

Abstract
Antiphospholipid syndrome (APS) is an autoimmune disease characterized by venous, arterial, or small‐vessel thrombosis and/or pregnancy‐related morbidity, associated with persistent positivity of antiphospholipid antibodies (aPL). Pregnancy‐related morbidity in APS patients is characterized by unexplained fetal deaths, premature birth of morphologically normal newborns, and/or consecutive pregnancy losses before the 10th week of gestation. Beta 2‐glycoprotein 1 (ß2GP1) is the main antigen recognized by aPL and plays an essential role in the pathogenesis of APS. Antibodies against ß2GP1 (aß2GP1) are involved in damage‐generating mechanisms in APS due to their interaction with trophoblasts, decidua, and endothelial cells. aß2GP1 might be used as a prognostic tool for obstetric risk stratification and ß2GP1 could be a target for molecular‐targeted treatment to prevent pregnancy morbidity in APS. This review describes these aspects of aß2GP1, including effects on different cellular targets, its association with the severity of obstetric manifestations and the potential of ß2GP1‐targeted therapies for APS.
Keywords: antiphospholipid antibodies, antiphospholipid syndrome, beta 2‐glycoprotein 1, pregnancy outcome
1. INTRODUCTION
Antiphospholipid syndrome (APS) is a chronic autoimmune disease characterized by persistent positivity ( 12 weeks) of moderate or high titers of antiphospholipid antibodies (aPL) against beta 2‐glycoprotein 1 (aß2GP1) or cardiolipin (aCL), and/or a positive lupus anticoagulant test (LA). 1 In turn, there are subtypes of antibodies against different domains of beta 2‐glycoprotein 1 (ß2GP1) and aCL that are dependent on ß2GP1 binding. 2 , 3 APS is defined as either primary APS 4 or secondary to other diseases like systemic lupus erythematosus (SLE), rheumatoid arthritis or cancer. 5 , 6 , 7 , 8
APS clinical manifestations include venous, arterial, or small‐vessel thrombosis (vascular APS), and pregnancy‐related morbidity (obstetric APS). 1 , 9 Moreover, obstetric APS has different clinical patterns: only with pregnancy‐related morbidity (purely obstetric form) or combined with other vascular clinical manifestations. 9 , 10 Deep vein thrombosis and early fetal loss (< 10 weeks of pregnancy) are the most common vascular and obstetric manifestations, respectively. 7 Pregnancy‐related morbidity in APS patients is furthermore characterized by one or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation, one or more premature births of a morphologically normal newborn before the 34th week of gestation because of eclampsia or severe preeclampsia, or three or more unexplained consecutive spontaneous fetal losses before the 10th week of gestation. 1 Another clinical variant of APS is the catastrophic APS (CAPS) which is characterized by rapid multiorgan failure (≥ 3 organs) due to small‐vessel microthrombosis. 11
ß2GP1 is a five‐domain plasma protein and considered the main antigen recognized by aß2GP1 and some aCL. 12 , 13 The interaction between aß2GP1 and ß2GP1 could help understand the etiology of APS, specifically APS‐related pregnancy morbidity. 14 , 15 Why aß2GP1 are formed is not known precisely; there seems to be a genetic predisposition as significant associations between the formation of aß2GP1 and the apolipoprotein H (APOH) gene on chromosome 17, and between formation of aß2GP1 directed against domain I and the mono‐ADP ribosylhydrolase 2 gene (MACROD2) on chromosome 20 which was found in a genome‐wide association study (GWAS). 16 In addition, a polymorphism that leads to an exchange valine for leucine in 247 position of domain 5 of the protein is related to formation and reactivity of IgG aß2GP1. 17 , 18
Molecular mimicry of ß2GP1 with microorganisms has also been proposed as a mechanism of formation of aß2GP1. In a murine model, mice immunized with Haemophilus influenzae, Neisseria gonorrhoeae, or tetanus toxoid developed antibodies against the hexapeptide TLRVYK. 19 This amino acid sequence is in the third domain of ß2GP1, and it is homologous with peptide domains of viruses such as Epstein‐Barr and bacteria such as Streptococcus pneumoniae. Moreover, these specific antibodies against the hexapeptide were associated with adverse obstetric effects. 19
The role of ß2GP1 itself in the pathogenesis of APS has been widely described, regardless of clinical manifestations. 20 , 21 , 22 , 23 aPL promotes antiangiogenic effects, complement activation, inflammatory response and inhibition of proliferation and migration of trophoblasts in the placenta. 9 , 24 Furthermore, it has been demonstrated that the aPL profile – defined as which types of aPL are present, their titers and the persistence of their positivity – is a prognostic tool in APS patients. 25 Patients with aPL triple positivity – meaning positive for aß2GP1, aCL, and LA – have the highest risk of fetal loss and thromboembolic events. 26 , 27 , 28 High titers of aß2GP1 have also been identified as an additional risk factor to have adverse pregnancy outcomes even using conventional treatment with low dose aspirin (LDA) and low molecular weight heparin (LMWH). 25 , 28 Moreover, next to aß2GP1, IgM aCL has been associated with placenta‐mediated complications in APS. However, it is not specified whether these aCL were ß2GP1‐dependent antibodies, as it is known that some aCL are dependent on binding of ß2GP1. 10 , 29
In conclusion, aß2GP1 have many cellular effects, positivity to aß2GP1 might be a useful tool for obstetric risk stratification and focusing on ß2GP1 might reveal new molecular‐targeted treatments in APS patients. 22 , 30 In this review these aspects of aß2GP1 are described, and the need for additional studies to determine the clinical importance of these antibodies in APS patients will be discussed.
2. BETA 2‐GLYCOPROTEIN 1: THE MAIN ANTIGEN
ß2GP1 is an apolipoprotein family phospholipid‐binding protein, with a molecular weight between 43 kDa and 50 kDa. 31 This range is due to different results obtained by techniques like sedimentation equilibrium (43 kDa) and sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) in the presence of reducing agents (50 kDa). 31 , 32 It has a plasma concentration of 0.2 mg/ml, and it is constituted by 326 amino acids arranged in five protein domains (I‐V). 33 Each protein domain has ∼ 60 amino acids, except domain V which has 82. 34 , 35 These domains belong to the complement control protein (CCP) family, which is involved in complement regulation during protein‐protein interactions. 36 , 37
ß2GP1 is mainly synthesized in hepatocytes but can be found in human endothelial cells, astrocytes, neurons, placenta and immune cells like monocytes, neutrophils, lymphocytes and macrophages in which the binding with the phosphatidylserine‐ß2GP1 complex have been observed. 36 , 38 , 39 , 40 , 41 , 42 , 43 , 44 In a murine model, ß2GP1 was detected in trophoblasts and uterine vessels endothelial cells, ß2GP1 was exhibited as well in the gut and brain in mice treated with lipopolysaccharide (LPS). 45 Furthermore, ß2GP1 is expressed in several pathological conditions, which was demonstrated by detecting the protein in myocardial cells after acute myocardial infarction. 15 , 46 ß2GP1 expression is not dependent on aPL presence, and it was detected in placenta tissue from complicated pregnancies, but also from normal pregnancies. 43 , 44 However, there is an increased expression of ß2GP1 on trophoblast surfaces and placentas in patients with elevated aPL titers. 43 , 44
ß2GP1 molecular structure allows binding with negative charge molecules and surfaces like lipoproteins, heparin, membrane of endothelial cells, cardiolipin or anionic phospholipids. 12 , 13 , 47 , 48 , 49 These anionic phospholipids are in the inner surface of the cell membrane and could be externalized during cell apoptosis or immune cell senescence and then interact with ß2GP1. Then, ß2GP1‐anionic phospholipids complex acts as an antigen for aPL. 34
Like other apolipoproteins, ß2GP1 has high conformational flexibility and responds to environmental variations. 32 Conformational changes of ß2GP1 have been described, and there are two structure organization types: closed or circular and open or linear. 50 The closed‐form is established by an interaction between the protein domain I and V and corresponds to 91% of plasma circulating ß2GP1. 50 In vitro, aCL binding to ß2GP1 immobilized on oxidized irradiated polystyrene enzyme‐linked immunosorbent assay (ELISA) plates even without phospholipids was shown. This demonstrated that ß2GP1 binding to negative charge surfaces is important to induce the subsequent aCL‐binding. 3 , 51
In vivo, LPS or anionic phospholipids like cardiolipin on the surface of the cell membrane bind ß2GP1. 48 , 50 , 52 , 53 Then, ß2GP1 binding to negatively charged surfaces induces a conformational change to the open form that expresses an epitope in the protein that allows the binding with aPL and encourages the subsequent formation of the aPL‐ß2GP1 complex that is essential in the pathophysiology of APS. 3 , 37 , 50
It is unknown how ß2GP1 conformational changes are induced. Some authors proposed that this is a consequence of electrostatic interactions of the molecule. 54 At high pH, the change from closed to open conformation is induced, while at low pH, the opposite‐way conformation change occurs. 50 Moreover, protein reduction by thiol‐oxidoreductases and disulfide isomerase has been proposed as mechanisms to induce conformational changes of ß2GP1. In turn, ß2GP1 oxidation increase its immunogenicity inducing human dendritic cell maturation and a lymphocyte T helper 1 response. 55 , 56 , 57
Furthermore, ß2GP1 interacts with different molecules involved in coagulation and immunological pathways. 15 ß2GP1 has an anticoagulant or procoagulant effect according to environmental characteristics to which it is exposed; however, these specific determinants have not been identified yet. 15 ß2GP1 procoagulant effect participates in the pathophysiology of APS, and it is characterized by inhibiting protein C and anticoagulant function of annexin V, because the binding of annexin V to cardiolipin and phosphatidylserine on procoagulant surfaces of trophoblast cell, platelets and the endothelial cells are impeded. 58 , 59 , 60
ß2GP1 could also have a direct immunological function as it is a mediator of the innate immune system. 52 ß2GP1 inhibition of complement activation has been documented. In an assay with non‐pathologic human serum, the binding of C3 to ß2GP1 creates binding sites for factor H, a mediator in the process of degradation of C3 by factor I. The inhibition of this mechanism by aß2GP1 could be an underlying cause for accumulation of C3 activation products in the placenta of APS patients. 23
3. PATHOGENESIS: RELEVANCE OF ANTIBODIES AGAINST ß2GP1 IN APS‐RELATED PREGNANCY MORBIDITY
It has been demonstrated that aPL recognized phospholipid‐binding plasma proteins and not phospholipids directly, which was an advance to understand the pathophysiology of APS. 13 , 61 , 62 , 63 aß2GP1 are the main antibodies related to pregnancy morbidity in APS patients due to their interaction with the trophoblasts, the decidua and the endothelium of uterine vessels, 24 , 45 , 64 in which ß2GP1 functions like an intermediary between these aPL and target cells starting the damaging processes in APS. 65
Although placental vessel thrombosis was previously considered the most important pathological process of adverse pregnancy outcomes in APS patients, recently it has been proposed that purely vascular form and obstetric APS are two syndromes with different pathophysiology, which is explained by a predominant pro‐inflammatory state related to obstetric APS. 9 , 65 , 66 , 67 , 68 , 69 , 70 The greater expression of ß2GP1 in decidual endothelial cells in contrast to other vascular endothelium has been associated with a pro‐inflammatory response mediated by aß2GP1 that encourages defects in placenta growth, and it has been proposed that this is the leading cause of pregnancy morbidity in APS patients. 9 , 45 , 66
IgG polyclonal antibodies with confirmed aCL and aß2GP1 activity purified from serum samples from patients with only pregnancy morbidity induced a reduction in the HTR8 cell (first‐trimester human trophoblast) migration, conversely IgG polyclonal antibodies purified from patients with vascular thrombosis, did not have this effect. 71
Furthermore, aß2GP1 have a wide range of effects in different cellular targets that are associated with pregnancy morbidity in APS patients. These main cellular targets and their downstream effects are summarized in Figure 1 and are described in the following paragraphs.
FIGURE 1.
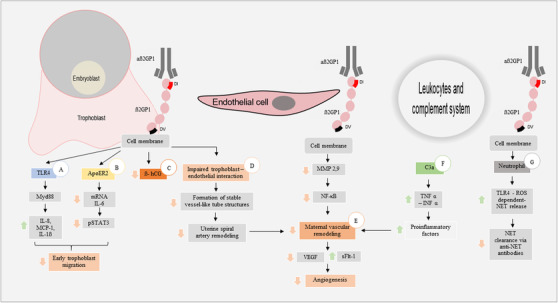
Effects of beta 2‐glycoprotein 1 antibodies on different cellular targets and their damage‐generating pathways (A) aß2GP1 pro‐inflammatory effect via TLR4/Myd88. (B) aß2GP1 induced a reduction in the promigratory effect of IL6 and STAT3. (C) aß2GP1‐mediated ß‐hCG decrease (D) Altered trophoblast‐endothelium interaction. (E) Dysfunction in maternal vascular remodeling. (F) Complement system early activation reduces maternal vascular remodeling. (G) aß2GP1 increases the release of NETs. Arrow up. Upregulation. Arrow down. Downregulation. Abbreviations. aß2GPI, anti‐beta 2‐glycoprotein‐1 antibodies; ß2GPI, beta 2‐ glycoprotein‐1; TLR4, Toll‐like receptor type 4; MyD88, Myeloid differentiation primary response 88; IL, interleukin; MCP‐1, monocyte chemoattractant protein 1; ApoER2, apolipoprotein E receptor 2; pSTAT3, phosphorylated signal transducer and activator of transcription 3; ß‐hCG, ß human chorionic gonadotropin hormone; MMP, matrix metalloproteinases; NF‐κB, nuclear factor kappa of activated B cells; VEGF, vascular endothelial growth factor; sFlt‐1 placental soluble fms like tyrosine kinase 1, TNF‐α, tumor necrosis factor alpha; INF‐ α, interferon alpha; ROS, reactive oxygen species; NET, neutrophil extracellular trap
3.1. aß2GP1‐mediated placental dysfunction
In a systematic review by Viall and Chamley, placental dysfunction was described as a cause of pregnancy morbidity in APS. 24 Decidual inflammation, placental infarction, alteration in the remodeling of the uterine spiral arteries, an increase in syncytial knots, a decrease in the vasculo‐syncytial membranes and the activation of the complement system (C4d) were the leading processes involved in this placental dysfunction. 24 Nevertheless, this review only included six studies with aß2GP1‐positive patients and did not differentiate between ß2GP1‐dependent and independent aCL effect. Future research should be performed to analyze the histopathological changes produced only by aß2GP1. 24
In vitro and in vivo extravillous trophoblast invasion was reduced by aß2GP1, this altered the remodeling of the uterine spiral arteries and caused a decreased blood flow to the placenta, increased production of pro‐inflammatory cytokines and antiangiogenic factors like the placental soluble fms like tyrosine kinase 1 (sFlt‐1). 70 In an in vitro study with a toll‐like receptor type 4/myeloid differentiation primary response 88 (TLR4/MyD88) dependent model, monoclonal aß2GP1 caused an inflammatory response in trophoblastic cells, which leads to an increase in interleukin (IL) 8, IL1ß and monocyte chemoattractant protein 1 (MCP‐1) production. 72 Another in vitro TLR4/MyD88 independent model demonstrated that trophoblastic cells treated with monoclonal aß2GP1 had lower mRNA IL6 levels, which was correlated with low levels of phosphorylated signal transducer and activator of transcription 3 (STAT3). These changes in pro‐inflammatory molecules generated a decrease in early trophoblast migration. 73
Furthermore, aPL are internalized into the syncytiotrophoblast via a low‐density lipoprotein receptor (LDLR). 74 After internalization into the syncytiotrophoblast, aPL affected the inner mitochondrial membrane, increasing the Cytochrome c release to the cytosol and the ROS production by trophoblast cells. 74 , 75 Mitochondrial dysfunction altered cell death processes of the syncytiotrophoblast, leading to an increase in the release of necrotic syncytiotrophoblast debris that encourages maternal endothelial cell activation. 74 , 76 In vitro, syncytiotrophoblast debris increased the endothelial cell surface expression of Intercellular Adhesion Molecule 1 (ICAM‐1), and it could be avoided with RAP addition, an inhibitor of LDLR receptors. 74 , 76 Binding of aß2GP1‐ß2GP1 complex to the apolipoprotein E receptor 2 (ApoER2) in the trophoblast also induced a reduction in cell migration in a murine model. 77 Moreover, ß human chorionic gonadotropin hormone (ß‐hCG) trophoblast production decline is another aß2GP1 mediated mechanism that results in placental dysfunction in APS patients. 78 , 79
Thus, placental dysfunction is a leading cause of APS‐related pregnancy morbidity, and aß2GP1 play an important role in the dysfunction by promoting different pathophysiological pathways that lead to a placental pro‐inflammatory state, decidual endothelial cell dysfunction and altered trophoblast cell migration (Figure 1).
3.2. aß2GP1‐mediated endothelial dysfunction
Endothelial dysfunction has been demonstrated in APS patients with different clinical manifestations. 24 , 80 This dysfunction and imbalance between proangiogenic factors, like the vascular endothelial growth factor (VEGF), and antiangiogenic factors, like sFlt‐1, promote obstetric complications like preeclampsia. 81 In vitro serum from women with APS‐related pregnancy morbidity (with or without previous thrombosis) decreased trophoblast‐endothelium interaction and disturbed formation of stable vessel‐like tube structures. 82 , 83 Furthermore, in vivo and in vitro aPL decreased angiogenesis, reduced VEGF production and interfered with matrix metalloproteinases (MMPs) activity which is necessary for placental angiogenesis. 84
In a human microvascular endothelial cell line (HMEC‐1), trophoblastic debris derived from healthy first trimester placentas induced proteomic and transcriptomic changes characterized by an upregulated release of pro‐inflammatory cytokines like IL8 and negative gene regulation of apoptosis. 85 This interaction has been considered a maternal adaptive mechanism and a part of feto‐maternal communication during healthy pregnancy. 85 aß2GP1 produced mitochondrial changes that lead to cellular death with the consequent release of syncytial nuclear aggregates that could interact with maternal endothelial cells and set off vascular pathologic changes. 70 Moreover, high aß2GP1 concentration induced trophoblastic cell apoptosis via caspases 3, 8 and 9. 72 Despite this, there is a lack of evidence about aß2GP1‐mediated endothelial dysfunction in purely obstetric APS form in clinical studies, considering the lack of a clear description of the APS patients clinical classification in previous studies. 80
3.3. Role of complement and neutrophil interaction in pregnancy‐related morbidity
During pregnancy, the complement helps in extensive tissue remodeling caused by trophoblast invasion, especially in remodeling of spiral uterine arteries. 86 Early complement activation (especially the alternative pathway) and hypocomplementemia during gestation have been identified as predictors of adverse pregnancy outcomes in aPL‐positive women. 87 , 88 , 89 Besides, the Bb and sC5b‐9 serum levels were higher in aPL positive patients with adverse pregnancy outcomes. 87
Complement activation allowed leucocyte recruitment, and these produced pro‐inflammatory molecules like tumor necrosis factor alpha (TNF‐α) and interferon alpha (INF‐α) that hindered the vascular remodeling. 90 Complement activation was associated with fetal resorption and fetal growth restriction in a murine model; however, there was no demonstration of specific aß2GP1‐ mediated damage. 91 Furthermore, these adverse pregnancy outcomes were avoided using the C3 convertase inhibitor complement receptor 1‐related gene/protein γ (Crrγ)‐Ig, and C3 deficient mice were resistant to aPL‐mediated pregnancy morbidity. 91
The C5a key role in the aPL‐related fetal injury was described by Girardi, et al. 92 C5a activity encourages the recruitment of tissue factor‐expressing neutrophils to trophoblast tissue, which leads to respiratory burst and subsequent trophoblast injury. 93 , 94 Nevertheless, these studies used human IgG‐containing aPL antibodies, and there was no demonstration of specific aß2GP1 – mediated damage. To our knowledge, there still are no studies investigating specific aß2GP1‐mediated effects in complement function and its role in pregnancy‐related morbidity in APS patients.
Also, neutrophils have been associated with fetal damage in mice treated with aPL. 92 Yalavarthi et al. demonstrated that aß2GP1 binds to the neutrophil surface and increased the neutrophil extracellular trap (NET) release, a form of neutrophil death recognized as a leading mediator in arterial and venous thrombosis. 42 , 95 ß2GP1‐mediated upregulation of the beta‐2 integrin Mac‐1 leaded to a TLR‐4 dependent NET release due to increased neutrophil interaction with the endothelium. 96 Moreover, a decrease in NET clearance by circulating deoxyribonucleases was shown in APS patients. 97 , 98
In vitro the IgG serum from APS pregnant women without previous thrombosis induced increase NET release via reactive oxygen species (ROS) production. 99 Furthermore, these APS‐IgG stimulated NETs reduced the invasion and migration of HTR8 cells and migration and tube formation of Human Umbilical Vein Endothelial Cells (HUVEC). Interestingly, all included patients were positive for IgG aß2GP1. 99
4. ANTIBODIES AGAINST ß2GP1 ARE IMPORTANT DETERMINANTS OF ADVERSE PREGNANCY OUTCOMES IN APS
It has been proposed that aß2GP1 positivity is a useful tool for obstetric risk stratification in aPL‐positive patients. 22 Adverse pregnancy outcomes have been more strongly associated with aß2GP1 than with aCL and LA. 9 , 22 , 100 All aß2GP1 are associated with adverse pregnancy outcomes across all of gestation; however, differences have been reported in pathophysiology mechanisms between APS patients with early and late pregnancy morbidity. 22 , 101 , 102 aß2GP1 against domain I are related to an increased risk of thrombotic events and, to a lesser extent, APS‐related pregnancy morbidity. 103 , 104 , 105 It has been proposed that the presence of this aß2GP1 subtype is a leading predictor of late‐onset pregnancy morbidity due to the role of placental thrombotic infarction in its pathogenesis. 22 , 106 , 107
aß2GP1 are determinants of pregnancy outcomes in APS patients using conventional treatment. 15 Specifically, high levels of aß2GP1 are a risk factor to fetal loss and pregnancy morbidity in this patient's group. 28 , 108 The PREGNANTS study including pregnant women with APS using conventional treatment demonstrated that patients only positive for aß2GP1 had a higher incidence of pregnancy morbidities like preeclampsia, fetal growth restriction, preterm delivery and stillbirth, and a lower rate of live births than patients positive only for aCL or LA. 26 As mentioned above, the study did not specify whether these aCL were ß2GP1‐dependent antibodies, which could affect the results.
Furthermore, by using the EUREKA algorithm, a tool to define the magnitude of the obstetric risk including any titer of aPL (diagnostic or not), it was observed that patients only positive for aß2GP1 IgG had a higher risk of adverse pregnancy outcomes than those only positive for aCL. Moreover, patients positive for LA and aß2GP1 IgG had the highest risk for these adverse outcomes. 30
Most of the available studies concerning obstetric risk stratification are retrospective and did not include different clinical manifestations, representing a barrier to extrapolating these promising results directly to the clinical practice and to possibly changing the treatment in women with the highest risk. Thereby, there is a need for prospective studies that evaluate aPL profile (and especially aß2GP1) in relation to the severity of adverse pregnancy outcomes and used treatment in pregnant APS patients.
5. ß2GP1 AS NOVEL MOLECULAR‐THERAPEUTIC TARGET
European League Against Rheumatism (EULAR) APS treatment recommendations were published in 2019. Different treatment regimens depending on previous pregnancy morbidity, aPL profile, and response to conventional treatment were proposed. 25 LDA and prophylactic or therapeutic doses (according to the clinical scenario) of LMWH are the conventional treatment for APS‐related pregnancy morbidity, and with this treatment a live birth is achieved in more than 70% of pregnancies. 109 To improve outcome, ß2GP1 have been proposed as a molecular‐therapeutic target due to their role in APS pathogenesis, and aß2GP1 association with more severe clinical manifestations despite conventional treatment. Below we describe some of the proposed strategies, that are summarized in Table 1.
TABLE 1.
Beta 2‐glycoprotein 1 as a molecular‐therapeutic target in APS‐related pregnancy morbidity
| Therapy | ß2GP1 relation | Mechanism of action | Effect | Model | References |
|---|---|---|---|---|---|
| TIFI peptide | Mimics Vth domain of ß2GP1. | Competitive inhibition: prevents binding of ß2GP1 to target cells. | Dose ‐ dependent inhibition of ß2GP1 binding to trophoblast. | In vitro. Cytotrophoblast cell culture. | [95, 96, 97] |
| Reduce growth retardation and fetal loss rate induced by aPL. | In vivo. Murine model, pregnant C57BL/6 mice. | ||||
| 1N11 monoclonal antibody | Monoclonal antibody to ß2GP1. | Decreases antibody binding to ß2GP1 and impede interaction between ß2GP1 and apoER2. | Prevents alterations of early trophoblast migration and proliferation. | In vitro. Trophoblast cell line HTR‐8SV neo. | [98] |
| Reduce increase in fetal resorption induced by aPL. | In vivo. Murine model, female Balb/c. | ||||
| MBB2ΔCH2 | Non‐complement fixing antibody to ß2GP1. | Competitive inhibition: prevents binding of aß2GP1 to ß2GP1 domain I. | Reduce fetal resorption frequency and increase fetal weight. |
In vitro. BeWo and HUVECs In vivo. Murine model‐ female BALB/c mice. |
[99] |
Abbreviations: aPL, antiphospholipid antibodies; apoER2, apolipoprotein E receptor 2; aß2GP1, antibodies anti beta 2‐glycoprotein‐1; HTR‐8SV. First‐trimester human trophoblast cells; HUVEC, human umbilical cord vein endothelial cells.;ß2GP1, beta 2‐ glycoprotein‐1.
5.1. TIFI synthetic peptide
TIFI is a 20 amino acid synthetic peptide that shares similarity with the Vth domain of ß2GP1. It is a competitive inhibitor of this domain and prevents ß2GP1 binding to cell surfaces and phospholipids. It was shown to prevent aPL‐mediated thrombosis and binding of ß2GP1 to endothelial cells and macrophages in a murine model. 110 Another study in mice demonstrated TIFI‐mediated inhibition of binding of the protein to trophoblast cells in a dose‐dependent scheme, resulting in less fetal growth restriction and fetal losses. 111 Moreover, in vitro TIFI prevented antiangiogenic effect of aß2GP1 in human endometrial endothelial cells. 112
Despite these promising results, the mechanism of action of TIFI peptide could interfere with the physiological functions of ß2GP1 in different tissues. ß2GP1 belongs to the LPS‐neutralizing proteins family, and it has a direct role in the innate immune system. 52 , 113 Avoiding the binding of LPS‐ß2GP1 complex to the cell surface of monocytes could be deleterious during infectious diseases in APS patients. Even though LPS binds to domain V of ß2GP1, and TIFI peptide shares similarities, there are still no studies investigating TIFI LPS‐neutralizing function. Besides, ß2GP1 facilitate phagocytosis of apoptotic cells and platelets microvesicles. 114 , 115 TIFI peptide could avoid the binding of these protein complexes to macrophages and leading to dysfunction in coagulation pathways.
5.2. Monoclonal antibodies
1N11 monoclonal antibody against ß2GP1 was shown to decrease aPL binding to the protein, inhibiting its interaction with ApoER2, prevents the adverse effects of aPL on endothelial cell migration and proliferation, and reduce the prothrombotic function of aPL. 116 Furthermore, a decrease in thrombotic and pregnancy adverse effects of aPL was shown using the non‐complement fixing antibody (MBB2ΔCH2) against ß2GP1 that prevents protein domain I binding to aß2GP1. 117
5.3. Recombinant domain I molecule
This recombinant domain I molecule is able to bind to aPL and thus prevents their adverse effects. 118 It had had promising results in murine models. However, the studies did not evaluate adverse pregnancy outcomes. 119 , 120 Although aß2GP1 directed against the domain I was associated with thrombosis and adverse pregnancy outcomes in APS patients, their prevalence in this population is around 45%. 105 Therefore, the contribution of antibodies with another specificity must be considering, and the new therapeutics focused only on aß2GP1 directed against domain I could be insufficient to prevent all aPL effects. 103 , 104 , 105 , 106
Research is going concerning these promising therapeutic strategies, but still in preliminary phases. Therefore, the Rheumatology and Obstetrics associations do not recommend these drugs yet as current treatment of APS patients. 25 , 121
6. CLINICALLY USED ALTERNATIVE TREATMENT OPTIONS NOT SPECIFICALLY TARGETING ß2GP1
6.1. Statins
Statins have been widely used to prevent and treat cardiovascular diseases, and new evidence is emerging about their use to treat placental insufficiency. 22 Pravastatin preliminary safety was shown in high‐risk of preeclampsia pregnant women. 122 Furthermore, no major congenital abnormalities have been identified in pregnant women treated with statins during the first trimester. 123
Previous murine models demonstrated that pravastatin improves vascular reactivity, decreasing sFlt‐1 levels and its antiangiogenic effects. 124 A reduction in adverse pregnancy outcomes in APS patients under conventional treatment plus pravastatin compared to conventional treatment alone was described. 125 Besides, the pravastatin plus therapy increases endothelial nitric oxide synthase (eNOS) synthesis leading to an increase in nitric oxide (NO) generation and improving placental vascular function. 126 Despite these promising results, statins safety and effectiveness in preventing adverse pregnancy outcomes need to be proved in large patients cohorts.
6.2. Hydroxychloroquine
Hydroxychloroquine (HCQ) disintegrates aß2GP1‐phospholipids complexes, diminishes complement activation, and has anticoagulation effects maintaining the annexin V shield in endothelial and trophoblast cell surfaces. 25 , 127 , 128 HCQ prevents platelet activation induced by aPL, and chloroquine could inhibit the internalization of aPL into the syncytiotrophoblast, reducing their mitochondrial deleterious effects. 74 , 75 , 129 HCQ treatment is approved for use in APS patients with adverse pregnancy outcomes despite conventional treatment. 25
6.3. Intravenous immunoglobulin
Intravenous immunoglobulin (IVIG) has been proposed for APS patients with adverse pregnancy outcomes despite conventional treatment. 130 , 131 IVIG treatment decreases LA activity and interferes in the aCL binding to cardiolipin. 132 , 133 Besides, anti–idiotypic antibodies to aPL in IVIG, which reduces aPL effects, have been documented. Nevertheless, effectiveness rates are controversial, IVIG therapy carries high costs, and there is a lack of evidence about clinical outcomes in APS‐related pregnancy morbidity. 130 , 134
7. CONCLUSION
ß2GP1 has a key role in the pathogenesis of APS‐related pregnancy morbidity, which is different compared to the pathogenesis of vascular APS manifestations. Pro‐inflammatory state and an imbalance between proangiogenic and antiangiogenic factors disturb uterine spiral artery remodeling and trophoblast proliferation and migration, resulting in adverse pregnancy outcomes like preeclampsia in APS patients. The effects of ß2GP1‐dependent aCL are not determined sufficient in previous studies, therefore is poorly understood the role of these specific aCL in the pathogenesis of APS.
Furthermore, aß2GP1 are more often associated with severe adverse clinical outcomes despite conventional treatment compared to other aPL in patients with APS‐related pregnancy morbidity. Therefore, aß2GP1 positivity is becoming a useful tool for obstetric risk stratification. However, prospective studies including cohorts of APS patients are required to determine the usefulness of aß2GP1 based risk stratification. At last, ß2GP1 might be a possible target of molecular‐targeted treatment in the future to prevent obstetric complications in APS.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGMENTS
This work was supported by Ministerio de Ciencia, Tecnología e Innovación (MinCiencias, Colombia) [Grant numbers 1115800762949, 930‐2019 ASC and 757‐2016]
Fierro JJ, Velásquez M, Cadavid AP, de Leeuw K. Effects of anti‐beta 2‐glycoprotein 1 antibodies and its association with pregnancy‐related morbidity in antiphospholipid syndrome. Am J Reprod Immunol. 2022;87:e13509. 10.1111/aji.13509
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Miyakis S, Lockshin MD, Atsumi T, , et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295‐306. [DOI] [PubMed] [Google Scholar]
- 2. Kelchtermans H, Chayoua W, Laat B. The significance of antibodies against domain i of beta‐2 glycoprotein i in antiphospholipid syndrome. Semin Thromb Hemost. 2018;44(5):458‐465. [DOI] [PubMed] [Google Scholar]
- 3. Chamley LW, Duncalf AM, Konarkowska B, Mitchell MD, Johnson PM. Conformationally altered beta 2‐glycoprotein I is the antigen for anti‐cardiolipin autoantibodies. Clin Exp Immunol. 1999;115(3):571‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mackworth‐Young CG, Loizou S, Walport MJ. Primary antiphospholipid syndrome: features of patients with raised anticardiolipin antibodies and no other disorder. Ann Rheum Dis. 1989;48(5):362‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pugliese L, Bernardini I, Pacifico E, Viola‐Magni M, Albi E. Antiphospholipid antibodies in patients with cancer. Int J Immunopathol Pharmacol. 2006;19(4):879‐888. [DOI] [PubMed] [Google Scholar]
- 6. Pham C, Shen YM. Antiphospholipid antibodies and malignancy. Hematol Oncol Clin N Am. 2008;22(1):121‐130. vii. [DOI] [PubMed] [Google Scholar]
- 7. Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46(4):1019‐1027. [DOI] [PubMed] [Google Scholar]
- 8. Pons‐Estel GJ, Andreoli L, Scanzi F, Cervera R, Tincani A. The antiphospholipid syndrome in patients with systemic lupus erythematosus. J Autoimmun. 2017;76:10‐20. [DOI] [PubMed] [Google Scholar]
- 9. Meroni PL, Borghi MO, Grossi C, Chighizola CB, Durigutto P, Tedesco F. Obstetric and vascular antiphospholipid syndrome: same antibodies but different diseases?. Nat Rev Rheumatol. 2018;14(7):433‐440. [DOI] [PubMed] [Google Scholar]
- 10. Bouvier S, Cochery‐Nouvellon E, Lavigne‐Lissalde G, Mercier E, Marchetti T, Balducchi JP, et al. Comparative incidence of pregnancy outcomes in treated obstetric antiphospholipid syndrome: the NOH‐APS observational study. Blood. 2014;123(3):404‐413. [DOI] [PubMed] [Google Scholar]
- 11. Asherson RA, Cervera R, de Groot PG, Erkan D, Boffa MC, Piette JC, et al. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12(7):530‐534. [DOI] [PubMed] [Google Scholar]
- 12. George J, Gilburd B, Hojnik M, Levy Y, Langevitz P, Matsuura E, et al. Target recognition of beta2‐glycoprotein I (beta2GPI)‐dependent anticardiolipin antibodies: evidence for involvement of the fourth domain of beta2GPI in antibody binding. J Immunol. 1998;160(8):3917‐3923. [PubMed] [Google Scholar]
- 13. Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda‐Vriesman PJ, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335(8705):1544‐1547. [DOI] [PubMed] [Google Scholar]
- 14. Pierangeli SS, Chen PP, Raschi E, Scurati S, Grossi C, Borghi MO, et al. Antiphospholipid antibodies and the antiphospholipid syndrome: pathogenic mechanisms. Semin Thromb Hemost. 2008;34(3):236‐250. [DOI] [PubMed] [Google Scholar]
- 15. McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, et al. The role of beta‐2‐glycoprotein I in health and disease associating structure with function: more than just APS. Blood rev. 2019:100610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muller‐Calleja N, Rossmann H, Muller C, Wild P, Blankenberg S, Pfeiffer N, et al. Antiphospholipid antibodies in a large population‐based cohort: genome‐wide associations and effects on monocyte gene expression. Thromb Haemost. 2016;116(1):115‐123. [DOI] [PubMed] [Google Scholar]
- 17. Atsumi T, Tsutsumi A, Amengual O, Khamashta MA, Hughes GR, Miyoshi Y, et al. Correlation between beta2‐glycoprotein I valine/leucine247 polymorphism and anti‐beta2‐glycoprotein I antibodies in patients with primary antiphospholipid syndrome. Rheumatology. 1999;38(8):721‐723. [DOI] [PubMed] [Google Scholar]
- 18. Yasuda S, Atsumi T, Matsuura E, Kaihara K, Yamamoto D, Ichikawa K, et al. Significance of valine/leucine247 polymorphism of beta2‐glycoprotein I in antiphospholipid syndrome: increased reactivity of anti‐beta2‐glycoprotein I autoantibodies to the valine247 beta2‐glycoprotein I variant. Arthritis Rheum. 2005;52(1):212‐218. [DOI] [PubMed] [Google Scholar]
- 19. Blank M, Krause I, Fridkin M, Keller N, Kopolovic J, Goldberg I, et al. Bacterial induction of autoantibodies to beta2‐glycoprotein‐I accounts for the infectious etiology of antiphospholipid syndrome. J Clin Invest. 2002;109(6):797‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011;7(6):330‐339. [DOI] [PubMed] [Google Scholar]
- 21. Corban MT, Duarte‐Garcia A, McBane RD, Matteson EL, Lerman LO, Lerman A. Antiphospholipid syndrome: role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. J Am Coll Cardiol. 2017;69(18):2317‐2330. [DOI] [PubMed] [Google Scholar]
- 22. Beltagy A, Trespidi L, Gerosa M, Ossola MW, Meroni PL, Chighizola CB. Anti‐phospholipid antibodies and reproductive failures. Am J Reprod Immunol. 2020:e13258. [DOI] [PubMed] [Google Scholar]
- 23. Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallstrom T, et al. beta(2)‐glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. 2011;118(10):2774‐2783. [DOI] [PubMed] [Google Scholar]
- 24. Viall CA, Chamley LW. Histopathology in the placentae of women with antiphospholipid antibodies: a systematic review of the literature. Autoimmunity Rev. 2015;14(5):446‐471. [DOI] [PubMed] [Google Scholar]
- 25. Tektonidou MG, Andreoli L, Limper M, Amoura Z, Cervera R, Costedoat‐Chalumeau N, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. 2019;78(10):1296‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saccone G, Berghella V, Maruotti GM, Ghi T, Rizzo G, Simonazzi G, et al. Antiphospholipid antibody profile based obstetric outcomes of primary antiphospholipid syndrome: the PREGNANTS study. Am J Obstet Gynecol. 2017;216(5):525. e1‐.e12. [DOI] [PubMed] [Google Scholar]
- 27. Rottenstreich A, Arad A, Terespolsky H, Elchalal U, Amsalm H, Roth B, et al. Antiphospholipid antibody profile‐based outcome of purely vascular and purely obstetric antiphospholipid syndrome. J Thromb Thrombolysis. 2018;46(2):166‐173. [DOI] [PubMed] [Google Scholar]
- 28. Latino JO, Udry S, Aranda FM, Peres Wingeyer SDA, Fernandez Romero DS, de Larranaga GF. Pregnancy failure in patients with obstetric antiphospholipid syndrome with conventional treatment: the influence of a triple positive antibody profile. Lupus. 2017;26(9):983‐988. [DOI] [PubMed] [Google Scholar]
- 29. Pengo V, Bison E, Denas G, SP Jose, Zoppellaro G, Banzato A. Laboratory diagnostics of antiphospholipid syndrome. Semin Thromb Hemost. 2018;44(5):439‐444. [DOI] [PubMed] [Google Scholar]
- 30. Pregnolato F, Gerosa M, Raimondo MG, Comerio C, Bartoli F, Lonati PA, et al. EUREKA algorithm predicts obstetric risk and response to treatment in women with different subsets of anti‐phospholipid antibodies. Rheumatology. 2020;0:1‐11. [DOI] [PubMed] [Google Scholar]
- 31. Cai G, Guo Y, Shi J. Purification of apolipoprotein H by polyethylene glycol precipitation. Protein Expr Purif. 1996;8(3):341‐346. [DOI] [PubMed] [Google Scholar]
- 32. Lee NS. beta 2‐Glycoprotein I. Molecular properties of an unusual apolipoprotein, apolipoprotein H. J Biol Chem. 1983;258(8):4765‐4770. [PubMed] [Google Scholar]
- 33. Lozier J, Takahashi N, Putnam FW. Complete amino acid sequence of human plasma beta 2‐glycoprotein I. Proc Natl Acad Sci U S A. 1984;81(12):3640‐3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwarzenbacher R, Zeth K, Diederichs K, et al. Crystal structure of human beta2‐glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. 1999;18(22):6228‐6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Laat B, Derksen RHWM, Urbanus RT, de Groot PG. IgG antibodies that recognize epitope Gly40‐Arg43 in domain I of β2–glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood. 2005;105(4):1540‐1545. [DOI] [PubMed] [Google Scholar]
- 36. Steinkasserer A, Estaller C, Weiss EH, Sim RB, Day AJ. Complete nucleotide and deduced amino acid sequence of human beta 2‐glycoprotein I. Biochem J. 1991;277(2):387‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bouma B, de Groot PG, van den Elsen JM, et al. Adhesion mechanism of human beta(2)‐glycoprotein I to phospholipids based on its crystal structure. EMBO J. 1999;18(19):5166‐5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balasubramanian K, Schroit AJ. Characterization of phosphatidylserine‐dependent beta2‐glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J Biol Chem. 1998;273(44):29272‐29277. [DOI] [PubMed] [Google Scholar]
- 39. Caronti B, Calderaro C, Alessandri C, et al. Beta2‐glycoprotein I (beta2‐GPI) mRNA is expressed by several cell types involved in anti‐phospholipid syndrome‐related tissue damage. Clin Exp Immunol. 1999;115(1):214‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. George J, Harats D, Gilburd B, et al. Immunolocalization of beta2‐glycoprotein I (apolipoprotein H) to human atherosclerotic plaques: potential implications for lesion progression. Circulation. 1999;99(17):2227‐2230. [DOI] [PubMed] [Google Scholar]
- 41. Conti F, Sorice M, Circella A, et al. Beta‐2‐glycoprotein I expression on monocytes is increased in anti‐phospholipid antibody syndrome and correlates with tissue factor expression. Clin Exp Immunol. 2003;132(3):509‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yalavarthi S, Gould TJ, Rao AN, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheum. 2015;67(11):2990‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. La Rosa L, Meroni PL, Tincani A, et al. Beta 2 glycoprotein I and placental anticoagulant protein I in placentae from patients with antiphospholipid syndrome. J Rheumatol. 1994;21(9):1684‐1693. [PubMed] [Google Scholar]
- 44. Chamley LW, Allen JL, Johnson PM. Synthesis of beta2 glycoprotein 1 by the human placenta. Placenta. 1997;18(5‐6):403‐410. [DOI] [PubMed] [Google Scholar]
- 45. Agostinis C, Biffi S, Garrovo C, et al. In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood. 2011;118(15):4231‐4238. [DOI] [PubMed] [Google Scholar]
- 46. Niessen HW, Lagrand WK, Rensink HJ, et al. Apolipoprotein H, a new mediator in the inflammatory changes ensuring in jeopardised human myocardium. J Clin Pathol. 2000;53(11):863‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Del Papa N, Sheng YH, Raschi E, et al. Human beta 2‐glycoprotein I binds to endothelial cells through a cluster of lysine residues that are critical for anionic phospholipid binding and offers epitopes for anti‐beta 2‐glycoprotein I antibodies. J Immunol. 1998;160(11):5572‐5578. [PubMed] [Google Scholar]
- 48. Hunt J, Krilis S. The fifth domain of beta 2‐glycoprotein I contains a phospholipid binding site (Cys281‐Cys288) and a region recognized by anticardiolipin antibodies. J Immunol. 1994;152(2):653‐659. [PubMed] [Google Scholar]
- 49. Manganelli V, Capozzi A, Recalchi S, et al. Altered traffic of cardiolipin during apoptosis: exposure on the cell surface as a trigger for "Antiphospholipid Antibodies". J Immunol. 2015:847985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Agar C, van Os GM, Morgelin M, et al. Beta2‐glycoprotein I can exist in 2 conformations: implications for our understanding of the antiphospholipid syndrome. Blood. 2010;116(8):1336‐1343. [DOI] [PubMed] [Google Scholar]
- 51. Matsuura E, Igarashi Y, Yasuda T, Triplett DA, Koike T. Anticardiolipin antibodies recognize beta 2‐glycoprotein I structure altered by interacting with an oxygen modified solid phase surface. J Exp Med. 1994;179(2):457‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Agar C, de Groot PG, Morgelin M, et al. beta(2)‐glycoprotein I: a novel component of innate immunity. Blood. 2011;117(25):6939‐6947. [DOI] [PubMed] [Google Scholar]
- 53. Agar Ç, de Groot PG, Marquart JA, Meijers JC. Evolutionary conservation of the lipopolysaccharide binding site of β₂‐glycoprotein I. Thromb Haemost. 2011;106(6):1069‐1075. [DOI] [PubMed] [Google Scholar]
- 54. Buchholz I, Nestler P, Koppen S, Delcea M. Lysine residues control the conformational dynamics of beta 2‐glycoprotein I. PhysChemChemPhys. 2018;20(42):26819‐26829. [DOI] [PubMed] [Google Scholar]
- 55. Passam FH, Rahgozar S, Qi M, et al. Redox control of beta2‐glycoprotein I‐von Willebrand factor interaction by thioredoxin‐1. J Thromb Haemost. 2010;8(8):1754‐1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Passam FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular pathophysiology of the antiphospholipid syndrome: the role of oxidative post‐translational modification of beta 2 glycoprotein I. J Thromb Haemost. 2011;9(1):275‐282. [DOI] [PubMed] [Google Scholar]
- 57. Buttari B, Profumo E, Mattei V, et al. Oxidized beta2‐glycoprotein I induces human dendritic cell maturation and promotes a T helper type 1 response. Blood. 2005;106(12):3880‐3887. [DOI] [PubMed] [Google Scholar]
- 58. Hanly JG, Smith SA. Anti‐beta2‐glycoprotein I (GPI) autoantibodies, annexin V binding and the anti‐phospholipid syndrome. Clin Exp Immunol. 2000;120(3):537‐5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keeling DM, Wilson AJ, Mackie IJ, Isenberg DA, Machin SJ. Role of beta 2‐glycoprotein I and anti‐phospholipid antibodies in activation of protein C in vitro. J Clin Pathol. 1993;46(10):908‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Reutelingsperger CP, van Heerde WL. Annexin V, the regulator of phosphatidylserine‐catalyzed inflammation and coagulation during apoptosis. Cell Mol Life Sci. 1997;53(6):527‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Laat B, Mertens K, de Groot PG. Mechanisms of disease: antiphospholipid antibodies‐from clinical association to pathologic mechanism. Nat Clin Pract Rheumatol. 2008;4(4):192‐199. [DOI] [PubMed] [Google Scholar]
- 62. McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti‐phospholipid antibodies are directed against a complex antigen that includes a lipid‐binding inhibitor of coagulation: beta 2‐glycoprotein I (apolipoprotein H). Proc Natl Acad Sci U S A. 1990;87(11):4120‐4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;336(8708):177‐178. [DOI] [PubMed] [Google Scholar]
- 64. Tong M, Viall CA, Chamley LW. Antiphospholipid antibodies and the placenta: a systematic review of their in vitro effects and modulation by treatment. Hum Reprod. 2015;21(1):97‐118. [DOI] [PubMed] [Google Scholar]
- 65. Abrahams VM. Mechanisms of antiphospholipid antibody‐associated pregnancy complications. Thromb Res. 2009;124(5):521‐525. [DOI] [PubMed] [Google Scholar]
- 66. D'Ippolito S, Meroni PL, Koike T, Veglia M, Scambia G, Di Simone N. Obstetric antiphospholipid syndrome: a recent classification for an old defined disorder. Autoimmunity Rev. 2014;13(9):901‐908. [DOI] [PubMed] [Google Scholar]
- 67. De Wolf F, Carreras LO, Moerman P, Vermylen J, Van Assche A, Renaer M. Decidual vasculopathy and extensive placental infarction in a patient with repeated thromboembolic accidents, recurrent fetal loss, and a lupus anticoagulant. Am J Obstet Gynecol. 1982;142(7):829‐834. [DOI] [PubMed] [Google Scholar]
- 68. Out HJ, Kooijman CD, Bruinse HW, Derksen RH. Histopathological findings in placentae from patients with intra‐uterine fetal death and anti‐phospholipid antibodies. Eur J Obstet Gynecol Reprod Biol. 1991;41(3):179‐186. [DOI] [PubMed] [Google Scholar]
- 69. Stone S, Pijnenborg R, Vercruysse L, et al. The placental bed in pregnancies complicated by primary antiphospholipid syndrome. Placenta. 2006;27(4‐5):457‐467. [DOI] [PubMed] [Google Scholar]
- 70. Abrahams VM, Chamley LW, Salmon JE. Emerging treatment models in rheumatology: antiphospholipid syndrome and pregnancy: pathogenesis to translation. Arthritis Rheum. 2017;69(9):1710‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Poulton K, Ripoll VM, Pericleous C, et al. Purified IgG from patients with obstetric but not IgG from non‐obstetric antiphospholipid syndrome inhibit trophoblast invasion. Am J Reprod Immunol. 2015;73(5):390‐3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mulla MJ, Brosens JJ, Chamley LW, et al. Antiphospholipid antibodies induce a pro‐inflammatory response in first trimester trophoblast via the TLR4/MyD88 pathway. Am J Reprod Immunol. 2009;62(2):96‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mulla MJ, Myrtolli K, Brosens JJ, et al. Antiphospholipid antibodies limit trophoblast migration by reducing IL‐6 production and STAT3 activity. Am J Reprod Immunol. 2010;63(5):339‐348. [DOI] [PubMed] [Google Scholar]
- 74. Viall CA, Chen Q, Liu B, et al. Antiphospholipid antibodies internalised by human syncytiotrophoblast cause aberrant cell death and the release of necrotic trophoblast debris. JAutoimmun. 2013;47:45‐57. [DOI] [PubMed] [Google Scholar]
- 75. Zussman R, Xu LY, Damani T, et al. Antiphospholipid antibodies can specifically target placental mitochondria and induce ROS production. JAutoimmun. 2020;111:102437. [DOI] [PubMed] [Google Scholar]
- 76. Chen Q, Viall C, Kang Y, Liu B, Stone P, Chamley L. Anti‐phospholipid antibodies increase non‐apoptotic trophoblast shedding: a contribution to the pathogenesis of pre‐eclampsia in affected women?. Placenta. 2009;30(9):767‐773. [DOI] [PubMed] [Google Scholar]
- 77. Ulrich V, Gelber SE, Vukelic M, et al. ApoE receptor 2 mediation of trophoblast dysfunction and pregnancy complications induced by antiphospholipid antibodies in mice. Arthritis Rheum. 2016;68(3):730‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marchetti T, Ruffatti A, Wuillemin C, de Moerloose P, Cohen M. Hydroxychloroquine restores trophoblast fusion affected by antiphospholipid antibodies. J Thromb Haemost. 2014;12(6):910‐920. [DOI] [PubMed] [Google Scholar]
- 79. Di Simone N, Meroni PL, de Papa N, et al. Antiphospholipid antibodies affect trophoblast gonadotropin secretion and invasiveness by binding directly and through adhered beta2‐glycoprotein I. Arthritis Rheum. 2000;43(1):140‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Velasquez M, Rojas M, Abrahams VM, Escudero C, Cadavid AP. Mechanisms of endothelial dysfunction in antiphospholipid syndrome: association with clinical manifestations. Front Physiol. 2018;9:1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms‐like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111(5):649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alvarez AM, Mulla MJ, Chamley LW, Cadavid AP. Abrahams VM. Aspirin‐triggered lipoxin prevents antiphospholipid antibody effects on human trophoblast migration and endothelial cell interactions. Arthritis Rheum. 2015;67(2):488‐497. [DOI] [PubMed] [Google Scholar]
- 83. Velásquez BM, Álvarez GÁM, Cadavid JÁP. Cuantificación sistematizada de la remodelación vascular in vitro en la morbilidad gestacional asociada al síndrome antifosfolípido. J Revista chilena de obstetricia y ginecología. 2016;81:455‐464. [Google Scholar]
- 84. Di Simone N, Di Nicuolo F, D'Ippolito S, et al. Antiphospholipid antibodies affect human endometrial angiogenesis. Biology of reproduction. 2010;83(2):212‐219. [DOI] [PubMed] [Google Scholar]
- 85. Wei J, Lau SY, Blenkiron C, et al. Trophoblastic debris modifies endothelial cell transcriptome in vitro: a mechanism by which fetal cells might control maternal responses to pregnancy. Sci Rep. 2016;6:30632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Agostinis C, Bulla R, Tripodo C, et al. An alternative role of C1q in cell migration and tissue remodeling: contribution to trophoblast invasion and placental development. J Immunol. 2010;185(7):4420‐4429. [DOI] [PubMed] [Google Scholar]
- 87. Kim MY, Guerra MM, Kaplowitz E, et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis. 2018;77(4):549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. De Carolis S, Botta A, Santucci S, et al. Complementemia and obstetric outcome in pregnancy with antiphospholipid syndrome. Lupus. 2012;21(7):776‐778. [DOI] [PubMed] [Google Scholar]
- 89. Chaturvedi S, Brodsky RA, McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol. 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Alijotas‐Reig J. The complement system as a main actor in the pathogenesis of obstetric antiphospholipid syndrome. Med Clin. 2010;134(1):30‐34. [DOI] [PubMed] [Google Scholar]
- 91. Holers VM, Girardi G, Mo L, et al. Complement C3 activation is required for antiphospholipid antibody‐induced fetal loss. J Exp Med. 2002;195(2):211‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Girardi G, Berman J, Redecha P, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112(11):1644‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Redecha P, Tilley R, Tencati M, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110(7):2423‐2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118(10):3453‐3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tambralli A, Gockman K, Knight JS. NETs in APS: current knowledge and future perspectives. Curr Rheumatol Rep. 2020;22(10):67. [DOI] [PubMed] [Google Scholar]
- 96. Sule G, Kelley WJ, Gockman K, et al. Increased adhesive potential of antiphospholipid syndrome neutrophils mediated by β2 integrin Mac‐1. Arthritis Rheum. 2020;72(1):114‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol. 2014;32(1):66‐70. [PubMed] [Google Scholar]
- 98. Zuo Y, Yalavarthi S, Gockman K, et al. Anti‐neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheum. 2020;72(12):2130‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lu Y, Dong Y, Zhang Y, et al. Antiphospholipid antibody‐activated NETs exacerbate trophoblast and endothelial cell injury in obstetric antiphospholipid syndrome. J Cell Mol Med. 2020;24(12):6690‐6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Meroni PL. Anti‐beta‐2 glycoprotein I epitope specificity: from experimental models to diagnostic tools. Lupus. 2016;25(8):905‐910. [DOI] [PubMed] [Google Scholar]
- 101. Antovic A, Sennstrom M, Bremme K, Svenungsson E. Obstetric antiphospholipid syndrome. Lupus Sci Med. 2018;5(1):e000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Andreoli L, Chighizola CB, Nalli C, et al. Clinical characterization of antiphospholipid syndrome by detection of IgG antibodies against beta2 ‐glycoprotein i domain 1 and domain 4/5: ratio of anti‐domain 1 to anti‐domain 4/5 as a useful new biomarker for antiphospholipid syndrome. Arthritis Rheum. 2015;67(8):2196‐2204. [DOI] [PubMed] [Google Scholar]
- 103. de Laat B, Pengo V, Pabinger I, et al. The association between circulating antibodies against domain I of beta2‐glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009;7(11):1767‐1773. [DOI] [PubMed] [Google Scholar]
- 104. Iwaniec T, Kaczor MP, Celinska‐Lowenhoff M, Polanski S, Musial J. Clinical significance of anti‐domain 1 beta2‐glycoprotein I antibodies in antiphospholipid syndrome. Thromb Res. 2017;153:90‐94. [DOI] [PubMed] [Google Scholar]
- 105. Radin M, Cecchi I, Roccatello D, Meroni PL, Sciascia S. Prevalence and thrombotic risk assessment of anti‐beta2 glycoprotein I domain I antibodies: a systematic review. Semin Thromb Hemost. 2018;44(5):466‐474. [DOI] [PubMed] [Google Scholar]
- 106. Chighizola CB, Pregnolato F, Andreoli L, et al. Beyond thrombosis: anti‐beta2GPI domain 1 antibodies identify late pregnancy morbidity in anti‐phospholipid syndrome. J Autoimmun. 2018;90:76‐83. [DOI] [PubMed] [Google Scholar]
- 107. Liu T, Gu J, Wan L, et al. Anti‐β2GPI domain 1 antibodies stratify high risk of thrombosis and late pregnancy morbidity in a large cohort of Chinese patients with antiphospholipid syndrome. Thromb Res. 2020;185:142‐149. [DOI] [PubMed] [Google Scholar]
- 108. Chighizola C, Raimondo M, Comerio C, et al. The risk of obstetric complications and the effects of treatment in women with low titer and medium‐high titer anti‐phospholipid antibodies. ACR abstracts. 2016. [Google Scholar]
- 109. Schreiber K, Sciascia S, de Groot PG, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4(1):17103. [DOI] [PubMed] [Google Scholar]
- 110. Ostertag MV, Liu X, Henderson V, Pierangeli SS. A peptide that mimics the Vth region of beta‐2‐glycoprotein I reverses antiphospholipid‐mediated thrombosis in mice. Lupus. 2006;15(6):358‐365. [DOI] [PubMed] [Google Scholar]
- 111. de la Torre YM, Pregnolato F, D'Amelio F, et al. Anti‐phospholipid induced murine fetal loss: novel protective effect of a peptide targeting the beta2 glycoprotein I phospholipid‐binding site. Implications for human fetal loss. J Autoimmun. 2012;38(2‐3):J209‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Di Simone N, D'Ippolito S, Marana R, et al. Antiphospholipid antibodies affect human endometrial angiogenesis: protective effect of a synthetic peptide (TIFI) mimicking the phospholipid binding site of β(2) glycoprotein I. Am J Reprod Immunol. 2013;70(4):299‐308. [DOI] [PubMed] [Google Scholar]
- 113. de Groot PG, Meijers JC. β(2) ‐Glycoprotein I: evolution, structure and function. J Thromb Haemost. 2011;9(7):1275‐1284. [DOI] [PubMed] [Google Scholar]
- 114. Balasubramanian K, Chandra J, Schroit AJ. Immune clearance of phosphatidylserine‐expressing cells by phagocytes. The role of beta2‐glycoprotein I in macrophage recognition. J Biol Chem. 1997;272(49):31113‐31117. [DOI] [PubMed] [Google Scholar]
- 115. Nomura S, Komiyama Y, Matsuura E, Kokawa T, Takahashi H, Koike T. Binding of beta 2‐glycoprotein I to platelet‐derived microparticles. Br J Haematol. 1993;85(3):639‐640. [DOI] [PubMed] [Google Scholar]
- 116. Mineo C, Lanier L, Jung E, et al. Identification of a monoclonal antibody that attenuates antiphospholipid syndrome‐related pregnancy complications and thrombosis. PloS one. 2016;11(7):e0158757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Agostinis C, Durigutto P, Sblattero D, et al. A non‐complement‐fixing antibody to beta2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. 2014;123(22):3478‐3487. [DOI] [PubMed] [Google Scholar]
- 118. Ioannou Y, Rahman A. Domain I of beta2‐glycoprotein I: its role as an epitope and the potential to be developed as a specific target for the treatment of the antiphospholipid syndrome. Lupus. 2010;19(4):400‐405. [DOI] [PubMed] [Google Scholar]
- 119. Pericleous C, Ruiz‐Limon P, Romay‐Penabad Z, et al. Proof‐of‐concept study demonstrating the pathogenicity of affinity‐purified IgG antibodies directed to domain I of beta2‐glycoprotein I in a mouse model of anti‐phospholipid antibody‐induced thrombosis. Rheumatology. 2015;54(4):722‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. McDonnell TCR, Willis R, Pericleous C, et al. PEGylated domain i of beta‐2‐glycoprotein I inhibits the binding, coagulopathic, and thrombogenic properties of IgG from patients with the antiphospholipid syndrome. Front Immunol. 2018;9:2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sammaritano LR, Bermas BL, Chakravarty EE, et al. 2020 American college of rheumatology guideline for the management of reproductive health in rheumatic and musculoskeletal diseases. Arthritis Rheum. 2020;72(4):529‐556. [DOI] [PubMed] [Google Scholar]
- 122. Costantine MM, Cleary K, Hebert MF, et al. Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high‐risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214(6):720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bateman BT, Hernandez‐Diaz S, Fischer MA, et al. Statins and congenital malformations: cohort study. BMJ (Clinical research ed). 2015;350:h1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Costantine MM, Tamayo E, Lu F, et al. Using pravastatin to improve the vascular reactivity in a mouse model of soluble fms‐like tyrosine kinase‐1‐induced preeclampsia. Obstet Gynecol. 2010;116(1):114‐120. [DOI] [PubMed] [Google Scholar]
- 125. Lefkou E, Mamopoulos A, Dagklis T, Vosnakis C, Rousso D, Girardi G. Pravastatin improves pregnancy outcomes in obstetric antiphospholipid syndrome refractory to antithrombotic therapy. J Clin Invest. 2016;126(8):2933‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lefkou E, Varoudi K, Pombo J, et al. Triple therapy with pravastatin, low molecular weight heparin and low dose aspirin improves placental haemodynamics and pregnancy outcomes in obstetric antiphospholipid syndrome in mice and women through a nitric oxide‐dependent mechanism. Biochem Pharmacol. 2020;182:114217. [DOI] [PubMed] [Google Scholar]
- 127. Liu Z, Sun S, Xu H, et al. Prognostic analysis of antibody typing and treatment for antiphospholipid syndrome‐related recurrent spontaneous abortion. Int J Gynaecol Obstet. 2021;00:1‐6. [DOI] [PubMed] [Google Scholar]
- 128. Rand JH, Wu XX, Quinn AS, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood. 2010;115(11):2292‐2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Espinola RG, Pierangeli SS, Gharavi AE, Harris EN. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost. 2002;87(3):518‐522. [PubMed] [Google Scholar]
- 130. Ruffatti A, Hoxha A, Favaro M, et al. Additional treatments for high‐risk obstetric antiphospholipid syndrome: a comprehensive review. Clin Rev Allergy Immunol. 2017;53(1):28‐39. [DOI] [PubMed] [Google Scholar]
- 131. Carreras LD, Perez GN, Vega HR, Casavilla F. Lupus anticoagulant and recurrent fetal loss: successful treatment with gammaglobulin. Lancet. 1988;2(8607):393‐394. [DOI] [PubMed] [Google Scholar]
- 132. McVerry BA, Spearing R, Smith A. SLE anticoagulant: transient inhibition by high dose immunoglobulin infusions. Br J Haematol. 1985;61(3):579‐580. [DOI] [PubMed] [Google Scholar]
- 133. Caccavo D, Vaccaro F, Ferri GM, Amoroso A, Bonomo L. Anti‐idiotypes against antiphospholipid antibodies are present in normal polyspecific immunoglobulins for therapeutic use. J Autoimmun. 1994;7(4):537‐548. [DOI] [PubMed] [Google Scholar]
- 134. Sherer Y, Levy Y, Shoenfeld Y. Intravenous immunoglobulin therapy of antiphospholipid syndrome. Rheumatology. 2000;39(4):421‐426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.