

Abstract
Background
Microvesicles are vesicles shed by plasma membranes following cell activation and apoptosis. The role of lymphocyte‐derived microvesicles in endothelial function remains poorly understood.
Methods
CD4+ T cells isolated from peripheral blood of healthy human donors were stimulated using anti‐CD3/anti‐CD28‐coated beads. Proteomic profiling of microvesicles was performed using linear discriminant analysis (LDA) from activated T cells (MV.Act) and nonactivated T cells (MV.NAct). In addition, data processing analysis was performed using MaxQUANT workflow. Differentially expressed proteins found in MV.Act or MV.NAct samples with identification frequency = 100%, which were selected by both LDA (p < .01) and MaxQUANT (p < .01) workflows, were defined as “high‐confidence” differentially expressed proteins. Functional effects of MV.Act on human primary microvascular endothelial cells were analysed.
Results
T cells released large amounts of microvesicles upon stimulation. Proteomic profiling of microvesicles using LDA identified 2279 proteins (n = 2110 and n = 851 proteins in MV.Act and MV.NAct, respectively). Protein–protein interaction network models reconstructed from both differentially expressed proteins (n = 594; LDA p ≤ .01) and “high‐confidence” differentially expressed proteins (n = 98; p ≤ .01) revealed that MV.Act were enriched with proteins related to immune responses, protein translation, cytoskeleton organisation and TNFα‐induced apoptosis. For instance, MV.Act were highly enriched with IFN‐γ, a key proinflammatory pathway related to effector CD4+ T cells. Endothelial cell incubation with MV.Act induced superoxide generation, apoptosis, endothelial wound healing impairment and endothelial monolayer barrier disruption.
Conclusions
T cell receptor‐mediated activation of CD4+ T cells stimulates the release of microvesicles enriched with proteins involved in immune responses, inflammation and apoptosis. T cell‐derived microvesicles alter microvascular endothelial function and barrier permeability, potentially promoting tissue inflammation.
Keywords: endothelial cell, extracellular vesicle, microvesicle, proteomics, T cells
1. INTRODUCTION
Cell–cell communication is key to maintaining the homoeostasis of living organisms. Secreted extracellular vesicles are a class of intercellular communicators that can activate surface receptors and deliver their cargoes—which are comprised of proteins, nucleic acids and lipids—into recipient cells. Extracellular vesicles have been traditionally categorised into three subclasses: Small vesicles including exosomes (30–120 nm in diameter); microvesicles (MVs; previously referred to as microparticles or ectosomes; 100–1300 nm in diameter) and apoptotic bodies (>1000 nm in diameter). As apparent, vesicle subclasses overlap in size; however, they differ in their biogenesis: Exosomes are released by an endosomal pathway via the formation of multivesicular bodies, whereas MVs are directly shed from blebbing plasma membranes of various cell types including immune cells in response to stress stimuli (e.g. shear stress, oxidative stress and pharmacological agents). 1 , 2 By contrast, apoptotic bodies arise from dying cells.
Circulating vesicles are mainly derived from blood and vascular cells. Increased concentrations of circulating vesicles have been reported in patients with various conditions (e.g. atherosclerosis, sepsis, and immune diseases 3 ). Activated immune cells release MVs that regulate inflammation and vascular permeability. 4 In this regard, lymphocyte‐derived MVs have been less studied than neutrophil‐ and monocyte‐derived MVs. An early study 5 showed that the human lymphoid CEM T cell line stimulated with phytohaemagglutinin (PHA) released microparticles that altered protein expression and function in endothelial cells. Another study 6 described the secretion of immunoregulatory MVs from a mixed nontumoral T cell population stimulated with PHA. A subsequent study 7 showed that human T cells purified from peripheral blood and activated with immobilised anti‐CD3 mAb through the T cell receptor (TCR) released MVs. By contrast, another mitogenic signal, such as PMA and ionomycin, did not induce any release. More recently, proteomics of exosomes produced by normal human T cell blasts was compared to those produced by tumoral Jurkat cells with the objective of identifying proteins associated with tumoral exosomes that could have a role in malignancy, showing that only around 40% of proteins was shared by the two groups. 8
Here, we aimed to investigate the release of MVs from human CD4+ T cells purified from peripheral blood and activated with anti‐CD3/anti‐CD28‐coated beads. This model closely mimics physiological TCR‐mediated T cell activation by antigen‐presenting cells. 9 Our proteomic analysis revealed that MVs released from activated T cells (MV.Act) were enriched with proteins related to various biological processes such as immune responses and apoptosis. Given the regulatory role of microvascular endothelium in inflammation and vessel barrier function, we investigated the functional effects of MV.Act on human primary cardiac‐derived microvascular endothelial cells (HMVECs). Our results indicate that MV.Act regulate functional activities in HMVECs.
2. MATERIAL AND METHODS
Reporting of the study conforms to broad EQUATOR guidelines. 10
2.1. T cell isolation and stimulation
Buffy coats were obtained from healthy donors who gave informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated using Lympholyte (Cederlane, CL5020) density gradient separation. CD4+ T cells were MACsorted from PBMCs using human anti‐CD4 magnetic beads (Miltenyi Biotech, 130–045–101), stained with carboxyfluorescein succinimidyl ester (CFSE; Life Technologies, C34554). and plated in RPMI Medium 1640 (Gibco, A10491‐01) supplemented with 10% FBS, 1% penicillin/streptomycin, 1% Minimum Essential Medium Nonessential Amino Acid (MEM‐NAA; Gibco, 11140–035) and 0.1% 2‐mercaptoethanol. For stimulation, T cells were incubated with anti‐CD3/anti‐CD28 beads (Dynabeads Human T‐Activator, Gibco, 11131D) for 5 days. T cell proliferation was measured by flow cytometric CFSE dilution assay using LSRII Fortessa analyzer. T cell phenotype was assessed using anti‐CD45RA, anti‐CD45RO, anti‐CCR7, and anti‐CD27 antibodies (all from eBioscience).
2.2. MV isolation and characterisation
Microvesicles were purified from T cell‐conditioned media using a published MV isolation protocol. 11 Briefly, cells and debris were removed by centrifugation at 450 × g for 7 min, followed by 2000 × g for 20 min, supernatant filtration with 1‐µm filters (Acrodisc) and centrifugation at 16,000 × g at 4 °C for 30 min. Pellets were re‐suspended in PBS. MV concentrations were measured by flow cytometry and shown as CFSE+ particles/ml. Nanoparticle tracking analysis was performed using Nanosight NS300 (Malvern Instruments). Mean particle size was determined from 3 sequential recordings using NTA 3.2 software.
2.3. Transmission electron microscopy
Samples were fixed with 2.5% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.35) in cellulose capillary tubes washed in cacodylate buffer and contrasted with 1% osmium tetroxide for 30 min in 0.1 M cacodylate buffer, and in 1% aqueous uranyl acetate for 1 h. Samples were dehydrated in an ethanol series, followed by 1 hr in propylene oxide, prior to embedding in Epon/Araldite (Sigma). Ultrathin (70 nm) sections were poststained with lead citrate and analysed using a transmission electron microscope (Tecnai G2 Spirit, Thermo Fisher Scientific) at an acceleration voltage of 120 KV using a Gatan Orius digital camera.
2.4. Western blotting
Total proteins were extracted by RIPA buffer supplemented with SIGMAFAST™ Protease Inhibitors and Phosphatase Inhibitor Cocktails 3 and 2 (Sigma), boiled with Laemmli SDS sample buffer (VWR Int.) for 5 min, separated on 4–20% Mini‐PROTEAN®TGX™ Precast Gel and transferred onto a PVDF membrane using a semi‐dry transfer system (Bio‐Rad). Membranes were blocked with LI‐COR Blocking Buffer (LI‐COR Biosciences) and incubated with the following primary antibodies at the indicated dilutions: Anti‐VE‐cadherin (CellSignaling, 2500; 1:1000), anti‐mitofilin (Invitrogen, AB‐2547893; 1:500), anti‐vinculin (ThermoFisher, VLN01; 1:1000), anti‐tumour susceptibility gene‐101 (TSG101; Abcam, ab125011; 1:1000), anti‐synthenin‐I (Abcam, ab133257; 1:1000), anti‐GRP94 (Abcam, ab238126; 1:1000), anti‐claudin‐5 (Abcam, ab15106; 1:500), anti‐GAPDH (Abcam, ab181602; 1:1000). Actin polymerization was assessed using Biochem Kit (Cytoskeleton).
2.5. ImageStreamX imaging flow cytometry
Cell or MV suspensions labelled with fluorescent dyes or antibodies were analysed using Amnis® Mark II ImageStreamX imaging flow cytometer (Merck Millipore). 12 Cells were FACsorted using an Aria III system (BD Bioscience). Phosphatidylserine staining was performed using Annexin V detection kit (BioLegend).
2.6. Liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) analysis and proteomic data processing
A total of 8 samples (n = 4 for MV.Act and n = 4 for MV from nonactivated T cells; MV.NAct) were analysed using Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer at Functional Genomics Centre, University of Zurich. Raw files were processed by the Sequest HT algorithm contained in the Proteome Discoverer 2.5 software (ThermoFisher). Experimental MS/MS spectra were compared with the theoretical mass spectra obtained by in silico digestion of a Homo sapiens protein database containing 75,776 sequences (UNIPROT, November 2021; www.uniprot.org). Searching criteria were as follows: Trypsin enzyme, three missed cleavages per peptide, mass tolerances of ±50 ppm for precursor ions and ± 0.8 Da for fragment ions. Percolator node was used with a target‐decoy strategy to give a final false discovery rate of ≤0.01 (strict) based on q‐values, considering maximum delta‐CN = 0.05. Only peptides with a minimum peptide length of six amino acids, "high‐level” confidence and rank 1 were considered. Spearman's rank correlation coefficient was used for assessing correlations among replicate analyses. The spectral count (SpC) values of the identified proteins were normalised using a total signal normalisation method and compared using a label‐free quantitation approach. 13 The data matrix dimensionality (n = 4/condition) was reduced by linear discriminant analysis (LDA). A pairwise comparison (MV.Act vs. MV.NAct) was performed. Only proteins with an F ratio ≥5 and p‐value ≤.01 were retained. Fold changes were estimated using the natural logarithm of the average Peptide Spectrum Matches spectral count (avPSM) ratio (avPSM. Act/avPSM.NAct). The fold‐change value of a protein identified in only one condition was conventionally set to ±10. Most abundant proteins in samples were estimated by normalising the PSM of each protein for the respective molecular weight. 14 Proteins selected by LDA were processed by Hierarchical Clustering applying the Ward's method and the Euclidean distance metric. Data processing was performed using JMP 15.1 SAS software. To increase the significance of protein identification and quantification, raw data was additionally processed by MaxQuant (version 1.6.2.3). 15 Scaffold (Proteome Software) was used for visualisation of protein and peptide identification results. Data were searched against the human database (fgcz_9606_reviewed_cnl_20190709). Fold‐change log2 (FC log2) was computed as the difference between the means in the two groups (with MV.NAct as the reference group). The regulated proteins were reported using predefined filtering settings (adjusted p‐value <.05, |FC log2| ≥1). Pseudo.log2FC and pseudo.adj.P. Val. were estimated for the proteins present in a single condition using the mean expression level of the bottom 10% proteins as a substitute for enrichment analysis.
2.7. Functional and network proteomic analysis
Functional Annotation Tool of DAVID database 16 was used to characterise the most highly enriched biological processes. Specifically, background = Homo Sapiens, count >5 and EASE p < .001 were set. Enrichment profiles were compared by LDA (p < .01). A Homo sapiens MV protein‐protein interaction (PPI) network model was reconstructed starting from differentially expressed proteins (DEPs; p ≤ .01; n = 594 for MV.Act vs. MV.NAct). The network was reconstructed using PescaApp 17 of Cytoscape. 18 In addition to stringApp, proteins were grouped in functional modules using BINGO 2.44. 19 Homo sapiens organism, hypergeometric test, Benjamini–Hochberg FDR correction and a significance level ≤0.01 were set. The reconstructed network was visualised and handled by Cytoscape. Node colour code indicated upregulated (red) and down‐regulated (blue) proteins based on average Spectral count (avSpC) normalisation. “High‐confidence” DEPs were defined as those selectively found in either MV.Act or MV.NAct samples with an identification frequency = 100% and selected by data processing analyses using both Discoverer 2.5 and MaxQUANT v1.6.2.3. Starting from “high‐confidence” DEPs, a PPI network model was reconstructed considering their first neighbours as putative protein targets, using PescaApp. 17 The reconstructed network was processed at the topological level by Cytoscape's plugin Centiscape 2.2, 20 as reported. 21 Specifically, Betweenness and Degree were calculated to define protein hubs. 22 Statistical significance of topological results was tested by considering randomised network models. They were reconstructed and analysed by an in‐house R script based on VertexSort (to build random models), igraph (to compute centralities) and ggplot2 (to plot results) libraries.
2.8. Endothelial cells
Cardiac HMVECs (Lonza; CC‐7030) were cultured in EC Growth Medium‐2 (EGM‐2, supplemented EBM‐2; SingleQuots, Lonza, CC‐4176), 10% FBS and 1% penicillin/streptomycin in a 5% CO2 air‐humidified atmosphere at 37°C. HMVECs were stained with CellTrace Violet (LifeTechnologies, C34557).
2.9. Immunofluorescence microscopy
Human primary cardiac‐derived microvascular endothelial cells were incubated with MV.Act (40,000 CFSE+ particles/ml) for 24 h. Phalloidin staining was performed using cytoskeleton Rhodamine Phalloidin (14 microM in methanol; PHDR1). Vinculin immunostaining was performed using a specific antibody (ThermoFisher, VLN01; 1:1000). Fluorescence microscopy was performed using an Olympus DP50 microscope. Fluorescent cellular areas and mean fluorescence intensity (MFI) were measured using Fuji software.
2.10. Superoxide generation and apoptosis
Human primary cardiac‐derived microvascular endothelial cells were incubated with varying MV.Act concentrations for different time periods, processed with MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermofisher, M36008) and analysed by flow cytometry. Apoptosis was assessed using Annexin V detection kit (BioLegend).
2.11. Endothelial scratch assay
Confluent HMVEC monolayers were preincubated with MV.Act, activated T cells, nonactivated T cells (20,000 cells/well each) or rhTNF‐α (10 ng/ml) for 24 h. Scratch was induced mechanically. Gap closure fractions were measured microscopically.
2.12. Electric cell‐substrate impedance sensing (ECIS)
Human primary cardiac‐derived microvascular endothelial cells (150,000 cells/well) were plated in ECIS culture ware type 8W10E PET (Ibidi, 72010) stabilised according to manufacturer's instructions (BioPhysics, ECIS Zθ) and cultured in EGM‐2 medium for 24 h in an incubator connected to the ECIS system. Cell confluence was confirmed by a plateau in trans‐endothelial electrical resistance recordings. MV.Act, activated or nonactivated T cells or rhTNFα were added to the wells. Measurements were obtained in multiple frequencies over 24 h. 23
2.13. PBMC adhesion assay
Confluent HMVEC monolayers on BioFlux plates were preincubated with MV.Act, activated or nonactivated T cells or rhTNFα. Freshly isolated PBMCs were labelled with CFSE. PBMC adhesion to HCMVECs was measured in a BioFlux™ 200 system (IL‐biosystems) under a constant flow (1 dyn).
2.14. Statistical analysis
Data were analysed by the unpaired, two‐tailed Student's t‐test, two‐tailed one‐way ANOVA (for normally distributed data), Mann–Whitney test (for nonparametric data) or two‐way ANOVA for multiple comparison analyses. All analyses were performed with GraphPad Prism 8 software. Values are shown as mean ±SEM. Differences were considered statistically significant for p < .05.
3. RESULTS
3.1. T cell stimulation triggers MV secretion
CD4+ T cells exposed to anti‐CD3/anti‐CD28 beads exhibited >90% division rates (Figure 1A) and a CD45RA lo CD45RO+CCR7+CD27+ main phenotype (Figure 1B,C). This immunophenotype has been traditionally associated with central memory T cells. 24 The concentration of MV.Act preparations ranged from 14*108 to 17*108 CFSE+ particles/ml. The concentration of MV.NAct preparations was lower by 2–3 orders of magnitude compared to that of MV.Act preparations. Flow cytometry, nanoparticle tracking analysis, and TEM indicated MV.Act particle diameters in the ranges of 0.3–1.5 µm, 0.1–0.8 µm and 0.1–1.0 µm, respectively (Figure 1D–F). Methodological factors may account for differences in particle size measured using different techniques. Western analysis of MV.Act demonstrated expression of TSG101, synthenin‐I and mitofilin. Low levels of GRP94, used as a marker for cytosolic content and cell debris, were detected (Figure 1G). CD3/CD4 expression and Annexin V–positive staining on MV.Act was demonstrated by ImageStreamX flow cytometry (Figure 1H). These results indicated that the isolated preparations were largely comprised of MVs.
FIGURE 1.

Characterisation of human naive CD4+ T cells stimulated by TCR engagement, and of their secreted MVs (MV.Act). (A) Left panel: FACS plot showing human PBMC‐derived CD4+ naïve T cells 5 days after TCR stimulation by anti‐CD3/anti‐CD28 beads. Cells were labelled with CFSE before stimulation. Mid panel: Representative CFSE dilution profiles of activated and nonactivated T cells, day 5. Right panel: Quantitative analysis of T cell proliferation 5 days after TCR stimulation. (B) FACS plot showing gated CD4+‐activated T cells stained with anti‐CD45RO/CD45RA. CD45RA lo CD45RO+ and CD45RA+CD45RO lo phenotypes correspond to activated and nonactivated T cells, respectively. Quantitative analysis of CD45RA lo CD45RO+ cells in activated T cells and nonactivated T cells are shown. (C) Representative FACS plot gated CD4+ CD45RA lo CD45RO+ showing subpopulation distribution of TCR‐stimulated T cells, day 5. Quantitative analysis of phenotypic T cell subsets: central memory (TCM) ‐ CD45RA lo CD45RO+CCR7+CD27+, effector memory (TEM) ‐ CD45RA lo CD45RO+CCR7−CD27−, effector (TEFF) ‐ CD45RA lo CD45RO+CCR7−CD27+. TCM was the predominant T cell phenotype. (D) Flow cytometric representative plots showing CFSE‐labelled MV.Act and MV.NAct. Red, blue and green circles indicate calibration beads (0.5 µm, 0.9 µm and 3.0 µm, respectively). A CFSE+ population of a size traditionally associated with MVs is indicated. (E) Nanoparticle Tracking Analysis (NTA) of MV.Act. Upper panel: Particle diameter distribution (peak, 260 nm). (F) Electron microphotographs of activated T cells shedding MVs and isolated MV.Act. (G) Western blot analysis of TSG101, GRP94, synthenin‐I and mitofilin in activated T cells and MV.Act. (H) ImageStreamX imaging flow cytometry of MV.Act from CFSE‐labelled T cells (green). Both single‐channel images and merged images of Annexin V (yellow) and CD3/CD4 (red)‐stained cells are shown. A schematic depicting CFSE‐labelled, Annexin V‐positive, MVs expressing CD3 and CD4 is shown (data shown in panels A and B were analysed with the Student's t‐test for two independent groups, data shown in panel C were analysed using one‐way ANOVA, and data shown in panel D were analysed using the Mann–Whitney test *p < .05)
3.2. T cell stimulation regulates MV protein composition
A total of 2279 proteins were identified in MV.Act or MV.NAct by LC‐MS/MS analysis (Figure 2 and Table S1). Of these, 2110 and 851 were found in MV.Act and MV.NAct, respectively. Reproducibility of the data was demonstrated by replicate analyses (Figure 2A). Approximately 40% of the identified proteins were characterised by an average PSMs >1 (Figure 2B and Table S2). MV.Act were enriched with proteins related to the immune system, signal transduction, protein and vesicle transport, RNA processing/splicing and TNF/apoptosis. Regarding the TNF/apoptosis family/function, TNF was identified in MV.Act, but not in MV.NAct, whereas Fas (CD95) ligand (FasL) and APO2 ligand (APO2L)/TNF‐related apoptosis‐inducing ligand (TRAIL) were not identified in either MV type. Most DEPs were upregulated in MV.Act vs. MV.NAct (Figure 3A). MV.NAct were enriched with proteins related to intermediate filaments/extracellular matrix, peptidase inhibitors and blood coagulation (Figure S1). DEPs identified with high confidence (see Methods) included 98 proteins (Table S3). By function/family analysis, such “high‐confidence” DEPs were grouped into the following function/families: immune system, protein translation, cytoskeleton organisation/microtubule‐based motility, TNF/apoptosis, solute carrier family and phosphatidylinositol signalling (Figure 3B). Based on the assumption that DEPs upregulated in MV.Act participate in signal transduction pathways, their potential protein targets were identified by processing at a topological level a PPI network model reconstructed by selecting their first neighbours using centrality parameters. 25 Based on centrality and betweenness compared to random networks, 37 proteins (not found in MV.Act) were identified as best targets for DEPs upregulated in MV.Act. These proteins included ubiquitin‐related, transcription/translation, receptor/signalling and cell cycle/microtubule‐related proteins (Figure S2 and Table S4).
FIGURE 2.
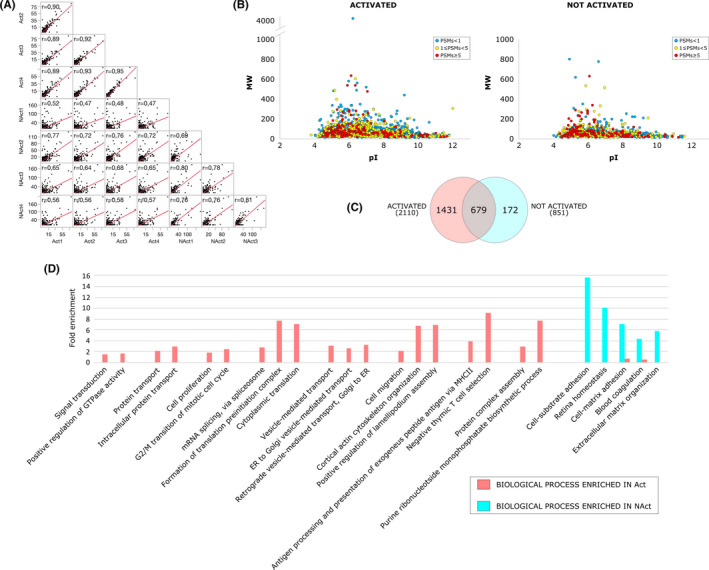
Proteomic analysis of MVs secreted by activated T cells (MV.Act) and by nonactivated T cells (MV.NAct). (A) Spearman's correlation comparing protein profiles from replicate analyses of MV.Act and MV.NAct. (B) Virtual 2DMap (pI vs. MW) of proteins identified in MV.Act or MV.NAct. (C) Venn diagram of the identified proteins. (D) Biological processes (BPs) differentially enriched in MV.Act (red bars) or MV.NAct (blue bars)
FIGURE 3.
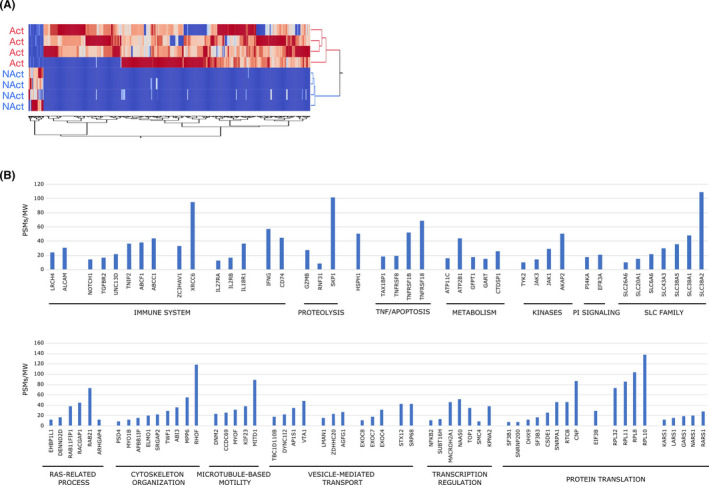
Label‐free protein quantification comparison of proteins enriched in MV.Act or MV.NAct. (A) Hierarchical clustering was performed by computing the PSMs of differentially expressed proteins (DEPs; n = 592). (B) “High‐confidence” DEPs exclusively found in MV.Act and selected by both Discoverer 2.5 and MaxQUANT. Proteins were grouped in functional modules. The corresponding PSMs/MW values are shown
3.3. MV.Act establish physical contacts to HMVECs
We first asked whether activated or nonactivated T cells physically interacted with cultured HMVECs. Incubation of VioletTrace‐labelled HMVECs with CFSE‐labelled, activated T cells was associated with the appearance of a CFSE+VioletTrace+ subpopulation on flow cytometry plots (Figure 4A). Co‐localisation of CFSE+ T cell content and VioletTrace+ HMVECs was demonstrated by ImageStreamX (Figure 4B). These findings were not observed using nonstimulated T cells. We then asked whether MV.Act could reproduce these effects of their parent cells. After HMVEC incubation with MV.Act, and subsequent extensive washout of unbound MV.Act, a CFSE+VioletTrace+ subpopulation appeared on flow cytometry plots (Figure 4C). ImageStreamX demonstrated co‐localisation of MV.Act and HMVECs (Figure 4D). These results indicated that MV.Act established physical contacts with HMVECs.
FIGURE 4.
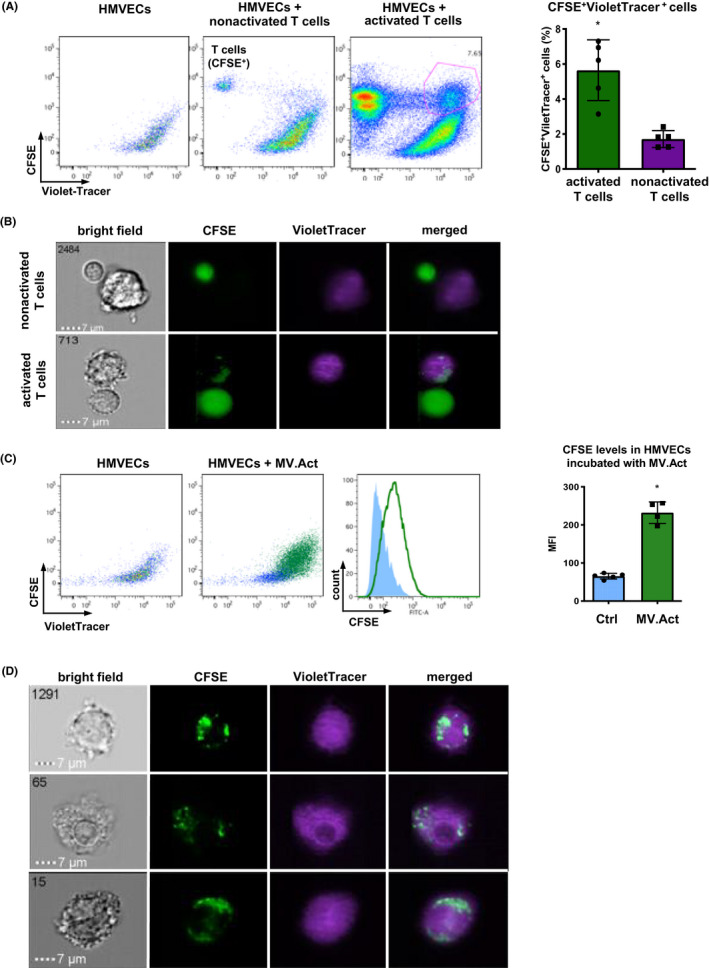
Physical contacts of both T cells and MV.Act with HMVECs. (A) FACS representative plots showing VioletTracer‐labelled HCMVECs incubated with CFSE‐labelled activated or nonactivated T cells. Nontreated HCMVECs are shown as a control. Analyses were performed on day 3. Quantitative analysis of CFSE+VioletTracer+ population in co‐cultures. (B) ImageStreamX imaging flow cytometry of HCMVECs incubated with either activated or nonactivated T cells. Co‐localisation of CFSE+ T cell content (green) and VioletTracer+ HCMVECs was selectively observed using activated T cells. (C) Flow cytometry plots showing VioletTracer‐labelled HCMVECs incubated with CFSE‐labelled MV.Act. Quantitative analysis of CFSE levels in HCMVECs incubated with MV.Act and control HCMVECs (MFI, mean fluorescence intensity). (D) ImageStreamX imaging flow cytometry of VioletTracer‐labelled HCMVECs incubated with CFSE‐labelled MV.Act. Co‐localisation of CFSE+ T cell content (green) and VioletTracer+ HCMVECs can be seen (A, C graphs were analysed with the Student's t‐test for two independent groups, *p < .05)
3.4. MV.Act induce cytoskeleton rearrangement in HMVECs
After 24 h‐incubation with MV.Act, HMVECs acquired a spindle‐shaped morphology. These changes were quantitated as FSC/SSC ratios by flow cytometry (Figure 5A). MV.Act regulated F‐actin expression in HMVECs, as assessed using phalloidin staining (Figure 5B), resulting in increased F‐actin/G‐actin ratios (Figure 5C), consistent with actin polymerisation. Vinculin expression was unaffected (Figure 5B). These findings were consistent with MV.Act‐mediated cytoskeleton rearrangement.
FIGURE 5.
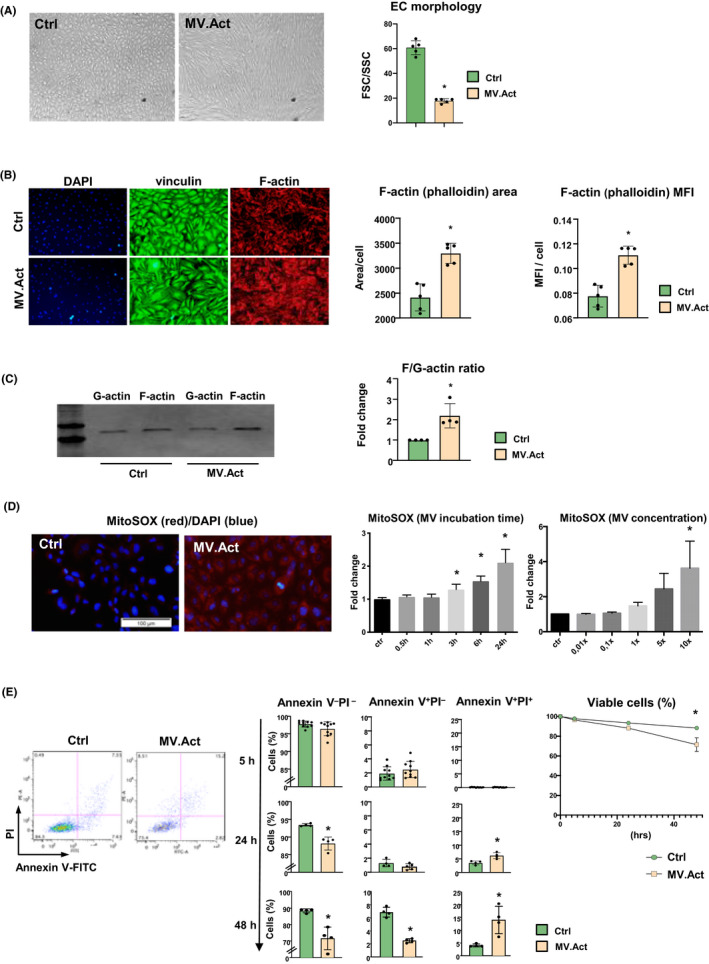
MV.Act‐mediated morphological and functional changes in HMVECs. (A) Bright‐field microphotographs of HCMVECs cultured at 90% confluence, incubated with either MV.Act or no MVs (control) for 24h. HMVECs incubated with MV.Act exhibit a spindle‐shaped morphology. Quantitative analysis of flow cytometric FSC/SCC plots is shown. (B) Vinculin immunostaining and F‐actin staining using phalloidin in HCMVECs incubated with either MV.Act or no MVs (control). Quantitative analyses of F‐actin‐positive area/cell and F‐actin mean fluorescence intensity (MFI). (C) Western blot analysis of G‐actin and F‐actin expression in HCMVECs incubated with either MV.Act or no MVs (control). Quantitative analysis of F/G‐actin ratio. (D) HMVEC life staining with MitoSox (red); nuclear staining with DAPI (blue). Quantitative analysis of MitoSox fluorescence intensity using varying MV concentrations and incubation times (data are fold increases over control, n = 8). (E) Representative FACS plots of HMVECs incubated with MV.Act and stained with Annexin V and PI. Quantitative analysis of early apoptotic cells (Annexin V+PI−) and late apoptotic/dead cells (Annexin V+PI+) at the indicated time points (data from n ≥4 experiments are shown). Right panel: Percentages of viable HMVECs (Annexin V−PI−) at different time points (data in A, B, E graphs were analysed with the Student's t‐test, data in C graph were analysed using the Mann–Whitney test, and data in D graph were analysed using one‐way ANOVA, *p < .05)
3.5. MV.Act induce superoxide generation and apoptosis in HMVECs
MV.Act induced time‐ and concentration‐dependent increases in superoxide levels in HMVECs (Figure 5D), along with increased numbers of Annexin V+PI+ cells (i.e. late apoptotic and dead cells; Figure 5E). These results indicated that MV.Act promoted oxidative stress and apoptosis in HMVECs.
3.6. MV.Act inhibit endothelial wound healing
The ability of an endothelial monolayer to restore its physical integrity after mechanical injury reflects an important functional activity of these cells. Using a scratch assay, HMVECs incubated with MV.Act exhibited a lower endothelial gap closure activity than untreated cells. Similar effects were observed using activated T cells, but not using nonactivated T cells or rhTNF‐α (Figure 6A,B).
FIGURE 6.
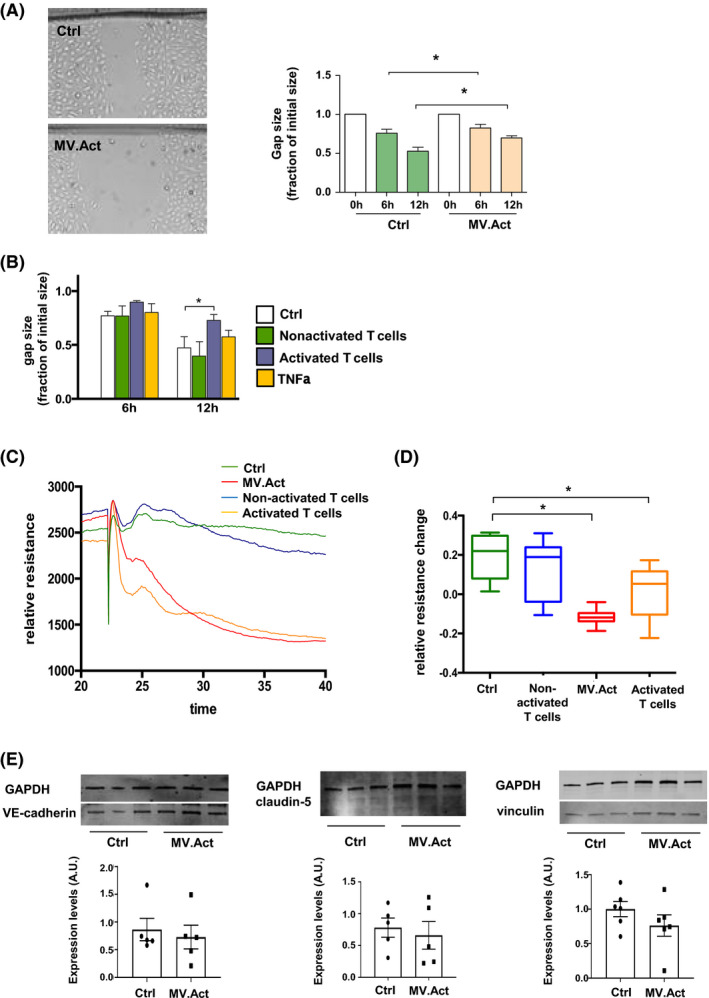
Effects of T cells and MV.Act on endothelial wound healing and endothelial barrier function. (A) Representative photomicrographs of HMVECs incubated with either MV.Act or no MVs (control) using a scratch assay. Quantitative analysis of endothelial monolayer gap closure at the indicated time points after scratch injury, normalised to 0 timepoint (data from n ≥4 experiments are shown). (B) Quantitative analysis of endothelial monolayer gap closure after cell incubation with either activated or nonactivated T cells, or after pretreatment with rhTNFα (data from n ≥4 experiments). (C) Trans‐endothelial electrical resistance (TEER) measured by ECIS technology in HMVECs incubated with either activated or nonactivated T cells, MV.Act, or in the absence of treatment (control). Relative resistance changes normalised for baseline conditions are shown. Representative individual real‐time TEER measurements in the different group are shown. (D) Quantitative analysis of TEER data (n = 8/group). (E) Western blot analysis and quantification of VE‐cadherin, claudin‐5 and vinculin in MV.Act‐treated conditions vs. control (data shown in A, B graphs were analysed using two‐way ANOVA, data shown in C graph were analysed using one‐way ANOVA, and data shown in E graph were analysed with the Student's t‐test, *p < .05)
3.7. MV.Act break down endothelial barrier function
Microvesicles can affect endothelial barrier integrity, a key regulator of tissue inflammation. 8 We investigated the role of MV.Act in endothelial barrier disruption. HMVECs incubated with either MV.Act or activated T cells exhibited a reduced trans‐endothelial electrical resistance (TEER), measured by ECIS. 23 TEER was unaffected by By contrast, HMVEC incubation with nonactivated T cells (Figure 6C,D). Western analysis of VE‐cadherin, claudin‐5 and vinculin suggested that MV.Act did not regulate the expression of these intercellular junction proteins in HMVECs (Figure 6E). These results indicated that MV.Act disrupted HMVEC barrier function. Further studies aiming at elucidating the underlying molecular mechanisms are warranted.
3.8. MV.Act do not regulate PBMC adhesion to HMVECs
Adhesion of circulating leukocytes to the endothelium initiates tissue inflammation. HMVEC preincubation with activated T cells or rhTNF‐α, but not with MV.Act, upregulated ICAM‐1, VCAM‐1 and E‐selectin (Figure 7A,B). These effects were paralleled by changes in PBMC adhesion to HMVECs under flow conditions (Figure 7C). These findings suggested that activated T cells might facilitate PBMC adhesion to HMVECs through MV‐independent mechanisms.
FIGURE 7.
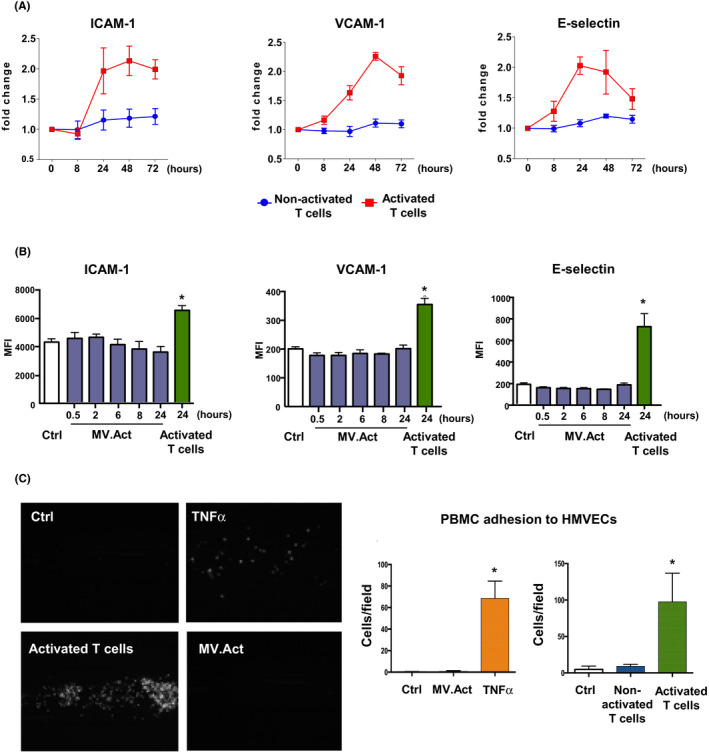
Adhesion molecule expression and leukocyte adhesion to by HMVECs. (A) Quantitative analysis of ICAM‐1, VCAM‐1 and E‐selectin expression on HMVECs incubated with either activated or nonactivated T cells for varying periods of time (n > 3). (B) Quantitative flow cytometric analysis of adhesion molecule expression on HMVECs incubated with MV.Act for varying time periods. Data on HMVECs incubated with activated T cells are shown as a positive control. (C) Left panel: Representative microphotographs of PBMC adhesion to HMVECs preincubated with either activated T cells or MV.Act, or pretreated with rhTNF‐α using a BioFlux system. Right panel: Quantitative analysis of PBMC adhesion (graphs were analysed with one‐way ANOVA, *p < .05)
4. DISCUSSION
Activated immune cells secrete MVs that regulate inflammation and endothelial function. Here, we characterised MVs released by primary human CD4+ T cells upon TCR activation using anti‐CD3/anti‐CD28‐coated beads. Proteomic analysis showed that these MVs (i.e. MV.Act) were enriched with proteins participating in immune and inflammatory processes. Moreover, these MVs appeared to regulate various aspects of microvascular endothelial function.
Among extracellular vesicle subclasses, we specifically investigated MVs, as this vesicle subclass is typically released in response to cell activation by extracellular stimuli. 1 , 2 Using a published MV isolation protocol, 11 the isolated vesicle preparation was predominantly, though not exclusively, comprised of MVs. Several pieces of data supported this assumption: i) Particle size was largely in the range that has been traditionally associated with MVs 1 , 2 ; ii) Particles stained positive for Annexin V; iii) Particles expressed mitofilin, which has been specifically associated with large vesicles. 11 Our results are in line with previous data showing that human T cells purified from peripheral blood release MVs upon TCR activation. 7
As microvascular endothelium regulates tissue inflammation, we addressed the functional effects of MV.Act on HMVECs. ImageStreamX analysis revealed that MV.Act added to cultured HMVECs established physical contacts with the cells. Similar findings were obtained upon incubation of HMVECs with activated T cells, but not with nonactivated T cells. Physical interactions between MV.Act and HMVECs were not characterised more precisely. In this regard, recent data suggested that mesenchymal stromal cell‐derived membrane particles might be internalised by endothelial cells through receptor‐mediated endocytosis and phagocytosis. 26 Accordingly, membrane particle uptake by endothelial cells involved the actin cytoskeleton and phosphoinositide 3‐kinase, which are implicated in macropinocytosis and phagocytosis. In the present study, MV.Act induced an increase in the F/G‐actin ratio in HMVECs consistent with actin polymerisation and cytoskeleton rearrangement.
Furthermore, MV.Act stimulated superoxide generation in a dose‐dependent manner while also inducing apoptosis in HMVECs. In addition, MV.Act affected the ability of endothelial monolayers to heal mechanical wounds, as determined using a scratch assay. More important, MV.Act disrupted endothelial barrier integrity, as measured by ECIS biosensor technology. This effect has not been reported previously. Increased paracellular permeability of the endothelial barrier can facilitate the migration of leukocytes across the endothelium. Changes observed in the paracellular space using ECIS technology have been associated with changes in junctional proteins regulating the overall strength of the junctional communication between neighbouring endothelial cells. 23 Here, MV.Act did not significantly regulate expression levels of selected junctional proteins (VE‐cadherin, claudin‐5, vinculin) in HMVECs; however, other proteins have also been associated with the regulation of endothelial barrier function. 23 Finally, stimulated T cells, but not MV.Act, regulated adhesion molecule expression in HMVECs and PBMC adhesion to them, as assessed using a BioFlux system. These findings suggest that activated T cells may induce PBMC adhesion to HMVECs through MV‐independent mechanisms (e.g. proinflammatory cytokines).
To improve the specificity of the proteomic analysis, two different data processing systems —Discoverer 2.5 and MaxQUANT — were used. This approach allowed for the identification of 98 DEPs in MV.Act vs. MV.NAct with high confidence. By functions/families analysis, these DEPs could be grouped into families related to the immune system, protein translation and cytoskeleton organisation, proteolysis, TNF/apoptosis, solute carrier family and phosphatidylinositol signalling. Best targets of DEPs enriched in MV.Act (and never detected in MV.NAct) included 37 proteins related to receptor/signalling, ubiquitination, transcription/translation and cell cycle/microtubule‐related processes. Overall, differences in protein composition between MV.Act and MV.NAct nicely matched the functional effects of MV.Act on HMVECs. For instance, MV.Act were highly enriched with IFN‐γ, a key proinflammatory cytokine released from effector CD4+ T cells, which inhibits endothelial cell proliferation, triggers endothelial cell apoptosis and induces VE‐cadherin–directed vascular barrier disruption in inflammatory bowel disease. 27 Our protein target analysis identified TNF‐α, JAK1 and STAT3 among the best protein targets of IFN‐γ (Figure S3). TNF‐α and related family members promote endothelial cell apoptosis via NF‐κB and JAK1/STAT3 activation. An earlier study 6 showed that MVs released from normal human T cell blasts upon PHA stimulation contained bioactive death ligands such as FasL and TRAIL. These molecules are involved in activation‐induced cell death, a process whereby once a cellular immune response has taken place, most activated T cells are eliminated to prevent potential autoimmune damage. We identified TNF‐α, but not FasL and TRAIL (two members of the TNF superfamily), in MV.Act. These findings may reflect methodological differences including the use of TCR‐activated CD4+ T cells in our study versus PHA‐stimulated T cell blasts in the previous study. 6 Major differences in the release of T cell‐derived MVs upon TCR activation versus treatment with mitogenic agents have been described. 7
We also found MV.Act to be enriched with additional TNF/apoptosis‐related proteins including TNF‐receptor superfamily‐18 (TNFRSF18/glucocorticoid‐induced TNF‐receptor family‐related protein [GITR]) and TNF‐receptor superfamily‐1B (TNFRSF1B/TNF‐receptor–2 [TNFR2]). TNFRSF18 is expressed on regulatory T cells (Treg) and effector T cells and is activated by its ligand, GITRL, which is expressed on antigen‐presenting cells and endothelial cells. The TNFRSF18/GITRL system exerts proinflammatory roles by regulating the extravasation process and by activating innate immunity cells and effector T cells. 28 Another MV.Act‐enriched protein was a C‐type natriuretic peptide, which was recently associated with increased blood‐brain barrier permeability. 29 These examples indicate that, apart from IFN‐γ, MV.Act are enriched with multiple proteins that can mediate endothelial barrier disruption.
The present study did not include an analysis of MVs released from CD8+ T cells. Differential characteristics of MVs from CD4+ T cells and CD8+ T cells are poorly known. A previous study 30 showed that T cell‐derived microparticles were increased in the blood from patients with active hepatitis C, whereby microparticles from activated and apoptotic CD8+ T cells showed a higher ex vivo fibrolytic activity in hepatic stellate cells, as compared to those from activated CD4+ T cells. CD147/Emmprin was identified as a candidate transmembrane molecule in the fibrolytic activity of CD8+ T cell‐derived microparticles. Comparative studies of MVs from CD4+ and CD8+ T cells are warranted.
In conclusion, our results demonstrate that MVs released from stimulated T cells selectively affect microvascular endothelial functions. It should be noted that while in vivo T cell activation primarily takes place in lymph nodes and tissues, MVs secreted by activated T cells, regardless of their antigen specificity and their site of activation, are released into the circulation and can break down the endothelial barrier in distant organs (Figure S4).
CONFLICT OF INTEREST
The authors declare no conflicts of interest. G.G.C. is co‐inventor on the International Patent WO/2020/226993 filed in April 2020, which relates to the use of antibodies specifically binding IL‐1α to reduce CNS ischaemia‐reperfusion injury, and a consultant to Sovida solutions limited. T.F.L. has received educational and research grants from Abbott, Amgen, Boehringer‐Ingelheim, Daichi‐Sankyo, Novartis, Servier and Vifor unrelated to this work.
AUTHOR CONTRIBUTION
D.V. designed and performed research and analysed data; C.B. performed selected experiments and participated in data analysis; D.d.S. performed proteomic analysis; G.P. assisted in proteomic analysis; Y.M.P. assisted in selected experiments; M.Z.C. assisted in selected experiments; P.M. contributed to proteomic analysis, G.G.C. contributed to data analysis; T.F.L. contributed to study design and writing assistance; U.E. contributed to study design and writing assistance; G.V. contributed to study design, data analysis and wrote the paper.
Supporting information
Fig S1
Fig S2
{kind=link}
Fig S3
{kind=link}
Fig S4
{kind=link}
Tab S1
Tab S2
Tab S3
Tab S4
ACKNOWLEDGEMENTS
The Center for Microscopy and Image Analysis and Flow Cytometry Facility of the University of Zurich and Marta Bachmann provided expert technical support. Anton Schirg assisted in graphical design. Open Access Funding provided by Universitat Zurich. [Correction added on 31 May 2022, after first online publication: CSAL funding statement has been added.]
Vdovenko D, Balbi C, Di Silvestre D, et al. Microvesicles released from activated CD4+ T cells alter microvascular endothelial cell function. Eur J Clin Invest. 2022;52:e13769. doi: 10.1111/eci.13769
Urs Eriksson and Giuseppe Vassalli contributed equally to this work.
Funding information
D.V. was supported by a Swiss National Science Foundation grant (no. 169194) to G.V., and a Swiss Heart Foundation grant to U.E. and the Zurich Heart house—Foundation of Cardiovascular Research
REFERENCES
- 1. Stahl PD, Raposo G. Extracellular vesicles: exosomes and microvesicles, integrators of homeostasis. Physiology (Bethesda). 2019;34(3):169‐177. [DOI] [PubMed] [Google Scholar]
- 2. Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ridger VC, Boulanger CM, Angelillo‐Scherrer A, et al. Microvesicles in vascular homeostasis and diseases. Position paper of the European society of cardiology (ESC) working group on atherosclerosis and vascular biology. Thromb Haemost. 2017;117(7):1296‐1316. [DOI] [PubMed] [Google Scholar]
- 4. Chatterjee V, Yang X, Ma Y, Wu MH, Yuan SY. Extracellular vesicles: new players in regulating vascular barrier function. Am J Physiol Heart Circ Physiol. 2020;319(6):H1181‐H1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin S, Tesse A, Hugel B, et al. Shed membrane particles from T lymphocytes impair endothelial function and regulate endothelial protein expression. Circulation. 2004;109(13):1653‐1659. [DOI] [PubMed] [Google Scholar]
- 6. Martínez‐Lorenzo J, Anel A, Gamen S, et al. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J Immunol. 1999;163:1274‐1281. [PubMed] [Google Scholar]
- 7. Blanchard J, Lankar D, Faure F, et al. TCR Activation of human T cells induces the production of exosomes bearing the TCR/CD3/x complex. J Immunol. 2002;168:3235‐3241. [DOI] [PubMed] [Google Scholar]
- 8. Bosque A, Dietz L, Gallego‐Lleyda A, et al. Comparative proteomics of exosomes secreted by tumoral Jurkat T cells and normal human T cell blasts unravels a potential tumorigenic role for valosin‐containing protein. Oncotarget. 2016;7(20):29287‐29305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trickett A, Kwan YL. T cell stimulation and expansion using anti‐CD3/CD28 beads. J Immunol Methods. 2003;275(1–2):251‐255. [DOI] [PubMed] [Google Scholar]
- 10. Simera I, Moher D, Hoey J, Schulz K f, Altman DG. A catalogue of reporting guidelines for health research. Eur J Clin Invest. 2010;40(1):35‐53. [DOI] [PubMed] [Google Scholar]
- 11. Crescitelli R, Lässer C, Jang SC, et al. Subpopulations of extracellular vesicles from human metastatic melanoma tissue identified by quantitative proteomics after optimized isolation. J Extracell Vesicles. 2020;9(1):1722433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Headland SE, Jones HR, D'Sa Adelina SV, Perretti M, Norling LV. Cutting‐edge analysis of extracellular microparticles using ImageStream(X) imaging flow cytometry. Sci Rep. 2014;4:5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bari E, Di Silvestre D, Mastracci L, et al. GMP‐compliant sponge‐like dressing containing MSC lyo‐secretome: proteomic network of healing in a murine wound model. Eur J Pharm Biopharm. 2020;155:37‐48. [DOI] [PubMed] [Google Scholar]
- 14. Motta S, Vecchietti D, Martorana AM, et al. The landscape of pseudomonas aeruginosa membrane‐associated proteins. Cells. 2020;9(11):2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc. 2016;11(12):2301‐2319. [DOI] [PubMed] [Google Scholar]
- 16. da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scardoni G, Tosadori G, Pratap S, Spoto F, Laudanna C. Finding the shortest path with PesCa: a tool for network reconstruction. F1000Res. 2015;4:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:8.13.1‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448‐3449. [DOI] [PubMed] [Google Scholar]
- 20. Scardoni G, Tosadori G, Faizan M, Spoto F, Fabbri F, Laudanna C. Biological network analysis with CentiScaPe: centralities and experimental dataset integration. F1000Res. 2014;3:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Silvestre D, Vigani G, Mauri P, Hammadi S, Morandini P, Murgia I. Network topological analysis for the identification of novel hubs in plant nutrition. Front Plant Sci. 2021;12:629013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Silvestre D, Bergamaschi A, Bellini E, Mauri PL. Large scale proteomic data and network‐based systems biology approaches to explore the plant world. Proteomes. 2018;6(2):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robilliard LD, Kho D, Johnson R, et al. The importance of multifrequency impedance sensing of endothelial barrier formation using ECIS technology for the generation of a strong and durable paracellular barrier. Biosensors (Basel). 2018;8(3):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geginat J, Sallusto F, Lanzavecchia A. Cytokine‐driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med. 2001;194(12):1711‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koschützki D, Schreiber F. Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene Regul Syst Bio. 2008;2:193‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. da Costa Gonçalves F, Korevaar SS, Ortiz Virumbrales M, et al. Mesenchymal stromal cell derived membrane particles are internalized by macrophages and endothelial cells through receptor‐mediated endocytosis and phagocytosis. Front Immunol. 2021;15(12):651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langer V, Vivi E, Regensburger D, et al. IFN‐γ drives inflammatory bowel disease pathogenesis through VE‐cadherin‐directed vascular barrier disruption. J Clin Invest. 2019;129(11):4691‐4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shami A, Atzler D, Bosmans LA, et al. Glucocorticoid‐induced tumour necrosis factor receptor family‐related protein (GITR) drives atherosclerosis in mice and is associated with an unstable plaque phenotype and cerebrovascular events in humans. Eur Heart J. 2020;41(31):2938‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bohara M, Kambe Y, Nagayama T, Tokimura H, Arita K, Miyata A. C‐type natriuretic peptide modulates permeability of the blood‐brain barrier. J Cereb Blood Flow Metab. 2014;34(4):589‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kornek M, Popov Y, Libermann TA, Afdhal NH, Schuppan D. Human T cell microparticles circulate in blood of hepatitis patients and induce fibrolytic activation of hepatic stellate cells. Hepatology. 2011;53(1):230‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Tab S1
Tab S2
Tab S3
Tab S4