

Abstract
Alzheimer’s disease (AD) is usually manifested in patients with dementia, accompanied by anxiety and other mental symptoms. Emerging evidence from humans indicates that people who suffer from anxiety in their early life are more likely to develop AD in later life. Mitochondria, the prominent organelles of energy production in the brain, have crucial physiological significance for the brain, requiring considerable energy to maintain its normal physiological activities. Net reactive oxygen species (ROS) was produced by mitochondrial impairment, in which oxidative stress is also included, and the production of ROS is mostly more than that of removal. In this paper, we propose that as a critical process in brain pathology, mitochondrial dysfunction caused by anxiety triggering oxidative stress might be a possible mechanism that links early life anxiety to AD in later life. Several pivotal physiological roles of mitochondria are reviewed, including functions regulating glucose homeostasis, which may disrupt in oxidative stress. Increased levels of oxidative stress are constantly shown in anxiety disorder patients, and antioxidant drugs have promise in treating anxiety. In the early stages of AD, mitochondrial dysfunction is concentrated around senile plaques, a landmark lesion composed of aggregated Aβ and Tau protein. In turn, the accumulated Aβ and Tau disrupts mitochondrial activity, and the tricky physiological processes of mitochondria might be significant to the course of AD. In the end, we conclude that mitochondria might present as one of the novel therapeutic targets to block oxidative stress in patients with anxiety disorders to prevent AD in the early stage.
Keywords: Alzheimer’s disease, anxiety, early life, mitochondrial dysfunction, oxidative stress
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia in later life. The pathophysiological mechanism of AD progression to dementia is extracellular neuroinflammatory plaque and fiber accumulation (β amyloid aggregation, neuronal fiber tangles and hyperphosphorylated tau protein aggregation, etc.), leading to extensive neuronal loss and altered neurotransmitter systems. β amyloid (Aβ) plaque-like deposition results from incomplete clearance or overproduction of peptides and is the most critical aspect of AD pathogenesis, which further leads to neurofibrillary tangles and cell death, culminating in cognitive dysfunction [1]. Recently, growing evidence shows that early life anxiety has enduring effects on the central nervous system which has matured and, consequently, on behaviors in later life [2-6]. One of the meta-analyses on the association between anxiety and risk of AD indicated that anxiety is slightly involved with an increased risk of AD based on a total of 24,528 participants, yet a high degree of heterogeneity across the studies and the inconsistency in the way of anxiety measuring might lead to the differences to be insignificant [7]. Another recent meta-analysis confirmed that anxiety significantly increases the risk of dementia by 29% [8]. Mitochondrion-centered hypometabolism is a key feature of brain aging and AD [9]. Aging is a process characterized by the gradual loss of tissue and organ function. The oxidative stress theory of aging is based on the hypothesis that age-associated functional losses are due to the accumulation of reactive oxygen species (ROS) -induced damages [10]. Given the long-term association between anxiety disorder and dementia, brain mitochondrial dysfunction and oxidative stress have a causal link between anxiety and AD.
Compared to other organs, the brain is susceptible to oxidative stress. Considering the brain not only has a high rate of oxygen consumption, but also has high requirements for transition metal ions and antioxidant enzymes that are involved in redox reactions [11]. There is growing evidence that free radical oxygen damage, mitochondrial dysfunction, accumulation of oxidative condensates, inflammation and defects in scavenging proteins constitute a complex system of pathological damage that act together on brain nerve cells, a pathological mechanism that has similarities in neurodegenerative diseases (e.g., AD, Parkinson’s disease). On the other hand, the differences of the pathology of neurodegenerative diseases are recognized as the result of complex interactions among genetic, environmental, aging, lifestyle and psychosocial factors. AD is the most common neurodegenerative disease, accounting for an estimated 60-70% of all dementia cases worldwide [12]. Therefore, here we attempt to elaborate the role played by oxidative stress in the development of AD in later life in patients with early life anxiety. A study has shown that oxidative stress plays a significant part in the progression of AD [13]. Mitochondrial and oxidative damage will cause an unbalance both in the production and removal the ROS, thereby increasing the net ROS [14]. As we know, the energy supply of neurons entirely relies on mitochondrial oxidative phosphorylation (OXPHOS). When OXPHOS is impaired, neurons have limited ability to obtain energy through glycolysis, which makes them particularly vulnerable to mitochondrial dysfunction [15]. Excessive stimulation of the electron transport chain can promote the overproduction of mitochondrial reactive oxygen species, leading to oxidative stress. Many disease mechanisms involve oxidative stress. Mitochondrial proteins, lipids, and DNA will undergo various oxidation reactions, and nerve cells will also be impaired [16]. Several studies pointed anxiety might be associated with decreased antioxidant defense capabilities and up-regulated oxidative damage to proteins, lipids, and nucleic acids. Notably, the oxidative modification of proteins is considered a potential factor in the occurrence and progression of anxiety [17].
In the following sections, we summarized several mechanisms that may account for this connection, focusing on brain mitochondrial dysfunction, supposed to be possible mechanisms that link early life anxiety to AD in later life.
2. Early life anxiety proved a risk factor for AD clinically
The prevalence of anxiety symptoms in AD is about 40%, and it can be a prelude to AD. Anxiety often occurs early in the course of AD, especially among patients with mild cognitive impairment (MCI), mild dementia, or early-onset forms of the disease, and can promote progression and conversion from MCI to dementia (Table 1) [18].
Table 1.
Anxiety as a risk factor of Alzheimer’s disease in clinical trials.
| Year/Study design/Group | Mean age (SD) |
Country/Source of data/Study setting | Diagnostic criteria: Anxiety/AD | Main findings | Conclusions | Ref. |
|---|---|---|---|---|---|---|
| 1999/None/Community-dwelling AD patients (n=523) | \ | USA/University of Washington and Group Health Cooperative of Puget Sound/Clinic | DSM-IV/CT | Anxiety symptoms were common, occurring in 70% of subjects. | Anxiety symptoms were common and significantly related to ADL and additional neuropsychiatric problems in this sample. | [149] |
| 2003/Cross-sectional study/Probable AD (n=115), VaD (n=43), FTD (n=33), Controls(n=40) | 77.2(7.6); 75.1(9.8); 65.8(8.5); 73.6(6.1) | USA/UCLA Alzheimer’s Disease Center database/Clinic | NPI/NINCDS-ADRDA | In AD, anxiety was more prevalent among patients with a younger age at onset (under age 65). | In AD, anxiety is common in those with more severe cognitive deterioration and an earlier age at onset. | [150] |
| 2010/None/a-MCI (n=19), Ade (n=15), Adm (n=12), HS (n=23) | 73.3(6.9), 75.5(7.0), 70.0(8.2), 63.9(9.5) | Italy/Pecialist dementia clinic of Santa Lucia Foundation/Clinic | NPI-12, VBM/MRI | Anxiety was present in both a-MCI and AD. | Anxiety is present since the earliest AD stages. | [151] |
| 2013/Prospective pilot study/Early stage of AD patients (n=54), Healthy controls (n= 64) | 76.9(8.5); 69.3(8.7) | Switzerland/Memory Clinic of the Old Age Psychiatry Service of the Lausanne University /Clinic | NPI-Q/NINCDS-ADRDA | Behavioral and psychological symptoms, in particular apathy, anxiety, are frequent occurrences in early-stage AD. | Premorbid personality was not associated with BPS in early stage of AD, although complex and non-linear relationships between the two are not excluded. | [152] |
| 2014/None/EOAD patients (n=23), LOAD patients (n=22) | 57.68(4.19); 80.32(5.89) | USA/Departments of Neurology and Geriatric Psychiatry at the Veterans Affairs Greater Los Angeles Healthcare Center /Clinic | NPI/- | EOAD patients had significantly more anxiety symptoms than LOAD patients. | Among LOAD patients, anxiety was associated with psychotic and activating psychiatric symptoms. | [153] |
| 2015/Prospective cohort study/Healthy, older adults (n=333) | 70.0(6.8) | Australia/Australian Imaging, Biomarkers, and Lifestyle Study/Clinic | HADS/PET, APOE genotyping | A positive Aβ status at baseline was associated with elevated anxiety symptoms; Compared with the Aβ+, low-anxiety group, slopes of cognitive decline were significantly more pronounced in the Aβ+ high-anxiety group. | Elevated anxiety symptoms moderate the effect of Aβ on cognitive decline in preclinical AD, resulting in more rapid decline in several cognitive domains. | [154] |
| 2015/Cross-sectional study/Mild Dementia (n = 55), Moderate Dementia (n = 17), Severe Dementia (n = 20) | 58.8(4.1); 58.8(3.7); 59.7(3.1) | Japan/Kumamoto University Hospital/Clinic | NPI/MRI | Scores of the anxiety increased significantly with increased dementia severity. | Hallucinations, depression, and anxiety showed different patterns in EOAD. | [155] |
| 2017/None /EOAD (n = 16), NCs (n = 19) | 57.6 (4.2); 55.9(8.9) | USA/Greater Los Angeles Healthcare Center/Clinic | NPI/- | On the Neuropsychiatric Inventory, the ORs among the EOAD patients significantly correlated with anxiety scores. | Anxiety in mild EOAD may be associated with widening attentional refocusing to socioemotional stimuli, possibly reflecting decreased sensorimotor gating in the entorhinal cortex. | [156] |
| 2018/Longitudinal study/Community-dwelling, cognitively normal elderly individuals (n=270) | 73.6(6.1) | USA/Harvard Aging Brain Study/Community sample | Anxiety-concentration cluster/PiB-PET, Hollingshead score, AMNART | Higher PiB binding also predicted steeper rates of increase for anxiety-concentration scores. | A direct or indirect association of elevated amyloid beta levels with worsening anxious-depressive symptoms and support the hypothesis that emerging neuropsychiatric symptoms represent an early manifestation of preclinical Alzheimer's disease. | [157] |
| 2019/Longitudinal study/No AD (n=3968), Incident AD(n=87) | 72.83(9.03); 83.72(7.13) | Spain/Zaragoza Dementia and Depression project/Population-based | GMS-AGECAT/- | Significant association between anxiety cases at baseline and AD risk in the univariate analysis that persisted in the fully adjusted model; No significant association between 'subcases' of anxiety at baseline and AD risk was found. | Late-life, clinically significant anxiety (but not subclinical anxiety) seems to increase the risk of AD, independently of the effect of several confounders, including depression. | [158] |
| 2019/None/EOAD(n=24), LOAD(n=56) | 59.3(6.0), 82.3(4.9) | UK/Memory services of the South London and Maudsley NHS Foundation Trust /Clinic | NPI/- | Participants with EOAD were significantly worse on anxiety subscales. | The NPS severity was similar between EOAD and LOAD although EOAD had higher symptom prevalence and career distress. | [159] |
| 2020/Longitudinal study/CU(n=104), MCI(n=53) | 52(50.0); 22(41.5) |
Sweden/Swedish BioFINDER study/Clinic | HADS/MRI, Amyloid PET scanning | Apathy and anxiety were shown related to Aβ deposition and predicted cognitive decline; Anxiety also interacted with amyloid status to predict faster cognitive deterioration. | The associations between apathy and anxiety with Aβ deposition and cognitive decline point to these symptoms as early clinical manifestations of Alzheimer's disease. | [160] |
DSM-IV: the diagnostic and statistical manual of mental disorders, 4th ed; CT: computed tomography; ADL: activities of daily living; VaD: vascular dementia; FTD: frontotemporal dementia; NPI: neuropsychiatric inventory; NINCDS-ADRDA: The National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association; a-MCI: amnestic mild cognitive impairment; Ade: early Alzheimer’s disease; Adm: moderate Alzheimer’s disease; VBM: voxel-based-morphometry; MRI: Magnetic resonance imaging; NPI-Q: neuropsychiatric inventory questionnaire; BPS: behavioral and psychological symptoms; EOAD: early-onset Alzheimer disease; LOAD: late-onset Alzheimer disease; HADS: hospital anxiety and depression scale; PET: positron emission tomography; AMNART: American national adult reading test; GMS-AGECAT: Geriatric mental state schedule- automated geriatric examination for computer assisted taxonomy.
Although there is a consensus on the positive association between dementia and anxiety, it is noted that results vary considerably from study to study, with a higher probability of dementia of up to 46.7% reported recently [19]. The presence of cognitive impairment, reduced ability to perceive symptoms and verbal communication disorders can make the diagnosis of elderly patients more complex [20]. Anxiety in AD patients tends to present differently from anxiety in non-demented elderly, which differs from that of younger patients [21]. This discrepancy is due to the different thresholds set by the diagnostic criteria [7,22]. In the third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III), anxiety neurosis was split into generalized anxiety disorder (GAD) and panic disorder. Since the revision of DSM-III, worries about various life events have been gradually emphasized as the distinctive symptom of GAD. Thus, the cognitive aspect of anxiety has become a core criterion for GAD [23]. 41.73% of the registered psychiatric inpatients were diagnosed with dementia in which AD was the dominant subtype [24]. The prevalence of anxiety disorder is also disproportionally high among patients diagnosed with MCI, which presents as a relatively mild form of the cognitive deficit between normal aging and dementia [25]. It is found that neuropsychiatric symptoms can be risk factors for cognitive decline in clinically normal older adults [26]. The reported prevalence of anxiety in MCI patients ranged between 9.9%-52% [27]. Even before MCI or other cognitive symptoms, anxiety disorder appears to predict the progression of AD [28]. Gulpers et al. found that anxiety is associated with an increased risk for cognitive impairment and dementia in the community. Stronger associations were driven by higher age, suggesting a prodromal symptom [29]. Other studies, however, indicate that anxiety disorder is not a risk factor. Still, an early symptom of the neurodegenerative process of AD, advocating an overlap between the neuropathological events causing cognitive deficits and those causing anxiety disorder symptoms in dementia [30,31]. A large body of structural and functional imaging data show that a neuropathological basis in cognition correlates with the progression of anxiety disorder.
The advent of cerebrospinal fluid (CSF) and fluorodeoxyglucose-positron emission tomography (FDG-PET) methods for the assessment of amyloid and tau has allowed the characterization of these more direct AD biomarkers to identify early stages and high-risk populations [32]. In the case of amyloid, its rise in the brain is thought to precede structural magnetic resonance imaging and FDG-PET changes [33]. Therefore, several studies have attempted to determine the relationship between anxiety and these in vivo measures of AD pathology (Table 1). Overall, anxiety has been strongly associated with direct biomarkers of amyloid and tau in vivo, in the same way as FDG-PET abnormalities. Furthermore, as with hypometabolism on FDG-PET, there is evidence that anxiety appears to enhance the effect of brain amyloid to progression, although its role in normal subjects seems less clear. The regional distribution of amyloid affects anxiety also needs to be further explored, as it seems to vary based on stage. Such work would also benefit from the advent of specialized tau PET imaging, as regional tau distribution has been shown to correlate better with symptoms than amyloid plaques [34].
3. Mitochondrial function in the physiological and pathological brain
There are roughly two sources of mitochondria in the brain. One is called non-synaptic mitochondria, mainly from neurons and glial cell bodies, and the other is called synaptic mitochondria, mainly from nerve terminals [35]. Using two-dimensional differential gel electrophoresis and mass spectrometry to detect synaptic proteins, the data show significant differences in superoxide dismutase [Mn] (Sod2) and complement component 1Q subcomponent binding protein (C1qbp), which supports the idea that synapses are highly sensitive to oxidative stress[36]. ROSs are small biological molecules, including superoxide (O2-), hydrogen peroxide (H2O2), and hydroxyl (OH-) free radicals, which are continuously and naturally produced in aerobic organisms [37]. The role of mitochondria as an organelle of energy metabolism is more prominent. Compared with nonneuronal cells, oxidative damage caused by mitochondria had a greater influence on the central neuron [38]. It is found that mitochondria provide energy for messaging by focusing on presynaptic terminals through active transport. Mitochondria target metabolic fluxes to generate power by interacting with glial cells around the synapse [36]. Most of the brain's energy supply is consumed by neuronal postsynaptic currents. It is essential to describe how neurons control the mitochondria where it distributes or displays the understanding of how synapses communicate with each other [39].
The neuronal activity requires the mitochondrial OXPHOS system at all times. Respiratory complexes I to IV and complex V form the electron transport chain (ETC). The coordinated operation of the ETC makes the mitochondria release energy usually. These redox reactions generate energy in the form of ATP, and release ROS, which can participate in cell signal transduction and ultimately cause oxidative stress [40]. Metabolic coordination between neurons and astrocytes is critical for the brain’s health [41]. In the brain, mROS derived from astrocytes are produced at an order of magnitude faster than neurons [42]. When the mitochondrial membrane-associated type 1 cannabinoid receptor (mtCB1) is activated by cannabinoids, phosphorylation of NDUFS4, a subunit of mitochondrial complex I, is reduced, further leading to instability and low activity of complex I. As a result, the ROS generated by astrocytes occurs to a reduction, which impacted the lactate levels produced by the glycolytic process through the hypoxia-inducible factor 1 pathway. All these interactions would act on neuronal redox stress [43]. Thus, when the stability of the mitochondrial complex is disrupted, the mROS initially released by astrocytes is transmitted to the neurons, thus subjecting the neurovascular unit to an energetic crisis and ultimately leading to neurodegeneration (Table 2) [44].
Table 2.
Mitochondrial dysfunctions in AD.
| Experiment models | Mitochondria dysfunctions | Mechanisms | Ref. |
|---|---|---|---|
| ApoE KO mice; Aβ1-40 | Oxidative stress, mitochondrial dysfunction and caspase activation are up-regulated. | Thiobarbituric acid-reactive substances were at a higher level, which is in accordance with the situation of ApoE KO mice synapses occurred to lipid peroxidation. | [161] |
| Tg2576 mice | Mitochondrial stress response; altered the respiratory chain complexes I and III's protein subunit; down-regulated the state 3 respiration and noncontinuous brain mitochondria respiration; reduced glucose metabolism. | The changes in mitochondrial proteome and function in Tg2576 mice brain precede plaque pathology. | [162] |
| Tg mice | Synaptic mitochondria accumulate the Aβ; mitochondrial alterations; up-regulate the transform of mitochondrial permeability up-regulate the mitochondrial oxidative stress; down-regulate the ECT function and the COX activity; | Synaptic mitochondria rich in Aβ would rather have Aβ-induced damage, and synaptic mitochondrial dysfunction is linked with the development of synaptic degeneration in AD. | [91] |
| Overexpressing ABAD Tg mice | Neuronal oxidative stress damage and memory loss. | The active site used to inhibit NAD binding has a substantial deformation shown in the crystal structure of Aβ-bound ABAD. ABAD peptide specifically prevents ABAD-Aβ from interaction and further restraints apoptosis triggered by Aβ and neuron free-radical generation. | [163] |
| CypD-deficient mAPP mice | Cortical mitochondria lack of CypD leads to having immunity in mitochondrial swelling and permeability transition induced by Aβ and Ca2+. | The synaptic function can be improved by CypD deficiency. | [164] |
| CypD-deficient mAPP mice | Down-regulated calcium caused by mitochondrial swelling; up-regulated the uptake ability of mitochondrial calcium; improved mitochondrial respiratory function. | Neuronal and synaptic stress that CypD mediated mPTP can be triggered by mAPP and oxidative stress. | [165] |
| CypD-deficient mice (primary cortical neurons and astrocytes) | The reason for synaptic versus nonsynaptic mitochondria has a difference in the Ca2+ handling is that in synaptic mitochondria, the levels of CypD are detected higher. | The neuronal mitochondria had a high level of CypD makes it vulnerable to mPTP. | [166] |
| Tg mAPP/ABAD mice | Spontaneous generation of hydrogen peroxide and superoxide anion, and decreased ATP, the release of cytochrome c from mitochondria and induction of caspase-3-like activity followed by DNA fragmentation and loss of cell viability. | ABAD-induced oxidant stress is related to cellular dysfunction accociated with AD. | [167] |
| Hippocampal neurons from embryonic day 18 rats; Aβ35-25 | Impaired mitochondrial transport; morphological changes; inhibited mitochondrial transport by acting through GSK3β. | To determine the important acute effect of Aβ molecules on nerve cells, which may lead to various abnormalities of neuronal function under AD conditions. | [168] |
| Human neuroblastoma M17 cells | Abnormal mitochondrial distribution pattern; reduced mitochondrial density. | The overexpression of DLP1 may be through the process of repopulating neurons with mitochondria to prevent ADDL-induced synapse loss, indicating that abnormal mitochondrial dynamics are fragile for the synaptic abnormal induced by ADDL. | [169] |
| Human neuroblastoma M17 cells (APP overexpression) | The perinuclear area is surrounded by fragmented structure and abnormal distribution; upgraded ROS levels, decreased mitochondrial potential difference, and reduced ATP releasement. | Fragmented mitochondria and abnormal distribution account for the mitochondrial and neuronal loss. | [170] |
| sAD patients’ fibroblasts | The characteristic of abnormal mitochondrial distribution is that slender mitochondria accumulate in the pernuclear area of 19.3% of sporadic AD (sAD) fibroblasts; decreased DLP1. | The reason why the levels of DLP1 reduced and mitochondrial allocation is unusual is that up-regulated oxidative stress and amyloid production in AD cells. | [171] |
| AD postmortem brain tissues, AβPP tg mice (primary hippocampal neurons) | Abnormal mitochondrial dynamics increase as AD progresses. | Crucial factors, including the increased production of Aβ and the interaction of Aβ with Drp1, lead to mitochondrial fragmentation, abnormal mitochondrial dynamics and synaptic damage. | [172] |
| Hippocampal neuron from C57BL/6day 1 pup; Aβ25-35 peptide | Less level of motile mitochondria; less average speed of motile mitochondria; decreased mitochondrial length; less synaptic immunoreactivity. | In neurons of AD models, toxic Aβ can impair mitochondrial movement, shorten the length of mitochondria, and endanger synaptic loss. | [173] |
| AD postmortem brain tissues; APP, APP/PS1 and 3XTg.AD mice | Elevated mitochondrial fission-linked GTPase activity. | Aβ and phosphorylated tau and Drp1 are entangled with each other, causing damage to mitochondria and synapses, which in turn leads to cognitive memory deficits. | [92] |
| AD postmortem brain tissues; CaMKIIα-tTA and tet-APPswe/ind mice | Enhanced mitophagy; depletion of cytosolic Parkin; reduced anterograde and increased retrograde transport of axonal mitochondria. | Chronic mitochondrial stress associated with AD under pathophysiological conditions in vitro and in vivo effectively triggers Parkin-dependent mitochondrial autophagy. | [174] |
| AD postmortem brain tissues; APP/PS1 mice | Down-regulated mitophagy. | Mitophagy enhancement eliminates AD-related tau hyperphosphorylation in human neuronal cells and reverses the memory impairment of genetically modified tau nematodes and mice. | [86] |
mPTP: mitochondrial permeability transition pore; ABAD: amyloid protein binding of alcohol dehydrogenase; COX: cytochrome c oxidase; GSK3β: glycogen synthase kinase 3β; DLP1: dynamin-like protein; OPA1: mitochondrial dynamin-like GTPase; Mfn1/2: mitofusin 1/2; ADDLs: amyloid-β-derived diffusible ligands; Opa1: mitochondrial dynamin-like GTPase 1; TOMM40: translocase of outer mitochondrial membrane 40.
4. Brain mitochondrial dysfunction in anxiety
The relationship between oxidative stress and anxiety symptoms was first described in 2001. In a non-clinical sample of healthy adults aimed at the levels of 8-hydroxy-2'-deoxyguanosine (8-OH-dG) in peripheral blood leukocytes, female subjects showed a positive relationship between the amount of 8-OH-dG and the psychosocial factors, such as anxiety [45]. In 2004, Sklan hypothesized that anxiety results from a complex, incompletely understood genome and external environment. According to his research, the acetyl-cholinesterase protein controls the termination of stress-enhanced acetylcholine signaling. In contrast, paraoxonase protein exhibits peroxidase-like activity, protecting blood proteins from oxidative stress. In addition, the instantaneous score of state anxiety and the susceptibility score of trait anxiety seem to be related to enzyme activity [46]. Olsen et al. tested the water maze, the situational fearfulness experiment and the elevated maze in the overexpressing mitochondrial catalase mice and concluded that overexpression of catalase may be sufficient to improve cognitive performance and reduce anxiety in the absence of changes in oxidative levels [47]. In 2014, a series of behavioral tests carried out on transgenic mice expressing the COXIV zip code uncovered an “anxiety-like” behavioral phenotype, indicating that the axon transport of nuclear-encoded mitochondrial mRNAs plays a pivotal part in neuronal physiology and rodent behavior [48]. Several studies confirm this connection, contributing to a better understanding of the intimated interaction between brain mitochondria and anxiety [49].
4.1. Mitochondrial oxidative stress acts as a core of mitochondrial dysfunction, illustrating the mechanisms of anxiety
Some scholars have incorporated antioxidants with potential neuroprotective effects, such as ascorbic acid, into their treatment strategies when studying mental illness. In the face of mood and anxiety disorders that are currently difficult to treat, ascorbic acid supplements have been presumed to be candidate drugs because of their fast onset, low toxicity, and high tolerability treatment response [50]. There are two main types of drugs most commonly used in the treatment of recurrent anxiety disorder in the world, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors (SNRIs) [51]. The above preparations may have an underestimated antioxidant effect that could underly the potential mechanisms [52]. Thioredoxin (Trx) is an antioxidant protein that reverses the oxidation of protein cysteines and promotes the scavenging of reactive oxygen species. Incubation of hippocampal cells from HT22 mice with fluoxetine for five consecutive days revealed upregulation of Trx protein levels, followed by inhibition of protein sulphonation and nitrosylation, suggesting that fluoxetine may protect against oxidative stress[53]. The genetic fragility of mitochondria leads to a calcium imbalance and leads to hyperexcitability of serotonergic neurons, which is thought to be susceptible to oxidative stress [54]. Basu et al. used the three-photon microscope to image serotonin in living preimplantation mouse embryos. Live embryos pre-incubated with serotonin showed higher mitochondrial potential, indicating the potential to regulate mitochondria [55]. In a word, these observations suggest an exciting link between the regulation of mitochondrial oxidative stress by serotonin and the anxiolytic effects of SSRIs.
4.2. Brain mitochondrial regulation of glucose homeostasis
The hippocampus, amygdala, and prefrontal cortex are brain regions associated with memory and emotion. After repeated stressful stimuli, structural remodeling is induced, which decreases memory and increases anxiety-like behavior and aggression. Structural and functional magnetic resonance imaging studies of anxiety disorders have demonstrated that the human brain may be affected similarly [56]. Another coherent connection linked oxidative stress to anxiety is brain mitochondria regulation of glucose homeostasis. High anxiety can lead to the release of sympathetic hormones, thereby increasing cortisol and glucose levels, reducing the insulin release, or affecting the sensitivity and resistance of insulin hormones [57]. From September 2013 to May 2015, Zhou et al. conducted a study on 646 anxiety patients who met the revised standards of the 10th edition of the International Statistical Classification of Diseases and Related Health Problems. It is found that the general probability of abnormal blood glucose regulation in anxiety patients accounts for 24.61%. The results of this article partially confirm the great quantity of impaired glucose regulation in patients with anxiety. Impaired glucose in the young group is closely related to the hypothalamic-pituitary-adrenal axis. By contrast, glucose impairment in the elderly group is closely associated with changes in the hypothalamic-pituitary-thyroid axis [58]. Impaired glucose homeostasis can, in turn, affect the normal function of insulin. Early studies on insulin focused on the periphery, where insulin takes up glucose by transporting and activating glucose transporter proteins in cell membranes. Later it is found that the central nervous system can also take up glucose through insulin-mediated transduction. Therefore the brain insulin signaling process in anxious patients should also be noticed. The improved well-being and self-confidence in normal-weight individuals after eight weeks of intranasal insulin treatment suggests that brain insulin modulates mood-related neural signaling pathways [59]. Brain insulin receptors are widely distributed in the olfactory bulb, cerebral cortex, hippocampus, hypothalamus, amygdala and vomeronasal nucleus, and their number decreases with age [1,33,59,60]. Studies have shown that anxiety, fear and stress are correlated. This chronic stress can cause amygdala hyperactivity and damage to the prefrontal cortex (PFC) and hippocampal structures, thereby impairing emotion regulation [61].
4.3. Translocator protein 18 kDa (TSPO)
Prior to 1977, little was known about the transporter protein 18 kDa (TSPO), a protein-coding gene known as an alternative binding site for the benzodiazepine diazepam. It is an evolutionarily conserved, tryptophan-rich protein that, together with voltage-dependent anion channels and the adenine nucleotide transporter (ANT), forms the mitochondrial permeability transition pore (MPTP) [62]. Lower levels of TSPO are expressed in healthy brain tissue. Disorders associated with TSPO include hepatic encephalopathy and generalized anxiety disorder. In addition, TSPO is overexpressed in different types of neuroinflammatory diseases (e.g., AD) but at downregulated levels in psychiatric disorders (e.g., anxiety disorders). [63]. The finding that the rs6971 polymorphism of the TSPO gene leads to amino acid substitutions in the fifth transmembrane loop of human proteins also supports the anxiety-related disorder TSPO abnormality [64]. When administered a series of TSPO ligands, the rodent models showed relief of anxiety symptoms. Recent progress has focused on the clinical progression of TSPO drug ligands. For example, recent research has found that rodent models and human volunteers administered by the selective TSPO ligand XBD173 (AC-5216, Emapunil) have a better anxiolytic effect [65].
5. Mitochondrial oxidative stress and the dysfunction of cerebral mitochondria in AD
Globally, 47 million people have dementia, and this number is estimated to increase to 131 million by 2050. AD is the commonest cause of dementia, with approximately 5 million new cases occurring annually [66,67]. Advances in diagnostic methods, such as biomarkers and computer-aided diagnosis based on image analysis, have been a significant step forward [68,69]. The current "biological" conception of AD is based on consideration of three biomarkers: amyloid, tau, and "neurodegeneration" [70]. Aggregation of the misfolded protein, such as β-amyloid and tau, is always accompanied by oxidative stress [71]. A large number of studies have demonstrated the important role of mitochondrial dysfunction and oxidative damage in the pathogenesis of AD. Oxidative stress shows in the early stages of the AD brain, before the occurrence of Aβ and Tau deposition [72]. Recent literature uncovered by researchers revealed the relationship between oxidative stress and AD pathology. Oxidative stress may influence the process of APP or tau metabolism. For instance, when the c-Jun amino-terminal kinase and p38 mitogen-activated protein kinase (MAPK) became activated, β-secretase was released caused by oxidative stress [73]. Meikeqi et al. isolated 8-OH-dG, a marker of nucleic acid oxidation, from mitochondrial DNA in the cerebral cortex. Further, 8-OH-dG gradually increases with age and appears to be more in the AD brain, which supports the hypothesis of oxidative stress [74]. However, there is still doubt whether oxidative stress should be a cause or a consequence of AD [75,76]. Birnbaum et al. detected elevated ROS levels in postmortem brain samples and rodent models. They believe that the increase in ROS may act an indispensable part in the occurrence of sporadic AD before the appearance of amyloid and tau protein pathology [77]. Sivandzade et al. hypothesized that overproduction of ROS and electrophiles is one of the main precursors to the occurrence and progression of AD [78]. Mitochondria are an essential source of H2O2, and their dysfunction is usually related to changes in the redox state. Yin et al. showed that chronic oxidative stress is also a significant factor in the cognitive decline associated with AD [9].
5.1. Mitochondrial oxidative stress links brain ROS production to AD
In preclinical models or human patients, cigarette smoke/smoking is consistent with AD neuropathology. Smoking-related brain oxidative stress is a potential mechanism that promotes the pathological process of AD [79]. Emerging evidence suggests that sleep deprivation and circadian rhythm disorder may interact and increase the risk of AD progression by increasing local brain oxidative stress and reducing circulating melatonin levels [80]. People who suffer from Type 2 diabetes and obesity are easier developed to AD than usual. Owning the habit of a Mediterranean diet can alleviate the above situations and so does other activities, such as strengthening physical exercise. Mediterranean diet is rich in vitamins, polyphenolic compounds, and partial fatty acids, which can be absorbed into the body to reduce oxidative stress [81]. Insufficiency of cerebral perfusion and hypo-glycemia can cause neuroinflammation and oxidative stress, leading to brain synaptic dysfunction and neuronal degeneration/loss, leading to gray and white matter atrophy, cognitive dysfunction, and AD [82]. The central nervous system communicates with peripheral blood through cells and factors circulating to the brain, and vice versa. Microarray and metabolomics suggest that central and peripheral glycolytic abnormalities and dysfunction of oxidative pathways are similar [83]. The short-term or sustained destruction of the integrity of the blood-brain barrier (BBB) is related to decreased CNS protection and increased permeability of pro-inflammatory (such as cytokines, ROS) substances from peripheral blood into the brain [84]. Chronic neuroinflammation has been well studied so far, and it's reported that inflammatory cytokines released by highly sensitive microglia can further trigger oxidative stress, which could illustrate the pathology of AD [85].
5.2. Aβ deposition, Tau aggregation and brain mitochondria
Mitochondrial dysfunction is shown in brain tissues from clinical samples and AD transgenic rodent models [86]. Numbers of evidence have shown multiple defects in mitochondria at the early stage of AD, such as impaired mitochondrial bioenergetics impairment, damaged mitophagy, decreased mitochondrial biosynthesis, and abnormal mitochondrial oxidative stress [87-90]. Aβ aggregates at synaptic terminals and can also enter synaptic mitochondria to cause synaptic impairment and abnormal mitochondrial function. It is found that Aβ can bind to mitochondrial proteins, such as mitochondrial splitting protein Drp1, mitochondrial outer membrane protein VDAC, etc [91,92]. These abnormal effects cause excessive free radical production, enhance mitochondrial splitting and affect the mitochondria biogenesis, ultimately leading to mitochondrial dysfunction. Mitochondrial DNA is close to the site of oxidative reactions. It is highly susceptible to damage due to the absence of histones and repair mechanisms in mitochondrial DNA, and this oxidative stress damage is most evident at AD synapses [73]. Tau loses control of microtubules and neurons accumulate as neurofibrillary tangles [93]. Hyperphosphorylated and overexpressed tau proteins accumulate around mitochondria and cause abnormal mitochondrial distribution. Moreover, pathological tau takes part mitochondrial dynamics by regulating mitochondrial fission/fusion proteins. All that damage leads to neuronal loss and mitochondrial disorder [94]. The reduced/oxidized form of nicotinamide adenine dinucleotide plays an important role in redox homeostasis and energy requirements [95,96]. The first molecule that enters the ETC of the mitochondria, is nicotinamide dinucleotide. The ETC comprises four major protein-metal complexes (I-IV), which trigger ATP production since the electrons flow. ATP synthase, also termed complex V, which is linked with OXPHOS. When the electron flow through the ETC, Complex V comes to the synthesis of ATP [97]. Repairing the disabled mitochondria by supplementing nicotinamide adenine dinucleotide (NAD) has been confirmed as a promising strategy for the treatment of AD and other dementia [98].
6. Oxidative stresses triggered by mitochondria dysfunction is a possible mechanism that links early life anxiety to AD in later life
The source of ROS may mainly date back to brain mitochondrial dysfunction, which is seen as a therapeutic target in the CNS. In oxidative stress, mitochondrial dysfunction is of great importance, and now literatures on the pathogenesis of AD and anxiety disorder are mainly underlain on them. A large amount of literature, including clinical research and rodent model research, have determined that up-regulated releasement of oxidative stress is related to AD and anxiety.
6.1. Akt1
Akt is related to neurological diseases. It has three isoforms, Akt1/Akt2/Akt3, with specific brain cell types that may have different effects on behavior [99]. An essential aspect of the Akt1 function is cellular metabolism and energy production. In some cases, activation of Akt1 strongly increases oxidative stress and causes apoptosis when cells gradually accumulate excess free radicals [100]. Accordingly, ROS can be involved in the oxidative modification of Akt1, which causes synaptic dysfunction in AD. Therefore, there is a growing number of therapeutic strategies dedicated to promoting synaptic Akt1-mTOR signaling [101]. Arvanitakis et al. conducted a clinicopathological study, originating from a community-based cohort study. In the second analysis, normalized pT308 Akt1 was positively correlated with amyloid load and tau tangles density. In addition, normalized pT308 Akt1 is linked with lower levels of overall cognition [102]. Another report showed that Akt1-KO mice had down-regulated tau phosphorylation at the familiar site of microtubule affinity-regulated kinase 2 (PAR1/MARK2) [103]. William et al. used different ratios of glucose and insulin to culture human stromal vascular cells and differentiated adipocytes and found increased Aβ secretion in the conditioned medium. Adipocytes were cultured with Aβ leading to decreased insulin receptor substrate-2 and reduced Akt1 phosphorylation [104]. The associations between the 17-item Hamilton Depression Rating Scale, total score, four factors, and the Akt1 rs2494746 and rs3001371 polymorphisms were analyzed through UNPHASED software. It seemed that Akt1 polymorphisms are connected with anxiety symptoms in patients suffering from depressive disorder [105].
6.2. Insulin
Insulin acts on the brain to affect cognition and mood. Brain insulin resistance and reduced insulin sensitivity in the central nervous system are standard features of dementia and aging [106]. Patients with AD have been found clinically to exhibit hyperinsulinemia on both fasting glucose and oral glucose tolerance tests. However, chronic hyperinsulinemia decreases the number of insulin receptors and thus the amount of insulin entering the brain [107,108]. The researchers suggest that finding targets of insulin resistance in the central nervous pathway may be an essential aspect of improving cognitive impairment [109]. Specifically, activated insulin signaling had brain glucose homeostasis in control and was resistant to the damage caused by synaptic loss, (neuro)inflammation, and oxidative stress [110-113]. Astrocytes take control of the metabolic rate of brain glucose level as well as neuronal activity. The activation of insulin signaling by astrocytes in the hypothalamus is explained by the fact that astrocytes readily co-control glucose and circulate glucose metabolism in the CNS by regulating the transport of glucose into the BBB [114]. Revealing the role of the astrocyte GLP-1R pathway could study the integrity of mitochondria. Adaptive stress response caused by GLP-1R pathway disorder further mounts an increase of memory forming and systemic glucose moderation [115]. Intranasal and peripheral insulin administration can be turned into a potential therapeutic target for AD, showing that it can improve the memory of AD patients [110,116,117]. Furthermore, it is confirmed that brain insulin has a good effect on anxiolytic both in patients and rodent models. The critical pathological symptoms of AD, including impaired memory, inflammation markers, energy utilization of neuronal and brain activity, have been facilitated by insulin when carrying out the first clinical trials in MCI/AD patients [118]. A study of albino vom strain mice in an elevated maze test found that insulin at a dose of 1.0 IU/kg was found to exert anxiolytic effects [119]. Soto and co-workers knocked out insulin receptors and IGF-1 receptors in the hippocampus or central amygdala of mice, which in turn led to defective insulin signaling, as evidenced by decreased levels of GluA1 subunits of the glutamate AMPA receptor on the one hand and increased levels of anxiety-like behavior, cognitive impairment, and metabolic disturbances (e.g., glucose intolerance) on the other. [120].
6.3. Serotonin and brain glucose homeostasis
The search for therapeutic targets to treat anxiety through the angiotensin-converting enzyme [ACE] type 2 (ACE2), angiotensin [Ang]-(1-7), and Mas receptor pathways of the renin-angiotensin system is promising [121]. SSRIs and SNRIs are first-line drugs to treat anxiety disorders [122]. The serotonin system is of great importance to anxiety pharmacology and molecular imaging [123]. Dopa decarboxylase is a crucial enzyme that participates in synthesizing both dopamine and serotonin [124]. It has been established that excitation mediated by dopaminergic (DA) neurons in the ventral tegmental area (VTA) and the mesocortical border pathway is associated with the progression of anxiety. Current research indicates that the dopamine D2 receptor signaling pathway that connects the VTA to the basolateral amygdala regulates fear and anxiety [123,125]. The dorsal medial part of the dorsal raphe nucleus is preferentially innervated by important forebrain structures related to anxiety state regulation, which can project the distributed nervous system that regulates anxiety. And partial serotonergic neurons in this brain area are activated by anxiety stimulation [126]. More and more importance is attached to the relationship between anxiety plots and other alterations, including changed γ-aminobutyric acid levels [127], increased insulin secretion and insulin resistance [128], and oxidative stress [129-131], which provides a promising target for anxiolytics. An extensive study on the serotonergic system has revealed a possible target for treating anxiety disorders considering its relationship with memory and oxidative stress, including behavioral/psychological symptoms of AD and dementia [132-134]. Furthermore, one of several mechanisms by which serotonin may directly affect the pathogenesis of AD is to up-regulate the α-secretase activity through 5-hydroxytryptamine (HT)4 receptors [135]. Ma et al. showed that fluoxetine effectively attenuated tau hyperphosphorylation at Ser396 [136].
Serotonin, also termed 5-hydroxytryptamine (5-HT), is of great importance in neurotransmitters, growth factors, and hormones, which account for a series of physiological functions [137]. Another way that 5-HT is involved with anxiety disorder and AD is to modulate brain glucose metabolism. There is some evidence that SSRIs have a beneficial effect on glucose homeostasis [138]. In addition, fluoxetine has been shown to reverse apoptosis and mitochondrial dysfunction by modulating mitochondrial respiratory chain components and Krebs cycle enzymes, and to affect mitochondria-related redox parameters [139]. Hydrogen sulfide targets modulate oxidative stress and neuroplasticity to treat pathological anxiety [129]. Researchers believe that the anxiety-related brain area involves the dorsal raphe nucleus. The serotonergic neurons in the dorsal/tail part can project to the limbic region of the forebrain [140]. The gut microbiome is also an effective medium for synthesizing intestinal-derived serotonin. The peripheral source of serotonin is itself a regulator of glucose homeostasis [141]. Knockout of 5-HT2CR on pro-opiomelanocortin neurons in hypothalamus arcuate nucleus (ARC) of mice did not affect body weight but showed hyperglycemia, hyperinsulinemia and insulin resistance, suggesting a clear role of central serotonin signaling in the regulation of glucose metabolism [142]. The clinical efficacy of fluoxetine plays a new mechanism through fluoxetine targeting glucose metabolism by regulating the palmitoylation of the glucose transporter [143]. To figure out how the shell of the nucleus accumbens (sNAc) serotonin affects systemic glucose metabolism, Deepenbroek et al. inserted microdialysis in the bilateral sNAc of male Wistar rats. One hour later, they detected upregulated blood glucose concentrations but no change in glucoregulatory hormones in the sNAc. [144]. It is worth noting that some experiments have addressed the effectiveness of using SSRIs to promote 5-HT-mediated synaptic communication to control anxiety in patients with dementia [145]. Other trials targeting SSRIs seem to be necessary to earn their potential value in treating AD (Table 3) [146].
Table 3.
Current studies on pharmacological treatment of agitation and psychosis in AD with anxiolytics.
| Mechanism of action | Name | Study population | Treatment | Results/status of the study | Study ID (ClinicalTrial.gov) |
|---|---|---|---|---|---|
| A selective 5-hydroxytryptamine (HT)2A receptor inverse agonist/antagonist | Pimavanserin | AD psychosis | Pimavanserin 34 mg vs. PLC | Significant improvement for pimavanserin Primary endpoint (week 6): Mean change in the Neuropsychiatric Inventory-Nursing Home version psychosis score; Pimavanserin versus PLC: -3.76 points (SE = 0.65) vs -1.93 points (0.63) (mean difference -1.84 [95% CI -3.64 to -0.04], Cohen’s d = -0.32; p = 0.045); No significant advantage for pimavanserin vs PLC at week 12 (treatment difference -0.51 [95% CI -2.23 to 1.21]; p = 0.561); |
ACP-103-019 |
| AD psychosis | Pimavanserin 34 mg vs. PLC | Significant efficacy in patients with higher baseline severity of psychotic symptoms (delta = -4.43, Cohen’s d = -0.73, p = 0.011); Pimavanserin vs PLC: ≥30% improvement was 88.9% vs. 43.3% (p < 0.001); ≥50% improvement was 77.8% vs. 43.3% (p = 0.008); | ACP-103-019 | ||
| A potent 5-HT2A antagonist, an inhibitor of serotonin reuptake | Lumateperone (ITI-007) | Agitation in patients with dementia, including AD | A phase 3, 4-week, randomized, double-blind, placebo-controlled, multi-center study (ITI-007, 9 mg/d vs. PLC) | No results posted | NCT02817906 |
| Serotonin and norepinephrine reuptake inhibitor | Dextromethorphan | Agitation in patients with AD | A phase 3, 12-week, multicenter, randomized, double-blind, placebo-controlled, parallel-design study (AVP-786 (dose 1) vs AVP-786 (dose 2) vs. PLC) | Ongoing (study is recruiting participants) | NCT03393520 |
| Selective serotonin reuptake inhibitor | Escitalopram | Agitation in AD | A phase 3, 12-week, randomized, double-blind, placebo-controlled trial (5-15 mg/day (target: 15mg/day if tolerated) of escitalopram vs. PLC) | Ongoing (study is recruiting participants) | NCT03108846 |
| A partial 5-HT1A receptor agonist | Tandospirone | AD psychosis | A phase 4, 12-week, randomized, open-label, parallel-group study (Tandospirone, 30-60 mg/d + Donepezil, 10 mg/d vs Donepezil, 10 mg/d.) | No results posted | NCT03151382 |
| Serotonin receptor antagonists and reuptake inhibitors | Trazodone | AD psychosis | A phase 3, 14-days, randomized, double-blind, placebo-controlled study (Trazodone tablets, 50 mg/d vs. PLC) | No results posted | NCT01142258 |
| An analog of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) | Gabapentin Enacarbil | Agitation in AD | A phase 4, pilot, 8-week, double-blind, placebo-controlled randomized clinical trial (GEn, 300 mg/d vs. PLC.) | Ongoing (study is recruiting participants) | NCT03082755 |
data available at: ClinicalTrials.gov (accessed February 12, 2022); filters used: agitation, psychosis, anxiolytics, and AD; Studies: recruiting; not yet recruiting; active, not recruiting; enrolling by invitation; PLC, placebo; AD, Alzheimer’s disease; GEn, Gabapentin Enacarbil.
7. Conclusion and prospect
As we all know, throughout the life cycle, early exposure to anxiety controls lifelong sensitivity to subsequent stress. Animals susceptible to anxiety disorders exhibit behaviors related to cognitive impairment. Although mitochondria have electron transport chains capable of producing ROS, the network of antioxidant defense systems is also developed [73]. Mitochondrial oxidative stress is caused by an imbalance between ROS generation and ROS detoxification. The observation that enhanced mitochondrial antioxidant defense reduces anxiety symptoms supports the importance of net mitochondrial ROS in producing anxiety. Discover the key molecular mechanisms that mediate the effects of anxiety in AD from mitochondrial dysfunction and make feasible biological targets that used to treat anxiety-induced sensitivity in early life, which may be the control of potential AD-related neurodegeneration diseases of this pharmacological target on possible clinical research has opened the door (Fig. 1).
Figure 1.
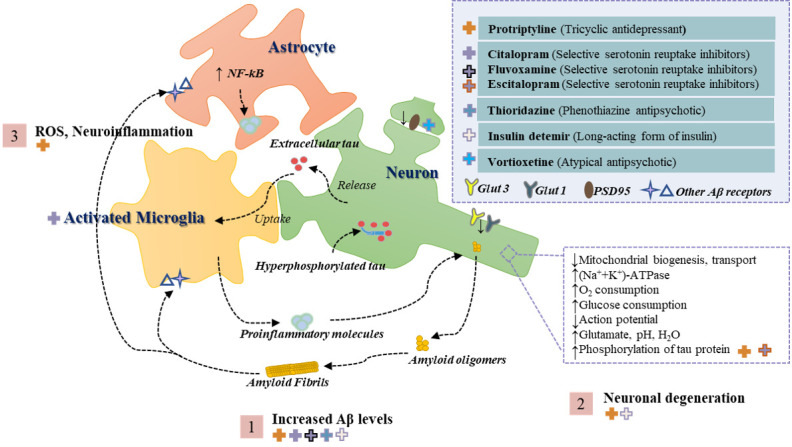
Overview of the pathogenesis of AD triggered by extracellular and intravascular Aβ deposition. Step 1 shows the interaction of Aβ oligomers and fibronectin with neuronal cells through several receptors which in turn triggers an overall increase in brain Aβ levels. Step 2 shows that neurons suffer from impaired mitochondrial bioenergetics and glucose homeostasis and thus degeneration. Step 3 illustrates that the neuroinflammatory environment and oxidative stress to which the neuronal cells are exposed can exacerbate the pathological process of AD. In addition, the top right corner of the figure shows the anxiolytic drugs reported in the literature for the treatment of AD models, with the crosses on the left corresponding to the respective antipathogenic mechanism in the figure and the classification of the drug in parentheses. Abbreviations: Aβ, amyloid peptide; GLUT, glucose transporter; NF-κB, nuclear factor kappa light chain enhancer of activated B cells; ROS, reactive oxygen species; PSD95, postsynaptic density protein-95.
The lifetime prevalence of anxiety is a known relatively decisive risk factor for AD, and it is reflected in many behavioral models of AD. Previous studies have shown that anxiety in the early stages of life can lead to a folding increase in the risk of adult AD, depending on its timing, intensity, and specific characteristics. It is also known that anxiety in the early stages of life will increase behavioral sensitivity, making people more affected by anxiety in the later stages of life. It will have a particularly strong effect on the nucleus accumbens (an important component of the brain's reward system). Through two functional magnetic resonance imaging studies, Li et al. found that anxiety affects processing efficiency far greater than performance efficiency, and the effect on processing efficiency of brain areas related to spatial working memory is much greater than that of verbal working memory, and the central brain area involved in the medial prefrontal lobe and anterior cuneiform lobe. At the same time, the activities of the medial frontal and subparietal lobules related to working memory of individuals with high trait anxiety are enhanced, the connection between the insula and the cingulate gyrus related to self-awareness is enhanced, but the connection between the cingulate gyrus and the amygdala responsible for emotion regulation is weakened. This part of the results reveals the unique emotions of anxious individuals and the brain networks related to working memory. In short, these results suggest an important cognitive basis for anxiety disorders and provide unique evidence for the neural mechanism of the interaction between anxiety and working memory [147,148].
In the future, it will be important to understand how mitochondrial changes in the neural circuits of anxiety and working memory. The importance of determining the characterization of mitochondrial function in different disease processions is much more than that only to consider the progress of research methods on mitochondria. Of note, anxiety phenotype shaped by brain mitochondrial dysfunction could contribute to the outcome of mitochondrial biogenesis, mitochondrial dynamics. Attaching more importance to the way how mitochondria shape the early life anxiety phenotypes that further leads to AD holds great promise among the efforts in the early prevention of AD of the future.
Acknowledgements
This work was supported by Natural Science Foundation of Shanghai, No. 21ZR1460900 to M Zhou, Innovation Project for Undergraduates of Shanghai University of Traditional Chinese Medicine (202110268240), and also sponsored by the General Program of National Natural Science Foundation of China (81773927).
Footnotes
Competing interests
All authors report no biomedical financial interests or potential conflicts of interest.
References
- [1].Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring HU (2016). Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev, 96:1169-1209. [DOI] [PubMed] [Google Scholar]
- [2].McClelland S, Korosi A, Cope J, Ivy A, Baram TZ (2011). Emerging roles of epigenetic mechanisms in the enduring effects of early-life stress and experience on learning and memory. Neurobiol Learn Mem, 96:79-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bland ST, Beckley JT, Young S, Tsang V, Watkins LR, Maier SF, et al. (2010). Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav Immun, 24:329-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kosten TA, Kim JJ, Lee HJ (2012). Early life manipulations alter learning and memory in rats. Neurosci Biobehav Rev, 36:1985-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Van den Bergh BR (2011). Developmental programming of early brain and behaviour development and mental health: A conceptual framework. Dev Med Child Neurol, 53Suppl 4:19-23. [DOI] [PubMed] [Google Scholar]
- [6].Lähdepuro A, Savolainen K, Lahti-Pulkkinen M, Eriksson JG, Lahti J, Tuovinen S, et al. (2019). The impact of early life stress on anxiety symptoms in late adulthood. Sci Rep, 9:4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Santabárbara J, Lipnicki DM, Bueno-Notivol J, Olaya-Guzmán B, Villagrasa B, López-Antón R (2020). Updating the evidence for an association between anxiety and risk of Alzheimer's disease: A meta-analysis of prospective cohort studies. J Affect Disord, 262:397-404. [DOI] [PubMed] [Google Scholar]
- [8].Santabárbara J, Lipnicki DM, Villagrasa B, Lobo E, Lopez-Anton R (2019). Anxiety and risk of dementia: Systematic review and meta-analysis of prospective cohort studies. Maturitas, 119:14-20. [DOI] [PubMed] [Google Scholar]
- [9].Yin F, Sancheti H, Patil I, Cadenas E (2016). Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic Biol Med, 100:108-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. (2018). Oxidative stress, aging, and diseases. Clin Interv Aging, 13:757-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Floyd RA (1999). Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med, 222:236-245. [DOI] [PubMed] [Google Scholar]
- [12].Leng F, Edison P (2021). Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol, 17:157-172. [DOI] [PubMed] [Google Scholar]
- [13].Zhu M, Gu F, Shi J, Hu J, Hu Y, Zhao Z (2008). Increased oxidative stress and astrogliosis responses in conditional double-knockout mice of Alzheimer-like presenilin-1 and presenilin-2. Free Radic Biol Med, 45:1493-1499. [DOI] [PubMed] [Google Scholar]
- [14].Andreyev AY, Kushnareva YE, Starkov AA (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc), 70:200-214. [DOI] [PubMed] [Google Scholar]
- [15].Streck EL, Gonçalves CL, Furlanetto CB, Scaini G, Dal-Pizzol F, Quevedo J (2014). Mitochondria and the central nervous system: Searching for a pathophysiological basis of psychiatric disorders. Braz J Psychiatry, 36:156-167. [DOI] [PubMed] [Google Scholar]
- [16].Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, et al. (2015). Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother, 74:101-110. [DOI] [PubMed] [Google Scholar]
- [17].Fedoce ADG, Ferreira F, Bota RG, Bonet-Costa V, Sun PY, Davies KJA (2018). The role of oxidative stress in anxiety disorder: Cause or consequence? Free Radic Res, 52:737-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mendez MF (2021). The relationship between anxiety and Alzheimer's disease. J Alzheimers Dis Rep, 5:171-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu S, Li C, Shi Z, Wang X, Zhou Y, Liu S, et al. (2017). Caregiver burden and prevalence of depression, anxiety and sleep disturbances in Alzheimer's disease caregivers in China. J Clin Nurs, 26:1291-1300. [DOI] [PubMed] [Google Scholar]
- [20].Wilkinson P, Ruane C, Tempest K (2018). Depression in older adults. BMJ, 363:k4922. [DOI] [PubMed] [Google Scholar]
- [21].Wolitzky-Taylor KB, Castriotta N, Lenze EJ, Stanley MA, Craske MG (2010). Anxiety disorders in older adults: A comprehensive review. Depress Anxiety, 27:190-211. [DOI] [PubMed] [Google Scholar]
- [22].Teng E, Ringman JM, Ross LK, Mulnard RA, Dick MB, Bartzokis G, et al. (2008). Diagnosing depression in Alzheimer disease with the national institute of mental health provisional criteria. Am J Geriatr Psychiatry, 16:469-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Crocq MA (2017). The history of generalized anxiety disorder as a diagnostic category. Dialogues Clin Neurosci, 19:107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shao Y, Xu H, Wang J, Dai X, Liang W, Ren L, et al. (2021). Agitation and apathy increase risk of dementia in psychiatric inpatients with late-onset psychiatric symptoms. BMC Psychiatry, 21:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Culpepper L, Lam RW, McIntyre RS (2017). Cognitive impairment in patients with depression: Awareness, assessment, and management. J Clin Psychiatry, 78:1383-1394. [DOI] [PubMed] [Google Scholar]
- [26].Burhanullah MH, Tschanz JT, Peters ME, Leoutsakos JM, Matyi J, Lyketsos CG, et al. (2020). Neuropsychiatric symptoms as risk factors for cognitive decline in clinically normal older adults: The Cache county study. Am J Geriatr Psychiatry, 28:64-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma L (2020). Depression, anxiety, and apathy in mild cognitive impairment: Current perspectives. Front Aging Neurosci, 12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuring JK, Mathias JL, Ward L (2020). Risk of dementia in persons who have previously experienced clinically-significant depression, anxiety, or PTSD: A systematic review and meta-analysis. J Affect Disord, 274:247-261. [DOI] [PubMed] [Google Scholar]
- [29].Gulpers B, Ramakers I, Hamel R, Köhler S, Oude Voshaar R, Verhey F (2016). Anxiety as a predictor for cognitive decline and dementia: A systematic review and meta-analysis. Am J Geriatr Psychiatry, 24:823-842. [DOI] [PubMed] [Google Scholar]
- [30].Burke SL, Cadet T, Alcide A, O'Driscoll J, Maramaldi P (2018). Psychosocial risk factors and Alzheimer's disease: The associative effect of depression, sleep disturbance, and anxiety. Aging Ment Health, 22:1577-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, et al. (2018). Analysis of shared heritability in common disorders of the brain. Science, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Patel P, Masurkar AV (2021). The relationship of anxiety with Alzheimer's disease: A narrative review. Curr Alzheimer Res, 18:359-371. [DOI] [PubMed] [Google Scholar]
- [33].Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. (2013). Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol, 12:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol, 71:362-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhuravliova E, Barbakadze T, Jojua N, Zaalishvili E, Shanshiashvili L, Natsvlishvili N, et al. (2012). Synaptic and non-synaptic mitochondria in hippocampus of adult rats differ in their sensitivity to hypothyroidism. Cell Mol Neurobiol, 32:1311-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Völgyi K, Gulyássy P, Háden K, Kis V, Badics K, Kékesi KA, et al. (2015). Synaptic mitochondria: A brain mitochondria cluster with a specific proteome. J Proteomics, 120:142-157. [DOI] [PubMed] [Google Scholar]
- [37].Ebrahimpour S, Zakeri M, Esmaeili A (2020). Crosstalk between obesity, diabetes, and alzheimer's disease: Introducing quercetin as an effective triple herbal medicine. Ageing Res Rev, 62:101095. [DOI] [PubMed] [Google Scholar]
- [38].Yang P, Sheng D, Guo Q, Wang P, Xu S, Qian K, et al. (2020). Neuronal mitochondria-targeted micelles relieving oxidative stress for delayed progression of Alzheimer's disease. Biomaterials, 238:119844. [DOI] [PubMed] [Google Scholar]
- [39].MacAskill AF, Atkin TA, Kittler JT (2010). Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci, 32:231-240. [DOI] [PubMed] [Google Scholar]
- [40].Giachin G, Bouverot R, Acajjaoui S, Pantalone S, Soler-López M (2016). Dynamics of human mitochondrial complex I assembly: Implications for neurodegenerative diseases. Front Mol Biosci, 3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. (2019). Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell, 177:1522-1535.e1514. [DOI] [PubMed] [Google Scholar]
- [42].Vicente-Gutierrez C, Bonora N, Jimenez-Blasco D, Lopez-Fabuel I, Bates G, Murphy MP, et al. (2021). Abrogating mitochondrial ROS in neurons or astrocytes reveals cell-specific impact on mouse behaviour. Redox Biol, 41:101917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jimenez-Blasco D, Busquets-Garcia A, Hebert-Chatelain E, Serrat R, Vicente-Gutierrez C, Ioannidou C, et al. (2020). Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature, 583:603-608. [DOI] [PubMed] [Google Scholar]
- [44].Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S (2019). Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J Clin Invest, 129:34-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Irie M, Asami S, Nagata S, Ikeda M, Miyata M, Kasai H (2001). Psychosocial factors as a potential trigger of oxidative DNA damage in human leukocytes. Jpn J Cancer Res, 92:367-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sklan EH, Lowenthal A, Korner M, Ritov Y, Landers DM, Rankinen T, et al. (2004). Acetylcholinesterase/paraoxonase genotype and expression predict anxiety scores in health, risk factors, exercise training, and genetics study. Proc Natl Acad Sci U S A, 101:5512-5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Olsen RH, Johnson LA, Zuloaga DG, Limoli CL, Raber J (2013). Enhanced hippocampus-dependent memory and reduced anxiety in mice over-expressing human catalase in mitochondria. J Neurochem, 125:303-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kar AN, Sun CY, Reichard K, Gervasi NM, Pickel J, Nakazawa K, et al. (2014). Dysregulation of the axonal trafficking of nuclear-encoded mitochondrial mRNA alters neuronal mitochondrial activity and mouse behavior. Dev Neurobiol, 74:333-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Misiewicz Z, Iurato S, Kulesskaya N, Salminen L, Rodrigues L, Maccarrone G, et al. (2019). Multi-omics analysis identifies mitochondrial pathways associated with anxiety-related behavior. PLoS Genet, 15:e1008358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moritz B, Schmitz AE, Rodrigues ALS, Dafre AL, Cunha MP (2020). The role of vitamin C in stress-related disorders. J Nutr Biochem, 85:108459. [DOI] [PubMed] [Google Scholar]
- [51].Craske MG, Stein MB (2016). Anxiety. Lancet, 388:3048-3059. [DOI] [PubMed] [Google Scholar]
- [52].Gałecki P, Mossakowska-Wójcik J, Talarowska M (2018). The anti-inflammatory mechanism of antidepressants - SSRIs, SNRIs. Prog Neuropsychopharmacol Biol Psychiatry, 80:291-294. [DOI] [PubMed] [Google Scholar]
- [53].Bharti V, Tan H, Deol J, Wu Z, Wang JF (2020). Upregulation of antioxidant thioredoxin by antidepressants fluoxetine and venlafaxine. Psychopharmacology (Berl), 237:127-136. [DOI] [PubMed] [Google Scholar]
- [54].Kato T (2019). Current understanding of bipolar disorder: Toward integration of biological basis and treatment strategies. Psychiatry Clin Neurosci, 73:526-540. [DOI] [PubMed] [Google Scholar]
- [55].Basu B, Desai R, Balaji J, Chaerkady R, Sriram V, Maiti S, et al. (2008). Serotonin in pre-implantation mouse embryos is localized to the mitochondria and can modulate mitochondrial potential. Reproduction, 135:657-669. [DOI] [PubMed] [Google Scholar]
- [56].McEwen BS (2006). Sleep deprivation as a neurobiologic and physiologic stressor: Allostasis and allostatic load. Metabolism, 55:S20-23. [DOI] [PubMed] [Google Scholar]
- [57].Wong H, Singh J, Go RM, Ahluwalia N, Guerrero-Go MA (2019). The effects of mental stress on non-insulin-dependent diabetes: Determining the relationship between catecholamine and adrenergic signals from stress, anxiety, and depression on the physiological changes in the pancreatic hormone secretion. Cureus, 11:e5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhou Y, Dong Z, A R, Liao Z, Guo J, Liu C, et al. (2019). The prevalence of impaired glucose regulation in anxiety disorder patients and the relationship with hypothalamic-pituitary-adrenal axis and hypothalamic-pituitary-thyroid axis activity. J Evid Based Med, 12:51-55. [DOI] [PubMed] [Google Scholar]
- [59].Hallschmid M, Benedict C, Schultes B, Born J, Kern W (2008). Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J Obes (Lond), 32:275-282. [DOI] [PubMed] [Google Scholar]
- [60].Böni-Schnetzler M, Méreau H, Rachid L, Wiedemann SJ, Schulze F, Trimigliozzi K, et al. (2021). IL-1beta promotes the age-associated decline of beta cell function. iScience, 24:103250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mah L, Szabuniewicz C, Fiocco AJ (2016). Can anxiety damage the brain? Curr Opin Psychiatry, 29:56-63. [DOI] [PubMed] [Google Scholar]
- [62].Barresi E, Robello M, Costa B, Da Pozzo E, Baglini E, Salerno S, et al. (2021). An update into the medicinal chemistry of translocator protein (TSPO) ligands. Eur J Med Chem, 209:112924. [DOI] [PubMed] [Google Scholar]
- [63].Scarf AM, Kassiou M (2011). The translocator protein. J Nucl Med, 52:677-680. [DOI] [PubMed] [Google Scholar]
- [64].Owen DR, Fan J, Campioli E, Venugopal S, Midzak A, Daly E, et al. (2017). TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem J, 474:3985-3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Costa B, Da Pozzo E, Martini C (2012). Translocator protein as a promising target for novel anxiolytics. Curr Top Med Chem, 12:270-285. [DOI] [PubMed] [Google Scholar]
- [66].Arvanitakis Z, Shah RC, Bennett DA (2019). Diagnosis and management of dementia: Review. JAMA, 322:1589-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tobore TO (2019). On the central role of mitochondria dysfunction and oxidative stress in Alzheimer's disease. Neurol Sci, 40:1527-1540. [DOI] [PubMed] [Google Scholar]
- [68].Santos LE, Beckman D, Ferreira ST (2016). Microglial dysfunction connects depression and Alzheimer's disease. Brain Behav Immun, 55:151-165. [DOI] [PubMed] [Google Scholar]
- [69].Mendoza-Léon R, Puentes J, Uriza LF, Hernández Hoyos M (2020). Single-slice Alzheimer's disease classification and disease regional analysis with supervised switching autoencoders. Comput Biol Med, 116:103527. [DOI] [PubMed] [Google Scholar]
- [70].Salardini A (2019). Interpretation of biomarker data in diagnosis of primary dementias. Semin Neurol, 39:200-212. [DOI] [PubMed] [Google Scholar]
- [71].Abramov AY, Potapova EV, Dremin VV, Dunaev AV (2020). Interaction of oxidative stress and misfolded proteins in the mechanism of neurodegeneration. Life (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol, 60:759-767. [DOI] [PubMed] [Google Scholar]
- [73].Lin MT, Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443:787-795. [DOI] [PubMed] [Google Scholar]
- [74].Mecocci P, Boccardi V, Cecchetti R, Bastiani P, Scamosci M, Ruggiero C, et al. (2018). A long journey into aging, brain aging, and Alzheimer's disease following the oxidative stress tracks. J Alzheimers Dis, 62:1319-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Andersen JK (2004). Oxidative stress in neurodegeneration: Cause or consequence? Nat Med, 10Suppl:S18-25. [DOI] [PubMed] [Google Scholar]
- [76].Luque-Contreras D, Carvajal K, Toral-Rios D, Franco-Bocanegra D, Campos-Peña V (2014). Oxidative stress and metabolic syndrome: Cause or consequence of Alzheimer's disease? Oxid Med Cell Longev, 2014: 497802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Birnbaum JH, Wanner D, Gietl AF, Saake A, Kündig TM, Hock C, et al. (2018). Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer's disease patients. Stem Cell Res, 27:121-130. [DOI] [PubMed] [Google Scholar]
- [78].Sivandzade F, Prasad S, Bhalerao A, Cucullo L (2019). NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol, 21:101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Durazzo TC, Mattsson N, Weiner MW (2014). Smoking and increased Alzheimer's disease risk: A review of potential mechanisms. Alzheimers Dement, 10:S122-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wu H, Dunnett S, Ho YS, Chang RC (2019). The role of sleep deprivation and circadian rhythm disruption as risk factors of Alzheimer's disease. Front Neuroendocrinol, 54:100764. [DOI] [PubMed] [Google Scholar]
- [81].Miranda A, Gómez-Gaete C, Mennickent S (2017). Role of Mediterranean diet on the prevention of Alzheimer disease. Rev Med Chil, 145:501-507. [DOI] [PubMed] [Google Scholar]
- [82].Daulatzai MA (2017). Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer's disease. J Neurosci Res, 95:943-972. [DOI] [PubMed] [Google Scholar]
- [83].Macchi B, Marino-Merlo F, Frezza C, Cuzzocrea S, Mastino A (2014). Inflammation and programmed cell death in Alzheimer's disease: Comparison of the central nervous system and peripheral blood. Mol Neurobiol, 50:463-472. [DOI] [PubMed] [Google Scholar]
- [84].Patel JP, Frey BN (2015). Disruption in the blood-brain barrier: The missing link between brain and body inflammation in bipolar disorder? Neural Plast, 2015: 708306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009). Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener, 4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci, 22:401-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Reddy PH, Oliver DM (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's disease. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. (2017). Mitophagy and Alzheimer's disease: Cellular and molecular mechanisms. Trends Neurosci, 40:151-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Agrawal I, Jha S (2020). Mitochondrial dysfunction and Alzheimer's disease: Role of microglia. Front Aging Neurosci, 12:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shoshan-Barmatz V, Nahon-Crystal E, Shteinfer-Kuzmine A, Gupta R (2018). VDAC1, mitochondrial dysfunction, and Alzheimer's disease. Pharmacol Res, 131:87-101. [DOI] [PubMed] [Google Scholar]
- [91].Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010). Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A, 107:18670-18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Manczak M, Reddy PH (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet, 21:2538-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gorantla NV, Chinnathambi S (2021). Autophagic pathways to clear the tau aggregates in Alzheimer's disease. Cell Mol Neurobiol, 41:1175-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cheng Y, Bai F (2018). The association of tau with mitochondrial dysfunction in Alzheimer's disease. Front Neurosci, 12:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dong Y, Sameni S, Digman MA, Brewer GJ (2019). Reversibility of age-related oxidized free NADH redox states in Alzheimer's disease neurons by imposed external Cys/CySS redox shifts. Sci Rep, 9:11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Dong Y, Digman MA, Brewer GJ (2019). Age- and AD-related redox state of NADH in subcellular compartments by fluorescence lifetime imaging microscopy. Geroscience, 41:51-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cadonic C, Sabbir MG, Albensi BC (2016). Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol Neurobiol, 53:6078-6090. [DOI] [PubMed] [Google Scholar]
- [98].Xie X, Gao Y, Zeng M, Wang Y, Wei TF, Lu YB, et al. (2019). Nicotinamide ribose ameliorates cognitive impairment of aged and Alzheimer's disease model mice. Metab Brain Dis, 34:353-366. [DOI] [PubMed] [Google Scholar]
- [99].Wong H, Levenga J, LaPlante L, Keller B, Cooper-Sansone A, Borski C, et al. (2020). Isoform-specific roles for AKT in affective behavior, spatial memory, and extinction related to psychiatric disorders. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Tang Y, Halvarsson C, Nordigården A, Kumar K, Åhsberg J, Rörby E, et al. (2015). Coexpression of hyperactivated AKT1 with additional genes activated in leukemia drives hematopoietic progenitor cells to cell cycle block and apoptosis. Exp Hematol, 43:554-564. [DOI] [PubMed] [Google Scholar]
- [101].Ahmad F, Singh K, Das D, Gowaikar R, Shaw E, Ramachandran A, et al. (2017). Reactive oxygen species-mediated loss of synaptic Akt1 signaling leads to deficient activity-dependent protein translation early in Alzheimer's disease. Antioxid Redox Signal, 27:1269-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Arvanitakis Z, Wang HY, Capuano AW, Khan A, Taïb B, Anokye-Danso F, et al. (2020). Brain insulin signaling, Alzheimer disease pathology, and cognitive function. Ann Neurol, 88:513-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Dickey CA, Koren J, Zhang YJ, Xu YF, Jinwal UK, Birnbaum MJ, et al. (2008). Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci U S A, 105:3622-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tharp WG, Gupta D, Smith J, Jones KP, Jones AM, Pratley RE (2016). Effects of glucose and insulin on secretion of amyloid-β by human adipose tissue cells. Obesity (Silver Spring), 24:1471-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yang C, Sun N, Ren Y, Sun Y, Xu Y, Li A, et al. (2012). Association between AKT1 gene polymorphisms and depressive symptoms in the Chinese Han population with major depressive disorder. Neural Regen Res, 7:235-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Banks WA, Owen JB, Erickson MA (2012). Insulin in the brain: There and back again. Pharmacol Ther, 136:82-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hong S, Han K, Park CY (2021). The insulin resistance by triglyceride glucose index and risk for dementia: Population-based study. Alzheimers Res Ther, 13:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Martín-Segura A, Ahmed T, Casadomé-Perales Á, Palomares-Perez I, Palomer E, Kerstens A, et al. (2019). Age-associated cholesterol reduction triggers brain insulin resistance by facilitating ligand-independent receptor activation and pathway desensitization. Aging Cell, 18:e12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kellar D, Craft S (2020). Brain insulin resistance in Alzheimer's disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol, 19:758-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, Page G, Duarte AI, Giusti P, et al. (2019). Current and emerging avenues for Alzheimer's disease drug targets. J Intern Med, 286:398-437. [DOI] [PubMed] [Google Scholar]
- [111].Oh Y, Lai JS, Mills HJ, Erdjument-Bromage H, Giammarinaro B, Saadipour K, et al. (2019). A glucose-sensing neuron pair regulates insulin and glucagon in Drosophila. Nature, 574:559-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Jin Z, Jin Y, Kumar-Mendu S, Degerman E, Groop L, Birnir B (2011). Insulin reduces neuronal excitability by turning on GABA(A) channels that generate tonic current. PLoS One, 6:e16188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Newsholme P, Keane KN, Carlessi R, Cruzat V (2019). Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am J Physiol Cell Physiol, 317:C420-c433. [DOI] [PubMed] [Google Scholar]
- [114].García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. (2016). Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell, 166:867-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Timper K, Del Río-Martín A, Cremer AL, Bremser S, Alber J, Giavalisco P, et al. (2020). GLP-1 receptor signaling in astrocytes regulates fatty acid oxidation, mitochondrial integrity, and function. Cell Metab, 31:1189-1205.e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Tumminia A, Vinciguerra F, Parisi M, Frittitta L (2018). Type 2 diabetes mellitus and Alzheimer's disease: Role of insulin signalling and therapeutic implications. Int J Mol Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zemva J, Schubert M (2014). The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer's disease: Possible therapeutic implications. CNS Neurol Disord Drug Targets, 13:322-337. [DOI] [PubMed] [Google Scholar]
- [118].Hölscher C (2020). Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin Investig Drugs, 29:333-348. [DOI] [PubMed] [Google Scholar]
- [119].Akanmu MA, Nwabudike NL, Ilesanmi OR (2009). Analgesic, learning and memory and anxiolytic effects of insulin in mice. Behav Brain Res, 196:237-241. [DOI] [PubMed] [Google Scholar]
- [120].Soto M, Cai W, Konishi M, Kahn CR (2019). Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc Natl Acad Sci U S A, 116:6379-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].De Melo LA, Almeida-Santos AF (2020). Neuropsychiatric properties of the ACE2/Ang-(1-7)/Mas pathway: A brief review. Protein Pept Lett, 27:476-483. [DOI] [PubMed] [Google Scholar]
- [122].Pillay NS, Stein DJ (2007). Emerging anxiolytics. Expert Opin Emerg Drugs, 12:541-554. [DOI] [PubMed] [Google Scholar]
- [123].Brandão ML, Coimbra NC (2019). Understanding the role of dopamine in conditioned and unconditioned fear. Rev Neurosci, 30:325-337. [DOI] [PubMed] [Google Scholar]
- [124].Blum K, Cadet JL, Baron D, Badgaiyan RD, Brewer R, Modestino EJ, et al. (2020). Putative covid- 19 induction of reward deficiency syndrome (RDS) and associated behavioral addictions with potential concomitant dopamine Depletion: Is covid-19 social distancing a double edged sword? Subst Use Misuse, 55:2438-2442. [DOI] [PubMed] [Google Scholar]
- [125].Brandão ML, Lovick TA (2019). Role of the dorsal periaqueductal gray in posttraumatic stress disorder: Mediation by dopamine and neurokinin. Transl Psychiatry, 9:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Lowry CA, Hale MW, Evans AK, Heerkens J, Staub DR, Gasser PJ, et al. (2008). Serotonergic systems, anxiety, and affective disorder: Focus on the dorsomedial part of the dorsal raphe nucleus. Ann N Y Acad Sci, 1148:86-94. [DOI] [PubMed] [Google Scholar]
- [127].Jembrek MJ, Vlainic J (2015). GABA receptors: Pharmacological potential and pitfalls. Curr Pharm Des, 21:4943-4959. [DOI] [PubMed] [Google Scholar]
- [128].Byrne ME, Tanofsky-Kraff M, Kelly NM, Grammer AC, Jaramillo M, Mi SJ, et al. (2019). Pediatric loss-of-control eating and anxiety in relation to components of metabolic syndrome. J Pediatr Psychol, 44:220-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chen M, Pritchard C, Fortune D, Kodi P, Grados M (2020). Hydrogen sulfide: A target to modulate oxidative stress and neuroplasticity for the treatment of pathological anxiety. Expert Rev Neurother, 20:109-121. [DOI] [PubMed] [Google Scholar]
- [130].Bouayed J, Rammal H, Soulimani R (2009). Oxidative stress and anxiety: Relationship and cellular pathways. Oxid Med Cell Longev, 2:63-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Boldrini P, Fusco A, Nicoletti F, Badiani A, Saso L (2018). Potential use of modulators of oxidative stress as add-on therapy in patients with anxiety disorders. Curr Drug Targets, 19:636-650. [DOI] [PubMed] [Google Scholar]
- [132].Afshar S, Shahidi S, Rohani AH, Soleimani Asl S, Komaki A (2019). Protective effects of 5-HT(1A) receptor antagonist and 5-HT(2A) receptor agonist on the biochemical and histological features in a rat model of Alzheimer's disease. J Chem Neuroanat, 96:140-147. [DOI] [PubMed] [Google Scholar]
- [133].Geldenhuys WJ, Van der Schyf CJ (2011). Role of serotonin in Alzheimer's disease: A new therapeutic target? CNS Drugs, 25:765-781. [DOI] [PubMed] [Google Scholar]
- [134].Rodríguez JJ, Noristani HN, Verkhratsky A (2012). The serotonergic system in ageing and Alzheimer's disease. Prog Neurobiol, 99:15-41. [DOI] [PubMed] [Google Scholar]
- [135].Bockaert J, Claeysen S, Compan V, Dumuis A (2008). 5-HT(4) receptors: History, molecular pharmacology and brain functions. Neuropharmacology, 55:922-931. [DOI] [PubMed] [Google Scholar]
- [136].Ma J, Gao Y, Tang W, Huang W, Tang Y (2020). Fluoxetine protects against dendritic spine loss in middle-aged APPswe/PSEN1dE9 double transgenic Alzheimer's disease mice. Curr Alzheimer Res, 17:93-103. [DOI] [PubMed] [Google Scholar]
- [137].Jones LA, Sun EW, Martin AM, Keating DJ (2020). The ever-changing roles of serotonin. Int J Biochem Cell Biol, 125:105776. [DOI] [PubMed] [Google Scholar]
- [138].Hennings JM, Schaaf L, Fulda S (2012). Glucose metabolism and antidepressant medication. Curr Pharm Des, 18:5900-5919. [DOI] [PubMed] [Google Scholar]
- [139].de Oliveira MR (2016). Fluoxetine and the mitochondria: A review of the toxicological aspects. Toxicol Lett, 258:185-191. [DOI] [PubMed] [Google Scholar]
- [140].Paul ED, Lowry CA (2013). Functional topography of serotonergic systems supports the Deakin/Graeff hypothesis of anxiety and affective disorders. J Psychopharmacol, 27:1090-1106. [DOI] [PubMed] [Google Scholar]
- [141].Martin AM, Yabut JM, Choo JM, Page AJ, Sun EW, Jessup CF, et al. (2019). The gut microbiome regulates host glucose homeostasis via peripheral serotonin. Proc Natl Acad Sci U S A, 116:19802-19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Xu Y, Berglund ED, Sohn JW, Holland WL, Chuang JC, Fukuda M, et al. (2010). 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate insulin sensitivity in liver. Nat Neurosci, 13:1457-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Stapel B, Gorinski N, Gmahl N, Rhein M, Preuss V, Hilfiker-Kleiner D, et al. (2019). Fluoxetine induces glucose uptake and modifies glucose transporter palmitoylation in human peripheral blood mononuclear cells. Expert Opin Ther Targets, 23:883-891. [DOI] [PubMed] [Google Scholar]
- [144].Diepenbroek C, Rijnsburger M, Eggels L, van Megen KM, Ackermans MT, Fliers E, et al. (2017). Infusion of fluoxetine, a serotonin reuptake inhibitor, in the shell region of the nucleus accumbens increases blood glucose concentrations in rats. Neurosci Lett, 637:85-90. [DOI] [PubMed] [Google Scholar]
- [145].Chakraborty S, Lennon JC, Malkaram SA, Zeng Y, Fisher DW, Dong H (2019). Serotonergic system, cognition, and BPSD in Alzheimer's disease. Neurosci Lett, 704:36-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Chow TW, Pollock BG, Milgram NW (2007). Potential cognitive enhancing and disease modification effects of SSRIs for Alzheimer's disease. Neuropsychiatr Dis Treat, 3:627-636. [PMC free article] [PubMed] [Google Scholar]
- [147].Pan DN, Wang Y, Li X (2019). Strategy bias in the emotion regulation of high trait anxiety individuals: An investigation of underlying neural signatures using ERPs. Neuropsychology, 33:111-122. [DOI] [PubMed] [Google Scholar]
- [148].Geng H, Li X, Chen J, Li X, Gu R (2015). Decreased intra- and inter-salience network functional connectivity is related to trait anxiety in adolescents. Front Behav Neurosci, 9:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Teri L, Ferretti LE, Gibbons LE, Logsdon RG, McCurry SM, Kukull WA, et al. (1999). Anxiety of Alzheimer's disease: prevalence, and comorbidity. J Gerontol A Biol Sci Med Sci, 54:M348-352. [DOI] [PubMed] [Google Scholar]
- [150].Porter VR, Buxton WG, Fairbanks LA, Strickland T, O'Connor SM, Rosenberg-Thompson S, et al. (2003). Frequency and characteristics of anxiety among patients with Alzheimer's disease and related dementias. J Neuropsychiatry Clin Neurosci, 15:180-186. [DOI] [PubMed] [Google Scholar]
- [151].Serra L, Perri R, Cercignani M, Spanò B, Fadda L, Marra C, et al. (2010). Are the behavioral symptoms of Alzheimer's disease directly associated with neurodegeneration? J Alzheimers Dis, 21:627-639. [DOI] [PubMed] [Google Scholar]
- [152].Pocnet C, Rossier J, Antonietti JP, von Gunten A (2013). Personality traits and behavioral and psychological symptoms in patients at an early stage of Alzheimer's disease. Int J Geriatr Psychiatry, 28:276-283. [DOI] [PubMed] [Google Scholar]
- [153].Kaiser NC, Liang LJ, Melrose RJ, Wilkins SS, Sultzer DL, Mendez MF (2014). Differences in anxiety among patients with early- versus late-onset Alzheimer's disease. J Neuropsychiatry Clin Neurosci, 26:73-80. [DOI] [PubMed] [Google Scholar]
- [154].Pietrzak RH, Lim YY, Neumeister A, Ames D, Ellis KA, Harrington K, et al. (2015). Amyloid-β, anxiety, and cognitive decline in preclinical Alzheimer disease: A multicenter, prospective cohort study. JAMA Psychiatry, 72:284-291. [DOI] [PubMed] [Google Scholar]
- [155].Tanaka H, Hashimoto M, Fukuhara R, Ishikawa T, Yatabe Y, Kaneda K, et al. (2015). Relationship between dementia severity and behavioural and psychological symptoms in early-onset Alzheimer's disease. Psychogeriatrics, 15:242-247. [DOI] [PubMed] [Google Scholar]
- [156].Joshi A, Jimenez E, Mendez MF (2017). Initial heart rate reactivity to socioemotional pictures in early-onset Alzheimer's disease. J Alzheimers Dis, 60:1325-1332. [DOI] [PubMed] [Google Scholar]
- [157].Donovan NJ, Locascio JJ, Marshall GA, Gatchel J, Hanseeuw BJ, Rentz DM, et al. (2018). Longitudinal association of amyloid beta and anxious-depressive symptoms in cognitively normal older adults. Am J Psychiatry, 175:530-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Santabárbara J, Villagrasa B, López-Antón R, Olaya B, Bueno-Notivol J, de la Cámara C, et al. (2019). Clinically relevant anxiety and risk of Alzheimer's disease in an elderly community sample: 4.5 years of follow-up. J Affect Disord, 250:16-20. [DOI] [PubMed] [Google Scholar]
- [159].Baillon S, Gasper A, Wilson-Morkeh F, Pritchard M, Jesu A, Velayudhan L (2019). Prevalence and severity of neuropsychiatric symptoms in early- versus late-onset Alzheimer's disease. Am J Alzheimers Dis Other Demen, 34:433-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Johansson M, Stomrud E, Lindberg O, Westman E, Johansson PM, van Westen D, et al. (2020). Apathy and anxiety are early markers of Alzheimer's disease. Neurobiol Aging, 85:74-82. [DOI] [PubMed] [Google Scholar]
- [161].Keller JN, Lauderback CM, Butterfield DA, Kindy MS, Yu J, Markesbery WR (2000). Amyloid beta-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem, 74:1579-1586. [DOI] [PubMed] [Google Scholar]
- [162].Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, et al. (2007). Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics, 7:605-616. [DOI] [PubMed] [Google Scholar]
- [163].Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science, 304:448-452. [DOI] [PubMed] [Google Scholar]
- [164].Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med, 14:1097-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Du H, Guo L, Zhang W, Rydzewska M, Yan S (2011). Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging, 32:398-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Naga KK, Sullivan PG, Geddes JW (2007). High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci, 27:7469-7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, et al. (2005). ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J., 19:597-598. [DOI] [PubMed] [Google Scholar]
- [168].Rui Y, Tiwari P, Xie Z, Zheng JQ (2006). Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci, 26:10480-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci, 29:9090-9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. (2008). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A, 105:19318-19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Wang X, Su B, Fujioka H, Zhu X (2008). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol, 173:470-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Manczak M, Calkins MJ, Reddy PH (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet, 20:2495-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Calkins MJ, Reddy PH (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta, 1812:507-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Ye X, Sun X, Starovoytov V, Cai Q (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet, 24:2938-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]