

Abstract
Chronic spontaneous urticaria (CSU) is characterized by the spontaneous development of wheals, itching, and/or angioedema, for ≥6 weeks. In China, non‐sedating H1‐antihistamines (H1AH) are the recommended first‐line treatment, with escalation up to 4× the standard dose in symptomatic patients to achieve control. Treatment options for Chinese patients who remain symptomatic on H1AH treatment are limited. This 20‐week randomized, double blind, placebo‐controlled, parallel‐group study investigated the efficacy and safety of omalizumab as an add‐on therapy for the treatment of patients with CSU who remained symptomatic despite H1AH treatment in China. Adult patients (N = 418) diagnosed with refractory CSU for ≥6 months were randomized (2:2:1) to receive omalizumab 300 mg (OMA300), omalizumab 150 mg (OMA150) or placebo, subcutaneously, every 4 weeks. Primary outcome was change from baseline to week 12 in weekly itch severity score (ISS7). Safety was assessed by rates of adverse events (AEs). Demographic and disease characteristics at baseline were comparable across treatment groups. At week 12, statistically significant greater decreases from baseline were observed in ISS7 with OMA300 (least square mean difference [LSM]: −4.23; 95% confidence interval [CI]: −5.70, −2.77; p < 0.001) and OMA150 (LSM: −3.79; 95% CI: −5.24, −2.33; p < 0.001) versus placebo. Incidence of treatment‐emergent AEs over 20 weeks was slightly higher with OMA300 (71.3%) compared to OMA150 and placebo groups (64.7% and 63.9%, respectively). The incidences of serious AEs were balanced between groups. This study demonstrated the efficacy and safety of omalizumab in Chinese adult patients with CSU who remained symptomatic despite H1AH therapy.
Keywords: Chinese, chronic spontaneous urticaria, efficacy, omalizumab, safety
1. INTRODUCTION
Chronic spontaneous urticaria (CSU) is a skin disorder characterized by the spontaneous development of daily, or almost daily, wheals (hives), itching, and/or angioedema, for ≥6 weeks. 1 , 2 , 3
CSU is estimated to affect 0.5%–1% of the world at any given time. 4 Although national data for the prevalence of CSU in China are unavailable, prevalence is seen to be on the rise with increasing urbanization. 5 According to an epidemiological survey of urticaria conducted by the Third Military Medical University in China, CSU accounts for about half of the total urticaria population. 6 A recent hospital‐based, multicenter, epidemiological study in the Chinese population found CSU to be the most common subtype (61%) in patients diagnosed with chronic urticaria. 7
Currently, non‐sedating H1 antihistamines (H1AH) are the recommended first‐line treatment for CSU in Europe and US as well as in China, with treatment at doses up to 4× the standard dose in patients who remain symptomatic. 3 , 8 However, up to 50% of CSU patients remain symptomatic despite high‐dose H1AH therapy in China. 9 Treatment options for those who remain symptomatic on treatment with H1AH are limited. A new, effective, and safe therapy is urgently needed.
The European Academy of Allergy and Clinical Immunology/Global Allergy and Asthma European Network/European Dermatology Forum/World Allergy Organization (EAACI/GA 2 LEN/EDF/WAO) and American Academy of Allergy, Asthma, and Immunology/American College of Allergy, Asthma, and Immunology (AAAAI/ACAAI) guideline recommend omalizumab as add‐on therapy for patients who remain symptomatic even with high‐dose H1AH treatment 10 based on the clinical efficacy demonstrated in multiple trials globally (ASTERIA I, ASTERIA II, GLACIAL), as well as in the Asian population (POLARIS). 1 , 11 , 12 , 13
While the POLARIS study was conducted in the Japanese and Korean population, there is a lack of data about the efficacy and safety of omalizumab in the Chinese population with CSU.
This study reports the efficacy and safety of omalizumab, compared with placebo, as an add‐on to H1AH therapy in Chinese adult patients who remained symptomatic despite H1AH treatment.
2. METHODS
2.1. Study design
This was a randomized, multicenter, double blind, placebo‐controlled, parallel‐group study conducted in China. Omalizumab was given as an add‐on therapy for the treatment of patients with refractory CSU who remained symptomatic on treatment with H1AH.
This study was designed, implemented, executed, and reported in accordance with the International Council for Harmonization (ICH) Harmonized Tripartite Guidelines for Good Clinical Practice, with applicable local regulations, and with the ethical principles laid down in the Declaration of Helsinki. Written consent from the patients was obtained.
The study consisted of three distinct epochs (screening, randomized treatment, and post‐treatment follow‐up) over 24 weeks (Figure 1). Patients were randomized (2:2:1) to receive subcutaneous injections of omalizumab 300 mg (OMA 300), omalizumab 150 mg (OMA 150), or placebo. Patients were required to stay on stable H1AH treatment during the screening epoch. The screening epoch was extended for patients who required treatment for latent tuberculosis to allow for a 4‐week latent tuberculosis treatment period prior to randomization.
FIGURE 1.
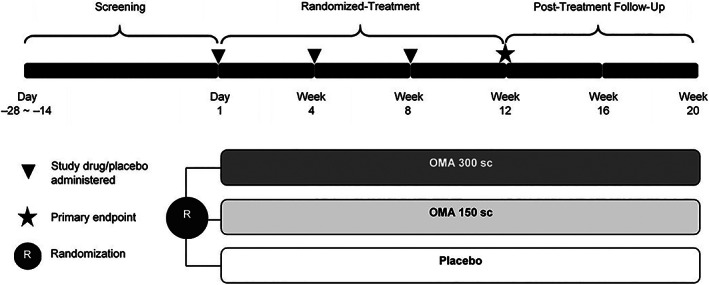
Study design OMA 150/300, omalizumab 150/300 mg; sc, subcutaneous
Eligible patients received their respective injection every 4 weeks (on day 1, week 4, and week 8) during the 12‐week, double blind, randomized‐treatment epoch. They were instructed to stay on the same CSU H1AH treatment at stable dose as per prerandomization period. Diphenhydramine was allowed as rescue medication. The primary efficacy assessment was done at week 12.
After the completion of the 12‐week randomized‐treatment epoch, all patients entered the 8‐week post‐treatment follow‐up epoch to allow for further characterization of the efficacy and safety data and collection of additional pharmacokinetics and pharmacodynamics of omalizumab. No study drug treatment was given during the post‐treatment follow‐up epoch.
2.2. Study population
Patients aged 18–75 years who were diagnosed with refractory CSU for ≥6 months, with weekly Urticaria Score (UAS7, range: 0–42) ≥16 and itch component of UAS7 (0–21) ≥8 during 7 days prior to randomization, and in‐clinic UAS ≥4 on at least one of the screening visit days (day −14, day −7, or day 1), remained symptomatic (presence of itch and hives for ≥6 consecutive weeks) at any time prior to randomization despite conventional H1AH treatment were included in this study. Included patients were on approved dose of a non‐sedating H1AH for CSU for at least 3 consecutive days immediately prior to the day −14 screening visit and should have documented current use on the day of the initial screening visit.
Patient who had a clearly defined underlying etiology for chronic urticaria other than CSU (e.g., solar, cholinergic, heat, cold, etc.) and those who had skin disease other than CSU with chronic itching that could confound the results of the study were excluded.
2.3. Endpoints
The primary efficacy endpoint was the change from baseline in the weekly Itch Severity Score (ISS7) at week 12. Secondary efficacy endpoints collected included change from baseline in the UAS7 at week 12, change from baseline in the NHS7 at week 12, proportion of patients who achieved UAS7 ≤ 6 at week 12, complete UAS7 response (UAS7 = 0) at week 12, ISS7 MID response (defined as reduction from baseline in ISS7 of ≥5 points) at week 12, and the change from baseline in overall Dermatology Life Quality Index (DLQI) score at week 12 and time to ISS7 MID by week 12.
Safety assessments consisted of collecting all adverse events (AEs), serious adverse events (SAEs), with their severity and relationship to the study treatment.
2.4. Analysis set
Efficacy assessment was performed on full analysis set (FAS), defined as all‐randomized patients who received at least one dose of the study drug with the exception of those who had inadvertently been randomized into the study. Safety assessment was performed on safety set (SAF), which consisted of all patients who took at least 1 dose of study medication.
2.5. Statistical analysis
The total sample size of 420 (including 10% dropout) with 2:2:1 assignment ratio was determined to allow at least 93% power to achieve the primary objective and at least 80% power to achieve all the first 3 secondary objectives (change from baseline in UAS7, NHS7, and proportion of patients with USA7 ≤ 6 at week 12) for both OMA 300 and OMA 150. The power was examined according to the hierarchy order of the multiplicity type I error control plan with the overall α level controlled at 0.05 (2‐sided).
Analysis was performed with a mixed‐effect linear model with repeated measures (MMRM) approach to obtain the least square (LS) mean estimate for each treatment group for change from baseline in ISS7 (primary endpoint), UAS7, NHS7, or DLQI at week 12. The MMRM model included terms of treatment group, week (1–12) (or visit, in the model for DLQI), baseline score, baseline score‐by‐week (or visit) interaction, and treatment‐by‐week (or visit) interaction as fixed effects. The within‐patient correlation was modeled using the unstructured covariance matrix; compound symmetry covariance structure was used if the model did not converge. The difference in LS mean estimates between OMA 300/OMA 150 versus placebo, together with a 95% confidence interval (CI), were presented.
Between group comparisons of the proportion of patients with UAS7 ≤ 6, UAS7 = 0, ISS7 MID at week 12 was conducted using a logistic regression model with treatment group as a factor and their respective baseline value as a covariate. Between group comparisons of time to ISS7 MID response by week 12 were made using a Cox proportional hazard model, with treatment group as a factor and baseline value as a covariate.
The hypotheses testing on the primary and all secondary endpoints between OMA 300/OMA 150 versus placebo were included in the testing strategy to ensure that the family‐wise type I error was kept at an overall level of less than 5%, using a flexible gate‐keeping procedure. 14
Descriptive summary statistics were presented for safety assessments.
3. RESULTS
3.1. Patient disposition and baseline characteristics
A total of 632 patients were screened, of which 418 patients from 27 sites in China were randomized in the study (Figure 2). The majority of the patients completed the randomized‐treatment epoch (94.5%). Of the 23 patients (5.5%) who discontinued during the randomized‐treatment epoch, the majority (n = 12) cited patient/guardian decision. Discontinuations were numerically higher in the OMA 300 group (n = 16, 9.5%) versus OMA 150 (n = 5, 3.0%) and placebo group (n = 2, 2.4%).
FIGURE 2.
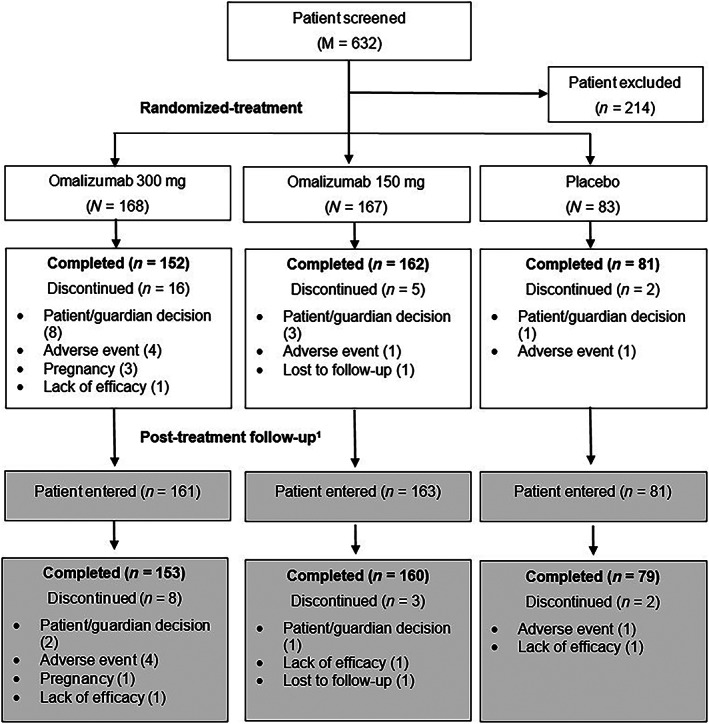
Patient disposition. 1 All randomized patients were to enter the post‐treatment follow‐up irrespective of completion status in randomized treatment epoch. M, total number of patients screened; N, total number of patients randomized; n, number of patients
Of the randomized patients, 13 (7 in OMA 300, 4 in OMA 150, and 2 in placebo) did not enter the follow‐up epoch. In total, 392/405 (93.8%) patients completed the follow‐up epoch. AEs, patient/guardian decision, and lack of efficacy were the most commonly reported reasons for discontinuation across the treatment groups. Higher discontinuations were reported in the OMA 300 group versus OMA 150 and placebo groups. One patient in the OMA 300 group did not receive any dose of the study drug and was excluded from the FAS and the SAF sets.
The baseline demographics and disease characteristics were comparable across treatment groups. The majority of patients randomized in the study were females (66.0%) and < 65 years (97.1%) with the median age of 38 years (Table 1). Patients randomized in the study had a mean body mass index (BMI) of 23.49 kg/m2, a mean disease duration of 4.27 years, and evidence of severe disease as noted by a mean UAS7 score of 31.77. Over half of the patients (69.9%) had been on >3 CSU medications prior to inclusion and 61.2% reported previous use of systemic steroid and/or immunosuppressant treatment for CSU.
TABLE 1.
Demographics and baseline disease characteristics
| Demographics | OMA 300 N = 168 | OMA 150 N = 167 | Placebo N = 83 | Total N = 418 |
|---|---|---|---|---|
| Age (years) | ||||
| Median (range) | 38.0 (20–67) | 36.0 (18–67) | 42.0 (22–72) | 38.0 (18–72) |
| Age group in years, n (%) | ||||
| <65 | 164 (97.6) | 163 (97.6) | 79 (95.2) | 406 (97.1) |
| ≥65 | 4 (2.4) | 4 (2.4) | 4 (4.8) | 12 (2.9) |
| Sex female, n (%) | 115 (68.5) | 108 (64.7) | 53 (63.9) | 276 (66.0) |
| BMI (kg/m2), mean (SD) | 23.35 (3.26) | 23.53 (3.10) | 23.72 (2.81) | 23.49 (3.11) |
| Baseline disease characteristics | ||||
| Duration of CSU (years), mean (SD) | 4.18 (6.16) | 4.11 (5.26) | 4.79 (6.46) | 4.27 (5.87) |
| Prior medication for CSU, n (%) | ||||
| ≤3 | 49 (29.2) | 52 (31.1) | 25 (30.1) | 126 (30.1) |
| >3 | 119 (70.8) | 115 (68.9) | 58 (69.9) | 292 (69.9) |
| Prior systemic treatment for CSU yes, n (%) | 101 (60.1) | 103 (61.7) | 52 (62.7) | 256 (61.2) |
| Total IgE (ng/ml), median (range) |
230 (6.6–3170) |
272 (0–3770) |
232 (12.1–2810) |
256 (0–3770) |
| In‐clinic UAS, mean (SD) | 4.4 (1.25) | 4.6 (1.21) | 4.7 (1.13) | 4.6 (1.21) |
| ISS7, mean (SD) | 14.38 (3.87) | 14.92 (3.90) | 16.11 (3.76) | 14.94 (3.90) |
| UAS7, mean (SD) | 30.52 (7.38) | 31.94 (7.10) | 33.96 (6.81) | 31.77 (7.25) |
| NHS7, mean (SD) | 16.14 (4.33) | 17.02 (4.04) | 17.84 (3.91) | 16.83 (4.17) |
| Angioedema yes, n (%) | 29 (17.3) | 43 (25.7) | 23 (27.7) | 95 (22.7) |
Note: Duration of CSU is calculated from the date of diagnosis of CSU recorded on the visit 1. Previous numbers of CSU medication and systemic treatment for CSU are collected at visit 1. Baseline related to eDiary data is defined over the 7 days prior to the first treatment date (or prior the randomization date when a patient did not take any study medication). Other baseline disease characteristics are defined as the last non‐missing assessment collected before or on the first treatment date (or before or on the randomization date when a patient did not take any study medication). Total IgE values below the lower limit of quantification (4.80 ng/ml) were set to 0.
Abbreviations: BMI, body mass index; CSU, chronic spontaneous urticaria; IgE, immunoglobulin E; ISS7, weekly Itch Severity Score; NHS7, weekly Number of Hives Score; OMA, omalizumab; UAS7, weekly Urticaria Activity Score.
In all treatment groups, the most frequent newly started concomitant medication was non‐sedating H1AHs, as this study allowed one additional non‐sedating H1AH during the follow‐up period. Three patients used corticosteroids and/or immunosuppressant during the follow‐up period. Prior medications were balanced across groups, mainly with antihistamine treatment (95%) followed by herbal preparation (42.2%) and traditional medicine (23.7%).
3.2. Efficacy
OMA 300 and OMA 150 demonstrated clinically and statistically significant superiority over placebo in improving ISS7 score in patients with CSU refractory to H1AH. At week 12, the LS mean difference (95% CI) of change from baseline in ISS7 was −4.23 (−5.70, −2.77) for OMA 300 (p < 0.001) and − 3.79 (−5.24, −2.33) for OMA 150 (p < 0.001) compared to placebo, respectively (Table 2).
TABLE 2.
Primary and secondary efficacy endpoints
| Comparison | LS mean (SE) | Comparison of LS mean | ||||
|---|---|---|---|---|---|---|
| MMRM analysis of change from baseline in ISS7 at week 12 | ||||||
| Test | Reference | Difference (SE) | 95% CI | Adjusted p value | ||
| OMA 300 (n = 167) versus placebo (n = 83) | −10.11 (0.43) | −5.87 (0.60) | −4.23 (0.75) | (−5.70, −2.77) | <0.001 | |
| OMA 150 (n = 166) versus placebo (n = 83) | −9.66 (0.42) | −5.87 (0.60) | −3.79 (0.74) | (−5.24, −2.33) | <0.001 | |
| MMRM analysis of change from baseline in UAS7 at week 12 | ||||||
| Test | Reference | Difference (SE) | 95% CI | Adjusted p value | ||
| OMA 300 (n = 167) versus placebo (n = 83) | −21.82 (0.90) | −11.62 (1.26) | −10.19 (1.56) | (−13.25, −7.14) | <0.001 | |
| OMA 150 (n = 166) versus placebo (n = 83) | −20.74 (0.88) | −11.62 (1.26) | −9.12 (1.54) | (−12.14, −6.10) | <0.001 | |
| MMRM analysis of change from baseline in NHS7 at week 12 | ||||||
| Test | Reference | Difference (SE) | 95% CI | Adjusted p value | ||
| OMA 300 (n = 167) versus placebo (n = 83) | −11.68 (0.49) | −5.76 (0.69) | −5.92 (0.85) | (−7.59, −4.24) | <0.001 | |
| OMA 150 (n = 166) versus placebo (n = 83) | −11.11 (0.49) | −5.76 (0.69) | −5.35 (0.84) | (−7.00, −3.69) | <0.001 | |
| Logistic regression analysis of proportion of patients with UAS7 ≤ 6 at week 12 | ||||||
| n/M (%) | Comparison | OR | 95% CI | Adjusted p value | ||
| OMA 300 | 81/167 (48.5) | Versus placebo | 7.02 | (3.27, 15.06) | <0.001 | |
| OMA 150 | 79/167 (47.3) | Versus placebo | 7.03 | (3.29, 15.06) | <0.001 | |
| Placebo | 9/83 (10.8) | – | – | – | – | |
| Logistic regression analysis of proportion of patients with UAS7 = 0 at week 12 | ||||||
| n/M (%) | Comparison | OR | 95% CI | Adjusted p value | ||
| OMA 300 | 62/167 (37.1) | Versus placebo | 11.21 | (3.88, 32.37) | <0.001 | |
| OMA 150 | 39/167 (23.4) | Versus placebo | 5.88 | (2.01, 17.17) | 0.002 | |
| Placebo | 4/83 (4.8) | – | – | – | – | |
| Logistic regression analysis of proportion of patients with ISS7 MID at week 12 | ||||||
| n/M (%) | Comparison | OR | 95% CI | Adjusted p value | ||
| OMA 300 | 125/167 (74.9) | Versus placebo | 2.73 | (1.51, 4.95) | 0.002 | |
| OMA 150 | 125/167 (74.9) | Versus placebo | 2.53 | (1.41, 4.56) | 0.002 | |
| Placebo | 49/83 (59.0) | – | – | – | – | |
| MMRM analysis of change from baseline in overall DLQI at week 12 | ||||||
| Test | Reference | Difference (SE) | 95% CI | Adjusted p value | ||
| OMA 300 (n = 165) versus placebo (n = 83) | −10.4 (0.50) | −6.5 (0.69) | −4.0 (0.85) | (−5.7, −2.3) | 0.002 | |
| OMA 150 (n = 166) versus placebo (n = 83) | −9.9 (0.49) | −6.5 (0.69) | −3.5 (0.85) | (−5.1, −1.8) | 0.002 | |
| Cox regression analysis of time to first ISS7 MID response by week 12 | ||||||
| n/M (%) | Comparison | HR | 95% CI | Adjusted p value | ||
| OMA 300 | 142/167 (85.0) | Versus placebo | 1.71 | (1.25, 2.33) | 0.002 | |
| OMA 150 | 144/167 (86.2) | Versus placebo | 1.66 | (1.22, 2.25) | 0.002 | |
| Placebo | 59/83 (71.1) | – | – | – | – | |
Abbreviations: CI, confidence interval; DLQI, Dermatology Life Quality Index; HR, hazard ratio; ISS7, weekly Itch Severity Score; LS mean, least squares mean; M, total number of patients in the analysis; MID, minimally important difference; MMRM, mixed model with repeated measures; n (in the MMRM model results), total number of patients in the analysis; n (in the Logistic regression results), number of patients who achieved the investigated response at week 12 (after imputation); n (in the Cox regression results), total number of events included in the analysis; NHS7, weekly Number of Hives Score; OMA, omalizumab; OR, odds ratio; SE, standard error; UAS7, weekly Urticaria Activity Score.
Mean ISS7 score decreased from Baseline in all treatment groups during the study period. A greater mean decrease in ISS7 score was observed for patients in the OMA 300 and OMA 150 from week 1 through week 12, compared to patients in the placebo group. Mean change from baseline in ISS7 score for the omalizumab treatment groups started to approach those in the placebo group after the treatment phase. The mean change from baseline in ISS7 for all treatment groups remained below Baseline through the end of assessment, week 20 (Figure 3A). Similarly at week 12, the change from baseline was statistically significant for both of the omalizumab doses compared to placebo in reducing the UAS7 and NHS7 scores. The LS mean difference (95% CI) of UAS7 was −10.19 (−13.25, −7.14) for OMA 300 (p < 0.001) and −9.12 (−12.14, −6.10) for OMA 150 (p < 0.001) compared to placebo, respectively (Table 2). Mean UAS7 score decreased from Baseline in all treatment groups during the study period. Patients in the OMA 300 and OMA 150 achieved a greater mean decrease in UAS7 score from week 1 through week 12, compared to patients in the placebo group. Similar to those observed in ISS7, mean UAS7 score for the omalizumab treatment groups approached those in the placebo group after week 12 (end of treatment epoch) but remained below baseline through week 20 (Figure 3B).
FIGURE 3.
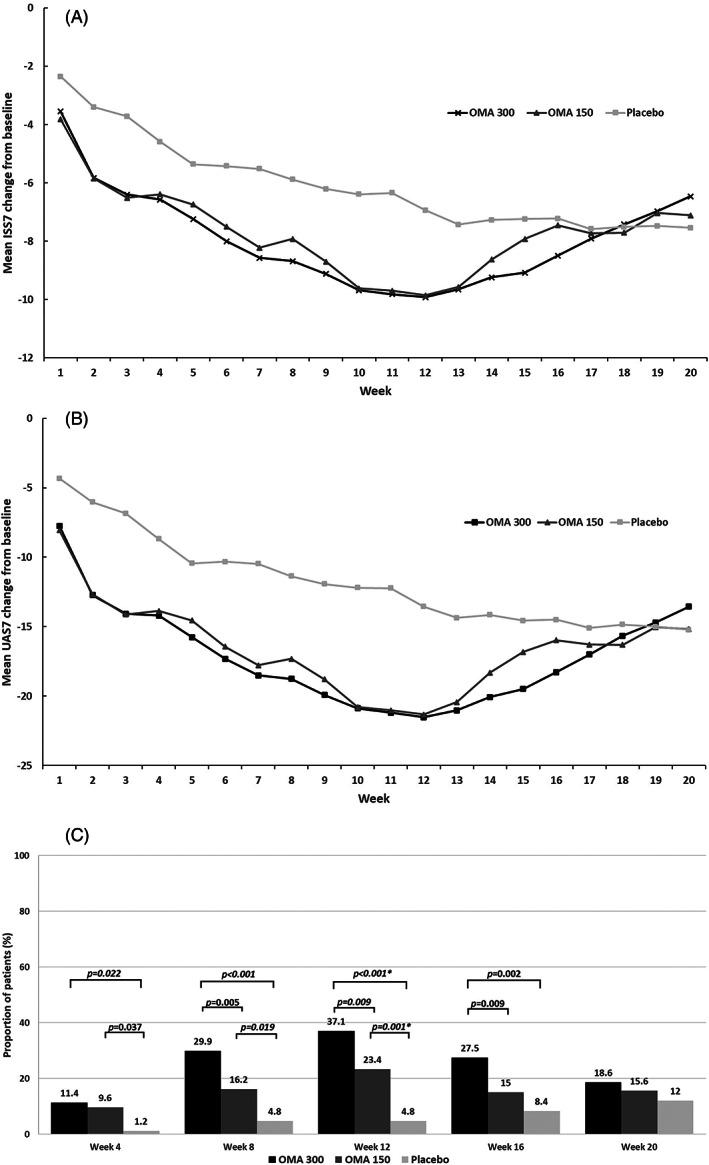
Clinical response over the study period as measured by (A) mean change from baseline in ISS7, (B) mean change from baseline in UAS7, and (C) proportion of patients with UAS7 = 0†. ISS7, weekly Itch Severity Score; OMA, omalizumab; UAS7, weekly Urticaria Activity Score. †All comparisons are post hoc analysis, except for week 12 omalizumab (both doses) versus placebo, which are predefined secondary endpoint; All p values presented are nominal p values, and need to be interpreted with caution. †is to note that the analysis for Figure 3C are post‐hoc analysis
At week 12, a statistically significantly greater proportion of patients achieved UAS7 = 0 (complete responders) in the OMA 300 (62/167, 37.1%) and OMA 150 groups (39/167, 23.4%) than in the placebo group (4/83, 4.8%). The odds ratio (ORs, 95% CI) for OMA 300 and OMA 150 were 11.21 (3.88, 32.37; p < 0.001) and 5.88 (2.01, 17.17; p = 0.002) compared to placebo, respectively (Table 2). A post hoc analysis was conducted to further analyze the dose‐dependent profile in proportion of complete responders over time. Results showed that omalizumab demonstrated a dose‐dependent improvement in proportion of patients achieving UAS7 = 0 starting at week 8 and maintained through week 16 (Figure 3C).
The LS mean difference (95% CI) of NHS7 was −5.92 (−7.59, −4.24) for OMA 300 (p < 0.001) and − 5.35 (−7.00, −3.69) for OMA 150 (p < 0.001) compared to placebo, respectively (Table 2). At week 12, statistically significant greater proportion of patients achieved UAS7 ≤ 6 (responders) in the OMA 300 (81/167, 48.5%) and OMA 150 groups (79/167, 47.3%) than in the placebo group (9/83, 10.8%). The ORs (95% CI) for OMA 300 and OMA 150 were 7.02 (3.27, 15.06; p < 0.001) and 7.03 (3.29, 15.06; p < 0.001) compared to placebo, respectively (Table 2).
Statistically significant greater proportion of patients achieved ISS7 MID response (reduction from baseline in ISS7 ≥ 5 points) at week 12 in the OMA 300 (125/167, 74.9%) and OMA 150 groups (125/167, 74.9%) than in the placebo group (49/83, 59.0%). The ORs (95% CI) for OMA 300 and OMA 150 groups were 2.73 (1.51, 4.95; p = 0.002) and 2.53 (1.41, 4.56; p = 0.002) compared to placebo, respectively (Table 2). Patients in the OMA 300 (142/167, 85.0%) and OMA 150 groups (144/167, 86.2%) had a shorter median time to achieve ISS7 MID response (2.0 weeks for both the omalizumab groups) than did patients in the placebo group (59/83, 71.1%; 4 weeks) with hazard ratio (95% CI) of 1.71 (1.25, 2.33) for OMA 300 (P = 0.002) and 1.66 (1.22, 2.25) for OMA 150 (p = 0.002) compared to placebo.
OMA 300 and OMA 150 showed statistical significant improvement in DLQI at week 12 compared to placebo. The LS mean of treatment difference (95% CI) was −4.0 (−5.7, −2.3) for OMA 300 (p= 0.002) and −3.5 (−5.1, −1.8) for OMA 150 (p= 0.002) compared to placebo, respectively (Table 2).
3.3. Safety
In the study, patients had a median duration of exposure of 12 weeks among the treatment groups, ranging from 4.0 to 14.3 weeks for omalizumab treatment groups and 4.0–13.3 weeks for the placebo group. Treatment‐emergent AEs over 20 weeks were slightly higher in OMA 300 group (71.3%) compared to OMA 150 and placebo groups (64.7% and 63.9%, respectively). SAE incidence was similar between treatment groups. Only 1 SAE of pelvic inflammatory disease was suspected to be related to study drug (OMA 150), and for which study drug was discontinued. No death was reported (Table 3).
TABLE 3.
Summary of AE
| Preferred term | OMA 300 (N = 167) | OMA 150 (N = 167) | Placebo (N = 83) |
|---|---|---|---|
| Any AE | 119 (71.3) | 108 (64.7) | 53 (63.9) |
| Treatment‐related AE | 30 (18.0) | 29 (17.4) | 7 (8.4) |
| Severe AE | 5 (3.0) | 3 (1.8) | 2 (2.4) |
| SAE | 5 (3.0) | 5 (3.0) | 3 (3.6) |
| Treatment related | 0 | 1 (0.6) | 0 |
| Deaths | 0 | 0 | 0 |
| AE leading to discontinuation of study drug | 7 (4.2) | 1 (0.6) | 1 (1.2) |
| Treatment related | 3 (1.8) | 1 (0.6) | 0 |
| Most commonly reported AE (≥3% incidences in any group) | |||
| Upper respiratory tract infection | 37 (22.2) | 25 (15.0) | 12 (14.5) |
| Cough | 11 (6.6) | 3 (1.8) | 2 (2.4) |
| Influenza | 7 (4.2) | 7 (4.2) | 1 (1.2) |
| Nasopharyngitis | 7 (4.2) | 8 (4.8) | 7 (8.4) |
| Arthralgia | 6 (3.6) | 4 (2.4) | 0 |
| Blood uric acid increased | 5 (3.0) | 5 (3.0) | 4 (4.8) |
| Eczema | 5 (3.0) | 4 (2.4) | 1 (1.2) |
| Hypertension | 5 (3.0) | 3 (1.8) | 0 |
| Pyrexia | 5 (3.0) | 4 (2.4) | 1 (1.2) |
| Oropharyngeal pain | 4 (2.4) | 1 (0.6) | 3 (3.6) |
| Dermatitis | 2 (1.2) | 1 (0.6) | 3 (3.6) |
| Alanine aminotransferase increased | 1 (0.6) | 6 (3.6) | 1 (1.2) |
| Blood creatine phosphokinase increased | 1 (0.6) | 5 (3.0) | 0 |
| Pharyngitis | 1 (0.6) | 4 (2.4) | 3 (3.6) |
| Hepatic function abnormal | 0 | 2 (1.2) | 3 (3.6) |
Note: Preferred terms are sorted by descending frequency in the OMA 300 group. A patient with multiple occurrences of an AE under one treatment is counted only once in that AE category for that treatment. MedDRA Version 22.1 has been used for reporting.
Abbreviations: AE, adverse event; OMA, omalizumab; SAE, serious adverse event.
The most commonly reported AEs were upper respiratory tract infection, cough, nasopharyngitis, influenza, and increased blood uric acid (Table 3). Of all the reported treatment‐emergent AEs during the whole duration of the study, five patients in the OMA 300 group, three patients in the OMA 150 group, and two patients in the placebo group reported AEs of severe intensity (Table 3). No events for anaphylaxis were reported in either of the treatment groups. Other AEs identified as risks associated with omalizumab were infrequently reported in both the omalizumab groups and none of them was of severe intensity.
4. DISCUSSION
In this study, omalizumab demonstrated significant clinical benefits in CSU patients refractory to H1 antihistamines, including statistically significant superiority over placebo in improving ISS7, UAS7, and NHS7 at week 12. Treatment effect was seen as early as week 1 (Figure 3A), which was sustained throughout the treatment period. After week 12 (end of treatment epoch), symptom scores gradually increased, however, there was no evidence of any rebound effect as symptom scores did not reach pretreatment levels. The clinical efficacy results were paralleled by improvements in the quality of life as seen in the improvement in DLQI scores.
Results from this study were consistent with other phase 3 studies of omalizumab 1 , 11 , 12 , 13 in demonstrating efficacy of omalizumab in CSU. Overall efficacy was comparable between the OMA 300 and OMA 150 groups, except in the proportion of patients achieving UAS7 = 0 at week 12. Unlike prior studies, this study did not show clear dose‐dependent effect in most efficacy endpoints. However, a small numerical treatment difference was observed between both dose groups favoring OMA 300 for the primary and most secondary efficacy endpoints. This difference was particularly evident in the higher percentage of complete responders (patients who achieved UAS7 = 0 at week 12) seen in the OMA 300 group. The observation of similar efficacy appears to be driven by a higher response seen in the OMA 150 group in this study compared with previous phase 3 studies. 1 , 12 , 13 However, it should be noted that the study was not designed to show any superiority of doses between 300 mg and 150 mg dose groups. These study results in Chinese patients with CSU, who remained symptomatic despite H1AH therapy are overall in alignment with those from the earlier POLARIS study conducted in Japan and Korea. 1 Dose‐dependent efficacy of omalizumab is unlikely to be an ethnic effect.
In this study, omalizumab showed no new or unexpected safety signals. The overall safety profile of omalizumab is consistent with other CSU phase 3 studies and consistent with well‐established profile of omalizumab. 1 , 11 , 12 , 13 The incidence rates of SAEs were low and similar across the three treatment groups.
The current study had a lower proportion of patients with angioedema at baseline (range: 17.3%–27.7%) than those observed in global studies (range: 38.0%–55.0%), 11 , 12 , 13 and observed greater reduction in ISS7 in omalizumab‐treated groups than in the placebo‐treated group. The current findings validated the results from POLARIS study in demonstrating efficacy of omalizumab in the treatment of CSU regardless of severity and complication with angioedema. 1 The authors are aware that the current study was conducted only in the Chinese population, a direct comparison of efficacy between Chinese and Western populations cannot be made.
5. CONCLUSION
In conclusion, this study demonstrated significant clinical benefits in Chinese adult CSU patients who remained symptomatic despite H1AH therapy and were treated with OMA 300 and 150 mg and confirmed the well‐established safety profile.
CONFLICT OF INTEREST
Weiru Yuan (MD, PhD), Min Li (MD, PhD), Lijia Yang (MD), Lingling Liu (MD), Min Zheng (MD, PhD), Zaipei Guo (MD), Zhiqiang Song (MD, PhD), Chunlei Zhang (MD, PhD), Qingchun Diao (MD, PhD), Jinhua Xu (MD, PhD), and Jie Zheng (MD, PhD) declare no potential conflicts of interest. Robert Fogel (MD), Monica Ligueros‐Saylan (MD), and Alkaz Uddin (PhD) report being employees and shareholders of Novartis Pharmaceuticals Corporation (USA). Alexia Richard (MSc) being employee and shareholder of Novartis Pharma AG (Switzerland). Tianmeng Lyu (PhD) is an employee of Novartis Pharmaceuticals Corporation (USA). Shuling Hu (MD, PhD) is an employee of Novartis Institutes for BioMedical Research (China). Moreshwar Patwardhan (MPharm) is an employee of Novartis Healthcare Pvt Ltd (India).
AUTHOR CONTRIBUTIONS
Weiru Yuan, Min Li, Lijia Yang, Lingling Liu, Min Zheng, Zaipei Guo, Zhiqiang Song, Chunlei Zhang, Qingchun Diao, and Jinhua Xu's role in the study includes investigation, methodology, project administration, formal analysis, and writing (review and editing). Jie Zheng's role in the study includes investigation, methodology, project administration, supervision, formal analysis, and writing (review and editing). Robert Fogel's role in the study includes investigation, methodology, supervision, formal analysis, and writing (review and editing). Monica Ligueros‐Saylan's role in the study includes supervision, formal analysis, and writing (review and editing). Alkaz Uddin and Tianmeng Lyu's role in the study include methodology, supervision, formal analysis, and writing (review and editing). Moreshwar Patwardhan and Alexia Richard's role in the study includes methodology, supervision, validation, and writing (review and editing). Shuling Hu's role in the study includes investigation, methodology, project administration, supervision, formal analysis, and writing (review and editing).
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. Authors also take complete responsibility for the integrity of the data and accuracy of the data analysis. All authors take responsibility for the accuracy of the results. All authors reviewed, provided feedback on subsequent versions, and approved the submission of the manuscript for publication.
ETHICS STATEMENT
This study was designed, implemented, executed, and reported in accordance with the International Council for Harmonization (ICH) Harmonized Tripartite Guidelines for Good Clinical Practice, with applicable local regulations, and with the ethical principles laid down in the Declaration of Helsinki.
ACKNOWLEDGMENTS
Medical writing, editorial, and other assistance: The authors would like to thank Poh Sien Ooi (Novartis Corporation Sdn Bhd, Malaysia) and Ashwini KM Kumar (Novartis Healthcare Pvt Ltd, India) for providing medical writing assistance in accordance with the Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Yuan W, Hu S, Li M, et al. Efficacy and safety of omalizumab in Chinese patients with anti‐histamine refractory chronic spontaneous urticaria. Dermatologic Therapy. 2022;35(4):e15303. doi: 10.1111/dth.15303
Funding information The study was funded by Novartis Pharma AG.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Hide M, Park H, Igarashi I, et al. Efficacy and safety of omalizumab in Japanese and Korean patients with refractory chronic spontaneous urticaria. J Dermatol Sci. 2017;87(1):70‐78. [DOI] [PubMed] [Google Scholar]
- 2. Min TK, Saini SS. Emerging therapies in chronic spontaneous urticaria. Allergy Asthma Immunol Res. 2019;11(4):470‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zuberbier T, Aberer W, Asero R, et al. The EAACI/GA2LEN/EDF/WAO guideline for the definition, classification, diagnosis and management of urticaria. Allergy. 2018;73(7):1393‐1414. [DOI] [PubMed] [Google Scholar]
- 4. Metz M, Vadasz Z, Kocaturk E, Gimenéz‐Arnau A. Omalizumab updosing in chronic spontaneous urticaria: an overview of real‐world evidence. Clin Rev Allergy Immunol. 2020;59:38‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xiao Y, Huang X, Jing D, et al. The prevalence of atopic dermatitis and chronic spontaneous urticaria are associated with parental socioeconomic status in. Acta Derm Venereol. 2019;99:321‐326. [DOI] [PubMed] [Google Scholar]
- 6. Luo J, Song Z, Zhong H, et al. Clinical epidemiological characteristics of chronic urticaria: report of 535 cases. J Third Mil Med Univ. 2011;33(22):2421‐2424. [Google Scholar]
- 7. Zhong H, Song Z, Chen W, et al. Chronic urticaria in Chinese population: a hospital‐based multicenter epidemiological study. Allergy Eur J Allergy Clin Immunol. 2014;69:359‐364. [DOI] [PubMed] [Google Scholar]
- 8. Xu J, Hao F, Tang H. Guideline for diagnosis and treatment of urticaria in China (2018). Chin J Dermatol. 2019;52(1):1‐5. [Google Scholar]
- 9. Li L, Wang K. Diagnosis and treatment strategy for chronic refractory urticaria. Chin J Dermatol. 2017;50(1):8‐11. [Google Scholar]
- 10. Choi W, Lim E, Ban G, et al. Disease‐specific impairment of the quality of life in adult patients with chronic spontaneous urticaria. Korean J Intern Med. 2018;33(1):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaplan A, Ledford D, Ashby M, et al. Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol. 2013;132(1):101‐109. [DOI] [PubMed] [Google Scholar]
- 12. Maurer M, Rosén K, Hsieh H‐J, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N Engl J Med. 2013;368(10):924‐935. [DOI] [PubMed] [Google Scholar]
- 13. Saini SS, Bindslev‐Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on h 1 antihistamines: a randomized, placebo‐controlled study. J Invest Dermatol. 2015;135(1):67‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4):586‐604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.