

Abstract
Alzheimer’s disease (AD) is a primary neurological disease with no effective cure. A hallmark of AD is the presence of intracellular tangles and extracellular plaques derived from the aberrant aggregation of tau- and beta-amyloid (Aβ). Aβ presents in the brain as well as in cerebrospinal fluid and the circulation, and Aβ toxicity has been attributed to amyloidosis and inflammation, among other causes. In this study, the effects of the plasma protein corona have been investigated with regard to the blood cell association and cytokine secretion of oligomeric (Aβo) and fibrillar Aβ1–42(Aβf), two major forms of the peptide aggregates. Aβo displayed little change in membrane association in whole blood or washed blood (i.e., cells in the absence of plasma proteins) at 37 °C, while Aβf showed a clear preference for binding with all cell types sans plasma proteins. Immune cells exposed to Aβo, but not to Aβf, resulted in significant expression of cytokines IL-6 and TNF measured in real-time by a localized surface plasmon resonance sensor. These observations indicate greater immune cell association and cytokine stimulation of Aβo than Aβf and shed new light on the contrasting toxicities of Aβo and Aβf resulting from their differential capacities in acquiring a plasma protein corona. These results further implicate a close connection between Aβ amyloidosis and immunopathology in AD.
Graphical Abstract
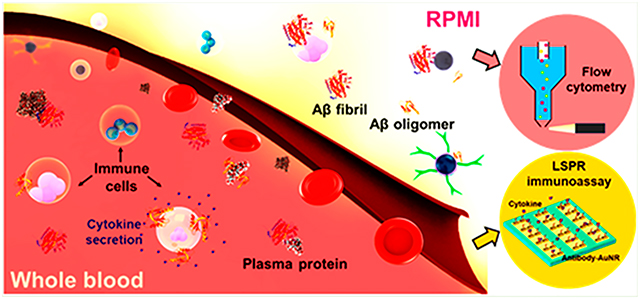
INTRODUCTION
Beta amyloid (Aβ) originates from amyloid precursor protein (APP), an integral membrane protein expressed in tissues and especially in the brain. The APP is cleaved off by β and γ secretases, yielding two major peptide products Aβ1–40 and Aβ1–42.1 Among the two peptides, Aβ1–42 is more hydrophobic due to the two additional amino acids of isoleucine and alanine at the C-terminus and is considerably more cytotoxic due to its higher tendency in aberrant aggregation. The extracellular amyloid deposits of Aβ and the intracellular tangles of tau are two histopathological hallmarks of Alzheimer’s disease (AD), a primary form of neurological disorder in aging populations.2
While Aβ plaques are often located in the brain tissues of AD patients postmortem, the peptide itself can also be traced in cerebrospinal fluid (CSF) and the circulation.3 The relationship between the peptide levels in the brain, CSF, and plasma in healthy and diseased individuals, however, remains unclear.4 Compelling evidence has shown the transport of Aβ by serum albumin and apolipoproteins in the blood plasma,5,6 likely due to the poor solubility of the peptide as well as the chaperone-like capacity of serum albumin7–9 against the conformational changes of Aβ. Furthermore, while specific receptors on microglia and monocytes/macrophages in the brain may determine the clearance of extracellular Aβ peptides through noninflammatory phagocytosis or pro-inflammatory cytokine secretion,10–14 adherence of complement C3b to complement receptor 1 (CR1) of erythrocytes is a mechanism hypothesized for the peripheral clearance of Aβ.15
The literature has suggested the use of Aβ in the blood as an effective indicator for the early diagnosis of AD.16 The effect of human serum albumin (HSA) on the reduction of α-synuclein aggregation (associated with Parkinson’s disease) has been reported recently.17 However, the associations of plasma proteins and blood cells with Aβ in its major aggregation forms, namely, oligomers and amyloid fibrils (abbreviated as AβO and Aβf hereafter, to refer to the chiefly oligomeric and fibrillar forms of the peptide), remain unclear. Furthermore, the immune responses of blood cells to amyloidogenic peptides and their aggregates have not been systematically investigated. To understand the transformation of Aβ in circulation, here we examined the binding of human blood cells with AβO and Aβf with a special attention to plasma proteins, using a high-throughput blood association assay.18,19 We further characterized real-time secretion of cytokines, i.e., interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF), by human monocytes and lymphocytes exposed to AβO and Aβf, using a localized surface plasmon resonance (LSPR) immunoassay.20 Both our blood cell and LSPR immune assays implicated a significant role of plasma protein corona in shaping the cell binding affinity and toxicity of amyloid-protein aggregates and demonstrated a convoluted relationship between amyloidosis and inflammation in AD.
MATERIALS AND METHODS
Aβ preparation.
Hexafluoro-2-propanol (HFIP)-treated human Aβ1–42 (AnaSpec, sequence, AIAEGDSHVLKEGAY-MEIFDVQGHVFGGKIFRVVDLGSHNVA; purity, HPLC ≥ 95%; abbreviated as Aβ hereafter) was used in preparation of the two aggregating states of Aβ. Specifically, AβO was rendered by incubating the freshly dissolved Aβ in 0.003% NH4OH buffer at room temperature for 30 h, while Aβf was obtained by incubating the peptide at 37 °C for more than 60 h. The two aggregation states were confirmed by a ThT kinetic assay and TEM imaging.
Transmission Electron Microscopy (TEM).
For TEM imaging, 5 μL of AβO and Aβf (each of 50 μM), plasma proteins, Aβo, and Aβf in interaction with plasma proteins were pipetted onto copper grids (400 mesh, glow-discharged for 15 s; Formvar film, ProSciTech) and let to adsorb for 1 min. After removing unbound samples by filter paper the grids were rinsed with 10 μL of Milli-Q water. The grids were then negatively stained with 5 μL of 1% uranyl acetate (UA) for 30 s and blown dry. The samples were imaged by a transmission electron microscope (Tecnai G2 F20, FEI, Eindhoven, The Netherlands) under an electric potential of 200 kV. Images were acquired with a CCD camera (UltraScan 1000, Gatan).
Thioflavin T (ThT) Kinetic Assay.
A kinetic assay on peptide fibrillization was conducted using 50 μM Aβ and 100 μM ThT dye pipetted into a 96-well plate (Costar). Changes in ThT fluorescence, indicating the β-sheet content in the sample, were recorded at 37 °C until the saturation phase after 60 h by a plate reader (PerkinElmer EnSight HH33400; Ex/Em, 440/485 nm). The assay was done with triplicate for each sample condition.
Cell Culture and Toxicity Assay.
SH-SY5Y neuroblastoma cells were cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMED/F12, ATCC) with 10% fetal bovine serum (FBS). For the viability assay, a 96-well plate (Costar) was pretreated with poly-l-lysine (Sigma, 0.01%), incubated at 37 °C (5% CO2) for 30 min, and washed with phosphate buffered saline (PBS) thrice. Approximately 60 000 cells were added to each well and incubated at 37 °C and 5% CO2 to reach 80% confluency. Propidium iodide (PI, 1 μM) dye in fresh DMEM/F12 was added and incubated with the cells for 30 min. After optimization of concentrations, samples of 20 μM Aβ in the form of oligomers or fibrils were added to the wells. After 15 h of treatment, the cell viability was read by an Operetta instrument (PerkinElmer, 20× lens; numerical aperture, 0.7) at 37 °C with 5% CO2. The PI-positive apoptotic cells were counted by the mapping function of the instrument. Nine reads per well for samples of triplicate were acquired. Untreated cells were imaged as the control.
Helium Ion Microscopy (HIM).
Whole and washed blood cells were treated with 10 μM AβO and Aβf with and without plasma proteins. The samples were then treated by 2.5% paraformaldehyde and incubated at 4 °C for 10 h. After the incubation, the samples were centrifuged, and paraformaldehyde/medium was replaced every 2 h with gradient concentrations of ethanol (20%, 40%, 60%, 80%, and 95%). A 30 μL portion of each sample was transferred to a carbon tape and air-dried. The cell morphologies were visualized by Orion NanoFab (Zeiss).
Association of Aβ Species with Human Immune Cells.
Fresh blood was drawn from a healthy donor into sodium heparin Vacuettes (Greiner Bio-One) in accordance with the University of Melbourne Human ethics approval 1443420 and the Australian National Health and Medical Research Council Statement on Ethical Conduct in Human Research. The blood cells were counted using a CELL-DYN Emerald analyzer (Abbott). To prepare washed blood cells, 10 mL of whole blood was topped up to 50 mL with PBS and spun down at 950g, for 10 min, with slow brake. The process was repeated four more times to collect washed blood cells. The removal of plasma proteins from the cells was indicated using a UV–vis spectrophotometer (Nanodrop 2000, Thermo Fisher), where the protein absorbance at 280 nm was absent to confirm their removal. The cells were resuspended in serum-free RPMI 1640 (Gibco) to keep the cell concentration of whole and washed blood consistent. ThT-labeled AβO or Aβf was added to 100 μL of blood in triplicate at 5, 10, and 15 μM and incubated at 4 and 37 °C for 1 h. Pharm Lyse buffer (BD) was added to lyse red blood cells at 40× blood volume, followed by washing twice with 4 mL of PBS (500g, 7 min). The cells were phenotyped for 1 h on ice by titrating antibodies against CD3 AF700 (SP34-2, BD), CD14 APC-H7 (MΦP9, BD), CD56 PE (B159, BD), lineage-1 cocktail FITC (BD), HLA-DR PerCP-Cy5.5 (G46–6, BD), and CD19 BV650 (HIB19, Biolegend). Free antibodies were washed and removed by centrifugation (500g, 7 min) with a PBS buffer containing 0.5% w/v BSA and 2 mM EDTA at 4 °C. The cells were fixed by formaldehyde in PBS at 1% weight to volume ratio. The samples were analyzed for cell association using flow cytometry (LSRFortessa, BD Biosciences) and software FlowJo V10.
LSPR Detection of Immune Cell Responses to Aβ Species.
Cell Culture.
Jurkat human T cells (ATCCCRL-2901) were cultured in RPMI-1640 medium (ATCC) with 200 μg/mL G428 and 10% FBS (ATCC). The cells were incubated at 37 °C with 5% CO2 (Thermo Scientific). Epstein–Barr virus transformed human B lymphoblasts (ATCC) were cultured in RPMI-1640 medium with 10% FBS. Human monocytic THP-1 cells (ATCC) were cultured in RPMI-1640 medium with 50 μM mercaptoethanol and 10% FBS. The cell culture was maintained by replenishing the medium every 2–3 days at 1 × 105 to 1 × 106 cells/mL. The cells were collected by centrifugation (125g, 5 min) and resuspended in fresh culture medium.
LSPR Immunoassay.
The human immune cell lines (T cells, B cells, or THP-1 cells) were resuspended in human plasma proteins (Innovative Research) and RPMI-1640 medium at 1 × 106 cells/mL, respectively. The immune cells were incubated with Aβo and Aβf of 5, 10, and 15 μM final concentrations for 2 h at 4 and 37 °C. The immune responses of human THP-1 cells, human Jurkat T cells, and human B cells were investigated after stimulations with AβO and Aβf. After 2 h of incubation, the culture medium was extracted and pipetted into an LSPR immunoassay chip (for fabrication and measurement details, see the description in the Supporting Information, as well as Figure S1) for the detection of secretory cytokines from the immune cells. A total of more than 100 chips were used for this assay.
RESULTS AND DISCUSSION
Interactions of Aβ Aggregates with Plasma Proteins.
Due to the kinetic nature of amyloidosis, which is a convolution of both primary and secondary nucleation followed by elongation and saturation,1 the AβO and Aβf samples were not pure oligomers or fibrils but were inclusive of a collection of minor heterogeneous aggregates. Indeed, the reverse phase HPLC (RP-HPLC; for the method, see the Supporting Information) revealed formation of slightly hydrophobic Aβo from its hydrophilic monomeric origin (centered on the retention time of ~20 min). As for the Aβf sample, the monomeric and oligomeric species were depleted and converted into fibrillar structures of varying hydrophobicity (Figure S2).
The interaction of Aβ with blood components is of intense interest as it offers a model to understand the immune response to amyloid proteins. Previously, Kuo et al. reported the interactions of fresh Aβ1–40 and Aβ1–42 with plasma proteins.21 Here, our TEM imaging of plasma proteins, and Aβo and Aβf with and without plasma proteins confirmed the two aggregation states as globules and fibrils (Figure 1a,b), respectively. TEM imaging showed the morphologies of plasma proteins (Figure 1c) and their associations with the Aβ species (Figure 1d,e). Specifically, in the case of Aβf, a protein “corona” was rendered upon the peptide incubation with plasma proteins, mediated by H-bonding and electrostatic interactions between the Aβf surface moieties and the amphiphilic plasma proteins.22
Figure 1.
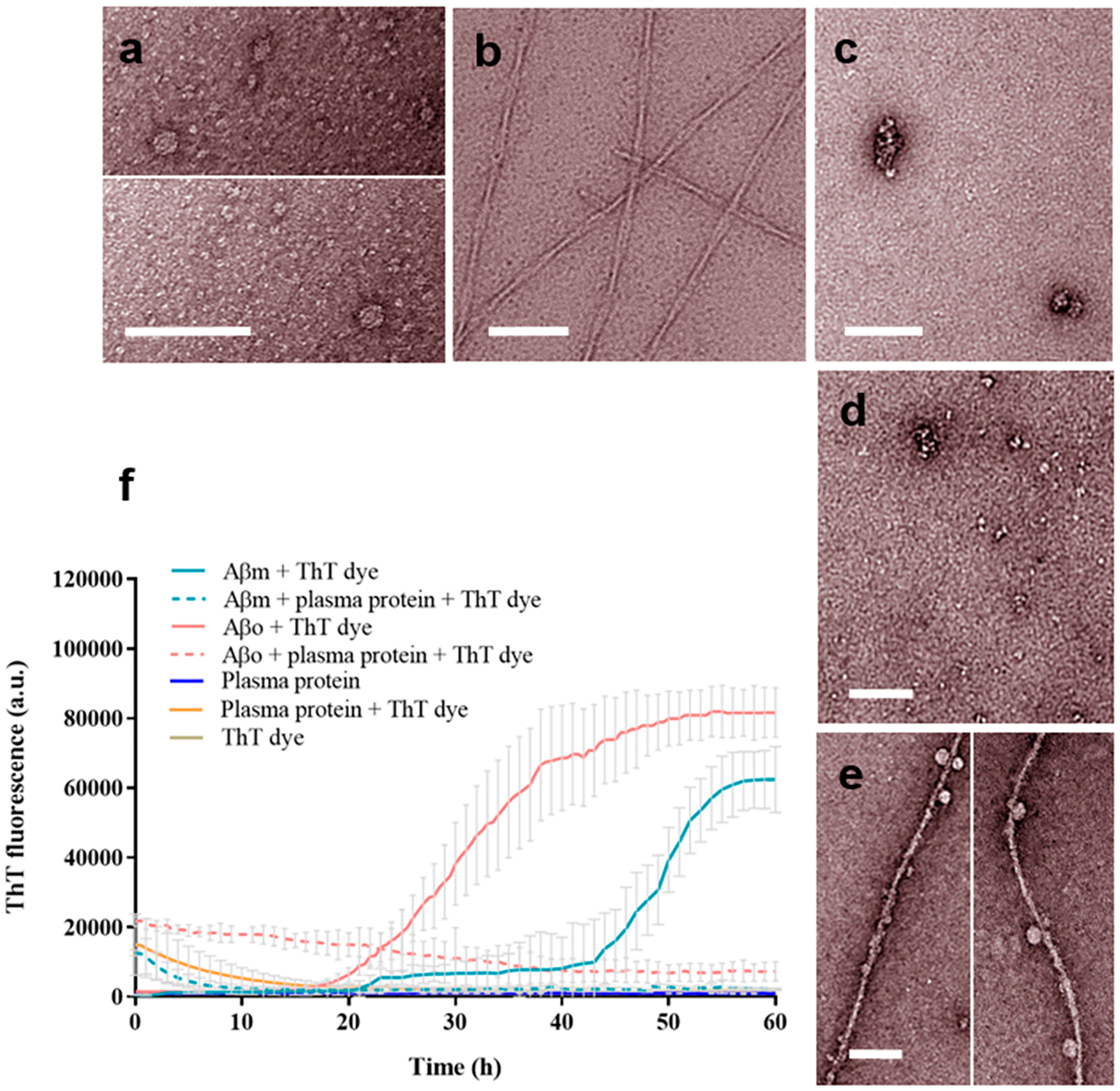
TEM imaging and ThT kinetic assay of Aβ fibrillization. TEM images show the following structures: (a) Aβo, (b) Aβf, (c) plasma proteins, (d) Aβo with plasma proteins, and (e) Aβf with plasma proteins. The experiments were performed in triplicate. The error bars indicate the standard deviations of averaged data sets. Aβ concentration: 50 μM (ThT assay, 37 °C) and 20 μM (TEM, at room temperature). Scale bars: 100 nm. (f) ThT kinetic assay of Aβm and Aβo fibrillization with and without plasma proteins.
Aβ Aggregation Kinetics and Cytotoxicity.
The ThT kinetics of Aβm and AβO displayed a nucleation phase, followed by an elongation phase to a saturation phase of the peptide after ~60 h (Figure 1f). This result is consistent with the Aβ fibrillization in the literature.23–25 In the presence of plasma proteins, interactions of both Aβm and AβO with the proteins blocked the addition of Aβ monomers to inhibit the further assembly of the oligomers into amyloid fibrils.
Aggregation of amyloid proteins is a hallmark of neuro-degenerative diseases and type 2 diabetes. Aβ aggregation is associated with neuronal cell degeneration,26–28 and the Aβ oligomers are considered to be the most toxic species.29,30 The cytotoxicity of Aβ in this study was consistent with the literature and with the aggregation inhibition of Aβ in interaction with plasma proteins (Figure 2a), revealing suppressed toxicity of AβO with plasma proteins (39 ± 2.6% without plasma proteins down to 30 ± 2% with plasma proteins). In comparison, Aβf induced a modest 13% cytotoxicity which was not affected by plasma proteins.
Figure 2.
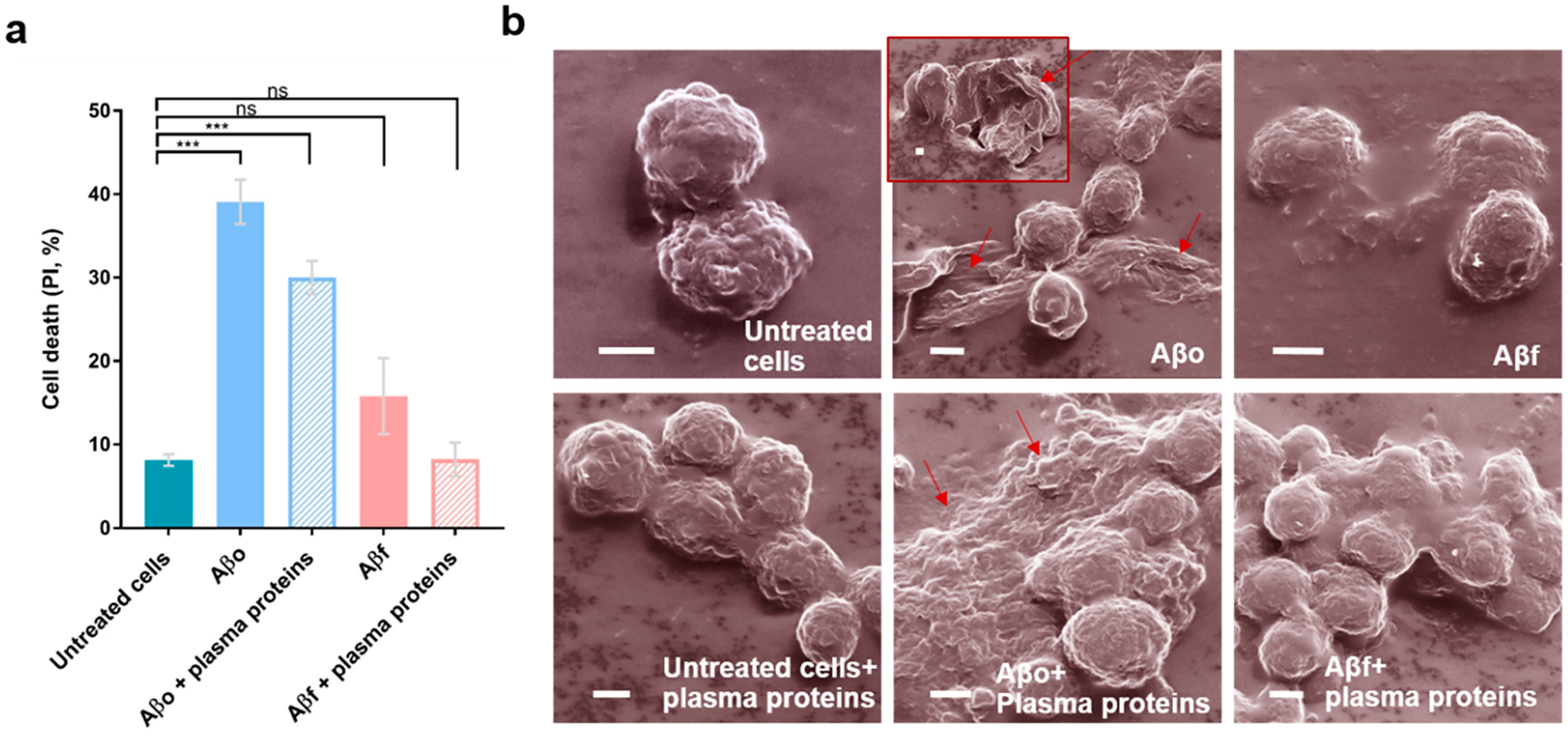
Viability and morphology of neuronal cells exposed to the Aβ species with or without plasma proteins. (a) SY5Y cell toxicities exposed to Aβo and Aβf. PI: propidium iodide. (b) Helium ion microscopy indicates the toxicity of Aβ species with and without plasma proteins. Arrows indicate the deformation of cell membranes induced by Aβo. Cells were treated with the Aβ species for 2 h, with or without plasma proteins. The experiment was performed in triplicate, and error bars indicate standard deviations (ns, P > 0.05; and ***, P ≤ 0.001). Aβ concentration: 20 μM. Scale bars: 2 μm.
Aβ Aggregation-Induced Membrane Damage.
The morphologies of SH-SY5Y neuronal cells exposed to the two Aβ aggregating species were examined with helium ion microscopy (HIM). HIM utilizes a helium ion source to excite a small sample volume with a large depth and is advantageous to conventional scanning electron microscopy in both resolution and image brightness. Consistent with the viability assay, significant damage including membrane deformation and blebbing of the SH-SY5Y cells was observed upon their exposure to AβO (Figure 2b), while such damage was reduced in the presence of Aβo incubated with plasma proteins. In comparison, no damage was evident when the cells were exposed to Aβf with or without plasma proteins.
Cell Associations with Aβ Aggregates.
In recent research, AD is considered as more than a neural-centric disease but has its origin in the immune system and inflammation.31,32 To better understand the interaction between Aβ and blood immune cells, the association of fresh human blood phagocytes (granulocytes, monocytes, and dendritic cells) and lymphocytes (T cells, B cells, and natural killer/NK cells) with AβO and Aβf in the presence and absence of plasma proteins was assessed by flow cytometry (Figures 3 and 4). The Aβ structures displayed no apparent association with any type of the immune cells at 4 °C in either whole blood or washed blood. An increment of temperature from 4 to 37 °C, which elevated biomolecular diffusion as well as the biological processes of endocytosis and phagocytosis uptake,19 gave rise to increased association of Aβ with all types of the immune cells. Although the most prominent association of the Aβ proteins was with blood immune cells of phagocytic nature (granulocytes, monocytes, and dendritic cells), substantial association was also observed for B and T lymphocytes (Figure 4).
Figure 3.
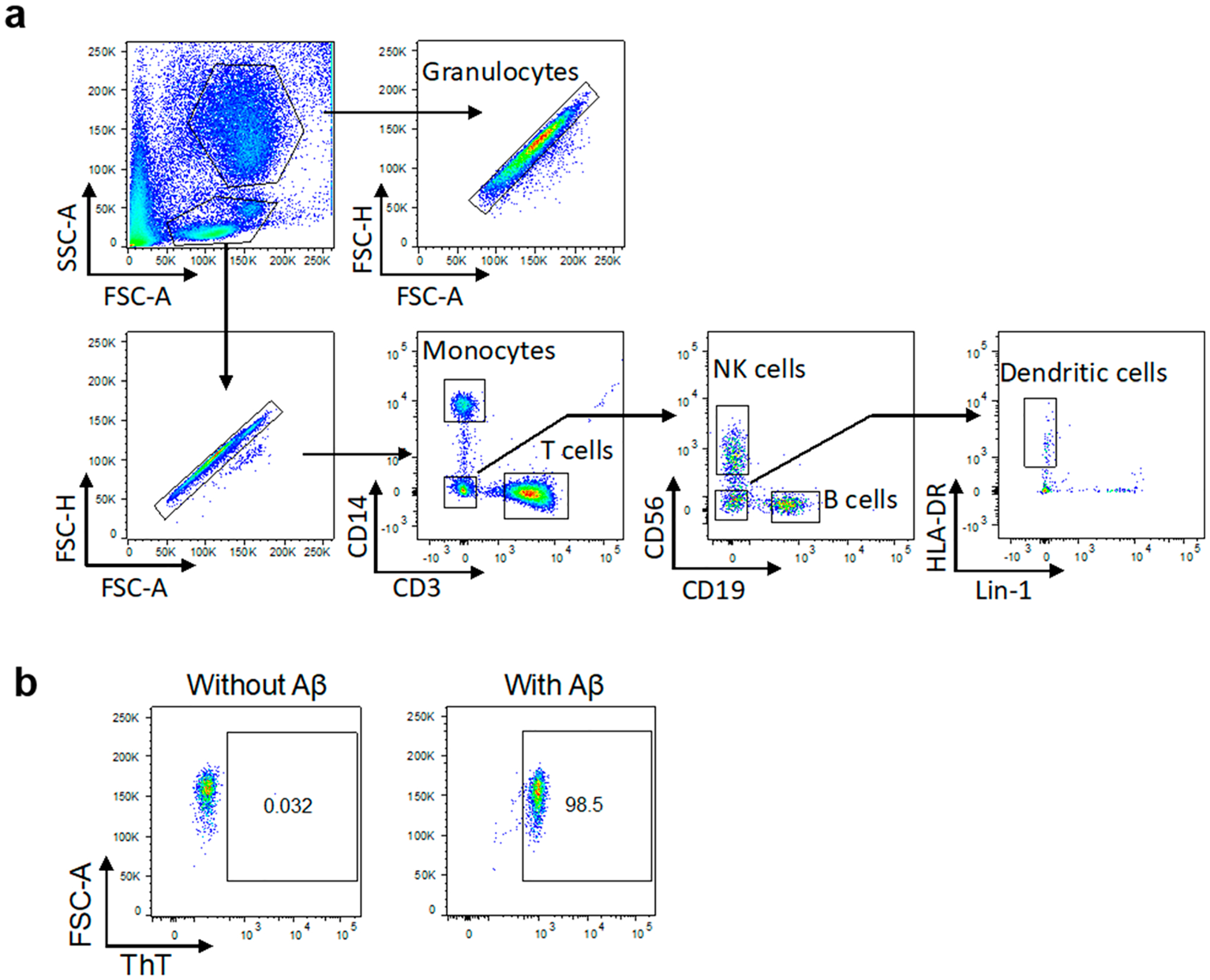
Gating strategy used to determine white blood cell populations and amyloid association. (a) Representative gating strategy used to determine white blood cell populations. Forward scatter area (FSC-A) and side scatter area (SSC-A) were first analyzed to locate white blood cells. Doublets were excluded based on the FSC-A vs FSC-H of single cells gated. The following cell types were identified using sequential gating based on expression of surface markers or light scatter: high SSC-A granulocytes; CD3+ T cells; CD14+ monocytes; CD56+ NK cells; CD19+ B cells; and Lin1-veHLA-DR+ dendritic cells. (b) Cell association with ThT-labeled Aβ was then measured, and an example of the gating is shown for the monocyte population. Further examples can be found in the Supporting Information, Figure S3.
Figure 4.
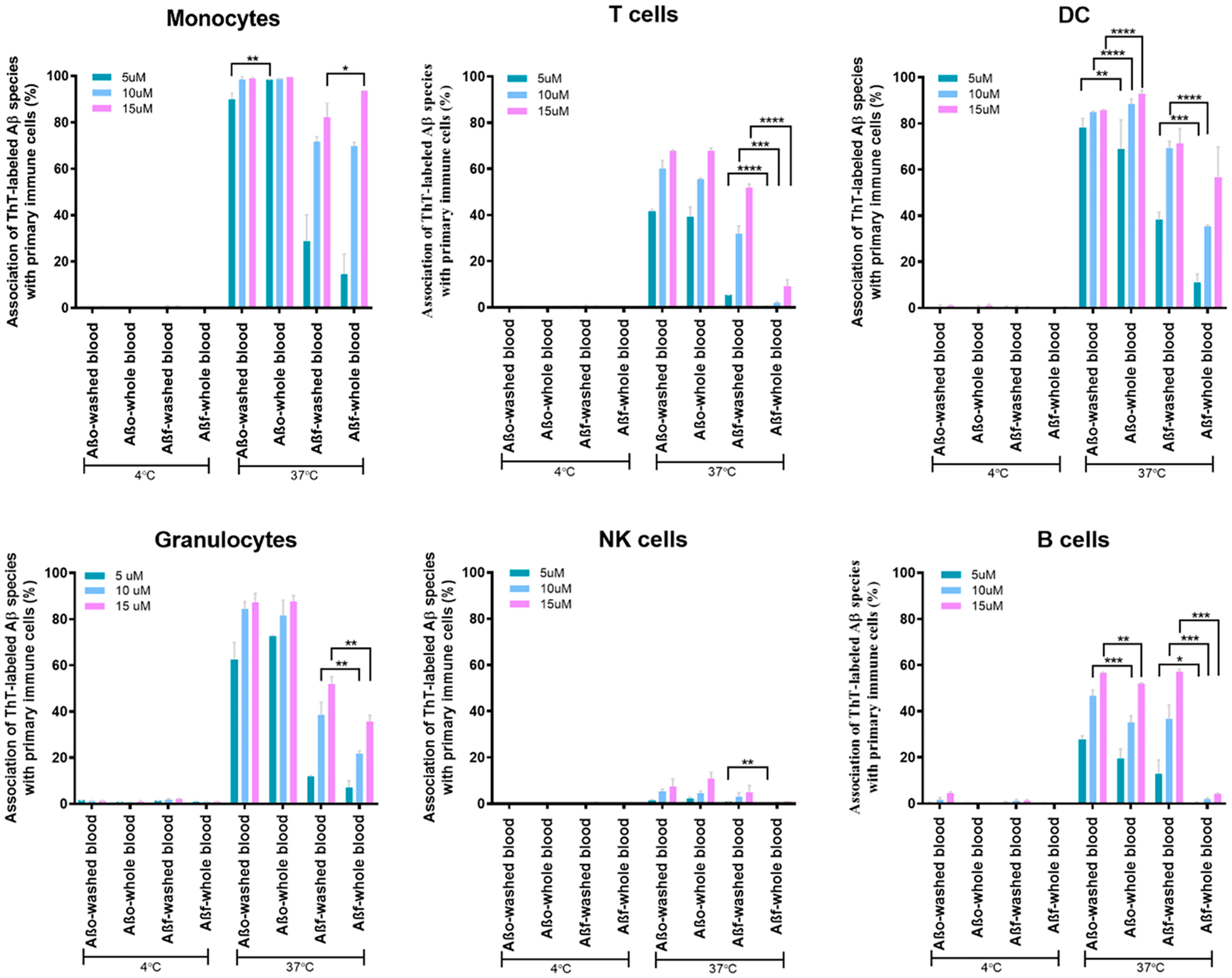
Association of the Aβ species with immune cells. The graphs show the association of immune cells as monocytes, T cells, dendritic cells, granulocytes, NK cells, and B cells with ThT-labeled Aβo and Aβf in 3 different concentration of 5, 10, and 15 μM. The treatments with Aβo and Aβf were applied at 4 and 37 °C in whole blood (with plasma proteins) and washed blood (plasma proteins removed). Association of Aβ with cell membranes was significantly lower in whole blood than washed blood. The assay was performed in triplicate. The error bars show standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001).
The amphiphilic structures of oligomers and protofibrils, known as the most toxic species of Aβ,1 are less prone to interact with the hydrophilic surfaces of plasma proteins and biomolecules. On the other hand, amyloid peptides and proteins show a high propensity for cell membranes by initiating contact via the N termini of their functional monomers, which triggers their structural transition from monomers to alpha helices, toxic oligomers, and protofibrils, and, eventually, β-sheet rich amyloid fibrils.1 Consistent with these known biochemical properties, we found that AβO displayed strong and comparable associations with the neuronal cells in both whole blood and washed blood cells(i.e., with plasma proteins removed, Figure 4). In contrast, Aβf showed less association with the immune cells compared to AβO. We hypothesized that this is likely due to the high capacity of Aβf to establish hydrogen bonds with free plasma proteins, forming a nonspecific protein corona around the Aβf when incubated with whole blood.33 Indeed, we observed that Aβf association with immune cells in whole blood (with plasma proteins) was generally greater than that observed in washed blood (without plasma proteins). This was particularly evident in the lymphocyte population (T, B, NK cells) where association was almost completely abrogated, with a less marked effect on granulocytes and dendritic cells.
The association of Aβ aggregates with plasma proteins entails both physical and toxicological implications. Serum albumin, the most abundant protein in the plasma, has been shown to inhibit Aβ1–40 aggregation.34,35 The higher level of toxic Aβ association with cell membranes can result in an elevated response from monocytes among immune cells.34 Previous studies showed that the accumulations of Aβ, microglia, as well as blood monocyte macrophages were significantly involved in anti-inflammatory response to excess Aβ.36,37 In this study, we noted a high level of interaction between the Aβ species and monocytes for all conditions (i.e., 90–100% for all concentrations of AβO and 20–40% and 60–80% for low and high concentrations of Aβf, respectively; Figure 4). Overall, the monocytes displayed the highest association with Aβo compared to other types of blood cells.
In addition to monocytes, recruitment of T cells at Aβ plaques has been reported.38 T cells play a major role in the pathophysiology of AD,39 where the cellular response induced a near complete clearance of Aβ.38 In addition, research has shown that AD patients exhibit an elevated T cell response to Aβ as compared to middle-aged healthy individuals.40 The telomere length of T cells is dysregulated in AD patients, which may have consequential effects on the immune system and the brain.41 In the present study, strong associations of AβO (i.e., 37–41%, 55–60%, and 66–67% for the three chosen concentrations) with T cells, independent of the presence of plasma proteins, were observed. In comparison, Aβf showed lower interactions with the T cells (5.2 ± 0.1%, 38.2 ± 3.2%, and 51.8 ± 1.5% for the three chosen concentrations) without plasma proteins, and no significant interactions (<10%) with plasma proteins (Figure 4).
Dendritic cells (DCs), an initiator of adaptive immune responses, were reported to endocytose amyloid fibrils.14 While the age-related behavior of DCs in enhancing peripheral inflammation has been documented, no significant changes in the secretion of chemokines or cytokines have been recorded with Aβ fibrils.42 Here, association of 78–85% of AβO in the absence of plasma proteins and stronger association of 69–93% of Aβo with DCs were recorded in whole blood cells. Aβf, in contrast, showed weaker interactions with DCs than Aβo at 11.1 ± 3.7%, 35 ± 0.7%, and 56.5 ± 13% and 38.2 ± 3.3%, 69.2 ± 3.1%, and 71.2 ± 6.4% for the three concentrations with and without plasma proteins, respectively (Figure 4).
Previous studies revealed hindered circulation of low-density granulocytes in AD-type dementia patients, suggesting a damaging effect of AD on inflammatory cells in the periphery.43 In the current study, a strong association of AβO with granulocytes (80–90%) was recorded with no considerable difference with and without plasma proteins. In the case of Aβf, the associations were determined to be 7.0 ± 2.9%, 21.6 ± 1.2%, and 35.7 ± 2.6% and 11.7 ± 0.4%, 38.5 ± 5.4%, and 51.8 ± 3.1% with and without plasma proteins, respectively (Figure 4). In addition to the blood cells characterized, a very low association of NK cells with AβO and no association with Aβf, especially with plasma proteins, were observed.
The production of antibodies by B cells toward Aβ42 protofibrils has been shown to be much enhanced in AD patients as compared to healthy individuals.44 In the present study, both Aβ species displayed interactions with B cells. Specifically, association of Aβo with B cells slightly decreased from whole blood to washed cells for the 3 concentrations of Aβo and significantly dropped from 12.8 ± 5.9%, 36.6 ± 6.0%, and 57.1 ± 0.9% to below 5% for the 3 concentrations of Aβf, respectively (Figure 4).
Aβo- and Aβf-Induced Immune Responses with and without Plasma Proteins.
The deposition of Aβ peptide has been revealed to trigger a range of inflammation responses from immune cells to express cytokines and chemokines.45 Laboratory and clinical investigations have shown evidence of increased release of pro-inflammatory cytokines in both the brain and plasma of AD patients.46 In addition, plasma proteins including HSA have been demonstrated to mitigate Aβ amyloidosis and participate in AD-incited inflammatory responses in the brain. HSA binds 90% of plasma Aβ and could potentially affect their molecular distribution and pharmacokinetics.47 Unveiling the influence of plasma proteins on the Aβ peptide-induced immune response and understanding the pro-inflammatory cytokine release profiles upon stimulation may offer new insights into Aβ-induced cytotoxicity to facilitate the development of AD therapy. Conventional cytokine detection methods such as enzyme-linked immunosorbent assay (ELISA) usually require laborious regent processing procedures, including multiple steps of staining, washing, and blocking, and posing challenges in real-time analysis of dynamic molecular release processes.20 In this study, we performed a label-free microfluidic-based LSPR immunoassay for real-time detection of multiple cytokines secreted by three types of immune cells, after coculturing the cells with AβO or Aβf. The immune responses induced by AβO and Aβf with and without plasma proteins were assessed by the levels of cytokine secretion.
Immune cell lines (T cells, B cells, or monocytic cells) were incubated with the two types of Aβ aggregates in human plasma and RPMI medium at 4 and 37 °C, respectively. LSPR immunoassay chips consisting of antibody-functionalized gold nanorods (AuNRs) were used to detect TNF and IL-6 secreted by the immune cells (Figure 5a). Cytokine binding with AuNR–antibody conjugate altered the localized-refractive index and enlarged the scattering cross section of the nanostructure. The plasmon resonance gave rise to a red shift in the scattering spectrum, coupled with an increased scattering intensity (Figure 5b). Images of the sensing spot arrays were acquired in real-time by an ultrasensitive electron multiplying CCD (EMCCD, Photometrics). The scattering intensities of the sensing spots were processed by MATLAB and converted to cytokine concentrations based on pre-established calibrations (Figure 5c). More pronounced inflammatory responses were observed for immune cells exposed to the Aβ species at 37 °C than at 4 °C due to higher cell activity and metabolism (Figure 6). Specifically, AβO induced overall heightened immune responses as compared to Aβf at 37 °C, suggesting stronger interaction and toxicity of the former with the immune cells. THP-1 cells (as a model system for monocytes), in particular, displayed a prominent pro-inflammatory cytokine secretion upon exposure to AβO, consistent with the observed monocyte association with the Aβ aggregate (Figure 4).
Figure 5.
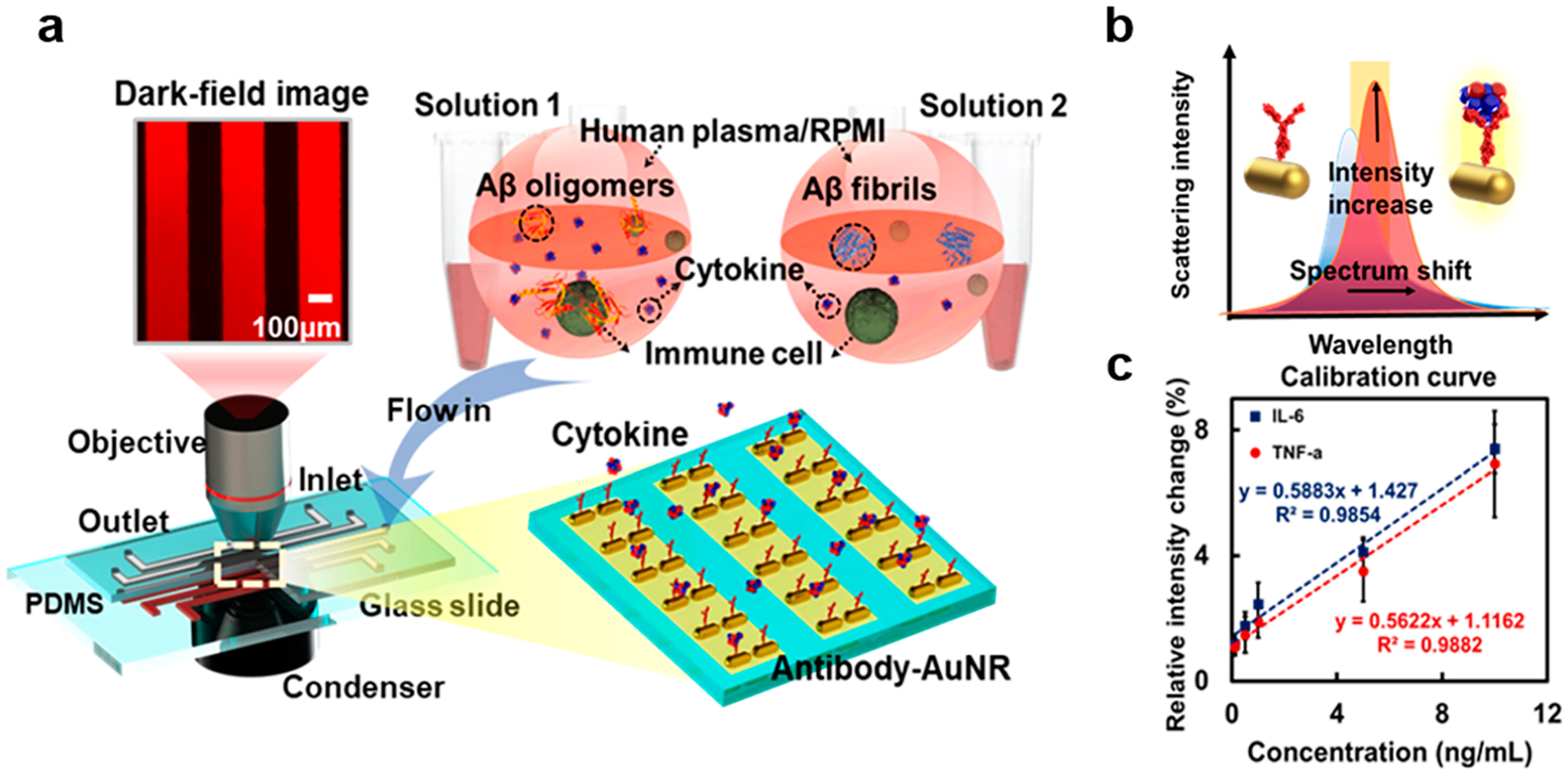
Schematics of the LSPR immunoassay. (a) LSPR immunoassay on Aβo- and Aβf-induced immune responses. Human immune cells (T cells, B cells, or THP-1 cells) were incubated with Aβo and Aβf in human plasma or RPIM medium, respectively. The cellular responses were compared by detecting secreted concentrations of TNF and IL-6 in the medium using LSPR microfluidic chips in dark field. (b) Cytokine binding with AuNR–antibody conjugates altered the local refractive index and scattering cross section of the nanostructure, giving rise to a red shift in the SRP peak coupled with an elevated scattering intensity. The cytokines secreted were determined by changes to the scattering light intensity using an EMCCD. (c) Calibrations of cytokines TNF and IL-6 showing the relative intensity changes of the LSPR barcode with varied cytokine concentrations. The trendline equations and R2 values are indicated. AuNR: gold nanorod.
Figure 6.
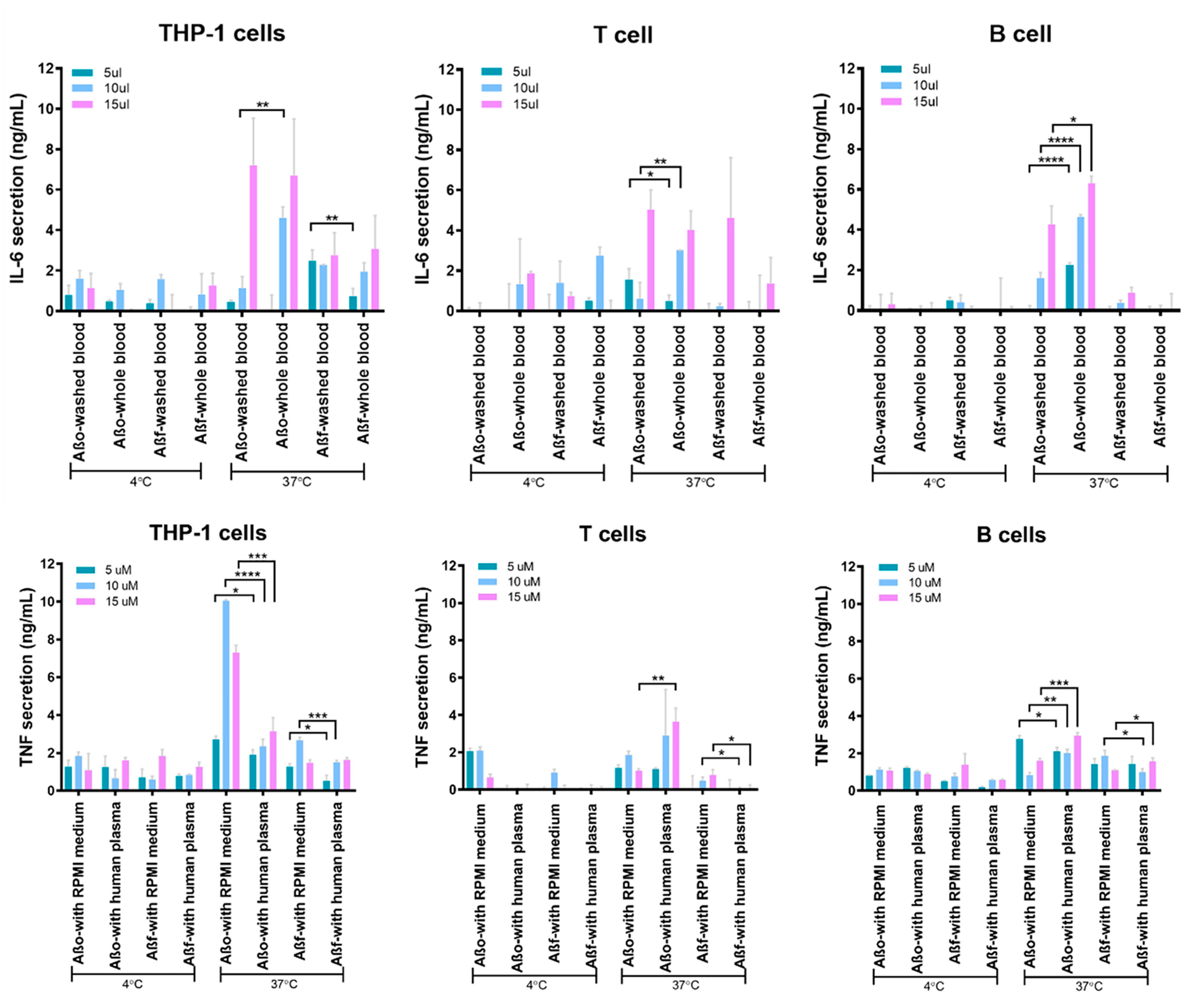
Aβ-induced immune responses of human immune cells. Cytokine secretion profiles for THP-1 cells, T cells, and B cells incubated with Aβo and Aβf in human plasma or RPMI-1640 medium. The assay was performed in triplicate. The error bars indicate standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001).
Contrasting effects of plasma proteins on AβO- and Aβf-induced immune responses were observed at 37 °C. Whereas Aβf displayed overall suppressed immune responses after interacting with plasma proteins for all three types of immune cells, AβO-associated immune toxicity was mostly promoted in the presence of plasma proteins, except for TNF expression from the THP-1 cells. Such differential immune toxicity of Aβ depositions can be attributed to two main factors: size and hydrophobicity, correlating to the capacities of the Aβ species and their protein-coronae to bind and penetrate the immune cell membranes. It was reported that amylin oligomers of smaller size and higher hydrophobicity elicited a stronger immune response than their counterparts with similar size but less solvent-exposed hydrophobic residues.48,49 As such, binding of AβO with serum albumin in plasma proteins inhibited the peptide aggregation to maintain their small size and surface hydrophobicity, facilitating immune cell interactions to elevate immune responses.35 In contrast, Aβf with lower structural plasticity and less exposed hydrophobic residues tends to bind to plasma proteins via charged and polar groups, resulting in a larger colloidal-like amyloid-protein corona while displaying weaker toxicity. It should be noted that TNF expression from THP-1 cells could be promoted through receptor-specific signaling pathways such as the Mac-1 receptor.50 Hence, the formation of an AβO-plasma protein corona could potentially shield the exposed AβO activation motifs and drastically prohibit TNF secretion. Such a phenomenon was not observed for Mac-1 receptor-negative T-cells and B-cells.
CONCLUSION
Amyloidosis has been extensively investigated for the past decades, driven by the urgent need to find a cure for amyloid diseases.1 Much of this research emphasis has been centered on the amyloid hypothesis51 and its pathological implications. The oligomers are widely viewed as the most toxic products of protein aggregation as established by extensive in vitro and in vivo data,52,53 while amyloid fibrils are generally considered as benign despite experimental discrepancies.54 Here, we have revealed a convoluted relationship between the amyloidosis and immunogenicity of Aβ1–42, using a high-throughput blood assay and a label-free microfluidic-based LSPR immunoassay. Specifically, AβO displayed strong blood cell association independent of plasma proteins. In contrast, binding of Aβf with the cells was weaker and was further dampened in whole blood as a result of a protein corona. Consistently, the LSPR immunoassay revealed elevated cytokine secretion of immune cells against both AβO and Aβf, especially more so for the oligomers. The Aβf-elicited immune response was screened by plasma proteins, via a protein corona mediated by nonspecific forces. The oligomers, on the other hand, maintained their capacity for immune cell association due to their small size and finite hydrophobicity. This study has implicated the intertwined relationship between the amyloidosis and immunogenicity of Aβ, two aspects underlining the pathological cascade and therapeutic solution of AD.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by ARC Grant CE140100036 (T.P.D.), the National Science Foundation Grant CBET-1701363 (P.C.), the National Institutes of Health Grant R35GM133795 (P.C.), and AFTAM Research Collaboration Award (T.P.D. and P.C.K.). HIM imaging was performed by Anders Barlow at the MCFP platform, University of Melbourne.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-mac.9b01116.
Fabrication and measurement details of LSPR immunoassay; RP-HPLC elution of Aβm, Aβo, and Aβf structures; and examples of gating related to cell association with ThT-labeled Aβ (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Ke PC; Sani MA; Ding F; Kakinen A; Javed I; Separovic F; Davis TP; Mezzenga R Implications of Peptide Assemblies in Amyloid Diseases. Chem. Soc. Rev 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ballard C; Gauthier S; Corbett A; Brayne C; Aarsland D; Jones E Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- (3).Tublin JM; Adelstein JM; Del Monte F; Combs CK; Wold LE Getting to the Heart of Alzheimer Disease. Circ. Res 2019, 124, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mackic JB; Bading J; Ghiso J; Walker L; Wisniewski T; Frangione B; Zlokovic BV Circulating Amyloid-Beta Peptide Crosses the Blood-Brain Barrier in Aged Monkeys and Contributes to Alzheimer’s Disease Lesions. Vasc. Pharmacol 2002, 38, 303–313. [DOI] [PubMed] [Google Scholar]
- (5).Biere AL; Ostaszewski B; Stimson ER; Hyman BT; Maggio JE; Selkoe DJ Amyloid Beta-Peptide is Transported on Lipoproteins and Albumin in Human Plasma. J. Biol. Chem 1996, 271, 32916–32922. [DOI] [PubMed] [Google Scholar]
- (6).Matsubara E; Sekijima Y; Tokuda T; Urakami K; Amari M; Shizuka-Ikeda M; Tomidokoro Y; Ikeda M; Kawarabayashi T; Harigaya Y; Ikeda S; Murakami T; Abe K; Otomo E; Hirai S; Frangione B; Ghiso J; Shoji M Soluble Abeta Homeostasis in AD and DS: Impairment of Anti-Amyloidogenic Protection by Lipoproteins. Neurobiol. Aging 2004, 25, 833–841. [DOI] [PubMed] [Google Scholar]
- (7).Finn TE; Nunez AC; Sunde M; Easterbrook-Smith SB Serum Albumin Prevents Protein Aggregation and Amyloid Formation and Retains Chaperone-like Activity in the Presence of Physiological Ligands. J. Biol. Chem 2012, 287, 21530–21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Algamal M; Milojevic J; Jafari N; Zhang W; Melacini G Mapping the Interactions between the Alzheimer’s Abeta-peptide and Human Serum Albumin beyond Domain Resolution. Biophys. J 2013, 105, 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kakinen A; Javed I; Faridi A; Davis TP; Ke PC Serum Albumin Impedes the Amyloid Aggregation and Hemolysis of Human Islet Amyloid Polypeptide and Alpha Synuclein. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1803–1809. [DOI] [PubMed] [Google Scholar]
- (10).Ramanathan A; Nelson AR; Sagare AP; Zlokovic BV Impaired Vascular-Mediated Clearance of Brain Amyloid Beta in Alzheimer’s Disease: the Role, Regulation and Restoration of LRP1. Front. Aging Neurosci 2015, 7, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Marsh SE; Abud EM; Lakatos A; Karimzadeh A; Yeung ST; Davtyan H; Fote GM; Lau L; Weinger JG; Lane TE; Inlay MA; Poon WW; Blurton-Jones M The Adaptive Immune System Restrains Alzheimer’s Disease Pathogenesis by Modulating Microglial Function. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E1316–E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rogers J; Strohmeyer R; Kovelowski CJ; Li R Microglia and Inflammatory Mechanisms in the Clearance of Amyloid Beta Peptide. Glia 2002, 40, 260–269. [DOI] [PubMed] [Google Scholar]
- (13).Wang ZT; Zhong XL; Tan MS; Wang HF; Tan CC; Zhang W; Zheng ZJ; Kong LL; Tan L; Sun L Association of Lectin-like Oxidized Low Density Lipoprotein Receptor 1 (OLR1) Polymorphisms with Late-Onset Alzheimer Disease in Han Chinese. Ann. Transl. Med 2018, 6, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Marx F; Blasko I; Pavelka M; Grubeck-Loebenstein B The Possible Role of the Immune System in Alzheimer’s Disease. Exp. Gerontol 1998, 33, 871–881. [DOI] [PubMed] [Google Scholar]
- (15).Rogers J; Li R; Mastroeni D; Grover A; Leonard B; Ahern G; Cao P; Kolody H; Vedders L; Kolb WP; Sabbagh M Peripheral Clearance of Amyloid Beta Peptide by Complement C3-Dependent Adherence to Erythrocytes. Neurobiol. Aging 2006, 27, 1733–1739. [DOI] [PubMed] [Google Scholar]
- (16).Pesini P; Perez-Grijalba V; Monleon I; Boada M; Tarraga L; Martinez-Lage P; San-Jose I; Sarasa M Reliable Measurements of the Beta-Amyloid Pool in Blood Could Help in the Early Diagnosis of AD. Int. J. Alzheimer’s Dis 2012, 2012, 604141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bellomo G; Bologna S; Cerofolini L; Paciotti S; Gatticchi L; Ravera E; Parnetti L; Fragai M; Luchinat C Dissecting the Interactions between Human Serum Albumin and α-Synuclein: New Insights on the Factors Influencing α-Synuclein Aggregation in Biological Fluids. J. Phys. Chem. B 2019, 123, 4380–4386. [DOI] [PubMed] [Google Scholar]
- (18).Weiss ACG; Kelly HG; Faria M; Besford QA; Wheatley AK; Ang CS; Crampin EJ; Caruso F; Kent SJ Link between Low-Fouling and Stealth: A Whole Blood Biomolecular Corona and Cellular Association Analysis on Nanoengineered Particles. ACS Nano 2019, 13, 4980–4991. [DOI] [PubMed] [Google Scholar]
- (19).Glass JJ; Chen L; Alcantara S; Crampin EJ; Thurecht KJ; De Rose R; Kent SJ Charge Has a Marked Influence on Hyperbranched Polymer Nanoparticle Association in Whole Human Blood. ACS Macro Lett. 2017, 6, 586–592. [DOI] [PubMed] [Google Scholar]
- (20).Javed I; He J; Kakinen A; Faridi A; Yang W; Davis TP; Ke PC; Chen P Probing the Aggregation and Immune Response of Human Islet Amyloid Polypeptides with Ligand-Stabilized Gold Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 10462–10471. [DOI] [PubMed] [Google Scholar]
- (21).Kuo YM; Kokjohn TA; Kalback W; Luehrs D; Galasko DR; Chevallier N; Koo EH; Emmerling MR; Roher AE Amyloid-Beta Peptides Interact with Plasma Proteins and Erythrocytes: Implications for Their Quantitation in Plasma. Biochem. Biophys. Res. Commun 2000, 268, 750–756. [DOI] [PubMed] [Google Scholar]
- (22).Pilkington EH; Gustafsson OJR; Xing Y; Hernandez-Fernaud J; Zampronio C; Kakinen A; Faridi A; Ding F; Wilson P; Ke PC; Davis TP Profiling the Serum Protein Corona of Fibrillar Human Islet Amyloid Polypeptide. ACS Nano 2018, 12, 6066–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sudhakar S; Kalipillai P; Santhosh PB; Mani E Role of Surface Charge of Inhibitors on Amyloid Beta Fibrillation. J. Phys. Chem. C 2017, 121, 6339–6348. [Google Scholar]
- (24).Knowles TP; Vendruscolo M; Dobson CM The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- (25).Shozawa H; Oguchi T; Tsuji M; Yano S; Kiuchi Y; Ono K Supratherapeutic Concentrations of Cilostazol Inhibits Beta-Amyloid Oligomerization in Vitro. Neurosci. Lett 2018, 677, 19–25. [DOI] [PubMed] [Google Scholar]
- (26).Wiltzius JJ; Sievers SA; Sawaya MR; Eisenberg D Atomic structures of IAPP (amylin) Fusions Suggest a Mechanism for Fibrillation and the Role of Insulin in the Process. Protein Sci. 2009, 18, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang X; St. Clair JR; London E; Raleigh DP Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry 2017, 56, 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pilkington EH; Lai M; Ge X; Stanley WJ; Wang B; Wang M; Kakinen A; Sani MA; Whittaker MR; Gurzov EN; Ding F; Quinn JF; Davis TP; Ke PC Star Polymers Reduce Islet Amyloid Polypeptide Toxicity via Accelerated Amyloid Aggregation. Biomacromolecules 2017, 18, 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Haataja L; Gurlo T; Huang CJ; Butler PC Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev 2008, 29, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Faridi A; Sun Y; Okazaki Y; Peng G; Gao J; Kakinen A; Faridi P; Zhao M; Javed I; Purcell AW; Davis TP; Lin S; Oda R; Ding F; Ke PC Mitigating Human IAPP Amyloidogenesis In Vivo with Chiral Silica Nanoribbons. Small 2018, 14, No. 1802825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cao W; Zheng H Peripheral Immune System in Aging and Alzheimer’s Disease. Mol. Neurodegener 2018, 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Solana C; Tarazona R; Solana R Immunosenescence of Natural Killer Cells, Inflammation, and Alzheimer’s Disease. Int. J. Alzheimer’s Dis 2018, 2018, 3128758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cedervall T; Lynch I; Lindman S; Berggård T; Thulin E; Nilsson H; Dawson KA; Linse S Understanding the Nano-particle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci. U. S.A 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Reyes Barcelo AA; Gonzalez-Velasquez FJ; Moss MA Soluble Aggregates of the Amyloid-Beta Peptide are Trapped by Serum Albumin to Enhance Amyloid-Beta Activation of Endothelial Cells. J. Biol. Eng 2009, 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Milojevic J; Esposito V; Das R; Melacini G Understanding the Molecular Basis for the Inhibition of the Alzheimer’s Abeta-Peptide Oligomerization by Human Serum Albumin Using Saturation Transfer Difference and Off-resonance Relaxation NMR Spectroscopy. J. Am. Chem. Soc 2007, 129, 4282–4290. [DOI] [PubMed] [Google Scholar]
- (36).Wang MM; Miao D; Cao XP; Tan L; Tan L Innate Immune Activation in Alzheimer’s Disease. Ann. Trans. Med 2018, 6, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Michaud JP; Bellavance MA; Prefontaine P; Rivest S Real-Time in Vivo Imaging Reveals the Ability of Monocytes to Clear Vascular Amyloid Beta. Cell Rep 2013, 5, 646–653. [DOI] [PubMed] [Google Scholar]
- (38).Fisher Y; Nemirovsky A; Baron R; Monsonego A T Cells Specifically Targeted to Amyloid Plaques Enhance Plaque Clearance in a Mouse Model of Alzheimer’s Disease. PLoS One 2010, 5, No. e10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bondy B; Hofmann M; Muller-Spahn F; Witzko M; Hock C Reduced Beta-Amyloid Response in Lymphocytes of Patients with Alzheimer’s Disease. Pharmacopsychiatry 1995, 28, 143–146. [DOI] [PubMed] [Google Scholar]
- (40).Monsonego A; Zota V; Karni A; Krieger JI; Bar-Or A; Bitan G; Budson AE; Sperling R; Selkoe DJ; Weiner HL Increased T Cell Reactivity to Amyloid Beta Protein in Older Humans and Patients with Alzheimer Disease. J. Clin. Invest 2003, 112, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Panossian LA; Porter VR; Valenzuela HF; Zhu X; Reback E; Masterman D; Cummings JL; Effros RB Telomere Shortening in T Cells Correlates with Alzheimer’s Disease Status. Neurobiol. Aging 2003, 24, 77–84. [DOI] [PubMed] [Google Scholar]
- (42).Agrawal S; Abud EM; Snigdha S; Agrawal A IgM Response against Amyloid-Beta in Aging: a Potential Peripheral Protective Mechanism. Alzheimer’s Res. Ther 2018, 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jaremo P; Milovanovic M; Buller C; Nilsson S; Winblad B Alzheimer’s Disease and Granulocyte Density Diversity. Eur. J. Clin. Invest 2013, 43, 545–548. [DOI] [PubMed] [Google Scholar]
- (44).Sollvander S; Ekholm-Pettersson F; Brundin RM; Westman G; Kilander L; Paulie S; Lannfelt L; Sehlin D Increased Number of Plasma B Cells Producing Autoantibodies Against Abeta42 Protofibrils in Alzheimer’s Disease. J. Alzheimer’s Dis 2015, 48, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cao W; Zheng H Peripheral Immune System in Aging and Alzheimer’s Disease. Mol. Neurodegener 2018, 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zheng C; Zhou XW; Wang JZ The Dual Roles of Cytokines in Alzheimer’s Disease: Update on Interleukins, TNF-alpha, TGF-beta and IFN-gamma. Transl. Neurodegener 2016, 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lexa KW; Dolghih E; Jacobson MP A Structure-based Model for Predicting Serum Albumin Binding. PLoS One 2014, 9, No. e93323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mannini B; Mulvihill E; Sgromo C; Cascella R; Khodarahmi R; Ramazzotti M; Dobson CM; Cecchi C; Chiti F Toxicity of Protein Oligomers is Rationalized by A Function Combining Size and Surface Hydrophobicity. ACS Chem. Biol 2014, 9, 2309–2317. [DOI] [PubMed] [Google Scholar]
- (49).Campioni S; Mannini B; Zampagni M; Pensalfini A; Parrini C; Evangelisti E; Relini A; Stefani M; Dobson CM; Cecchi C; Chiti F A Causative Link between the Structure of Aberrant Protein Oligomers and Their Toxicity. Nat. Chem. Biol 2010, 6, 140–147. [DOI] [PubMed] [Google Scholar]
- (50).Deng ZJ; Liang M; Monteiro M; Toth I; Minchin RF Nanoparticle-induced Unfolding of Fibrinogen Promotes Mac-1 Receptor Activation and Inflammation. Nat. Nanotechnol 2011, 6, 39–44. [DOI] [PubMed] [Google Scholar]
- (51).Hardy J; Selkoe DJ The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- (52).Kayed R; Head E; Thompson JL; McIntire TM; Milton SC; Cotman CW; Glabe CG Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- (53).Benilova I; Karran E; De Strooper B The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- (54).Krotee P; Rodriguez JA; Sawaya MR; Cascio D; Reyes FE; Shi D; Hattne J; Nannenga BL; Oskarsson ME; Philipp S; Griner S; Jiang L; Glabe CG; Westermark GT; Gonen T; Eisenberg DS Atomic Structures of Fibrillar Segments of hIAPP Suggest Tightly Mated β-sheets are Important for Cytotoxicity. eLife 2017, 6, No. e19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.