

Summary
Increasing evidence points to a relation between increased glucocorticoid (GC) exposure and weight gain. In support, long‐term cortisol measurements using hair analysis revealed that many individuals with obesity appear to have cortisol values in the high physiological range. The mechanisms behind this relationship need to be determined in order to develop targeted therapy to reach sustainable weight loss in these subgroups.
The effect of GCs is not only determined by the plasma concentration of GCs but also by individual differences in GC sensitivity and the target tissue, which can be analyzed by functional GC assays. GC sensitivity is influenced by multiple genetic and acquired (e.g., disease‐related) factors, including intracellular GC availability, hormone binding affinity, and expression levels of the GC receptors and their isoforms, as well as factors involved in the modulation of gene transcription. Interindividual differences in GC sensitivity also play a role in the response to exogenous GCs, with respect to both therapeutic and adverse effects.
Accordingly, in this review, we summarize current knowledge on mechanisms that influence GC sensitivity and their relationships with obesity and discuss personalized treatment options targeting the GC receptor.
Keywords: cortisol, glucocorticoid sensitivity, glucocorticoids, obesity
1. INTRODUCTION
Glucocorticoids (GCs) are steroid hormones and essential for life. They are involved in reproduction, metabolism, growth, coagulation, bone turnover, stress, cognition, water and electrolyte homeostasis, and inflammatory and immune responses. 1 , 2 Synthetic derivatives of GCs are widely used in clinical practice because of their immunosuppressive and anti‐inflammatory properties, for example, in the treatment of inflammatory diseases, autoimmune disorders, and hematological cancers. 3
Dysregulation of GC production results in distinct clinical disease phenotypes, with features resembling Cushing's syndrome (characterized by GC excess) or adrenal insufficiency (characterized by GC deficiency). 4 GCs follow secretion patterns showing circadian and ultradian rhythms and are also induced in response to inflammation and other stressors, be it emotional or physical. 4 However, if the stressor exceeds a temporal threshold or certain severity, GC secretion can become, in part, maladaptive, leading to adverse effects, such as anxiety and cognitive dysfunction, mood alteration, immune suppression, and insulin resistance associated with central obesity. 5 , 6 Chronically increased cortisol levels, exemplified by Cushing's, are consistently associated with weight gain.
The metabolic effects of GCs are many and involve many organs and tissues. For example, elevated cortisol levels can modulate adipose tissue functioning. 7 GCs seem to have tissue specific effects, whereby high levels promote preferential expansion of unfavorable visceral adipose depots while impairing favorable brown adipose tissue function and promote a “brown” to “white” phenotypic conversion. 7 , 8 Similarly, studies in rodent models of Cushing's syndrome have demonstrated that systemic high levels of endogenous corticosterone lead to white adipose tissue growth, reduced insulin tolerance, and hyperglycemia. 9 This can eventually stimulate development of (components of the) metabolic syndrome (MetS), including abdominal obesity, hypertension, dyslipidemia, and insulin resistance. 8 , 10 The correspondence of MetS to the signs and symptoms of the Cushing's syndrome suggests that GC signaling also plays a role in obesity in general. 11 , 12 , 13 In this respect, it is interesting that long‐term GC measurements in scalp hair revealed that around half of the individuals with obesity of our Dutch patient cohort appear in the high physiological range, whereas the other half is in the normal physiological range. 8 , 14
However, the ultimate response to GCs is not only determined by the serum/blood concentration of GCs but also by individual differences in tissue‐specific GC sensitivity, influenced by genetic and acquired (e.g., disease‐related) factors. 15 , 16 GCs act mainly by modulating gene expression via transactivation and transrepression mechanisms. The beneficial or harmful effects of GCs are dependent on the specific cellular signaling pathways that are activated. 17 Many of the molecular mechanisms underlying the interindividual variation in GC sensitivity in different tissues/processes are still unknown. Insights in such mechanisms might offer personalized therapy and diagnostic options in patients with obesity.
In this article, we will summarize the current knowledge, primarily focusing on human studies, on mechanisms behind interindividual differences in GC sensitivity in relation to obesity. The review includes differences in GC receptor‐mediated signaling, association of genetic polymorphisms and mutations and assays to measure GC sensitivity. Gaining insight into the signaling mechanisms is essential to understand the contribution of subtle increases in endogenous GCs and/or exogenous GC use to increased cardiometabolic risks in obesity. In addition, it may also help in understanding how to improve the balance between the beneficial and harmful effects of exogenous GCs, as many patients rely on such treatments. Altered sensitivity to GCs may exacerbate metabolic sequelae in vulnerable individuals, such as those with obesity, possibly necessitating personalized dosing strategies. 15
2. ENDOGENOUS GC MECHANISMS OF ACTION
The production of endogenous GCs is under control of the hypothalamus–pituitary–adrenal (HPA) axis. The hypothalamic corticotropin‐releasing hormone (CRH) stimulates the pituitary gland to release adrenocorticotropic hormone (ACTH). Thereafter, ACTH activates the cAMP (adenosine 3′5′‐cyclic monophosphate) pathway in cells within the zona fasciculata of the adrenal cortex, which leads to production and secretion of GCs. 18 , 19 The most important GC in humans is cortisol. An increase in levels of cortisol inhibits the release of CRH and ACTH via negative feedback mechanisms at the level of the hypothalamic paraventricular nucleus (PVN) (inhibition of CRH) and the pituitary gland (inhibition of ACTH). 15 , 20
Cortisol is highly lipophilic and therefore mostly bound to corticosteroid‐binding globulin (CBG) when transported via blood. Approximately 90% of circulating cortisol is bound to CBG, 6% is bound with a low affinity to albumin, and only 4% is free and biologically active. 21 Cortisone is the biologically inactive form of cortisol and can act as a reservoir of inactive precursor steroid. The ratio of cortisone/cortisol is tissue specific and is regulated by expression levels and activity of the 11β‐hydroxysteroid dehydrogenase (11β‐HSD) enzymes. 11β‐HSD type 1 (11β‐HSD1) can (locally) convert cortisone to active cortisol, whereas 11β‐HSD2 can inactivate cortisol and locally prevent GC action in specific tissues. 21 , 22 11β‐HSD1 is mainly expressed in metabolic target tissues including liver (highest expression and affecting systemic cortisone/cortisol ratios), adipose tissue, and central nervous system, whereas 11β‐HSD2 is expressed mainly in epithelial tissues such as the kidney (highest expression), colon, salivary, and sweat glands. 23 , 24 , 25
2.1. The glucocorticoid receptor
Unbound GCs can diffuse across the plasma membrane of many types of cells and exert their effect upon binding to the glucocorticoid receptor (GR) or mineralocorticoid receptor (MR). 3 , 18 , 22 The GR and MR are expressed in many tissues, where they can interact with each other in synergy or in opposition. 26 The affinity of cortisol for the GR is about 10‐fold lower compared with the MR; hence, GR is only partially occupied when GC levels are low. 27 , 28 The MR is occupied substantially even under basal conditions (late evening), whereas at stress levels/or the circadian peak of GC secretion (morning), the GR is gradually activated. 26 , 29 , 30 Cortisol and aldosterone have the same binding affinity for MR. However, under normal conditions, 11β‐HSD2 can inactivate cortisol, which allows aldosterone to bind the MR in target cells, for example, epithelial cells. 22 The GR is expressed in nearly all organs and cells in the body, including the pituitary gland and PVN. Accordingly, the GR has a central role in negative feedback regulation of GC production, via action at the (core of the) HPA axis. 4 Because both elevated cortisol levels and synthetic GCs affect predominantly the GR, below we will focus on mechanisms and processes that relate to GR, but very similar principles apply to MR.
The human GR (hGR), encoded by the NR3C1 gene located on chromosome 5q31–32 and consisting of nine exons, belongs to the nuclear hormone receptor subfamily 3 (Figure 1). 31 , 32 It consists of an N‐terminal transactivation domain (NTD), a central DNA‐binding domain (DBD), and a C‐terminal ligand‐binding domain (LBD). 2 , 33 The NTD is involved in the transcriptional activation of GR target genes; the DBD recognizes and binds DNA target sequences, also known as GC response elements (GREs), and the LBD is responsible for the binding of GCs and transcriptional activity. The DBD and LBD are separated by the hinge region, of which the function is still unclear, but which is subject to posttranslational modification (suggesting a role beyond linking the other domains). 2 , 32
FIGURE 1.
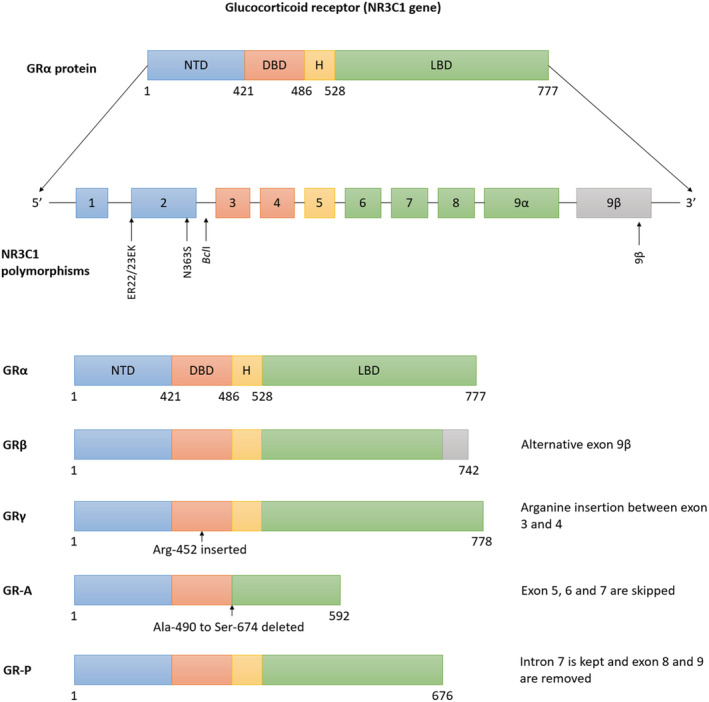
Glucocorticoid receptor (GR) isoforms and NR3C1 polymorphisms. GRα is known as the classical GR, consists of nine exons, and has three main domains, N‐terminal transactivation domain (Exon 2) (NTD), a central DNA‐binding domain (Exons 3 and 4) (DBD), and a C‐terminal ligand‐binding domain (Exons 5–9) (LBD). Alternative splicing, translation initiation mechanisms, and posttranslational modifications result in multiple isoforms: GRβ, GRγ, GR‐A, and GR‐P. The location of four NR3C1 polymorphisms, N363S and BclI, associated with glucocorticoid hypersensitivity, and exon 9β and ER22/23EK, associated with glucocorticoid hyposensitivity, is illustrated. DBD, central DNA‐binding domain; H, hinge region; LBD, C‐terminal ligand‐binding domain; NTD, N‐terminal transactivation domain
The best characterized signaling mode of the GR is the genomic pathway (Figure 2). The GR, in its quiescent state, is located in the cytoplasm bound to a chaperone complex. This complex consists of, among others, the heat shock protein 90 (hsp90), hsp70, and FK506 binding protein 5 (FKBP5). 34 On binding of GCs, the GR undergoes conformational changes, leading to its dissociation from the chaperone complex and translocation of the ligand‐receptor complex into the nucleus. Within the nucleus, the complex can bind to GREs and to other transcription factors and stimulate and repress expression of many different target genes. 2 , 32
FIGURE 2.
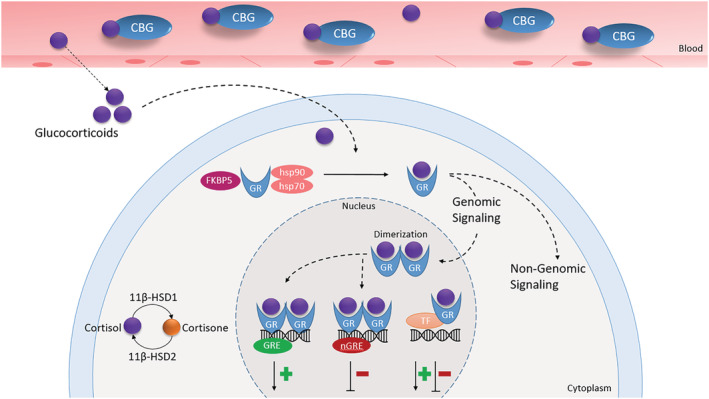
Schematic overview of glucocorticoid receptor (GR) signaling pathway. Glucocorticoids can bind to the GR and exert their effects by genomic (main pathway) and non‐genomic mechanisms. When inside the nucleus, the GR can cause either transactivation or transrepression of target genes. CBG, corticosteroid‐binding globulin; GRE, glucocorticoid response elements; hsp, heat shock protein; nGRE, negative glucocorticoid response elements; TF, transcription factors
Transactivation by the GR via GREs is thought to play a major role in most unfavorable metabolic effects of GCs. 35 In addition, the GR can cause transrepression of target genes by binding to other specific “negative GREs” (nGREs). 2 , 32 The accessibility of GREs and nGREs contributes to the tissue and cell specific effects of GCs. 4 On the other hand, the GR can also transrepress GC‐mediated reactions independent of nGREs by protein–protein interaction with transcription factors, for example, AP‐1 (activating protein‐1) and NF‐κB (nuclear factor‐κB). 36 GCs can also exert their effects via membrane GR, a non‐genomic signaling mechanism that is more rapid, but much less understood than the genomic signaling mode. 37
Both the therapeutic and adverse effects of GCs are mediated by genomic and non‐genomic mechanisms. 38 The GR can increase the expression of anti‐inflammatory proteins (transactivation) or decrease the production of pro‐inflammatory proteins (transrepression). 38 The interindividual and tissue‐specific responses after GR activation are partly due to different GRE accessibility, different repertoires of interacting proteins, and GR isoforms. 32
2.2. GR isoforms
The GR exists as multiple protein isoforms (Figure 1), all derived from the NR3C1 gene. 32 The multiple isoforms are formed by alternative splicing and translation initiation mechanisms and posttranslational modifications. 39 , 40 The levels of expression of the different GR isoforms vary among tissues. 39
Alternative splicing of the GR generates two main isoforms defined at the mRNA level, GRα and GRβ, which are similar through amino acid 1–727 but differ in their C‐termini. 32 The GRα is known as the classical GR and is mainly located in the cytoplasm in the absence of hormone. It binds GCs, translocates into the nucleus, and activates gene expression. The GRβ can be located either in the cytoplasm or nucleus and is a dominant negative inhibitor of GRα. 41 , 42 In addition to negatively regulating the actions of GRα, GRβ also exerts its own independent effects. 42 It has been reported that the ratio of GRα and GRβ expression in various cells is critical for GC sensitivity, in particular in inflammatory contexts. 43 Under normal conditions, the expression of GRα is much greater than the expression of GRβ in most cells and tissues, making cells sensitive for GCs. 42 However, it is hypothesized that an increase in expression of GRβ can lead to GC resistance in immune cells. 43
Other isoforms generated by alternative splicing are GR‐P, GRγ, and GR‐A. The GRγ is widely expressed and able to bind GCs and DNA, and it is often associated with GC resistance. The isoforms GR‐P and GR‐A miss large regions of the LBD and do not bind GCs. 34 Little is known about GR‐A, but GR‐P was reported to increase the activity of GRα. The interaction of GR‐P with GRα may therefore also play an important role in GC sensitivity. 44 Strikingly, alternative translation initiation starting sites in GRα mRNA produce an additional eight GR N‐terminal translational isoforms (GRα‐A, GRα‐B, GRα‐C1, GRα‐C2, GRα‐C3, GRα‐D1, GRα‐D2, and GRα‐D3) that may be present for all splice variants. 39 These translational isoforms are less well characterized, but also differ in the expression patterns, and are known to be involved in GC sensitivity. 34
3. GENETIC MUTATIONS AND POLYMORPHISMS IN ASSOCIATION WITH GC SENSITIVITY
3.1. GC sensitivity
Multiple factors influence individual GC effects at the tissue level, including intracellular GC availability (dependent on, e.g., 11β‐HSD activity and metabolic clearance rate), hormone binding affinity, expression levels of the GC receptors and isoforms and factors involved in the modulation of gene transcription. 41 , 45 , 46 The diversity of these mechanisms also causes interindividual variability in GC sensitivity to be tissue specific. 47
3.2. GR resistance
Generalized hyposensitivity or resistance to GCs is a rare, familial, or isolated condition with only a few dozen cases previously reported. 2 , 15 , 48 , 49 , 50 To compensate for the resistance to GCs, levels of circulating cortisol and ACTH concentrations are elevated. 51 , 52 Despite biochemical hypercortisolism, no classical phenotype of Cushing's syndrome is observed. 50 However, there is a large variation in clinical manifestations that can range from completely asymptomatic to hyperandrogenism, fatigue, hypertension, hypokalemic alkalosis, and/or (apparent) mineralocorticoid excess. 50 , 51 , 52 , 53
Generalized GC resistance is most often the result of mutations in the GR LBD or—in its milder forms—polymorphisms in the NR3C1 gene (discussed later in the article). 2 , 15 , 48
3.3. GR hypersensitivity
Similarly, GC hypersensitivity syndrome, also referred to as cortisol hyperreactive syndrome or normocortisolemic Cushing's syndrome, is also rare. It is characterized by the appearance of Cushingoid features and normal to low serum cortisol levels. 54 , 55 Cultured skin fibroblasts from the earliest reported patient showed hyperreactivity to GCs in vitro. 56 Another case showed increased numbers of GRs per cell (observed in peripheral lymphocytes) with normal binding affinity. 57 Interestingly, GC hypersensitivity seems to predominantly affect the peripheral tissues but not the hypothalamus and pituitary glands; hence, the negative feedback loop is largely intact, and suppression of the HPA axis is disproportional to the clinical signs. 54 These cases illustrate the need for clinical awareness of GC hypersensitivity in patients suspected of Cushing's syndrome, when cortisol levels are low and exogenous GC use is excluded.
GR gain‐of‐function mutations in the NR3C1 gene are observed in congenital GC hypersensitivity. To our knowledge, only few gain‐of‐function mutations have been described. 41 , 58 A heterozygous D401H mutation detected in Exon 2 of the GR gene showed increased transcriptional activity, normal ligand‐binding affinity, and normal nuclear translocation. The D401H mutation is associated with an increased risk of obesity, hypertension, and other characteristics of the MetS. 58
3.4. Polymorphisms associated with GC sensitivity
Polymorphisms in the GR gene are associated with differential GC sensitivity, altered metabolic profiles, and differences in body composition, although causality has not been unambiguously demonstrated (Figure 1) (Table 1). 48 , 59 , 60 , 61 , 62 , 63 Two well‐characterized polymorphisms in GRα that are associated with hypersensitivity to GCs are N363S and BclI, with allele frequencies in the general (Caucasian) population of 4.5% and 38% (6% and 47% in heterozygous carriers), respectively. 15 , 48 Both polymorphisms are associated with an adverse lipid profile, abdominal obesity, hyperinsulinemia, and hypertension. 59 , 60 , 64
TABLE 1.
Polymorphisms in the NR3C1 gene associated with glucocorticoid (GC) sensitivity
| Polymorphism | GC sensitivity | Effects on body composition and metabolic profile | References |
|---|---|---|---|
| N363S | Increased | Increased LDL cholesterol levels in elderly, higher BMI, and hypertension | 64 , 65 , 67 , 68 |
| BclI | Increased | Increased insulin resistance, BMI, abdominal obesity, total body fat, and hypertension | 64 , 71 , 72 |
| 9β | Decreased | Reduction in central obesity and a beneficial lipid profile | 76 |
| ER22/23EK | Decreased | Lower fasting insulin levels and LDL cholesterol concentrations. Sex‐specific beneficial body compositional changes | 77 , 79 |
Abbreviations: BMI, body mass index; BMI, body mass index; LDL, low‐density lipoprotein; LDL, low‐density lipoprotein.
The N363S polymorphism (rs56149945), located in Exon 2, causes an increase in transactivation capacity of the GR, which is thought to be responsible for most metabolic effects of GCs, but has no apparent effect on the transrepression capacity. 61 , 65 , 66 The N363S polymorphism is associated with an increase in low‐density lipoprotein (LDL) cholesterol levels in elderly and a higher body mass index (BMI). 67 , 68 It is speculated that heterozygous carriers of the N363S polymorphism who develop obesity store fat more effectively via activation of lipogenesis. 64 , 69
The BclI polymorphism (rs41423247), located in Intron 2, influences GR expression and modulates the GR transcriptional activity of genes responsible for glucose and insulin homeostasis. 70 Homozygous carriers of the BclI polymorphism have, compared with noncarriers, more total body fat and insulin resistance, as well as higher blood pressure. 71 , 72 , 73 Carriers of both the BclI and N363S polymorphisms had higher blood pressure and higher LDL cholesterol compared with carriers of the N363S polymorphism alone. 64
In addition, several polymorphisms in the GR gene are correlated with a relative GC resistance. Polymorphisms related to GC hyposensitivity are the 9β and the ER22/23EK variants, with allele frequencies in the general (Caucasian) population of 16.5% and 2.5% (28% and 5% in heterozygous carriers), respectively. 15
The polymorphism in exon 9β (rs6198) of the GR gene increases stability of the mRNA such as the GRβ mRNA. 74 Because the GRβ is a dominant negative inhibitor of GRα, the 9β polymorphism may contribute to GC hyposensitivity as has been demonstrated in vitro. 41 , 74 In the Caucasian population, the rs6198 polymorphism is associated with a more favorable phenotype including less central obesity in women and a better lipid profile in men, although this may be different in other ethnic groups. However, a haplotype comprising this polymorphism was found to be associated with a more pro‐inflammatory system and cardiovascular disease. 75 , 76
The reduction in sensitivity to GCs in carriers of the ER22/23EK (rs6189 and rs6190) polymorphism is reflected by a smaller decrease in cortisol concentrations and higher serum concentrations of cortisol after a 1 mg dexamethasone (DEX) suppression test. 77 In contrast to the N363S polymorphism, the ER22/23EK polymorphism causes a reduction in transactivation capacity of the GR, but has no effect on transrepression. 65 Interestingly, Russcher et al. showed in vitro that the ER22/23EK polymorphism is associated with an increase in the expression of the translational isoform GRα‐A at the expense of GRα‐B, which can lead to an overall decrease in transcriptional activity. 65 , 78 This seems a plausible mechanism to explain the association between the ER22/23EK polymorphism and GC resistance. In vivo studies have shown that heterozygous carriers of the ER22/23EK polymorphism have lower fasting and non‐fasting insulin, slightly lower fasting glucose levels, and lower total and LDL cholesterol concentrations compared with noncarriers. These findings suggests that ER22/23EK carriers are more resistant to the effects of GCs and have better metabolic profiles. 77 In addition, beneficial body composition was observed in ER22/23EK carriers around young‐adult age compared with noncarriers. Male carriers were taller and had more muscle strength and female carriers were likely to have smaller waist and hip circumferences. 79
The aforementioned polymorphisms N363S, BclI, 9β, and ER22/23EK seem to influence GC sensitivity and have differential effects on the metabolic profile (Table 1). A profile of the NR3C1 gene polymorphisms may provide an indication of risks for diseases related to obesity and adverse cardiometabolic profiles, leading to more targeted therapy.
4. ASSAYS TO MEASURE GC SENSITIVITY
In addition to inter‐ and intraindividual differences, GC sensitivity may also be tissue specific. 80 GC sensitivity can be measured in vivo and in vitro. It is therefore important to select the appropriate test, depending on the clinical need.
4.1. In vivo evaluation of GC sensitivity
GC sensitivity of the GR can be measured in vivo by oral or intravenous DEX suppression test (DST). This test assesses the negative feedback action mediated by DEX, a synthetic GC, via the GR as indicated by the suppression of HPA axis activity. 81 Insufficient suppression of cortisol after DST (in the absence of signs of Cushing's syndrome) suggests GC resistance. A modification of this test using very low dose DEX (0.25 mg) is often used to assess hypersensitivity. 53
The DST is limited to the indication of GC sensitivity that is either genetically determined or specific at the level of the pituitary gland and—for the higher dose—the hypothalamus, whereas local (acquired) GC resistance may not be sufficiently represented. It has been amply demonstrated that GC sensitivity results may vary between tissues as observed in the reported differences between in vivo and in vitro measurements in the same individual. 15 , 80
4.2. In vitro evaluation of GC sensitivity
Differences in GR activity can result from a decrease in GR affinity for GCs or number of GRs, impaired translocation of the GR into the nucleus, receptor thermolability, or a change in protein–protein interaction with coactivators. 15 , 82 , 83 , 84 The relative expression of the different GR isoforms can be assessed in various tissues or peripheral blood mononuclear cells (PBMCs) by quantitative reverse transcription polymerase chain reaction (qRT‐PCR). 42 , 82 It has been suggested that the number of GR in PBMCs represents individual responsiveness to GCs in vivo and in vitro. 85
Several in vitro functional assays have been developed to assess GR sensitivity to GCs in different target tissues such as in PBMCs. The DEX Inhibition of Lymphocyte Proliferation Assay (DILPA) assessed the extent of inhibition of phytohemagglutinin (PHA) induced T‐lymphocyte proliferation by DEX. 86 , 87 These assays have been superseded by nonradioactive modifications such as the BLISS (BrdU Lymphocyte Incorporation Steroid Sensitivity) assay, performed in PBMCs. 88 Despite showing promise in the identification of GC nonresponders, this assay needs further validation of its efficacy.
Another in vitro bioassay measured the GR‐mediated transactivation or transrepression of GC‐responsive genes, such as GC‐induced leucine zipper (GILZ) or interleukin (IL)‐2, respectively, in PBMCs. 82 , 89 , 90 , 91 This assay demonstrated an association between in vitro GC sensitivity and clinical GC therapy outcome in rheumatoid arthritis. 90 However, other studies using this assay have reported a lack of correlation between GILZ and IL‐2 expression and also with the outcome of the DST. 89 In vivo, GCs can inhibit the expression of pro‐inflammatory mediators such as IL‐2. 15 , 38 , 92 On the other hand, transactivation of regulatory molecules such as GILZ has also been demonstrated to have beneficial effects by indirectly repressing the expression of cytokines. 93 , 94 This suggests that transactivation, transrepression, and regulation of the HPA axis are complex, partially independent, processes. Nevertheless, these bioassays might be useful in developing prediction models to optimize the type and dosage of GCs to balance their therapeutic and adverse effects in patients.
Expression of a single or few genes does not fully recapitulate the complexity of the molecular response to physiological triggers, such as cortisol. Other studies have performed transcriptome analysis of DEX‐treated PBMCs to capture the dynamic transcriptional response to GR activation. 95 RNA sequencing in cultured lung carcinoma cells identified several new and potentially important genes (e.g., PER1, ZFP36, and BIRC1) that are responsive to GCs. Importantly, dose and time after DEX treatment were crucial determinants of GR‐mediated transcriptional changes. 95 , 96 Thus, whereas transcriptomic analysis of DEX‐stimulated PBMCs may identify key genes involved in altered GC signaling, the results emphasize the importance of the dose and time after treatment underscoring the complexity of the effects induced by GC.
5. INDIVIDUAL DIFFERENCES IN GC SENSITIVITY AND THEIR RELATIONSHIPS TO BODY WEIGHT AND METABOLIC FEATURES
The associations between increased fasting plasma cortisol levels and high blood pressure and insulin resistance and lipid levels, including increased triglycerides, LDL, and high‐density lipoprotein (HDL) cholesterol levels have been well described. 13 , 15 However, the relation between cortisol levels as measured in blood, saliva and urine, and obesity is inconsistent. 14 , 97 , 98 This can be attributed to the circadian secretion pattern of cortisol as well as the variation caused by circumstances such as acute stress. 99 In contrast, the correlation between cortisol levels measured in hair (that are assumed to represent long‐term exposure to cortisol) and BMI and waist–hip ratio is demonstrated to be more robust across different populations. 14 , 98 , 100 , 101 , 102 , 103 An increment of 9.8% in hair cortisol level was associated with 2.5 kg/m2 higher BMI, cross‐sectionally. 100 , 103 In addition, elevated hair cortisol levels were also related to a higher prevalence of MetS. 12 We previously reported that about half of the individuals with obesity have an elevated long‐term cortisol level compared with individuals with overweight or normal weight. 8 , 14 , 104 The elevated cortisol, at systemic (e.g., in Cushing's syndrome) or local levels (e.g., overactivity of 11β‐HSD1), may result in preferential expansion of visceral fat depots. 7 , 105 Furthermore, previously described GR gene variations might enhance sensitivity of individuals for the biological effects of cortisol increasing their propensity to weight gain and obesity. Moreover, poor feedback regulation of the HPA axis activity is associated with obesity. 106 On the other hand, obesity itself can also lead to increased chronic stress. 107 Social stigma related to obesity is associated with increased stress and higher long‐term cortisol levels, which might contribute to a vicious circle. 108 This effect may be exacerbated by the use of exogenous GCs prescribed for comorbidities related to obesity, such as arthrosis and asthma. In this context, we have previously reported significantly higher use of medication containing corticosteroids in individuals with obesity. 107 , 109 Paradoxically, half of the individuals with obesity in our previously described study had normal long‐term cortisol levels. Although this dichotomy in cortisol levels in obesity is under investigation, the strong association of elevated cortisol with metabolic dysfunction in human and animal studies suggests that at least a proportion of the individuals may benefit from cortisol‐reducing therapies.
6. EXOGENOUS SYNTHETIC GC USE AND OBESITY
Exogenous GCs are widely used potent anti‐inflammatory drugs with multiple indications and many administration forms. 110 , 111 Increased levels of endogenous cortisol and chronic exposure to certain exogenous GCs have more or less similar biological effects, because exogenous corticosteroids also bind to the GR with varying affinity. However, in contrast to endogenous GCs, exogenous GCs (except hydrocortisone and prednisolone) are not bound to CBG and are not metabolized by 11β‐HSD2. 112
Side effects of GC treatment are common: abdominal weight gain, metabolic side effects, diabetes mellitus, neuropsychiatric (e.g., depression), musculoskeletal, gastrointestinal, cardiovascular, dermal, ocular, or immunological in nature. 111 , 113 , 114 Therefore, it is not surprising that associations have been found between obesity and GC use. 107 In our large study comprising 140,879 adult participants from the Lifelines Cohort from the northern Netherlands, we found that use of locally applied GCs, particularly inhaled types, and/or systemic GCs was associated with higher likelihood of having MetS, higher BMI, and other adverse cardiometabolic traits, especially among women. 111 In addition, we found in the same cohort associations between, in particular, inhaled and systemic corticosteroids and reduced executive cognitive functioning and a higher likelihood of mood and anxiety disorders. 115 Nevertheless, unlike systemic use and prolonged use of higher dosages of locally applied GCs, where a causal role in the manifestation of adverse effects have been demonstrated, such a relation is yet to be shown for local administration of GCs.
Interestingly, long‐term cortisone levels measured in scalp hair were suppressed in users of inhaled, nasal, or topical corticosteroids, which suggests mild adrenal suppression due to exogenous GCs. 116 This warrants caution with the use of exogenous GCs, in particular also in individuals with increased GR sensitivity where it may lead to substantial weight gain and other adverse effects. 107 , 109
6.1. Innovative strategies for exogenous synthetic GC use
In the past, novel synthetic derivatives of GCs have been developed that distinguish between GR signaling modes. These compounds have been termed “dissociated ligands,” selective GR modulators, or more recently selective GR agonists (SEGRAs). 117 , 118 , 119 , 120 , 121 These compounds are currently being investigated for improvement in therapeutic efficacy, that is, anti‐inflammatory effects coupled with minimal adverse side effects. Whereas the side effects of GCs are thought to be mainly due to transactivation of genes, their desirable therapeutic effects have been attributed to transrepression. 17 The original notion has been to favor transrepression over transactivation, but current insights suggest that this is likely overly simplistic. The role of transactivation in suppressing inflammation is increasingly evident from animal models. 122 Transactivation of anti‐inflammatory genes may also be necessary for full anti‐inflammatory efficacy. 119 This may change the view on developing new GC therapies with reduced side effects.
The adverse side effects from prolonged and/or high dosage treatment with GCs are not only caused by the broad pharmacological activity but also by the variation in biodistribution. In order to get more insight into this variation, there is a clinical need for biomarkers. To balance efficacy versus toxicity of GC treatment, liposomes are now being studied as targeted drug delivery systems to enhance the biodistribution and target site accumulation of GCs. 123 , 124 Also, coadministration of other drugs may be a way to prevent side effects: A promising recent study showed that metformin administration can improve some of the metabolic profile and clinical outcomes for GC‐treated patients with inflammatory disease. 125
7. CONCLUDING REMARKS
The net effect of endogenous and exogenous GCs on various aspects of physiology is determined by individual differences in GC sensitivity brought about by genetic and acquired (e.g., disease‐related) factors including intracellular GC availability, hormone binding affinity, expression levels of the GR and isoforms, factors involved in the modulation of gene transcription, 11β‐HSD activity, and metabolic clearance rate. 15 , 16 , 41 , 45 , 46
The complex relation between GCs and obesity is most likely bidirectional. However, the central question that is still under investigation is to what extent an increased sensitivity to GCs leads to (abdominal) obesity or vice versa whether obesity leads to a self‐sustaining vicious circle characterized by high cortisol levels leading to fat redistribution and associated metabolic sequelae. It would be interesting to assess the relation of GC sensitivity with long‐term cortisol levels measured in scalp hair in the context of obesity. Furthermore, transcriptome analysis of patient tissues, to capture the dynamic transcriptional response to GR activation, might shed further light on the mechanisms of GC sensitivity in obesity and help to design improved personalized treatment options.
CONFLICTS OF INTEREST
All authors declare that they have no conflict of interest.
ACKNOWLEDGMENTS
EFCvR is supported by a Vidi grant from the Netherlands Organisation for Scientific Research (NWO) (grant number: 91716453). EFCvR and BvdV are also funded by the Elisabeth Foundation.
Lengton R, Iyer AM, van der Valk ES, et al. Variation in glucocorticoid sensitivity and the relation with obesity. Obesity Reviews. 2022;23(3):e13401. doi: 10.1111/obr.13401
Funding information Elisabeth Foundation; Netherlands Organisation for Scientific Research, Grant/Award Number: 91716453
REFERENCES
- 1. Nicolaides NC, Kyratzi E, Lamprokostopoulou A, Chrousos GP, Charmandari E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation. 2015;22(1–2):6‐19. [DOI] [PubMed] [Google Scholar]
- 2. Vitellius G, Lombes M. GENETICS IN ENDOCRINOLOGY: glucocorticoid resistance syndrome. Eur J Endocrinol. 2020;182(2):R15‐R27. [DOI] [PubMed] [Google Scholar]
- 3. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132(5):1033‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. 2017;17(4):233‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papadopoulou A, Siamatras T, Delgado‐Morales R, et al. Acute and chronic stress differentially regulate cyclin‐dependent kinase 5 in mouse brain: implications to glucocorticoid actions and major depression. Transl Psychiatry. 2015;5(6):e578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374‐381. [DOI] [PubMed] [Google Scholar]
- 7. Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842(3):473‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Rossum EFC. Obesity and cortisol: new perspectives on an old theme. Obesity (Silver Spring). 2017;25(3):500‐501. 10.1002/oby.21774 [DOI] [PubMed] [Google Scholar]
- 9. Dumontet T, Sahut‐Barnola I, Septier A, et al. PKA signaling drives reticularis differentiation and sexually dimorphic adrenal cortex renewal. JCI Insight. 2018;3(2):e98394. 10.1172/jci.insight.98394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Constantinopoulos P, Michalaki M, Kottorou A, et al. Cortisol in tissue and systemic level as a contributing factor to the development of metabolic syndrome in severely obese patients. Eur J Endocrinol. 2015;172(1):69‐78. [DOI] [PubMed] [Google Scholar]
- 11. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stalder T, Kirschbaum C, Alexander N, et al. Cortisol in hair and the metabolic syndrome. J Clin Endocrinol Metab. 2013;98(6):2573‐2580. 10.1210/jc.2013-1056 [DOI] [PubMed] [Google Scholar]
- 13. Walker BR. Cortisol—cause and cure for metabolic syndrome? Diabet Med. 2006;23(12):1281‐1288. 10.1111/j.1464-5491.2006.01998.x [DOI] [PubMed] [Google Scholar]
- 14. Wester VL, Staufenbiel SM, Veldhorst MAB, et al. Long‐term cortisol levels measured in scalp hair of obese patients. Obesity. 2014;22(9):1956‐1958. [DOI] [PubMed] [Google Scholar]
- 15. Quax RA, Manenschijn L, Koper JW, et al. Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol. 2013;9(11):670‐686. [DOI] [PubMed] [Google Scholar]
- 16. Quax RA, van Laar JA, van Heerebeek R, et al. Glucocorticoid sensitivity in Behcet's disease. Endocr Connect. 2012;1(2):103‐111. 10.1530/EC-12-0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schacke H, Schottelius A, Docke WD, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci U S A. 2004;101(1):227‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol. 2009;300(1–2):7‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gomez MT, Magiakou MA, Mastorakos G, Chrousos GP. The pituitary corticotroph is not the rate limiting step in the postoperative recovery of the hypothalamic‐pituitary‐adrenal axis in patients with Cushing syndrome. J Clin Endocrinol Metab. 1993;77(1):173‐177. [DOI] [PubMed] [Google Scholar]
- 20. Gjerstad JK, Lightman SL, Spiga F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress. 2018;21(5):403‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gathercole LL, Lavery GG, Morgan SA, et al. 11beta‐Hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. Endocr Rev. 2013;34(4):525‐555. 10.1210/er.2012-1050 [DOI] [PubMed] [Google Scholar]
- 22. Gomez‐Sanchez E, Gomez‐Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol. 2014;4(3):965‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garbrecht MR, Klein JM, McCarthy TA, Schmidt TJ, Krozowski ZS, Snyder JM. 11‐Beta hydroxysteroid dehydrogenase type 2 in human adult and fetal lung and its regulation by sex steroids. Pediatr Res. 2007;62(1):26‐31. 10.1203/PDR.0b013e3180676cf3 [DOI] [PubMed] [Google Scholar]
- 24. Paulsen SK, Pedersen SB, Fisker S, Richelsen B. 11Beta‐HSD type 1 expression in human adipose tissue: impact of gender, obesity, and fat localization. Obesity (Silver Spring). 2007;15(8):1954‐1960. [DOI] [PubMed] [Google Scholar]
- 25. Yakirevich E, Morris DJ, Tavares R, et al. Mineralocorticoid receptor and 11beta‐hydroxysteroid dehydrogenase type II expression in renal cell neoplasms: a tissue microarray and quantitative RT‐PCR study. Am J Surg Pathol. 2008;32(6):874‐883. 10.1097/PAS.0b013e31815f2362 [DOI] [PubMed] [Google Scholar]
- 26. Spijker AT, van Rossum EFC. Glucocorticoid sensitivity in mood disorders. Neuroendocrinology. 2012;95(3):179‐186. [DOI] [PubMed] [Google Scholar]
- 27. Le Billan F, Amazit L, Bleakley K, et al. Corticosteroid receptors adopt distinct cyclical transcriptional signatures. FASEB J. 2018;32(10):5626‐5639. 10.1096/fj.201800391RR [DOI] [PubMed] [Google Scholar]
- 28. Joëls M, Sarabdjitsingh RA, den Boon FS, Karst H. Rapid and slow effects of corticosteroid hormones on hippocampal activity. In: Fink G, ed. Stress: Neuroendocrinology and Neurobiology. Handbook of Stress Series. Vol. 2. Academic Press; 2017:327‐341. 10.1016/B978-0-12-802175-0.00033-4 [DOI] [Google Scholar]
- 29. Finsterwald C, Alberini CM. Stress and glucocorticoid receptor‐dependent mechanisms in long‐term memory: from adaptive responses to psychopathologies. Neurobiol Learn Mem. 2014;112:17‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE. The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition. Brain Cogn. 2007;65(3):209‐237. [DOI] [PubMed] [Google Scholar]
- 31. Yudt MR, Cidlowski JA. Molecular identification and characterization of a and b forms of the glucocorticoid receptor. Mol Endocrinol. 2001;15(7):1093‐1103. [DOI] [PubMed] [Google Scholar]
- 32. Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue‐specific actions of glucocorticoids. J Biolumin Chemilumin. 2011;286(5):3177‐3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol. 2005;94(5):383‐394. [DOI] [PubMed] [Google Scholar]
- 34. Liu B, Zhang TN, Knight JK, Goodwin JE. The glucocorticoid receptor in cardiovascular health and disease. Cell. 2019;8(10):1227. 10.3390/cells8101227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wester VL, Lamberts SW, van Rossum EFC. Advances in the assessment of cortisol exposure and sensitivity. Curr Opin Endocrinol Diabetes Obes. 2014;21(4):306‐311. 10.1097/MED.0000000000000077 [DOI] [PubMed] [Google Scholar]
- 36. Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998;93(4):487‐490. [DOI] [PubMed] [Google Scholar]
- 37. Ayrout M, Simon V, Bernard V, et al. A novel non genomic glucocorticoid signaling mediated by a membrane palmitoylated glucocorticoid receptor cross talks with GnRH in gonadotrope cells. Sci Rep. 2017;7(1):1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4(10):525‐533. [DOI] [PubMed] [Google Scholar]
- 39. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N‐terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18(3):331‐342. [DOI] [PubMed] [Google Scholar]
- 40. Lu NZ, Cidlowski JA. The origin and functions of multiple human glucocorticoid receptor isoforms. Ann N Y Acad Sci. 2004;1024(1):102‐123. [DOI] [PubMed] [Google Scholar]
- 41. Santen RJ, Jewell CM, Yue W, et al. Glucocorticoid receptor mutations and hypersensitivity to endogenous and exogenous glucocorticoids. J Clin Endocrinol Metab. 2018;103(10):3630‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34(9):518‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lewis‐Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069(1):1‐9. 10.1196/annals.1351.001 [DOI] [PubMed] [Google Scholar]
- 44. de Lange P, Segeren CM, Koper JW, et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor‐mediated effects in transfected cells. Cancer Res. 2001;61(10):3937‐3941. [PubMed] [Google Scholar]
- 45. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17(3):245‐261. [DOI] [PubMed] [Google Scholar]
- 46. Melo MR, Faria CDC, Melo KC, Rebouças NA, Longui CA. Real‐time PCR quantitation of glucocorticoid receptor alpha isoform. BMC mol Biol. 2004;5(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ebrecht M, Buske‐Kirschbaum A, Hellhammer D, et al. Tissue specificity of glucocorticoid sensitivity in healthy adults. J Clin Endocrinol Metab. 2000;85(10):3733‐3739. [DOI] [PubMed] [Google Scholar]
- 48. Manenschijn L, van den Akker ELT, Lamberts SW, van Rossum EFC. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci. 2009;1179(1):179‐198. 10.1111/j.1749-6632.2009.05013.x [DOI] [PubMed] [Google Scholar]
- 49. Ruiz M, Lind U, Gafvels M, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf). 2001;55(3):363‐371. 10.1046/j.1365-2265.2001.01323.x [DOI] [PubMed] [Google Scholar]
- 50. Huizenga NA, de Lange P, Koper JW, et al. Five patients with biochemical and/or clinical generalized glucocorticoid resistance without alterations in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 2000;85(5):2076‐2081. [DOI] [PubMed] [Google Scholar]
- 51. Charmandari E, Kino T, Chrousos GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci. 2004;1024(1):168‐181. [DOI] [PubMed] [Google Scholar]
- 52. Vingerhoeds AC, Thijssen JH, Schwarz F. Spontaneous hypercortisolism without Cushing's syndrome. J Clin Endocrinol Metab. 1976;43(5):1128‐1133. 10.1210/jcem-43-5-1128 [DOI] [PubMed] [Google Scholar]
- 53. van Rossum EFC, Lamberts SW. Glucocorticoid resistance syndrome: a diagnostic and therapeutic approach. Best Pract Res Clin Endocrinol Metab. 2006;20(4):611‐626. 10.1016/j.beem.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 54. Ugwu E. Glucocorticoid hypersensitivity syndrome resulting from inhaled corticosteroid: a case report. Egypt J Intern Med. 2017;29(4):201‐203. 10.4103/ejim.ejim_49_17 [DOI] [Google Scholar]
- 55. Krysiak R, Okopien B. Glucocorticoid hypersensitivity syndrome‐‐a case report. West Indian Med J. 2012;61(8):844‐846. [PubMed] [Google Scholar]
- 56. Iida S, Nakamura Y, Fujii H, et al. A patient with hypocortisolism and Cushing's syndrome‐like manifestations: cortisol hyperreactive syndrome. J Clin Endocrinol Metab. 1990;70(3):729‐737. 10.1210/jcem-70-3-729 [DOI] [PubMed] [Google Scholar]
- 57. Newfield RS, Kalaitzoglou G, Licholai T, et al. Normocortisolemic Cushing's syndrome initially presenting with increased glucocorticoid receptor numbers. J Clin Endocrinol Metab. 2000;85(1):14‐21. 10.1210/jcem.85.1.6220 [DOI] [PubMed] [Google Scholar]
- 58. Charmandari E, Ichijo T, Jubiz W, et al. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR‐mediated gene expression. J Clin Endocrinol Metab. 2008;93(12):4963‐4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Rossum EFC, Lamberts SW. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59(1):333‐357. 10.1210/rp.59.1.333 [DOI] [PubMed] [Google Scholar]
- 60. Roerink SH, Wagenmakers MA, Smit JW, et al. Glucocorticoid receptor polymorphisms modulate cardiometabolic risk factors in patients in long‐term remission of Cushing's syndrome. Endocrine. 2016;53(1):63‐70. 10.1007/s12020-016-0883-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wester VL, Koper JW, van den Akker ELT, Franco OH, Stolk RP, van Rossum EFC. Glucocorticoid receptor haplotype and metabolic syndrome: the Lifelines cohort study. Eur J Endocrinol. 2016;175(6):645‐651. [DOI] [PubMed] [Google Scholar]
- 62. Koper JW, van Rossum EFC, van den Akker ELT. Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids. 2014;92:62‐73. 10.1016/j.steroids.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 63. Majer‐Lobodzinska A, Adamiec‐Mroczek J. Glucocorticoid receptor polymorphism in obesity and glucose homeostasis. Adv Clin Exp Med. 2017;26(1):143‐148. [DOI] [PubMed] [Google Scholar]
- 64. Di Blasio AM, van Rossum EFC, Maestrini S, et al. The relation between two polymorphisms in the glucocorticoid receptor gene and body mass index, blood pressure and cholesterol in obese patients. Clin Endocrinol (Oxf). 2003;59(1):68‐74. 10.1046/j.1365-2265.2003.01798.x [DOI] [PubMed] [Google Scholar]
- 65. Russcher H, Smit P, van den Akker ELT, et al. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid‐regulated gene expression. J Clin Endocrinol Metab. 2005;90(10):5804‐5810. 10.1210/jc.2005-0646 [DOI] [PubMed] [Google Scholar]
- 66. Charmandari E. Primary generalized glucocorticoid resistance and hypersensitivity. Horm Res Paediatr. 2011;76(3):145‐155. [DOI] [PubMed] [Google Scholar]
- 67. Kuningas M, Mooijaart SP, Slagboom PE, Westendorp RG, van Heemst D. Genetic variants in the glucocorticoid receptor gene (NR3C1) and cardiovascular disease risk. The Leiden 85‐plus study. Biogerontology. 2006;7(4):231‐238. 10.1007/s10522-006-9021-2 [DOI] [PubMed] [Google Scholar]
- 68. Marti A, Ochoa MC, Sanchez‐Villegas A, et al. Meta‐analysis on the effect of the N363S polymorphism of the glucocorticoid receptor gene (GRL) on human obesity. BMC Med Genet. 2006;7(1):50. 10.1186/1471-2350-7-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lin RC, Wang XL, Dalziel B, Caterson ID, Morris BJ. Association of obesity, but not diabetes or hypertension, with glucocorticoid receptor N363S variant. Obes Res. 2003;11(6):802‐808. 10.1038/oby.2003.111 [DOI] [PubMed] [Google Scholar]
- 70. Weaver JU, Hitman GA, Kopelman PG. An association between a Bc1I restriction fragment length polymorphism of the glucocorticoid receptor locus and hyperinsulinaemia in obese women. J Mol Endocrinol. 1992;9(3):295‐300. [DOI] [PubMed] [Google Scholar]
- 71. Geelen CC, van Greevenbroek MMJ, van Rossum EFC, et al. BclI glucocorticoid receptor polymorphism is associated with greater body fatness: the Hoorn and CODAM studies. J Clin Endocrinol Metab. 2013;98(3):E595‐E599. [DOI] [PubMed] [Google Scholar]
- 72. Rosmond R, Holm G. A 5‐year follow‐up study of 3 polymorphisms in the human glucocorticoid receptor gene in relation to obesity, hypertension, and diabetes. J Cardiometab Syndr. 2008;3(3):132‐135. [DOI] [PubMed] [Google Scholar]
- 73. van Moorsel D, van Greevenbroek MM, Schaper NC, et al. BclI glucocorticoid receptor polymorphism in relation to cardiovascular variables: the Hoorn and CODAM studies. Eur J Endocrinol. 2015;173(4):455‐464. 10.1530/EJE-15-0381 [DOI] [PubMed] [Google Scholar]
- 74. Derijk RH, Schaaf MJ, Turner G, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta‐isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28(11):2383‐2388. [PubMed] [Google Scholar]
- 75. van den Akker ELT, Koper JW, van Rossum EFC, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168(1):33‐39. [DOI] [PubMed] [Google Scholar]
- 76. Syed AA, Irving JA, Redfern CP, et al. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity (Silver Spring). 2006;14(5):759‐764. 10.1038/oby.2006.86 [DOI] [PubMed] [Google Scholar]
- 77. van Rossum EFC, Koper JW, Huizenga NA, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes. 2002;51(10):3128‐3134. 10.2337/diabetes.51.10.3128 [DOI] [PubMed] [Google Scholar]
- 78. Russcher H, van Rossum EFC, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. Increased expression of the glucocorticoid receptor‐a translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19(7):1687‐1696. 10.1210/me.2004-0467 [DOI] [PubMed] [Google Scholar]
- 79. van Rossum EFC, Voorhoeve PG, te Velde SJ, et al. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with a beneficial body composition and muscle strength in young adults. J Clin Endocrinol Metab. 2004;89(8):4004‐4009. 10.1210/jc.2003-031422 [DOI] [PubMed] [Google Scholar]
- 80. Chriguer RS, Elias LL, da Silva IM Jr. Glucocorticoid sensitivity in young healthy individuals: in vitro and in vivo studies. J Clin Endocrinol Metab. 2005;90(11):5978‐5984. 10.1210/jc.2005-0067 [DOI] [PubMed] [Google Scholar]
- 81. Dogra P, Vijayashankar NP. Dexamethasone suppression test. 2019; Accessed May 12, 2020. https://www.ncbi.nlm.nih.gov/books/NBK542317/ [PubMed]
- 82. Russcher H, Smit P, van Rossum EFC, et al. Strategies for the characterization of disorders in cortisol sensitivity. J Clin Endocrinol Metab. 2006;91(2):694‐701. 10.1210/jc.2005-2212 [DOI] [PubMed] [Google Scholar]
- 83. Koper JW, Stolk RP, de Lange P, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99(5):663‐668. [DOI] [PubMed] [Google Scholar]
- 84. Bronnegard M, Werner S, Gustafsson JA. Primary cortisol resistance associated with a thermolabile glucocorticoid receptor in a patient with fatigue as the only symptom. J Clin Invest. 1986;78(5):1270‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Grasso G, Lodi L, Lupo C, Muscettola M. Glucocorticoid receptors in human peripheral blood mononuclear cells in relation to age and to sport activity. Life Sci. 1997;61(3):301‐308. [DOI] [PubMed] [Google Scholar]
- 86. Creed TJ, Lee RW, Newcomb PV, di Mambro AJ, Raju M, Dayan CM. The effects of cytokines on suppression of lymphocyte proliferation by dexamethasone. J Immunol. 2009;183(1):164‐171. [DOI] [PubMed] [Google Scholar]
- 87. Hearing SD, Norman M, Probert CS, Haslam N, Dayan CM. Predicting therapeutic outcome in severe ulcerative colitis by measuring in vitro steroid sensitivity of proliferating peripheral blood lymphocytes. Gut. 1999;45(3):382‐388. 10.1136/gut.45.3.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Williams EL, Stimpson ML, Collins PL, et al. Development and validation of a novel bioassay to determine glucocorticoid sensitivity. Biomark Res. 2016;4(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Smit P, Russcher H, de Jong FH, Brinkmann AO, Lamberts SWJ, Koper JW. Differential regulation of synthetic glucocorticoids on gene expression levels of glucocorticoid‐induced leucine zipper and interleukin‐2. J Clin Endocrinol Metab. 2005;90(5):2994‐3000. 10.1210/jc.2004-2298 [DOI] [PubMed] [Google Scholar]
- 90. Quax RAM, Koper JW, de Jong PHP, et al. In vitro glucocorticoid sensitivity is associated with clinical glucocorticoid therapy outcome in rheumatoid arthritis. Arthritis Res Ther. 2012;14(4):R195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. van Oosten MJM, Dolhain RJEM, Koper JW, et al. Polymorphisms in the glucocorticoid receptor gene that modulate glucocorticoid sensitivity are associated with rheumatoid arthritis. Arthritis Res Ther. 2010;12(4):R159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. de Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor‐kappaB or activator protein‐1: molecular mechanisms for gene repression. Endocr Rev. 2003;24(4):488‐522. [DOI] [PubMed] [Google Scholar]
- 93. Ronchetti S, Migliorati G, Riccardi C. GILZ as a mediator of the anti‐inflammatory effects of glucocorticoids. Front Endocrinol (Lausanne). 2015;6:170. 10.3389/fendo.2015.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ayroldi E, Riccardi C. Glucocorticoid‐induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009;23(11):3649‐3658. [DOI] [PubMed] [Google Scholar]
- 95. Breen MS, Bierer LM, Daskalakis NP, et al. Differential transcriptional response following glucocorticoid activation in cultured blood immune cells: a novel approach to PTSD biomarker development. Transl Psychiatry. 2019;9(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Reddy TE, Pauli F, Sprouse RO, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19(12):2163‐2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Incollingo Rodriguez AC, Epel ES, White ML, Standen EC, Seckl JR, Tomiyama AJ. Hypothalamic‐pituitary‐adrenal axis dysregulation and cortisol activity in obesity: a systematic review. Psychoneuroendocrinology. 2015;62:301‐318. [DOI] [PubMed] [Google Scholar]
- 98. Savas M, Wester VL, de Rijke YB, et al. Hair glucocorticoids as a biomarker for endogenous Cushing's syndrome: validation in two independent cohorts. Neuroendocrinology. 2019;109(2):171‐178. [DOI] [PubMed] [Google Scholar]
- 99. Tsigos C, Chrousos GP. Hypothalamic‐pituitary‐adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53(4):865‐871. [DOI] [PubMed] [Google Scholar]
- 100. Stalder T, Steudte‐Schmiedgen S, Alexander N, et al. Stress‐related and basic determinants of hair cortisol in humans: a meta‐analysis. Psychoneuroendocrinology. 2017;77:261‐274. [DOI] [PubMed] [Google Scholar]
- 101. Manenschijn L, Koper JW, Lamberts SW, van Rossum EFC. Evaluation of a method to measure long term cortisol levels. Steroids. 2011;76(10–11):1032‐1036. 10.1016/j.steroids.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 102. Wester VL, van Rossum EFC. Clinical applications of cortisol measurements in hair. Eur J Endocrinol. 2015;173(4):M1‐M10. 10.1530/EJE-15-0313 [DOI] [PubMed] [Google Scholar]
- 103. Van der Valk ES, Abawi O, Mohensi M, et al. The relation between cortisol and anthropometric measurements throughout lifespan: a systematic review and meta‐analysis. J Endocr Soc. 2021;5(Supplement_1):A30‐A30. 10.1210/jendso/bvab048.057 [DOI] [Google Scholar]
- 104. Noppe G, van den Akker ELT, de Rijke YB, Koper JW, Jaddoe VW, van Rossum EFC. Long‐term glucocorticoid concentrations as a risk factor for childhood obesity and adverse body‐fat distribution. Int J Obes (Lond). 2016;40(10):1503‐1509. 10.1038/ijo.2016.113 [DOI] [PubMed] [Google Scholar]
- 105. Pickering RT, Lee MJ, Karastergiou K, Gower A, Fried SK. Depot dependent effects of dexamethasone on gene expression in human omental and abdominal subcutaneous adipose tissues from obese women. PLoS ONE. 2016;11(12):e0167337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rosmond R, Chagnon YC, Holm G, et al. A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic‐pituitary‐adrenal axis. Obes Res. 2000;8(3):211‐218. [DOI] [PubMed] [Google Scholar]
- 107. van der Valk ES, Savas M, van Rossum EFC. Stress and obesity: are there more susceptible individuals? J Adv Model Earth Syst. 2018;7(2):193‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jackson SE, Kirschbaum C, Steptoe A. Perceived weight discrimination and chronic biochemical stress: a population‐based study using cortisol in scalp hair. Obesity (Silver Spring). 2016;24(12):2515‐2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Savas M, Wester VL, Staufenbiel SM, et al. Systematic evaluation of corticosteroid use in obese and non‐obese individuals: a multi‐cohort study. Int J Med Sci. 2017;14(7):615‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fardet L, Petersen I, Nazareth I. Prevalence of long‐term oral glucocorticoid prescriptions in the UK over the past 20 years. Rheumatology (Oxford). 2011;50(11):1982‐1990. [DOI] [PubMed] [Google Scholar]
- 111. Savas M, Muka T, Wester VL, et al. Associations between systemic and local corticosteroid use with metabolic syndrome and body mass index. J Clin Endocrinol Metab. 2017;102(10):3765‐3774. [DOI] [PubMed] [Google Scholar]
- 112. Song QQ, Xie WY, Tang YJ, Zhang J, Liu J. Genetic variation in the glucocorticoid pathway involved in interindividual differences in the glucocorticoid treatment. Pharmacogenomics. 2017;18(3):293‐316. [DOI] [PubMed] [Google Scholar]
- 113. Oray M, Samra KA, Ebrahimiadib N, Meese H, Foster CS. Long‐term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457‐465. 10.1517/14740338.2016.1140743 [DOI] [PubMed] [Google Scholar]
- 114. Judd LL, Schettler PJ, Brown ES, et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am J Psychiatry. 2014;171(10):1045‐1051. [DOI] [PubMed] [Google Scholar]
- 115. Savas M, Vinkers CH, Rosmalen JGM, et al. Systemic and local corticosteroid use is associated with reduced executive cognition, and mood and anxiety disorders. Neuroendocrinology. 2020;110(3–4):282‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wester VL, Noppe G, Savas M, van den Akker ELT, de Rijke YB, van Rossum EFC. Hair analysis reveals subtle HPA axis suppression associated with use of local corticosteroids: the lifelines cohort study. Psychoneuroendocrinology. 2017;80:1‐6. [DOI] [PubMed] [Google Scholar]
- 117. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2012;134(1):54‐67. [DOI] [PubMed] [Google Scholar]
- 118. Wang JC, Shah N, Pantoja C, et al. Novel arylpyrazole compounds selectively modulate glucocorticoid receptor regulatory activity. Genes Dev. 2006;20(6):689‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Potamitis C, Siakouli D, Papavasileiou KD, et al. Discovery of new non‐steroidal selective glucocorticoid receptor agonists. J Steroid Biochem Mol Biol. 2019;186:142‐153. [DOI] [PubMed] [Google Scholar]
- 120. Chirumamilla CS, Palagani A, Kamaraj B, et al. Selective glucocorticoid receptor properties of GSK866 analogs with cysteine reactive warheads. Front Immunol. 2017;8:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Meijer OC, Koorneef LL, Kroon J. Glucocorticoid receptor modulators. Ann Endocrinol (Paris). 2018;79(3):107‐111. [DOI] [PubMed] [Google Scholar]
- 122. Hubner S, Dejager L, Libert C, Tuckermann JP. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. 2015;396(11):1223‐1231. [DOI] [PubMed] [Google Scholar]
- 123. Ozbakir B, Crielaard BJ, Metselaar JM, Storm G, Lammers T. Liposomal corticosteroids for the treatment of inflammatory disorders and cancer. J Control Release. 2014;190:624‐636. [DOI] [PubMed] [Google Scholar]
- 124. Voorzaat BM, van der Bogt KEA, Bezhaeva T, et al. A randomized trial of liposomal prednisolone (LIPMAT) to enhance radiocephalic fistula maturation: a pilot study. Kidney Int Rep. 2020;5(8):1327‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pernicova I, Kelly S, Ajodha S, et al. Metformin to reduce metabolic complications and inflammation in patients on systemic glucocorticoid therapy: a randomised, double‐blind, placebo‐controlled, proof‐of‐concept, phase 2 trial. Lancet Diabetes Endocrinol. 2020;8(4):278‐291. [DOI] [PubMed] [Google Scholar]