

Abstract
In silico toxicology is an emerging field. It gains increasing importance as research is aiming to decrease the use of animal experiments as suggested in the 3R principles by Russell and Burch. In silico toxicology is a means to identify hazards of compounds before synthesis, and thus in very early stages of drug development. For chemical industries, as well as regulatory agencies it can aid in gap‐filling and guide risk minimization strategies. Techniques such as structural alerts, read‐across, quantitative structure–activity relationship, machine learning, and deep learning allow to use in silico toxicology in many cases, some even when data is scarce. Especially the concept of adverse outcome pathways puts all techniques into a broader context and can elucidate predictions by mechanistic insights.
This article is categorized under:
Structure and Mechanism > Computational Biochemistry and Biophysics
Data Science > Chemoinformatics
Keywords: adverse outcome pathway, computational toxicology, in silico toxicology, machine learning, read across
How computers can help toxicologists' decision‐making.
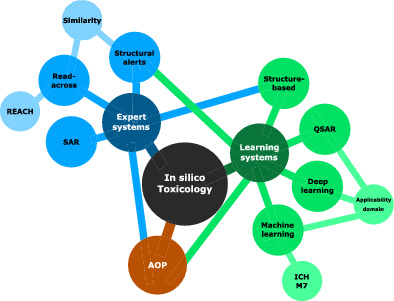
1. INTRODUCTION
Risk assessment is crucial and inevitable for pharmaceutical, cosmetics, and chemical industries as well as regulatory agencies. The goals of such assessments, however, are most diverse. In the pharmaceutical industries, risk assessment is conducted throughout the whole drug discovery and development process. Starting with the first evaluation of a compound, even before synthesis, it carries on until clinical trials where risk assessment is crucial for human health. Despite rigorous safety assessment of novel compounds, attrition still is high during the development process, often due to safety concerns.1 This is one of the reasons which prompted the European Federation of Pharmaceutical Industries and Associations (EFPIA; efpia.eu) and the European Commission to team up and found the Innovative Medicines Initiative (imi.europa.eu).2 This resulted in the launch of several large scale industry‐academia collaborative projects aiming at in silico prediction of toxicity, such as eTOX3 and eTRANSAFE (etransafe.eu).
In the chemical industries safety assessment is conducted as well, however, often with the primary goals being worker safety and environmental risk assessment. The cosmetics industry's risk assessments mostly revolve around consumer safety. Current safety testing in all industries is trying to implement the 3R principles which propose the reduction, refinement, and replacement of safety studies performed on animals.4, 5, 6 The announcement of EU regulation (EC) No 1907/2006 (widely known as REACH) and the European Union's Regulation (EC) No 1223/2009 (ban on animal testing in the cosmetics industry) shifted the focus from in vivo studies towards in vitro and in silico procedures for risk assessment. Another major step towards this goal was the European Pharmaceuticals Agency's (EPA) announcement of abolishing all funding for mammalian safety testing, as well as to eliminate mammalian safety testing itself from the approval processes by 2035.7
The aim of in silico toxicology is to predict certain hazards, such as mutagenicity or organ toxicities, based on computational models. These models can be created by experts (e.g., structural alerts or read across) or created automatically (e.g., machine learning techniques). The applicability of models ranges from the earliest stages of drug development—where a compound needs only to exist virtually to be testable—to risk assessment in time‐critical cases where in vitro or in vivo testing is not feasible, thus allowing for guidance on timely needed decisions. For example, the REACH legislation, where large amounts of chemicals needed to be tested for various apical toxicities, aimed towards in silico toxicology for gap filling techniques to deal with the impossible task of testing reams of chemicals within the given time frame. With in silico methods, hazards can be estimated early on, eliminating the need for synthesis. This can be especially important during drug development, where the in silico prediction of a compound can direct the choice for the lead compounds, not only concerning efficacy but also safety. Testing can, therefore, be directed towards specific hazards before a compound is taken forward to the preclinical safety testing. Furthermore, in silico toxicology could serve as a predictive tool for rare idiosyncratic events which can neither be seen in preclinical nor clinical safety assessments. For chemical safety assessment in silico models can be used to predict hazards for the users of such compounds, as well as for environmental safety.
The underlying assumption of all in silico toxicology approaches is the relationship between a chemical structure and its biological activity. This relationship states that the physiological action is a function of the chemical constitution, which was proposed by Brown and Fraser as early as 1868.8 A consequence of this is the “molecular similarity principle”,9 which states that similar structures will exhibit similar biological activities. This axiom was broadened over the last years by incorporating similarities based on gene expression or bioactivities.10, 11
While in silico toxicology has advanced significantly in the last years, in times of “big data” there remain plenty of opportunities for even greater leaps forward. Especially within regulatory frameworks its use still is quite limited and could be improved with the availability and FAIRification of new data.12 One example is the eTRANSAFE project, where preclinical toxicity data is integrated with clinical data (etransafe.eu). However, regulatory agencies currently only accept in silico predictions within the ICH M7 guideline13 for impurity testing and within gap filling for the REACH legislation. This is mainly due to the fact that until now not enough evidence and trust could be built up to reach the necessary confidence in such models.
In this review, we will give an overview of the different methods of in silico toxicology, especially with a focus on newly developed technology. We will discuss pitfalls and caveats for model building and frame our vision of the future of in silico toxicology.
2. TOXICITY TESTING
The goal of in silico toxicology is to help in risk identification, compound prioritization and ultimately, in combination with in vitro testing, to replace in vivo testing. Toxicological testing can be divided into general toxicity testing and mechanistic toxicity testing.14
2.1. General toxicology—saftey screening
Safety screening of novel drugs, chemicals, or other substances of concern (such as food contaminants) is generally conducted using in vitro and in vivo tests where a compound is administered in different doses and an effect has to be observed. For regulatory safety documents mostly in vivo tests need to be conducted. Such tests are conducted for two purposes: first, to determine the acute toxicity of a compound and second, to determine the repeated dose effects. Acute toxicity tests are usually used to determine the starting doses for organ toxicity testing.15 Repeated dose studies usually determine the toxicity of a compound. Mostly subacute repeated dose studies (28 days) and subchronic repeated dose studies (90 days) to determine organ (OECD TG 407 and 408),16, 17 inhalative (OECD TG 412 and 413)18, 19 or dermal toxicity (OECD TG 410 and 411)20, 21 are conducted in rodents. In some cases (e.g., for the REACH annex X22) long‐term toxicity studies (52 weeks) have to be conducted, however, this can be done in combination with a carcinogenicity study (OECD TG 452 and 453).23, 24
These tests are used to evaluate different exposure levels: The no observed adverse effect level which is a dose where the compound does not affect the organism and the lowest observed adverse effect level, which is the dose where first effects can be observed. These levels are then used to estimate either the first doses for clinical trials or the dose‐limits for human safety. However, such doses can only be estimated for noncarcinogenic and nongenotoxic substances. In addition to the required in vivo tests, there are many in vitro tests such as the Ames mutagenicity test or the skin sensitization assay, which can serve as a replacement for the in vivo tests. Further for specific chemical classes, such as agrochemicals, additional studies, like ecotoxicological assessment, have to be conducted. Additional information on regulatory toxicology can be found in References 25 and 26.
2.2. Mechanistic toxicology
Mechanistic toxicology revolves around investigating the reasons for substances to cause toxicity. This involves mechanistic in vitro as well as in vivo studies. Elucidation of mechanistic information involves illumination of the target as well as the pathways which lead to the apical toxicity endpoint. This knowledge can be used to gain deeper insights into the mechanisms of toxic substances. Furthermore, such knowledge can be utilized to prevent toxicity by avoiding specific structural features. Following mechanistic studies, cellular receptors and transporters, which can lead to certain types of toxicities, were identified. Such targets are called off‐targets, two of the most prominent examples are the human Ether‐à‐go‐go Related Gene (hERG) channel and the bile salt export pump (BSEP) transporter. Substances binding to hERG can lead to severe torsades des pointes27 and BSEP blockers often lead to cholestasis.28 Alongside (off)‐targets, (toxico)kinetics, especially in junction with genetic components, also plays an important role in the elucidation of toxicity mechanisms.29 Using kinetics, the exposure defines whether a hazard becomes an imminent risk. Similarly, the metabolism, and genetic variations thereof, such as fast and rapid metabolizers, can determine whether a patient develops severe side effects. In addition, genetic variants of the immune system might be responsible for severe idiosyncratic drug reactions.30 In the future, toxicogenomics might be able to help clarify such mechanisms further and develop a better understanding of pathways related to different apical toxicity endpoints.31 More detailed information on mechanistic toxicology can be found in Reference 32.
3. EXPERT METHODS
Expert methods use the knowledge and experience of experts to deduce or explain toxicity mechanisms. This can be done for single compounds as well as whole compound classes. Whereas read‐across is used to infer toxicity from other related compounds, structural alerts are a means to highlight potential hazards and help understanding the underlying mechanism. Expert methods are aimed at helping to assess compounds where no previous knowledge is available, therefore they can use mechanistic evidence as well as data from safety screenings to provide an assessment.
3.1. Read‐across
Read‐across is a method aiming at filling data gaps for a novel or previously uncharacterized compound.33 It relies on the formation of chemical categories from structures. The underlying assumption is that similar structures will have similar bioactivities.9 Therefore, read‐across is conducted by identifying similar molecules and evaluating their bioactivity, thus inferring the (non) toxicity of the parent compound. In the best case, the occurrence of a common scaffold makes it possible to analyze the influence of different substituents on the activity, allowing for a thorough assessment. Chemical similarity can be defined by calculating the similarity of feature vectors such as chemical properties or fingerprints using similarity metrics such as the Euclidean or Tanimoto similarity, opening up numerous possibilities for the calculation.34
Another part of the read‐across approach is the interpretation of available data, which is highly subjective and therefore there is no impartial solution. Due to the problem of chemical similarity and the subjective interpretation of the available data, read‐across was being regarded as an “ugly duckling” for methods that “originate from the idea that any information is better than no information in situations where there is no budget”.35 However, available data, as well as the use of the method is increasing. Especially the REACH initiative, which required large amounts of testing to be conducted, allowed read‐across for gap filling. The good news is that with “big data” a manifold of data sources is available to guide read‐across. Recently, Pawar et al. conducted a meta‐review identifying more than 900 databases that contain data relevant for read‐across.36
Several recent case studies demonstrated that a careful read‐across can be utilized to estimate the toxicities of various compound classes.37, 38, 39, 40, 41 However, all case‐studies show that the approach is highly dependent on the available data and the definition of similarity between the parent compound and the analogs. This is summarized in a final review of the conducted case studies by Schultz and Cronin who identified primarily transparency and uncertainties as being the main factor for a successful read‐across.42 In particular, they note “Today ‘chemical similarity’ means more than proving similarity in chemistry; it requires the category formation and RA [read‐across] process to be transparent, reproducible and clearly documented. Specifically, key principles of biological, as well as chemical, similarity need to be supported, where possible, by data and scientific evidence.”42 which emphasizes the high complexity of read‐across. To create a framework and guidelines for read‐across, the Organisation for Economic Co‐operation and Development (OECD), European Chemicals Agency (ECHA), and the European Centre for Ecotoxicology and Toxicology of Chemicals put efforts into guidelines for read‐across assessment.43, 44, 45 Especially many efforts were taken towards a reproducible and objective read‐across using mathematical models and quantifying the uncertainty of the prediction. The overarching conduction of read‐across consists of seven steps (based on Reference 46):
Decision context: what is the question to be answered.
Data gap analysis: where are knowledge gaps.
Overarching similarity rationale: wow is similarity defined.
Analog identification: based on the definition, which compounds are similar.
Analog evaluation: which similar compounds can be used.
Data gap filling: combine the knowledge gained from the analogs.
Uncertainty assessment: how clear is the evidence, where are still gaps, is the extrapolation adequate.
Thus far, many tools have been developed for category formation or read across (see also Reference 47). The ECHA made an effort to create a tool to guide, as well as asses, read‐across approaches and identify gaps and uncertainties. This framework is called the read‐across assessment framework (RAAF).48 It establishes different scenarios for the assessment of similarity and the subsequent deduction of evidence. This is important because especially compound similarities, evidence, used data and lack of plausibility were identified by Ball and coworkers as main reasons for rejected read‐across studies.49
Recently, the focus was shifted from purely chemical similarity towards using auxiliary bioactivity fingerprints for a better characterization of the underlying mechanisms.10, 50, 51, 52, 53 One approach, called GenRA, uses a bioactivity based similarity derived from different toxicity endpoints. Helman and coworkers could show that GenRA achieves a better predictivity when applied to structurally‐related clusters of chemicals.54 Recently, GenRA was also implemented in the EPAs ToxDashboard.55
The most important step of a read‐across, besides the assessment of the compound similarity, is the assessment of the remaining uncertainty. For uncertainty assessment, many analyses were done defining different sources such as the similarity of the analogs, confidence in the underlying data or the weight of the available evidence. Schultz and coworkers made a great effort to characterize those various sources.56 Their analysis resulted in 30 questions that target all the uncertainties and thus allow a very thorough estimate. In addition, they evaluated existing read‐across frameworks and conclude that currently, the RAAF incorporates most of these questions. New approaches for read‐across include the use of gene expression data57 or the implementation of local quantitative structure–activity relationships (QSARs) for the prediction of the compounds.58 However, especially the QSARs suffer from having a low explainability and thus contradict the concept of the transparency of a read‐across. Nevertheless, local QSARs, especially with explainable algorithms, can help in assessing the toxicity in addition to the read‐across if sufficient data is available. For more information see Reference 59.
3.2. Structural alerts
Structural alerts, like read‐across, are based on the idea that chemicals can be grouped into clusters of molecules exhibiting similar toxicity and, moreover, having a similar mode of action. A structural alert is a common substructure that can be linked to a certain type of toxicity. Such alerts can be derived through expert knowledge60 or by statistical evaluation of fragmented datasets (e.g., References 61, 62, 63, 64, for a comparison see Reference 65). The statistical evaluation of a dataset is usually done by utilizing a fragmentation algorithm and subsequently deriving structures that are associated with certain toxicities. For this, the user assumes that, if a substructure is present in a higher percentage of toxic than nontoxic molecules, the structure might be responsible for the toxicity. Floris and coworkers complemented this approach by laying out a basis for the statistical analysis of newly found alerts, which is using the chi‐squared test on an alert's contingency table.66
However, often no mode of action can be directly assigned to such statistically found alerts. Derivation of alerts by expert knowledge yields the advantage of an explanation for an alert. This explanation often results in mechanistic insights that subsequently can be used to suggest structural changes, where applicable.67 Independent of their source, structural alerts can help in the identification of hazards. Alerts can cluster datasets according to a potential common mechanism of such compounds.68 In addition, they yield a starting point for mechanistic studies69 or structural changes67 to minimize the risk of toxicity. However, as alerts only see one part of a structure, they fail at analyzing the different effects of multiple groups occurring at the same molecules. Those effects might be influencing each other and thus influence the toxicity. This is, for example, known for the induction of mutagenicity of nitro groups. Although widely known as a structural alert, the nitro group is often used in therapeutics (e.g., cancer therapeutics) as a functional group providing reactivity towards the target.70 However, it is also known that by structural modification of such compounds the genotoxicity can be reduced, if not abolished.71, 72
A new method, which is able to partly incorporate such modifications is the generation of structural alerts through scaffold trees.73 The generated trees show how the hazard of a molecule changes when the parent fragment is modified. When assessing a compound with structural alerts it is important to understand the absence of an alert does not denote a molecule to be nontoxic, rather it states no known alert could be found. However, with sufficient available data this can be made possible.74 It was also shown that the potency of an alert might be corresponding to the daily dose, therefore alerts might not be relevant for low doses of drugs.75 Alves and coworkers therefore proposed that structural alerts should not be seen as a ”yes” or ”no” concerning toxicity but rather as a hypothesis about the mode of action and subsequently trigger closer mechanistic studies.67, 76 Similarly, Limban and coworkers propose the replacement of structural alerts by functional groups that counteract the supposed toxicity mechanism rather than directly rejecting such compounds.67 Kalgutkar also highlighted, that, although structural alerts should not be ignored, especially in the case of toxicity via metabolic activation, alerts should be used with care77 as metabolic activation might be heavily influenced by existing residues of a scaffold. Myden and coworkers also showed that structural alerts can be highly sensitive, leading to many false‐positive results.78 Overall, structural alerts serve as a means to group chemical structures and to derive common mode of actions. However, as alerts do not consider the whole molecule and do not allow for truly negative predictions, they should only be used to indicate a hazard which can be used to guide future studies.
4. MACHINE LEARNING BASED PREDICTIONS
4.1. 2D and 3D quantitative structure–activity relationships
Quantitative structure–activity relationship (QSAR) analysis is the advancement of SAR analysis. SAR analysis dates back to a thesis of Cros who discovered that toxicity increases with diminishing water solubility.79, 80 By SAR analysis of congeneric series, it can be determined how substituents influence the bioactivity of a compound. In 1963 Hansch and Fujita published a first mathematical method termed p − σ − π analysis to correlate changes in biological activity with changes of structural properties.81 This was complemented by Free and Wilson's mathematical contribution to structure–activity relationships.82 With both methods, it became possible to have a quantitative hypothesis for the contribution of different substituents to the biological activity. However, it needs to be noted that in traditional QSAR only distinct congeneric series are analyzed. This is mainly because QSAR was intended to provide information on the physicochemical features required at specific positions of a chemical scaffold in order to enhance binding affinity. Basic requirements for a traditional QSAR analysis thus require a concrete ligand–receptor interaction, presumably in the same binding mode. Only then changes in for example, electronic properties of an R group can be related to changes in the biological activity of the compound.
In the early times of QSAR, descriptors and fingerprints derived from the 2D structure of the compounds were used as an input feature vector. With the increasing computational power available, also the use of methods based on three‐dimensional representations of molecules became possible. Nevertheless, this comes with the cost of intense conformational sampling requirements. In alignment‐dependent 3D‐QSAR methods such as CoMFA83 and CoMSIA,84 each compounds' conformational space needs to be sampled to derive the energetically most favorable conformation, and these need to be aligned. In alignment independent methods, such as GrIND,85 VolSurf,86 Pentacle,87 and 3D autocorrelation vectors,88 descriptors are calculated from distinct 3D conformations of the individual molecules and used for correlation analysis. Although in this case no alignment is necessary, the methods still require a thorough conformational analysis, as the descriptor values are conformation‐dependent.
Considering all this, it becomes evident that classical 2D‐QSAR and 3D‐QSAR are not considered to be a valuable method for predicting toxicity, especially in case of complex in vivo endpoints such as hepatotoxicity, cardiotoxicity, or neurotoxicity. In these cases, multiple physicochemical (solubility, crossing barriers) and molecular (interaction with a transporter, cytochromes, off‐targets) events are involved, which preclude any traditional QSAR method other than correlation with very global physicochemical features such as logP, TPSA, or number of H‐bond donors, ‐acceptors, or rotatable bonds. However, for very specific endpoints, and if congeneric series are available, QSAR and 3D QSAR can be used to determine properties decreasing the toxicity.89, 90, 91, 92
4.2. Structure‐based approaches
With the increasing amount of protein structures deposited in the Protein Data Bank (pdb; http://www.rcsb.org/),93 also structure‐based approaches are used to predict toxicity. However, in this case there needs to be a clear, causal link between a protein (in this case called off‐target) and the adverse effect. One of the prototype examples in this respect is the hERG potassium channel and its link to the long QT syndrome. Although recent work demonstrates that blocking hERG not necessarily leads to Torsades de pointes,94 affinity to hERG is routinely tested in the drug discovery and development process. This need also triggered the development of respective in silico methods. Besides numerous ligand‐based models applying basically all methods used in machine learning,95, 96, 97, 98, 99 also structure‐based approaches such as docking into protein homology models of hERG were explored. These studies comprise, for example, induced fit docking of a small set of hERG blockers,100 insights into the molecular basis of drug trapping of propafenones in hERG,101 up to calculation of absolute binding free energies between hERG and structurally diverse ligands.102
Also, for other off‐targets such as ABC‐transporter structure‐based methods were applied to get a deeper understanding of the molecular basis of compound/off‐target interaction. In case of P‐glycoprotein, SAR guided docking103 led to a docking protocol which allowed to perform structure‐based classification of a large set of P‐gp inhibitors and noninhibitors.104 Also for the BSEP, an ABC‐transporter linked to cholestasis, structure‐based classification was successfully implemented.105 Finally, we would like to mention the off‐target safety assessment (OTSA) framework, which combines a number of 2D target prediction methods with 3D protein‐based approaches. Briefly, the combination of six different cheminformatic methods with 3D‐methods such as pocket descriptors allows to predict off‐target interactions for small molecules.106 While OTSA basically predicts potential interactions, proteochemometric modeling tries to predict quantitative compound‐target interaction values by including target‐based descriptors into the data matrix for classical QSAR analyses.107
4.3. Traditional machine learning
Over the years the original QSAR analysis was expanded by different groups. Today, in the field of in silico toxicology, the term QSAR is used very widely to describe all forms of predictive modeling, including more complex (and less quantitative) machine learning models. Machine learning is a term comprising supervised model building for classification and regression, unsupervised techniques such as clustering, and reinforcement learning techniques mostly applied to sequential decision making tasks such as molecular design.108 In the following, we will use the term machine learning to refer to supervised learning for classification or regression models if not stated otherwise. Machine learning is split into two ”worlds.” Traditional machine learning refers to techniques such as k‐nearest neighbors (kNNs), random forests, or support vector machines, which are separated from neural networks as the central algorithm of deep learning (which will be discussed in the next section).
To train traditional machine learning models, datasets of 100 compounds or more should be used. For training, the molecules have to be transformed into a suitable representation such as descriptors or fingerprints. Descriptors usually constitute molecular properties such as the log P or the number of hydrogen bond donors, but can also involve more complex 3‐dimensional properties such as atom distances or surfaces. Fingerprints are bit‐vectors or count vectors which report the presence or absence or the number of occurrences of a structural feature. These representations are then used as input for model generation. By trying to predict the training set molecules correctly, and reporting the errors made back to the model, the model is improving its prediction.
A model can either be a regression model, predicting a continuous variable such as the LD50, or a classification model, such as a model for mutagenicity. Classification models are usually predicting a binary outcome or a continuous value between 0 and 1. Such continuous values can either be transformed into a binary outcome using a threshold (commonly 0.5) or can be transformed into a probability. Lately, many developments focus on thorough validation procedures to increase regulatory acceptance and to build trust. In this respect the main concerns are related to the limited chemical information used for building the model, which reduces the confidence in prediction of dissimilar molecules. Frenzel and coworkers showed that freely available Ames models can especially help in the prioritization for mutagenic or carcinogenic heat‐induced food contaminants. However, they concluded that the evaluated models are not yet predictive enough to use them as a stand‐alone tool.109 In contrast, a large international study revealed that existing and commonly used models for Ames mutagenicity are working well and the predictivity can compete with the in vitro Ames test.110 In this context it has to be noted that, depending on the dataset used, it might be that the contaminants Frenzel and coworkers used are too specialized to be caught by models in the public domain, which usually also only rely on public data. Thus, their molecules might not be in the applicability domain (see also Section 4.5). Another important finding by Honma and coworkers was that incorporating newly available data also increases the performance and thus models should be updated regularly.110
Another important step is leveraging available (big) data to build better machine learning models. This was shown by Luechtefeld et al. who mined available data from regulatory agencies and developed RASAR, reporting to outperform animal test accuracy.111 Although the methodology was criticized later,112 this approach highlights the need to leverage the available data and combine available datasets. Besides that, data mining, especially from nontabular data such as study reports, can increase the availability of data and thus improve existing models.
Unfortunately, developed models often lack an explanation of the prediction. Yet, this is one of the important prerequisites in the OECD's QSAR validation guidelines (see Section 4.6). The last point states that models should have a (mechanistic) explanation to be widely accepted. Usually, models are black boxes and, apart from the input and the output, the user does not know what is happening internally. Some models (such as regression or random forests) can at least explain the most relevant input features. Therefore, the focus has shifted from the mere model building towards establishing trust in a model's prediction, by giving confidence estimates for a prediction. In particular, conformal predictions can yield such an estimate of the certainty of the model for a specific prediction.113, 114 In brief, a conformal predictor is built using an existing model along with a calibration dataset. Predictions of this dataset are then used to determine the p‐value of a new prediction. This p‐value is calculated for both classes (i.e., toxic or nontoxic) separately. If the p‐value is above the chosen significance level the compound belongs the class. Possible outcomes of conformal predictions are a compound belonging to (a) only one class, meaning the compound is classified with high confidence, (b) both classes, denoting the compound cannot be predicted with high confidence, or (c) it belongs to no class, denoting that the compound is too dissimilar and cannot be predicted.
The last option, therefore, represents a way to get confidence scores and using them as an applicability domain for the models.115 In addition, it was shown that conformal predictions can also be used to deal with imbalanced data, which is a common problem for toxicological datasets.114 Another method to convert predictions into real probabilities is Platt scaling, which uses a learned log transformation to calculate the class probabilities from existing predictions.116 A very interesting approach to interpretability was introduced by Alves and coworkers. Their approach, Multi‐Descriptor Read Across, uses a kNN approach to classify compounds. The novelty lies in using four different types of descriptors to determine the nearest neighbors.117 Using the kNN method a prediction can be intuitively understood by inspecting the nearest neighbors used by the algorithm. The drawback of all traditional machine learning methods is that missing data has to be imputed to be able to use incomplete data matrices.
4.4. Neural networks and deep learning
Neural networks are a technique dating back to the 1940s and 1950s.118, 119 They have long been used to predict different tasks. However, it was not until 2006 that their real breakthrough started.120, 121, 122 With the availability of graphical processing units, training of neural networks has been hugely facilitated yielding new opportunities.120 Since neural networks now contain much higher numbers of hidden layers the technique was re‐branded as deep learning. For bioactivity predictions, the Merck Kaggle challenge and the Tox21 challenge showed that neural networks might be superior to traditional machine learning, yielding higher predictive performance and thus winning the challenge.116, 123, 124, 125
The biggest advantages of deep learning for bioactivity/toxicity predictions are (a) the flexibility with regard to the structural representation and (b) the possibility of multi‐task predictions (i.e., predicting multiple toxicities in the same model). In fact, deep learning is always seen as a method able to extract meaningful features by itself, thus not needing any feature generation beforehand. This can be impressively seen in the image recognition field.126 For the prediction of chemicals, this implies, instead of using descriptors, the molecules themselves should be used as an input. Effectively, this opens doors for predictive toxicology (see Figure 1). By providing images, molecular graphs, 3D grids, or SMILES strings, the network can learn the necessary properties or patterns by itself, based on the assumption that all information needed is encoded in the structure.127 Especially in the case of graphs, smiles or 3D grids, the model is not biased by the user's selection of descriptors, which might not be suitable for the task at hand. Such approaches have yet to be extensively studied.
Figure 1.
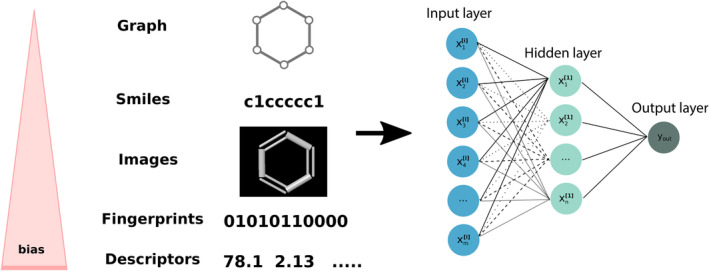
Scheme of a neural network with possible inputs. The order of the input from up to down, is in concordance with the bias introduced by the user
Nevertheless, a few pioneers already obtained interesting results. Goh et al. developed Chemception, a network predicting toxicity from 2D images of chemical structures, and later asked: “How much chemistry does a network need to know?”.128, 129 The results showed that training can be conducted with images but encoding additional chemical knowledge is helpful for the predictive performance. Similarly Fernandez et al. trained a network solely on images and reported comparable performances to other state‐of‐the‐art models.130 Although training on structural images might not be the goal for predictive toxicity, image training could purposefully be used in the detection of toxicities from pathological or image‐based techniques. Jimenez‐Carretero et al. showed that convolutional neural networks can be used to predict toxicity from cell staining images and even classify them by their mode of action.131 Another study was able to predict assay outcomes from microscopy images from high throughput screening studies.132
Apart from images, SMILES can also be used to predict the toxicity of a molecule, as implemented in SmilesNet developed by Gini and coworkers. It uses SMILES strings as an input, transforms them into a feature vector, and subsequently predicts the mutagenicity of a compound.133 A representation which seems to be the most natural in light of representing molecules as atoms and bonds is the molecule representation by graphs. In such graphs, the atoms are represented by the vertices and the bonds are represented by the edges between them. Xu et al. showed that the prediction of acute oral toxicity can benefit from a graph representation.134
Two very interesting approaches which do not use chemical structures as an input, but interpret in vitro assay outcomes are the prediction of seizure induction by a compound135 and the prediction of liver toxicity endpoints by outcomes from transcriptomic data.136
Another discovery which has highly impacted bioactivity prediction is the use of multitask networks.137, 138, 139 These networks predict multiple toxicities at once. It was shown that they benefit from the multitask setting by increasing the performance and at the same time regularizing the network to prevent overfitting. Tasks used in such approaches do not have to be closely related, however, a commonality of such tasks, like for example the same mode of action, should be suspected to benefit from multitasking.140 Without any commonality or similar structures, the model cannot make use of a shared representation for the tasks which is without benefit for training.
This is of great interest to the toxicological community since it is known that compounds can have multiple targets and mode of actions and thus might not be constricted to causing one single toxicity. Moreover, the method does not need to be used on a completely filled outcome matrix, but compounds can have missing activities for some endpoints and nonetheless be used for the predictions. This yields much larger datasets which can be used for better training of models. Many studies already highlighted that multitask networks exhibit better predictivity than single task networks. The application so far was done for modeling of acute toxicity,134, 141 reactivity,142 and ADME‐Tox properties.143 All this work highlights, that without the need to use descriptors or fingerprints to transform a molecule into a suitable input of machine learning models, in silico toxicology can benefit greatly. The possibility to combine related endpoints to form larger datasets is also a very important step towards larger, and hopefully more generalized, datasets.
Although all methods are very promising, the problem of the interpretability still exists, especially for multitask networks. However, interesting conclusions were drawn by Mayr et al., Gini et al., and Xu and coworkers who could show that networks can learn representations which are comparable to structural alerts.116, 133, 134 Wenzel and coworkers introduced response maps to highlight important features used by the network.143
4.5. Applicability domain
Models are trained on a dataset representing a certain part of the chemical space. The aim is to generalize a model such that it can possibly predict all available chemical compounds. However, looking at the currently known organic chemical space which was estimated at 3.4 × 109 144 or even more compounds and the dataset size, which usually lies between a few hundred to a few thousand molecules, it becomes clear that even training with a large dataset of 10,000 compounds means that only 0.00003% of the chemical space is covered. Therefore, a model can never reliably predict the whole chemical space and predictions should be confined to the chemical space used for model building. The concept of comparing newly predicted molecules to the training set, as well as giving confidence about the new prediction, is called the applicability domain. This important criterion is also included in the OECD guideline for the validation of QSAR models (see Section 4.6).
In their very comprehensive review of applicability domain methods, Mathea and coworkers divided the concept of applicability domain into novelty detection and confidence estimation.145 They define novelty detection as checking whether an unseen compound is part of the chemical space the model was trained on. Confidence estimation is defined as the reliability of a prediction, therefore denoting how confident a model is that the predicted class is correct. The separation into the descriptor and predictive domain make an interpretation of a prediction for an unseen compound comprehensible and traceable.
Hanser and coworkers expand this concept into three stages: they term the novelty detection as applicability and split the concept of confidence estimation into a reliability and decidability estimation.146 Reliability estimation uses the compound neighborhood to determine if there are data points close enough and how well the model predicts these data points. The decidability is the weight of evidence that the prediction of the novel compound is correct. This could, for example, be a posterior probability or the evidence from single decision trees in a random forest. Both publications highlight the importance of dividing the applicability domain into two areas, namely the descriptor space and the predictive space. Both add valuable information about the prediction and can inform the user how trustworthy a prediction is. Thus, aligning with the aforementioned methodology, the modeler should give users an estimate of how close the predicted molecule is to the training dataset. Only if the predicted molecule shares a certain similarity, a model can reliably extrapolate from the seen values (see Figure 2). For example, a model trained on a quinone dataset will be able to predict quinone‐like molecules, however, it might fail to predict benzopyrene due to the limited structural similarity.
Figure 2.
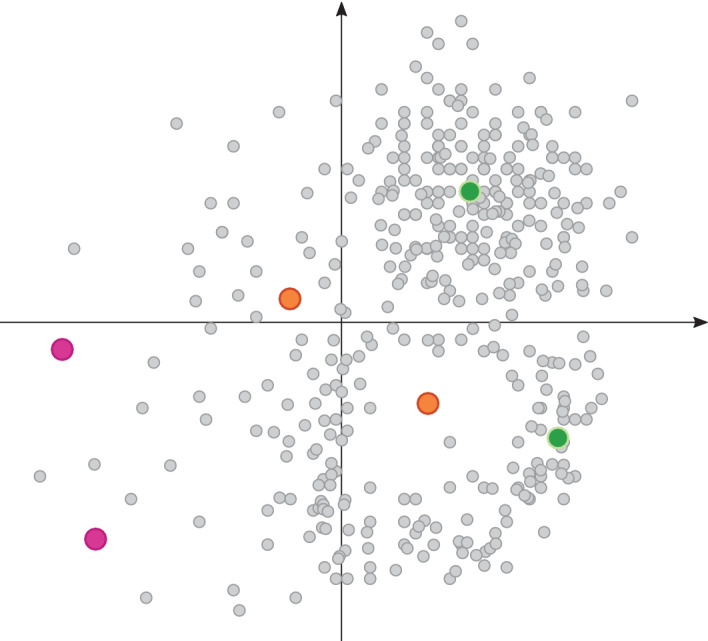
The chemical space of a QSAR model. The grey dots represent compounds from a hypothetical training dataset. The colored dots represent compounds which should be predicted by the model. The green dots represent compounds that are within the applicability domain. The pink dots represent compounds that would be predicted as out of domain as they lie on the borders of the chemical space which is very little populated. For the orange compounds, it is not clear from visual inspection how confident the model is in the extrapolation of such compounds as the surrounding area is populated, the compound, however, lies in a gap in the models' chemical space. Different applicability domain calculation might give different results here
This again leads to the question of the definition of structural similarity. As already noted, assessing structural similarity is highly complex and always dependent on the molecular representation. For small datasets, a trained medicinal chemist might be able to define the represented chemical space by visual inspection, whereas for large datasets and newly predicted molecules this needs to be automated. Independent of the method, the chemical similarity of a compound to the training dataset should always be measured based on the model input such as fingerprints or descriptors. As secondary information, the modeler should give the user as much information as possible on the closest compounds in the training set146 as well as a confidence score, which is inherent in most used classifiers.145
Roy and coworkers recently published an applicability domain calculation that implements all three criteria proposed by Hanser and coworkers. They use the prediction error of the 10 nearest neighbors in cross‐validation, the similarity to the training dataset, and the proximity of the predicted activity to the mean of the training data.147 Although they finally develop a combined score, their findings are in line with the conclusion from Hanser and coworkers, that a combination of applicability, reliability, and decidability is beneficial as all contribute to a successful prediction. Conformal predictors as introduced before are using their means of generating out of domain prediction. They can fail to confidently assign a class to a prediction, therefore, the user knows the compound is out of domain.115 This estimate can be considered as information about reliability.
In addition to the question of how an applicability domain can be successfully implemented, it is also interesting to see which models need a defined applicability domain. Liu and coworkers showed that, while it is generally accepted that traditional machine learning models need an applicability domain, deep learning models also cannot generalize much better.148 However, this is not very surprising given that until now most deep learning models are not built on much larger datasets than traditional models. In the future, with the availability of bigger datasets, this might change in favor of a more relaxed definition of the applicability domain.
4.6. Regulatory frameworks for QSAR
In general, QSAR is not only an old but still evolving field as can be seen by the more extensive use and thus the efforts to harmonize and increase the use. In 2007 the OECD published guidelines on the validation of QSAR models.149 The guidelines demand models which conform with the following principles:
a defined endpoint,
an unambiguous algorithm,
a defined domain of applicability,
appropriate measures of goodness‐of‐fit, robustness, and predictivity,
mechanistic interpretation, if possible.
These guidelines also highlight one of the biggest pitfalls of QSAR modeling: While it is easy to obtain a predictive model, it is much harder to ensure that the predictive model is useful. Whereas the first two principles—the endpoint and the algorithm—can be managed with little effort, the last three points need thorough consideration. The applicability domain, as discussed earlier, is important since the chemical space is too large for a model to be wholly generalized. Thus, a measure is needed to evaluate the similarity of a compound to the training set as well as a possibility to obtain a reliability for the prediction. Point number four—the performance—is probably the most important point. Model evaluation should not only be conducted thoroughly, that is using an external test set to obtain the performance on a nonrelated structural dataset, but also should use appropriate measures. The last point—a mechanistic explanation—is not obligatory for a model. However, in addition to the applicability domain, mechanistic information can not only inform a user but also help to evaluate a developed model further.
With these principles available for model development, in 2014 the ICH M7 guideline was published. The revised ICH M7 guideline for the mutagenicity assessment was a big step towards the use of in silico approaches for regulatory documents. This guideline was a milestone for in silico toxicology as it represents the first guideline to allow the replacement of an in vitro test with an in silico prediction. Furthermore, this guideline is accepted by many regulatory agencies.150 It allows the assessment of manufacturing impurities of low quantity employing two orthogonal approaches. These approaches usually are one expert tool, relying on structural alerts, and one machine learning model. Both models have to be complementary and have to comply with the OECD validation principles.151 If both systems are predicting the compound as negative, no in vitro Ames test has to be conducted. For an ambiguous outcome—for example, contradicting predictions, an out of domain or inconclusive prediction—the expert review plays a crucial role for further decisions.152, 153 The expert review can, by adding additional evidence, clarify the prediction and make a final decision. In addition, it can also be used to refute a positive prediction, but substantial evidence is needed.152 This application of in silico prediction also highlights the need for certain negative predictions and the applicability domain. For instance, the absence of a structural alert does not necessarily denote that a compound is inactive. In combination with the applicability or novelty detection, for example, by looking at the closest negative compounds, this negative prediction can, however, be justified as a true negative.74
Apart from being a milestone, the ICH M7 guideline has also influenced other guidelines such as the toxic substances control act154 or the International Standard ISO 10993‐1 for the biological evaluation of medical devices, which both implement the possibility for in silico assessments.
5. ADVERSE OUTCOME PATHWAYS
The goal in toxicology has shifted from a mere determination of the hazard of a compound towards a mechanistic explanation. This explanation enables structural changes as well as an extrapolation of the risk of a compound (e.g., is the mechanism relevant only if a compound was ingested or can skin contact lead to sensitization, etc.) and thus a better safety profile of drugs as well as chemicals. Thus far we have explained how different in silico techniques can be used to determine hazards emanating from a compound. In principle, two events usually are described by such models: first, the direct interaction of the compound with cellular organelles or compartments—like for example the binding of a compound to DNA—or second, the resulting apical endpoint, such as carcinogenicity, which is commonly used for hazard evaluation. The first concept is also termed molecular initiating event (MIE), and the second is termed adverse outcome. The event cascade which is following the MIE is referred to as an adverse outcome pathway (AOP), the events are referred to as key events (see Figure 3).
Figure 3.
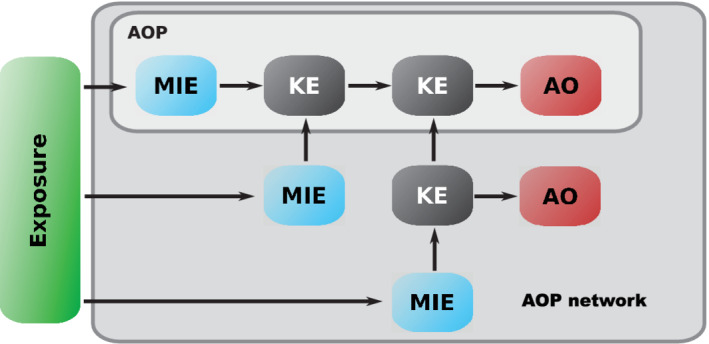
Schematic drawing of an adverse outcome pathway. The pathway consists of a molecular initiating event (MIE), several key events (KE), and an adverse outcome (AO). The light grey area depicts a scheme for a linear, single adverse outcome pathway. The dark grey area depicts an adverse outcome pathways network with multiple initiating events and multiple adverse outcomes. The green square with exposure highlights that this information is not yet part of the AOP framework, however, it is highly interlinked
These pathways were first introduced for ecotoxicology.155 1 The AOP framework allows the assessment of chemical hazards through the incorporation of mechanistic information. This information can elucidate species differences or account for causal evidence of found statistical correlations. Very useful resources to discover known AOPs are the AOP Wiki (aopwiki.org) and the AOP‐KB (aopkb.org) or the OECD series on AOPs.156
An important property of AOPs is their independence from a specific substance. Therefore, an AOP can be used for all compounds where mechanistic evidence is available. AOPs can either be generated manually by literature review or mechanistic studies or by automated data mining.157, 158, 159 As mentioned earlier, high throughput screening data is a valuable tool to advance in silico toxicology. Fay and coworkers showed that high throughput screening data can also be used to identify MIE or key events for AOPs and help prioritization of testing resources.160 Incorporating mechanistic information can highlight data gaps or pave the way towards specific testing. Such tests can then be utilized in early phases of drug development to test a compound for the activation of AOPs, which might be a crucial factor in the decision process for the advancement of a novel compound.
To a certain extent, this is already done by specific testing for off‐target activities such as hERG‐channel blocking, which is known to increase the risk of QT‐elongation in drug‐users. With the availability of more MIEs, this safety panel could largely expand and focused in vitro testing approaches could be developed. Furthermore, general toxicogenomics data is already used (although infrequently) in mechanistic toxicology studies to elucidate the mode of action.161, 162 Thus it could be used to elucidate the mechanisms and help the development of AOPs, as well as, AOP networks.163 AbdulHameed and coworkers mined several toxicogenomics datasets and integrated the information in their assessment of the AOP for cholestasis and thereby identified mitochondrial toxicity as a, so far, overlooked cause of cholestasis.164
Yet, information of an AOP is limited to a potential hazard. For risk assessment, the AOP framework can only be used when linked with toxicokinetic data165 or as suggested by Escher and coworkers linking the AOP to the exposome and aggregate exposure pathways (AEPs)166 (see Figure 3). In a case study, Hines and coworkers showed that AEPs indeed can be beneficial to human risk assessment.167 Quantitative AOPs were proposed by Conolly and coworkers and extend the concept of AEPs by including dose‐ and time‐response information.168
Apart from being a self‐contained method, AOPs can also be linked to the above mentioned in silico methods. Structural alerts can, first of all, categorize compounds and subsequently link those groups to specific MIEs if the alerts mode of action is known.68, 169 In a read‐across analysis, AOPs can be incorporated as an additional layer of information. In the assessment of compound toxicity via traditional machine learning or deep learning, the AOP concept can also be very useful. Machine learning can be used to predict the MIE or the AOP of a compound, and in principle all intermediate steps as well, to guide the safety assessment.170 However, it has to be noted that the MIE, as well as the adverse outcome, have large differences in the model quality. The MIE is a very specific and well‐defined event, where usually structural determinants are known or can be deduced by knowledge of the target structure. The adverse outcomes are often much harder to model. One reason is the lacking clarity of the endpoints, as data is often based on the interpretation of an outcome. For example, in the case of drug‐induced liver injury, the data is based on case reports, which, for example, might be biased by the examiner or via unknown multidrug use. In animals, the outcome is based on the histopathologist's interpretation of the seen lesion which might vary between individual assessments. The second reason is that one adverse outcome, such as drug‐induced liver injury, can be caused by a multitude of MIEs. Thus, the model needs to be able to interlink all possible causes with the outcome, which is more complex as the structural variability is much higher.
Nevertheless, both events can be modeled, however, with different purposes. Whereas the MIE can guide early hazard assessment towards specific assays or tests, the adverse outcome can be used, in combination with toxicokinetics, to conduct a risk assessment. This assessment could be used for gap filling as well as the prediction of potential mechanisms to guide further investigations. A useful guideline on how AOPs can be integrated into risk assessment was published by Sakuratani and coworkers.171 Arzuaga and coworkers showed that the AOP framework can be used to identify and describe possible species‐specific effects.172 By introducing information about genetic screens Mortensen et al. could also show that AOPs can be used to gain information about possible susceptibility introduced through genetic variation.173 The biggest strength and drawback of AOPs is their linearity. On one hand it introduces simplicity and facilitates usability, on the other hand most AOPs are not limited to a single event chain but can be much more interlinked through common MIEs, modulators or key events. To analyze such dependencies AOP networks were proposed, which can add additional mechanistic information174, 175 (see Figure 3). Oki and coworkers demonstrated that such networks can be created by mining high throughput screening data in combination with toxicogenomics data, applying frequent itemset mining.176
Systems toxicology (following systems biology) is a relatively new discipline, which especially aims to integrate toxicological assessment with novel techniques monitoring changes in the whole organism, such as omics approaches and leverage this information to gain a more complete understanding of toxicological mechanism. Sturla and coworkers aptly describe, “It [Systems Toxicology] will enable the gradual shift from toxicological assessment using solely apical end points toward understanding the biological pathways perturbed by active substances. Systems Toxicology is expected, therefore, to create knowledge regarding both the dynamic interactions between biomolecular components of a complex biological system and how perturbing these interactions with active substances alters homeostasis and leads to adverse reactions and disease.”.177 Consequently, systems toxicology will also enable the development of more complex and detailed AOPs and AOP networks that will guide in silico toxicology assessments in the future.
6. PITFALLS AND CAVEATS
All methods described in this review have a commonality: all require datasets that can be analyzed or used for model training. Depending on the dataset size some methods might not be feasible. Especially deep learning requires large amounts of data to prevent overfitting. However, if the datasets for the derivation of structural alerts, AOPs or traditional machine learning are small, their applicability also becomes very limited. Read‐across requires the least amount of data with only a few high similarity compounds being sufficient for an extrapolation of the activity. Nevertheless, this specific type of data might be even harder to find, especially for novel structural classes. As all methods build upon data, it is of utmost importance to ascertain that this data contain as few errors as possible, since they have a direct impact on the modeling outcome. Therefore, data curation is essential. Data curation, on one hand, is the removal of uncertain or inconclusive activity values,178 and on the other hand, it involves structural curation.179, 180, 181 The removal of data points always is a balancing act between having very few reliable data points or a large dataset with more (less reliable) data points. It always has to be considered carefully and adapted to the method. Whereas deep learning can handle—and might even profit from—noise in the dataset,182 for read‐across the activities need to be highly curated and reliable to allow a high certainty in the extrapolation. Structural curation is also necessary to remove duplicates and ensure a similar representation of functional groups.179 This, in turn, is needed to ensure a unified calculation of molecular descriptors.
For machine and deep learning, the model evaluation is also crucial. In any case overtraining has to be avoided and the model should be carefully evaluated. Models can be validated by (a) cross‐validation, (b) a train‐test split of the dataset, or (c) an external evaluation dataset (see Figure 4). Cross‐validation is the easiest to use and can be used for cases where data availability is very limited. However, one has to be aware that it only gives an estimate for the model performance and it can be biased by highly similar cross‐validation folds. Therefore, it might not reflect the performance in real‐world scenarios. Train‐test splits are commonly used when the dataset size is large enough to allow for the retention of data solely for testing. With this method, the “real world” performance can be much better estimated, with the drawback that parts of the available data are not used for training. This implies that the model does not benefit from all available information. Just like cross‐validation, it can also be biased by a high similarity between the splits. Thus, the best training method is the evaluation with a true external test set which was compiled independently from the training dataset. Naturally, the external test set has to be within the applicability domain of the model. An interesting approach to first asses the real‐world performance and secondly evaluate the impact of different external datasets is the nested cross‐validation suggested by Baumann and Baumann.183 In addition, with the clustered cross‐validation suggested by Mayr et al. in their challenge‐winning DeepTox pipeline,116 this could lead to more stable and generalized training of QSAR and deep learning models.
Figure 4.
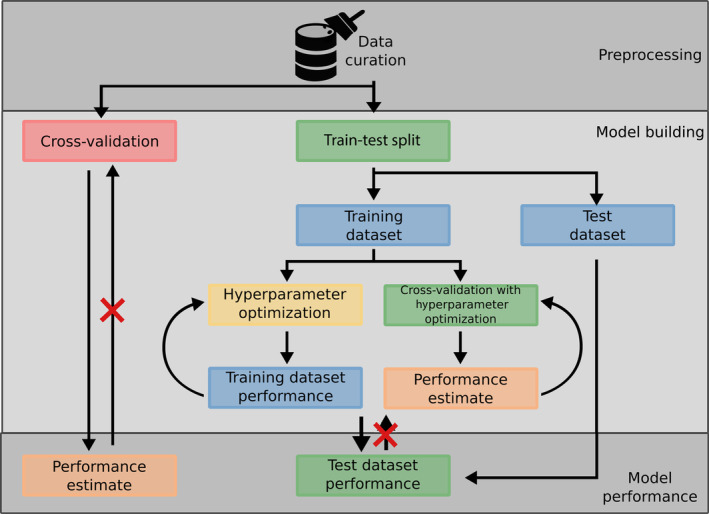
Schematic drawing of possible ways to train a machine learning model. The left path highlights that cross‐validation is possible without splitting the dataset; however, the obtained performance is only an estimate. The left path highlights that a train and test split is needed for example, if hyper‐parameters need to be tuned (in this case second inner cross‐validation can be used) or, in general, if a “real world” situation should be mimicked. It is important to note that, once the test set was used to evaluate the model performance, one should never go back to adjusting parameters with the training set to obtain a better performance on the test set as this would introduce bias into the model
Special care has to be taken when training is conducted with imbalanced datasets. Due to training with greedy algorithms, many machine learning algorithms (including neural networks) tend to ignore the minority class, as the error becomes quite small when all labels are predicted as the majority class. Therefore, the training procedure might have to be modified by the use of cost‐sensitive learning or by over‐ or undersampling techniques.184 Examples for such techniques are meta‐cost,185 bagging,186 or SMOTE.187 A model‐independent method to overcome imbalance is conformal predictions as shown by Sun and coworkers.114 Lately, it also was shown that for neural networks SMILES enumeration could be a viable method to oversample datasets.188 Despite such methods, appropriate evaluation metrics need to be used to determine the models performance. For imbalanced datasets the confusion matrix itself, the Matthews correlation coefficient,189, 190 the balanced accuracy or the positive or negative predictive value, are appropriate, with the final choice depending on the purpose of the model. The most important measure is always the confusion matrix itself. It already highlights model biases towards a class, which is important especially for imbalanced data. Since from the confusion matrix all other measures can be derived, it just seems logical to always report this for predictive models, along with the modeler's favorite choice of metrics. This allows the reader to in turn calculate their favorite metrics and compare their models to the reported one.
One exception is the area under the receiver operating curve (AUC). This metric cannot be derived from the confusion matrix. However, this already highlights the biggest drawback. It does not evaluate the model at its current state, rather the model's capability regarding different thresholds. However, although the performance at one point might be high, the respective classification threshold is not necessarily useful—just imagine a model with a sensitivity and specificity of 0.8, but a classification threshold of 0.01—here the threshold already highlights the models bias towards one class. Therefore, the AUC should only be used with great care and concomitantly with the confusion matrix to allow an unbiased model evaluation. This brings us back to the OECD validation guidelines for QSAR models. So far, points (1) to (4) were discussed and their importance stressed. The last point—a mechanistic explanation—is probably the hardest to achieve, but might be the most important point to establish confidence in predictions. Whereas structural alerts derived by an expert will mostly have some rational explanation, most automated methods are black‐boxes. For statistically generated structural alerts a literature search can highlight the importance of different structural classes or substructures, while for machine learning methods there is no easy explanation. Although some methods can provide feature importances, no method can yet give a human‐like explanation of a prediction. This is one of the reasons why in silico toxicology is often used as an indicator, but the final call will always be made by expert review, in vitro or in vivo studies. A very comprehensive work, defining how in silico toxicology should be used to become the powerful tool it could be, was published by Myatt and coworkers.191
In general, it is important to know how a model was built and especially for which purpose. A model for toxicity prediction in an early stage should not be overly sensitive. In this stage, hazards should be specific to be able to decide if a compound should be advanced. However, a missed alert is not crucial as further tests for safety are conducted. For example, for consumer safety, a missed hazard might lead to subsequent risks. Similarly, the absence of structural alerts never indicates any hazard at all, unless the model was developed to indicate specifically negatives. AOPs are also an example: while they are able to indicate events leading to an apical toxicity endpoint, they are not complete pathways and thus should not be used as such. Especially without any known exposure or kinetic modeling, they only indicate hazards. In fact, different exposures, such as repeated low doses or one‐time high‐dose, could lead to different AOPs. Thus, modifying or compensatory reactions, which are not (yet) part of the AOP framework, can diminish or even abolish the apical toxicity thus negate a possible hazard.
Overall, it can be concluded that all methods have benefits and drawbacks. It is important for modelers and users to specify those and to be aware of limitations, especially during result interpretation.
7. WHERE ARE WE HEADED?
A lot has been achieved in the field of in silico toxicology. Emerging technologies are boon and bane. While the modeling gets much more complex with deep learning technologies, it might offer new paths for higher predictivity of models. Especially for ”black‐box” models, which do not allow the users to understand a prediction, the modelers need to build trust. An emerging technology is explainable AI which tries to open the black box. There are many already existing possibilities to explain model prediction, as shown by Polishchuk.192 Newer technologies are emerging, especially with regard to neural networks. Two model‐independent approaches are often used: The LIME193 and SHAP194 frameworks utilize local explainable models to assess the importance of the input feature. In contrast to random forests or regression, this importance is on a per prediction, instead of a per‐model basis. For deep learning, layer wise relevance propagation195 is a method that can calculate the contributions of the input features. Many more methods are emerging which could be beneficial for in silico toxicology.196 Such methods could generate insights into the structural features or properties which are used by the model. Informing a user about such choices could also improve the trustability. Especially with new techniques such as graph‐based training of neural networks we could gain novel insights into the genesis of a prediction, and maybe leverage such information by discovering novel structural patterns relevant for in silico toxicology.
In addition to better interpretability, studies to evaluate models on a large scale (i.e., across multiple companies, as pursued in the eTOX and eTRANSAFE projects) would not only enhance the confidence, but also improve the models.110 However, even with such approaches the current lack of large datasets makes it hard to provide generalized assumptions for modeling or risk assessment. To generate models, as well as read across, or structural alerts with higher generalization potential, data mining and thus a compilation of larger datasets is essential. A drawback is that mined data is never as reliable as manually curated datasets.197 However, as stated previously there will always be a trade‐off between reliable data and large datasets. Certainly, with the availability of more data, the amount of reliable data will also increase. An interesting approach could also be the utilization of high throughput screening outcomes as biological fingerprints.198, 199 Such data is already generated in early drug screening and should, therefore, be leveraged as much as possible.
High throughput data will also enable studies for the combination of structure based and deep learning methods. Lenselink and coworkers showed that proteochemometric modeling, in combination with deep learning, can outperform other approaches for target bioactivity predictions.200 Consequently systems toxicology will be able to generalize the modeling approaches not only to singular endpoints but towards an network based prediction. Such approaches could lead to dynamic and computable biological networks, able to predict outcomes based on network perturbances.177 The possibilities of such networks are highlighted by Yepiskoposyan and coworkers, who used networks for mucociliary clearance to assess the network perturbance as well as the related mechanism for pyocynanine and IL‐13 treatment.201 The relating datasets and information gained from systems toxicology need to be stored in a useful format. In contrast to traditional relational databases, pathway related data is best stored in graph databases which are able to encode the relevant structures such as proteins or genes in the vertices and the relevant relationships such as binding or upregulation, in the edges of the graph.202
While we have large public resources for chemical datasets, sadly a plethora of data is only available in proprietary environments. Gedeck and coworkers published an exciting study where models were trained with proprietary data, without the necessity to enclose the structure.203 This, in combination with the development of distributed learning,204 can also pose a big opportunity to leverage available information in spite of sensitive data.
The AOP framework is also a powerful tool, not only to complement read across or structural alerts, but also to bridge the gaps between all in silico disciplines. If an AOP (or parts of it) is known, respective machine learning models for the MIE(s) or key events could be further used to guide the decision‐making process.
8. CONCLUSIONS
In silico toxicology is a cheap and fast tool to detect hazards. Although it is already used within regulatory frameworks, there is still a lot of uncertainty concerning the application and especially the interpretation of such approaches. Once protocols are established and the users get more confident, in silico, along with in vitro tests certainly will in some cases be used as a replacement for in vivo testing. In addition, modelers have to adhere to the OECD guidelines and make sure that the user is informed about the process and intended use of a model. This will establish more trust in models and may overcome the often too rigorous assessment of computational models, as they are expected to outperform the in vitro or in vivo test predictivity. The more we use in silico models the more we can learn about their benefits and limitations and understand where further research could offer new paths.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
AUTHOR CONTRIBUTIONS
Jennifer Hemmerich: Conceptualization; writing‐original draft; writing‐review and editing. Gerhard Ecker: Conceptualization; supervision; writing‐original draft; writing‐review and editing.
RELATED WIREs ARTICLES
Computational toxicology: A tool for all industries
In silico toxicology: Computational methods for the prediction of chemical toxicity
Hemmerich J, Ecker GF. In silico toxicology: From structure–activity relationships towards deep learning and adverse outcome pathways. WIREs Comput Mol Sci. 2020;10:e1475. 10.1002/wcms.1475
Funding information Austrian Science Fund, Grant/Award Number: W1232; Innovative Medicines Initiative, Grant/Award Number: 777365
Endnote
Here it has to be noted that there were mechanistic toxicology approaches, such as the mode of action concept, which elucidated the mechanisms for specific compounds or compound groups, however, the AOP concept allowed the unification of such approaches into a common framework.
References
REFERENCES
- 1. Waring MJ, Arrowsmith J, Leach AR, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14(7):475–486. Available from: https://www.nature.com/articles/nrd4609. [DOI] [PubMed] [Google Scholar]
- 2. Goldman M. The innovative medicines initiative: A European response to the innovation challenge. Clin Pharmacol Ther. 2012;91(3):418–425. Available from: https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1038/clpt.2011.321. [DOI] [PubMed] [Google Scholar]
- 3. Sanz F, Pognan F, Steger‐Hartmann T, et al. Legacy data sharing to improve drug safety assessment: The eTOX project. Nat Rev Drug Discov. 2017;16(12):811–812. [DOI] [PubMed] [Google Scholar]
- 4. Russell WMS, Burch RL. The principles of humane experimental technique. London: Methuen, 1959. Available from: http://books.google.com/books?id=j75qAAAAMAAJ. [Google Scholar]
- 5. Zurlo J, Rudacille D, Goldberg AM. The three Rs: The way forward. Environ Health Perspect. 1996;104(8):878–880. Available from: https://ehp.niehs.nih.gov/doi/10.1289/ehp.96104878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Executive Committee of the Congress . Background to the three Rs declaration of Bologna, as adopted by the 3rd World Congress on Alternatives and Animal Use in the Life Sciences, Bologna, Italy, on 31 August 1999. Altern Lab Anim. 2009;37(3):286–289. [PubMed] [Google Scholar]
- 7. Wheeler AR. Directive to prioritize efforts to reduce animal testing. US Environmental Protection Agency (EPA), 2019. Available from: https://www.epa.gov/sites/production/files/2019-09/documents/image2019-09-09-231249.pdf. [Google Scholar]
- 8. Brown AC, Fraser TR. On the connection between chemical constitution and physiological action; with special reference to the physiological action of the salts of the ammonium bases derived from Strychnia, Brucia, Thebaia, Codeia, Morphia, and Nicotia. J Anat Physiol. 1868;2(2):224–242. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1318606/. [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson MA, Maggiora GM, Society AC, editors. Concepts and applications of molecular similarity. New York: Wiley, 1990. [Google Scholar]
- 10. Shah I, Liu J, Judson RS, Thomas RS, Patlewicz G. Systematically evaluating read‐across prediction and performance using a local validity approach characterized by chemical structure and bioactivity information. Regul Toxicol Pharmacol. 2016;79:12–24. Available from: http://www.sciencedirect.com/science/article/pii/S0273230016301118. [DOI] [PubMed] [Google Scholar]
- 11. Wu Y, Wang G. Machine learning based toxicity prediction: From chemical structural description to transcriptome analysis. Int J Mol Sci. 2018;19(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2016;3(1):1–9. Available from: https://www.nature.com/articles/sdata201618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. ICH . Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk M7(R1). International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), 2017. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M7/M7_R1_Addendum_Step_4_2017_0331.pdf. [Google Scholar]
- 14. Barile FA. Principles of toxicology testing. CRC Press, 2007. Available from: https://www-taylorfrancis-com.uaccess.univie.ac.at/books/9780429075056. [Google Scholar]
- 15. Erhirhie EO, Ihekwereme CP, Ilodigwe EE. Advances in acute toxicity testing: Strengths, weaknesses and regulatory acceptance. Interdiscip Toxicol. 2018;11(1):5–12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6117820/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. OECD . Test no. 407: Repeated dose 28‐day oral toxicity study in rodents. OECD Publishing, 2008. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070684-en. [Google Scholar]
- 17. OECD . Test no. 408: Repeated dose 90‐day oral toxicity study in rodents. OECD Publishing, 2018. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070707-en. [Google Scholar]
- 18. OECD . Test no. 412: Subacute inhalation toxicity: 28‐day study. OECD Publishing, 2018. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070783-en. [Google Scholar]
- 19. OECD . Test no. 413: Subchronic inhalation toxicity: 90‐day study. OECD Publishing, 2018. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070806-en. [Google Scholar]
- 20. OECD . Test no. 410: Repeated dose dermal toxicity: 21/28‐day study. OECD Publishing, 1981. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070745-en. [Google Scholar]
- 21. OECD . Test no. 411: Subchronic dermal toxicity: 90‐day study. OECD Publishing, 1981. Available from: https://www.oecd-ilibrary.org/content/publication/9789264070769-en. [Google Scholar]
- 22.Document 32006R1907 – Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02006R1907-20200101.
- 23. OECD . Test no. 452: Chronic toxicity studies. OECD Publishing, 2018. Available from: https://www.oecd-ilibrary.org/content/publication/9789264071209-en. [Google Scholar]
- 24. OECD . Test no. 453: Combined chronic toxicity/carcinogenicity studies. OECD Publishing, 2018. Available from: https://www.oecd-ilibrary.org/content/publication/9789264071223-en. [Google Scholar]
- 25. Gad SC. Regulatory toxicology. 3rd ed.: CRC Press, 2018. Available from: https://www-taylorfrancis-com.uaccess.univie.ac.at/books/e/9780429464737. [Google Scholar]
- 26. Robinson L. A practical guide to toxicology and human health risk assessment. 1st ed. Hoboken, NJ: Wiley, 2019. [Google Scholar]
- 27. Lester RM, Olbertz J. Early drug development: Assessment of proarrhythmic risk and cardiovascular safety. Expert Rev Clin Pharmacol. 2016;9(12):1611–1618. 10.1080/17512433.2016.1245142. [DOI] [PubMed] [Google Scholar]
- 28. Jetter A, Kullak‐Ublick GA. Drugs and hepatic transporters: A review. Pharmacol Res. 2019;104234 Available from: http://www.sciencedirect.com/science/article/pii/S1043661819300325. [DOI] [PubMed] [Google Scholar]
- 29. Lauschke VM, Ingelman‐Sundberg M. Prediction of drug response and adverse drug reactions: From twin studies to next generation sequencing. Eur J Pharm Sci. 2019. Mar;130:65–77. Available from: http://www.sciencedirect.com/science/article/pii/S0928098719300326. [DOI] [PubMed] [Google Scholar]
- 30. Chen CB, Abe R, Pan RY, et al. An updated review of the molecular mechanisms in drug hypersensitivity. J Immunol Res. 2018;2018:6431694 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5830968/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z, Huang R, Roberts R, Tong W. Toxicogenomics: A 2020 vision. Trends Pharmacol Sci. 2019;40(2):92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boelsterli UA. Mechanistic toxicology: The molecular basis of how chemicals disrupt biological targets. 2nd ed. Boca Raton, FL: CRC Press, 2007. [Google Scholar]
- 33. Kv L, Schultz TW, Henry T, Diderich B, Veith GD. Using chemical categories to fill data gaps in hazard assessment. SAR QSAR Environ Res. 2009;20(3–4):207–220. 10.1080/10629360902949179. [DOI] [PubMed] [Google Scholar]
- 34. Mellor CL, Marchese Robinson RL, Benigni R, et al. Molecular fingerprint‐derived similarity measures for toxicological read‐across: Recommendations for optimal use. Regul Toxicol Pharmacol. 2019;101:121–134. [DOI] [PubMed] [Google Scholar]
- 35. Teubner W, Landsiedel R. Read‐across for hazard assessment: The ugly duckling is growing up. Altern Lab Anim. 2015;43(6):P67–P71. [DOI] [PubMed] [Google Scholar]
- 36. Pawar G, Madden JC, Ebbrell D, Firman JW, Cronin MTD. In silico toxicology data resources to support read‐across and (Q)SAR. Front Pharmacol. 2019;10:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pradeep P, Mansouri K, Patlewicz G, Judson R. A systematic evaluation of analogs and automated read‐across prediction of estrogenicity: A case study using hindered phenols. Comput Toxicol. 2017;4:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schultz TW, Przybylak KR, Richarz AN, et al. Read‐across of 90‐day rat oral repeated‐dose toxicity: A case study for selected n‐alkanols. Comput Toxicol. 2017;2:12–19. Available from: http://www.sciencedirect.com/science/article/pii/S2468111317300117. [Google Scholar]
- 39. Schultz TW, Przybylak KR, Richarz AN, Mellor CL, Bradbury SP, Cronin MTD. Read‐across of 90‐day rat oral repeated‐dose toxicity: A case study for selected 2‐alkyl‐1‐alkanols. Comput Toxicol. 2017;2:28–38. Available from: http://www.sciencedirect.com/science/article/pii/S2468111317300130. [Google Scholar]
- 40. Przybylak KR, Schultz TW, Richarz AN, Mellor CL, Escher SE, Cronin MTD. Read‐across of 90‐day rat oral repeated‐dose toxicity: A case study for selected β‐olefinic alcohols. Comput Toxicol. 2017. Feb;1:22–32. Available from: http://www.sciencedirect.com/science/article/pii/S2468111316300032. [Google Scholar]
- 41. Mellor CL, Schultz TW, Przybylak KR, Richarz AN, Bradbury SP, Cronin MTD. Read‐across for rat oral gavage repeated‐dose toxicity for short‐chain mono‐alkylphenols: A case study. Comput Toxicol. 2017;2:1–11. Available from: http://www.sciencedirect.com/science/article/pii/S2468111317300178. [Google Scholar]
- 42. Schultz TW, Cronin MTD. Lessons learned from read‐across case studies for repeated‐dose toxicity. Regul Toxicol Pharmacol. 2017;88:185–191. Available from: http://www.sciencedirect.com/science/article/pii/S0273230017301800. [DOI] [PubMed] [Google Scholar]
- 43. ECETOC . TR 116 – Category approaches, read‐across, (Q)SAR. Available from: http://www.ecetoc.org/publication/tr-116-category-approaches-read-across-qsar/.
- 44. ECHA . Guidance on information requirements and chemical safety assessment, R.6 QSAR and grouping of chemicals. ECHA; 2008. Available from: https://echa.europa.eu/documents/10162/13632/information_requirements_r6_en.pdf/77f49f81-b76d-40ab-8513-4f3a533b6ac9.
- 45. OECD . Grouping of chemicals: Chemical categories and read‐across. Organisation for Economic Co‐operation and Development (OECD), 2014;p. 194 Available from: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2014)4&doclanguage=en. [Google Scholar]
- 46. Schultz TW, Amcoff P, Berggren E, et al. A strategy for structuring and reporting a read‐across prediction of toxicity. Regul Toxicol Pharmacol. 2015;72(3):586–601. Available from: http://www.sciencedirect.com/science/article/pii/S0273230015001154. [DOI] [PubMed] [Google Scholar]
- 47. Grace P, George H, Prachi P, Imran S. Navigating through the minefield of read‐across tools: A review of in silico tools for grouping. Comput Toxicol. 2017;3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. ECHA . Read‐across assessment framework (RAAF). Helsinki: European Chemicals Agency (ECHA), 2017. [Google Scholar]
- 49. Ball N, Cronin MTD, Shen J, et al. Toward good read‐across practice (GRAP) guidance. ALTEX. 2016;33(2):149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hartung T. Making big sense from big data in toxicology by read‐across. ALTEX. 2016;33(2):83–93. [DOI] [PubMed] [Google Scholar]
- 51. Zhu H, Bouhifd M, Donley E, et al. Supporting read‐across using biological data. ALTEX. 2016;33(2):167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo Y, Zhao L, Zhang X, Zhu H. Using a hybrid read‐across method to evaluate chemical toxicity based on chemical structure and biological data. Ecotoxicol Environ Saf. 2019;178:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Benigni R. Towards quantitative read across: Prediction of Ames mutagenicity in a large database. Regul Toxicol Pharmacol. 2019;108:104434. [DOI] [PubMed] [Google Scholar]
- 54. Helman G, Patlewicz G, Shah I. Quantitative prediction of repeat dose toxicity values using GenRA. Regul Toxicol Pharmacol. 2019;109:104480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Helman G, Shah I, Williams AJ, Edwards J, Dunne J, Patlewicz G. Generalized read‐across (GenRA): A workflow implemented into the EPA CompTox chemicals dashboard. ALTEX. 2019;36(3):462–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schultz TW, Richarz AN, Cronin MTD. Assessing uncertainty in read‐across: Questions to evaluate toxicity predictions based on knowledge gained from case studies. Comput Toxicol. 2019;9:1–11. Available from: http://www.sciencedirect.com/science/article/pii/S2468111318300811. [Google Scholar]
- 57. De Abrew KN, Shan YK, Wang X, et al. Use of connectivity mapping to support read across: A deeper dive using data from 186 chemicals, 19 cell lines and 2 case studies. Toxicology. 2019;423:84–94. Available from: http://www.sciencedirect.com/science/article/pii/S0300483X19301623. [DOI] [PubMed] [Google Scholar]
- 58. Helma C, Vorgrimmler D, Gebele D, et al. Modeling chronic toxicity: A comparison of experimental variability with (Q)SAR/read‐across predictions. Front Pharmacol. 2018;9:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kovarich S, Ceriani L, Fuart Gatnik M, Bassan A, Pavan M. Filling data gaps by read‐across: A mini review on its application, developments and challenges. Mol Inform. 2019;38(8–9): Available from: https://onlinelibrary-wiley-com.uaccess.univie.ac.at/doi/10.1002/minf.201800121. [DOI] [PubMed] [Google Scholar]
- 60. Marchant CA, Briggs KA, Long A. In silico tools for sharing data and knowledge on toxicity and metabolism: Derek for windows, meteor, and vitic. Toxicol Mech Methods. 2008;18(2–3):177–187. [DOI] [PubMed] [Google Scholar]
- 61. Ferrari T, Cattaneo D, Gini G, Golbamaki Bakhtyari N, Manganaro A, Benfenati E. Automatic knowledge extraction from chemical structures: The case of mutagenicity prediction. SAR QSAR Environ Res. 2013;24(5):365–383. [DOI] [PubMed] [Google Scholar]
- 62. Ahlberg E, Carlsson L, Boyer S. Computational derivation of structural alerts from large toxicology data sets. J Chem Inf Model. 2014;54(10):2945–2952. [DOI] [PubMed] [Google Scholar]
- 63. Métivier JP, Lepailleur A, Buzmakov A, et al. Discovering structural alerts for mutagenicity using stable emerging molecular patterns. J Chem Inf Model. 2015;55(5):925–940. 10.1021/ci500611v. [DOI] [PubMed] [Google Scholar]
- 64. Cortes‐Ciriano I. Bioalerts: A python library for the derivation of structural alerts from bioactivity and toxicity data sets. Journal of Cheminformatics. 2016;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang H, Li J, Wu Z, Li W, Liu G, Tang Y. Evaluation of different methods for identification of structural alerts using chemical Ames mutagenicity data set as a benchmark. Chem Res Toxicol. 2017;30(6):1355–1364. [DOI] [PubMed] [Google Scholar]
- 66. Floris M, Raitano G, Medda R, Benfenati E. Fragment prioritization on a large mutagenicity dataset. Mol Inform. 2017;36(7). [DOI] [PubMed] [Google Scholar]
- 67. Limban C, Nuţă DC, Chiriţă C, et al. The use of structural alerts to avoid the toxicity of pharmaceuticals. Toxicol Rep. 2018;5:943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. TEH A, Goodman JM, Gutsell S, Russell PJ. Using 2D structural alerts to define chemical categories for molecular initiating events. Toxicol Sci. 2018;165(1):213–223. [DOI] [PubMed] [Google Scholar]
- 69. Garcia‐Serna R, Vidal D, Remez N, Mestres J. Large‐scale predictive drug safety: From structural alerts to biological mechanisms. Chem Res Toxicol. 2015;28(10):1875–1887. [DOI] [PubMed] [Google Scholar]
- 70. Nepali K, Lee HY, Liou JP. Nitro‐group‐containing drugs. J Med Chem. 2019;62(6):2851–2893. [DOI] [PubMed] [Google Scholar]
- 71. Boechat N, Carvalho AS, Salomão K, et al. Studies of genotoxicity and mutagenicity of nitroimidazoles: Demystifying this critical relationship with the nitro group. Mem Inst Oswaldo Cruz. 2015;110(4):492–499. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4501412/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Klein M, Erdinger L, Boche G. From mutagenic to non‐mutagenic nitroarenes: Effect of bulky alkyl substituents on the mutagenic activity of nitroaromatics in Salmonella typhimurium: Part II. Substituents far away from the nitro group. Mutat Res. 2000;467(1):69–82. Available from: http://www.sciencedirect.com/science/article/pii/S1383571800000139. [DOI] [PubMed] [Google Scholar]
- 73. Hsu KH, Su BH, Tu YS, Lin OA, Tseng YJ. Mutagenicity in a molecule: Identification of core structural features of mutagenicity using a scaffold analysis. PLoS One. 2016;11(2):e0148900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Williams RV, Amberg A, Brigo A, et al. It's difficult, but important, to make negative predictions. Regul Toxicol Pharmacol. 2016;76:79–86. [DOI] [PubMed] [Google Scholar]
- 75. Kalgutkar AS. Should the incorporation of structural alerts be restricted in drug design? An analysis of structure–toxicity trends with aniline‐based drugs. Curr Med Chem. 2015;22(4):438–464. [DOI] [PubMed] [Google Scholar]
- 76. Alves V, Muratov E, Capuzzi S, et al. Alarms about structural alerts. Green Chem. 2016;18(16):4348–4360. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5423727/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kalgutkar AS. Designing around structural alerts in drug discovery. J Med Chem. 2019. [DOI] [PubMed] [Google Scholar]
- 78. Myden A, Guesne SJ, Cayley A, Williams RV. Utility of published DNA reactivity alerts. Regul Toxicol Pharmacol. 2017;88:77–86. [DOI] [PubMed] [Google Scholar]
- 79. Cros AFA. Action de l'alcool amylique sur l'organisme. Faculté de médecine de Strasbourg; 1863.
- 80. Kubinyi H. From narcosis to hyperspace: The history of QSAR. Quant Struct Act Relationships. 2002;21(4):348–356. Available from: http://onlinelibrary.wiley.com/doi/10.1002/1521-3838(200210)21:4¡348::AID-QSAR348¿3.0.CO;2-D/abstract. [Google Scholar]
- 81. Hansch C, Fujita T. p–σ–π analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc. 1964;86(8):1616–1626. [Google Scholar]
- 82. Free SM, Wilson JW. A mathematical contribution to structure–activity studies. J Med Chem. 1964;7(4):395–399. 10.1021/jm00334a001. [DOI] [PubMed] [Google Scholar]
- 83. Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110(18):5959–5967. 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 84. Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37(24):4130–4146. 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 85. Pastor M, Cruciani G, McLay I, Pickett S, Clementi S. GRid‐INdependent descriptors (GRIND): A novel class of alignment‐independent three‐dimensional molecular descriptors. J Med Chem. 2000;43(17):3233–3243. [DOI] [PubMed] [Google Scholar]
- 86. Cruciani G, Crivori P, Carrupt PA, Testa B. Molecular fields in quantitative structure–permeation relationships: The VolSurf approach. J Mol Struct THEOCHEM. 2000;503(1):17–30. Available from: http://www.sciencedirect.com/science/article/pii/S0166128099003607. [Google Scholar]
- 87. Durán Á, Martínez GC, Pastor M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in molecular interaction fields. J Chem Inform Model. 2008;48(9):1813–1823. [DOI] [PubMed] [Google Scholar]
- 88. Devillers J, Domine D, Guillon C. Autocorrelation modeling of lipophilicity with a back‐propagation neural network. Eur J Med Chem. 1998;33(7):659–664. Available from: http://www.sciencedirect.com/science/article/pii/S022352349880024X. [Google Scholar]
- 89. Tong L, Guo L, Lv X, Li Y. Modification of polychlorinated phenols and evaluation of their toxicity, biodegradation and bioconcentration using three‐dimensional quantitative structure–activity relationship models. J Mol Graph Model. 2017;71:1–12. [DOI] [PubMed] [Google Scholar]
- 90. Fengxian C, Reti H. Analysis of positions and substituents on genotoxicity of fluoroquinolones with quantitative structure–activity relationship and 3D pharmacophore model. Ecotoxicol Environ Saf. 2017. Feb;136:111–118. [DOI] [PubMed] [Google Scholar]
- 91. Stoyanova‐Slavova IB, Slavov SH, Buzatu DA, Beger RD, Wilkes JG. 3D‐SDAR modeling of hERG potassium channel affinity: A case study in model design and toxicophore identification. J Mol Graph Model. 2017;72:246–255. [DOI] [PubMed] [Google Scholar]
- 92. Nagai J, Shi H, Kubota Y, et al. Quantitative structure–cytotoxicity relationship of pyrano[4,3‐b]chromones. Anticancer Res. 2018;38(8):4449–4457. [DOI] [PubMed] [Google Scholar]
- 93. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. Available from: https://academic.oup.com/nar/article/28/1/235/2384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang S, Liu W, Wu J, Cao L, Meng Q, Kennedy PJ. Training deep neural networks on imbalanced data sets. 2016 International Joint Conference on Neural Networks (IJCNN), 2016; p. 4368–4374. 10.1021/ci100423z. [DOI] [Google Scholar]
- 95.Obiol‐Pardo C, Gomis‐Tena J, Sanz F, Saiz J, Pastor M. A multiscale simulation system for the prediction of drug‐induced cardiotoxicity. J Chem Inf Model. 2011;51(2):483–492. [DOI] [PubMed] [Google Scholar]
- 96. Wacker S, Noskov SY. Performance of machine learning algorithms for qualitative and quantitative prediction drug blockade of hERG1 channel. Comput Toxicol. 2018;6:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang Y, Zhao J, Wang Y, et al. Prediction of hERG K+ channel blockage using deep neural networks. Chem Biol Drug Des. 2019;94(5):1973–1985. [DOI] [PubMed] [Google Scholar]
- 98. Lee HM, Yu MS, Kazmi SR, et al. Computational determination of hERG‐related cardiotoxicity of drug candidates. BMC Bioinform. 2019;20(suppl 10):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ogura K, Sato T, Yuki H, Honma T. Support vector machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA‐II. Sci Rep. 2019;9(1):12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Farid R, Day T, Friesner RA, Pearlstein RA. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem. 2006;14(9):3160–3173. [DOI] [PubMed] [Google Scholar]
- 101. Thai KM, Windisch A, Stork D, et al. The hERG potassium channel and drug trapping: Insight from docking studies with propafenone derivatives. ChemMedChem. 2010;5(3):436–442. [DOI] [PubMed] [Google Scholar]
- 102. Negami T, Araki M, Okuno Y, Terada T. Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci Rep. 2019. Nov;9(1):1–12. Available from: https://www.nature.com/articles/s41598-019-53120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Klepsch F, Chiba P, Ecker GF. Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P‐glycoprotein. PLoS Comput Biol. 2011;7(5): Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3093348/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Klepsch F, Vasanthanathan P, Ecker GF. Ligand and structure‐based classification models for prediction of P‐glycoprotein inhibitors. J Chem Inf Model. 2014. Jan;54(1):218–229. 10.1021/ci400289j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jain S, Grandits M, Richter L, Ecker GF. Structure based classification for bile salt export pump (BSEP) inhibitors using comparative structural modeling of human BSEP. J Comput Aided Mol Des. 2017;31(6):507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rao MS, Gupta R, Liguori MJ, et al. Novel computational approach to predict off‐target interactions for small molecules. Front Big Data. 2019;2: Available from: https://www.frontiersin.org/articles/10.3389/fdata.2019.00025/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cortés‐Ciriano I, Ain QU, Subramanian V, et al. Polypharmacology modelling using proteochemometrics (PCM): Recent methodological developments, applications to target families, and future prospects. MedChemComm. 2015;6(1):24–50. Available from: https://pubs.rsc.org/en/content/articlelanding/2015/md/c4md00216d. [Google Scholar]
- 108. Elton DC, Boukouvalas Z, Fuge MD, Chung PW. Deep learning for molecular design—A review of the state of the art. Mol Syst Des Eng. 2019;4(4):828–849. Available from: http://pubs.rsc.org/en/content/articlelanding/2019/me/c9me00039a. [Google Scholar]
- 109. Frenzel F, Buhrke T, Wenzel I, Andrack J, Hielscher J, Lampen A. Use of in silico models for prioritization of heat‐induced food contaminants in mutagenicity and carcinogenicity testing. Arch Toxicol. 2017;91(9):3157–3174. [DOI] [PubMed] [Google Scholar]
- 110. Honma M, Kitazawa A, Cayley A, et al. Improvement of quantitative structure–activity relationship (QSAR) tools for predicting Ames mutagenicity: Outcomes of the Ames/QSAR International Challenge Project. Mutagenesis. 2019;34(1):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Luechtefeld T, Marsh D, Rowlands C, Hartung T. Machine learning of toxicological big data enables read‐across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicol Sci. 2018;165(1):198–212. Available from: https://academic.oup.com/toxsci/article/165/1/198/5043469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Alves VM, Borba J, Capuzzi SJ, et al. Oy Vey! A comment on “Machine learning of toxicological big data enables read‐across structure activity relationships outperforming animal test reproducibility”. Toxicol Sci. 2019;167(1):3–4. Available from: https://academic.oup.com/toxsci/article/167/1/3/5220777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Norinder U, Carlsson L, Boyer S, Eklund M. Introducing conformal prediction in predictive modeling. A transparent and flexible alternative to applicability domain determination. J Chem Inf Model. 2014. Jun;54(6):1596–1603. 10.1021/ci5001168. [DOI] [PubMed] [Google Scholar]
- 114. Sun J, Carlsson L, Ahlberg E, Norinder U, Engkvist O, Chen H. Applying Mondrian cross‐conformal prediction to estimate prediction confidence on large imbalanced bioactivity data sets. J Chem Inf Model. 2017;57(7):1591–1598. 10.1021/acs.jcim.7b00159. [DOI] [PubMed] [Google Scholar]
- 115. Norinder U, Rybacka A, Andersson PL. Conformal prediction to define applicability domain – A case study on predicting ER and AR binding. SAR QSAR Environ Res. 2016;27(4):303–316. [DOI] [PubMed] [Google Scholar]
- 116. Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: Toxicity prediction using deep learning. Front Environ Sci. 2016;3: Available from: http://journal.frontiersin.org/article/10.3389/fenvs.2015.00080/full. [Google Scholar]
- 117. Alves VM, Golbraikh A, Capuzzi SJ, et al. Multi‐descriptor read across (MuDRA): A simple and transparent approach for developing accurate quantitative structure–activity relationship models. J Chem Inf Model. 2018;58(6):1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. McCulloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys. 1943;5(4):115–133. Available from: https://link.springer.com/article/10.1007/BF02478259. [PubMed] [Google Scholar]
- 119. Rosenblatt F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol Rev. 1958;65(6):386–408. [DOI] [PubMed] [Google Scholar]
- 120. Chellapilla K, Puri S, Simard P.. High performance convolutional neural networks for document processing; 2006. Available from: https://hal.inria.fr/inria-00112631.
- 121. Hinton GE, Osindero S, Teh YW. A fast learning algorithm for deep belief nets. Neural Comput. 2006;18(7):1527–1554. 10.1162/neco.2006.18.7.1527. [DOI] [PubMed] [Google Scholar]
- 122. Schmidhuber J. Deep learning in neural networks: An overview. Neural Netw. 2015;61:85–117. Available from: http://www.sciencedirect.com/science/article/pii/S0893608014002135. [DOI] [PubMed] [Google Scholar]
- 123. MerckKaggle . Merck molecular activity challenge; 2012. Available from: https://www.kaggle.com/c/MerckActivity.
- 124. Team K. Deep learning how i did it: Merck 1st place interview; 2012. Available from: http://blog.kaggle.com/2012/11/01/deep-learning-how-i-did-it-merck-1st-place-interview/.
- 125. Tox21 . Tox21 data challenge 2014; 2014. Available from: https://tripod.nih.gov/tox21/challenge/.
- 126. Lee H, Grosse R, Ranganath R, Ng AY. Convolutional deep belief networks for scalable unsupervised learning of hierarchical representations. Proceedings of the 26th Annual International Conference on Machine Learning. ICML '09. New York, NY: ACM, 2009; p. 609–616. 10.1145/1553374.1553453. [DOI] [Google Scholar]
- 127. Hessler G, Baringhaus KH. Artificial intelligence in drug design. Molecules. 2018;23(10):2520 Available from: https://www.mdpi.com/1420-3049/23/10/2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Goh GB, Siegel C, Vishnu A, Hodas NO, Baker N. How much chemistry does a deep neural network need to know to make accurate predictions? arXiv:171002238 [cs, stat]; 2017. Available from: http://arxiv.org/abs/1710.02238.
- 129. Goh GB, Siegel C, Vishnu A, Hodas NO, Baker N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert‐developed QSAR/QSPR models. arXiv:170606689 [cs, stat]; 2017. Available from: http://arxiv.org/abs/1706.06689.
- 130. Fernandez M, Ban F, Woo G, et al. Toxic colors: The use of deep learning for predicting toxicity of compounds merely from their graphic images. J Chem Inf Model. 2018;58(8):1533–1543. [DOI] [PubMed] [Google Scholar]
- 131. Jimenez‐Carretero D, Abrishami V, Fernández‐de Manuel L, et al. Tox_(R)CNN: Deep learning‐based nuclei profiling tool for drug toxicity screening. PLoS Comput Biol. 2018;14(11):e1006238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hofmarcher M, Rumetshofer E, Clevert DA, Hochreiter S, Klambauer G. Accurate prediction of biological assays with high‐throughput microscopy images and convolutional networks. J Chem Inf Model. 2019;59(3):1163–1171. [DOI] [PubMed] [Google Scholar]
- 133. Gini G, Zanoli F, Gamba A, Raitano G, Benfenati E. Could deep learning in neural networks improve the QSAR models? SAR QSAR Environ Res. 2019;1–26. [DOI] [PubMed] [Google Scholar]
- 134. Xu Y, Pei J, Lai L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. 2017;57(11):2672–2685. 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- 135. Gao M, Igata H, Takeuchi A, Sato K, Ikegaya Y. Machine learning‐based prediction of adverse drug effects: An example of seizure‐inducing compounds. J Pharmacol Sci. 2017;133(2):70–78. [DOI] [PubMed] [Google Scholar]
- 136. Wang H, Liu R, Schyman P, Wallqvist A. Deep neural network models for predicting chemically induced liver toxicity endpoints from transcriptomic responses. Front Pharmacol. 2019;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Unterthiner T, Mayr A. Deep learning as an opportunity in virtual screening. Deep learning and representation learning workshop (NIPS 2014), 2014; p. 9. [Google Scholar]
- 138. Dahl GE, Jaitly N, Salakhutdinov R. Multi‐task neural networks for QSAR predictions. arXiv:14061231 [cs, stat]; 2014. Available from: http://arxiv.org/abs/1406.1231.
- 139. Ramsundar B, Kearnes S, Riley P, Webster D, Konerding D, Pande V. Massively multitask networks for drug discovery. arXiv:150202072 [cs, stat]; 2015. Available from: http://arxiv.org/abs/1502.02072.
- 140. Xu Y, Ma J, Liaw A, Sheridan RP, Svetnik V. Demystifying multitask deep neural networks for quantitative structure–activity relationships. J Chem Inf Model. 2017;57(10):2490–2504. 10.1021/acs.jcim.7b00087. [DOI] [PubMed] [Google Scholar]
- 141. Sosnin S, Karlov D, Tetko IV, Fedorov MV. Comparative study of multitask toxicity modeling on a broad chemical space. J Chem Inf Model. 2019;59(3):1062–1072. 10.1021/acs.jcim.8b00685. [DOI] [PubMed] [Google Scholar]
- 142. Hughes TB, Dang NL, Miller GP, Swamidass SJ. Modeling reactivity to biological macromolecules with a deep multitask network. ACS Cent Sci. 2016;2(8):529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wenzel J, Matter H, Schmidt F. Predictive multitask deep neural network models for ADME‐Tox properties: Learning from large data sets. J Chem Inf Model. 2019;59(3):1253–1268. [DOI] [PubMed] [Google Scholar]
- 144. Drew KLM, Baiman H, Khwaounjoo P, Yu B, Reynisson J. Size estimation of chemical space: How big is it? J Pharm Pharmacol. 2012;64(4):490–495. 10.1111/j.2042-7158.2011.01424.x. [DOI] [PubMed] [Google Scholar]
- 145. Mathea M, Klingspohn W, Baumann K. Chemoinformatic classification methods and their applicability domain. Mol Inform. 2016;35(5):160–180. 10.1002/minf.201501019. [DOI] [PubMed] [Google Scholar]
- 146. Hanser T, Barber C, Marchaland JF, Werner S. Applicability domain: Towards a more formal definition. SAR QSAR Environ Res. 2016;27(11):893–909. [DOI] [PubMed] [Google Scholar]
- 147. Roy K, Ambure P, Kar S. How precise are our quantitative structure–activity relationship derived predictions for new query chemicals? ACS Omega. 2018;3(9):11392–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu R, Wang H, Glover KP, Feasel MG, Wallqvist A. Dissecting machine‐learning prediction of molecular activity: Is an applicability domain needed for quantitative structure–activity relationship models based on deep neural networks? J Chem Inf Model. 2019;59(1):117–126. [DOI] [PubMed] [Google Scholar]
- 149. OECD . Guidance document on the validation of (quantitative)structure–activity relationships [(Q)SAR] models. OECD Publishing, 2007. Available from: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono%282007%292&doclanguage=en. [Google Scholar]
- 150. Hasselgren C, Ahlberg E, Akahori Y, et al. Genetic toxicology in silico protocol. Regul Toxicol Pharmacol. 2019;107:104403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Barber C, Hanser T, Judson P, Williams R. Distinguishing between expert and statistical systems for application under ICH M7. Regul Toxicol Pharmacol. 2017;84:124–130. [DOI] [PubMed] [Google Scholar]
- 152. Amberg A, Beilke L, Bercu J, et al. Principles and procedures for implementation of ICH M7 recommended (Q)SAR analyses. Regul Toxicol Pharmacol. 2016;77:13–24. [DOI] [PubMed] [Google Scholar]
- 153. Amberg A, Andaya RV, Anger LT, et al. Principles and procedures for handling out‐of‐domain and indeterminate results as part of ICH M7 recommended (Q)SAR analyses. Regul Toxicol Pharmacol. 2019;102:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.114th Congress. S. Rept. 114‐67 – Frank R. Lautenberg chemical safety for the 21st century act. Available from: https://www.congress.gov/congressional-report/114th-congress/senate-report/67/1?overview=closed.
- 155. Ankley GT, Bennett RS, Erickson RJ, et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 2010;29(3):730–741. [DOI] [PubMed] [Google Scholar]
- 156. Farhat A, Kennedy SW. Adverse Outcome Pathway on Aryl hydrogen receptor activation leading to early life stage mortality, via reduced VEGF. Paris; 2019. p. 16. Available from: https://www.oecd-ilibrary.org/environment/adverse-outcome-pathway-on-aryl-hydrogen-receptor-activation-leading-to-early-life-stage-mortality-via-reduced-vegf_063e1bf4-en.
- 157. Nymark P, Rieswijk L, Ehrhart F, et al. A data fusion pipeline for generating and enriching adverse outcome pathway descriptions. Toxicol Sci. 2018;162(1):264–275. [DOI] [PubMed] [Google Scholar]
- 158. Carvaillo JC, Barouki R, Coumoul X, Audouze K. Linking bisphenol S to adverse outcome pathways using a combined text mining and systems biology approach. Environ Health Perspect. 2019;127(4):47005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Oki NO, Farcal L, Abdelaziz A, et al. Integrated analysis of in vitro data and the adverse outcome pathway framework for prioritization and regulatory applications: An exploratory case study using publicly available data on piperonyl butoxide and liver models. Toxicol In Vitro. 2019;54:23–32. [DOI] [PubMed] [Google Scholar]
- 160. Fay KA, Villeneuve DL, Swintek J, et al. Differentiating pathway‐specific from nonspecific effects in high‐throughput toxicity data: A foundation for prioritizing adverse outcome pathway development. Toxicol Sci. 2018;163(2):500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Vahle JL, Anderson U, Blomme EAG, Hoflack JC, Stiehl DP. Use of toxicogenomics in drug safety evaluation: Current status and an industry perspective. Regul Toxicol Pharmacol. 2018;96:18–29. [DOI] [PubMed] [Google Scholar]
- 162. Buesen R, Chorley BN, da Silva LB, et al. Applying 'omics technologies in chemicals risk assessment: Report of an ECETOC workshop. Regul Toxicol Pharmacol. 2017;91(suppl):S3–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Brockmeier EK, Hodges G, Hutchinson TH, et al. The role of omics in the application of adverse outcome pathways for chemical risk assessment. Toxicol Sci. 2017;158(2):252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. AbdulHameed MDM, Ippolito DL, Wallqvist A. Predicting rat and human pregnane X receptor activators using bayesian classification models. Chem Res Toxicol. 2016;29(10):1729–1740. [DOI] [PubMed] [Google Scholar]
- 165. Zhang Q, Li J, Middleton A, Bhattacharya S, Conolly RB. Bridging the data gap from in vitro toxicity testing to chemical safety assessment through computational modeling. Front Public Health. 2018;6:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Escher BI, Hackermüller J, Polte T, et al. From the exposome to mechanistic understanding of chemical‐induced adverse effects. Environ Int. 2017;99:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hines DE, Edwards SW, Conolly RB, Jarabek AM. A case study application of the aggregate exposure pathway (AEP) and adverse outcome pathway (AOP) frameworks to facilitate the integration of human health and ecological end points for cumulative risk assessment (CRA). Environ Sci Technol. 2018;52(2):839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Conolly RB, Ankley GT, Cheng W, et al. Quantitative adverse outcome pathways and their application to predictive toxicology. Environ Sci Technol. 2017;51(8):4661–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mellor CL, Steinmetz FP, Cronin MTD. Using molecular initiating events to develop a structural alert based screening workflow for nuclear receptor ligands associated with hepatic steatosis. Chem Res Toxicol. 2016;29(2):203–212. [DOI] [PubMed] [Google Scholar]
- 170. Wittwehr C, Aladjov H, Ankley G, et al. How adverse outcome pathways can aid the development and use of computational prediction models for regulatory toxicology. Toxicol Sci. 2017;155(2):326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sakuratani Y, Horie M, Leinala E. Integrated approaches to testing and assessment: OECD activities on the development and use of adverse outcome pathways and case studies. Basic Clin Pharmacol Toxicol. 2018;123(S5):20–28. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/bcpt.12955. [DOI] [PubMed] [Google Scholar]
- 172. Arzuaga X, Walker T, Yost EE, Radke EG, Hotchkiss AK. Use of the adverse outcome pathway (AOP) framework to evaluate species concordance and human relevance of dibutyl phthalate (DBP)‐induced male reproductive toxicity. Reprod Toxicol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Mortensen HM, Chamberlin J, Joubert B, et al. Leveraging human genetic and adverse outcome pathway (AOP) data to inform susceptibility in human health risk assessment. Mamm Genome. 2018;29(1–2):190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Villeneuve DL, Crump D, Garcia‐Reyero N, et al. Adverse outcome pathway (AOP) development I: Strategies and Principles. Toxicol Sci. 2014;142(2):312–320. Available from: https://academic.oup.com/toxsci/article/142/2/312/1621273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Knapen D, Angrish MM, Fortin MC, et al. Adverse outcome pathway networks I: Development and applications. Environ Toxicol Chem. 2018;37(6):1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Oki NO, Edwards SW. An integrative data mining approach to identifying adverse outcome pathway signatures. Toxicology. 2016;350–352:49–61. [DOI] [PubMed] [Google Scholar]
- 177. Sturla SJ, Boobis AR, FitzGerald RE, et al. Systems toxicology: From basic research to risk assessment. Chem Res Toxicol. 2014;27(3):314–329. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3964730/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Steinmetz FP, Enoch SJ, Madden JC, et al. Methods for assigning confidence to toxicity data with multiple values – Identifying experimental outliers. Sci Total Environ. 2014;482–483:358–365. [DOI] [PubMed] [Google Scholar]
- 179. Fourches D, Muratov E, Tropsha A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model. 2010;50(7):1189–1204. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2989419/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inform. 2010;29(6–7):476–488. 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 181. Mansouri K, Grulke CM, Richard AM, Judson RS, Williams AJ. An automated curation procedure for addressing chemical errors and inconsistencies in public datasets used in QSAR modelling. SAR QSAR Environ Res. 2016;27(11):911–937. 10.1080/1062936X.2016.1253611. [DOI] [PubMed] [Google Scholar]
- 182. Noh H, You T, Mun J, Han B. Regularizing deep neural networks by noise: Its interpretation and optimization. Proceedings of the 31st International Conference on Neural Information Processing Systems. NIPS'17. California: Curran Associates Inc., 2017; p. 5115–5124 Available from: http://dl.acm.org/citation.cfm?id=3295222.3295264. [Google Scholar]
- 183. Baumann D, Baumann K. Reliable estimation of prediction errors for QSAR models under model uncertainty using double cross‐validation. J. Cheminform. 2014;6(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. He H, Garcia EA. Learning from imbalanced data. IEEE Trans Knowl Data Eng. 2009. Sep;21(9):1263–1284. [Google Scholar]
- 185. Domingos P. MetaCost: A general method for making classifiers cost‐sensitive. Proceedings of the fifth ACM SIGKDD international conference on Knowledge discovery and data mining – KDD'99. San Diego, CA: ACM Press, 1999; p. 155–164 Available from: http://portal.acm.org/citation.cfm?doid=312129.312220. [Google Scholar]
- 186. Breiman L. Bagging predictors. Mach Learn. 1996;24(2):123–140. Available from: https://link.springer.com/article/10.1007/BF00058655. [Google Scholar]
- 187. Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: Synthetic minority over‐sampling technique. J Artif Intell Res. 2002;16:321–357. Available from: https://jair.org/index.php/jair/article/view/10302. [Google Scholar]
- 188. Bjerrum EJ. SMILES enumeration as data augmentation for neural network modeling of molecules. arXiv:170307076 [cs]; 2017. Available from: http://arxiv.org/abs/1703.07076.
- 189. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405(2):442–451. Available from: http://www.sciencedirect.com/science/article/pii/0005279575901099. [DOI] [PubMed] [Google Scholar]
- 190. Baldi P, Brunak S, Chauvin Y, CAF A, Nielsen H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics. 2000. May;16(5):412–424. Available from: https://academic.oup.com/bioinformatics/article/16/5/412/192336. [DOI] [PubMed] [Google Scholar]
- 191. Myatt GJ, Ahlberg E, Akahori Y, et al. In silico toxicology protocols. Regul Toxicol Pharmacol. 2018. Jul;96:1–17. Available from: http://www.sciencedirect.com/science/article/pii/S0273230018301144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Polishchuk P. Interpretation of quantitative structure–activity relationship models: Past, present, and future. J Chem Inf Model. 2017. Nov;57(11):2618–2639. 10.1021/acs.jcim.7b00274. [DOI] [PubMed] [Google Scholar]
- 193. Ribeiro MT, Singh S, Guestrin C “Why should I trust you?” Explaining the predictions of any classifier. arXiv:160204938 [cs, stat]; 2016. Available from: http://arxiv.org/abs/1602.04938.
- 194. Lundberg SM, Lee SI. A unified approach to interpreting model predictions. In: Guyon I, Luxburg UV, Bengio S, et al., editors. advances in neural information processing systems. Volume 30. Curran Associates, Inc., 2017; p. 4765–4774 Available from: http://papers.nips.cc/paper/7062-a-unified-approach-to-interpreting-model-predictions.pdf. [Google Scholar]
- 195. Bach S, Binder A, Montavon G, Klauschen F, Müller KR, Samek W. On pixel‐wise explanations for non‐linear classifier decisions by layer‐wise relevance propagation. PLoS One. 2015;10(7):e0130140 Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0130140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Adadi A, Berrada M. Peeking inside the black‐box: A survey on explainable artificial intelligence (XAI). IEEE Access. 2018;6:52138–52160. [Google Scholar]
- 197. Pinches MD, Thomas R, Porter R, Camidge L, Briggs K. Curation and analysis of clinical pathology parameters and histopathologic findings from eTOXsys, a large database project (eTOX) for toxicologic studies. Regul Toxicol Pharmacol. 2019;107:104396 Available from: http://www.sciencedirect.com/science/article/pii/S0273230019301497. [DOI] [PubMed] [Google Scholar]
- 198. Svensson F, Norinder U, Bender A. Modelling compound cytotoxicity using conformal prediction and PubChem HTS data. Toxicol Res. 2017;6(1):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sturm N, Sun J, Vandriessche Y, et al. Application of bioactivity profile‐based fingerprints for building machine learning models. J Chem Inf Model. 2019;59(3):962–972. [DOI] [PubMed] [Google Scholar]
- 200. Lenselink EB, ten Dijke N, Bongers B, et al. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J Cheminform. 2017;9(1):45. 10.1186/s13321-017-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Yepiskoposyan H, Talikka M, Vavassori S, et al. Construction of a suite of computable biological network models focused on mucociliary clearance in the respiratory tract. Front Genet. 2019;10: Available from: https://www.frontiersin.org/articles/10.3389/fgene.2019.00087/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Duran‐Frigola M, Fernández‐Torras A, Bertoni M, Aloy P. Formatting biological big data for modern machine learning in drug discovery. WIREs Comput Mol Sci. 2019;9(6):e1408 Available from: http://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1408. [Google Scholar]
- 203. Gedeck P, Skolnik S, Rodde S. Developing collaborative QSAR models without sharing structures. J Chem Inf Model. 2017;57(8):1847–1858. [DOI] [PubMed] [Google Scholar]
- 204. Dean J, Corrado G, Monga R, et al. Large scale distributed deep networks. In: Pereira F, Burges CJC, Bottou L, Weinberger KQ, editors. Advances in neural information processing systems. Volume 25. Curran Associates, Inc., 2012; p. 1223–1231 Available from: http://papers.nips.cc/paper/4687-large-scale-distributed-deep-networks.pdf. [Google Scholar]
FURTHER READING
- Cronin M, Madden J. (eds.) In silico toxicology: Principles and applications; London, UK: Royal Society of Chemistry, 2010. ISBN: 978‐1‐84973‐004‐4. 10.1039/9781849732093 [DOI] [Google Scholar]
- Ekins S (editor), Computational toxicology: Risk assessment for pharmaceutical and environmental chemicals. Hoboken, NJ: John Wiley & Sons, 2007. ISBN: 978‐0‐470‐04962‐4. 10.1002/97804701458902006 [DOI] [Google Scholar]
- Pfannkuch F, Suter‐Dick L. (eds.) Predictive toxicology: From vision to reality, Hoboken, NJ: John Wiley & Sons, 2015. ISBN: 978‐3‐527‐33608‐1. 10.1002/9783527674183 [DOI] [Google Scholar]