

Abstract
A novel platform of dendritic nanogels is herein presented, capitalizing on the self‐assembly of allyl‐functional polyesters based on dendritic‐linear‐dendritic amphiphiles followed by simple cross‐linking with complementary monomeric thiols via UV initiated off‐stoichiometric thiol‐ene chemistry. The facile approach enabled multigram creation of allyl reactive nanogel precursors, in the size range of 190–295 nm, being readily available for further modifications to display a number of core functionalities while maintaining the size distribution and characteristics of the master batch. The nanogels are evaluated as carriers of a spread of chemotherapeutics by customizing the core to accommodate each individual cargo. The resulting nanogels are biocompatible, displaying diffusion controlled release of cargo, maintained therapeutic efficacy, and decreased cargo toxic side effects. Finally, the nanogels are found to successfully deliver pharmaceuticals into a 3D pancreatic spheroids tumor model.
Keywords: cancer treatment, dendritic nanogel, drug delivery, nanomedicine
A novel dendritic nanogel system is created from the self‐assembly of allyl functional 2,2‐bis(hydroxymethyl)propionic acid polyesters with dendritic‐lineardendritic configuration followed by simple cross‐linking with complementary monomeric thiols via UV initiated offstoichiometric thiol‐ene chemistry. The resulting nanogels are biocompatible and capable of loading and releasing cargo, maintain therapeutic efficacy, and decrease the cargos toxic side effects.

1. Introduction
With astronomical costs associated to the introduction of new drugs, the research focus has been redirected toward improving the efficacy and reducing the side effect of already approved pharmaceuticals.1 An attractive strategy that improves the therapeutic outcome of drugs is their formulation in nanosized delivery vehicles, commonly known as a drug delivery systems (DDSs). These are classified according to their inherent building blocks, e.g., inorganic nanoparticles,2 self‐assembled low molecular weight lipids,3 and self‐assembled polymeric DDSs.4 In the case of the latter, the inherent modularity of polymers, available in infinite chemical compositions and an array of configurations, makes them ideal carriers for targeted delivery.[[qv: 4a,5]] Increased drug concentration at tumor sites due to the enhanced permeability and retention (EPR), cargo protection from degradation and improved therapeutic efficacy are typical features that have reported.[[qv: 4b,6]]
While amphiphilic self‐assembled DDSs provide enhanced therapeutic outcome, their apparent steady state between single polymeric chains and assembled micelles, causes stability concerns. Especially in diluted environments, i.e., bloodstream with large shear forces present that may disassemble the micelles and lead to premature release of drugs resulting in undesired systemic effects. Such mechanisms are micellar specific, being strongly dictated by the interaction forces between the amphiphilic polymers and thereof defined critical micelle concentration (CMC).7 To overcome the CMC limitation, attention has been directed toward cross‐linked or unimolecular systems which aim to provide large and stable constructs through cross‐linking or sophisticated design.8
Unimolecular micelles, pioneered by Newkome et al.,9 are designed as single molecules displaying both hydrophobic compartments and hydrophilic solubilizing domains. Such frameworks can be achieved by a “grafting from” or “grafting to” approach that amalgamate hydrophilic polymers to hydrophobic cores.10 In contrast to self‐assembled systems, unimolecular‐micelles are often of smaller hydrodynamic volume and release their cargo through either degradation or diffusion. Most recent advances in unimolecular micelle strategies include the exploitation of redox sensitive constituents relying on disulfides bridges that selectively disassembly intracellularly by enzymatic mechanisms.11
Larger DDSs capitalizes on cross‐linked particles, i.e., hydrogel‐like constructs, which are accurately synthesized in sizes ranging from micro‐ down to the nanoscale. These micro‐ and nanogels, commonly produced through emulsion polymerizations or self‐assembly strategies, deliver aqueous stable particles.12 While emulsion polymerization yield cross‐linked particles in one step,13 the self‐assembled strategies include a subsequent crosslinking step. The latter are of more modular nature as the crosslinking can be predestined to either the hydrophobic core14 or the hydrophilic corona, essentially creating large and stable polymeric nanoparticles. For instance, cross‐linked knedel‐like nanoparticles and their applications as DDSs have during the past decade been extensively explored by Wooley and co‐workers.15
Micro‐ and nanogels are typically fabricated by batch specific strategy to display a desired functionality. This limitation causes inherent batch‐to‐batch variations such as size, cross‐linking density, and core functionality. To overcome this issue, Klinger and co‐workers proposed a novel approach in which a master batch of core functionalized microgels was produced with the capacity to postfunctionalize the core while maintaining the size distribution and cross‐linking density of the gels.[[qv: 14b]] Independently, a major drawback that arises for DDSs of larger configuration, built from physiologically stable bonds, is their potential bioaccumulation upon delivery of their therapeutic load.16 To counteract bioaccumulation, cross‐linked DDSs encompassing building blocks with intrinsic ester bonds are envisaged to facilitate hydrolytic and enzymatic degradation. Upon their introduction, time‐dependent degradation and disassembly of the DDSs will efficiently occur assisting secretion of the fragments via the renal system.
To this end, polyester dendritic scaffolds based on 2,2‐bis(hydroxymethyl)propionic acid (bis‐MPA) cover all the necessary prerequisites to generate next generation DDSs including biocompatibility and biodegradability features as well multiple representation of functional groups for efficient crosslinking mechanisms.17 However, a system that amalgamates the unique features of linear (L) polyethylene glycol (PEG) and bis‐MPA dendritic (D) components in dendritic‐linear hybrid configuration has yet not been proposed for the development of nanogels and thereof use as potential drug delivery carriers.
In this work, a novel platform of nanogel was sought out and that capitalizes on a self‐assembly strategy using amphiphilic block copolymers that comprises linear hydrophilic PEGs and hydrophobic dendritic bis‐MPA components displaying multitude of allylic end‐groups. The hydrophobic dendritic constituents, with dense representation of multiple allylic end‐groups, were postulated to provide three important elements to generate cross‐linked nanogels: i) directing the self‐assembly of the block copolymers; ii) core crosslinking via UV‐initiated thiol‐ene coupling chemistry with complementary hydrophobic thiol monomers to generate stable nanoparticles and; iii) provide readily accessible allyl for postfunctionalization. To accomplish the creation functional nanogels, off‐stoichiometric thiol‐ene (OSTE) chemistry,18 between the reactive enes allocated in the hydrophobic core and molar deficient amounts of multifunctional thiol monomer was a natural and straightforward route to target. If successful, an inherently simple strategy would be unfolded based on OSTE chemistry and that permits large scale production of nanogel precursors with residual allyls being readily available for facile postfunctionalization and thereof tunable core nanoenvironment with emphasis on encapsulation and delivery of a multitude potent drugs.
2. Results and Discussion
2.1. Material Preparation
2.1.1. Material Synthesis
A library of amphiphilic dendritic‐linear‐dendritic (DLD) and linear‐dendritic (LD) hybrids were synthesized through divergent growth approach from bifunctional or monofunctional PEGs and bis‐MPA monomer, as can be seen in Figure 1 .[[qv: 4d]] Both DLD and LD architectures were produced, to evaluate their influence on self‐assembly and two different hydrophobic to hydrophilic ratios were targeted, either 13 or 23 wt% hydrophobicity. To attain the described ratios a PEG length of either 5 or 10 kDa was used for the LD hybrids and a 10 or 20 kDa was used for the DLD‐hybrids. All PEGs were hybridized with dendritic constituents of a third generation (G3). As a complement to the perfectly branched dendron segments, DLD and LD with hyperbranched (hb) dendritic segments were synthesized via pseudo polycondensation reactions.19 Subsequently, the hydroxyl end‐groups for all hybrids was successfully modified with hydrophobic allyls, using anhydride activated 4‐pentenoic acid, which introduced the desired amphiphilic features required for self‐assembly.
Figure 1.
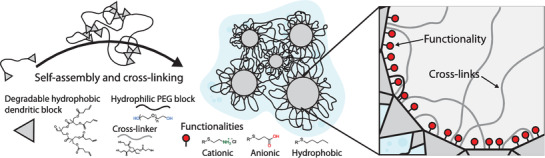
Purposed structure of dendritic nanogels (DNGs) constructed from amphiphilic linear dendritic hybrids between poly (ethylene glycol) (PEG) and 2,2‐bis(hydroxymethyl)propionic acid (bis‐MPA). Created DNGs can further be functionalized to display both anionic, cationic, and hydrophobic interior while maintaining size and hydrophilic PEG exterior. This enables DNGs to be tailor‐made to load and deliver different cargos.
2.1.2. Self‐Assembly Assessment
Prior to crosslinking, a micellar self‐assembly study was performed for all hybrids using dialysis, micro precipitation, evaporation, and thin film absorption methods as described in the electronic supplementary information (ESI). The most consistent results were attained using thin film absorption. Independent of architecture, structural perfection or molecular weight no stable assemblies were attained for the 23 wt% hydrophobic ratio. The DLD hyperbranched scaffold PEG20K‐hbG3‐allyl with 13 wt% hydrophobicity ratio yielded stable assemblies while their structurally perfect counterpart, i.e., DLD with perfect dendrons, seemed to precipitate without any indication of generating micelles. The stable assembles obtained for PEG20K‐hbG3‐allyl could be due to the more irregular conformation of the hyperbranched segments having the allyl functional groups homogenously displayed along the hydrophobic dendritic block. A concentration dependence study, from 1 to 10 mg mL−1, on the self‐assembly of PEG20K‐hbG3‐allyl was conducted using DLS as a characterization technique and as a mean to understand the effect on micelle size and polydispersity index (PDI). The size of the self‐assembled constructs appeared to increase with increased concentration of PEG20K‐hbG3‐allyl (164–295 nm), however, the effect appeared to correlate in a stepwise manner rather than linearly (see Figure 2 a). For the highest concentration evaluated, 10 mg mL−1, traces of larger aggregates were observed. The lowest PDI was noted for the concentration of 5 mg mL−1 indicating the most uniform self‐assembly (see Figure 2a,b) with particle size of 220 nm and PDI value of 0.07. Notably, the large size and uniform nature of the formed particle assemblies indicate the presence of several interconnected micelles rather than one single micelle (see Figure 1).
Figure 2.

Investigation of self‐assembly and cross‐linking into DNG for PEG‐20K‐hbG3‐allyl. a) Size evaluation by DLS of self‐assembled adducts at different concentrations. b) PDI evaluation by DLS of self‐assembled adducts at different concentrations. c) Graphical representation of degree of cross‐linking and remaining functionalization of DNGs 1–5 (theoretical). d) Size evaluation by DLS of DNG 1–5 at 5 mg mL−1. e) Solution stability for DNG 2 both before cross‐linking and after. f) Size and PDI evaluation by DLS of DNG1‐5 in PBS at room temperature over time. Values were normalized against initial value and represented as percent based on initial size.
2.1.3. Nanogel Formation
As the PEG20K‐hbG3‐allyl displayed all the elements to generate stable nanosized particles with low PDI, it was selected as the prime candidate for further crosslinking to generate novel nanogel precursors. The crosslinking was achieved by the incorporation of trimethylolpropane tris(3‐mercaptopropionate) (TMP‐SH) during the thin film adsorption step of the self‐assembly along with 2,2‐dimethoxy‐2‐phenylacetophenone (DMPA) as an UV‐initiator.
Both the TMP‐SH and DMPA are sufficiently hydrophobic to be encapsulated in the hydrophobic regions where the ene‐unsaturations are located. Several different OSTE [SH:ene] ratios were assesses with excess of enes, i.e., [10:16], [6:16], [5:16], [4:16], and [3:16]. The crosslinking resulted in 5 dendritic nanogel systems denoted DNG1, DNG2, DNG3, DNG4, and DNG5, Figure 2c and Table S1 (Supporting Information). During the self‐assembly with TMP‐SH and DMPA, the particles increased slightly in size, from 220 to 295 nm. However, upon UV exposure the obtained DNGs contracted down to the expected value of 220 nm, as can be seen for DNG2 in Figure 2e. Generally, a higher degree of crosslinking resulted in a smaller DNGs, again variation appears step wise rather than linear, Figure 2d. A formulated DNG2, prior to cross‐linking, appears less stable in which the assembly is jeopardized within 48 h while the cross‐linked DNG maintain their stability over time, see Figure 2e. Additionally, five different batches of DNG2 were produced to evaluate the potential batch‐to‐batch variations. In all cases, the produced DNG2 were found consistent having similar size and PDI, Figure S1 in the Supporting Information. The same reproducibility was obtained for DNG1, DNG3, and DNG4 and their overall stability at room temperature in PBS was confirmed for at least 250 days, Figure 2f. On the other hand, the DNG5 having the lowest cross‐linking density, with an OSTE ratio of 3:16 between the thiols and enes, was noted to disassemble after ≈100 days with changes in relative size and PDI.
One of important benefits of using the cross‐linked micelle approach to nanogels is the facile and modular internal modification. While the allyl functional DNGs were straightforwardly obtain via two‐step strategy, self‐assembly and OSTE crosslinking, their further manipulations with desired functions could be achieved in a pseudo one‐pot manner. This is simplistically accomplished by the addition of a thiol with desired functionality and DMPA into the cross‐linked DNG mixture followed by exposure to UV‐light. To facilitate the access to the allyl groups in the core of the DNGs, the reactions were conducted in a H2O: THF solution of 4:1 in volume. As a proof‐of‐concept, carboxylic, primary amine, and hydrophobic n‐alkyl functionalities were introduced to the DNG3 using 3‐mercaptopropionic acid, 2‐mercaptoethylamine, and 1‐hexanethiol, respectively. By using the same batch of reactive nanogel precursor, DNG3‐allyl with an average size of 295 nm, core functionalized DNG3 were obtained with similar but contracted size of 190 nm, Figure 3 a. The pH responsiveness of the carboxylic functional DNG3 was also measured until reaching a study‐state in size. In this study, the carboxylated DNG3 was found to contract at acidic conditions of pH 4–6 (95% and 180 nm) and swell under basic conditions of pH 8–9 (110% and 208 nm), Figure 3b. For all DNGs, the amount of allyls the production was monitored and measured by 1H NMR spectroscopy. Indeed, excess of allyls were detected during the OSTE cross‐linking, see Figure 3c. Some degree of shielding was expected due to the core–shell nature of the DNGs,20 however, complete consumption of the allyls upon functionalization was noted corroborating on successful covalent bonding of the desired functionalities to the DNGs.
Figure 3.

Investigation of functionalized DNG3 based on PEG20K‐hbG3‐allyl. a) Size evaluation by DLS. b) Swelling evaluation of DNG3‐COOH at pH 4–8 size measured by DLS and divided by initial size (295 nm). c) Allyls during formation of DNG3 as measured by 1H NMR (400 MHz, CDCl3) signal associated with CH2 of bis‐MPA at δ(ppm): 4.09 was kept constant while an average of signals corresponding to the allyl δ(ppm): 5.84 and 5.06 was used to asses remaining allyls. d) SEM analysis including size distribution.
2.2. Evaluation as a Drug Delivery System
2.2.1. Particle Size
DNGs produced in this work were sought out as universal DDSs based on degradable dendritic bis‐MPA components. Their apparent facile customization with intrinsically different cores, i.e., hydrophobic, anionic or cationic functionalities enables the delivery of chemotherapeutics of different properties. Notably, all three types of functional DNGs have similar hydrodynamic size of 190 nm, as determined by DLS in Figure 3a, being within the optimal size range for the EPR effect to facilitate accumulation in solid tumors in vivo.[[qv: 5a,6]] The morphology of DNGs were observed by scanning electron microscope (SEM), showing that DNGs with hydrophobic modification formed spherical particles while the charged DNGs tend to form elongated or irregular shaped particles, which might reflect the existence of electrostatic repulsive force, Figure 3d. However, the size distribution measured by SEM tends to be smaller compared to DLS. This is reasonable since the size measured by SEM technique is dry size while DLS size is hydrodynamic size.
2.2.2. Drug Loading Capabilities
To assess the DNGs as potential DDS, doxorubicin (DOX) was selected as a standard hydrophobic chemotherapeutic agent. All three types of DNG3 with hydrophobic, anionic or cationic core were subjected to DOX to explore the loading and release profile. Regardless of the core functionality, all DNGs possess an inherently hydrophobic core due to their dendritic polyester based backbone, enabling them to load hydrophobic drugs via simple physical encapsulation. For all DOX‐DNGs, any free DOX was dialyzed off and the final concentration of DOX encapsulated were measurement by UV–vis. To confirm DOX encapsulation, free DOX solutions were prepared in the same concentration as fresh DOX‐DNG3‐Hexyl (100 µg mL−1), DOX‐DNG3‐NH2 (75 µg mL−1), and DOX‐DNG3‐COOH (89 µg mL−1), respectively, then fluorescent intensity of drug were compared and a significant change (due to the shielding of carriers) observed in all three cases, indicating the success of DOX encapsulation (Figure S1b, Supporting Information).21 As expected, all the DNGs were able to encapsulate DOX with only minimal differences in loading efficiency, which could be due to differences in internal pH, considering DOX itself possess an amine functionality. Due to its more hydrophobic nature, the DOX‐DNG3‐Hexyl showed the highest loading of 23.0 ± 0.5% in drug yield or 5.7% in drug loading capacity (DLC) = m(drug)/m(DNG). The DOX‐DNG3‐COOH and DOX‐DNG3‐NH2 noted 22.4 ± 0.2% (or 5.6% in DLC) and 21.9 ± 0.4% (or 5.5% in DLC), respectively (Table S2, Supporting Information). The differences in DOX loading using different core functional DNGs indicate that core property can influence drug loading providing validity to our approach. More importantly, the size of DNGs after DOX loading remained consistent (Table S2, Supporting Information) and therefore still valid for the EPR effect. This can be compared to previous reported physically self‐assembled micellar systems built from similar polymers where the size increased significantly after DOX loading, probably due to incomplete internalization of DOX. For those systems, the DOX loaded micelles were reaching sizes of 370 nm, which might be beyond the optimal size range of the EPR effect.22
The cargo release profile for the DNGs following a slightly faster rate (Figure 4 a) to previously established cholesterol‐directed LD micellar systems within 12 h but have a similar release amount after 96 h.[[qv: 4d]]
Figure 4.

Evaluation of DNGs as DDSs for cancer treatment. a) In vitro DOX release in PBS for all three core functional DNGs. b) Cytotoxicity screening of pure DNGs on two noncancerous and pancreatic cancer cell lines. Several other cell lines were also tested see ESI Figure S3 in the Supporting Information. AlamarBlue assay was used and cells were with substrate for 72 h. c) Cytotoxicity induced by free DOX and DOX‐DNGs against a panel of cancer cell lines with 72 h incubation.
To better understand drug loading capabilities of the DNGs, especially with respect to the impact of the interior charge (i.e., the effect of electrostatic forces and/or interior environment), two additional chemotherapeutics, gemcitabine and methotrexate, were examined by encapsulation to DNG3‐COOH and DNG3‐NH2, respectively. The gemcitabine is a hydrophilic drug that contains one amine while methotrexate displays two amines and two carboxylic functionalities being insoluble in pure water and most organic solvents. An alternative loading strategy was used for the encapsulation of gemcitabine hydrochloride and included the neutralization of the drug followed by mixing with DNG3‐COOH in deionized water. After 2 h of dialysis for the removal unloaded gemcitabine, the loaded DNG3 was freeze dried and 1H NMR analysis indeed indicated successful drug encapsulation, and a concentration of drug of 950 µg mL−1 (Figure S2, Supporting Information). For the methotrexate, the only viable route to dissolve it is in a basic solution (e.g., 1 m NaOH). However, the drug may not be stable in the presence of a strong base23 and IV administration of a basic solution is not optimal to gain health benefits. Remarkably, the DNG3‐NH2 was found to overcome the formulation challenges associated with methotrexate. In here, a simple mixing procedure was unfolded in which the insoluble methotrexate was mixed in DI water with DNG3‐NH2. Due to the basic nature of the DNG, the methotrexate is encapsulated through interactions with the available amines. Any remaining insoluble drugs were straightforwardly removed by simple filtration. The successful loading of methotrexate was also confirmed by 1H NMR and approximated to 460 µg mL−1 (Figure S2, Supporting Information). The successful encapsulation of three different chemotherapeutics indeed validates the modular nature of the presented DNG platform. By straightforward manipulations of the interior, customization of desired cargo can be accomplished which ultimately opens new possibilities to utilize drugs that might normally be difficult to formulate and administer.
2.2.3. Bioevaluations
DDSs intended for clinical applications should be nontoxic as pure carriers. To assess this, the neat DNGs were screened against four cancer and two noncancerous cell lines. No obvious cytotoxicity was observed even at high dose of 500 µg mL−1, indicating good biocompatibility of the DNGs, Figure 4b and Figure S3: Supporting Information. Additionally, all the DOX‐DNGs demonstrated therapeutic efficacy similar to free DOX on tested cancer cell lines (Figure 4c), as well as relatively reduced toxicity to noncancerous cells found in Figure S4 in the Supporting Information. This suggests that the DNGs could potentially reduce side‐effects in cancer treatment, which is a crucial benefit of utilizing a DDS.24
Finally, to verify that the DNGs can deliver drugs into a tumor a 3D spheroids tumor model (PANC1 cells) was applied for in vitro drug tracking.[[qv: 21b]] A 3D model affords more useful information compared to a conventional 2D cell culture as each spheroid can be seen as a tiny solid tumor which has a relatively complex extracellular environment similar to the situation in vivo.25 All the DNGs were found to deliver the payload of DOX into the 3D tumors within 24 h, and delivered drugs were homogeneously distributed throughout the 3D sphere, Figure 5 a. Detailed colocalization study revealed that DOX signal covers nuclei and cytoplasm (lysosome labeling), Figure 5b and Figure S5: Supporting Information. As a mean to further understand the delivery mechanism in 3D tumors, the DNG3‐NH2 was internally labeled with NHS activated pacific blue dye, by covalent coupling to the internal amines, and incubated with 3D PANC1 spheroids. To no surprise, the fluorescence from DNG3‐NH‐PB was also found inside the tumor (Figure 5c), implying that DNGs can carry DOX into a tumors and continuously release DOX.
Figure 5.

Colocalization study on a 3D spheroid model. a,b) Colocalization study of DOX‐DNG3‐NH2 on 3D PANC1 spheroids. DOX signal was represented by red fluorescence; green fluorescence indicates (in a) Calcein‐AM labeled living cells or (in b) lysoTracker DND‐26 labeled lysosomes in cytoplasm; nuclei were stained with Hoechst 33342 and represented as blue signal. c) Colocalization study of pacific blue labeled DNG‐NH2 (DNG‐NH‐PB) on 3D PANC1 spheroids. Blue fluorescence represents DNG‐NH‐PB and green fluorescence indicates Calcein‐AM labeled living cells. Scale bar = 100 µm.
3. Conclusion
In summary, a novel platform of DNGs has been unfolded from DLD hybrids based on bis‐MPA monomers and linear PEGs. Capitalizing on UV initiated OSTE chemistry, allyl functional DNG scaffolds were straightforwardly produced in multigram scale. The core functionality was successfully altered to display hydrophobic, amine or carboxylic features and in parallel maintain size distribution and functionality. Doxorubicin, gemcitabine, and methotrexate were efficiently encapsulated, as potent therapeutic agents, in the interior of the DNGs. The DOX loaded DNGs were found to possess similar therapeutic efficacy as free DOX on breast cancer, pancreatic cancer, and bone cancer cell lines. Importantly, the therapeutic DNGs displayed a reduced cytotoxicity toward noncancerous cells. Finally, the DNGs can be internalized into 3D pancreatic tumors and deliver its cargo through diffusion controlled release.
4. Experimental Section
Materials Synthesis and Characterization: Synthesis and characterization details for the materials can be found in the Supporting Information.
Micelle Formation—Evaporation Method: LD‐hybrid was dissolved as desired concentration in dichloromethane (DCM) (0.5 mL) in a glass vial. PBS (1 mL, filtered through a 0.2 µm syringe filter) was added to the vial with lid along with a small magnetic bar, the mixture was allowed to homogenize for 30 min at 300 rpm. The lid was removed and replaced with a tissue paper and the stirring set to 100 rpm and the organic solvent was allowed to slowly evaporate overnight (14 h).
Micelle Formation—Thin Film Method: LD‐hybrid was dissolved at desired concentration in DCM (100 µL). The DCM was allowed to evaporate slowly while the vessel was rotated to create a thin polymeric film subsequently high vacuum was applied for 15 min ensuring complete removal of DCM. Milli‐Q water (1 mL) was added to the vessel to achieve final concentration of 1 mg mL−1; vigorous vortex for 15 s followed by ultrasound bath for 15 min.
Micelle Formation—Micro Precipitation Method: LD‐hybrid was dissolved at desired concentration in THF (100 µL), the solution was quickly added by micropipette to 1 mL of Milli‐Q water in a vial equipped with a magnetic stir bar under vigorous stirring (1500 rpm). Solution was left to evaporate for 12 h at 100 rpm stirring rate.
Micelle Formation—Dialysis Method: LD‐hybrid was dissolved at desired concentration in DMF (500 µL), the mixture was transferred to a dialysis bag diameter 1 cm, effective length 3 cm, cutoff 6000 Da. Dialysis against 100 mL Milli‐Q water was conducted over a period of 24 h with solvent exchanges at 1, 2, 4, 8, and 20 h.
Drug Loading: Three drugs with different properties were explored. DOX as the standard hydrophobic drug was loaded to all the three types of DNGs because of the hydrophobicity of dendritic compartments. DNGs was dissolved in DCM (5 mg mL−1, 1.6 mL) and mixed with DOX (in DCM 2 mg mL−1, 1 mL with 3 molar equivalents of triethylamine (TEA) vs DOX) in a small glass vial. The DCM was removed by auto evaporation and a dry film was form in each vial. Then DI water (8 mL, filtered through a 0.2 µm syringe filter) was added to each vial and allowed to homogenize in ultrasound bath for 15 min. Then the vials were transferred to a shaking table for gently mixing another 2 h. DOX and DNGs mixture were then transferred into dialysis bag (molecular weight cut off (MWCO) 12–14 kDa) immersed in 4 L DI water for 2 h (water changed every 20 min) to remove free and loosely attached DOX. The resulting DOX‐DNGs in DI water were collected and drug concentrations were determined by comparing UV absorbance to a standard curve (three replicates) at 490 nm. Gemcitabine hydrochloride 15 mg was neutralized with equal amount of sodium chloride in DI water (6 mL) containing 30 mg DNG3‐COOH. While methotrexate hydrate (16.8 mg, containing methotrexate 15 mg) was added directly to DNG3‐NH2 (30 mg in 6 mL DI water) solution. Both mixtures were allowed 15 min mixing in ultrasound following extra shaking 2 h on a shaking table. Extra free drugs were removed with the same dialysis procedure as DOX. The insoluble methotrexate powder was removed with 0.2 µm syringe filter. The resulting drug‐DNGs were analyzed with 1H NMR to confirm drug loading.
In Vitro DOX Release: 2.7 mL of free DOX or fresh prepared DOX‐DNGs solution were transferred to dialysis cassettes (MWCO 10 000 Slide‐A‐Lyzer G2, Thermo) individually and dialyze against 4 L PBS (pH 7.4) at room temperature. 10 µL (triplicates) of sample solution were collected from the inside of the dialysis cassettes into 96‐well plates (black, transparent, and flat bottom) at the following time points: 0, 2, 4, 8, 10, 12, 24, 48, 72, and 96 h. 100 µL of DMF:H2O (4:1) were added to each well to allow DOX‐DNGs to disaggregate. The fluorescent intensity of the resulting solution was measured with a plate reader (Tecan Infinite M200 Pro) at the wavelength of 485/595 (excitation/emission) nm.
Cell Culture: A panel of cancer cell lines (breast cancer: MDA‐MB‐231, MCF‐7; pancreatic cancer: PANC1; osteosarcoma: MG‐63.) and two noncancerous cell lines (mouse monocyte cell Raw 264.7 and human debris fibroblast cell (hDF)) were applied in this study. They were obtained from ATCC (American Tissue Culture Collection) initially and were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum FBS and 100 units mL−1 penicillin plus 100 mg mL−1 streptomycin under 5% CO2 at 37 °C.
Cytotoxicity Assessment: AlamarBlue assay was applied to evaluate the cytotoxicity induced by pure DNGs, DOX, and DOX‐DNGs. Cells were harvested and transferred into 96‐well plates at a concentration of 1 × 104 cells per well in 100 mL DMEM cultured 24 h before use. Old medium were removed and fresh DMEM containing testing samples were added. Six parallel wells were set for each sample. After 72 h incubation, 10 µL of AlamarBlue agents were added into each well and fluorescent intensity were measured 4 h later with a plate reader (Tecan Infinite M200 Pro) at the wavelength of 560/590 (excitation/emission) nm.
Localization Study in 3D Spheroids Model: 3D tumor spheroids were cultured from PANC1 cells as described before.[[qv: 21b,25]] Harvested PANC1 cells were suspended in complete DMEM (without rho) medium supplemented with 20% methylcellulose at a concentration of 3 × 104 cells mL−1. Then cell suspension was transferred into 96‐well plates (round bottom) at the 100 µL per well and incubated 3 days to generate 3D spheroids. On the day four, 3D tumors were exposed to 2.5 µg mL−1 (DOX concentration) of free DOX, DOX‐DNGs or 100 µg mL−1 pacific‐blue labeled DNG‐NH2 and incubated for 24 h. On the day five, 3D spheroids were incubated with other fluorescent probes Hoechst 33342 (5 µg mL−1) and Calcien‐AM (5 × 10−6 m) or LysoTracker Green DND‐26 (75 × 10−9 m) according to manuals and washed with PBS twice before observation. Fluorescent signal was detected and recorded by a Nikon Ti‐S fluorescent microscope with the following filter settings (ex/em nm): blue 350/460; green 480/510; red 560/635. Data was recorded with software Lumenera INFINITY ANALYZE and analyzed with ImageJ.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supplementary
Acknowledgements
Y.Z. and O.C.J.A. contributed equally to this work. The authors acknowledge Knut och Alice Wallenberg Foundation (KAW) (Grant No: 2012‐0196), Seventh Framework Programme (Grant No: 60418), Barncancerfonden (Grant No: TJ2017‐0009), and Swedish Research Council (VR) (Grant No: 2010‐453) for financial support.
Zhang Y., Andrén O. C. J., Nordström R., Fan Y., Malmsten M., Mongkhontreerat S., Malkoch M., Adv. Funct. Mater. 2019, 29, 1806693. 10.1002/adfm.201806693
References
- 1.a) DiMasi J. A., Grabowski H. G., Hansen R. W., J. Health Econ. 2016, 47, 20; [DOI] [PubMed] [Google Scholar]; b) Prausnitz M. R., Mitragotri S., Langer R., Nat. Rev. Drug Discovery 2004, 3, 115. [DOI] [PubMed] [Google Scholar]
- 2.a) Horcajada P., Chalati T., Serre C., Gillet B., Sebrie C., Baati T., Eubank J. F., Heurtaux D., Clayette P., Kreuz C., Chang J. S., Hwang Y. K., Marsaud V., Bories P. N., Cynober L., Gil S., Ferey G., Couvreur P., Gref R., Nat. Mater. 2010, 9, 172; [DOI] [PubMed] [Google Scholar]; b) Slowing I. I., Vivero‐Escoto J. L., Wu C. W., Lin V. S. Y., Adv. Drug Delivery Rev. 2008, 60, 1278; [DOI] [PubMed] [Google Scholar]; c) Rosenholm J. M., Meinander A., Peuhu E., Niemi R., Eriksson J. E., Sahlgren C., Linden M., ACS Nano 2009, 3, 197; [DOI] [PubMed] [Google Scholar]; d) Mamaeva V., Sahlgren C., Linden M., Adv. Drug Delivery Rev. 2013, 65, 689. [DOI] [PubMed] [Google Scholar]
- 3.a) Huynh N. T., Passirani C., Saulnier P., Benoit J. P., Int. J. Pharm. 2009, 379, 201; [DOI] [PubMed] [Google Scholar]; b) Matougui N., Boge L., Groo A. C., Umerska A., Ringstad L., Bysell H., Saulnier P., Int. J. Pharm. 2016, 502, 80. [DOI] [PubMed] [Google Scholar]
- 4.a) Duncan R., Nat. Rev. Drug Discovery 2003, 2, 347; [DOI] [PubMed] [Google Scholar]; b) Maeda H., Bharate G. Y., Daruwalla J., Eur. J. Pharm. Biopharm. 2009, 71, 409; [DOI] [PubMed] [Google Scholar]; c) Riess G., Prog. Polym. Sci. 2003, 28, 1107; [Google Scholar]; d) Andren O. C. J., Zhang Y. N., Lundberg P., Hawker C. J., Nystrom A. M., Malkoch M., Chem. Mater. 2017, 29, 3891. [Google Scholar]
- 5.a) Whitehead K. A., Langer R., Anderson D. G., Nat. Rev. Drug Discovery 2009, 8, 129; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Torchilin V. P., Cell. Mol. Life Sci. 2004, 61, 2549; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Maeda H., Kimura M., Sasaki I., Hirose Y., Konno T., in Poly(Ethylene Glycol) Chemistry (Ed: Harris J. M.), Springer, New York: 1992, Ch. 11; [Google Scholar]; d) Chandra S., Dietrich S., Lang H., Bahadur D., J. Mater. Chem. 2011, 21, 5729. [Google Scholar]
- 6. Matsumura Y., Maeda H., Cancer Res. 1986, 46, 6387. [PubMed] [Google Scholar]
- 7. Zana R., Langmuir 1996, 12, 1208. [Google Scholar]
- 8. Lee C. C., Gillies E. R., Fox M. E., Guillaudeu S. J., Frechet J. M. J., Dy E. E., Szoka F. C., Proc. Natl. Acad. Sci. USA 2006, 103, 16649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newkome G. R., Yao Z. Q., Baker G. R., Gupta V. K., J. Org. Chem. 1985, 50, 2003. [Google Scholar]
- 10.a) Piotti M. E., Rivera F., Bond R., Hawker C. J., Frechet J. M. J., J. Am. Chem. Soc. 1999, 121, 9471; [Google Scholar]; b) Liu M. J., Kono K., Frechet J. M. J., J. Controlled Release 2000, 65, 121. [DOI] [PubMed] [Google Scholar]
- 11.a) Porsch C., Zhang Y., Montanez M. I., Malho J. M., Kostiainen M. A., Nystrom A. M., Malmstrom E., Biomacromolecules 2015, 16, 2872; [DOI] [PubMed] [Google Scholar]; b) Abandansari H. S., Abuali M., Nabid M. R., Niknejad H., Polymer 2017, 116, 16; [Google Scholar]; c) Andren O. C. J., Fernandes A. P., Malkoch M., J. Am. Chem. Soc. 2017, 139, 17660. [DOI] [PubMed] [Google Scholar]
- 12.a) Oh J. K., Siegwart D. J., Lee H. I., Sherwood G., Peteanu L., Hollinger J. O., Kataoka K., Matyjaszewski K., J. Am. Chem. Soc. 2007, 129, 5939; [DOI] [PubMed] [Google Scholar]; b) Oh J. K., Drumright R., Siegwart D. J., Matyjaszewski K., Prog. Polym. Sci. 2008, 33, 448; [Google Scholar]; c) O'Reilly R. K., Hawker C. J., Wooley K. L., Chem. Soc. Rev. 2006, 35, 1068; [DOI] [PubMed] [Google Scholar]; d) Hamidi M., Azadi A., Rafiei P., Adv. Drug Delivery Rev. 2008, 60, 1638. [DOI] [PubMed] [Google Scholar]
- 13.a) Warren N. J., Armes S. P., J. Am. Chem. Soc. 2014, 136, 10174; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Karagoz B., Esser L., Duong H. T., Basuki J. S., Boyer C., Davis T. P., Polym. Chem. 2014, 5, 350. [Google Scholar]
- 14.a) Ryu J. H., Chacko R. T., Jiwpanich S., Bickerton S., Babu R. P., Thayumanavan S., J. Am. Chem. Soc. 2010, 132, 17227; [DOI] [PubMed] [Google Scholar]; b) Fleischmann C., Gopez J., Lundberg P., Ritter H., Killops K. L., Hawker C. J., Klinger D., Polym. Chem. 2015, 6, 2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Thurmond K. B., Kowalewski T., Wooley K. L., J. Am. Chem. Soc. 1996, 118, 7239; [Google Scholar]; b) Huang H. Y., Remsen E. E., Kowalewski T., Wooley K. L., J. Am. Chem. Soc. 1999, 121, 3805; [Google Scholar]; c) Li A., Luehmann H. P., Sun G. R., Samarajeewa S., Zou J., Zhang S. Y., Zhang F. W., Welch M. J., Liu Y. J., Wooley K. L., ACS Nano 2012, 6, 8970; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Thurmond K. B., Huang H. Y., Clark C. G., Kowalewski T., Wooley K. L., Colloids Surf., B 1999, 16, 45. [Google Scholar]
- 16. Markovsky E., Baabur‐Cohen H., Eldar‐Boock A., Omer L., Tiram G., Ferber S., Ofek P., Polyak D., Scomparin A., Satchi‐Fainaro R., J. Controlled Release 2012, 161, 446. [DOI] [PubMed] [Google Scholar]
- 17.a) Feliu N., Walter M. V., Montanez M. I., Kunzmann A., Hult A., Nystrom A., Malkoch M., Fadeel B., Biomaterials 2012, 33, 1970; [DOI] [PubMed] [Google Scholar]; b) Carlmark A., Malmstrom E., Malkoch M., Chem. Soc. Rev. 2013, 42, 5858. [DOI] [PubMed] [Google Scholar]
- 18.a) Mongkhontreerat S., Oberg K., Erixon L., Lowenhielm P., Hult A., Malkoch M., J. Mater. Chem. A 2013, 1, 13732; [Google Scholar]; b) Mongkhontreerat S., Andren O. C. J., Boujemaoui A., Malkoch M., J. Polym. Sci. Polym. Chem. 2015, 53, 2431. [Google Scholar]
- 19. Andren O. C. J., Walter M. V., Yang T., Hult A., Malkoch M., Macromolecules 2013, 46, 3726. [Google Scholar]
- 20. Webb A. G., Magn. Reson. Chem. 2005, 43, 688. [DOI] [PubMed] [Google Scholar]
- 21.a) Gillies E. R., Frechet J. M., Bioconjugate Chem. 2005, 16, 361; [DOI] [PubMed] [Google Scholar]; b) Zhang Y., Lundberg P., Diether M., Porsch C., Janson C., Lynd N. A., Ducani C., Malkoch M., Malmstrom E., Hawker C. J., Nystrom A. M., J. Mater. Chem. B 2015, 3, 2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hed Y., Zhang Y. N., Andren O. C. J., Zeng X. H., Nystrom A. M., Malkoch M., J. Polym. Sci. Polym. Chem. 2013, 51, 3992. [Google Scholar]
- 23. O'Neil M. J., The Merck index: An Encyclopedia of Chemicals, Drugs, and Biologicals, Merck, Whitehouse Station, NJ: 2001. [Google Scholar]
- 24. Schroeder A., Heller D. A., Winslow M. M., Dahlman J. E., Pratt G. W., Langer R., Jacks T., Anderson D. G., Nat. Rev. Cancer 2012, 12, 39. [DOI] [PubMed] [Google Scholar]
- 25. Longati P., Jia X. H., Eimer J., Wagman A., Witt M. R., Rehnmark S., Verbeke C., Toftgard R., Lohr M., Heuchel R. L., BMC Cancer 2013, 95, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary