

Abstract
Scope: Garlic is a source of bioactive phytonutrients that may have anti‐inflammatory or immunomodulatory properties. The mechanism(s) underlying the bioactivity of these compounds and their ability to regulate responses to enteric infections remains unclear.
Methods and Results: This study investigates if a garlic‐derived preparation (PTSO‐PTS) containing two organosulfur metabolites, propyl‐propane thiosulfonate (PTSO), and propyl‐propane thiosulfinate (PTS), regulate inflammatory responses in murine macrophages and intestinal epithelial cells (IEC) in vitro, as well as in a model of enteric parasite‐induced inflammation. PTSO‐PTS decreases lipopolysaccharide‐induced secretion of TNFα, IL‐6, and IL‐27 in macrophages. RNA‐sequencing demonstrates that PTSO‐PTS strongly suppresses pathways related to immune and inflammatory signaling. PTSO‐PTS induces the expression of a number of genes involved in antioxidant responses in IEC during exposure to antigens from the parasite Trichuris muris. In vivo, PTSO‐PTS does not affect T. muris establishment or intestinal T‐cell responses but significantly alters cecal transcriptomic responses. Notably, a reduction in T. muris‐induced expression of Tnf, Saa2, and Nos2 is observed.
Conclusion: Garlic‐derived organosulfur compounds exert anti‐inflammatory effects in macrophages and IEC, and regulate gene expression during intestinal infection. These compounds and related organic molecules may thus hold potential as functional food components to improve gut health in humans and animals.
Keywords: garlic, gut health, immune, inflammation, parasite infection
Garlic may contain several compounds with health‐promoting properties. Here, we examined if an extract containing two specific organosulfur compounds regulated inflammatory responses in macrophages, intestinal epithelial cells, and in the gut of mice during chronic infection with an enteric parasite. We show that these garlic‐derived compounds have anti‐inflammatory activity and hold promise as novel functional food components to promote gut health.
1. Introduction
The role of dietary components in gut health and immune function has attracted considerable attention in recent years. Plant‐derived compounds, such as polyphenols, inulin, and allicin, have been extensively studied for their potential roles in preventing inflammation and infectious diseases.[ 1 , 2 ] Moreover, the use of these phytonutrients in animal production has demonstrated several protective effects against numerous disorders by regulating the immune system or improving other properties.[ 3 ] Among these, garlic (Allium sativum), which has been used as a form of traditional medicine and food additive, has attracted increasing interest due to its antioxidant, antimicrobial, and antifungal activities.[ 4 ]
Garlic extracts or garlic‐derived compounds can induce immunomodulatory effects by regulating the secretion of inflammatory cytokines such as interleukin (IL)‐6, TNFα, and IL‐1β, indicating the capacity to affect immune homeostasis.[ 5 ] Consistent with this, garlic consumption can activate γδ‐T and natural killer cells, resulting in fewer cold symptoms and reduced flu severity, suggesting stronger immune function and lower inflammation in vivo.[ 6 ] The balance between T helper 1 and 2 (Th1 and Th2) responses, which are important for the adaptive immune system in terms of cytokine release and disease resistance, can be modulated in favor of a Th2 polarized response in phytohaemagglutinin‐stimulated spleen lymphocytes treated with garlic supplement.[ 7 ] The distinctive properties of garlic are related to active compounds, mainly including sulfur compounds, like allicin also called diallyl thiosulfinate, and non‐sulfur compounds such as phenols, flavonoids, and saponins.[ 8 , 9 ] Most studies have focused on sulfur‐containing compounds, to which the flavorful and medicinal properties of garlic are attributed. For example, propyl‐propane thiosulfonate (PTSO) and propyl‐propane thiosulfinate (PTS), garlic derived organosulfur compounds, are used as biopackaging materials for food due to their antimicrobial activity leading to enhanced food safety and longer shelf life.[ 10 ] Extracts containing PTSO and PTS have also been reported to affect intestinal mucosal thickness, and ileal villus morphology, which may be beneficial during Salmonella spp and Escherichia coli infection in broiler chickens.[ 11 ] Additionally, evidence from mouse studies with PTSO‐PTS (80% PTSO, 20% PTS) indicates PTSO‐PTS may directly interact with gastrointestinal cells to regulate mucosal immune responses by upregulating Reg3g, Ifng, and Il33 expression.[ 12 ] Furthermore, intestinal inflammatory responses induced by 2,4‐dinitrobenzene sulfonic acid (DNBS) and dextran sodium sulfate (DSS) can be ameliorated with PTSO, which is likely related to the regulation of intestinal epithelial barrier integrity as well as the modulation of intestinal microbiota.[ 13 ] The protective activities of PTSO‐PTS against inflammation suggests that PTSO‐PTS may be used as a natural therapy against infectious diseases but further investigations are still needed to understand the underlying mechanisms.
Intestinal pathogens such as parasites may cause dysbiosis, chronic inflammation, and altered gut function and nutrient metabolism.[ 14 ] The murine whipworm Trichuris muris is a natural gastrointestinal parasite of mice, and is a valuable experimental model for the closely related human parasite Trichuris trichiura.[ 15 ] Low doses of T. muris eggs in susceptible hosts induce a chronic infection characterized by a Th1 polarized cellular response, with increased expression of inflammatory cytokines such as interferon‐gamma (IFNу) and IL‐27. Thus, this model is a useful experimental system to study how dietary components may regulate intestinal immune function and inflammatory diseases in humans and animals.[ 16 ]
We hypothesized that PTSO‐PTS can exert protective effects on intestinal inflammation during chronic enteric parasite infection, with the potential effects related to immune cell or intestinal epithelial cell modulation. We show that PTSO‐PTS decreases cytokine release from murine macrophages, and regulates gene expression in macrophages and intestinal cells with or without either lipopolysaccharide (LPS)‐stimulation or activation with T. muris antigens. Furthermore, the immunoregulatory properties of PTSO‐PTS in vivo were evaluated using transcriptomic analysis of murine cecal tissue derived from T. muris‐infected mice. Our results shed light on how PTSO‐PTS regulates immune function and inflammatory responses, and encourage assessment of how compounds derived from garlic or related plants may be used to modulate enteric disease.
2. Experimental Section
2.1. Garlic Extract
The garlic extract was provided by Pancosma SA (Rolle, Switzerland). It was standardized to 40% PTSO‐PTS (of which 80% consists of PTSO, and 20% PTS). The remainder of the extract consisted of the solvent polysorbate‐80. The stated experimental concentrations referred to final PTSO‐PTS concentration used in assays or animal experiments. In all experiments, control cells or mice were treated with an equivalent amount of polysorbate‐80 as the PTSO‐PTS‐treated groups.
2.2. Parasites
T. muris parasites (strain E) were maintained and T. muris excretory/secretory (E/S) products prepared as described previously.[ 17 ] Eggs were purified from the feces of mice and embryonated for at least 2 months before being used for infection. Experimental mice were infected with 20 eggs suspended in water by oral gavage.
2.3. Cell Culture
Murine rectal epithelial CMT93 (ATCC‐CCL‐223) cells or RAW264.7 macrophages (ATCC‐TIB‐71) were cultured in DMEM (Sigma Aldrich, USA) medium with 10% fetal calf serum (Sigma Aldrich), 100 U mL−1 penicillin and 100 µ;g mL−1 streptomycin (Sigma Aldrich). The cell culture was maintained at 37 °C with 5% CO2. Cell viability was tested with prestoBlue cell viability reagent (Thermo Fisher Scientific) according to the manufacturer's protocols. Briefly, a concentration of 1 × 105 cells per mL were seeded into 96 well plate (Sigma‐Aldrich). After 24 h, the supernatant was removed and new medium was added into cells with 90 µ;L DMEM and 10 µ;L PrestoBlue reagent per well. The cells were incubated for further 2 h then the absorbance at 490 nm with a reference wavelength at 600 nm for PrestoBlue was measured.
2.4. Cell Stimulation
RAW264.7 cells were seeded into a 24‐well plate and 2 hours allowed for attachment. Following this, cells were stimulated with 20 µ;M PTSO‐PTS and 500 ng mL−1 LPS for either 6 h or 24 h. RNA was extracted from RAW264.7 cells cultured for 6 hours using RNeasy kits according to manufacturer's protocols (Qiagen, Denmark). After 24 h culture, supernatant from cells was collected for further analysis. For CMT93 cells, 20 µ;M PTSO‐PTS and T. muris E/S (50 µ;g mL−1) were added for 6 hours when cell confluence reached 80%. RNA extraction was conducted as for RAW264.7 cells.
2.5. ELISA
Levels of secreted IL‐6, TNFα, and IL‐27 were measured in RAW264.7 supernatants using Mouse paired ELISA kits (Duosets, R and D Systems, UK) according to the manufacturer's instructions.
2.6. Animals and Experimental Design
Female C57BL/6JOlaHsd mice (6–8 weeks of age, Envigo) were randomly divided into four groups (n = 5) and housed in ventilated cages with free access to standard murine diet (D30, SAFE) and water. Two untreated groups were included with vehicle water, the rest of groups received PTSO‐PTS‐supplemented water. PTSO‐PTS treatment commenced 7 days before infection and continued throughout the course of infection (35 days). Water was refreshed every 2 days. Mouse body weights were recorded weekly throughout all the experimental period. After 1 week of PTSO‐PTS intake, mice were infected with T. muris, and were sacrificed at 35 days post infection. Cecal tissues were collected in RNAlater (Thermo Fisher) and stored at −20 °C for further RNA extraction. RNA was extracted using miRNeasy mini kits after homogenization using a gentle MACS Dissociator (Miltenyi Biotec) in QIAzol lysis buffer. This study was approved by the Danish Animal Experimentation Inspectorate (Licence number 2015‐15‐0201‐00760) and conducted at the Experimental Animal Unit, University of Copenhagen.
2.6.1. Dosage Information
Water was prepared by solubilizing PTSO‐PTS to deliver a concentration 0.1 mg PTSO‐PTS per kg bodyweight (BW) per day or 1 mg kg−1 BW per day, assuming a water intake of 4 mL per day per mouse, and control water was normal drinking water with the same volume of polysorbate 80 alone.
2.7. Mesenteric Lymph Nodes (MLN) Cells Isolation
MLN tissues were collected from mice at 35 days post infection, then placed into 70 µ;M cell strainer for tissue disruption using a sterile syringe plunger to obtain a single cell suspension. RPMI media (Life Technologies) with 10% fetal calf serum (Sigma‐Aldrich) and 100 U mL−1 penicillin plus 100 mg mL−1 streptomycin (Sigma‐Aldrich) was added to the cells and cell counting was conducted by trypan blue staining. MLN cells were adjusted to 5.0×106 cells mL−1 for further analysis by flow cytometry.
2.8. Flow cytometry
MLN‐ derived T cell populations were phenotypically analyzed as described in Myhill et al.[ 17 ] Briefly, MLN cell suspensions were added to a 96 well plate for surface staining with anti‐mouse TCRβ‐FITC (clone H57‐597, BD Biosciences) and anti‐CD4‐PerCP‐Cy5.5 (clone RM4‐5, BD Biosciences). Anti‐T‐bet‐Alexafluor 647 (clone 4B10, BD Biosciences), anti‐GATA3‐PE (clone TWAJ, Thermo Fisher Scientific), and anti‐Foxp3‐FITC (clone FJK‐16s, Thermo Fisher Scientific) were used for intracellular staining. MLN cell analysis was carried out on a BD Accuri C6 flow cytometer, and data acquired using Accuri CFlow Plus software (Accuri Cytometers).
2.9. RNA‐Sequencing
RNA extracted from cultured cells or cecum tissue was used for RNA‐sequencing (150bp paired‐end Illumina NovaSeq6000 sequencing, Novogene, Cambridge, UK). Sequence data was subsequently mapped to the Mus muscularis (GRCm38) genome and read counts generated which were used to determine DEG using DEseq2.[ 18 ] Pathway analysis was done using the clusterProfile R package, utilizing both gene ontology and KEGG enrichment analysis. Volcano Plots were constructed using VolcaNoseR.[ 19 ] Pathways with correct p value of <0.05 were considered significant. Sequence data is available at GEO under the accession numbers GSE178282, GSE178499, and GSE178655.
2.10. Quantitative Real Time PCR
cDNA synthesis was conducted by QuantiTect Reverse Transcription Kit (Qiagen). qPCR was performed with PerfeCTa SYBR Green FastMIX Low ROX (Quanta Bioscience). The qPCR primers were shown in Table 1 . B2m was used as a reference gene for normalization, and fold changes calculated using the ΔΔCT method.
Table 1.
Primers used for qPCR
| Gene | Primer sequence (5’‐3’ forward/reverse) |
|---|---|
| Tnf |
F: CCACCACGCTCTTCTGTCTA R: AGGGTCTGGGCCATAGAACT |
| Nos2 |
F: GGTGAAGGGACTGAGCTGTT R: TGCACTTCTGCTCCAAATCCA |
| Saa2 |
F: AGTTCAGAAGGCTGTGTTGGG R: CCCAGAGAGCATCTTCAGTGTT |
| Mcpt1 |
F: CTCTGCATATGTGCCCTGGATTA R: GGGGGCAGACTGGGGATAGT |
| B2m |
F: GACCGGCCTGTATGCTATCC R: TTTCAATGTGAGGCGGGTGG |
2.11. Statistical Analysis
Statistical analysis was determined by one way ANOVA with Graph Pad Prism (Version 8.0). Principal component analysis was conducted using ClustVis.[ 20 ] All data were represented as means ± standard error of mean (SEM). p value <0.05 was considered significant.
3. Results
3.1. Effects of Garlic Organosulfur Compounds on Cytokine Production and Gene Expression from Murine Macrophages
To investigate the possible anti‐inflammatory effects of PTSO‐PTS, we first exposed RAW264.7 macrophages to either LPS alone or LPS combined with 20 µ;M PTSO‐PTS—the given dose was based on preliminary experiments that showed this amount was tolerated without any toxicity to cells (Figure S1, Supporting Information). After 24 h culture, quantification of IL‐6 and TNFα levels showed that they were markedly increased by LPS treatment alone, while concurrent PTSO‐PTS exposure significantly decreased the production of these two pro‐inflammatory cytokines (Figure 1A,B). Thus, PTSO‐PTS has a suppressive effect on inflammatory cytokine production following toll‐like receptor (TLR) stimulation.
Figure 1.
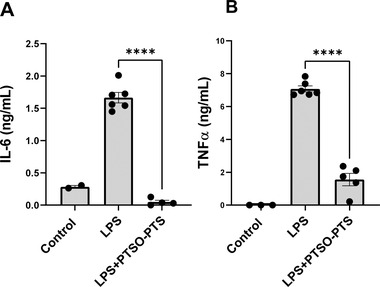
PTSO‐PTS suppressed proinflammatory cytokine secretion in RAW264.7 cells. A) IL‐6 production in RAW264.7 cells stimulated with lipopolysaccharide (LPS) or LPS combined with 20 µ;M PTSO‐PTS. B) Cytokine TNFα in RAW264.7 cells stimulated with LPS and LPS+PTSO‐PTS. Data bars represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Data are from at least two independent experiments.
To explore in detail the cellular activities modulated by PTSO‐PTS, RNA sequencing was conducted on RAW264.7 cells following PTSO‐PTS and/or LPS treatment. As shown in Figure 2A, in resting cells (no LPS exposure), PTSO‐PTS modulated the transcription of around 700 genes (fold change >2; adjusted P value <0.05). Notable upregulated genes included the transcription factors Jun and Fosl2, and the activation marker Cd14, suggesting that PTSO‐PTS had immunogenic properties and induced activation of resting macrophages (Figure 2B). Downregulated genes included many involved in cell adhesion and migration, such as Itga4, Tns3, and Nrp2 (Figure 2B). Gene ontology (GO) enrichment analysis revealed that in resting cells exposed to PTSO‐PTS, the main biological processes that were enriched were ubiquitin signaling, indicating an enhanced metabolic state of the cells. This was supported by KEGG pathway analysis showing that NF‐kB and MAPK‐related pathways, among others, were up‐regulated (Figure 2C,D). In contrast, GO and KEGG pathway analyses demonstrated a suppression of pathways relating to cell cycle and DNA replication, as well as fatty acid metabolism (Figure 2E,F). Interestingly, pathways related to cell cycle and fatty acid metabolism were also suppressed in RAW264.7 cells after exposure to LPS (Figure S2, Supporting Information), suggesting that PTSO‐PTS exposure had an immunostimulatory effect on the cells. However, we could not detect any secretion of TNFα or IL‐6 from PTSO‐PTS treated cells without concurrent LPS exposure (data not shown).
Figure 2.
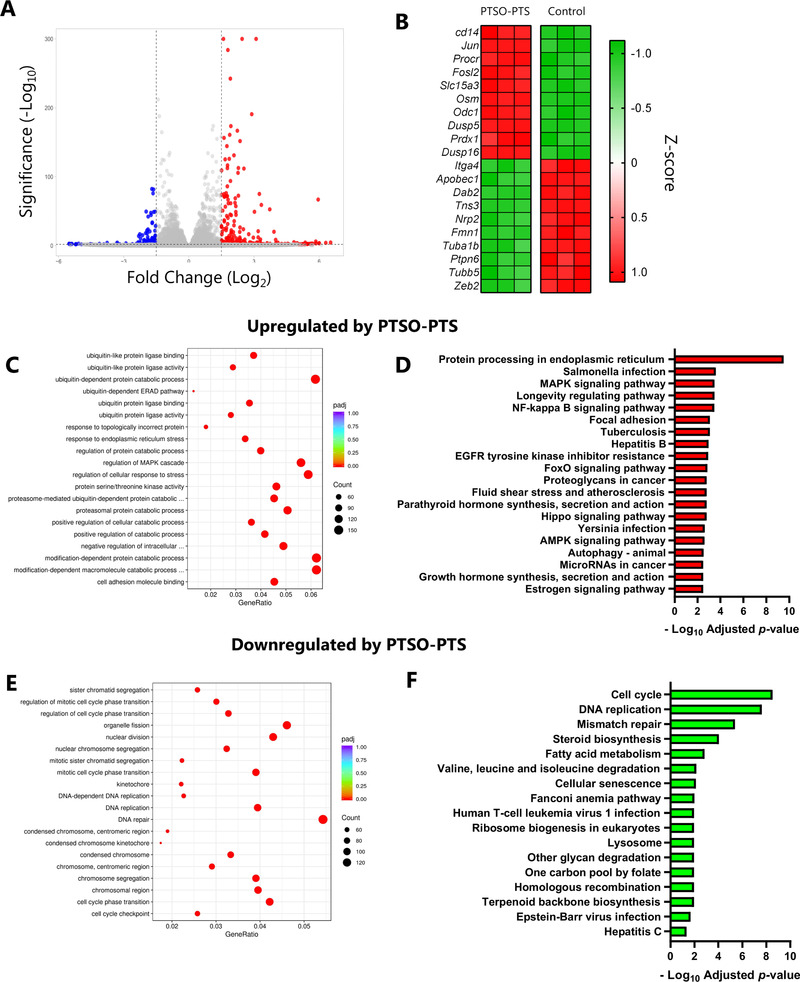
PTSO‐PTS modulates transcriptional pathways in cultured RAW264.7 cells. A) Volcano plot of gene expression fold changes of PTSO‐PTS treated cells (20 µ;M for 6 h) compared to non‐treated controls (Fold change >2; adjusted p value <0.05). B) Heat map of top 10 up‐ and down‐regulated genes form PTSO‐PTS treated RAW264.7 cells compared to non‐treated cells, C) Gene Ontology (GO) analysis of top upregulated pathways by PTSO‐PTS compared to non‐treated cells. D) Top upregulated KEGG pathways following exposure of cells to PTSO‐PTS (p adj < 0.05). E) GO analysis of top downregulated pathways by PTSO‐PTS. F) Significantly downregulated KEGG pathways following exposure of cells to PTSO‐PTS (p adj < 0.05) n = 3 per treatment group.
Given that LPS‐induced cytokine production was significantly impaired by PTSO‐PTS, we next investigated the transcriptomic response of LPS‐activated cells with or without concurrent PTSO‐PTS treatment. Around 1200 genes were significantly modulated by PTSO‐PTS treatment, relative to cells exposed only to LPS (Figure 3A). Principal component analysis indicated that the response to LPS was profoundly altered by PTSO‐PTS (Figure 3B). Down‐regulated genes include many related to immune function, such as Ccl22, but also genes related to lipid metabolism (Lpl) and G protein‐coupled receptor signaling (Rgs2). Notably, genes encoding inflammatory cytokines and chemokines (Tnf, Il27, Cxcl9) were suppressed. However, we also noted that other cytokine‐encoding genes (e.g., Il23a, Csf3) were increased in LPS‐treated cells exposed to PTSO‐PTS, suggesting a pleiotropic effect (Figure 3A,C). Interestingly, as observed in resting cells, expression of Cd14, Procr, and Osm was significantly increased by PTSO‐PTS in LPS‐activated cells, and GO analysis again revealed that a dominant biological response to PTSO‐PTS was ubiquitin activity (Figure 3D). KEGG pathway analysis showed many enriched pathways related to glutathione and lysosome activity, suggesting that xenobiotic metabolizing pathways were activated in response to PTSO‐PTS exposure (Figure 3E). Downregulated pathways identified by GO and KEGG analysis mainly related to immune defense and interferon signaling, consistent with the previously observed anti‐inflammatory activity (Figure 3F,G). To verify the anti‐inflammatory pathways identified in our transcriptomic analysis, we confirmed that LPS‐induced IL‐27 secretion was significantly impaired by PTSO‐PTS (Figure 3H). Collectively, these data show that resting macrophages adopt a semi‐activated state after exposure to PTSO‐PTS, but that TLR4‐induced inflammatory pathways are strongly suppressed, indicating that PTSO‐PTS is likely to demonstrate profound immunomodulatory properties in inflammatory settings.
Figure 3.
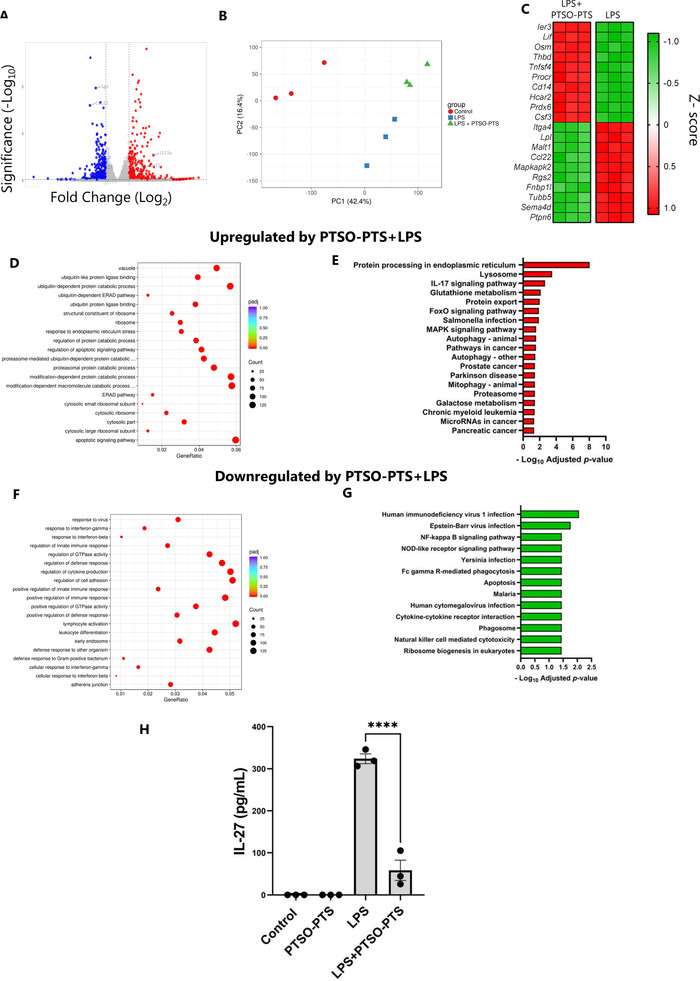
PTSO‐PTS regulates transcriptional pathways and inflammatory responses following lipopolysaccharide treatment in RAW264.7 cells. A) Volcano plot of genes expressed showing fold changes in PTSO‐PTS and lipopolysaccharide (LPS) treated cells compared to LPS‐only treated controls. B) Principal component analysis showing clustering of cells without stimulation, LPS stimulation, or LPS combined with 20 µ;M PTSO‐PTS. C) Heat map of top 10 up‐ and down‐regulated genes in PTSO‐PTS +LPS treated RAW264.7 cells compared to only LPS treated cells. D) Gene ontology (GO) analysis of top upregulated pathways in LPS‐treated cells exposed to PTSO‐PTS compared to LPS‐only treated controls. E) Top upregulated KEGG pathways in LPS‐treated cells following exposure to PTSO‐PTS, compared to LPS‐only treated controls (p adj < 0.05). F) GO analysis of top downregulated pathways in LPS‐treated cells exposed to PTSO‐PTS compared to LPS‐only treated controls. G) Significantly upregulated KEGG pathways in LPS‐treated cells following exposure to PTSO‐PTS, compared to LPS‐only treated controls (p adj < 0.05). H) IL‐27 secretion from LPS‐stimulated RAW264.7 macrophages cultured with PTSO‐PTS. Data bars represent mean ± SEM. ****p ≤ 0.0001. n = 3 per treatment group.
3.2. Effects of PTSO‐PTS Treatment on Murine Intestinal Epithelial Cells
As dietary components, the garlic‐derived compounds likely come into contact with the intestinal epithelium in relatively high concentrations before and during absorption. Therefore, we also investigated whether PTSO‐PTS modulated the activity of cultured intestinal epithelial cells (IEC) by exposing CMT‐93 cells to PTSO‐PTS for 6 h, followed by RNA sequencing. The dominant transcriptional response to PTSO‐PTS was related to cell cycle and DNA replication activity (Figure 4A,B). Moreover, consistent with the indications of xenobiotic metabolism‐related transcriptional responses in macrophages, the most significantly up‐regulated genes were involved in antioxidant responses (Gpx2, and the selenoprotein‐encoding genes Selenow and Selenoh; Figure 4C). Thus, PTSO‐PTS has a stimulatory effect on IEC under steady‐state conditions, and induces genes related to detoxification and antioxidant activity. To induce an inflammatory response in these cells, we exposed them to antigens from an enteric pathogen. T. muris causes chronic infections with colitis‐like inflammation, and its secreted antigens have been shown to induce the expression of pro‐inflammatory genes in IEC in vitro.[ 21 ] Thus, to explore the effect of PTSO‐PTS on IEC responses during exposure to pathogen antigens, we conducted RNA sequencing on T. muris excretory/secretory antigen (E/S)‐stimulated cells with or without concurrent PTSO‐PTS treatment. CMT‐93 cells reacted to T. muris E/S by up‐regulating transcriptional pathways related to cellular activity such as DNA replication and ribosome activity, and genes encoding nutrient transporters (Slc7a11, Tfrc) and cytokines/chemokines (Il24, Cxcl5) (Figure 4D–F). PTSO‐PTS–induced transcriptional changes in cell cycle pathways were maintained during exposure to T. muris E/S. These included MAPK signaling and cell cycle pathways (Figure 4H). Expression of the antioxidant‐related genes Gpx1 and Selenow was also upregulated by PTSO‐PTS in T. muris E/S‐treated cells (Figure 4I). Moreover, PTSO‐PTS ameliorated the upregulation of Cxcl5 in T. muris E/S treated cells. These data show that PTSO‐PTS treatment induced a varied transcriptomic response in IEC both alone and also in the presence of pathogen antigens, suggesting that PTSO‐PTS may exert immunomodulatory effects by contacting intestinal epithelium during enteric infection and inflammation.
Figure 4.
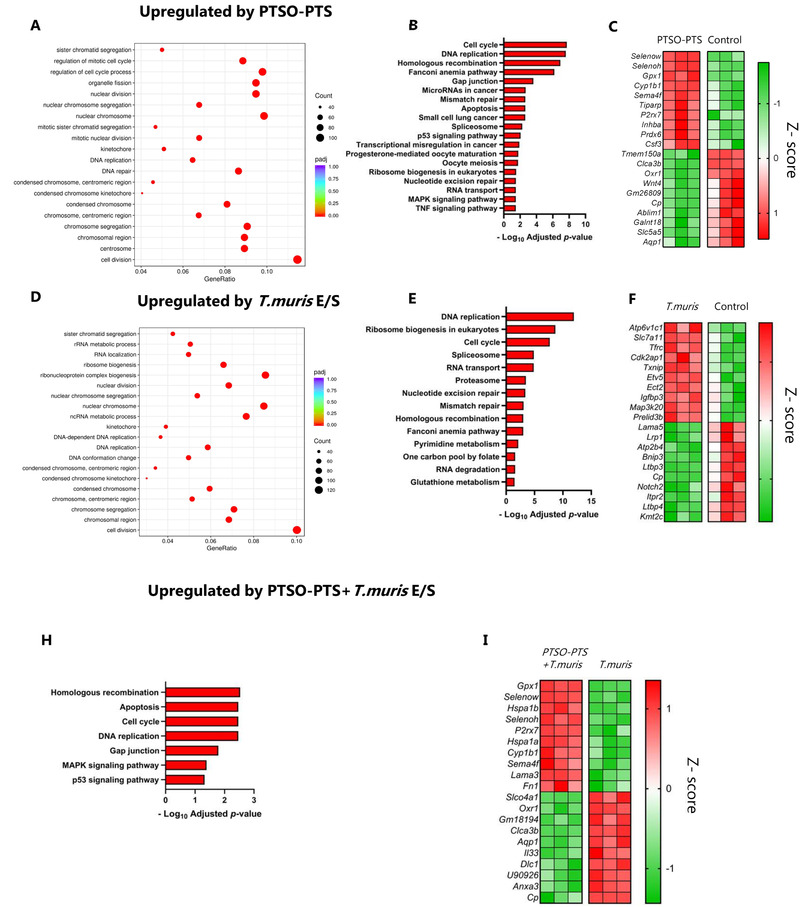
PTSO‐PTS modulates transcriptional pathways in cultured intestinal epithelial cells during Trichuris muris antigen stimulation . A) Gene ontology (GO) analysis of top upregulated pathways by PTSO‐PTS in CMT93 cells, compared to untreated cells. B) KEGG pathways upregulated following exposure of CMT93 cells to PTSO‐PTS, compared to untreated cells (p adj < 0.05). C) Heat map of top 10 up‐ and down‐regulated genes in CMT93 cells treated PTSO‐PTS, compared to untreated cells, D) GO analysis of top upregulated pathways following exposure of cells to T. muris E/S antigens, compared to untreated cells. E) KEGG pathways significantly upregulated by T. muris antigens in CMT93 cells, compared to untreated cells (p adj < 0.05). F) Heat map of top up‐ and down‐regulated genes by T. muris antigens, compared to untreated cells G) KEGG pathways upregulated in T. muris antigen‐treated cells exposed to by PTSO‐PTS, relative to only T. muris antigen treated cells (p adj < 0.05). H) Heat map of top up‐ and down‐regulated genes in T. muris antigen‐treated cells exposed to by PTSO‐PTS, relative to only T. muris antigen treated cells n = 3 per treatment group.
3.3. PTSO‐PTS Does Not Affect T. muris Burdens but Modulates Expression of Inflammation‐Related Genes
We next asked if oral treatment of mice with PTSO‐PTS could regulate immune function and inflammatory responses during T. muris infection. Pilot studies indicated that PTSO‐PTS was readily consumed by mice in the drinking water, with no effect on water consumption or daily weight gain (Figure S3, Supporting Information). In C57BL/6 mice, infection with low numbers of T. muris eggs (≤20) causes a chronic infection in the cecum, which is associated with the expression of numerous inflammation‐related genes and the development of sporadic colitis like‐pathology.[ 22 , 23 ] The ability of PTSO‐PTS to potentially reduce enteric infection and inflammation in vivo was studied by daily administration of 0.1 mg kg−1 or 1 mg kg−1 per body weight PTSO‐PTS in the drinking water during a chronic T. muris infection. For both concentrations of PTSO‐PTS, there was no effect on worm burdens compared to T. muris‐infected mice alone (Figure 5A). Although mice fed PTSO‐PTS had no changes in daily weight gain during the entire experiment, T. muris‐infected mice administered 1 mg kg−1 PTSO‐PTS displayed decreased weight gain compared to untreated, infected mice during infection (Figure 5B,C, respectively).
Figure 5.
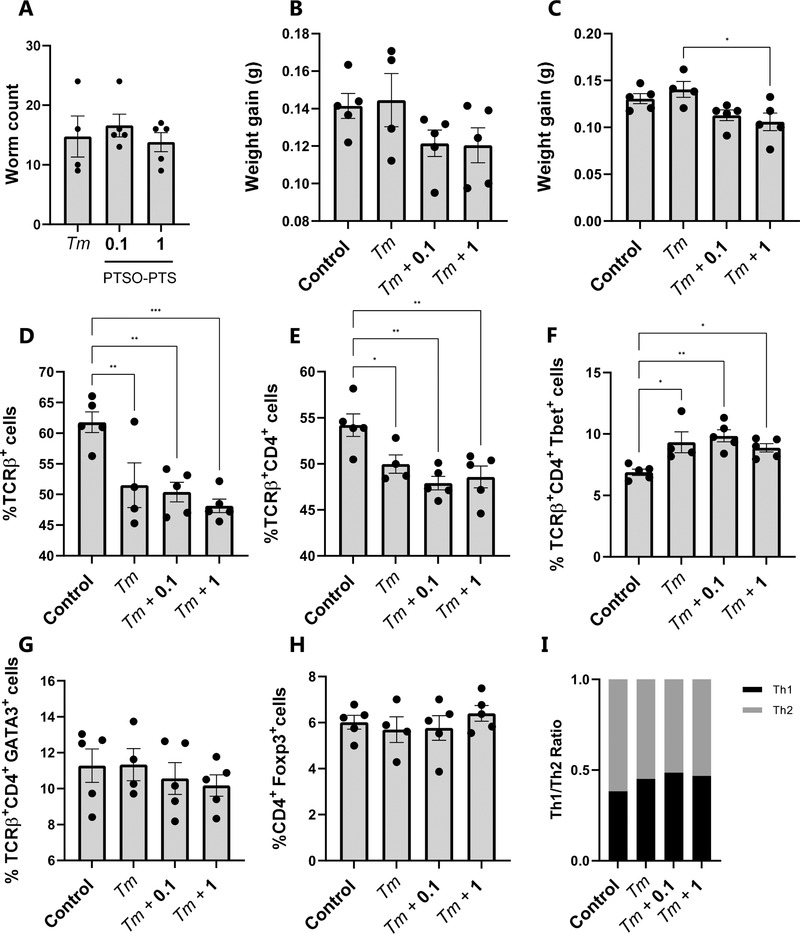
Trichuris muris infection regulates immune responses from mesenteric lymph node cells rather than PTSO‐PTS. A) T. muris burdens in mice inoculated with 20 eggs and given control drinking water, or water supplemented with either 0.1 mg kg−1 or 1 mg kg−1 PTSO‐PTS. Worm burdens were assessed at day 35 post‐infection. Weight gain throughout the entire experimental period B) and during infection with T. muris C). D) Proportions of TCRβ+ cells in mesenteric lymph nodes (MLN) following 35 days of T. muris infection in mice given control drinking water, or water supplemented with either 0.1 mg kg−1 or 1 mg kg−1 PTSO‐PTS. Shown also are uninfected control mice given normal drinking water. Proportions of CD4+ E), CD4+T‐bet+ F), CD4+GATA3+ G), and CD4+Foxp3+ T‐cells within the TCRβ+ population in MLN following 35 days of T. muris infection in mice given control drinking water, or water supplemented with either 0.1 mg kg or 1 mg kg PTSO‐PTS. Shown also are uninfected control mice given normal drinking water. I) Th2 (CD4+GATA3+) to Th1 (CD4+T‐bet+) ratio in MLN. * p < 0.05, ** p < 0.01, **p < 0.001 by ANOVA analysis. n = 4–5 per treatment group.
To explore the effect of PTSO‐PTS on host immune responses, mesenteric lymph node (MLN) cells were isolated for analysis of T‐cell populations. As shown in Figure 5D,E, T. muris infection clearly reduced the proportion of TCRβ+ T cells and TCRβ+CD4+ T cells, but there was no effect of PTSO‐PTS intake on the proportions of these cells within infected mice. Within the T‐cell population there was a significant increase in the proportion of Th1 cells (TCRβ+CD4+Tbet+) in infected mice, consistent with the type‐1 immune response known to be induced by chronic T. muris infection, but again PTSO‐PTS intake did not influence these parameters (Figure 5F). The proportions of Th2 (GATA3+) and T‐regulatory (Foxp3+) T‐cells were not altered by either infection or PTSO‐PTS intake (Figure 5G–I).
To explore in more detail whether PTSO‐PTS could modulate the mucosal inflammation induced by the infection, RNA sequencing was carried out on cecal tissue harvested from mice in each treatment group. As expected, T. muris infection altered the expression of a vast number of genes (>3500), relative to uninfected control mice. Amongst the top up‐regulated genes were the granzyme‐encoding genes Gzma and Gzmb, the interferon‐responsive gene Ifit2, and the inflammatory chemokine Ccl5 (Figure 6A). Consistent with this, enriched KEGG pathways were related to infection and inflammation, including TNF signaling (Figure S4, Supporting Information). Interestingly, numerous KEGG pathways related to nutrient metabolism (e.g., amino acids, fatty acid metabolism) were suppressed by infection (Figure S5, Supporting Information). Principal component analysis demonstrated that, within infected mice, PTSO‐PTS intake resulted in dose‐dependence divergence from control mice (Figure 6B). However, only a small number of genes were identified as being significantly (adjusted p value <0.05) regulated by PTSO‐PTS within infected mice. These included Upk3b (encoding an epithelial protein found in the intestinal and urogenital tracts), Lrrn4 (encoding a leucine‐rich protein involved in diverse cellular functions), and Myrf (encoding a myelin regulatory factor)—these genes were upregulated in both treatment groups (0.1 and 1 mg kg−1). In the higher dosage group, an additional three genes were regulated. These genes encoded a glutamate transporter (Slc17a8), an insulin‐like growth factor binding protein (Igfbp6), and an uncharacterized immunoglobulin variable region gene (Igkv5‐37). We also noted that both PTSO‐PTS treated groups induced a significant (adjusted p value <0.05) down‐regulation of several KEGG pathways related to protein metabolism, inflammation, and immune function. Interestingly, these pathways tended to be more strongly regulated in the 0.1 mg kg−1 group (Figure 6C,D). Further examination of the transcriptomic data showed that a number of genes related to inflammation (including Tnf, Nos2, and Saa2) were down‐regulated by PTSO‐PTS. Conversely, genes related to the development of type‐2 immunity (e.g., Mcpt1), were upregulated. As these genes were not significant after correcting for multiple testing, we verified their expression by qPCR. This confirmed a significant dose‐dependent suppression of Tnf, Nos2, and Saa2 expression (and an increase in Mcpt1 expression) (Figure 6E). Thus, in this model PTSO‐PTS intake was not sufficient to lower parasite burdens but did appear to attenuate the localized pro‐inflammatory transcriptional response in the cecum.
Figure 6.
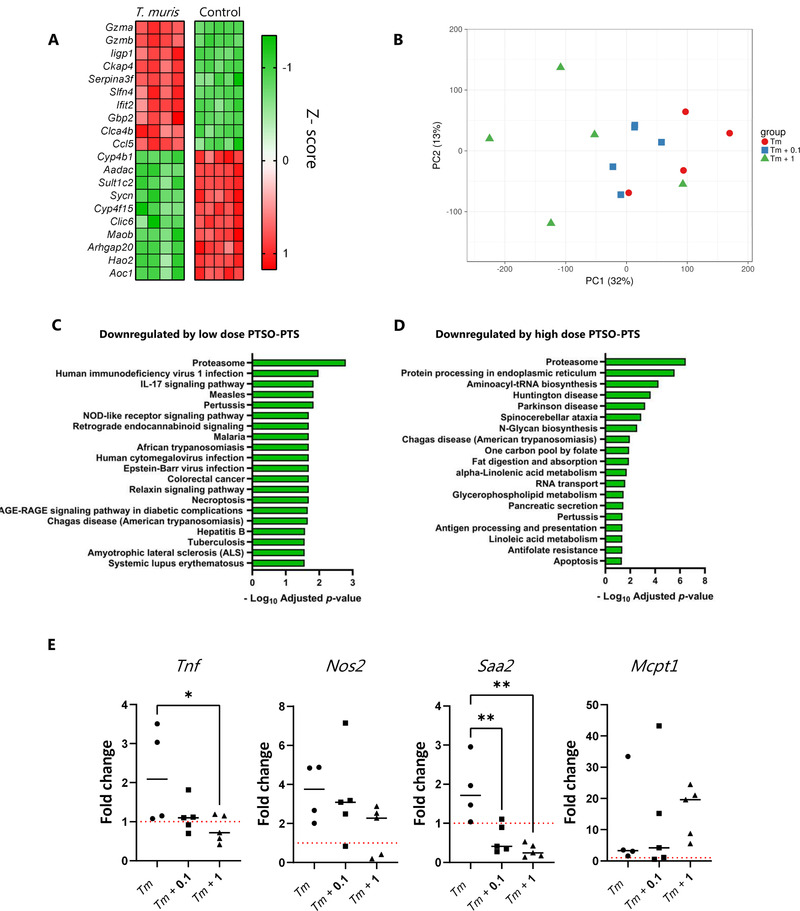
PTSO‐PTS modulates the pro‐inflammatory transcriptional response in the mouse cecum. A) Top 10 most up‐ and down‐regulated genes in cecal tissue of Trichuris muris‐infected mice, relative to control, uninfected mice. B) Principal component analysis showing clustering of cecal tissue in uninfected mice, and mice infected with T. muris for 35 days and given control drinking water, or water supplemented with either 0.1 mg kg−1 or 1 mg kg−1 PTSO‐PTS. KEGG biological pathways significantly down‐regulated in cecal tissue of T. muris‐infected mice given either 0.1 C) mg kg−1 or 1 mg kg−1 D) PTSO‐PTS, relative to T. muris‐infected mice given control drinking water. E) qPCR validation of the expression of Tnf, Nos2, Saa2, and Mcpt1 in cecal tissue following 35 days of T. muris infection in mice given control drinking water, or water supplemented with either 0.1 mg kg−1 or 1 mg kg−1 PTSO‐PTS. Shown also are uninfected control mice given normal drinking water. * p < 0.05, ** p < 0.01, by ANOVA analysis. n = 4–5 per treatment group.
4. Discussion
In recent years, garlic‐derived compounds have been increasingly studied as therapeutic agents. They have characteristic chemical structures, especially sulfur containing compounds, sulfides and alliin, which may influence their biological activities to protect animals and humans from diseases.[ 24 ] In most compounds of garlic, PTSO‐PTS show pungent odors, and PTS is produced by the interaction of alliinase and S‐propyl‐l‐cysteine sulfoxide, and then changes into PTSO due to its instability.[ 25 ] PTSO‐PTS is absorbed mainly in the intestine by amino acid transporter of cysteine, and the carrier of polysorbate 80 can increase its cellular permeability and bioavailability in digestion process compared with free PTSO.[ 24 , 26 , 27 , 28 ] Interestingly, PTSO‐PTS (0.01–0.1 mg kg−1) has also demonstrated immunomodulatory properties and can alleviate localized inflammation.[ 13 ] Additionally, PTSO with different doses (0.1, 0.5, and 1 mg kg−1) attenuate systemic inflammatory responses and weight gain related to obesity, showing its potential role for treatment of inflammation and metabolic syndrome.[ 29 ] This may perhaps be consistent with our results that mice administered PTSO‐PTS tended to gain less weight during T. muris infection (but not in the absence of infection). Further studies will be necessary to understand the complex effects of PTSO‐PTS on host metabolism during inflammation.
During transit throughout the digestive tract, PTSO‐PTS may modify intestinal responses by contacting intestinal enterocytes and immune cells, and we thus explored the PTSO‐PTS effects on murine intestine in vitro and in vivo to elucidate the underlying anti‐inflammatory mechanisms and whether these compounds may have protective effects during chronic enteric infection. Murine RAW264.7 macrophages are well‐known to secrete pro‐inflammatory mediators and thus mimic inflammation status during LPS activation.[ 30 ] We initially observed that PTSO‐PTS induced dramatic changes by inhibiting the secretion of the pro‐inflammatory cytokines TNF‐α and IL‐6 in RAW 264.7 cells, indicating potential anti‐inflammatory ability. In addition, the gene expression profile of macrophages was markedly affected by PTSO‐PTS treatment. Interestingly, PTSO‐PTS induced expression of Cd14, Jun, Procr, and Osm in resting cells, which are related to inflammation.[ 31 , 32 ] Cd14 has been associated with myeloid differentiation and subsequently involves in innate immune responses and clearance of apoptotic cells.[ 33 ] Similar to Cd14, Jun, coding c‐Jun as a member of AP‐1 complex, is required for different cell activities, especially apoptosis.[ 34 , 35 ] In Jun‐deficient mice, impaired liver regeneration can be found after partial hepatectomy, suggesting c‐Jun is responsible for hepatocyte proliferation.[ 36 ] Procr is expressed in various endothelial cells and Th17 cells.[ 37 , 38 ] Enhanced expression of Procr lead to the reduction of proinflammatory molecules, including IL1R and IL23R, while in Procr over‐expressing mice, significant reduction of cancer cell metastasis are observed.[ 38 , 39 ] Osm expression is associated with activated T cells, dendritic cells, monocytes, and neutrophils.[ 35 ] As a pleiotropic cytokine, Osm displays proinflammatory and anti‐inflammatory abilities as well as regulated apoptosis.[ 40 , 41 ] Notably, the suppressed pathways in PTSO‐PTS treated cells mainly refer to DNA replication and cell cycle, which is consistent with anticancer properties of garlic‐derived extracts.[ 42 ] Taken together, PTSO‐PTS in resting condition may have an immune‐activation effect and attenuate apoptosis development.
During exposure to LPS, PTSO‐PTS suppressed the induction of inflammatory markers in RAW264.7 cells, such as Ccl22, a chemokine related to cell migration and inflammatory function which can be induced to promote tissue damage, and is a main factor of pathogenesis of asthma and allergy.[ 43 ] Moreover, anti‐Ccl22 treatment has the ability to ameliorate CNS inflammation by modulating inflammatory macrophage accumulation and macrophage effector cytokine phenotype.[ 44 ] Genes encoding Il27, a cytokine belonging to the IL‐6 family, and Cxcl9, a chemokine involved in inflammation and tumor progression, were both significantly downregulated in LPS‐induced macrophages, confirming the strong anti‐inflammatory effect of PTSO‐PTS.[ 45 , 46 ] Interestingly, we noted that the glutathione activity was upregulated by PTSO‐PTS, which is related to oxidative response and detoxifying effects as shown by Anoush et al.[ 47 ] using garlic extracts, suggesting PTSO‐PTS is involved in detoxification and xenobiotic elimination. Taken together, PTSO‐PTS seems to exert immunostimulatory effects in resting cells and anti‐inflammatory properties towards TLR‐4 activated responses induced by LPS. This is consistent with previous studies on other bioactive plant molecules such as polyphenols, which may induce some immunostimulatory effects in the steady state but strongly inhibit acute inflammation induced by substances such as LPS.[ 48 ] The mechanisms underlying these pleiotropic effects remain to be fully elucidated, but seemingly the xenobiotic stress response induced in the cells after exposure to volatile natural compounds is sufficient to down‐regulate the TLR‐driven signaling responses that result in inflammatory cytokine production.
Allicin has been reported to produce metabolites that can contact and interact with intestinal cells.[ 24 ] However, whether PTSO‐PTS plays a similar role via contacting intestinal epithelial cells is unclear. The RNA‐seq of CMT cells with PTSO‐PTS indicates regulation of antioxidant responses. Gpx1 is associated with endothelial dysfunction.[ 49 ] Upregulation of Gpx1 is known to provide protection of ileum and colon mucosa from inflammation and oxidative stress with enhanced genes Selenow and Selenoh, which belong to intracellular selenium (Se)‐dependent glutathione peroxidase, contributing to glutathione activity as similarly observed in macrophages.[ 50 ] Notably, apoptosis pathways induced are PTSO‐PTS is consistent with the garlic compound ajoene which has been shown to limit tumor size in cancer.[ 51 ]
T. muris E/S products contain multiple proteins and small molecules secreted by T. muris, which are known to regulate host immunity.[ 52 ] Similarly, we found that treatment with T. muris E/S alone in vitro also tends to regulate energy metabolism including glycolysis, galactose metabolism, and glucagon signaling as previously reported.[ 53 ] PTSO‐PTS treatment with T. muris E/S modulated a variety of intestinal transcriptional responses. The significantly up‐regulated pathways were DNA replication and anabolic pathway, which were down‐regulated in macrophages, indicating PTSO‐PTS exerts pleiotropic effects in various cell lines. Furthermore, the significantly regulated genes Gpx1 and Selenow were also observed. Thus, our data suggests that antioxidant responses in intestinal epithelial cells may be important mechanism by which PTSO‐PTS limits inflammation.
Low dose T. muris inoculation in mice tends to drive a chronic infection, characterized by Th1 immune polarization, and intestinal inflammation.[ 54 ] Garlic has exhibited anticoccidial activity by impairing the development of parasites in a murine model, demonstrating the possibility of garlic as an anti‐parasite agent (in a Th1 dominated inflammatory state).[ 55 ] Our in vivo study showed that PTSO‐PTS supplement did not alter T. muris worm counts, which is in contrast to garlic extracts,[ 56 ] showing that PTSO‐PTS may not be a suitable anti‐parasite compound against T. muris, yet additional experiments are needed to fully investigate the effects of PTSO‐PTS on intestinal parasitic worms. Furthermore, we observed a distinct Th1‐polarized immune response towards T. muris, yet no changes in Th1, Th2, or Treg cell populations were induced by PTSO‐PTS treatment during T. muris infection. Thus, our data confirms that chronic T. muris infection causes the polarization of a type‐1 immune response as previously reported,[ 57 ] and PTSO‐PTS has no effect on MLN‐derived CD4+ T cell phenotypes.
In mice infected with low levels of T. muris, infection resulted in host defence transcriptional responses, especially upregulated T cell differentiation, TLR and TNF signaling, and IFN‐γ responses as reported in other studies.[ 58 ] Interestingly, T. muris infection suppressed metabolism of xenobiotics by cytochrome p450 response. The downregulation of cytochrome p450 reflects the host resistance mechanism against intestinal parasites, further supporting oxidative stress activity and pro‐inflammatory activity of the parasite.[ 59 ] Similarly, PTSO‐PTS treatment in infected mice induced significant changes to intestinal gene expression and pathways related to immune function and inflammation, which is consistent with protective effect of PTSO‐PTS against avian coccidiosis.[ 60 ] It is interesting to note the downregulated IL17 signaling pathway in 0.1 mg kg−1 PTSO‐PTS dosed mice rather than with the higher PTSO‐PTS concentration, as uncontrolled IL17 signaling can exacerbate autoimmune diseases and immunopathology.[ 61 ] Thus, the anti‐inflammatory property of PTSO‐PTS seems to be related to concentration, which could be investigated further in relation to treating autoimmune pathologies. During inflammation, the intestinal barrier is damaged as a result of excessive reactive nitrogen and oxygen (NO) species production, induced by the enzyme inducible nitric oxide synthase (NOS2).[ 62 ] The NOS2‐induced production of nitric oxide can be stimulated by pro‐inflammatory cytokines, such as TNFα and IFN‐γ, concomitant with selenoprotein inhibition.[ 62 ] Serum amyloid A2 (SAA2) is activated during infection and tissue injury as a marker of inflammation, and is involved in various chronic diseases.[ 63 ] Mucosal mast cell proteases (e.g., Mcpt1) are known to be induced by TGF‐β, which may be involved in degradation of pro‐inflammatory substances and thus exerts anti‐inflammatory activity.[ 64 , 65 ] Taken together, the downregulation of Nos2, Saa2 and upregulation of Mcpt1 further confirms the anti‐inflammatory ability of PTSO‐PTS during T. muris infection. The anti‐inflammatory activity of PTSO‐PTS in this model may derive from the antioxidant activity induced in immune cells at the mucosal barrier surface, as evidenced by our in vitro data, although future studies should also assess whether these compounds may interact with the gut microbiota to exert immunomodulatory effects, e.g., by the production of metabolites with systemic anti‐inflammatory properties.
This study clearly demonstrates the anti‐oxidant and anti‐inflammatory ability of PTSO‐PTS in vitro and in vivo, with these properties further demonstrated by the regulation of the gut environment resulting from the beneficial interplay between diet and helminth infection. Our results encourage further investigations to explore how bioactive compounds derived from common food sources may be used to prevent and/or treat enteric diseases.
Conflict of Interest
A.B. is an employee of Pancosma/ADM. The other authors declare no conflict of interests.
Author Contributions
L.Z. and A.R.W. designed the study. L.Z., L.J.M., and A.A.C. performed the experiments. S.M.T., A.B., and A.R.W. supervised the study and provided essential reagents. L.Z. and A.R.W. analyzed the data and wrote the paper. All authors reviewed and edited the final manuscript.
Supporting information
SUPPORTING INFORMATION
Acknowledgements
This work was supported by Independent Research Fund Denmark (Grant 7026‐0094B) and the Novo Nordisk Foundation (Grant 0052422). L.Z. was supported by the China Scholarship Council (Grant 201806910065). The authors thank Mette Schjelde and Stine Nielsen for assistance. Figures were created with the aid of BioRender.com.
Zhu L., Myhill L. J., Andersen‐Civil A. I. S., Thamsborg S. M., Blanchard A., Williams A. R., Garlic‐Derived Organosulfur Compounds Regulate Metabolic and Immune Pathways in Macrophages and Attenuate Intestinal Inflammation in Mice. Mol. Nutr. Food Res. 2022, 66, 2101004. 10.1002/mnfr.202101004
Data Availability Statement
RNA‐seq data is available at GEO under accession numbers GSE178282, GSE178499, and GSE178655. All other data are contained within the manuscript and it's supporting files.
References
- 1. Song J., Li Q., Everaert N., Liu R., Zheng M., Zhao G., Wen J., J. Anim. Sci. 2020, 98, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daglia M., Curr. Opin. Biotechnol. 2012, 23, 174. [DOI] [PubMed] [Google Scholar]
- 3. Sharifi‐Rad J., Hoseini‑Alfatemi S. M., Sharifi‐Rad M., Iriti M., Ann. Med. Health Sci. Res. 2014, 4, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruiz R., García M. P., Lara A., Rubio L. A., Vet. Microbiol. 2010, 144, 110. [DOI] [PubMed] [Google Scholar]
- 5. Arreola R., Quintero‐Fabián S., López‐Roa R. I., Flores‐Gutiérrez E. O., Reyes‐Grajeda J. P., Carrera‐Quintanar L., Ortuño‐Sahagún D., J. Immunol. Res. 2015, 7861207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Percival S. S., J. Nutr. 2016, 146, 433S. [DOI] [PubMed] [Google Scholar]
- 7. Zamani A., Vahidinia A., Ghannad M. S., Phyther. Res. 2009, 23, 579. [DOI] [PubMed] [Google Scholar]
- 8. El‐Sayed H. S., Chizzola R., Ramadan A. A., Edris A. E., Food Chem. 2017, 221, 196. [DOI] [PubMed] [Google Scholar]
- 9. Martins N., Petropoulos S., Ferreira I. C. F. R., Food Chem. 2016, 211, 41. [DOI] [PubMed] [Google Scholar]
- 10. Lira A. C., Prieto A. I., Baños A., Guillamón E., Moyano R., Jos A., Cameán A. M., Food Chem. Toxicol. 2020, 144, 111612. [DOI] [PubMed] [Google Scholar]
- 11. Peinado M. J., Ruiz R., Echávarri A., Rubio L. A., Poult. Sci. 2012, 91, 2148. [DOI] [PubMed] [Google Scholar]
- 12. Wlodarska M., Willing B. P., Bravo D. M., Finlay B. B., Sci. Rep. 2015, 5, 9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vezza T., Algieri F., Garrido‐Mesa J., Utrilla M. P., Rodríguez‐Cabezas M. E., Baños A., Guillamón E., García F., Rodríguez‐Nogales A., Gálvez J., Mol. Nutr. Food Res. 2019, 63, 1800653. [DOI] [PubMed] [Google Scholar]
- 14. Mohammadi R., Hosseini‐Safa A., Ardakani M. J. E., Rostami‐Nejad M., Gastroenterol. Hepatol. 2015, 8, 123. [PMC free article] [PubMed] [Google Scholar]
- 15. Klementowicz J. E., Travis M. A., Grencis R. K., Semin. Immunopathol. 2012, 34, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chenery A. L., Antignano F., Burrows K., Scheer S., Perona‐Wright G., Zaph C., Infect. Immun. 2016, 84, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Myhill L. J., Stolzenbach S., Mejer H., Jakobsen S. R., Hansen T. V. A., Andersen D., Brix S., Hansen L. H., Krych L., Nielsen D. S., P N., Thamsborg S. M., Williams A. R., J. Immunol. 2020, 204, 3042. [DOI] [PubMed] [Google Scholar]
- 18. Love M. I., Huber W., Anders S., Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goedhart J., Luijsterburg M. S., Sci. Rep. 2020, 10, 20560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Metsalu T., Vilo J., Nucleic Acids Res. 2015, 43, W566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deschoolmeester M. L., Manku H., Else K. J., Infect. Immun. 2006, 74, 6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holm J. B., Sorobetea D., Kiilerich P., Ramayo‐Caldas Y., Estellé J., Ma T., Madsen L., Kristiansen K., Svensson‐Frej M., PLoS One 2015, 10, e0125495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasnain S. Z., Thornton D. J., Grencis R. K., Parasite Immunol. 2011, 33, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ariga T., Seki T., BioFactors 2006, 26, 93. [DOI] [PubMed] [Google Scholar]
- 25. Sorlozano‐Puerto A., Albertuz‐Crespo M., Lopez‐Machado I., Gil‐Martinez L., Ariza‐Romero J. J., Maroto‐Tello A., Baños‐Arjona A., Gutierrez‐Fernandez J., Pharmaceuticals 2021, 14, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sorlozano‐Puerto A., Albertuz‐Crespo M., Lopez‐Machado I., Ariza‐Romero J. J., Baños‐Arjona A., Exposito‐Ruiz M., Gutierrez‐Fernandez J., Biomed. Res. Int. 2018, 7861207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abad P., Arroyo‐Manzanares N., Rivas‐Montoya E., Ochando‐Pulido J. M., Guillamon E., García‐Campaña A. M., Martinez‐Ferez A., Can. J. Anim. Sci. 2018, 99, 244. [Google Scholar]
- 28. Abad P., Arroyo‐Manzanares N., García‐Campaña A. M., Anal. Methods 2016, 8, 3730. [Google Scholar]
- 29. Vezza T., Garrido‐Mesa J., Diez‐Echave P., Hidalgo‐García L., Ruiz‐Malagón A. J., García F., Sánchez M., Toral M., Romero M., Duarte J., Guillamón E., Arjona A. B., Moron R., Galvez J., Rodríguez‐Nogales A., Rodríguez‐Cabezas M. E., Nutrients 2021, 13, 2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang M., Tian X., Wang Y., Wang D., Li W., Chen L., Pan W., Mehmood S., Chen Y., Int. J. Biol. Macromol. 2018, 107, 2679. [DOI] [PubMed] [Google Scholar]
- 31. Schnittker D., Kwofie K., Ashkar A., Trigatti B., Richards C. D., Mediators Inflamm. 2013, 317503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Triantafilou M., Triantafilou K., Trends Immunol. 2002, 23, 301. [DOI] [PubMed] [Google Scholar]
- 33. Gregory C. D., Curr. Opin. Immunol. 2000, 12, 27. [DOI] [PubMed] [Google Scholar]
- 34. Meng Q., Xia Y., Protein Cell 2011, 2, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Charron C. S., Dawson H. D., Albaugh G. P., Solverson P. M., Vinyard B. T., Solano‐Aguilar G. I., Molokin A., Novotny J. A., J. Nutr. 2015, 145, 2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stepniak E., Ricci R., Eferl R., Sumara G., Sumara I., Rath M., Hui L., Wagner E. F., Genes Dev. 2006, 20, 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yan Q., Xiaorong Z., Zhang Z., Bing W., Feng Y., Hong B., Pathol. Pract. 2017, 213, 1173. [DOI] [PubMed] [Google Scholar]
- 38. Kishi Y., Kondo T., Xiao S., Yosef N., Gaublomme J., Wu C., Wang C., Chihara N., Regev A., Joller N., J. Exp. Med. 2016, 213, 2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Sluis G. L., Niers T. M. H., Esmon C. T., Tigchelaar W., Richel D. J., Buller H. R., Van Noorden C. J. F., Spek C. A., Blood 2009, 114, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dumas A., Lagarde S., Laflamme C., Pouliot M., J. Cell. Mol. Med. 2012, 16, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamada T., Sato A., Hirano T., Yamamoto T., Son G., Onodera M., Torii I., Nishigami T., Tanaka M., Miyajima A., Nishiguchi S., Fujimoto J., Tsujimura T., Am. J. Pathol. 2007, 171, 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hahm E.‐R., Kim S.‐H., Mathan S. V., Singh R. P., Singh S. V., J. Cancer Prev. 2021, 26, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rapp M., Wintergerst M. W. M., Kunz W. G., Vetter V. K., Knott M. M. L., Lisowski D., Haubner S., Moder S., Thaler R., Eiber S., Meyer B., Röhrle N., Piseddu I., Grassmann S., Layritz P., Kühnemuth B., Stutte S., Bourquin C., Andrian U. H., Endres S., Anz D., J. Exp. Med. 2019, 216, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dogan R. E., Long N., Forde E., Dennis K., Kohm A. P., Miller S. D., Karpus W. J., J. Leukoc. Biol. 2011, 89, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flier J., Boorsma D. M., van Beek P. J., Nieboer C., Stoof T. J., Willemze R., Tensen C. P., J. Pathol. 2001, 194, 398. [DOI] [PubMed] [Google Scholar]
- 46. Carl Jr J. W., Bai X.‐F., Int. J. Clin. Exp. Pathol. 2008, 1, 117. [PMC free article] [PubMed] [Google Scholar]
- 47. Anoush M., Eghbal M. A., Fathiazad F., Hamzeiy H., Kouzehkonani N. S., Pak. J. Biol. Sci. 2009, 12, 765. [DOI] [PubMed] [Google Scholar]
- 48. Andersen‐Civil A., Leppa M. M., Thamsborg S. M., Salminen J.‐P., Williams A. R., Commun. Biol. 2021, 4, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spanier G., Xu H., Xia N., Tobias S., Deng S., Wojnowski L., Forstermann U., Li H., J. Physiol. Pharmacol. 2009, 60, 111. [PubMed] [Google Scholar]
- 50. Esworthy R. S., Yang L., Frankel P. H., Chu F.‐F., J. Nutr. 2005, 135, 740. [DOI] [PubMed] [Google Scholar]
- 51. Tilli C., Stavast‐Kooy A. J. W., Vuerstaek J. D. D., Thissen M., Krekels G. A. M., Ramaekers F. C. S., Neumann H. A. M., Arch. Dermatol. Res. 2003, 295, 117. [DOI] [PubMed] [Google Scholar]
- 52. Bancroft A. J., Grencis R. K., Parasitology 2021, 148, 1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tritten L., Tam M., Vargas M., Jardim A., Stevenson M. M., Keiser J., Geary T. G., Exp. Parasitol. 2017, 178, 30. [DOI] [PubMed] [Google Scholar]
- 54. Cliffe L. J., Potten C. S., Booth C. E., Grencis R. K., Infect. Immun. 2007, 75, 1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dkhil M. A., Abdel‐Baki A. S., Wunderlich F., Sies H., Al‐Quraishy S., Vet. Parasitol. 2011, 175, 66. [DOI] [PubMed] [Google Scholar]
- 56. Fesseha H., Goa E., Food Technol. Nutr. Sci. Open J. 2019, 5, 107. [Google Scholar]
- 57. Glover M., Colombo S. A. P., Thornton D. J., Grencis R. K., PLoS Pathog. 2019, 15, e1007926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cliffe L. J., Grencis R. K., Adv. Parasitol. 2004, 57, 255. [DOI] [PubMed] [Google Scholar]
- 59. Renton K. W., Curr. Drug Metab. 2004, 5, 235. [DOI] [PubMed] [Google Scholar]
- 60. Kim D. K., Lillehoj H. S., Lee S. H., Lillehoj E. P., Bravo D., Br. J. Nutr. 2013, 109, 76. [DOI] [PubMed] [Google Scholar]
- 61. Amatya N., Garg A. V., Gaffen S. L., Trends Immunol. 2017, 38, 310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Speckmann B., Pinto A., Winter M., Förster I., Sies H., Steinbrenner H., Radic F., Biol. Med. 2010, 49, 777. [DOI] [PubMed] [Google Scholar]
- 63. Jumeau C., Awad F., Assrawi E., Cobret L., Duquesnoy P., Giurgea I., Valeyre D., Grateau G., Amselem S., Bernaudin J.‐F., Karabina S. A., PloS One 2019, 14, e0217005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pejler G., Rönnberg E., Waern I., Wernersson S., Blood 2010, 115, 4981. [DOI] [PubMed] [Google Scholar]
- 65. Kakinoki A., Kameo T., Yamashita S., Furuta K., Tanaka S., Int. J. Mol. Sci. 2020, 21, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
Data Availability Statement
RNA‐seq data is available at GEO under accession numbers GSE178282, GSE178499, and GSE178655. All other data are contained within the manuscript and it's supporting files.