

Abstract
Interest in therapeutic discovery typically drives the preparation of natural product analogs, but these undertakings contribute significant advances for synthetic chemistry as well. The need for a highly efficient and scalable synthetic route to a complex molecular scaffold for diversification frequently inspires new methodological development or unique application of existing methods on structurally intricate systems. Additionally, synthetic planning with an aim toward late-stage diversification can provide access to otherwise unavailable compounds or facilitate preparation of complex molecules with diverse patterns of substitution around a shared carbon framework. For these reasons among others, programs dedicated to the diversification of natural product frameworks and other complex molecular scaffolds have been increasing in popularity, a trend likely to continue given their fruitfulness and breadth of impact. In this Perspective, we discuss our experience using late-stage diversification as a guiding principle for the synthesis of natural product analogs and reflect on the impact such efforts have on the future of complex molecule synthesis.
INTRODUCTION
Fine-tuned over thousands of centuries for specific biological roles,1 natural products have long served therapeutic purposes and continue to play a central role in drug development in the modern world.2 In addition to serving directly as pharmaceuticals, such as the analgesic morphine or the antimalarial artemisinin, natural products also provide inspiration for molecular design of many FDA-approved small molecule drugs.2,3 Interest in natural products as scaffolds for therapeutic development has been fueled by the discovery that the biological activities of small molecules are influenced by structural attributes such as ring system complexity, percentage of sp3-hybridized carbons, heteroatom content, and number of stereocenters.4 This realization has facilitated the study of the relationship of molecular structure with biological function, enabling the design of relevant molecular targets.5
The past few decades have witnessed a surge in efforts to create structurally complex, diverse molecules resembling natural products.6–8 In 2004, Danishefsky introduced the concept of “diverted total synthesis” (DTS),9 in which a late-stage synthetic intermediate is used to access a suite of non-natural complex molecules inspired by a natural product family, somewhat similar to the unified or collective synthesis strategy for accessing multiple natural products from a common core.10 This strategy is often preferable to direct modification of natural products because the synthetic scaffolds are often more accessible than the natural products themselves and can be designed for the purpose of diversification.11 Employing DTS, Danishefsky and co-workers prepared vast libraries of natural product analogs, many of which exhibited superior therapeutic behavior compared to the naturally occurring substances.12
Various other approaches for analog synthesis based on bioactivity have emerged and have been described as “function-oriented synthesis” (FOS),13 “biology-oriented synthesis” (BIOS),14 or “complexity to diversity” (CtD)15 based on the specifics of scaffold design and elaboration. An alternative strategy introduced by Schreiber called “diversity-oriented synthesis” (DOS)16 aims to screen for a wide variety of biological activity by producing as many different complex scaffolds as possible through modular combination of simple building blocks.17 Yet, another approach involves the preparation of hybrid molecules that contain structural elements of two or more natural product families with the aim of enhancing bioactivity.18 Despite the variations in these approaches, they all share a common goal: exploration of bioactive chemical space through synthesis and biological evaluation of novel complex organic compounds.
These strategies have been applied to the synthesis of vast libraries of natural product-inspired complex molecules, with notable examples arising from the research groups of Wender,19 Boger,20 Nicolaou,21 Myers,22 Carreira,23 Burke,24 Miller,25 and Baran,26 among others. These efforts have revealed important information about the mechanisms of activity among complex molecules, which, in turn, informs further synthetic design.27,28 This Perspective is not intended as a comprehensive review of the field of complex molecule diversification or natural product analog synthesis; several excellent reviews of this extensive research area have been published within the last several years.6–8 Instead, this piece describes our experiences with late-stage diversification as a guide for synthetic design and source of inspiration for methodological development.
To create libraries of diverse, structurally intricate compounds, many approaches involve the synthesis of a central molecular scaffold from which diversification can be achieved. This scaffold can be chosen strategically to maximize the number of useful functional handles available while retaining the structural framework of the natural product family and potentially associated bioactivity. Because the scaffold is intentionally designed for maximal synthetic accessibility, this strategy allows access to novel natural product analogs and proves more feasible than derivatizing the natural products themselves. The pathway to this critical synthetic intermediate often requires iterative refinement to generate derivatives efficiently. While this is also an important consideration in total synthesis efforts toward specific molecular targets, the amount of late-stage material required for a successful diversification project often exceeds what is needed for a typical total synthesis because the number of potential targets is essentially limitless.9 As such, continual optimization of the synthetic route to the main scaffold is a hallmark of diversification programs and often inspires the development of new methodologies or improvement of existing processes for transformations of complex frameworks.
Overall, diversification programs offer a unique synthetic perspective complementary to those of pure total synthesis and methods development. While a prominent goal of diversification efforts is to discover new biologically active compounds, the concomitant synthetic aims also deserve thoughtful discussion. The synthesis of a vast array of complex organic molecules represents a significant feat, especially considering the structural complexity of the natural products that inspire target design. Furthermore, the continual optimization required to diversify a complex scaffold offers repeated opportunities to showcase the synthetic utility of newly developed methodologies and generates a steady flow of widely applicable findings. Considering these synthetic benefits alongside the contributions made to medicinal chemistry, it is no wonder that complex molecule diversification has become a significant driving force among research efforts in organic synthesis.
LATE-STAGE DIVERSIFICATION OF THE CYANTHIWIGIN NATURAL PRODUCT CORE
The cyanthiwigin natural products have captivated the synthetic community for decades due to their intricate molecular architectures and intriguing bioactivities.29 Sharing a distinctive angularly fused 5–6–7 tricyclic framework with a larger family of more than 170 cyathane diterpenes, the cyanthiwigins feature a unique syn relative orientation of the two methyl substituents at the ring junctures instead of the anti configuration observed in most other cyathanes.30 Since their initial isolation in the early 1990s, a number of elegant syntheses of various cyanthiwigins have been reported.31 In 2008, our group disclosed a concise total synthesis of (−)-cyanthiwigin F (1) (Figure 1A).32 Exploitation of symmetry in early synthetic intermediates and application of a powerful double asymmetric allylic alkylation enabled preparation of tricyclic intermediate 6 in only seven steps from succinic acid (4, Figure 1B). Cyanthiwigins B (2), F (1), and G (3) were accessed from tricyclic diketone 6,33 and a similar strategy was employed toward the core of the structurally similar gagunin natural products (Figure 1C).34
Figure 1.

(A) Cyanthiwigin natural products accessible from 6, (B) synthetic approach toward tricycle 6 from succinic acid, and (C) structures of selected gagunin natural products.
Noting the apparent influence of oxygenated substituents on the biological activity of the gagunins as reported by Shin and co-workers,35 we sought to leverage our concise route to tricycle 6 to prepare a suite of oxygenated cyanthiwigin derivatives. Representing the framework of the cyanthiwigin natural products, tricycle 6 offered an ideal scaffold for this endeavor given the presence of multiple functional handles in the form of olefin and carbonyl moieties. We envisioned that installation of diverse oxygenated functionalities would give rise to cyanthiwigin–gagunin “hybrid” molecules possessing the carbocyclic framework characteristic of the cyanthiwigins and oxygenated substituents reminiscent of the gagunins. We anticipated that these efforts would generate novel complex molecules for biological study while contributing valuable synthetic insight into the reactivity of the cyanthiwigin framework under conventional strategies for oxidation and modern methods for C–H oxidation.
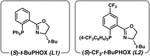
Because access to scaffold 6 was crucial for the success of our late-stage diversification plans, we began by critically re-examining our established synthesis of 6 despite its efficiency. We identified a few key transformations in need of further optimization for the large scale required for diversification studies. The first synthetic challenge arose at the double catalytic enantioselective allylic alkylation to prepare diketone (R,R)-5 from bis(β-ketoester) 9 and establish the requisite syn stereochemistry of the methyl substituents. This transformation required low reaction concentrations (0.01 M) and high loadings of Pd(dmdba)2 and PHOX ligand L1, both only accessible through multistep preparation (Table 1, Entry 1). To address these limitations, we explored alternate conditions and ultimately discovered that commercially available Pd(OAc)2 could be employed as a precatalyst with modified PHOX ligand L2 in toluene at 10× concentration (0.1 M) to achieve the desired transformation. Moreover, these reoptimized conditions generated (R,R)-5 in much higher yield and diastereoselectivity compared to the original conditions (Entry 2).36 Importantly, these conditions were also effective on a 10-g scale and required significantly less solvent, ligand, and Pd (by 20-fold) than the original protocol. This breakthrough streamlined the production of key enantioenriched diketone 5 on a multigram scale, constituting an important advance in the synthesis of tricyclic scaffold 6.
Table 1.
Comparison of Original (Entry 1) vs Modified Conditions (Entry 2) for Double Enantioselective Alkylation of 9
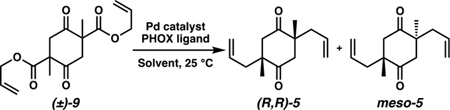
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol %) | Ligand (mol %) | Solvent | Cone. | Yield | dr | ee |
| 1 | Pd(dmdba)2 (5.0) | L1 (5.5) | Et20 | 0.01 M | 78% | 4.4:1 | 99% |
| 2 | Pd(OAc)2 (0.25) | L2 (2.5) | PhMe | 0.1 M | 97% | 4.9:1 | 99% |
|
| |||||||
| |||||||
After successful reoptimization of the critical stereodefining allylic alkylation, diketone 5 was converted to a vinyl triflate and subjected to Negishi coupling to afford tetraene 10 (Scheme 1). Ring-closing metathesis (RCM) to generate bicycle 11 proceeded smoothly, but the ensuing cross metathesis with vinylboronic acid pinacol ester (13) typically afforded aldehyde 12 in low yields after oxidative workup. Eager to continue refining the synthesis using recently developed methodologies, we applied the aldehyde-selective Tsuji–Wacker oxidation protocol reported by Grubbs and co-workers in 2013 to RCM product 11.37 To our delight, 11 proved to be a competent substrate for the nitrite-modified Tsuji–Wacker oxidation, despite the presence of a sterically encumbered quaternary carbon at the homoallylic position. This discovery enabled productive use of the accrued quantities of bicycle 11, thus increasing the amount of bicyclic aldehyde 12 available to undergo radical cyclization to generate the target cyanthiwigin natural product core (6).38
Scheme 1.

Preparation of Aldehyde 11, Facilitated by the Aldehyde-Selective Tsuji–Wacker Oxidation, and Completion of 5
Considering the challenges of forming aldehydes proximal to sterically demanding quaternary carbons, we were intrigued by the successful oxidation of bicycle 11 and decided to investigate the synthetic potential of the nitrite-modified Tsuji–Wacker oxidation in more detail.39 We began by examining its applicability to other sterically encumbered substrates given the ubiquity of such compounds as synthetic intermediates in the preparation of complex molecules. We were pleased to discover that terminal olefins bearing quaternary carbons at either the allylic or homoallylic position could be oxidized in high yields and aldehyde selectivity and with broad functional group tolerance (Scheme 2A). Considerably complex substrates were readily accommodated, as exemplified in the successful oxidation of aspewentin B derivative 16 (Scheme 2B), further underscoring the robustness of the nitrite-modified Tsuji–Wacker oxidation. Moreover, the aldehydes produced could be further transformed without purification to achieve direct conversion of terminal alkenes to a variety of functionalities. For instance, oxidation of alkene 18, followed by reductive amination of the crude aldehyde enabled formal anti-Markovnikov hydroamination in good yields (Scheme 3A). Extension of this protocol facilitated other synthetic transformations, including carbon chain homologation and other anti-Markovnikov hydrofunctionalizations requiring only one purification step (Scheme 3B). This strategy has since been employed in various synthetic efforts.40,41
Scheme 2.

(A) Aldehyde-Selective Tsuji–Wacker Oxidation of Hindered Terminal Alkene and (B) Including Aspewentin B Derivative 16
Scheme 3.

(A) Formal Anti-Markovnikov Hydroamination of 18 and (B) Synthetic Transformations of 18 Enabled by Aldehyde-Selective Tsuji–Wacker Oxidation
With access to ample quantities of the cyanthiwigin core from the reoptimized synthetic route, we were well-equipped to prepare non-natural oxygenated cyanthiwigin analogs42 and explore the reactivity of the cyanthiwigin core under various conditions for C–H oxidation.43 Noting the potent antileukemia activity of gagunin E (8, Figure 1C) and the diverse biological activities of the cyanthiwigins,29,30 we hypothesized that “hybrid” molecules possessing the core skeleton of the cyanthiwigins and the oxygenation pattern of the gagunins may exhibit heightened bioactivity.44 To this end, the C-ring olefin and A- and B-ring carbonyls offered convenient handles for installing oxygenated functionalities onto the tricyclic core (6). Dimethyldioxirane (DMDO) epoxidation of the C-ring olefin proceeded with strong facial selectivity, forming epoxide 26 as a single diastereomer in 99% yield (Scheme 4A).45 Although 26 was ultimately not employed as a synthetic precursor to cyanthiwigin–gagunin hybrids, the high stereoselectivity of the epoxidation indicated enhanced accessibility of the α-face of the C-ring of 6.
Scheme 4.

(A) Stereoselective DMDO Epoxidation of 6 and (B) Diversification of 6 to Access Cyanthiwigin–Gagunin Hybrids 29–31
In line with these findings, dihydroxylation of the C-ring olefin in 6 using catalytic dipotassium osmate in the presence of NMO occurred with the same facial selectivity, furnishing syn-diol 27 as a single diastereomer in good yield (Scheme 4B). Subsequent esterification using n-propyl anhydride followed by borohydride reduction of the A- and B-ring carbonyls with high facial selectivity afforded triol 28 in 80% yield (58% overall yield from 27). Triol 28 was subsequently employed as a further point of diversification; esterification using various anhydrides readily generated cyanthiwigin–gagunin hybrid molecules 29–31. Although these novel compounds did not exhibit significant antileukemia activity, these investigations revealed that tricycle 6 typically reacts preferentially at the α-face, as evidenced by the strong facial selectivity observed in epoxidation and dihydroxylation of the C-ring olefin in addition to hydride reduction of the A- and B-ring carbonyls. Given the concavity of the tricyclic framework, we were surprised to observe such strong preference for reactivity at the α-face. We surmised that the methyl substituents on the β-face of the cyanthiwigin core strongly influence reactivity, a conclusion supported by our C–H oxidation studies of the cyanthiwigin framework.
Considering the extensive interest in harnessing C–H functionalization as a robust strategy for complex molecule synthesis,7,46 we sought to employ tricycle 6 in a comparative analysis of C–H oxidation methodologies. We began by examining allylic C–H acetoxylation of the cyanthiwigin core. Interestingly, 6 was unreactive under various conditions for Pd-catalyzed allylic C–H acetoxylation47 and generated only low yields of acetoxylated product 32 in the presence of Oxone and Pd(OAc)2 (Scheme 5).48 In contrast, SeO2 readily oxidized 6, albeit in moderate yields. Allylic alcohol 33 was produced in the presence of catalytic selenium with tert-butyl hydroperoxide (TBHP) as the terminal oxidant, whereas aldehyde 34 was the major product when stoichiometric selenium was employed at elevated temperatures. These findings suggest that usually robust Pd-catalyzed methods for allylic C–H oxidation have further room for optimization when applied to complex systems, although conventional strategies for allylic oxidation meet this synthetic need. To explore the fate of the cyanthiwigin framework under conditions for 3° C–H oxidation, saturated tricycle 35 was prepared via Pt-catalyzed hydrogenation of the C-ring olefin. As observed in the cyanthiwigin–gagunin hybrid synthesis, H2 was added preferentially across the α-face of 6, corroborating our previous conclusions about facial selectivity in reactions of 6.
Scheme 5.

Allylic C–H Oxidation and Hydrogenation of the Cyanthiwigin Framework (6)
With substrate 35 in hand, we carried out a comparative study of 3° C–H hydroxylation, amination, and azidation reactions. Significant discrepancies in efficacy of 3° C–H hydroxylation49 were observed, with yields of tertiary alcohol 36 ranging from 15–64% (Scheme 6). Similarly varying results were observed in 3° amination50 and 3° azidation51 studies. While yields of tertiary azide 39 as high as 90% were achievable, most methods generated 39 in nearly 1:1 dr, an outcome with important implications for employing 3° C–H azidation in complex molecule synthesis. In all cases, 35 reacted exclusively at C12, indicating potential electronic deactivation of C4 and C5 by the proximal electron-withdrawing carbonyls. Oxidation of 2° C–H bonds was also achievable, albeit in lower yields. Investigation of 2° C–H oxygenation52 and C–H chlorination53 methods furnished ketone 37 and chloride 40, respectively, in modest yields. In both cases, oxidation was observed only at C13, likely due to steric and electronic deactivation at C10, C12, and C14. Notably, chlorination occurred preferentially on the α-face of 35, mirroring the facial selectivity observed in the reactivity of tricycle 6. Together, these findings showcase the influence of electronics, sterics, and intrinsic reactivities of C–H bonds in C–H oxidation reactions, highlighting the importance of methods that offer alternate regioselectivities.
Scheme 6.

2° and 3° C–H Oxidation of Hydrogenated Tricycle 35
Conditions for 3° C–H Hydroxylation.
a) RuCl3·xH2O (5 mol %), KBrO3, pyridine, MeCN, 60 °C, 42% yield; b) (Me3tacn)RuCl3 (2 mol %), CAN, AgClO4, t-BuOH/H2O, 23 °C, 64% yield; c) 6-chloro-4-trifluoromethyl-1,2,3-benzoxathiazine-2,2-dioxide (20 mol %), Oxone, HFIP/H2O, 70 °C, 21% yield; d) DMDO, acetone, 23 °C, 15% yield; e) Fe(S,S-PDP) (15 mol %), H2O2, AcOH, MeCN, 23 °C, 22% yield; f) Mn(OTf)2 (0.1 mol %), AcOOH, bipy, AcOH/H2O, 23 °C, 20% yield. Conditions for 3° C–H amination: g) (2,6-F2C6H3)OSO2NH2, [Rh2(esp)2] (10 mol %), PhI(OAc)2, PhMe2CCO2H, MgO, 5 Å MS, i-PrOAc, 30% yield; h) PhOSO2NH2, [Rh2(esp)2] (10 mol %), PhI(OPiv)2, Al2O3, t-BuCN, 70% yield; (i) (4-FC6H4)OSO2NH2, [Rh2(esp)2] (10 mol %), PhI(OPiv)2, Al2O3, t-BuCN, 72% yield. Conditions for 3° C–H azidation: j) methyl 2-(azidosulfonyl)benzoate, K2S2O8, NaHCO3, MeCN/H2O, 85 °C, 90% yield, 1.0:1.9 d.r.; k) 1-azido-1,2-benziodoxol-3-(1H)-one, Fe(OAc)2, i-Pr-Pybox, MeCN, 35 → 50 °C, 86% yield, 1.2:1.0 d.r.; l) 1-azido-1,2-benziodoxol-3-(1H)-one, BzOOBz, ABCN, DCE, 84 °C, 13% yield. Conditions for 2° C–H oxidation: m) Fe(R,R-CF3-PDP) (15 mol %), H2O2/H2O, MeCN, AcOH, 23 °C, 37% yield. Conditions for 2° C–H chlorination: n) ArCON(Cl)(t-Bu), Ar = 3,5-(CF3)2C6H3, Cs2CO3, hv (23W), PhH, 55 °C, 30% yield.
Overall, these investigations illustrate how pursuing a goal of diversifying a complex molecular scaffold can precipitate broadly applicable synthetic discoveries. In our quest to explore the reactivity of the cyanthiwigin core (6) and prepare structurally diverse analogs, we developed a revised synthetic route to 6 employing state-of-the-art methodologies. Beyond facilitating production of 6 on large scale, these efforts showcase the synthetic applicability of recently developed methodologies, expanding the known utility of powerful transformations by offering fresh contexts for their application.
SYNTHESES OF JORUNNAMYCIN A, JORUMYCIN, AND ANALOGS
The bis-tetrahydroisoquinoline (bis-THIQ) alkaloids have been extensively studied over the past 40 years due to their unique chemical structures and potent biological activities as antitumor antibiotics.54 Among these, jorumycin (41) and jorunnamycin A (42) feature a pentacyclic carbon skeleton, highly oxygenated ring termini, and a central pro-iminium ion that serves as an alkylating agent in vivo, resulting in covalent modification of DNA that ultimately leads to cell death (Figure 2).55 The potent bioactivity of these natural products yields promise as anticancer agents, such as Et 743 (43) (Yondelis, trabectedin) which has been approved for the treatment of advanced soft-tissue sarcoma and ovarian cancer in the United States and Europe.
Figure 2.

Bis-tetrahydroisoquinoline (bis-THIQ) alkaloids.
The highly electron-rich functional groups embedded in these natural products are key structural features in the biosynthetic pathways of the bis-THIQ alkaloids, which are forged by Pictet-Spenglerase enzymes.56 Previously reported chemical syntheses of jorumycin (41) and jorunnamycin A (42) feature biomimetic applications of electrophilic aromatic substitution (EAS) chemistry for the construction of one or more of the tetrahydroisoquinoline (THIQ) motifs (Figure 3).57 However, this approach is only limited to electron-rich groups appended to the phenyl ring, inhibiting the synthesis of non-natural analogs with substituents of differing electronic effects. Moreover, analogs possessing electron-withdrawing groups on these rings are inaccessible using biomimetic approaches, which is a commonly employed strategy to improve a drug molecule’s metabolic stability.58 Thus, to overcome the limitations of the current state of the art with respect to analog diversity, we implemented an alternative, nonbiomimetic route to access these natural products and their analogs.
Figure 3.

Conventional biomimetic approaches toward the bis-THIQ natural products versus our synthetic approach.
Toward diversification efforts of natural product analogs, we designed a nonbiomimetic route for the total syntheses of (−)-jorumycin (41) and (−)-jorunnamycin A (42) (Scheme 7).59 This unprecedented synthetic approach harnesses transition-metal catalysis to forge the two functionalized isoquinoline monomers 44 and 45 through a Pd-catalyzed cross-coupling reaction developed by Fagnou and co-workers, accessing bis-isoquinoline 46 in 93% yield on a 7-g scale.60 After installing the required oxidation levels of the natural product scaffold, an Ir-catalyzed enantioselective hydrogenation was performed to undergo reduction and subsequent cyclization to install pentacyclic intermediate 48. Inspired by the asymmetric ether-directed imine reduction using a chiral Ir catalyst developed by scientists at Ciba-Geigy (Syngenta) for the preparation of the herbicide metolachlor, we utilized the appended hydroxymethyl group as a directing group for hydrogenation.61 Using chiral Josiphos ligand L3, we initially observed reduction of the isoquinoline with the C1-appended hydroxy functionality, confirming the accelerating effects of the directing group under the hydrogenation conditions. Further optimization established the natural product scaffold 48 in 83% yield with >20:1 dr and 88% ee on greater than 1 mmol scale. Finally, late-stage C–H oxidation of the arenes enabled access to both (−)-jorunnamycin A (42) and (−)-jorumycin (41). The convergent coupling strategy allows for the preparation of a diverse set of isoquinoline monomers, wherein different permutations of partial and full oxygenations of the arene ring can be installed to explore structure–activity relationships of novel analogs.
Scheme 7.

Total Synthesis of (−)-Jorumycin (41) and (−)-Jorunnamycin A (42)
This nonbiomimetic synthetic route not only strategically leverages modern catalysis for the construction of the bis-THIQ natural products with high efficiency but also advances late-stage diversification with key intermediate 48 for the production of several jorunnamycin A analogs inaccessible through conventional EAS approaches. Using bis-THIQ 48 as a branching point for derivative synthesis, we sought to synthesize permutations of partial and full oxygenation patterns on the quinone rings to explore structure–activity relationships, which have previously been synthetically inaccessible. A late-stage C–H oxidation of 48 with 1.1 equiv of N-chlorosaccharine established the monochlorinated products 49 and 50 and dichlorinated product 51 in comparable yields (Scheme 8A). Hydroxylation of the aryl halides then established different oxygenation patterns on the western and eastern fragments of the molecule, furnishing analogs 52–54.
Scheme 8.

(A) Synthesis of Bis-THIQ Analogs and (B) Biological Evaluation of Non-Natural Analogs 52–55
Preliminary biological evaluations of these analogs were then conducted to probe the relative cytotoxicity against cancer cell lines (Scheme 8B). Interestingly, monohydroxylated products 52 and 53 show considerably different activity profiles depending on the relative location of oxygenation. Featuring only E-ring oxygenation, compound 53 displayed similar levels of cytotoxicity to fully oxygenated 54, while molecule 52 with A-ring oxygenation showed significantly diminished activity.59 Though further studies are needed to determine actual efficacy, comparing the activity of the series 52–55 highlights the significance of the location and degree of oxygenation on the A- versus E-rings.
In addition to creating a diverse library of non-natural analogs, this novel synthetic approach toward jorumycin and jorunnamycin A also inspired reaction development for further transformations of complex heterocyclic frameworks. Considering the limited reports on the asymmetric hydrogenation of isoquinolines,62 we drew inspiration from the hydrogenation of 47 in the total synthesis of jorumycin to develop a general method for the hydrogenation of 1,3-disubstituted isoquinolines. Using the hydroxymethyl group at the C1-position as a directing group, this synthetic method enabled access to a wide variety of enantioenriched tetrahydroisoquinolines (THIQs) and amino alcohols, both highly valuable pharmacophores.63 Using 1.25 mol % of [Ir(cod)Cl]2 and 3 mol % of Josiphos ligand L4, a broad scope of differentially substituted isoquinolines was well tolerated (Scheme 9).
Scheme 9.

General Asymmetric Hydrogenation of 1,3-Disubstituted Isoquinolines Inspired by the Total Synthesis of Jorumycin
Investigation of the hydrogenation methodology for a variety of 1,3-disubstituted isoquinolines required a simple and divergent synthetic route to access a wide range of 1-(hydroxymethyl)-3-arylisoquinoline substrates. To expand on the limited number of general methods for the synthesis of substituted isoquinolines, we developed an efficient synthetic approach toward isoquinoline diversification by accessing isoquinoline triflate 60 from a Pd-catalyzed enolate arylation and subsequent annulation and alcohol triflation (Scheme 10).64 At this stage, different aryl or heteroaryl groups could be coupled with intermediate 60 to deliver a wide range of 1,3-disubstituted isoquinolines, highlighting the divergent synthesis of this sequence. Finally, SeO2 oxidation to afford the aldehyde and subsequent NaBH4 reduction provided the desired isoquinoline substrate 62 to investigate the hydroxy-directed asymmetric hydrogenation.
Scheme 10.

Diversified Synthetic Approach toward a Wide Variety of 1,3-Disubstituted Isoquinolines
Ultimately, the development of this hydrogenation method provides access to a range of decorated THIQ analogs that are difficult to synthesize via biomimetic approaches. Furthermore, the hydroxymethyl directing group can also serve as a functional handle toward the synthesis of complex scaffolds from the hydrogenated products. The synthetic utility of this functionality was demonstrated by subjecting THIQ 63 to onestep protocols, accessing fused 6,6,5- and 6,6,6-tricyclic systems 64–66 (Figure 4A). Notably, scaffolds 64–65 are conserved structural motifs in a number of natural products such as quinocarcin, tetrazomine, and bioxalomycin (68–70, Figure 4B).65 Additionally, non-natural analog 67 of the tetrahydroprotoberberine alkaloids, a family of natural products with a tetracyclic bis-THIQ core, was synthesized via a 2-step sequence. After the reaction of 63 with glyoxal dimethyl acetal to access an oxazolidine-fused intermediate, a Pomeranz-Fritsch reaction using Eaton’s Reagent (7.7 wt % phosphorus pentoxide in methanesulfonic acid) delivers the fused pentacyclic THIQ scaffold 67 in 38% yield as a single diastereomer (Figure 4A). Overall, the application of this asymmetric hydrogenation technology toward the synthesis of complex THIQ structural motifs of several biologically active natural products represents an expansion of synthetic utility from the seminal hydrogenation sequence of the jorumycin synthesis.
Figure 4.

(A) Synthetic derivatizations of hydrogenated THIQ product 63 to complex THIQ scaffolds and (B) select examples of natural products with fused 6,6,5-tricyclic THIQ systems.
CONCLUDING REMARKS
The contributions of diversification studies to chemical synthesis are plentiful. Designing a synthesis to access an array of natural product analogs not only enables a concise, efficient synthetic route but also inspires new methodological development and motivates creative application of established methods in previously untested contexts. Once prepared, the diversification scaffold serves as a platform for creating libraries of structurally diverse complex molecules, a process which frequently reveals intriguing and unexpected patterns of reactivity. The outcomes of these investigations are broadly applicable, offering important synthetic insights and providing access to biosynthetically unavailable natural product analogs. In turn, the preparation of molecular libraries facilitates study of structure–activity relationships of privileged scaffolds with the potential to inform drug development. Given these considerable motivating factors, future synthetic endeavors will undoubtedly continue to feature diversification as a prominent aim, whether in the form of analog preparation or the demonstration of potential for access to derivatives. Distinct from traditional natural product total synthesis, the targeting of analog libraries reflects the modern age of chemical synthesis and increasingly calls on practitioners to address new research questions. Essentially, ruminations on whether or how a complex molecule can be synthesized are giving way to considerations of how the synthesis of a molecule can be designed for maximum impact. Interest in natural products as synthetic targets remains robust, but having cultivated the ability to prepare any molecule of interest with enough creativity and determination, synthetic chemists are reconceptualizing complex molecule synthesis. As evidenced by the last few decades, diversification will likely be a mainstay of organic synthesis research programs for many years to come.
ACKNOWLEDGMENTS
We acknowledge the NSF under the CCI Center for Selective C–H Functionalization (CCHF), CHE-1700982, NIH-NIGMS (R01GM127972A and R01GM080269), Caltech, and the University of Washington Tacoma for funding and support. We also thank current and former co-workers for helpful discussions and feedback on the preparation of this manuscript.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Kelly E. Kim, Sciences and Mathematics Division, School of Interdisciplinary Arts and Sciences, University of Washington, Tacoma, Washington 98402, United States
Alexia N. Kim, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States
Carter J. McCormick, Sciences and Mathematics Division, School of Interdisciplinary Arts and Sciences, University of Washington, Tacoma, Washington 98402, United States
Brian M. Stoltz, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States
REFERENCES
- (1).Paterson I; Anderson EA The Renaissance of Natural Products as Drug Candidates. Science 2005, 310, 451–453. [DOI] [PubMed] [Google Scholar]
- (2).(a) Newman DJ; Cragg GM Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [DOI] [PubMed] [Google Scholar]; (b) Butler MS Natural Products to drugs: natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [DOI] [PubMed] [Google Scholar]; (c) Danishefsky S On the potential of natural products in the discovery of pharma leads: A case for reassessment. Nat. Prod. Rep. 2010, 27, 1114–1116. [DOI] [PubMed] [Google Scholar]
- (3).(a) Lacaná E; Amur S; Mummanneni P; Zhao H; Frueh FW The Emerging Role of Pharmacogenomics in Biologics. Clin. Pharmacol. Ther. 2007, 82, 466–471. [DOI] [PubMed] [Google Scholar]; (b) Nelson AL; Dhimolea E; Reichert JM Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discovery 2010, 9, 767–774. [DOI] [PubMed] [Google Scholar]
- (4).Lachance H; Wetzel S; Kumar K; Waldmann H Charting, Navigating, and Populating Natural Product Chemical Space for Drug Discovery. J. Med. Chem. 2012, 55, 5989–6001. [DOI] [PubMed] [Google Scholar]
- (5).(a) Schreiber SL Chemical Genetics Resulting from a Passion for Synthetic Organic Chemistry. Bioorg. Med. Chem. 1998, 6, 1127–1152. [DOI] [PubMed] [Google Scholar]; (b) Schreiber SL The Small-Molecule Approach to Biology. Chem. Eng. News 2003, 81, 51–60. [Google Scholar]; (c) Stockwell BR Chemical Genetics: Ligand-Based Discovery of Gene Function. Nat. Rev. Genet. 2000, 1, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) O’Connor CJ; Laraia L; Spring DR Chemical genetics. Chem. Soc. Rev. 2011, 40, 4332–4345. [DOI] [PubMed] [Google Scholar]
- (6).For reviews covering approaches toward synthesis of natural product analogs, see: Robles O; Romo D Chemo-and site-selective derivatizations of natural products enabling biological studies. Nat. Prod. Rep. 2014, 31, 318–334. Maier ME Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. Bebbington MWP Natural product analogues: towards a blueprint for analogue-focused synthesis. Chem. Soc. Rev. 2017, 46, 5059–5109. Shugrue CR; Miller SJ Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev. 2017, 117, 11894–11951.
- (7).For reviews about the diversification of natural product scaffolds using C–H functionalization, see: Karimov RR; Hartwig JF Transition-Metal-Catalyzed Selective Functionalization of C(sp3)-H Bonds in Natural Products. Angew. Chem., Int. Ed. 2018, 57, 4234–4241. Fessner ND P450 Monooxygenases Enable Rapid Late-Stage Diversification of Natural Products via C-H Bond Activation. ChemCatChem 2019, 11, 2226–2242. Hong B; Luo T; Lei X Late-Stage Diversification of Natural Products. ACS Cent. Sci. 2020, 6, 622–635. Romero E; Jones BS; Hogg BN; Casamajo AR; Hayes MA; Flitsch SL; Turner NJ; Schnepel C Enzymatic Late-Stage Modifications: Better Late Than Never. Angew. Chem., Int. Ed. 2021, 60, 16824–16855.
- (8).(a) Karageorgis G; Foley DJ; Laraia L; Waldmann H Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [DOI] [PubMed] [Google Scholar]; (b) Truax NJ; Romo D Bridging the gap between natural product synthesis and drug discovery. Nat. Prod. Rep. 2020, 37, 1436–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karageorgis G; Foley DJ; Laraia L; Brakmann S; Waldmann H Pseudo Natural Products-Chemical Evolution of Natural Product Structure. Angew. Chem., Int. Ed. 2021, 60, 15705–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li L; Chen Z; Zhang X; Jia Y Divergent Strategy in Natural Product Total Synthesis. Chem. Rev. 2018, 118, 3752–3832. [DOI] [PubMed] [Google Scholar]
- (9).(a) Njardarson JT; Gaul C; Shan D; Huang X-Y; Danishefsky SJ Discovery of Potent Cell Migration Inhibitors through Total Synthesis: Lessons from Structure-Activity Studies of (+)-Migrastatin. J. Am. Chem. Soc. 2004, 126, 1038–1040. [DOI] [PubMed] [Google Scholar]; (b) Gaul C; Njardarson JT; Shan D; Dorn DC; Wu K-D; Tong WP; Huang X-Y; Moore MAS; Danishefsky SJ The Migrastatin Family: Discovery of Potent Cell Migration Inhibitors by Chemical Synthesis. J. Am. Chem. Soc. 2004, 126, 11326–11337. [DOI] [PubMed] [Google Scholar]
- (10).For some recent examples of collective/unified synthesis, see: Hirose A; Watanabe A; Ogino K; Nagatomo M; Inoue M Unified Total Syntheses of Rhamnofolane, Tigliane, and Daphnane Diterpenoids. J. Am. Chem. Soc. 2021, 143, 12387–12396. Löffler LE; Wirtz C; Fürstner A Collective Total Synthesis of Casbane Diterpenes: One Strategy, Multiple Targets. Angew. Chem., Int. Ed. 2021, 60, 5316–5322. Li G; Wang Q; Zhu J Unified divergent strategy towards the total synthesis of the three sub-classes of hasubanan alkaloids. Nat. Commun. 2021, 12, 36. Kim JH; Jeon H; Park C; Park S; Kim S Collective Asymmetric Total Synthesis of C-11 Oxygenated Cephalotaxus Alkaloids. Angew. Chem., Int. Ed. 2021, 60, 12060–12065. Anketell MJ; Sharrock TM; Paterson I A Unified Total Synthesis of the Actinoallolides, a Family of Potent Anti-Trypanosomal Macrolides. Angew. Chem., Int. Ed. 2020, 59, 1572–1576. Zou Y; Li X; Yang Y; Berritt S; Melvin J; Gonzales S; Spafford M; Smith AB III. Total Synthesis of (−)-Nodulisporic Acids D, C, and B: Evolution of a Unified Synthetic Strategy. J. Am. Chem. Soc. 2018, 140, 9502–9511. Yoritate M; Takahashi Y; Tajima H; Ogihara C; Yokoyama T; Soda Y; Oishi T; Sato T; Chida N Unified Total Synthesis of Stemoamide-Type Alkaloids by Chemoselective Assembly of Five-Membered Building Blocks. J. Am. Chem. Soc. 2017, 139, 18386–18391. Cheng H; Zhang Z; Yao H; Zhang W; Yu J; Tong R Unified Asymmetric Total Syntheses of (−)-Alotaketals A-D and (−)-Phorbaketal A. Angew. Chem., Int. Ed. 2017, 56, 9096–9100. Zi W; Zuo Z; Ma D Intramolecular Dearomative Oxidative Coupling of Indoles: A Unified Strategy for the Total Synthesis of Indoline Alkaloids. Acc. Chem. Res. 2015, 48, 702–711. Wagnières O; Xu Z; Wang Q; Zhu J Unified Strategy to Monoterpene Indole Alkaloids: Total Syntheses of (±)-Goniomitine, (±)-1,2-Dehydroaspidospermidine, (±)-Aspidospermidine, (±)-Vincadifformine, and (±)-Kopsihainanine A. J. Am. Chem. Soc. 2014, 136, 15102–15108. Jones SB; Simmons B; Mastracchio A; MacMillan DWC Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188.
- (11).Morrison KC; Hergenrother PJ Natural products as starting points for the synthesis of complex and diverse compounds. Nat. Prod. Rep. 2014, 31, 6–14. [DOI] [PubMed] [Google Scholar]
- (12).For an overview of these efforts, see: Wilson RM; Danishefsky SJ Small Molecule Natural Products in the Discovery of Therapeutic Agents: The Synthesis Connection. J. Org. Chem. 2006, 71, 8329–8351.
- (13).(a) Wender PA; Verma VA; Paxton TJ; Pillow TH Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]; (b) Wender PA Toward the ideal synthesis and transformative therapies: the roles of step economy and function oriented synthesis. Tetrahedron 2013, 69, 7529–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA Toward the ideal synthesis and molecular function through synthesis-informed design. Nat. Prod. Rep. 2014, 31, 433–440. [DOI] [PubMed] [Google Scholar]
- (14).(a) Kaiser M; Wetzel S; Kumar K; Waldmann H Biology-inspired synthesis of compound libraries. Cell. Mol. Life Sci. 2008, 65, 1186–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wetzel S; Bon RS; Kumar K; Waldmann H Biology-Oriented Synthesis. Angew. Chem., Int. Ed. 2011, 50, 10800–10826. [DOI] [PubMed] [Google Scholar]; (c) Rizzo S; Waldmann H Development of a Natural-Product-Derived Chemical Toolbox for Modulation of Protein Function. Chem. Rev. 2014, 114, 4621–4639. [DOI] [PubMed] [Google Scholar]
- (15).(a) Huigens RW III; Morrison KC; Hicklin RW; Flood T Jr.; Richter MF; Hergenrother PJ A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 2013, 5, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rafferty RJ; Hicklin RW; Maloof KA; Hergenrother PJ Synthesis of Complex and Diverse Compounds through Ring Distortion of Abietic Acid. Angew. Chem., Int. Ed. 2014, 53, 220–224. [DOI] [PubMed] [Google Scholar]
- (16).(a) Burke MD; Schreiber SL A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem., Int. Ed. 2004, 43, 46–58. [DOI] [PubMed] [Google Scholar]; (b) Galloway WRJD; Isidro-Llobet A; Spring DR Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [DOI] [PubMed] [Google Scholar]; (c) Tan DS Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [DOI] [PubMed] [Google Scholar]; (d) Spring DR Diversity-oriented synthesis; a challenge for synthetic chemists. Org. Biomol. Chem. 2003, 1, 3867–3870. [DOI] [PubMed] [Google Scholar]
- (17).Sun AW; Lackner S; Stoltz BM Modularity: Adding New Dimensions to Total Synthesis. Cell 2019, 1, 630–643. [Google Scholar]
- (18).(a) Mehta G; Singh V Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334. [DOI] [PubMed] [Google Scholar]; (b) Tietze LF; Bell HP; Chandrasekhar S Natural Product Hybrids as New Leads for Drug Discovery. Angew. Chem., Int. Ed. 2003, 42, 3996–4028. [DOI] [PubMed] [Google Scholar]; (c) Suzuki K Lessons from Total Synthesis of Hybrid Natural Products. Chem. Rec. 2010, 10, 291–307. [DOI] [PubMed] [Google Scholar]
- (19).Wender PA; Quiroz RV; Stevens MC Function through Synthesis-Informed Design. Acc. Chem. Res. 2015, 48, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Wu Z-C; Boger DL The quest for supernatural products: the impact of total synthesis in complex natural products medicinal chemistry. Nat. Prod. Rep. 2020, 37, 1511–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Z-C; Boger DL Maxamycins: Durable Antibiotics Derived by Rational Redesign of Vancomycin. Acc. Chem. Res. 2020, 53, 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nicolaou KC; Rigol S Perspectives from nearly five decades of total synthesis of natural products and their analogues for biology and medicine. Nat. Prod. Rep. 2020, 37, 1404–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Sun C; Wang Q; Brubaker JD; Wright PM; Lerner CD; Noson K; Charest M; Siegel DR; Wang Y-M; Myers AG A Robust Platform for the Synthesis of New Tetracycline Antibiotics. J. Am. Chem. Soc. 2008, 130, 17913–17927. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seiple IB; Zhang Z; Jakubec P; Langlois-Mercier A; Wright PM; Hog DT; Yabu K; Allu SR; Fukuzaki T; Carlsen PN; Kitamura Y; Zhou X; Condakes ML; Szczypinski FT; Green WD; Myers AG A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Volmer AA; Szpilman AM; Carreira EM Synthesis and biological evaluation of amphotericin B derivatives. Nat. Prod. Rep. 2010, 27, 1329–1349. [DOI] [PubMed] [Google Scholar]
- (24).Endo MM; Cioffi AG; Burke MD Our Path to Less Toxic Amphotericins. Synlett 2016, 27, 337–354. [Google Scholar]
- (25).(a) Pathak TP; Miller SJ Site-Selective Bromination of Vancomycin. J. Am. Chem. Soc. 2012, 134, 6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lewis CA; Merkel J; Miller SJ Catalytic site-selective synthesis and evaluation of a series of erythromycin analogs. Bioorg. Med. Chem. Lett. 2008, 18, 6007–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lewis CA; Longcore KE; Miller SJ; Wender PA An Approach to the Site-Selective Diversification of Apoptolidin A with Peptide-Based Catalysts. J. Nat. Prod. 2009, 72, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lewis CA; Miller SJ Site-Selective Derivatization and Remodeling of Erythromycin A by Using Simple Peptide-Based Chiral Catalysts. Angew. Chem., Int. Ed. 2006, 45, 5616–5619. [DOI] [PubMed] [Google Scholar]
- (26).(a) Mendoza A; Ishihara Y; Baran PS Scalable enantioselective total synthesis of taxanes. Nat. Chem. 2012, 4, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan C; Jin Y; Wilde NC; Baran PS Short, Enantioselective Total Synthesis of Highly Oxidized Taxanes. Angew. Chem., Int. Ed. 2016, 55, 8280–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kanda Y; Nakamura H; Umemiya S; Puthukanoori RK; Appala VRM; Gaddamanugu GK; Paraselli BR; Baran PS Two-Phase Synthesis of Taxol. J. Am. Chem. Soc. 2020, 142, 10526–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rossiter SE; Fletcher MH; Wuest WM Natural Products as Platforms To Overcome Antibiotic Resistance. Chem. Rev. 2017, 117, 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Goss RJM; Shankar S; Fayad AA The generation of “unNatural” products: Synthetic biology meets synthetic chemistry. Nat. Prod. Rep. 2012, 29, 870–889. [DOI] [PubMed] [Google Scholar]
- (29).Enquist JA Jr.; Stoltz BM Synthetic efforts toward cyathane diterpenoid natural products. Nat. Prod. Rep. 2009, 26, 661–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bailly C; Gao J-M Erinacine A and related cyathane diterpenoids: Molecular diversity and mechanisms underlying their neuroprotection and anticancer activities. Pharmacol. Res. 2020, 159, 104953. [DOI] [PubMed] [Google Scholar]
- (31).(a) Pfeiffer MWB; Phillips AJ Total Synthesis of (+)-Cyanthiwigin U. J. Am. Chem. Soc. 2005, 127, 5334–5335. [DOI] [PubMed] [Google Scholar]; (b) Pfeiffer MWB; Phillips AJ Conversion of cyanthiwigin U to related cyanthiwigins: total syntheses of cyanthiwigin W and cyanthiwigin Z. Tetrahedron Lett. 2008, 49, 6860–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reddy TJ; Bordeau G; Trimble L Total Synthesis of (+)-Cyanthiwigin AC. Org. Lett. 2006, 8, 5585–5588. [DOI] [PubMed] [Google Scholar]; (d) Wang C; Wang D; Gao S Total Synthesis of Cyanthiwigins A, C, G, and H. Org. Lett. 2013, 15, 4402–4405. [DOI] [PubMed] [Google Scholar]; (e) Chang Y; Shi L; Huang J; Shi L; Zhang Z; Hao H-D; Gong J; Yang Z Stereoselective Total Synthesis of (±)-5-epi-Cyanthiwigin I via an Intramolecular Pauson-Khand Reaction as the Key Step. Org. Lett. 2018, 20, 2876–2879. [DOI] [PubMed] [Google Scholar]
- (32).Enquist J Jr.; Stoltz BM The total synthesis of (−)-cyanthiwigin F by means of double catalytic enantioselective alkylation. Nature 2008, 453, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Enquist J Jr.; Virgil SC; Stoltz BM Total Syntheses of Cyanthiwigins B, F, and G. Chem. - Eur. J. 2011, 17, 9957–9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shibuya GM; Enquist J Jr.; Stoltz BM Enantioselective Synthesis of the 5–6-7 Carbocyclic Core of the Gagunin Diterpenoids. Org. Lett. 2013, 15, 3480–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).(a) Rho J-R; Lee H-S; Sim CJ; Shin J Gagunins, highly oxygenated diterpenoids from the sponge Phorbas sp. Tetrahedron 2002, 58, 9585–9591. [Google Scholar]; (b) Jang KH; Jeon J; Ryu S; Lee H-S; Oh K-B; Shin J Polyoxygenated Diterpenes from the Sponge Phorbas sp. J. Nat. Prod. 2008, 71, 1701–1707. [DOI] [PubMed] [Google Scholar]
- (36).Marziale AN; Duquette DC; Craig RA II; Kim KE; Liniger M; Numajiri Y; Stoltz BM An Efficient Protocol for the Palladium-Catalyzed Asymmetric Decarboxylative Allylic Alkylation Using Low Palladium Concentrations and a Palladium(II) Precatalyst. Adv. Synth. Catal. 2015, 357, 2238–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wickens ZK; Morandi B; Grubbs RH Aldehyde-Selective Wacker-Type Oxidation of Unbiased Alkenes Enabled by a Nitrite Co-Catalyst. Angew. Chem., Int. Ed. 2013, 52, 11257–11260. [DOI] [PubMed] [Google Scholar]
- (38).Kim KE; Stoltz BM A Second-Generation Synthesis of the Cyanthiwigin Natural Product Core. Org. Lett. 2016, 18, 5720–5723. [DOI] [PubMed] [Google Scholar]
- (39).Kim KE; Li J; Grubbs RH; Stoltz BM Catalytic Anti-Markovnikov Transformations of Hindered Terminal Alkenes Enabled by Aldehyde-Selective Wacker-Type Oxidation. J. Am. Chem. Soc. 2016, 138, 13179–13182. [DOI] [PubMed] [Google Scholar]
- (40).(a) Hirama N; Sakamoto R; Maruoka K Synthesis of a-Quaternary Aldehydes via a Stereoselective Semi-Pinacol Rearrangement of Optically Active Epoxy Alcohols. Asian J. Org. Chem. 2019, 8, 1390–1393. [Google Scholar]; (b) Cao M-Y; Ma B-J; Lao Z-Q; Wang H; Wang J; Liu J; Xing K; Huang Y-H; Gan K-J; Gao W; Wang H; Hong X; Lu H-H Optically Active Flavaglines-Inspired Molecules by a Palladium-Catalyzed Decarboxylative Dearomative Asymmetric Allylic Alkylation. J. Am. Chem. Soc. 2020, 142, 12039–12045. [DOI] [PubMed] [Google Scholar]; (c) Yuan Y; Zhang X; Qian H; Ma S Catalytic enantioselective allene-anhydride approach to b, c-unsaturated enones bearing an a-all-carbon-quarternary center. Chem. Sci. 2020, 11, 9115–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen Y; Dai Z-Y; Zheng C; Wang P-S Palladium-Catalyzed Allylic Alkylation via Photocatalytic Nucleophile Generation. ACS Catal. 2021, 11, 6757–6762. [Google Scholar]
- (41).Fernandes RA; Jha AK; Kumar P Recent advances in Wacker oxidation: from conventional to modern variants and applications. Catal. Sci. Technol. 2020, 10, 7448–7470. [Google Scholar]
- (42).Kim KE; Sakazaki Y; Stoltz BM Synthesis of non-natural cyanthiwigin-gagunin hybrids through late-stage diversification of the cyanthiwigin natural product core. Tetrahedron 2020, 76, 130755. [Google Scholar]
- (43).Kim KE; Adams AM; Chiappini ND; Du Bois J; Stoltz BM Cyanthiwigin Natural Product Core as a Complex Molecular Scaffold for Comparative Late-Stage C-H Functionalization Studies. J. Org. Chem. 2018, 83, 3023–3033. [DOI] [PubMed] [Google Scholar]
- (44).Fatta-Kassinos D; Vasquez MI; Kummerer K Transformation products of pharmaceuticals in surface waters and wastewater formed during photolysis and advanced oxidation processes-Degradation, elucidation of byproducts and assessment of their biological potency. Chemosphere 2011, 85, 693–709. [DOI] [PubMed] [Google Scholar]
- (45).The same stereoselectivity was observed when m-CPBA was used instead of DMDO, although lower yields of 21 were obtained using m-CPBA. Relative stereochemistry was assessed by NOE analysis.
- (46).(a) White MC; Zhao J Aliphatic C-H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Börgel J; Ritter T Late-Stage Functionalization. Chem. 2020, 6, 1877–1887. [Google Scholar]
- (47).(a) Campbell AN; White PB; Guzei IA; Stahl SS Allylic C-H Acetoxylation with a 4,5-Diazafluorenone-Ligated Palladium Catalyst: A Ligand-Based Strategy To Achieve Aerobic Catalytic Turnover. J. Am. Chem. Soc. 2010, 132, 15116–15119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen MS; White MC A Sulfoxide-Promoted, Catalytic Method for the Regioselective Synthesis of Allylic Acetates from Monosubstituted Olefins via C-H Oxidation. J. Am. Chem. Soc. 2004, 126, 1346–1347. [DOI] [PubMed] [Google Scholar]
- (48).Xing X; O’Connor NR; Stoltz BM Palladium(II)-Catalyzed Allylic C-H Oxidation of Hindered Substrates Featuring Tunable Selectivity Over Extent of Oxidation. Angew. Chem., Int. Ed. 2015, 54, 11186–11190. [DOI] [PubMed] [Google Scholar]
- (49).(a) McNeill E; Du Bois J Ruthenium-Catalyzed Hydroxylation of Unactivated Tertiary C-H Bonds. J. Am. Chem. Soc. 2010, 132, 10202–10204. [DOI] [PubMed] [Google Scholar]; (b) McNeill E; Du Bois J Catalytic C-H oxidation by a triazamacrocyclic ruthenium complex. Chem. Sci. 2012, 3, 1810–1813. [Google Scholar]; (c) Adams AM; Du Bois J Organocatalytic C-H hydroxylation with Oxone. enabled by an aqueous fluoroalcohol solvent system. Chem. Sci. 2014, 5, 656–659. [Google Scholar]; (d) Chen K; Baran PS Total synthesis of eudesmane terpenes by site-selective C-H oxidations. Nature 2009, 459, 824–828. [DOI] [PubMed] [Google Scholar]; (e) Chen MS; White MC A Predictably Selective Aliphatic C-H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]; (f) Adams AM; Du Bois J; Malik HA Comparative Study of the Limitations and Challenges in Atom-Transfer C-H Oxidations. Org. Lett. 2015, 17, 6066–6069. [DOI] [PubMed] [Google Scholar]
- (50).(a) Roizen JL; Zalatan DN; Du Bois J Selective Intermolecular Amination of C-H Bonds at Tertiary Carbon Centers. Angew. Chem., Int. Ed. 2013, 52, 11343–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chiappini ND; Mack JBC; Du Bois J Intermolecular C(sp3)-H Amination of Complex Molecules. Angew. Chem., Int. Ed. 2018, 57, 4956–4959. [DOI] [PubMed] [Google Scholar]
- (51).(a) Zhang X; Yang H; Tang P Transition-Metal-Free Oxidative Aliphatic C-H Azidation. Org. Lett. 2015, 17, 5828–5831. [DOI] [PubMed] [Google Scholar]; (b) Sharma A; Hartwig JF Metal-catalysed azidation of tertiary CH bonds suitable for late-stage functionalization. Nature 2015, 517, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karimov RR; Sharma A; Hartwig JF Late Stage Azidation of Complex Molecules. ACS Cent. Sci. 2016, 2, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang X; Bergsten TM; Groves JT Manganese-Catalyzed Late-Stage Aliphatic C-H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]
- (52).(a) Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]; (b) Gormisky PE; White MC Catalyst-Controlled Aliphatic C-H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. [DOI] [PubMed] [Google Scholar]
- (53).Quinn RK; Könst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ Site-Selective Aliphatic C-H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).(a) Newman DJ; Cragg GM Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]; (b) Scott JD; Williams RM Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev. 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]
- (55).Cuevas C; Francesch A Development of Yondelis. (Trabectedin, ET-743). A Semisynthetic Process Solves the Supply Problem. Nat. Prod. Rep. 2009, 26, 322–337. [DOI] [PubMed] [Google Scholar]
- (56).(a) Rath CM; Janto B; Earl J; Ahmed A; Hu FZ; Hiller L; Dahlgren M; Kreft R; Yu F; Wolff JJ; Kweon HK; Christiansen MA; Håkansson K; Williams RM; Ehrlich GD; Sherman DH Meta-omic Characterization of the Marine Invertebrate Microbial Consortium That Produces the Chemotherapeutic Natural Product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Song L-Q; Zhang Y-Y; Pu J-Y; Tang M-C; Peng C; Tang G-L Catalysis of Extracellular Deamination by a FAD-Linked Oxidoreductase after Prodrug Maturation in the Biosynthesis of Saframycin A. Angew. Chem., Int. Ed. 2017, 56, 9116–9120. [DOI] [PubMed] [Google Scholar]
- (57).For previous total syntheses of jorumycin, see: Lane JW; Chen Y; Williams RM Asymmetric Total Syntheses of (−)-Jorumycin, (−)-Renieramycin G, 3-epi-Jorumycin, and 3-epi-Renieramycin G. J. Am. Chem. Soc. 2005, 127, 12684–12690. Wu Y-C; Zhu J Asymmetric Total Syntheses of (−)-Renieramycin M and G and (−)-Jorumycin using Aziridine as a Lynchpin. Org. Lett. 2009, 11, 5558–5561. Liu W; Liao X; Dong W; Yan Z; Wang N; Liu Z Total Synthesis and Cytotoxicity of (−)-Jorumycin and its Analogues. Tetrahedron 2012, 68, 2759–2764. Chen R; Liu H; Chen X Asymmetric Total Synthesis of (−)-Jorunnamycins A and C and (−)-Jorumycin from L-Tyrosine. J. Nat. Prod. 2013, 76, 1789–1795.
- (58).Gunaydin H; Altman MD; Ellis JM; Fuller P; Johnson SA; Lahue B; Lapointe B Strategy for Extending Half-Life in Drug Design and its Significance. ACS Med. Chem. Lett. 2018, 9, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Welin ER; Ngamnithiporn A; Klatte M; Lapointe G; Pototschnig GM; McDermott MSJ; Conklin D; Gilmore CD; Tadross PM; Haley CK; Negoro K; Glibstrup E; Grunanger CU; Allan KM; Virgil SC; Slamon DJ; Stoltz BM Concise Total Syntheses of (−)-Jorunnamycin A and (−)-Jor-umycin Enabled by Asymmetric Catalysis. Science 2019, 363, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Campeau L-C; Schipper DJ; Fagnou K Site-Selective sp2 and Benzylic sp3 Palladium-Catalyzed Direct Arylation. J. Am. Chem. Soc. 2008, 130, 3266–3267. [DOI] [PubMed] [Google Scholar]
- (61).Dorta R; Broggini D; Stoop R; Ruegger H; Spindler F; Togni A Chiral Xyliphos Complexes for the Catalytic Imine Hydrogenation Leading to the Metolachlor Herbicide: Isolation of Catalyst-Substrate Adducts. Chem. - Eur. J. 2004, 10, 267–278. [DOI] [PubMed] [Google Scholar]
- (62).(a) Wang D-S; Chen Q-A; Lu S-M; Zhou Y-G Asymmetric Hydrogenation of Heteroarenes and Arenes. Chem. Rev. 2012, 112, 2557–2590. [DOI] [PubMed] [Google Scholar]; (b) Kim AN; Stoltz BM Recent Advances in Homogeneous Catalysts for the Asymmetric Hydrogenation of Heteroarenes. ACS Catal. 2020, 10, 13834–13851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kim AN; Ngamnithiporn A; Welin ER; Daiger MT; Grunanger CU; Bartberger MD; Virgil SC; Stoltz BM Iridium-Catalyzed Enantioselective and Diastereoselective Hydrogenation of 1,3-Disubstituted Isoquinolines. ACS Catal. 2020, 10, 3241–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Donohoe TJ; Pilgrim BS; Jones GR; Bassuto JA Synthesis of Substituted Isoquinolines Utilizing Palladium-Catalyzed a-Arylation of Ketones. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11605–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Scott JD; Williams RM Chemistry and Biology of Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev. 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]