

Abstract
Pemphigus vulgaris (PV) is an autoimmune disease characterized by the production of IgG autoantibodies owing to an imbalance in the Th1/Th2 and Th17/Tregs cell pathways. The role of gut microbiota in the development of immune system and autoimmune diseases has been unraveled in the last two decades. However, data pertaining to gut microbiota of PV patients is largely lacking. We aimed to compare the gut microbiota of PV patients and healthy controls and assessed potential correlation with circulating cytokines of Th1/Th2/Th17 cell. Faecal bacterial diversity was analysed in 18 PV patients and 14 age‐ and gender‐matched healthy individuals using hypervariable tag sequencing of the V3‐V4 region of the 16S rRNA gene. Plasma levels of 20 inflammatory cytokines were assessed using the Luminex screening system. As a result, we identified 10 differentially abundant taxa between patients and controls. At the genera level, Lachnospiracea_incertae_sedis and Coprococcus decreased, while Granulicatella, Flavonifractor enriched in PV. Plasma levels of C5a, interleukin (IL)‐2R, IL‐6, IL‐8, IL‐7, IL‐1β, IL17A, IL‐5 and IL‐21 were significantly increased in PV Flavonifractor exhibited a positive correlation with C5a, IL‐6, IL‐8, IL‐7, IL‐1β, IL17A and IL‐21. Lachnospiracea_incertae_sedis and Coprococcus showed a negative correlation with IL‐17A. Our results are consistent with the hypothesis that PV patients have gut microbial dysbiosis which might contribute to the immune disorder and the development of PV.
Keywords: cytokines, gut microbiota, pemphigus vulgaris
1. INTRODUCTION
Pemphigus vulgaris (PV) is a life‐threatening autoimmune blistering disease clinically characterized by intra‐epidermal blisters and acantholysis resulting from the formation of IgG autoantibodies against desmoglein (Dsg)3, which impairs epidermal cell‐cell adhesion.1 Despite recent advances in the diagnosis and treatment of PV, the mortality rate of patients approaches 6%; moreover, several gaps persist in our understanding of the pathogenesis of this disease.2, 3 Early studies focused on the role of humoral immune responses in the development of the disease, and more recent evidence further suggests that production of autoantibodies due to an imbalance in the Dsg3‐sensitized Th1 and Th2 cell pathways plays a key role in the pathogenesis of PV.4, 5 A number of recent studies have examined T cell and cytokine profiles in pemphigus patients with the hopes of clarifying the complex pathogenesis of this rare disease. Unfortunately, results have varied. Although a recent article reported higher serum levels of inflammatory Th1/Th17 cytokines in PV patients, most of studies proven to be a Th2 predominant response, along with concomitant suppression of Th1 response.6, 7 In addition, studies also presented the possibility of involvement of the unbalanced Th17/Tregs cells in the development and progression of autoimmune bullous diseases.8 Production of pro‐inflammatory cytokine production [especially, tumour necrosis factor (TNF)‐α, IL‐1 α and IL‐8] has also been implicated in the maintenance of pathogenic mechanisms of PV.9 While the contribution of T cell and cytokines in pemphigus is clear, the precise mechanisms by which these cytokines induce blister formation are not completely understood.
Additionally, the global incidence of autoimmune diseases is estimated at 3%‐5%, with an increasing trend observed over the past half‐century.10 Although multiple genetic factors are implicated in the development of these disorders, genetic drift alone cannot explain all phenomena involved in the pathogenesis of these diseases. The relationship between environmental factors especially gut microbiota and the increased prevalence of autoimmune disorders such as lupus erythematosus (SLE), rheumatoid arthriti, psoriasis, systemic sclerosis and Behcets disease has evoked considerable attention.11, 12, 13, 14, 15 The immune system co‐evolves with the microbiota with a complex interaction between the two. While the immune system affects microbial inhabitation and activity, the microbiota, in turn, modulates the innate and adaptive immune mechanisms, including peripheral differentiation of Th cells.16, 17, 18 For example, gut segmented filamentous bacteria (SFB) were shown to promote a Th17 response and autoimmunity in mouse models of arthritis and multiple sclerosis.11 In another study, high‐salt diet was shown to enhance the expression of pro‐inflammatory genes and suppress several anti‐inflammatory cytokines and chemokine genes, via reduction in the gut Lactobacillus sp. and protective short‐chain fatty acid production.19
Based on the above findings, we hypothesized that T cell and the associated cytokines may promote the pathogenesis of PV via interaction with the dysbiosis of gut microbiota. This study was designed to investigate the gut microbiome in faecal samples from PV patients and healthy controls. Circulating pro‐inflammatory markers and Th1/Th2/Th17/Treg‐related cytokines and chemokines were also assessed. This is the first study to report the dysbiosis of gut microbiota and to demonstrate the association between microbiome and cytokines in PV.
2. MATERIALS AND METHODS
2.1. Patients
Consecutive patients from the Affiliated Hospital of Southwest Medical University between January 2017 and May 2018 were screened for the presence of PV. After obtaining informed consent, skin biopsy for histopathology and direct immunofluorescence (DIF) examination were performed. The phenotype of PV, body mass index (BMI), age at onset, multiplicity of mucosal involvement, and relapse and remission rates were also recorded and reviewed. All PV patients who qualified the study criteria were offered enrolment. Age‐ and gender‐matched healthy subjects were enrolled as controls.
This research was approved by the Institutional Review Board of The Affiliated Hospital of Southwest Medical University. Written informed consent was obtained from all participants prior to their enrolment.
2.2. Inclusion and exclusion criteria
Patients with recent‐onset PV who had active PV skin lesions were prospectively enrolled. The diagnosis was based on clinical manifestations, histopathological evaluation and direct immunofluorescence.20 The inclusion criteria were age ≥18 years and BMI between 18 and 25 kg/m2.
The exclusion criteria were as follows: age <18 years old; BMI ≥25 or BMI <18; known history of chronic disease such as chronic dermatosis (eg psoriasis, seborrhoeic dermatitis, contact dermatitis, atopic dermatitis and pruritus), autoimmune or rheumatic diseases, metabolic diseases, chronic gastrointestinal diseases and malignant tumours; pregnant or breastfeeding women; presence of acute infection; history of gastrointestinal tract surgery; recent (<6 months) use of antibiotic or consumption of probiotics.
2.3. Sample collection
Fresh stool samples from each participant were collected in a sterile container, immediately homogenized, divided into ten aliquots of 220 mg, and frozen at −80°C within 30 minutes. An average of 2 mL of peripheral blood was collected from each participant into an anticoagulant tube. Plasma samples were obtained by centrifugation at 3000 rpm for 15 minutes and stored at −80°C. During the period of sample collection, specimens were kept at 4°C.
2.4. Detection of cytokines
Twenty‐one cytokines and chemokines (C5a, YKL‐40, IP‐10, CD163, IL‐2R, Osteopontin, ITAC‐1, TNF‐alpha, IL‐6, IL‐8/CXCL8, IL‐7, IL‐10, IL‐1beta, IFN‐gamma, IL‐4, IL‐17A, IL‐2, IL‐13, IL‐5, IL‐21, IL‐23) were tested by Human Magnetic Luminex Screening Assay (LXSAHM‐05P3/LXSAHM‐14P1, R&D Systems, Inc). Plasma levels of cytokines were determined using a Luminex 200 System (EMD Millipore) according to the manufacturer's instructions. The coefficient of variation between the duplicate wells was controlled within 10%, and R2 of the standard curve was at least 0.999.
2.5. DNA extraction, PCR amplification and sequencing
Microbial DNA was extracted from faecal samples using the QIAamp DNA Stool Minikit (Qiagen Ltd) according to manufacturer's protocols. The V3‐V4 region of the bacteria 16S ribosomal RNA genes were amplified by PCR using primers 341F 5’‐CCTACGGGRSGCAGCAG)‐3’ and 806R 5’‐GGACTACVVGGGTATCTAATC‐3. We used KAPA HiFi Hotstart ReadyMix PCR kit for high‐fidelity amplification and NanoDrop 2000 spectrophotometer and 2% agarose gel electrophoresis for assessing the quality of amplicons. After preparation of library, these tags were sequenced on Illumina Hiseq platform (Illumina, Inc) for paired end reads of 250 bp, which were overlapped on their 3 ends for concatenation into original longer tags. DNA extraction, library construction and sequencing were conducted at the Realbio Genomics Institute (Shanghai, China).
2.6. Sequence‐based microbiota analysis and statistical analysis
Paired End Reads were spliced through Overlap relationship between Reads after sequencing by Illumina platform. Spliced Reads were further checked with respect to their rest lengths and average base quality to obtain Clean Reads. 16S sequences were restricted between 220 and 500 bp such that the average Phred score of bases was no worse than 20 (Q20) and no more than 3 ambiguous N. Sequences were grouped into operational taxonomic units (OTUs) using the average neighbour algorithm; only the sequences with frequency >1, which tend to be more reliable, were clustered into OTUs, each of which had a representative sequence. OTUs were clustered with 97% similarity using UPARSE (http://drive5.com/uparse/), and chimeric sequences were identified and removed using USEARCH (version 7.0). Each representative tags was assigned to a taxa by RDP Classifier (http://rdp.cme.msu.edu/) against the RDP database (http://rdp.cme.msu.edu/) using a confidence threshold of 0.8.
OTU profiling table and alpha/beta diversity analyses were also performed using the python scripts of QIIME, while rarefaction curves were made. Each sample was randomized with sufficient sequencing depth to avoid deviations in the analysis due to different sample sizes; Alpha Diversity Index was used to measure the sequence depth. Principal Coordinates Analysis (PCoA) of faecal samples based on 16S rRNA sequences using both unweighted and weighted UniFrac to measure Beta Diversity, which was performed on the resulting matrix of distances between each pair of samples. LefSe analysis (LDA EffectSize) based on linear discriminant analysis (LDA) was applied to demonstrate differential abundance of bacterial taxa among the two groups. Only those taxa that achieved a log LDA score >2 were ultimately considered and verified by Wilcoxon test and Kruskal test using R3.1.0. Person correlation analysis was performed to assess the correlation between gut microbes and cytokines.
All P‐values reported are two‐sided, and P < 0.05 was considered to be statistically significant. We also applied the Benjamini and Hochberg false discovery rate test (FDR) or calculated the 95% confidence intervals (CI), if the FDR q value was >0.1.
2.7. Analysis of predicted metagenomes
PICRUSt was applied to analyse both 16S rRNA gene relative abundances and the predicted metabolic data. The protein sequences of genes in the merged gene catalogue were aligned to the Kyoto Encyclopedia of Genes and Genomes(KEGG) bioinformatics database (8th KEGG release, December 2014). The Statistical Analysis of Metagenomic Profiles (STAMP) software was used for data filtering and statistical analyses.
3. RESULTS
3.1. Patient characteristics
Eighteen patients with active PV and 14 age‐ and gender‐matched healthy controls were included in the study. The background characteristics are summarized in Table 1. All participants had a BMI <25 kg/m2. The clinical phenotype of PV patients was muco‐cutaneous (n = 10; 55.6%) and isolated cutaneous disease (n = 8; 44.4%). None of the patients had isolated mucosal disease. Half of the patients were treatment‐naive while others had received glucocorticoid and/or immunosuppressive therapy in the immediately preceding 3 months.
Table 1.
Background of PV patients and healthy controls
| Factor | PV (n = 18) | CN (n = 14) | P‐value |
|---|---|---|---|
| Gender (Female, n, (%)) | 9 (50%) | 5 (35.7%) | |
| Age (y, mean ± SD) | 45.78 ± 13.45 | 44.57 ± 14.72 | 0.811 |
| BMI (kg/m2) | 22.21 ± 2.26 | 22.46 ± 1.85 | 0.732 |
| Treatment history (untreated, n (%)) | 9 (50%) | ||
| Clinical phenotype, n (%) | |||
| Isolated mucosal | 0 | ||
| Muco‐cutaneous | 10 (55.6%) | ||
| Isolated cutaneous | 8 (44.4%) | ||
3.2. Diversity and structure of gut microbiota in PV patients and healthy controls
A total of 32 faecal samples were collected from patients and healthy controls for sequencing. Using the relative abundance in each sample of 97% identity (ID) OTU, a total of 607 OTUs were analysed. Among these, 418 OTUs were shared by the PV and control groups. PV samples had special 107 OUTs while control samples had 82 notable OTUs. Based on the results, the core bacteria identified in the study were Blautia, Baceroides, Escherichia/Shigella, Lachnospiraceae and Faecalibacterium.
There was no difference between patients and healthy controls with respect to gut microbiota diversity, as assessed by the Shannon diversity index (P = 0.72) and Simpson diversity index (P = 0.87) (Figure S1). When PCoA analysis was applied to assess discrepancies based on OTUs with different relative abundances, samples from healthy controls clustered in an intermediate position in a UniFrac PCoA plot but the pemphigus vulgaris individuals clustered in a disperse position (Figure 1A). UniFrac phylogenetic distance of the microbe composition among subjects was calculated to investigate the different structures of gut microbes between groups. MRPP analysis indicated a significant difference between PV and control samples (P = 0.051). Heat maps also showed different sample composition between the two groups (Figure 1B).
Figure 1.
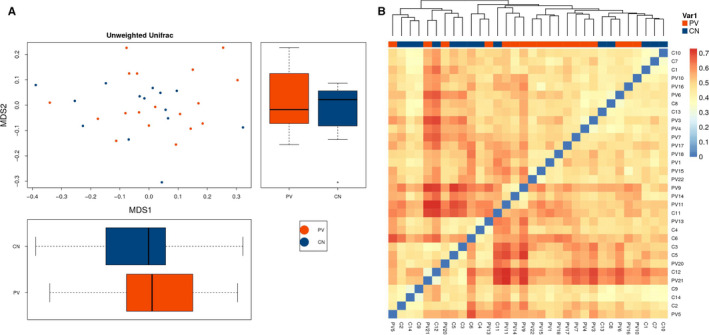
The structure of gut microbiota in PV patients and healthy controls. A, The microbiota of patients with pemphigus vulgaris differed from those of healthy controls based on OTUs with different relative abundances in a UniFrac PCoA plot. B, Heat map was applied to evaluate the similarity of each sample composition
3.3. Bacterial taxa difference between PV patients and healthy controls
LefSe analysis was applied to investigate the significantly different bacterial taxa among groups. We found that 19 different abundant taxa existed between the PV and CN groups, and all of them had a log LDA score >2 (Figure 2A,B). Subsequently, Wilcoxon test and Kruskal‐Wallis test were performed to verify the differential abundance at the phyla, family, class, order and genera levels, respectively. Normally, bacterial taxa associated with P < 0.05 and FDR q value < 0.1 were considered to be significantly different. Although all of the FDR q values were > 0.1, the mean abundance of taxa between the two groups was different (P < 0.05), and all of the 95% CIs of mean abundance difference of these taxa never spaned 0. Finally, only 10 taxa were found to be significantly different between the two groups as assessed by Kruskal‐Wallis test (P < 0.05) (Table 2). At the class level, the PV group had decreased Betaproteobacteria and increased Gammaproteobacteria as compared to the control group. Both family microbes Carnobacteriaceae and Enterobacteriaceae were more abundant in patients who also showed more abundance of Burkholderiales and decreased Enterobacteriales. At the genera level, Lachnospiracea_incertae_sedis and Coprococcus were decreased, while the Granulicatella and Flavonifractor were enriched in the PV group.
Figure 2.
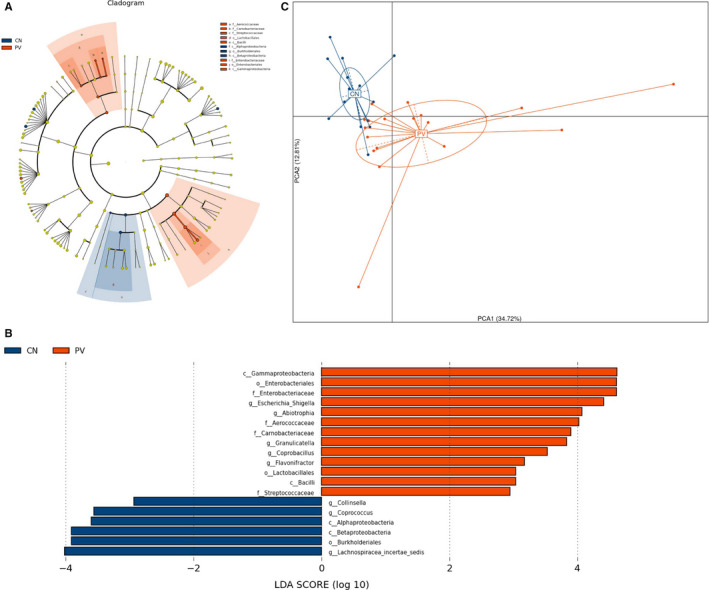
Bacterial taxa difference between PV patients and healthy controls. Different taxa were detected by LefSe (P < 0.05, linear discriminant analysis [LDA] >2 logs). A, Different microbes showed in Cladogram of LefSe analysis. B, Nineteen different microbes with LDA >2 logs between patients (red bars) and controls (blue bars) are displayed. C, Principle component analyses (PcoA) plot of different microbial taxa between groups
Table 2.
Relative abundance of PV and control samples. Only P‐values <0.05 are shown
| Taxon name | MAD (CN‐PV) | Lower (95% CIs) | Upper (95% CIs) | P‐value |
|---|---|---|---|---|
| c__Bacilli | 2.19 × 10−3 | −4.79 × 10−3 | 4.04 × 10−4 | 0.019 |
| c__Betaproteobacteria | 1.65 × 10−2 | 2.78 × 10−3 | 3.08 × 10−2 | 0.028 |
| c__Gammaproteobacteria | −8.78 × 10−2 | −1.61 × 10−1 | −1.45 × 10−2 | 0.034 |
| f__Carnobacteriaceae | −4.61 × 10−5 | −8.50 × 10−5 | −7.11 × 10−6 | 0.041 |
| f__Enterobacteriaceae | −8.57 × 10−2 | −1.57 × 10−1 | −1.48 × 10−2 | 0.012 |
| g__Collinsella | 1.11 × 10−3 | −1.77 × 10−3 | 3.98 × 10−3 | 0.047 |
| g__Coprococcus | 5.47 × 10−3 | −3.52 × 10−4 | −1.13 × 10−3 | 0.015 |
| g__Escherichia/Shigella | −5.00 × 10−2 | −1.01 × 10−1 | 7.70 × 10−4 | 0.002 |
| g__Flavonifractor | −2.38 × 10−3 | −4.45 × 10−3 | −3.11 × 10−4 | 0.012 |
| g__Granulicatella | −4.61 × 10−5 | −8.50 × 10−5 | −7.11 × 10−6 | 0.041 |
| g__Lachnospiracea_incertae_sedis | 2.04 × 10−2 | 1.52 × 10−3 | 4.23 × 10−2 | 0.003 |
| o__Burkholderiales | 1.66 × 10−2 | 2.40 × 10−3 | 3.08 × 10−2 | 0.027 |
| o__Enterobacteriales | −8.57 × 10−2 | −1.57 × 10−1 | −1.48 × 10−2 | 0.012 |
| o__Lactobacillales | −2.17 × 10−3 | −4.77 × 10−3 | 4.23 × 10−4 | 0.017 |
Indeed, according to the PcoA plot of different microbial taxa between groups, we could differentiate patients with PV from healthy controls using the relative abundance of 10 differential taxa at all levels (Figure 2C).
Further, we assessed the microbes in PV patients who had received treatment (PVT) and those in untreated PV patients (PVU). The results showed that with the exception of Granulicatella, the abundance of Lachnospiracea_incertae_sedis, Coprococcus and Flavonifractor genera was not different between the two subgroups (Figure S2).
3.4. Functional analysis
Functional profiling of microbial pathways was inferred from 16S sequences with PICRUSt; we annotated the gene catalogue by KEGG metabolic modules. We found 10 and 35 differently abundant pathways at 2 and 3 levels, which suggested a diverse change in the functions of the microbiota in PN subjects when compared to controls (Figure 3A, Figure S3).
Figure 3.
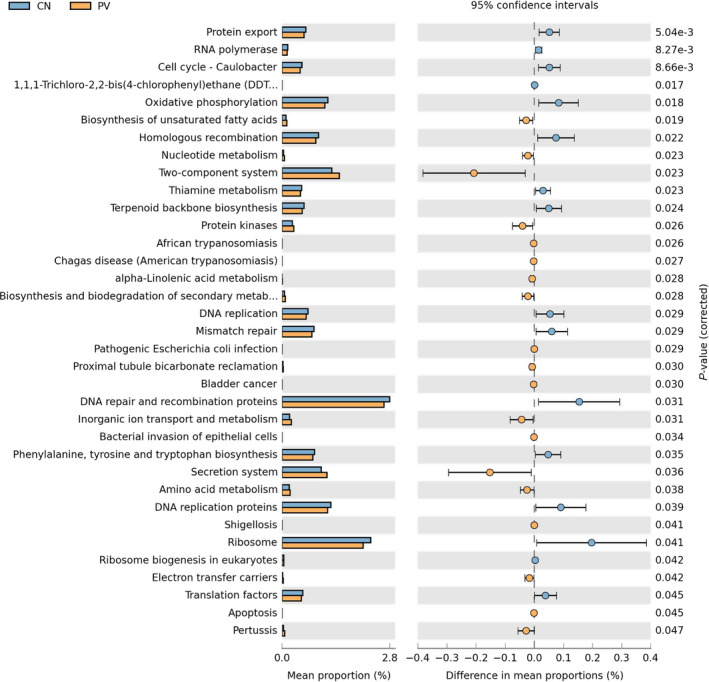
Distribution of Kyoto Encyclopedia of Genes and Genomes (KEGG) functional categories of KEGG Orthologs (KO) markers. Comparison between the healthy people‐enriched and PV patients‐enriched markers on level 3 of KEGG functional category. Abbreviation: LDA, linear discriminant analysis
Indeed, the upregulated pathways in PV subjects were those related to metabolism, signal transduction, excretory and infectious diseases. The downregulated pathways in PV patients were those related to cell growth and death, metabolism, replica and repair, folding, sorting, degradation and translation.
3.5. Plasma concentrations of cytokines in PV patients and healthy controls
Out of the 21 cytokines assessed, the levels of C5a (P = 0.02), YKL‐40 (P = 0.02), IL‐2R (P = 0.01), IL‐8 (P = 0.033), IL‐7 (P = 0.006) and IL‐1 beta (P = 0.038) in PV patients were significantly higher than those in healthy controls (Figure 4A). Additionally, plasma IL‐17A (P = 0.079), IL‐6 (P = 0.66), IL‐5 (P = 0.067) and IL‐21 (P = 0.072) showed an increasing trend in PV patients (Figure S4). Then, we compared the plasma level of cytokines between PVT and PVU groups and found that cytokines’ concentrations were not affected significantly by glucocorticoid and/or immunosuppressive therapy (Figures S5 and S6). In this study, all the patients include were screened for the presence of PV and the disordered immune and concentrations of cytokines were not inhibited effectively by such therapy, which could explain the result.
Figure 4.
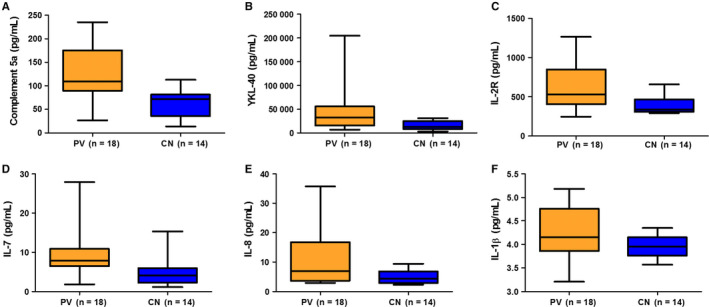
A‐F, Plasma concentrations of cytokines in patients and controls (P < 0.05 for all). The below and above lines indicate the minimum and maximum value. The middle lines represent the median values
3.6. Correlation between plasma cytokines and microbial taxa
We further assessed the correlation between plasma cytokines and the gut microbiota and presented the correlation efficiency through heat map (Figure 5). Most significantly, Flavonifractor at genus level showed a positive correlation with C5a (R = 0.631, P < 0.001), IL‐6 (R = 0.814, P < 0.001), IL‐8 (R = 0.441, P = 0.013), IL‐7 (R = 0.446, P = 0.012), IL‐1β (P = 0.482, P = 0.006) and IL‐21 (R = 0.518, P = 0.003). Lachnospiracea_incertae_sedis (R = −0.322, P = 0.056) and Coprococcus (R = −0.325, P = 0.074) were both negatively associated with IL‐17A Granulicatella and YKL‐40 showed a positive correlation (R = 0.416, P = 0.002).
Figure 5.
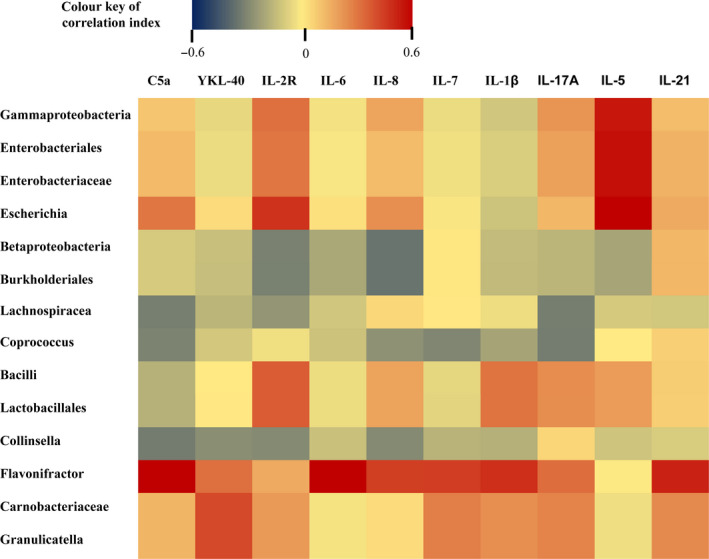
Correlation between plasma cytokines and microbial taxa was assessed by Person correlation test and displayed as a heat map
4. DISCUSSION
Metagenomic studies on gut microbiota have burst onto the scientific scene during the last decade. These studies have unraveled links between some disorders and dysbiosis in the gut microbial ecology. Remarkably, intestinal dysbiosis has also been associated with autoimmune diseases, such as rheumatoid arthritis, Behcets disease, SLE and inflammatory bowel disease (IBD).21, 22, 23, 24 Pemphigus refers to a group of IgG‐mediated autoimmune diseases; of these, a major subtype PV is associated with considerable morbidity and mortality.25 However, the pathogenesis of this disease remains to be fully elucidated. In addition, no studies have investigated the gut microbiota in patients with PV. This is the first study that reports the imbalance of gut microbiota in PV. At the genus level, the declined microbes were Lachnospiracea_incertae_sedis and Coprococcus and the enriched microbes were Granulicatella and Flavonifractor. In a previous study, the increment in Lachnospiracea abundance was reported to be consistent with microbiota changes of inhibited caspase‐1 in association with activation of pro‐inflammatory cytokines such as IL‐1β and IL‐18.26 Absence of Lachnospiracea_incertae_sedis and Coprococcus may cause a decline in short‐chain fatty acid production and is often associated with diseases such as asthma and inflammatory bowel disease.27 Additionally, decline in Coprococcus has also been demonstrated in patients with intestinal, neuropsychological, infectious, atopic and liver diseases.28, 29, 30, 31, 32 Lachnospiracea_incertae_sedis and Coprococcus, both butyrate producing bacteria, were shown to exert an anti‐inflammatory effect by inducing regulatory T cells (Tregs); consequently, these have the ability to modulate the immune system.28 In this study, both Lachnospiracea_incertae_sedis and Coprococcus showed a negative correlation with IL‐17A, which suggested that these two organisms may contribute to the pathogenesis of PV through regulation of T cell differentiation and the associated cytokines.
Flavonifractor has been shown to cleave quercetin, a flavonoid with anti‐oxidant and anti‐inflammatory properties.33, 34 Thus, presence of Flavonifractor may potentially, through cleaving the flavonoid C‐ring and degeneration of quercetin, induce oxidative stress and inflammation in the host.35 This is consistent with the observed positive correlation between Flavonifractor and circulating inflammatory markers (C5a, IL‐6, IL‐8, IL‐7, IL‐1β and IL‐21) in the present study. Indeed, increased Flavonifractor in patients with SLE, a complex autoimmune disease and patients with newly diagnosed bipolar disorder (BD) have been demonstrated.35, 36 Additionally, it was reported that specific Bifidobacterium strains could attenuate liver injury by downregulating the cytokines through modulating Flavonifractor.28 The other enriched gut microbe, Granulicatella, was originally known as a nutritional variant of Streptococcus and commonly reported to cause infective endocarditis.37 It was also shown to exhibit a positive association with severe diarrhoea.38 In this study, Granulicatella was increased in patients treated with systemic corticosteroids compared with treatment‐naive patients, which suggested its association with opportunistic infections caused by immune suppression.
T cells are mainly classified as CD4+ or CD8+ T cells. CD4+ T cells regulate antibody production by interacting with B cells and directly infiltrate the tissues expressing the target antigen, where they modulate inflammation through cytokines and surface molecules. Autoreactive CD4+ T cells have been implicated in anti‐Dsg3 antibody production.25 Although several recent studies have examined T cell and cytokine profiles in pemphigus patients, the results have been largely inconsistent. Overall, Th1 predominant and Th2 suppressant responses have been demonstrated in PV.4 Recent studies have shown that the activation of Th17 pathway may be involved in the pathogenesis of PV.8 In our study, we found a significant increase in the plasma concentrations of IL‐6, IL‐8, IL‐7, IL‐1β, IL17A, IL‐5 and IL‐21 in patients with PV, which is consistent with the results of previous studies. It was reported that Th cell subpopulations differentiate in gut associated lymphoid tissues and require specific gut microbes for their differentiation through the metabolites, short‐chain fatty acids.39, 40 Thus, whether the imbalanced Th1/Th2 or Th17/Tregs differentiationand abnormal cytokines in circulation were induced by changes in Lach‐nospiracea_incertae_sedis, Coprococcus and Flavonifractor in PV patients needs to be further studied.
Our study benefited from the inclusion of 18 well‐characterized patients with current presence of PV and 14 age‐ and gender‐matched healthy individuals. This is the first study that documents dysbiosis of gut microbiota in PV and also tries to explain the association between cellular immune and gut microbiota in this disease. However, some limitations of the study need to be acknowledged. First, the sample size of PV patients was small. However, this is largely attributable to the low incidence of this rare disease (range: 0.5‐50 cases per million population) 28; therefore, it is difficult to prospectively recruit a large cohort of PV patients. Second, half of our patients had started systemic corticosteroid therapy which is liable to affect the composition of gut microbiota. Nonetheless, all the included patients had current disease, which indicates that the treatment had not yet effectively induced immune suppression. Moreover, we also compared the microbes between patients undergoing treatment with systemic corticosteroids and treatment‐naive patients. Except Granulicatella, the abundance of Lachnospiracea_incertae_sedis, Coprococcus, Flavonifractor genus was not different between the two subgroups. This further suggests that the altered Lachnospiracea_incertae_sedis and Coprococcus, Flavonifractor in the PV group were not affected by systemic treatment.
In summary, our study employed a novel and comprehensive approach to investigate the symbiotic relationship between gut microbiota and cytokines in PV. We have identified declined Lachnospiracea_incertae_sedis and Coprococcus, and increased Flavonifractor in PV, and discussed its association with dyshemostasis of cytokines produced by Th1/Th2/Th17 cells. However the potential link between the gut microbiota, cytokines and PV still needs to be clarified. Further studies are required to establish a role of Lachnospiracea_incertae_sedis and Coprococcus, Flavonifractor in PV.
6. CONFLICT OF INTEREST
None.
7. AUTHOR CONTRIBUTIONS
Shuli Huang performed the experiments, collected the data and wrote the paper. Yongqiong Deng conceived and designed the study, analysed the data, interpreted the data and approved the final version of the manuscript. Xia Xiong collected the data, supervised the study and also approved the final version of the manuscript.
Supporting information
5. ACKNOWLEDGMENT
This study was supported by the Natural Science Fund of the Southwest Medical University.
Huang S, Mao J, Zhou L, Xiong X, Deng Y. The imbalance of gut microbiota and its correlation with plasma inflammatory cytokines in pemphigus vulgaris patients. Scand J Immunol. 2019;90:e12799. 10.1111/sji.12799
Contributor Information
Xia Xiong, Email: xiongxia789@126.com.
Yongqiong Deng, Email: dengyongqiong1@126.com.
REFERENCES
- 1. Cholera M, Chainani‐Wu N. Management of pemphigus vulgaris. Adv Ther. 2016;33:910‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kasperkiewicz M, Schmidt E, Zillikens D. Current therapy of the pemphigus group. Clin Dermatol. 2012;30:84‐94. [DOI] [PubMed] [Google Scholar]
- 3. Ljubojevic S, Lipozencic J, Brenner S, Budimcic D. Pemphigus vulgaris:a review of treatment over a 19‐year period. J Eur Acad Dermatol Venereol. 2002;16:599‐603. [DOI] [PubMed] [Google Scholar]
- 4. Giordano CN, Sinha AA. Cytokine networks in pemphigus vulgaris: an integrated viewpoint. Autoimmunity. 2012;45:427‐439. [DOI] [PubMed] [Google Scholar]
- 5. Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013;22:699‐704. [DOI] [PubMed] [Google Scholar]
- 6. Masjedi M, Esmaeil N, Saffaei A, et al. Cytokine indexes in pemphigus vulgaris: perception of its immunpathogenesis and hopes for non‐steroidal treatment. Iran J Pharm Res. 2017;16:1223‐1229. [PMC free article] [PubMed] [Google Scholar]
- 7. Timoteo RP, da Silva MV, Miguel CB, et al. Th1/Th17‐related cytokines and chemokines and their implications in the pathogenesis of pemphigus vulgaris. Mediators Inflamm. 2017;2017:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiang‐dong C, Hong C, et al. Roles of Th17 cells in patients with pemphigus vulgaris. TONGJI UNIVERSITY. 2009;4(106–110):112.** [Google Scholar]
- 9. Feliciani C, Toto P, Wang B, Sauder DN, Amerio P, Tulli A. Urokinase plasminogen activator mRNA is induced by IL‐1alpha and TNF‐alpha in in vitro acantholysis. Exp Dermatol. 2003;12:466‐471. [DOI] [PubMed] [Google Scholar]
- 10. Ramos‐Casals M, Brito‐Zeron P, Kostov B, et al. Google‐driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmun diseases. Autoimmun Rev. 2015;14:670‐679. [DOI] [PubMed] [Google Scholar]
- 11. Johnson BM, Gaudreau MC, Al‐Gadban MM, et al. Impact of dietary deviation on disease progression and gut microbiome composition in lupus‐prone SNF1 mice. Clin Exp Immunol. 2015;181:323‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brusca SB, Abramson SB, Scher JU. Microbiome and mucosal inflammation as extra art‐cular triggers for rheumatoid arthrisis and antoimmunity. Curr Opin Rheumatol. 2014;26:101‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alekseyenko AV, Perez‐Perez GI, De Souza A, et al. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome. 2013;1:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mielcarz DW, Kasper LH. The gut microbiomen multiple sclerosis. Curr Treat Options Neurol. 2015;17:344. [DOI] [PubMed] [Google Scholar]
- 15. Consolandi C, Turroni S, Emmi G, et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun Rev. 2015;14:269‐276. [DOI] [PubMed] [Google Scholar]
- 16. Shamriz O, Mizrahi H, Werbner M, Shoenfeld Y, Avni O, Koren O. Microbiota at the crossroads of autoimmunity. Autoimmun Rev. 2016;15:859‐869. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu J, Kubota T, Takada E, et al. Propionate‐producing bacteria in the intestine may associate with skewed responses of IL10‐producing regulatory T cells in patients with relapsing polychondritis. PLoS ONE. 2018;13:e0203657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Giorgi L, Sorini C, Cosorich I, Ferrarese R, Canducci F, Falcone M. Increased iNKT17 cell frequency in the intestine of non‐obese diabetic mice correlates with high bacterioidales and low clostridiales abundance. Front Immunol. 2018;9:1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miranda PM, De Palma G, Serkis V, et al. High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome. 2018;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hertl M, Jedlickova H, Karpati S, et al. Pemphigus. S2 guideline for diagnosis and treatment–guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405‐414. [DOI] [PubMed] [Google Scholar]
- 21. Edwards CJ, Costenbader KH. Epigenetics and the microbiome: developing areas in the understanding of the aetiology of lupus. Lupus. 2014;23:505‐506. [DOI] [PubMed] [Google Scholar]
- 22. Muñiz Pedrogo DA, Chen J, Hillmann B, et al. An increased abundance of Clostridiaceae characterizes arthritis in inflammatory bowel disease and rheumatoid arthritis: a cross‐sectional study. Inflamm Bowel Dis. 2018;25:902‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huttenhower C, Kostic AD, Xavier RJ. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shimizu J, Kubota T, Takada E, et al. Bifidobacteria abundance‐featured gut microbiota compositional change in patients with Behcet's disease. PLoS ONE. 2016;11:e0153746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasperkiewicz M, Ellebrecht CT, Takahashi H, et al. Masayuki Amagai Pemphigus. Nat Rev Dis Primers. 2017;11:17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong M‐L, Inserra A, Lewis MD, et al. Inflammasome signaling affects anxiety‐ and depressive‐like behavior and gut microbiome composition. Mol Psychiatry. 2016;21:797‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Velasquez‐Manoff M. Gut microbiome: the peacekeepers. Nature. 2015;518:S3‐11. [DOI] [PubMed] [Google Scholar]
- 28. Fang D, Shi D, Lv L, et al. Bifidobacterium pseudocatenulatum LI09 and Bifidobacterium catenulatum LI10 attenuate D‐galactosamine‐induced liver injury by modifying the gut microbiota. Sci Rep. 2017;7:8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen L, Wang W, Zhou R, et al. Characteristics of fecal and mucosa‐associated microbiota in Chinese patients with inflammatory bowel disease. Medicine (Baltimore). 2014;93:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keshavarzian A, Green SJ, Engen PA, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disord. 2015;30:1351‐1360. [DOI] [PubMed] [Google Scholar]
- 31. Kampmann C, Dicksved J, Engstrand L, Rautelin H. Composition of human faecal microbiota in resistance to Campylobacter infection. Clin Microbiol Infect. 2016;22:e61‐68. [DOI] [PubMed] [Google Scholar]
- 32. Nylund L, Nermes M, Isolauri E, Salminen S, de Vos WM, Satokari R. Severity of atopic disease inversely correlates with intestinal microbiota diversity and butyrate‐producing bacteria. Allergy. 2015;70:241‐244. [DOI] [PubMed] [Google Scholar]
- 33. Carlier JP, Bedora‐Faure M, K'Ouas G, et al. Proposal to unify Clostridium orbiscindens Winter et al. 1991 and Eubacterium plautii (Seguin 1928) Hofstad and Aasjord 1982, with description of Flavonifractor plautii gen. nov., comb. nov., and reassignment of Bacteroides capillosus to Pseudoflavonifractor capillosus gen. nov., comb. nov. Int J Syst Evol Microbiol. 2010;60:585‐590. [DOI] [PubMed] [Google Scholar]
- 34. Boots AW, Haenen GR, Bast A. Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol. 2008;585:325‐337. [DOI] [PubMed] [Google Scholar]
- 35. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog. 2016;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coello K, Hansen TH, Sørensen N, et al. Gut microbiota composition in patients with newly diagnosed bipolar disorder and their unaffected first‐degree relatives. Brain Behav Immun. 2018;75:112‐118. [DOI] [PubMed] [Google Scholar]
- 37. Shailaja T, Sathiavathy K, Unni G. Infective endocarditis caused by Granulicatella adiacens. Indian Heart J. 2013;65:447‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pop M, Walker AW, Paulson J, et al. Diarrhea in young children from low‐income countries leads to large‐scale alterations in intestinal microbiota composition. Genome Biol. 2014;15:R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232‐236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials