and new onset of depression. Results from the perinatal depression‐research & screening unit study. Compr Psychiatry. 2011;52(4):343‐351. [DOI] [PubMed] [Google Scholar]
- 16. de Tychey C, Spitz E, Briançon S, et al. Pre‐ and postnatal depression and coping: a comparative approach. J Affect Disord. 2005;85(3):323‐326. [DOI] [PubMed] [Google Scholar]
- 17. Josefsson A, Berg G, Nordin C, Sydsjö G. Prevalence of depressive symptoms in late pregnancy and postpartum. Acta Obstet Gynecol Scand. 2001;80(3):251‐255. [DOI] [PubMed] [Google Scholar]
- 18. Reck C, Struben K, Backenstrass M, et al. Prevalence, onset and comorbidity of postpartum anxiety and depressive disorders. Acta Psychiatr Scand. 2008;118(6):459‐468. [DOI] [PubMed] [Google Scholar]
- 19. Blom EA, Jansen PW, Verhulst FC, et al. Perinatal complications increase the risk of postpartum depression. The Generation R Study. BJOG. 2010;117(11):1390‐1398. [DOI] [PubMed] [Google Scholar]
- 20. Nielsen Forman D, Videbech P, Hedegaard M, Dalby Salvig J, Secher NJ. Postpartum depression: identification of women at risk. BJOG. 2000;107(10):1210‐1217. [DOI] [PubMed] [Google Scholar]
- 21. Klier CM, Rosenblum KL, Zeller M, Steinhardt K, Bergemann N, Muzik M. A multirisk approach to predicting chronicity of postpartum depression symptoms. Depress Anxiety. 2008;25(8):718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Hara MW, Swain AM. Rates and risk of postpartum depression—a meta‐analysis. Int Rev Psychiatry. 1996;8(1):37‐54. [Google Scholar]
- 23. Gavin NI, Gaynes BN, Lohr KN, Meltzer‐Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005;106(5 Pt 1):1071‐1083. [DOI] [PubMed] [Google Scholar]
- 24. Georgiopoulos AM, Bryan TL, Wollan P, Yawn BP. Routine screening for postpartum depression. J Fam Pract. 2001;50(2):117‐122. [PubMed] [Google Scholar]
- 25. Geier ML, Hills N, Gonzales M, Tum K, Finley PR. Detection and treatment rates for perinatal depression in a state Medicaid population. CNS Spectr. 2015;20(1):11‐19. [DOI] [PubMed] [Google Scholar]
- 26. Liu CH, Tronick E. Rates and predictors of postpartum depression by race and ethnicity: results from the 2004 to 2007 New York City PRAMS survey (Pregnancy Risk Assessment Monitoring System). Matern Child Health J. 2013;17(9):1599‐1610. [DOI] [PubMed] [Google Scholar]
- 27. Do T, Hu Z, Otto J, Rohrbeck P. Depression and suicidality during the postpartum period after first time deliveries, active component service women and dependent spouses, U.S. Armed Forces, 2007‐2012. MSMR. 2013;20(9):2‐7. [PubMed] [Google Scholar]
- 28. Luca DL, Margiotta C, Staatz C, Garlow E, Christensen A, Zivin K. Financial toll of untreated perinatal mood and anxiety disorders among 2017 births in the United States. Am J Public Health. 2020;110(6):888‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bauer A, Parsonage M, Knapp M, Iemmi A, Adelaja B. The costs of perinatal mental health problems. LSE & Centre for Mental Health; 2014. [Google Scholar]
- 30. U.S. Food and Drug Administration CfDEaR. Zulresso NDA 211371 approval letter, 2019. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211371Orig1s000Approv.pdf. Accessed April 12, 2021
- 31. ZULRESSO® (brexanolone) [package insert]. Sage Therapeutics, Inc; 2019. [Google Scholar]
- 32. De Crescenzo F, Perelli F, Armando M, Vicari S. Selective serotonin reuptake inhibitors (SSRIs) for post‐partum depression (PPD): a systematic review of randomized clinical trials. J Affect Disord. 2014;152–154:39‐44. [DOI] [PubMed] [Google Scholar]
- 33. Guille C, Newman R, Fryml LD, Lifton CK, Epperson CN. Management of postpartum depression. J Midwifery Womens Health. 2013;58(6):643‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Molyneaux E, Trevillion K, Howard LM. Antidepressant treatment for postnatal depression. JAMA. 2015;313(19):1965‐1966. [DOI] [PubMed] [Google Scholar]
- 35. Reinhart M, Patton C, Chawla A, Clemson C, Huang M, Bonthapally V. Treatment of postpartum depression: a systematic literature review. Value Health. 2017;20(9):A717. [Google Scholar]
- 36. Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22(9):410‐416. [DOI] [PubMed] [Google Scholar]
- 37. Selye H. Anesthetic effect of steroid hormones. Proc Soc Exp Biol Med. 1941;46(1):116‐121. [Google Scholar]
- 38. Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate‐like modulators of the GABA receptor. Science. 1986;232(4753):1004‐1007. [DOI] [PubMed] [Google Scholar]
- 40. Furukawa A, Miyatake A, Ohnishi T, Ichikawa Y. Steroidogenic acute regulatory protein (StAR) transcripts constitutively expressed in the adult rat central nervous system: colocalization of StAR, cytochrome P‐450SCC (CYP XIA1), and 3beta‐hydroxysteroid dehydrogenase in the rat brain. J Neurochem. 1998;71(6):2231‐2238. [DOI] [PubMed] [Google Scholar]
- 41. Agis‐Balboa RC, Pinna G, Zhubi A, et al. Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc Natl Acad Sci U S A. 2006;103(39):14602‐14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goetz T, Arslan A, Wisden W, Wulff P. GABA(A) receptors: structure and function in the basal ganglia. Prog Brain Res. 2007;160:21‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pařízek A, Hill M, Kancheva R, et al. Neuroactive pregnanolone isomers during pregnancy. J Clin Endocrinol Metab. 2005;90(1):395‐403. [DOI] [PubMed] [Google Scholar]
- 44. Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6(6):2311‐2322. [PubMed] [Google Scholar]
- 45. Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci. 2004;24(30):6667‐6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porcu P, Barron AM, Frye CA, et al. Neurosteroidogenesis today: novel targets for neuroactive steroid synthesis and action and their relevance for translational research. J Neuroendocrinol. 2016;28(2):12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uzunov DP, Cooper TB, Costa E, Guidotti A. Fluoxetine‐elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc Natl Acad Sci U S A. 1996;93(22):12599‐12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uzunova V, Sheline Y, Davis JM, et al. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci U S A. 1998;95(6):3239‐3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otte C, Gold SM, Penninx BW, et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2:16065. [DOI] [PubMed] [Google Scholar]
- 50. Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102(1):75‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Payne JL, Maguire J. Pathophysiological mechanisms implicated in postpartum depression. Front Neuroendocrinol. 2019;52:165‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yonkers KA, Ramin SM, Rush AJ, et al. Onset and persistence of postpartum depression in an inner‐city maternal health clinic system. Am J Psychiatry. 2001;158(11):1856‐1863. [DOI] [PubMed] [Google Scholar]
- 53. Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. 2000;157(6):924‐930. [DOI] [PubMed] [Google Scholar]
- 54. Silverman ME, Reichenberg A, Savitz DA, et al. The risk factors for postpartum depression: a population‐based study. Depress Anxiety. 2017;34(2):178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gilbert Evans SE, Ross LE, Sellers EM, Purdy RH, Romach MK. 3alpha‐reduced neuroactive steroids and their precursors during pregnancy and the postpartum period. Gynecol Endocrinol. 2005;21(5):268‐279. [DOI] [PubMed] [Google Scholar]
- 56. Hendrick V, Altshuler LL, Suri R. Hormonal changes in the postpartum and implications for postpartum depression. Psychosomatics. 1998;39(2):93‐101. [DOI] [PubMed] [Google Scholar]
- 57. Meltzer‐Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schiller CE, Meltzer‐Brody S, Rubinow DR. The role of reproductive hormones in postpartum depression. CNS Spectr. 2015;20(1):48‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59(2):207‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16(4):383‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159(4):663‐665. [DOI] [PubMed] [Google Scholar]
- 62. Bhagwagar Z, Wylezinska M, Taylor M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. Am J Psychiatry. 2004;161(2):368‐370. [DOI] [PubMed] [Google Scholar]
- 63. Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S. Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37(1):109‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Balan I, Beattie MC, O'Buckley TK, Aurelian L, Morrow AL. Endogenous neurosteroid (3α,5α)3‐hydroxypregnan‐20‐one inhibits toll‐like‐4 receptor activation and pro‐inflammatory signaling in macrophages and brain. Sci Rep. 2019;9(1):1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Faroni A, Magnaghi V. The neurosteroid allopregnanolone modulates specific functions in central and peripheral glial cells. Front Endocrinol (Lausanne). 2011;2:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bhat R, Axtell R, Mitra A, et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci USA. 2010;107(6):2580‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gregoire AJ, Kumar R, Everitt B, Henderson AF, Studd JW. Transdermal oestrogen for treatment of severe postnatal depression. Lancet. 1996;347(9006):930‐933. [DOI] [PubMed] [Google Scholar]
- 68. Yonkers KA, Lin H, Howell HB, Heath AC, Cohen LS. Pharmacologic treatment of postpartum women with new‐onset major depressive disorder: a randomized controlled trial with paroxetine. J Clin Psychiatry. 2008;69(4):659‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. O'Hara MW, Pearlstein T, Stuart S, Long JD, Mills JA, Zlotnick C. A placebo controlled treatment trial of sertraline and interpersonal psychotherapy for postpartum depression. J Affect Disord. 2019;245:524‐532. [DOI] [PubMed] [Google Scholar]
- 70. Hantsoo L, Ward‐O'Brien D, Czarkowski KA, Gueorguieva R, Price LH, Epperson CN. A randomized, placebo‐controlled, double‐blind trial of sertraline for postpartum depression. Psychopharmacology. 2014;231(5):939‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wisner KL, Sit DKY, Moses‐Kolko EL, et al. Transdermal estradiol treatment for postpartum depression: a pilot, randomized trial. J Clin Psychopharmacol. 2015;35(4):389‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wisner KL, Hanusa BH, Perel JM, et al. Postpartum depression: a randomized trial of sertraline versus nortriptyline. J Clin Psychopharmacol. 2006;26(4):353‐360. [DOI] [PubMed] [Google Scholar]
- 73. Lanza di Scalea T, Hanusa BH, Wisner KL. Sexual function in postpartum women treated for depression: results from a randomized trial of nortriptyline versus sertraline. J Clin Psychiatry. 2009;70(3):423‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Logsdon MC, Wisner K, Hanusa BH. Does maternal role functioning improve with antidepressant treatment in women with postpartum depression? J Womens Health (Larchmt). 2009;18(1):85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Logsdon MC, Wisner K, Hanusa BH, Phillips A. Role functioning and symptom remission in women with postpartum depression after antidepressant treatment. Arch Psychiatr Nurs. 2003;17(6):276‐283. [DOI] [PubMed] [Google Scholar]
- 76. Kashani L, Eslatmanesh S, Saedi N, et al. Comparison of saffron versus fluoxetine in treatment of mild to moderate postpartum depression: a double‐blind, randomized clinical trial. Pharmacopsychiatry. 2017;50(2):64‐68. [DOI] [PubMed] [Google Scholar]
- 77. Kanes S, Colquhoun H, Gunduz‐Bruce H, et al. Brexanolone (SAGE‐547 injection) in post‐partum depression: a randomised controlled trial. Lancet. 2017;390(10093):480‐489. [DOI] [PubMed] [Google Scholar]
- 78. Meltzer‐Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post‐partum depression: two multicentre, double‐blind, randomised, placebo‐controlled, phase 3 trials. Lancet. 2018;392(10152):1058‐1070. [DOI] [PubMed] [Google Scholar]
- 79. Sage Therapeutics Inc . A Multicenter, Randomized, Double‐Blind, Parallel‐Group, Placebo‐Controlled Study Evaluating the Efficacy, Safety, and Pharmacokinetics of SAGE‐547 Injection in the Treatment of Adult Female Subjects with Severe Postpartum Depression (data on file). 2018.
- 80. Sage Therapeutics Inc. A Multicenter, Randomized, Double‐Blind, Parallel‐Group, Placebo‐Controlled Study Evaluating the Efficacy, Safety, and Pharmacokinetics of SAGE‐547 Injection in the Treatment of Adult Female Subjects with Moderate Postpartum Depression (data on file). 2018.
- 81. Gerbasi ME, Meltzer‐Brody S, Acaster S, et al. Brexanolone in postpartum depression: post hoc analyses to help inform clinical decision‐making. J Womens Health (Larchmt). 2021;30(3):385‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cooper MC, Kilvert HS, Hodgkins P, Roskell NS, Eldar‐Lissai A. Using matching‐adjusted indirect comparisons and network meta‐analyses to compare efficacy of brexanolone injection with selective serotonin reuptake inhibitors for treating postpartum depression. CNS Drugs. 2019;33(10):1039‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Althaus AL, Ackley MA, Belfort GM, et al. Preclinical characterization of zuranolone (SAGE‐217), a selective neuroactive steroid GABA(A) receptor positive allosteric modulator. Neuropharmacology. 2020;181: 108333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gunduz‐Bruce H, Silber C, Kaul I, et al. Trial of SAGE‐217 in patients with major depressive disorder. N Engl J Med. 2019;381(10):903‐911. [DOI] [PubMed] [Google Scholar]
- 85. Deligiannidis KM, Meltzer‐Brody S, Gunduz‐Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psych. 2021. 10.1001/jamapsychiatry.2021.1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. ClinicalTrials.gov . A Study to Evaluate the Efficacy and Safety of SAGE‐217 in Participants With Severe Postpartum Depression (PPD). https://ClinicalTrials.gov/ct2/show/NCT04442503. Published 2021. Accessed April 12, 2021
- 87. Dichtel LE, Nyer M, Dording C, et al. Effects of open‐label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: a pilot study. J Clin Psychiatry. 2020;81(4):19m12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nohria V, Giller E. Ganaxolone. Neurotherapeutics. 2007;4(1):102‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ligsay A, Van Dijck A, Nguyen DV, et al. A randomized double‐blind, placebo‐controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J Neurodev Disord. 2017;9(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hernandez GD, Solinsky CM, Mack WJ, et al. Safety, tolerability, and pharmacokinetics of allopregnanolone as a regenerative therapeutic for Alzheimer's disease: a single and multiple ascending dose phase 1b/2a clinical trial. Alzheimers Dement (N Y). 2020;6(1):e12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. ClinicalTrials.gov . Allopregnanolone Regenerative Therapeutic for Mild Alzheimer's Disease (REGEN‐BRAIN©). https://ClinicalTrials.gov/ct2/show/NCT04838301. Published 2021. Accessed June 7, 2021.
- 92. ClinicalTrials.gov . Allopregnanolone Regenerative Therapeutic for Early Alzheimer's Disease: Intramuscular Study (Allo‐IM). https://ClinicalTrials.gov/ct2/show/NCT03748303. Published 2021. Accessed 7 June 2021
- 93. Carver CM, Reddy DS. Neurosteroid interactions with synaptic and extrasynaptic GABAA receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology. 2013;230(2):151‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]