

Abstract
The uncontrolled activation of endogenous enzymes may introduce both qualitative and quantitative artefacts when lipids are extracted from vegetal matrices. In the present study, a method based on hydrophilic interaction liquid chromatography coupled either to high‐resolution/accuracy Fourier‐transform mass spectrometry (HILIC‐ESI‐FTMS) or to linear ion trap multiple stage mass spectrometry (HILIC‐ESI‐MS n , with n = 2 and 3) with electrospray ionization was developed to unveil one of those artefacts. Specifically, the artificial generation of methyl esters of phosphatidic acids (MPA), catalysed by endogenous phospholipase D (PLD) during lipid extraction from five oleaginous microgreen crops (chia, soy, flax, sunflower and rapeseed), was studied. Phosphatidylcholines (PC) and phosphatidylglycerols (PG) were found to be the most relevant precursors of MPA among glycerophospholipids (GPLs), being involved in a transphosphatidylation process catalysed by PLD and having methanol as a coreactant. The combination of MS2 and MS3 measurements enabled the unambiguous recognition of MPA from their fragmentation pathways, leading to distinguish them from isobaric PA including a further CH2 group on their side chains. PLD was also found to catalyse the hydrolysis of PC and PG to phosphatidic acids (PAs). The described transformations were confirmed by the remarkable decrease of MPA abundance observed when isopropanol, known to inhibit PLD, was tentatively adopted instead of water during the homogenization of microgreens. The unequivocal identification of MPA might be exploited to assess if GPL alterations are actually triggered by endogenous PLD during lipid extractions from specific vegetal tissues.
Keywords: electrospray ionization mass spectrometry, hydrophilic interaction liquid chromatography, microgreens, phosphatidic acid methyl esters, phospholipase D
1. INTRODUCTION
The mass spectrometry‐based profiling of lipids in vegetal matrices is a fundamental aspect of plant lipidomics, contributing to unveil the multiple functions of lipids in plants. Besides their roles as major constituents of biomembranes or as energy reserves, lipids are involved in molecular signalling occurring during plant response to stress conditions, like wounding and temperature changes. 1 , 2 , 3 , 4 , 5 , 6 , 7
As for other branches of lipidomics, the coupling between liquid chromatography and mass spectrometry based on electrospray ionization (LC‐ESI‐MS) has been the most widely used approach also in plant lipidomics studies. 8 , 9 , 10 Among LC techniques, hydrophilic interaction liquid chromatography (HILIC) is known to provide a class‐related separation of amphiphilic lipid molecules, such as glycerolipids (GL), glycerophospholipids (GPLs) and sphingolipids (SL). 11 , 12 This feature, readily confirmed also in the context of plant lipidomics, 13 has been extensively exploited in our laboratory for the systematic characterization of complex lipid mixtures, including those obtained from edible fruits produced from different plants. 14 , 15 , 16 , 17
Recently, our attention was focused on lipids produced by microgreens, that is, immature greens, rich in micronutrients and bioactive compounds, produced from the seeds of vegetables and generally harvested at soil level until the appearance of the first pair of true leaves, when cotyledons are fully expanded and still turgid. 18 , 19 , 20 Specifically, lipids were extracted from microgreens of chia, soy, flax, sunflower and rapeseed, using a modified version of the Bligh and Dyer protocol. 21 The procedure was applied to previously lyophilized microgreens after homogenization into water. However, hints of artefacts in the original GPL profile were obtained during preliminary HILIC‐ESI‐MS analyses of microgreen lipid extracts. They appeared to be related to the action of one of the endogenous phospholipases of vegetal tissues 22 ; specifically, phospholipase D (PLD) 23 , 24 seemed to be involved.
When water is available, the reaction catalysed by PLD leads to the hydrolysis of GPL of other classes into the corresponding phosphatidic acids (PAs). However, if alcohols are also present, PLD may act as a transphosphatidylase, that is, catalyse the generation of PA esters from different GPL. 24 In the presence of methanol, like during the Bligh and Dyer extraction of lipids, PLD may thus catalyse the conversion of GPL into methyl esters of phosphatidic acids (MPA), also known as phosphatidylmethanols. MPA were indicated as extraction artefacts in early plant lipidomics studies based on thin‐layer chromatography (TLC) 25 and subsequently proposed as an indirect marker for PLD activity. 26 However, the extent of the enzymatic reaction, and then of the alteration of the GPL profile, has not been fully explored yet. This is likely due to the challenge posed by the discrimination between MPA and PA having one further methylene moiety along the side chains, that are perfectly isobaric species.
In the present paper, the use of HILIC, coupled either to high‐resolution Fourier‐transform (FTMS) or to multistage mass spectrometry (MS n ) with ESI, for the unequivocal recognition of MPA in plant lipid extracts will be described. Oleaginous microgreens will be considered as the target vegetal matrices and the preferential generation of MPA from phosphatidylcholines (PC) and phosphatidylglycerols (PG), confirmed by PLD‐catalysed transphosphatidylation reactions involving PC and PG lipid standards in the presence of methanol, will be emphasized.
2. MATERIALS AND METHODS
2.1. Chemicals
LC‐MS grade acetonitrile, water, isopropanol and methanol, used for HILIC‐ESI‐MS separations and/or lipid extraction, HPLC grade methanol and chloroform, used for lipid extraction, ammonium acetate (reagent grade), used as mobile phase additive, and PLD (Type IV) from white cabbage, were purchased from Sigma‐Aldrich (Milan, Italy). PG 12:0/12:0 (1,2‐dilauroyl‐sn‐glycero‐3‐phospho‐(1′‐rac‐glycerol)) and PC 16:0/14:0 (1‐palmitoyl‐2‐myristoyl‐sn‐glycero‐3‐phosphocholine) standard lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA).
2.2. Sample preparation
After a preliminary evaluation of the germinating ability, performed in Petri dishes, soy, chia, flax, rapeseed and sunflower seeds were sown, with a density of three seeds/cm2, on a substrate contained into a 150‐cm2 cultivation bowl. At the end of the procedure, the sowing bed was irrigated with nebulized distilled water, and each bowl was covered with a black polyethylene film, to keep internal humidity constant, and transferred into a growth chamber, at 20°C and 80% relative humidity. Total germination was assessed after 3 days; then, the black film was removed, relative humidity was lowered to 60% and neon lights were turned on to activate photosynthesis. Subirrigation with a Hoagland–Arnon nutrient solution 27 was performed by exploiting the holes on the bottom of the cultivation bowl. After 10 days, once the development of seedlings and the appearance of the first true leaves were assessed, harvesting was carried out by cutting the flax microgreens just above the surface of the growing medium. Seedlings were stored for 3 days at a −20°C temperature; then, they were freeze‐dried for 4 days in a ScanVac CoolSafe 55‐9 Pro freeze‐dryer (LaboGene ApS, Lynge, Denmark).
2.3. Lipid extraction
Lipid extraction from microgreens was performed using a slightly modified version of the Bligh and Dyer protocol 21 after homogenizing 0.2 g of lyophilized vegetal tissue in 1 ml of either water or isopropanol, with the latter tentatively used to inhibit endogenous PLD activity. In both cases, the dispersed plant tissue was subjected to manual homogenization at 25°C for 3 min into a Potter‐Elvehjem homogenizer. The resulting homogenate was suspended into 3 ml of a methanol/chloroform 2:1 (v/v) mixture, keep quiescent for 60 min in the dark at 25°C and then centrifuged at 4500 g for 10 min in a glass tube, to separate the solid component. Afterwards, a 1‐ml volume of the supernatant was withdrawn, and 1.25 ml of water and 1.25 ml of chloroform were added. The resulting mixture was vortexed and then centrifuged at 4500 g for 10 min to facilitate phase separation. The bottom organic layer was transferred to a glass vial and blown to dryness under a nitrogen stream. The residue was redissolved in 0.2 ml of methanol/chloroform 2:1 (v/v); then, the headspace of the tube was filled with nitrogen, and it was capped and stored at −20°C until use.
2.4. LC‐MS analysis
Microgreen lipid extracts were analysed using either a Q‐Exactive Hybrid Quadrupole‐Orbitrap mass spectrometer, for high‐resolution/accuracy FTMS acquisitions, or a Velos Pro linear ion trap mass spectrometer, for low‐resolution, collisionally induced dissociation (CID)–MS n analyses, each coupled with an Ultimate 3000 HPLC system (Thermo Fisher, West Palm Beach, CA, USA). In both cases, the chromatographic column effluent was transferred into the heated electrospray ionization (HESI) interface (Thermo Fisher, West Palm Beach, CA, USA) mounted on each mass spectrometer.
An Ascentis Express column (15‐cm length, 2.1 mm internal diameter) packed with core–shell 2.7‐μm silica particles (Supelco, Bellefonte, PA, USA) was adopted for HILIC separations, performed at a 0.3‐ml/min flow, with 5‐μl sample volumes injected. The following multistep binary elution gradient, based on an acetonitrile/water (97:3 v/v) mixture as phase A and a methanol/water (90:10 v/v) mixture as phase B, both containing ammonium acetate 2.5 mM, was adopted for lipid separation: 0–10 min linear increase of B from 2% to 20%; 10–15 min linear increase of B from 20% to 50%; 15–20 min isocratic at 50% B; 20–25 min return to 2% B; 25–30 min column reconditioning at 2% B.
The following values were set for the main parameters of the HESI interface and of the ion optics of the Q‐Exactive spectrometer: sheath gas flow rate, 35 a.u.; auxiliary gas flow rate, 15 a.u.; spray voltage, −2.5 kV (negative polarity)/3.5 kV (positive polarity); capillary temperature, 320°C; S‐lens RF level, 100. The spectrometer was operated at its maximum resolving power (140,000 at m/z 200) and high‐resolution spectra were acquired in a 100–2000 m/z interval. During MS measurements the Orbitrap fill time was set to 200 ms and the automatic gain control (AGC) level was set to 3 × 106. To achieve a ≤5‐ppm mass accuracy, the mass spectrometer was calibrated daily by infusing 5 μl/min of calibrant supplied by the instrument manufacturer before analyses.
The following values were set for the main operating parameters of the Velos Pro HESI source and ion optics: sheath gas flow rate, 35 a.u.; auxiliary gas flow rate, 5 a.u.; spray voltage, −2.5 kV (negative polarity)/3.5 kV (positive polarity); capillary temperature, 350°C; S‐lens RF level, 64.2. During MS experiments, the linear ion trap fill time was set to 10 ms, and the AGC level was set to 3 × 104. Multistage mass spectra (MS2 and MS3) were acquired in negative ion mode using a 1 m/z unit wide window for precursor ion isolation. The normalized collision energy (NCE) was set as 35% for both MS2 and MS3 experiments.
2.5. Generation of MPA from phospholipid standards catalysed by PLD
The generation of MPA from PC 14:0/16:0 and PG 12:0/12:0 standards was performed using white cabbage PLD in a two‐phase reaction system. First, 1 ml of a chloroform solution containing 500 mg/L of PC 14:0/16:0 and PG 12:0/12:0 was prepared. Afterwards, 2 ml of methanol were added, to replicate the proportion of the extracting solvent mixture employed in the first step of the extraction protocol. Finally, 1 ml of a 1‐g/L PLD aqueous solution was prepared, dissolving the enzyme into 0.1 M sodium acetate buffer (pH 5–6) containing calcium chloride 0.1 M, 23 and was gently added to the methanol/chloroform (2:1 v/v) mixture. The resulting reaction mixture was left under magnetic stirring for 24 h at 25°C; then, lipids in the reaction medium were extracted using the previously described protocol, and the resulting chloroform‐rich phase, dissolving extracted lipids, was subjected to solvent evaporation under a nitrogen flux. The dried residue was finally redissolved into 2 ml of chloroform and stored at −20°C in a screw cap glass vial with headspace saturated with nitrogen until HILIC‐ESI‐MS analysis was performed. The sample was diluted (1:50 v/v) in acetonitrile just before the analysis.
3. RESULTS AND DISCUSSION
3.1. GPL profile of microgreens and alterations induced by endogenous PLD
The HILIC‐ESI(−)‐MS total ion current (TIC) chromatogram referred to a lipid extract of chia microgreens, obtained using water during the homogenization step, is reported in Figure 1A. Sulfoquinovosyldiacylglycerols (SQDG), ceramides (Cer), hexosylceramides (HexCer), digalactosyldiacylglycerols (DGDG) and nonesterified fatty acids (NEFA) were eluted early from the HILIC column, due to their greater hydrophobicity. As for GPL, PG, phosphatidylinositols (PI), cardiolipins (CL) phosphatidylethanolamines (PE), PAs and PC were observed, in order of elution.
FIGURE 1.
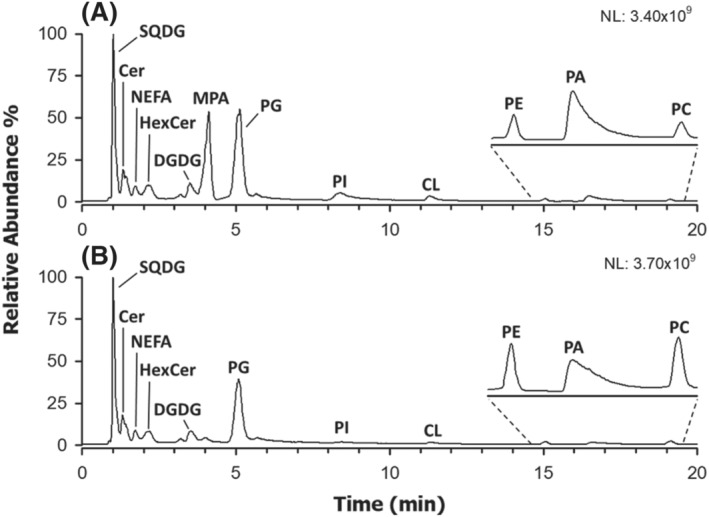
HILIC‐ESI(−)‐FTMS total ion current (TIC) chromatograms referred to lipid extracts of chia microgreens obtained using a slightly modified version of the Bligh and Dyer protocol after vegetal tissue homogenization in (A) water or (B) isopropanol. See the text for lipid classes labelling
Among GPL usually detected more favourably, or exclusively, as negative ions, the peak due to PG was found to prevail over those of PA, PI and CL, suggesting their intrinsically higher abundance in the lipid extracts of chia microgreens. It is worth noting, however, that the ionization yield of PG might be also favoured by their earlier elution, which, in this case, implies the presence in the mobile phase of a higher percentage of acetonitrile, a solvent generally improving the ESI yield. 28 As apparent from Figures 1A and S1A, a further peak was detected around 4 min when the homogenization of chia microgreens was performed in water. In order to unveil the identity of species related to this chromatographic feature, the corresponding ESI(−)‐FTMS spectrum was considered and is reported in Figure 2C. For comparison, spectra referred to the PC and PA classes for the same sample were also reported in Figure 2A,B, respectively. Accurate m/z values retrieved for monoisotopic peaks visible in all spectra of the figure were employed for lipid identification using the ALEX 123 lipid calculator (http://alex123.info/ALEX123/MS.php), setting a tolerance for m/z values of 0.005. As emphasized by peak labels in Figure 2, consistent sum compositions were retrieved for PC and PA, with prevailing numbers of carbon atoms on the acyl chains being 34 and 36, accompanied by a variable number of C═C bonds. When applied to the FTMS spectrum referred to the peak detected at approximately 4 min, the procedure returned systematically matches for PA apparently having an odd total number of carbon atoms on their side chains. However, the retention time was quite different from that of actual PA, eluting between 16 and 18 min (see Figures 1A and S1A).
FIGURE 2.
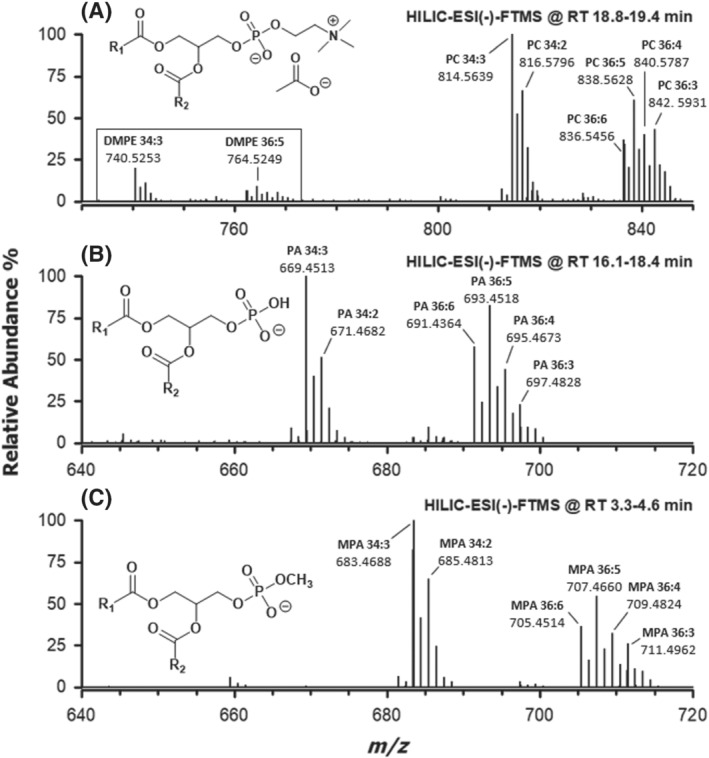
ESI(−)‐FTMS spectra averaged under the HILIC peaks (see retention time intervals) specific for (A) phosphatidylcholines, (B) phosphatidic acids and (C) phosphatidic acid methyl esters, referred to lipid extracts of chia microgreens. Sum compositions of side chains (number of carbon atoms: number of C═C bonds) are indicated for major monoisotopic peaks. The general structures of detected ions are shown in the corresponding panels. DMPE correspond to anions of dimethyl‐phosphatidylethanolamines (see text for details)
To account for the presence of a further carbon atom on the molecular structure, compared with PA recognized in the same sample (see Figure 2B), and for the decrease in retention time with respect to them, the methylation of the polar head of PA, that is, the generation of MPA, was conjectured, as depicted in the general structure reported in Figure 2C. Indeed, the methylation of the phosphate moiety of a PA is expected to decrease polarity and hydrogen bonding capability with respect to a normal PA, with a consequent weakening of the interaction with the silica stationary phase of the chromatographic column. Interestingly, the spectral profile of putative MPA was strikingly similar to that of PA, thus strengthening the hypothesis; moreover, a strong similarity was found also with the spectral profile of PC (see Figure 2). Notably, PC were detected mainly as acetate adducts of their zwitterionic forms (see the structure reported in Figure 2A). However, less abundant additional ions, resulting from acetate adducts upon in‐source detachment of a methyl group from the choline head and loss of the acetate anion (thus corresponding to the anions of dimethyl‐PE, i.e., DMPE), were also detected in the spectrum (see the emphasized region of the spectrum in Figure 2A). The origin of DMPE anions as fragments of acetate adducts of PC was confirmed by MS/MS acquisitions performed on the latter (data not shown).
As discussed before, MPA supposedly recognized in microgreen lipid extracts could be generated by a transphosphatidylation reaction involving GPL and methanol and catalysed by endogenous PLD. 22 , 23 , 24 To find an indirect confirmation of this hypothesis, an alternative lipid extraction procedure was attempted, using chia microgreens as matrix. In particular, isopropanol, a solvent known to inhibit PLD, 29 , 30 was adopted during the homogenization step; then, it was verified if the incidence of putative MPA in the lipid extract was decreased. TIC chromatograms shown in Figures 1B and S1B confirmed the hypothesis, as the peak tentatively assigned to MPA was almost undetectable when isopropanol was adopted for homogenization, even though methanol was subsequently adopted, with chloroform, for lipid extraction. Moreover, while peaks related to SQDG, NEFA, DGDG, Cer and HexCer were substantially unmodified, the PC peak was clearly increased, and the PA one was decreased (see Figures 1 and S1). The same outcome was generally observed when comparing the TIC chromatograms of lipid extracts of soy, flax, rapeseed and sunflower microgreens, obtained using water or isopropanol for homogenization (see Figure S2). The main differences commented so far between lipid extracts resulting from microgreen homogenization in water or isopropanol seemed thus to be related to the PLD activity, rather than to variations in extraction yields for lipid classes. As commented before, PLD has been reported to convert PC in PAs, in the presence of water, and in the corresponding esters, in this case MPA, if alcohols are also available 24 (see Scheme 1). Notably, also, the PE chromatographic peak seemed to be increased when isopropanol was adopted for microgreen homogenization, although the effect was much less evident than that found for PC (see Figures 1 and S1A). As shown in Figure S3, the profile of PE in chia microgreens was comparable with that of putative MPA. Consequently, although this has not been reported so far in the literature, the involvement of PE as precursors of MPA when endogenous PLD is active cannot be excluded.
SCHEME 1.
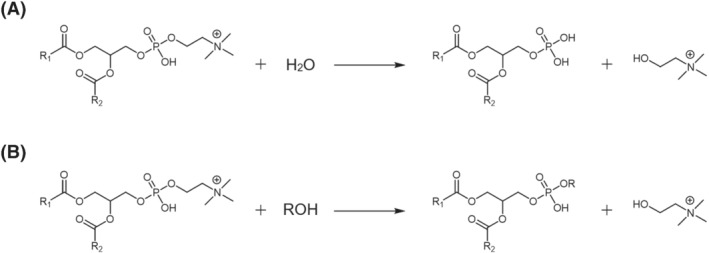
General schemes for (A) hydrolysis and (B) transphosphatidylation reactions of phosphatidylcholines catalysed by phospholipase D. Note that primary alcohols are expected to provide higher reaction yields for reaction B
To obtain a final confirmation of the identity of MPA and a further insight into their relationship with PA and PC, ESI(−)‐MS n (with n = 2 and 3) analyses were performed for these GPL on chia lipid extracts using the VelosPro linear ion trap mass spectrometer.
3.2. ESI(−)‐MS n characterization of MPA in lipid extracts of chia microgreens
The low‐resolution MS2 spectrum referred to the [M‐H]− ion of the most abundant MPA detected in chia microgreen lipid extracts obtained with homogenization in water, having a sum composition 34:3 (m/z 683.5), is reported in Figure 3C. In the figure, the spectrum is compared with those referred to the [M‐CH3]− ion (i.e., deprotonated DMPE) of the corresponding PC (m/z 740.5) and to the [M‐H]− ion of the corresponding PA (m/z 669.5), reported in Figure 3A,B, respectively.
FIGURE 3.
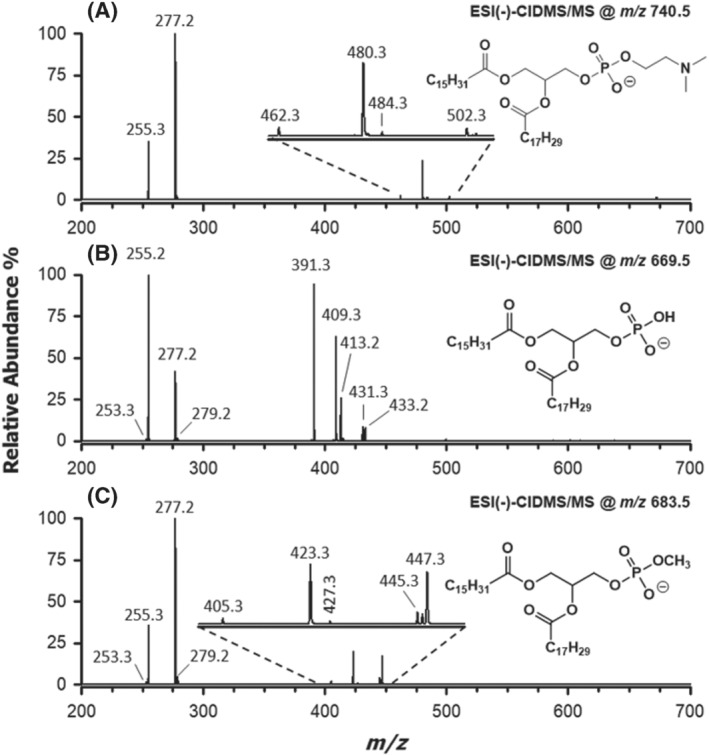
ESI(−)‐MS2 spectra related to the most abundant species (sum composition 34:3) detected for (A) phosphatidylcholines, (B) phosphatidic acids and (C) phosphatidic acid methyl esters in chia microgreen lipid extracts obtained after tissue homogenization in water (see Figure 2). Chemical structures indicated in each panel show the regiochemical attribution retrieved for the most abundant ion corresponding to the observed fragmentations. See text for details
The identity of the acyl chains associated with PC 34:3 was readily clarified by the carboxylate signals at m/z 255.3 (16:0) and 277.2 (18:3). Considering the preferential loss of ketene (m/z 480.3 and 502.3) over fatty acid (m/z 462.3 and 484.3) from the sn‐2 position, 31 , 32 the species PC 16:0/18:3 was assigned (Figure 3A), labelled in accordance with the sn‐1/sn‐2 regiochemical notation proposed by Liebisch et al. 33 The presence of acyl chains 16:0 and 18:3 were confirmed also for the [M‐H]− ion of PA 34:3 (see signals at m/z 255.2 and 277.2 in Figure 3B). Because acyl chains are preferentially lost as fatty acids and from the sn‐2 position of glycerol 34 in the case of PA, a 16:0/18:3 regiochemistry was inferred from signals detected at m/z 391.3 and 413.2. Signals due to ketene losses (m/z 409.3 and 431.3) were also detected for PA 34:3 but were less relevant, as expected. Incidentally, the presence of very week signals for two further carboxylates, having m/z 253.3 and 279.2 (see Figure 3B), suggested the cofragmentation of the [M‐H]− ion of a PA with side chains 16:1 and 18:2, that is, a compositional isomer of PA 16:0/18:3, partially coeluting with the latter from the HILIC column. A further, weak signal, related to the neutral loss of the 16:1 chain as ketene (m/z 433.2), was consistent with this hypothesis. Interestingly, no relevant signal was observed for the loss as ketene of 18:2 chain; a possible explanation for this outcome will be provided later in the paper.
Signals related to carboxylates of side chains 16:0 and 18:3 were easily recognized also in the MS/MS spectrum of the [M‐H]− ion of putative MPA 34:3 (m/z 683.5, see Figure 3C). This finding ruled out the possibility that the additional methylene moiety, compared with the formula of PA 34:3, was located on one of the side chains, thus leading to a PA 35:3. A first MS2‐related clue of esterification by methanol of the phosphatidyl moiety was thus obtained. Interestingly, carboxylates signals were more intense for MPA, in relative terms, than those detected for the corresponding PA. Moreover, signals related to ketene losses of 18:3 and 16:0 chains (m/z 423.3 and 445.3, respectively) were found to prevail over those related to fatty acid losses (m/z 405.3 and 427.3). They were consistent with a 16:0/18:3 regiochemistry if the preferential loss of ketene from the sn‐2 position of glycerol, like for [M‐CH3]− ions of PC, was hypothesized. This was a very interesting finding, because it suggested that the gas‐phase acidity related to the phosphate OH group of [M‐H]− ions of PA was removed by its esterification with methanol, thus making MPA more similar to a PC (or a PE) in terms of fragmentation pathways. Although indirectly, the observed change in the regioselectivity of fragmentation, compared with the PA one, was a further proof of the phosphatidyl group methylation.
Additional weak signals could be detected also in the MS2 spectrum of MPA 34:3. Among them, those at m/z 253.3 and 279.2 corresponded to the already cited carboxylates 16:1 and 18:2, respectively. This result indicated that even in the case of MPA 34:3, a partial coelution occurred between two compositional isomers, exactly as found for the corresponding PA. Moreover, as observed for the PA with chains 16:1 and 18:2, the loss of the 16:1 chain as ketene from the corresponding MPA (see the signal at m/z 447.3 in Figure 3C) was prevailing among neutral losses and was almost comparable with those of the 16:0 and 18:3 chains of the coeluting, more abundant, MPA. In order to clarify this feature, MS3 spectra were acquired on the three product ions arising from ketene losses (m/z 423.2, 445.2 and 447.2) detected in the MS2 spectrum of MPA 34:3. They are shown in Figure 4A–C, respectively. Notably, peak signals at m/z 423.2 and 445.2 correspond to the [M‐H]− ions of the lyso forms of MPA, including the 16:0 and the 18:3 chain, respectively (see Figure 4A,B and Scheme 2). The release of the single acyl chain as carboxylate was, by far, the main fragmentation event, yet weak signals could be detected at m/z values 166.9 and 184.9/185.0 (see insets of Figure 4A,B). As emphasized in Scheme 2, they could be easily interpreted as the result of the neutral loss of the only fatty acyl chain as free fatty acid and ketene, respectively. Interestingly, the signal related to the ketene loss was more intense when the fatty acyl chain was located on the sn‐2 position of glycerol (see Figure 4B). This was consistent with the fact that the ketene loss is a charge‐driven fragmentation (CDF) and that the sn‐2 chain is closer to the phosphate negative charge. On the other hand, when the fatty acyl chain was located on the sn‐1 position (see Figure 4A), a charge remote fragmentation (CRF) prevailed; thus, the fatty acid neutral loss was more likely. Interestingly, fragmentations observed for lyso‐MPA during MS3 analyses were consistent with those proposed by Hsu and Turk for the [M‐H]− ions of lyso‐PE. 32
FIGURE 4.
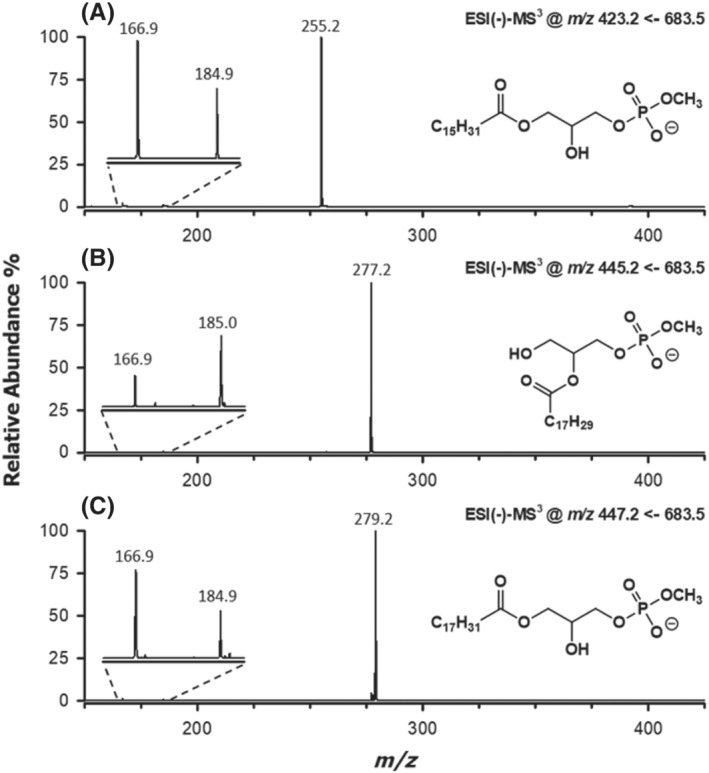
ESI(−)‐MS3 spectra obtained for the product ions generated upon neutral losses as ketene of (A) 18:3, (B) 16:0 and (C) 16:1 fatty acyl residues from the [M‐H]− ions of two partially coeluting isomeric MPA with a 34:3 sum composition (see Figure 3C). A magnification of the spectral region related to the neutral losses of the acyl chain as ketene or carboxylic acid is reported in the insets. See text for details
SCHEME 2.
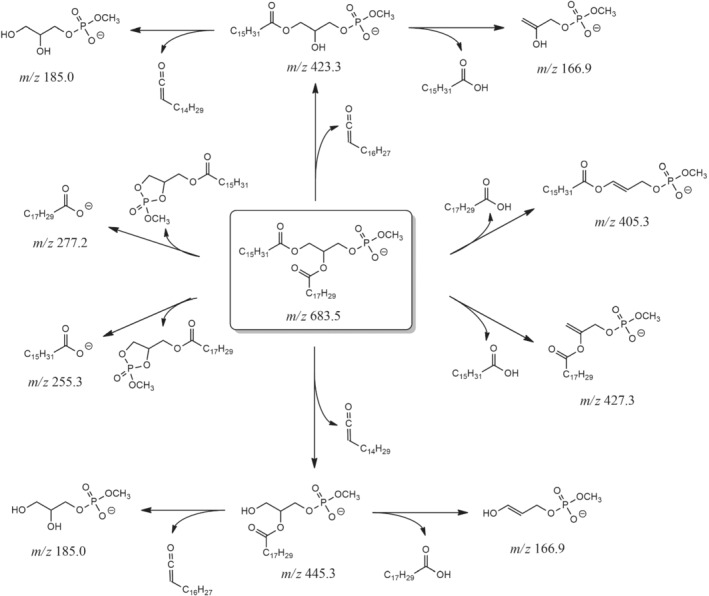
Main fragmentation pathways proposed for the [M‐H]− ion of MPA 16:0/18:3, the most abundant MPA detected in the lipid extract of chia microgreens (see Figure 2). The exact m/z ratio, rounded off to the first decimal figure, is reported for each ion
The presence of a prevailing peak related to the 18:2 carboxylate (m/z 279.2) in the MS3 spectrum referred to the m/z 447.2 ion (Figure 4C) confirmed that the precursor ion arose from a MPA having a 16:1 and a 18:2 chain after the loss of the 16:1 chain as ketene, occurring during the MS2 stage. Because this was the only loss of ketene observed for that MPA (see Figure 3C), the location of the 16:1 chain on the sn‐2 position of glycerol was hypothesized (see the structure reported in Figure 4C). This regiochemistry was nicely confirmed by the MS3 spectrum for the m/z 477.2 ion, because the remaining 18:2 chain was lost preferentially as fatty acid (m/z 166.9 ion), rather than as ketene (m/z 184.9 ion), thus confirming its location on the sn‐1 position of glycerol. Consequently, despite their partial coelution from the HILIC column and the consequent cofragmentation in the linear ion trap, isobaric MPA 16:0/18:3 and 18:2/16:1 could be distinctly recognized, and their respective regiochemistry could be assessed through a careful combination of MS2 and MS3 data. It is finally worth noting that a MS/MS spectrum was acquired for the MPA species with a 34:3 sum composition also using the HCD cell of the Q‐Exactive spectrometer, thus under high collisional energy conditions. In this case, the spectrum was dominated by signals due to side chain carboxylates; however, a weak but distinct signal was detected at a m/z ratio consistent with an exact value 110.9852 (data not shown). The latter correspond to the methyl‐hydrogen phosphate ion, whose detection provided a further, direct proof of the phosphate group methylation in MPA.
Interestingly, while the presence of compositional isomers MPA 16:0/18:3 and 18:2/16:1 was assessed also for PA, as evidenced by the MS/MS spectrum reported in Figure 3B, this was not the case of PC (see Figure 3A). This was surprising, as the latter was expected to be the main precursor of PA and MPA through, respectively, hydrolysis and transphosphatidylation reactions catalysed by PLD. The generation of PA and MPA 18:2/16:1 from a different GPL was thus hypothesized and a PG emerged as their putative source. Indeed, a negative ion corresponding to an accurate m/z 743.4880, compatible with a deprotonated PG with a 34:3 sum composition, was observed in the high‐resolution MS spectrum averaged under the PG peak for chia microgreens (see Figure S4A). The presence of carboxylate ions with m/z 253.3 and 279.3 in the corresponding MS2 spectrum (see Figure S4B) was an unequivocable evidence for the presence of a 16:1 and a 18:2 acyl chain. Notably, the ketene loss of the 16:1 chain (m/z 507.3) was totally prevailing, once again, over that related to the 18:2 chain, for which no signal at all was observed. Moreover, the m/z 415.3 ion was a fragment resulting from the loss of glycerol following that of the 16:1 chain (see Scheme S1A). These results indicated that the 16:1 chain was located on the sn‐2 position of glycerol and that there was a specific reason for the loss of that chain as a ketene to be so favoured.
Interestingly, a PG including a 16:1 chain on the sn‐2 position of glycerol was identified by Hsu et al. in lipid extracts of Arabidopsis thaliana leaves, and the chain was related specifically to trans‐3‐hexadecenoic fatty acid. 35 This carboxylic acid was claimed to be a building block for lipids embedded into the photosynthetic membranes of eukaryotic algae cells. 36 In the case of PG, its synthesis is catalysed by a specific enzyme, a phosphatidylglycerol desaturase 4 (FAD4), operating exclusively on fatty acyl chains linked to the sn‐2 position of glycerol. 36 It is important to point out that hydrogen atoms linked to the carbon atom adjacent to the carbonyl moiety in trans‐3‐hexadecenoic fatty acid (i.e., the C2 atom) are more acidic than those linked to the C2 atom in a totally saturated 16:0 chain. This effect is due to the stabilization of the carbanion resulting from proton abstraction, through resonance involving not just the C1═O bond but also the double bond between C3 and C4. Such a feature was expected to promote remarkably, through a CDF, the neutral loss of the trans‐3‐hexadecenoic fatty acyl chain as a ketene, during MS2 experiments on PG [M‐H]− ions including that chain on the sn‐2 position (see Scheme S1A). The PG 34:3 detected in chia microgreens was thus recognized as a PG 18:2/16:1. Due to the relevance of the loss of the 16:1 chain as ketene, observed previously for minor PA and MPA with a sum composition 36:3 and including that chain (see Figure 3B,C), the same fragmentation mechanism was proposed for them (see Scheme S1B for MPA 18:2/16:1). They were thus assigned as 18:2/16:1 species, generated upon transphosphatidylation of PG 18:2/16:1, and not of the corresponding PC.
A general summary of mass spectrometric and structural information retrieved for MPA, PA, PC, PG and PE detected in the chia microgreen lipid extract has been reported in Table 1. Note that the PE regiochemistry was inferred by using the same fragmentation rules adopted for PC and was consistent with that found for MPA and PA (see Table 1). This outcome strengthened the hypothesis that PE could also be involved in the PLD‐catalysed generation of those species, although at a lower extent compared with PG and PC. The ability of PLD to catalyse the reactions transforming PG and PC into MPA was confirmed using specific PC and PG standards.
TABLE 1.
Mass spectrometric and structural information obtained for major phospholipids belonging to MPA, PA, PC, PG and PE classes detected after HILIC‐ESI(−)‐FTMS analyses of lipid extracts from chia microgreens
| Lipid class | Accurate m/z a | Theoretical m/z | Accuracy (ppm) | Ion formula | Composition (sn‐1/sn‐2) b |
|---|---|---|---|---|---|
| MPA | 683.4688 | 683.4657 | 4.54 | C38H68O8P |
MPA 16:0/18:3 MPA 18:2/16:1 |
| 685.4813 | 685.4814 | −0.05 | C38H70O8P |
MPA 16:0/18:2 MPA 18:2/16:1 |
|
| 705.4514 | 705.4501 | 1.90 | C40H66O8P | MPA 18:3/18:3 | |
| 707.4660 | 707.4657 | 0.31 | C40H68O8P |
MPA 18:2/18:3 MPA 18:3/18:2 |
|
| 709.4824 | 709.4814 | 1.43 | C40H70O8P |
MPA 18:2/18:2 MPA 18:1/18:3 |
|
| 711.4962 | 711.4970 | −1.13 | C40H72O8P |
MPA 18:0/18:3 MPA 18:1/18:2 MPA 18:2/18:1 |
|
| PA | 669.4513 | 669.4501 | 1.83 | C37H66O8P |
PA 16:0/18:3 PA 18:2/16:1 |
| 671.4682 | 671.4657 | 3.65 | C37H68O8P |
PA 16:0/18:2 PA 18:1/16:1 |
|
| 691.4364 | 691.4344 | 2.90 | C39H64O8P | PA 18:3/18:3 | |
| 693.4518 | 693.4501 | 2.47 | C39H66O8P |
PA 18:2/18:3 PA 18:3/18:2 |
|
| 695.4673 | 695.4657 | 2.24 | C39H68O8P |
PA 18:2/18:2 PA 18:1/18:3 |
|
| 697.4828 | 697.4814 | 2.09 | C39H70O8P |
PA 18:0/18:3 PA 18:1/18:2 PA 18:2/18:1 PA 16:0/20:3 |
|
| PC | 814.5639 | 814.5604 | 4.40 | C44H81NO10P | PC 16:0/18:3 |
| 816.5796 | 816.5760 | 4.36 | C44H83NO10P | PC 16:0/18:2 | |
| 836.5456 | 836.5447 | 1.09 | C46H79NO10P | PC 18:3/18:3 | |
| 838.5628 | 838.5604 | 2.95 | C46H81NO10P |
PC 18:2/18:3 PC 18:3/18:2 |
|
| 840.5787 | 840.5760 | 3.18 | C46H83NO10P |
PC 18:2/18:2 PC 18:1/18:3 |
|
| 842.5931 | 842.5917 | 1.71 | C46H85NO10P |
PC 18:0/18:3 PC 18:1/18:2 PC 18:2/18:1 PC 16:0/20:3 |
|
| PG | 719.4898 | 719.4869 | 4.06 | C38H72O10P | PG 16:0/16:1 |
| 741.4733 | 741.4712 | 2.86 | C40H70O10P | PG 18:3/16:1 | |
| 743.4880 | 743.4869 | 1.52 | C40H72O10P |
PG 18:3/16:0 c PG 18:2/16:1 |
|
| 745.5036 | 745.5025 | 1.50 | C40H74O10P |
PG 18:2/16:0 c PG 18:1/16:1 |
|
| PE | 712.4950 | 712.4923 | 3.79 | C39H71NO8P | PE 16:0/18:3 |
| 714.5103 | 714.5079 | 3.37 | C39H73NO8P | PE 16:0/18:2 | |
| 734.4771 | 734.4766 | 0.70 | C41H69NO8P | PE 18:3/18:3 | |
| 736.4941 | 736.4923 | 2.52 | C41H71NO8P |
PE 18:2/18:3 PE 18:3/18:2 |
|
| 738.5099 | 738.5079 | 2.71 | C41H73NO8P |
PE 18:2/18:2 PE 18:1/18:3 |
|
| 740.5245 | 740.5236 | 1.19 | C41H75NO8P |
PE 18:0/18:3 PE 18:1/18:2 PE 18:2/18:1 PE 16:0/20:3 |
m/z ratios and formulas are referred to [M + CH3COO]− adducts for PC and to [M‐H]− ions for MPA, PA, PG and PE, with M representing the zwitterionic (PC) or the neutral (MPA, PA, PG and PE) form.
Regioisomers responsible for the most abundant signals are highlighted in bold character.
The couples of PG species detected at m/z 743.4880 and 745.5036 were well separated by the HILIC column; thus, their MS/MS spectra could be interpreted separately.
3.3. Characterization of MPA generated by PLD‐catalysed transphosphatidylation of PC and PG standards
The artificial generation of MPA from PC and PG during lipid extraction from microgreens in the presence of methanol was reproduced using PC 16:0/14:0 and PG 12:0/12:0 standards as precursors. They were dissolved in a methanol/chloroform 2:1 extraction mixture, to simulate the solvent mixture used during the lipid extraction protocol. Thereafter, an aqueous solution containing a commercial PLD obtained from white cabbage (1 g/L) was added, thus obtaining a two‐phase reaction mixture. In the present case, the amount of the aqueous solution was deliberately increased with respect to the typical lipid extraction protocol, to ensure the existence of a phase separation. This was useful to recreate the natural phase separation existing between the vegetal tissue including PLD and the extracting mixture, when homogenized microgreens were subjected to lipid extraction. As a result of the reaction, MPA 16:0/14:0 and 12:0/12:0 were generated and easily recognized upon analysis by HILIC‐ESI‐(−)‐MS/MS, as confirmed by MS2 spectra reported in Figure 5C,D for their precursor ions, having m/z 633.5 and 549.5, respectively. MS2 spectra obtained for negative ions of GPL from which the two MPA were generated, that is, [M‐CH3]− of PC 16:0/14:0 (m/z 690.5) and [M‐H]− of PG 12:0/12:0 (m/z 609.5), were also reported in Figure 5A,B for a comparative evaluation. The 16:0/14:0 regiochemistry was promptly inferred for the PC, based on the abundance ratio of ions related to the acyl chain loss as ketenes, detected at m/z 452.2 and 480.3. It was subsequently confirmed for the corresponding MPA, based on ions detected at m/z 395.2 and 423.2. Carboxylate ions for the 16:0 (m/z 255.1) and the 14:0 (m/z 227.1) chains and weak signals related to their loss as carboxylic acids (m/z 434.2 and 462.2 for PC, 377.1 and 405.1 for MPA) completed the set of product ions detected for PC and MPA with a side chain composition 16:0/14:0. As for PG and MPA with composition 12:0/12:0, apart from the release of the 12:0 carboxylate (m/z 199.1), that was the prevailing fragmentation pathway, the neutral loss of the 12:0 chain as ketene (m/z 427.1 and 367.1, respectively) was more relevant than that of its fatty acid (m/z 409.1 and 349.0, respectively). The m/z 409.1 ion seemed particularly abundant, yet this outcome was likely influenced by the loss of water from the glycerol moiety, following the loss of ketene, in the case of PG. Moreover, a signal related to the loss of the entire glycerol moiety, after the ketene loss, was observed (m/z 335.1).
FIGURE 5.
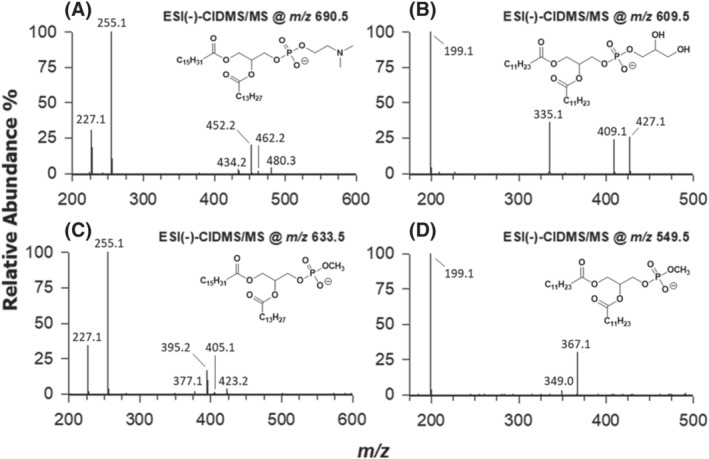
ESI(−)‐MS2 spectra of (A) PC 16:0/14:0 and (B) PG 12:0/12:0 standards, used as substrates for the simulation of PLD activity during lipid extraction, and of the corresponding transphosphatidylation reaction products: (C) MPA 16:0/14:0 and (D) MPA 12:0/12:0. See text for details
The experiment described in this section was thus able to confirm that, if not inhibited appropriately, endogenous PLD can catalyse the artificial generation of MPA from PC and PG during lipid extraction of vegetal tissues when methanol is adopted as a cosolvent with chloroform. As discussed before, the use of isopropanol at room temperature for vegetal tissue homogenization, before proceeding with lipid extraction involving water, methanol and chloroform, seemed to hinder the process without affecting significantly the extraction yield of lipids from different classes. Nonetheless, further approaches, like setting a low temperature during homogenization in water, or even using the three classical solvents of the Bligh and Dyer extraction already in the homogenization step, might result in a similar outcome. The MS‐based approach described in the present paper for the unequivocal recognition of MPA would enable a direct control of the transphosphatidylation extent in each case.
4. CONCLUSIONS
The use of HILIC coupled synergically to high‐resolution MS and to low‐resolution MS2/MS3 enabled the unambiguous identification of MPA generated artificially during methanol/chloroform‐based lipid extraction from five oleaginous microgreen crops (chia, soy, flax, rapeseed and sunflower), due to the activation of endogenous PLD during vegetal tissue homogenization in water. The strict similarity between the spectral profiles of MPA and of PAs and the one of PC, indicated PC to be the main precursors of MPA and also, through hydrolysis catalysed by PLD, of part of PA detected in microgreen lipid extracts. PE were also recognized as possible minor precursors of MPA and PA. A careful insight into the structures of the detected MPA, based on MS2 and MS3 analyses, suggested that also PG were possible precursors of PA and MPA, especially those including a 16:1 side chain. The generation of MPA was confirmed by a transphosphatidylation reaction involving standards of PC and PG and a commercial PLD.
The results reported in this paper suggest that, once recognized unequivocally by HILIC‐ESI‐MS, MPA can be used reliably to assess the presence of alterations in the original profile of GPLs induced by reactions catalysed by endogenous PLD during lipid extraction from vegetal sources. Their detection in lipid extracts can thus help in deciding which homogenization/extraction approach is more appropriate, according to the vegetal matrix, to minimize those alterations.
Supporting information
Figure S1. HILIC‐ESI(+)‐FTMS total ion current (TIC) chromatograms referred to lipid extracts of chia microgreens obtained using the Bligh & Dyer protocol after plant tissue homogenization in water (A) and isopropanol (B). Peak labels indicate the main lipid classes detected in ESI positive ion mode, namely hexosylceramides (HexCer), digalactosyldiacylglycerols (DGDG), phosphatidic acid methyl esters (MPA), phosphatidylglycerols (PG), phosphatidylethanolamines (PE), phosphatidic acids (PA) and phosphatidylcholines (PC). N.L. represents the Normalization Level of MS responses.
Figure S2. HILIC‐ESI(−)‐FTMS total ion current (TIC) chromatograms referred to lipid extracts of four oilseed microgreen crops, namely soy (A), flax (B), rapeseed (C) and sunflower (D), obtained using the Bligh & Dyer protocol after plant tissue homogenization in water. Line colour indicates the solvent in which lyophilized seedlings were homogenized, i.e., water (dark orange) and isopropanol (green). Each peak is labelled with the abbreviation of the corresponding lipid class: sulfoquinovosyldiacylglycerols (SQDG), non‐esterified fatty acids (NEFA), hexosylceramides (HexCer), digalactosyldiacylglycerols (DGDG), phosphatidic acid methyl esters (MPA), phosphatidylglycerols (PG), lyso‐N‐acylphosphatidylethanolamines (NAPE), phosphatidylinositols (PI), cardiolipins (CL), phosphatidylethanolamines (PE), phosphatidic acids (PA) and phosphatidylcholines (PC). MPA peak labels were highlighted in red to stress their remarkable reduction when lyophilized seedlings were homogenized in isopropanol. N.L. represents the Normalization Level of MS responses.
Figure S3. ESI(−)‐FTMS spectrum averaged under the HILIC peak (see retention time interval) specific for phosphatidylethanolamines (PE) detected during the analysis of the lipid extract of chia microgreens.
Figure S4. A) ESI(−)‐FTMS spectrum averaged under the HILIC band related to phosphatidylglycerols (PG) detected in the lipid extract of chia microgreens obtained using the Bligh & Dyer protocol from seedlings homogenized in aqueous medium. The general structure of PG [M‐H]− ions is also shown. B) ESI(−)‐MS/MS spectrum referred to the [M‐H]− ion of PG 18:2/16:1. The predominance of the signal related to the neutral loss as ketene of the 16:1 fatty acyl moiety (m/z 507.3) is typical of PG carrying a trans‐3‐hexadecenoic residue esterified in the sn‐2 position of glycerol (see the main text for details). The supposed fragmentation mechanisms responsible for m/z 507.3 and m/z 415.3 ions formation are shown in Scheme S1. In addition, the chemical structure of the considered PG 18:2/16:1 [M‐H]− ion is reported.
Scheme S1. Fragmentation mechanisms hypothesised for the neutral loss as ketene of the trans‐3‐hexadecenoic acyl chain located on the sn‐2 position of the glycerol backbone of PG 18:2/16:1 (A) and MPA 18:2/16:1 (B) [M‐H]− ions. For the former ion, the neutral loss of glycerol following that of ketene, leading to the m/z 415.2 product ion, is also shown.
ACKNOWLEDGEMENT
Financial support for the present study was received from the following project: PONa3_00395/1 “Bioscienze & Salute (B&H),” funded by the Italian Ministero per l'Istruzione, l'Università e la Ricerca (MIUR). Open Access Funding provided by Universita degli Studi di Bari Aldo Moro within the CRUI‐CARE Agreement. [Correction added on 19 May 2022, after first online publication: CRUI funding statement has been added.]
Castellaneta A, Losito I, Losacco V, et al. HILIC‐ESI‐MS analysis of phosphatidic acid methyl esters artificially generated during lipid extraction from microgreen crops. J Mass Spectrom. 2021;56(10):e4784. 10.1002/jms.4784
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Welti R, Li W, Li M, et al. Profiling membrane lipids in plant stress responses. Role of phospholipase Dα in freezing‐induced lipid changes in Arabidopsis. J Biol Chem. 2002;277(35):31994‐32002. 10.1074/jbc.M205375200 [DOI] [PubMed] [Google Scholar]
- 2. Welti R, Shah J, Li W, et al. Plant lipidomics: discerning biological function by profiling plant complex lipids using mass spectrometry. Front Biosci. 2007;12(1):2494‐2506. 10.2741/2250 [DOI] [PubMed] [Google Scholar]
- 3. Horn PJ, Chapman KD. Lipidomics in tissues, cells and subcellular compartments. Plant J. 2012;70(1):69‐80. 10.1111/j.1365-313X.2011.04868.x [DOI] [PubMed] [Google Scholar]
- 4. Shulaev V, Chapman KD. Plant lipidomics at the crossroads: from technology to biology driven science. Bioch Biophys Acta ‐ Mol Cell Biol Lipids. 2017;1862(8):786‐791. 10.1016/j.bbalip.2017.02.011 [DOI] [PubMed] [Google Scholar]
- 5. Rupasinghe TWT, Roessner U. Extraction of plant lipids for LC‐MS‐based untargeted plant lipidomics. Methods Mol Biol. 2018;1778:125‐135. 10.1007/978-1-4939-7819-9_9 [DOI] [PubMed] [Google Scholar]
- 6. Hou Q, Ufer G, Bartels D. Lipid signalling in plant responses to abiotic stress. Plant Cell Environ. 2016;39(5):1029‐1048. 10.1111/pce.12666 [DOI] [PubMed] [Google Scholar]
- 7. Buseman CM, Tamura P, Sparks AA, et al. Wounding stimulates the accumulation of glycerolipids containing oxophytodienoic acid and dinor‐oxophytodienoic acid in arabidopsis leaves. Plant Physiol. 2006;142(1):28‐39. 10.1104/pp.106.082115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hummel J, Segu S, Li Y, Irgang S, Jueppner J, Giavalisco P. Ultra performance liquid chromatography and high resolution mass spectrometry for the analysis of plant lipids. Front Plant Sci. 2011;2:1‐17. 10.3389/fpls.2011.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okazaki Y, Saito K. Plant lipidomics using UPLC‐QTOF‐MS. Methods Mol Biol. 2018;1778:157‐169. 10.1007/978-1-4939-7819-9_11 [DOI] [PubMed] [Google Scholar]
- 10. Herrfurth C, Liu YT, Feussner I. Targeted analysis of the plant lipidome by UPLC‐NanoESI‐MS/MS. In: Bartels D, Dormann P, eds. Plant Lipids. Methods in Molecular Biology. Vol.2295. Humana; 2021:135‐155. [DOI] [PubMed] [Google Scholar]
- 11. Zhu C, Dane A, Spijksma G, et al. An efficient hydrophilic interaction liquid chromatography separation of 7 phospholipid classes based on a diol column. J Chromatogr A. 2012;1220:26‐34. 10.1016/j.chroma.2011.11.034 [DOI] [PubMed] [Google Scholar]
- 12. Schwalbe‐Herrmann M, Willmann J, Leibfritz D. Separation of phospholipid classes by hydrophilic interaction chromatography detected by electrospray ionization mass spectrometry. J Chromatogr A. 2010;1217(32):5179‐5183. 10.1016/j.chroma.2010.05.014 [DOI] [PubMed] [Google Scholar]
- 13. Okazaki Y, Kamide Y, Hirai MY, Saito K. Plant lipidomics based on hydrophilic interaction chromatography coupled to ion trap time‐of‐flight mass spectrometry. Metabolomics. 2013;9(S1):121‐131. 10.1007/s11306-011-0318-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Losito I, Patruno R, Conte E, Cataldi TRI, Megli FM, Palmisano F. Phospholipidomics of human blood microparticles. Anal Chem. 2013;85(13):6405‐6413. 10.1021/ac400829r [DOI] [PubMed] [Google Scholar]
- 15. Granafei S, Losito I, Palmisano F, Cataldi TRI. Identification of isobaric lyso‐phosphatidylcholines in lipid extracts of gilthead sea bream (Sparus aurata) fillets by hydrophilic interaction liquid chromatography coupled to high‐resolution Fourier‐transform mass spectrometry. Anal Bioanal Chem. 2015;407(21):6391‐6404. 10.1007/s00216-015-8671-9 [DOI] [PubMed] [Google Scholar]
- 16. Bianco M, Calvano CD, Huseynli L, Ventura G, Losito I, Cataldi TRI. Identification and quantification of phospholipids in strawberry seeds and pulp (Fragaria × ananassa cv San Andreas) by liquid chromatography with electrospray ionization and tandem mass spectrometry. J Mass Spectrom. 2020;55:e4523. 10.1002/jms.4523 [DOI] [PubMed] [Google Scholar]
- 17. Calvano CD, Bianco M, Ventura G, Losito I, Palmisano F, Cataldi TRI. Analysis of phospholipids, lysophospholipids, and their linked fatty acyl chains in yellow lupin seeds (Lupinus luteus L.) by liquid chromatography and tandem mass spectrometry. Molecules. 2020;25:805. 10.3390/molecules25040805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kyriacou MC, Rouphael Y, Di Gioia F, et al. Micro‐scale vegetable production and the rise of microgreens. Trends Food Sci Technol. 2016;57:103‐115. 10.1016/j.tifs.2016.09.005 [DOI] [Google Scholar]
- 19. Paradiso VM, Castellino M, Renna M, et al. Nutritional characterization and shelf‐life of packaged microgreens. Food Funct. 2018;9(11):5629‐5640. 10.1039/c8fo01182f [DOI] [PubMed] [Google Scholar]
- 20. Kyriacou MC, De Pascale S, Kyratzis A, Rouphael Y. Microgreens as a component of space life support systems: a cornucopia of functional food. Front Plant Sci. 2017;8:8‐11. 10.3389/fpls.2017.01587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(1):911‐917. 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- 22. Dennis EA. Introduction to thematic review series: phospholipases: central role in lipid signaling and disease. J Lipid Res. 2015;56(7):1245‐1247. 10.1194/jlr.E061101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allegretti C, Denuccio F, Rossato L, Arrigo PD. Polar head modified phospholipids by phospholipase D‐catalyzed transformations of natural phosphatidylcholine for targeted applications: an overview. Catalysts. 2020;10:997–1015. 10.3390/catal10090997 [DOI] [Google Scholar]
- 24. Yang SF, Freer S, Benson AA. Transphosphatidylation by phospholipase D. J Biol Chem. 1967;242(3):477‐484. [PubMed] [Google Scholar]
- 25. Quarles RH, Dawson RMC. The distribution of phospholipase D in developing and mature plants. Biochem J. 1969;112(5):787‐794. 10.1042/bj1120787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de la Roche IA, Andrews CJ, Kates M. Changes in phospholipid composition of a winter wheat cultivar during germination at 2 C and 24 C. Plant Physiol. 1973;51(3):468‐473. 10.1104/pp.51.3.468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoagland DR, Arnon DI. The water‐culture method for growing plants without soil. Circ Calif Agric Exp Stn. 1950;347:1‐39. [Google Scholar]
- 28. Kostiainen R, Bruins AP. Effect of solvent on dynamic range and sensitivity in pneumatically‐assisted electrospray (ion spray) mass spectrometry. Rapid Commun Mass Spectrom. 1996;10(11):1393‐1399. [DOI] [Google Scholar]
- 29. Nichols BW. Separation of the lipids of photosynthetic tissues: Improvements in analysis by thin‐layer chromatography. Biochim Biophys Acta ‐ Spec Sect Lipids Relat Subj. 1963;70:417‐422. 10.1016/0926-6542(63)90060-X [DOI] [PubMed] [Google Scholar]
- 30. Shiva S, Enninful R, Roth MR, Tamura P, Jagadish K, Welti R. An efficient modified method for plant leaf lipid extraction results in improved recovery of phosphatidic acid. Plant Methods. 2018;14(1):1‐8. 10.1186/s13007-018-0282-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu FF, Turk J. Electrospray ionization with low‐energy collisionally activated dissociation tandem mass spectrometry of glycerophospholipids: mechanisms of fragmentation and structural characterization. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(26):2673‐2695. 10.1016/j.jchromb.2009.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsu FF, Turk J. Charge‐remote and charge‐driven fragmentation processes in diacyl glycerophosphoethanolamine upon low‐energy collisional activation: a mechanistic proposal. J Am Soc Mass Spectrom. 2000;11(10):892‐899. 10.1016/S1044-0305(00)00159-8 [DOI] [PubMed] [Google Scholar]
- 33. Liebisch G, Vizcaíno JA, Köfeler H, et al. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54(6):1523‐1530. 10.1194/jlr.M033506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsu FF, Turk J. Charge‐driven fragmentation processes in diacyl glycerophosphatidic acids upon low‐energy collisional activation. A mechanistic proposal. J Am Soc Mass Spectrom. 2000;11(9):797‐803. 10.1016/S1044-0305(00)00151-3 [DOI] [PubMed] [Google Scholar]
- 35. Hsu FF, Turk J, Williams TD, Welti R. Electrospray ionization multiple stage quadrupole ion‐trap and tandem quadrupole mass spectrometric studies on phosphatidylglycerol from Arabidopsis leaves. J Am Soc Mass Spectrom. 2007;18(4):783‐790. 10.1016/j.jasms.2006.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubertret G, Mirshahi A, Mirshahi M, Gerard‐Hirne C, Tremolieres A. Evidence from in vivo manipulations of lipid composition in mutants that the Δ3‐trans‐hexadecenoic acid‐containing phosphatidylglycerol is involved in the biogenesis of the light‐harvesting chlorophyll a/b‐protein complex of Chlamydomonas reinhardtii . Eur J Biochem. 1994;226(2):473‐482. 10.1111/j.1432-1033.1994.tb20072.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. HILIC‐ESI(+)‐FTMS total ion current (TIC) chromatograms referred to lipid extracts of chia microgreens obtained using the Bligh & Dyer protocol after plant tissue homogenization in water (A) and isopropanol (B). Peak labels indicate the main lipid classes detected in ESI positive ion mode, namely hexosylceramides (HexCer), digalactosyldiacylglycerols (DGDG), phosphatidic acid methyl esters (MPA), phosphatidylglycerols (PG), phosphatidylethanolamines (PE), phosphatidic acids (PA) and phosphatidylcholines (PC). N.L. represents the Normalization Level of MS responses.
Figure S2. HILIC‐ESI(−)‐FTMS total ion current (TIC) chromatograms referred to lipid extracts of four oilseed microgreen crops, namely soy (A), flax (B), rapeseed (C) and sunflower (D), obtained using the Bligh & Dyer protocol after plant tissue homogenization in water. Line colour indicates the solvent in which lyophilized seedlings were homogenized, i.e., water (dark orange) and isopropanol (green). Each peak is labelled with the abbreviation of the corresponding lipid class: sulfoquinovosyldiacylglycerols (SQDG), non‐esterified fatty acids (NEFA), hexosylceramides (HexCer), digalactosyldiacylglycerols (DGDG), phosphatidic acid methyl esters (MPA), phosphatidylglycerols (PG), lyso‐N‐acylphosphatidylethanolamines (NAPE), phosphatidylinositols (PI), cardiolipins (CL), phosphatidylethanolamines (PE), phosphatidic acids (PA) and phosphatidylcholines (PC). MPA peak labels were highlighted in red to stress their remarkable reduction when lyophilized seedlings were homogenized in isopropanol. N.L. represents the Normalization Level of MS responses.
Figure S3. ESI(−)‐FTMS spectrum averaged under the HILIC peak (see retention time interval) specific for phosphatidylethanolamines (PE) detected during the analysis of the lipid extract of chia microgreens.
Figure S4. A) ESI(−)‐FTMS spectrum averaged under the HILIC band related to phosphatidylglycerols (PG) detected in the lipid extract of chia microgreens obtained using the Bligh & Dyer protocol from seedlings homogenized in aqueous medium. The general structure of PG [M‐H]− ions is also shown. B) ESI(−)‐MS/MS spectrum referred to the [M‐H]− ion of PG 18:2/16:1. The predominance of the signal related to the neutral loss as ketene of the 16:1 fatty acyl moiety (m/z 507.3) is typical of PG carrying a trans‐3‐hexadecenoic residue esterified in the sn‐2 position of glycerol (see the main text for details). The supposed fragmentation mechanisms responsible for m/z 507.3 and m/z 415.3 ions formation are shown in Scheme S1. In addition, the chemical structure of the considered PG 18:2/16:1 [M‐H]− ion is reported.
Scheme S1. Fragmentation mechanisms hypothesised for the neutral loss as ketene of the trans‐3‐hexadecenoic acyl chain located on the sn‐2 position of the glycerol backbone of PG 18:2/16:1 (A) and MPA 18:2/16:1 (B) [M‐H]− ions. For the former ion, the neutral loss of glycerol following that of ketene, leading to the m/z 415.2 product ion, is also shown.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.