

Abstract
Mutations in the TECPR2 gene are the cause of an ultra-rare neurological disorder characterized by intellectual disability, impaired speech, motor delay, and hypotonia evolving to spasticity, central sleep apnea, and premature death (SPG49 or HSAN9; OMIM: 615031). Little is known about the biological function of TECPR2, and there are currently no available disease-modifying therapies for this disease. Here we describe implementation of an antisense oligonucleotide (ASO) exon-skipping strategy targeting TECPR2 c.1319delT (p.Leu440Argfs∗19), a pathogenic variant that results in a premature stop codon within TECPR2 exon 8. We used patient-derived fibroblasts and induced pluripotent stem cell (iPSC)-derived neurons homozygous for the p.Leu440Argfs∗19 mutation to model the disease in vitro. Both patient-derived fibroblasts and neurons showed lack of TECPR2 protein expression. We designed and screened ASOs targeting sequences across the TECPR2 exon 8 region to identify molecules that induce exon 8 skipping and thereby remove the premature stop signal. TECPR2 exon 8 skipping restored in-frame expression of a TECPR2 protein variant (TECPR2ΔEx8) containing 1,300 of 1,411 amino acids. Optimization of ASO sequences generated a lead candidate (ASO-005-02) with ∼27 nM potency in patient-derived fibroblasts. To examine potential functional rescue induced by ASO-005-02, we used iPSC-derived neurons to analyze the neuronal localization of TECPR2ΔEx8 and showed that this form of TECPR2 retains the distinct, punctate neuronal expression pattern of full-length TECPR2. Finally, ASO-005-02 had an acceptable tolerability profile in vivo following a single 20-mg intrathecal dose in cynomolgus monkeys, showing some transient non-adverse behavioral effects with no correlating histopathology. Broad distribution of ASO-005-02 and induction of TECPR2 exon 8 skipping was detected in multiple central nervous system (CNS) tissues, supporting the potential utility of this therapeutic strategy for a subset of patients suffering from this rare disease.
Keywords: Oligonucleotides: Therapies and Applications, antisense oligonucleotide, TECPR2, human induced pluripotent stem cells, SPG49, HSAN9, CNS disorder
Graphical abstract
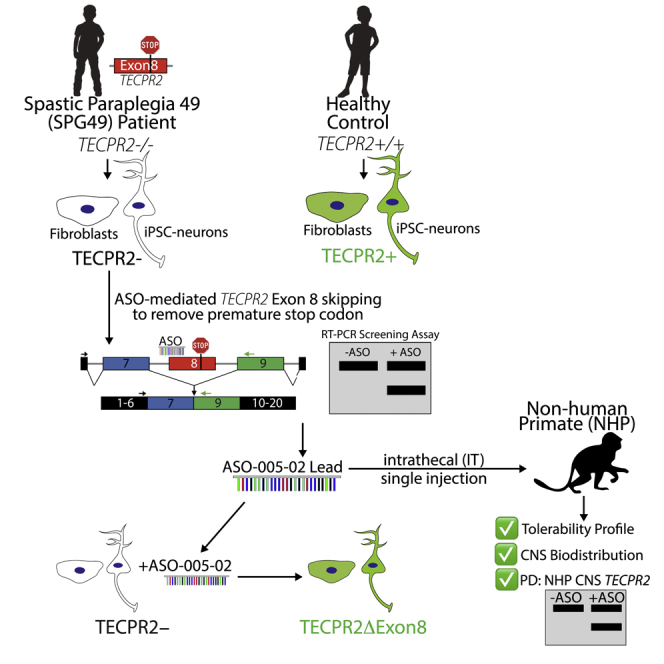
TECPR2 mutations result in an ultra-rare, life-threatening neurological disorder (SPG49/HSAN9). We identified an antisense oligonucleotide therapeutic candidate that induces TECPR2 exon 8 skipping to address a pathogenic frameshift mutation found in multiple SPG49/HSAN9 patients. The candidate showed activity in patient-derived cell lines and in vivo in non-human primate CNS tissues.
Introduction
Homozygous loss-of-function mutations in the TECPR2 gene, encoding tectonin beta-propeller repeat-containing protein 2, cause an ultra-rare, monogenic neurological disorder characterized by intellectual disability, impaired speech, hypotonia, spasticity and severe motor delay, gastroesophageal reflux, areflexia, central sleep apnea, and premature death.1,2 A homozygous pathogenic TECPR2 single-base-pair deletion mutation, c.3416delT; p.Leu1139Argfs∗75, was originally identified in five patients from three families of Bukharian Jewish origin.1 The TECPR2 mutation-induced disease was designated as an autosomal recessive form of complicated hereditary spastic paraparesis, spastic paraplegia 49 (SPG49), now classified as hereditary sensory and autonomic neuropathy type IX (HSAN9).
A second report identified two additional TECPR2 mutations (c.1319delT; p.Leu440Argfs19 and c.C566T; p.Thr189Ile) among three unrelated patients of non-Bukharian origin.2 Two of these patients were of Ashkenazi Jewish origin and had compound heterozygous c.1319delT/c.C566T and homozygous c.1319delT/c.1319delT TECPR2 genotypes, respectively. The third patient was of mixed Ashkenazi/Tunisian-Yamani/Kurdish origin and had the originally identified TECPR2 mutation and the c.1319delT mutation in compound heterozygous state, c.1319delT/c.3416delT. The c.1319delT; p.Leu440Argfs19 frameshift mutation in exon 8 of TECPR2 creates a premature termination codon and has estimated allele frequency of 0.003 in the Ashkenazi Jewish population,2 with substantially lower global frequency. The most perilous feature of SPG49/HSAN9 is progressive, life-threatening respiratory dysregulation reported to have complex multisystem pathophysiology.3
The human TECPR2 gene has 20 exons and encodes a full-length protein of 1,411 amino acids. There are two main structural annotations for TECPR2 in UniProt, an amino-terminal WD repeat domain with seven WD40 repeat motifs and a carboxy terminal TECPR domain containing six TECPR (tectonin beta-propeller repeat) motifs (Figure 1B). An LC3-interacting region has also been identified at the carboxy terminus of the protein.4 These three domains exhibit strong conservation even for TECPR2 orthologs found in distant species, suggesting important roles in TECPR2 function.4 The central region of the TECPR2 protein has no structural annotation. TECPR2 protein function is not well understood. TECPR2 has been proposed to act as a scaffold protein interacting with multiple partners.4,5 Decreased markers of autophagy were observed in SPG49 patient-derived fibroblasts, suggesting a role for TECPR2 in autophagy.1 A subsequent study, based on the TECPR2 protein interactome and functional analyses in human cell lines, reported a role of TECPR2 in endoplasmic reticulum (ER) export and autophagosome formation.4 Recently, impaired basal autophagic flux with accumulation of autophagosomes was observed in SPG49 patient fibroblasts, indicating a potential role for TECPR2 in targeting of autophagosomes to lysosomes.6 Expression of full-length TECPR2 or its TECPR domain was found to rescue aspects of the autophagy impairment, suggesting that the TECPR domain may be sufficient for regulating basal autophagy in fibroblasts.6
Figure 1.

TECPR2 gene and identification of ASO sequences inducing TECPR2 exon 8 skipping
(A) Map of TECPR2 gene showing exon/intron regions and highlighting exon 8 wild-type TECPR2+ and mutant TECPR2- (c.1319delT, p.Leu440Argfs∗19) alleles. The SPG49 patient is homozygous for the mutation (TECPR2-/-). (B) Diagram of TECPR2 protein, highlighting annotated WD domain (predicted for residues 23–343) and TECPR repeat domain (predicted for residues 945–1353), along with the protein region (in red) encoded by TECPR2 exon 8, which corresponds to 333 bp (111 amino acids). The premature stop codon in the SPG49 patient presumably results in truncated protein. ASO-mediated induction of TECPR2 exon 8 skipping could restore expression of a shorter form of TECPR2 protein lacking exon 8-encoded sequence (TECPR2ΔExon8). (C) Diagram of TECPR2 exon 8 and surrounding intron 7 and 8 regions showing location of initial set of 12 ASOs designed to induce exon 8 skipping. (D) Table listing ASO ID and sequence of initial set of 25-mer (25 nucleotides in length) ASOs used in primary screening. For these ASOs, all bases had 2′-O-methyl modification and all linkages were phosphorothioate chemistry. (E) Experimental outline for TECPR2 exon 8 skipping assay using TECPR2-/- patient-derived fibroblasts. Enrichment of a shorter (minus 333 bp) PCR amplicon of ∼399 bp is expected if TECPR2 exon 8 skipping is induced. (F) DNA gel inverse image with results of initial screening of 12 primary ASOs showing that treatment of TECPR2-/- patient fibroblasts with ASO-004, ASO-005, and ASO-007 can lead to significant enrichment of ∼399-bp PCR amplicon (highlighted in red rectangle and with red asterisks [∗∗]), corresponding to TECPR2 transcript minus exon 8. (−)PCR:H2O, water was used as negative control for PCR assay. (G) Sanger sequencing chromatogram of purified lower PCR amplicon (∼399 bp) obtained from samples treated with ASO-005, demonstrating TECPR2 exon 8 skipping with precise splicing of exon 7 and exon 9 sequences.
Two studies have characterized the effects of TECPR2 mutations in the central nervous system (CNS) in vivo. A spontaneous missense mutation (c.4009C>T or p.R1337W) affecting a highly conserved region in TECPR2 was identified in Spanish water dogs as the cause of an autosomal recessive neuroaxonal dystrophy marked by accumulation of spheroids resembling autophagosomes in the gray matter of the cerebral hemispheres, the cerebellum, the brain stem, and the spinal cord sensory pathways.7 Recently, Tecpr2 knockout mice were shown to exhibit neuroaxonal dystrophy with accumulation of autophagosomes in medulla oblongata of brain stem and dorsal tracts of spinal cord.8 These studies link TECPR2 mutation with perturbed autophagy and neurodegeneration in vivo. Despite these insights, neuronal disease mechanisms and the pathogenic basis of respiratory insufficiency in SPG49 remain poorly understood, and since TECPR2 is not a traditional drug target for small molecule modulation, therapeutic avenues for SPG49 are challenging.
Recently, antisense oligonucleotides (ASOs) that induce exon skipping have emerged as a direct therapeutic approach to address pathogenic frameshift and nonsense mutations.9,10 The clinical promise of this approach was reflected by the approval of Eteplirsen, an exon-skipping agent for Duchenne muscular dystrophy (DMD).9,11 The approval of nusinersen, a splice switching ASO for spinal muscular atrophy administered by intrathecal injection, has also validated this ASO therapeutic modality for CNS disorders.9,12, 13, 14
In its simplest form, splice-modulating ASO strategies can be used to target pathogenic genetic variants in exons that are dispensable and can be removed by in-frame skipping of exonic sequence. As TECPR2 exon 8 encodes a non-structured region of the protein and is 333 nucleotides in length, an ASO-mediated exon 8-skipping approach is a potentially feasible therapeutic approach for individuals with TECPR2 exon 8 mutations. The p.Leu440Argfs19 mutation is primarily present in individuals of Ashkenazi Jewish origin, with allele frequency estimated at 0.003 in that population.2 The exon 8-skipping strategy may be most effective for patients homozygous for this mutation that have two relevant target alleles. However, the strategy could benefit patients with the exon 8 mutation in compound heterozygous context since it could provide functional rescue from the single relevant target allele. It is important to note that other pathogenic TECPR2 mutations, including two additional exon 8 frameshift mutations reported in ClinVar, appear to be very rare. The patient population eligible for this specific therapeutic approach is thus expected to predominantly include individuals having the p.Leu440Argfs19 exon 8 mutation. Based on estimated carrier frequencies of exon 8 and other pathogenic TECPR2 alleles,15 and the shortened lifespan reported for SPG49 patients, this specific eligible patient population is expected to be up to a few hundred patients worldwide.
Here, we report the identification and optimization of TECPR2 exon 8-skipping ASOs. We show that a lead ASO rescues TECPR2 protein expression in dermal fibroblasts and induced pluripotent stem cell (iPSC)-derived neurons from an SPG49 patient. The rescued form of TECPR2 protein lacking exon 8 (TECPR2ΔEx8) exhibits the distinct, punctate neuronal distribution pattern of full-length TECPR2, providing a surrogate measure of its functionality. The candidate ASO induced TECPR2 exon 8 skipping throughout the CNS and had an acceptable tolerability profile in cynomolgus monkeys following a single intrathecal dose. Pending full safety evaluation, these results support the exon 8-skipping ASO as a promising therapeutic strategy for SPG49 patients with TECPR2 exon 8 mutation.
Results
Identification and optimization of ASO sequences inducing TECPR2 exon 8 skipping
A schematic of the TECPR2 gene and location of the exon 8 pathogenic c.1319delT (p.Leu440Argfs∗19) mutation is shown in Figure 1A. The human TECPR2 gene contains 20 exons and encodes a full-length protein of 1,411 amino acids. The mutation results in a frameshift and premature termination codon within exon 8. Since TECPR2 exon 8 is 333 nucleotides in length, ASO-mediated exon 8 skipping would preserve the TECPR2 reading frame and generate an mRNA transcript encoding a 1,300-amino acid form of TECPR2 protein (TECPR2ΔEx8) missing 111 amino acid (362–472) residues (Figure 1B). Importantly, TECPR2ΔEx8 retains the WD40 and TECPR repeat domains and lacks only a small portion of the central unstructured region, compatible with the concept that TECPR2ΔEx8 protein may retain wild-type function.
To identify candidate ASO sequences for exon 8 skipping, 12 25-mer ASOs spanning the TECPR2 exon 8 pre-mRNA region were designed, synthesized as 2′-O-methyl phosphorothioates, and selected for screening (Figures 1C and 1D). To test for exon 8-skipping activity, dermal fibroblasts derived from an SPG49 patient homozygous for the exon 8 TECPR2 mutation (TECPR2-/-) were transfected separately with each of the 12 ASOs at 1 μM and analyzed using an RT-PCR assay to detect the full-length (732 bp) and the exon 8-skipped (399 bp) TECPR2 mRNA amplicons (Figure 1E). After ASO treatment of the patient fibroblasts for 72 h, three ASOs, ASO-004, ASO-005, and ASO-007, yielded a clear PCR band of ∼399 bp, indicating potential exon 8-skipping activity (Figure 1F). Sequencing of the lower PCR product confirmed precise TECPR2 exon 8 skipping (Figure 1G). Subsequent analyses with optimized conditions (reduced ASO concentration, 300 nM versus 1 μM, and increased incubation time, 120 h versus 72 h) identified at least two additional ASOs, ASO-018 and ASO-020, targeting the 3′ end of exon 8 (Figure 1C) that induce robust exon 8 skipping (Figure S1).
The three most active ASOs (ASO-004, ASO-005, and ASO-007) target the first 64 nucleotides of TECPR2 exon 8 and define an active target region for exon 8 skipping (Figure 2A). We sought to identify an ASO lead molecule targeting this region with optimal features for therapeutic application, including shorter length, potency in the nanomolar range, clinically validated chemistry, and target sequence conservation to enable preclinical in vivo studies. The active target region was analyzed for sequence homology to animal species relevant for preclinical studies. The first 37 bases of the region and the last three bases of intron 7 have sequence identity with cynomolgus monkey (Figure 2A), and significantly lower sequence similarity to homologous regions in mouse and rat Tecpr2. To identify optimal ASO lead molecules with target sequence conservation in cynomolgus monkey, given its importance as a preclinical toxicology species, micro-tiling of the 40-base region was performed using sets of 18-mer and 21-mer ASOs, for a total of 41 ASO sequences (Figures S1 and S2) as well as shorter active derivatives of ASO-004 and ASO-005 (Figures S1 and S2). TECPR2-/- patient fibroblasts were transfected separately with the micro-tiling ASOs at 1 μM and analyzed using the patient fibroblast RT-PCR assay (Figure 1E). Multiple active ASOs yielding the 399-bp PCR product were identified (Figure 2B). Further prioritization of active ASOs was carried out based on bioinformatic analysis to eliminate those with potential off-target homologies. The initial ASO screening was performed using ASO chemistry with all RNA bases with 2′-O-methyl sugar modifications and all phosphorothioate linkages, based on efficiency and cost considerations. To assess selected ASOs using the clinically validated ASO chemistry to be applied in our clinical candidate, a set of nine prioritized leads among all ASOs were selected for conversion to chemistry with all RNA bases with 2′-O-methoxyethyl (2′-MOE) sugar modifications, 5-methyl cytosine and 5-methyl uracil bases, and all phosphorothioate linkages, the same chemistry employed in Spinraza (nusinersen).16 These nine ASOs share a core 13-base sequence (Figure 2C) and were tested in six-point concentration-response (300–1 nM, ∼3-fold dilutions) in the TECPR2-/- patient fibroblast RT-PCR assay (Figure 2D). Side-by-side comparisons of ASO chemistries demonstrated that the transition from 2′-O-methyl to 2′-MOE RNA bases did not change the ASO-mediated exon 8-skipping activity (Figure S3). ASO-005-02 (a 21-mer derivative of ASO-005) was the most potent ASO lead with a half maximal effective concentration (EC50) of ∼27.3 nM (Figure 2E). Two additional ASO sequences (ASO-059 and ASO-066, the latter being a 19-mer derivative of ASO-005) were also selected as back-up ASO leads based on robust RT-PCR assay results. ASO-005-02 was selected for further characterization in patient-derived cellular models.
Figure 2.

ASO tiling, optimization, and selection of ASO leads that induce TECPR2 exon 8 skipping
(A) TECPR2 exon 8 region showing binding locations of the three most active exon 8-skipping ASOs (ASO-004, ASO-005, and ASO-007, shown in green). Sub-region with 100% sequence identity to cynomolgus monkey is highlighted in light blue. (B) DNA gel inverse image with a subset of PCR results of additional screening after ASO micro-tiling and trimming of initial active sequences. Enrichment of lower PCR amplicon (∼399 bp) can be detected after treatment with several of these ASOs (highlighted in red rectangle). (C) Subset of nine ASO sequences of different length selected as priority leads. All sequences listed were then synthesized using 2′-O-methoxyethyl bases and 5-methyl cytosine and uracil bases, with phosphorothioate backbone. All ASO sequences share a core 13-base sequence highlighted in blue and underscored. (D) DNA gel inverse images of TECPR2 exon 8-skipping PCR results for ASO leads ASO-005-02, ASO-059, and ASO-066, assayed at different concentrations (300–1 nM). (E) Half maximal effective concentration (EC50) for ASO leads ASO-005-02, ASO-059, and ASO-066 calculated using percentage of TECPR2 exon 8 skipping (estimated as fraction of lower PCR amplicon over total PCR product) at different concentrations (each data point corresponds to n = 3 independent rounds of ASO treatment, each with n = 1 RT-PCR assay). ASO-005-02 (in blue) was the most potent (EC50 = ∼27.3 nM) and was selected for further studies in patient-derived cells.
Rescue of TECPR2 protein expression in TECPR2-/- patient fibroblasts and iPSC-derived neurons
TECPR2-/- patient fibroblasts do not express TECPR2 protein as assessed by immunocytochemistry when compared with fibroblasts obtained from healthy relatives of the SPG49 patient (Figure S4). The available antibody used for this analysis could detect overexpression of recombinant full-length TECPR2 and TECPR2 lacking exon 8-encoded sequence (TECPR2ΔEx8) in transfected cell lines by immunoblotting but was not effective with the same method at detecting endogenous TECPR2 in fibroblasts (Figure S5). Therefore, we utilized TECPR2 immunocytochemistry to assess ASO-mediated rescue of TECPR2 protein expression in patient-derived fibroblast cells. Transfection of patient fibroblasts with ASO-005-02 at 100 nM and 300 nM induced robust rescue of TECPR2 protein expression (Figures 3A–3C), indicating effective cellular expression of TECPR2 lacking exon 8-encoded sequence. Levels of protein expression in ASO-treated patient fibroblasts were similar to those detected in both TECPR2+/+ and TECPR2+/- fibroblasts (Figure 3C), although it was noted that only minimal differences in protein detection were found between the TECPR2+/+ and TECPR2+/- fibroblast samples, perhaps due to limitations in the detection efficiency of the antibody in this assay. These data indicated that ASO-induced exon 8 skipping could induce protein expression of TECPR2ΔEx8 to levels of endogenous TECPR2 detected in healthy control samples.
Figure 3.

ASO-005-02 can rescue TECPR2 protein expression in TECPR2-/- (SPG49) patient-derived fibroblasts
(A) Experimental outline for rescue of TECPR2 protein expression assay, using immunocytochemistry with a rabbit polyclonal antibody raised against human TECPR2 as the primary antibody. TECPR2 immunoreactivity in TECPR2-/- fibroblasts was assessed 5 days after treatment with ASO-005-02 using an IN Cell Analyzer 6000 platform. (B) Representative immunofluorescence images obtained from two fields of view (FOVs) for each of the different TECPR2 genotype and ASO treatment conditions, showing TECPR2 immunoreactivity with green signal and nuclear stain in blue. While untreated TECPR2-/- fibroblasts show no TECPR2 immunoreactivity, the same patient-derived cells treated with 100 nM or 300 nM lead ASO-005-02 show TECPR2 signal comparable with that of healthy control genotypes TECPR2+/+ and TECPR2+/- (scale bar, 20 μm). (C) Quantitative immunocytochemistry of the different TECPR2 genotype and ASO conditions. Each data point represents a single cell for which TECPR2 fluorescence signal has been estimated (n = 60–400 cells per condition, 16–32 FOVs, error bars = SEM, ∗∗∗p < 0.001 [pairwise t test FDR-corrected p value]).
Since SPG49 is primarily a neurological disorder,1,2 we sought to confirm the activity of the lead ASO candidate in neurons. Neurons were differentiated from iPSC lines derived from the SPG49 patient and unaffected TECPR2+/+ and TECPR2+/- relatives. Neurons were produced using a transcriptional programming approach whereby the proneuronal transcription factor NGN2 was overexpressed in iPSCs (Figure 4A) to generate a homogenous population of cortical excitatory neurons (NGN2 neurons).17,18 Differentiated neurons were treated at plating with ASO-005-02 at 5 μM, using gymnotic delivery. A scrambled, non-targeting ASO was used as a negative control. Immunocytochemistry analysis of TECPR2+/- NGN2 neurons treated with the scrambled control ASO indicated that all neurons identified by staining for MAP2 (microtubule-associated protein 2) were positive for TECPR2 staining (eight out of eight in field shown, Figures 4B and 4C). Patient-derived NGN2 neurons treated with control scrambled ASO were negative for TECPR2 immunoreactivity, indicating lack of TECPR2 protein expression (none out of seven in field shown, Figures 4D and 4E). Treatment of patient-derived neurons with ASO-005-02 resulted in TECPR2 protein expression (TECPR2ΔEx8) in a percentage of cells (five out of 15 in field shown; Figures 4F and 4G). Quantification across multiple fields of view indicated that 100% of TECPR2+/+ and TECPR2+/- NGN2 neurons treated with scrambled control ASO were positive for TECPR2 protein expression, while a background of ∼2.8% of patient-derived TECPR2-/- NGN2 neurons treated with scrambled control ASO showed some level of TECPR2 immunoreactivity (Figure 4H). Approximately 22.5% (15 out of 85) of patient-derived TECPR2-/- NGN2 neurons treated with ASO-005-2 showed clear TECPR2 immunoreactivity (Figures 4F–4H). Gymnotic delivery of ASOs to cell lines, including cultured human iPSC-derived neurons, is known to be inefficient, consistent with induction of measurable TECPR2 protein expression in only a fraction of the TECPR2-/- cells treated with ASO-005-02. Neuronal uptake of ASOs has been shown to be more efficient in vivo.19, 20, 21
Figure 4.

ASO-005-02 can rescue TECPR2 protein expression in TECPR2-/- (SPG49) patient iPSC-derived neurons
(A) Generation of SPG49 patient iPSC lines and cortical excitatory neurons via reprogramming of peripheral blood mononuclear cells (PBMCs) with pluripotency factors (OCT4, SOX2, KLF4, and cMYC) and transcriptional programming with pro-neuronal factor NEUROGENIN-2 (NGN2). TECPR2-/- patient and TECPR2+/- control iPSC-derived neurons were treated with either 5 μM ASO-005-02 or a scrambled (non-targeting) negative control ASO during plating (single treatment, gymnotic delivery) and cultured for 45 days to allow neuronal maturation. Effect of ASO treatment on TECPR2 expression was assessed using immunocytochemistry. (B and C) Control TECPR2+/-; NGN2 neurons show clear TECPR2 immunoreactivity in soma and neuronal processes. (B) TECPR2 signal only and (C) a merge of TECPR2 (in green) with the pan-neuronal marker MAP2 (in red). Individual neurons in these panels are indicated (numbers 1–8). (D and E) SPG49 patient TECPR2-/- NGN2 neurons treated with scrambled (negative control) ASO do not show clear TECPR2 immunoreactivity. Individual neurons marked by MAP2 staining are indicated (numbers 1–7). (F and G) A subset of SPG49 patient TECPR2-/- NGN2 neurons treated with 5 μM lead ASO-005-02 show clear TECPR2 immunoreactivity, indicating rescue of TECPR2 protein expression in the form of TECPR2ΔEx8. These patient-derived neurons positive for TECPR2 signal are indicated by numbers in green in (F). (B–G) Scale bar, 20 μm. (H) Quantification of several FOVs indicates that ∼22.5% of SPG49 TECPR2-/- patient iPSC neurons exhibited rescue of TECPR2 protein expression in culture, after single treatment (gymnotic delivery) with lead ASO-005-02.
TECPR2 and TECPR2ΔExon8 neuronal localization in patient iPSC-derived neurons
To examine the neuronal subcellular distribution of TECPR2 protein and TECPR2ΔEx8, high-resolution immunocytochemistry analysis was performed on iPSC-derived NGN2 neurons treated with ASOs by gymnotic delivery (5 μM) at plating and cultured for 45 days. Cells were co-stained for neuronal markers β-III TUBULIN (present in all neurites), MAP2 (present in soma and proximal dendrites), and for TECPR2. As shown in Figure 5A–5D′, scrambled control ASO-treated TECPR2+/+ NGN2 neurons exhibited a distinct, subcellular TECPR2 expression pattern with three discrete types of TECPR2-positive puncta: (1) broad, speckled puncta pattern within soma and proximal dendrites (Figure 5D, cyan arrowhead); (2) small discrete puncta in thick and thin β-III TUBULIN-positive neurites (Figure 5D, yellow arrowhead); and (3) rare large puncta in thin β-III TUBULIN-positive neurites (Figures 5D and 5D′, green arrowhead). TECPR2+/- iPSC-derived NGN2 neurons treated with scrambled control ASO showed a similar pattern of staining (Figures 5E–5H′). TECPR2-/- patient-derived NGN2 neurons treated with the scrambled control ASO showed no TECPR2 immunoreactivity and absence of the punctate expression pattern (Figures 5I–5L′). Three examples presented in Figures 5M–P′, 5Q–T′, and 5U–X′ show that treatment of patient TECPR2-/- NGN2 neurons with ASO-005-2 rescued all three types of neuronal TECPR2-positive puncta observed in TECPR2+/+ NGN2 neurons. The preserved punctate expression pattern is consistent with the functional integrity of TECPR2ΔEx8 and provides evidence that the TECPR2ΔEx8 protein is synthesized, folded, and transported to discrete subcellular domains similar to wild-type, full-length TECPR2.
Figure 5.

TECPR2 and TECPR2ΔExon8 neuronal localization in patient iPSC-derived neurons
(A–D′) High-resolution immunocytochemistry analyses of control TECPR2+/+ iPSC neurons (DIV45) treated with 5 μM scrambled non-targeting negative control ASO (scASO), single dose, gymnotic delivery at plating. Neurons were co-stained for MAP2 (A), β-III TUBULIN (B), and TECPR2 (D). β-III TUBULIN/TECPR2 merged signal (C), and an enlarged (∼2.1×) sub-region of the TECPR2 image (D′) are also presented. Distinct patterns of TECPR2 sub-cellular localization are highlighted in (D) and (D′): broad, speckled puncta pattern within soma and proximal dendrites (cyan arrowhead), small discrete puncta in neurites (yellow arrowhead), and rare large puncta in neurites (green arrowheads). (E–H′) Control TECPR2+/- iPSC neurons (DIV45) treated with 5 μM scASO, single dose, gymnotic delivery show similar pattern of TECPR2 subcellular localization to that of TECPR2+/+ (D and D′) with the three types of puncta highlighted by arrowheads in (H) and (H′): broad puncta pattern within soma and proximal dendrites (cyan arrowhead), small discrete puncta in neurites (yellow arrowhead), and rare large puncta in neurites (green arrowheads). (I–L′) SPG49 patient TECPR2-/- iPSC-derived NGN2 neurons (DIV45) treated with the 5 μM scASO show no TECPR2 immunoreactivity and absence of the punctate expression pattern. White arrowhead in (L) and (L′) indicates lack of TECPR2 immunoreactivity signal in an area of a MAP2+ soma, according to (I). (M–P′) SPG49 patient TECPR2-/- iPSC-derived NGN2 neurons (DIV45) treated with 5 μM ASO-005-02 (single dose, gymnotic delivery at plating) show rescue of broad puncta pattern of TECPR2 expression within soma and proximal dendrites, as highlighted by cyan arrowhead in (P) and enlarged (∼2.1×) in (P′). (Q–T′) SPG49 patient TECPR2-/- iPSC-derived NGN2 neurons (DIV45) treated with 5 μM ASO-005-02 (single dose, gymnotic delivery at plating) show rescue of expression of small discrete TECPR2-positive puncta in β-III TUBULIN-positive neurites, as highlighted by yellow arrowheads in (T) and enlarged (∼2.1×) (T′). (U–X′) SPG49 patient TECPR2-/- iPSC-derived NGN2 neurons (DIV45) treated with 5 μM ASO-005-02 (single dose, gymnotic delivery at plating) show rescue of expression of rare large TECPR2-positive puncta in neurites, as highlighted by green arrowhead in (X) and enlarged (∼2.1×) (X′). (A–X′) Scale bar, 20 μm.
Further definition of the subcellular structures represented by TECPR2-positive puncta could elucidate neuronal TECPR2 function. Preliminary data using immunocytochemistry for autophagy, ER export, and vesicle-mediated protein trafficking markers suggest involvement of VPS11 (vacuolar protein sorting-associated protein 11 homolog) as a potential TECPR2 interactor, as we observed significant overlap in TECPR2 and VPS11 subcellular localization in human iPSC-derived neurons (Figure S6). We did not detect significant co-localization of TECPR2 with a subset of markers for canonical endosomal, lysosomal, and autophagosomal compartments in neurons (Figure S7). Furthermore, in initial studies, we did not detect robust alterations in autophagy in patient-derived fibroblasts relative to healthy control fibroblasts (Figure S8) and consequently did not pursue autophagy assays as a basis for further assessment of TECPR2ΔEx8 functionality. In an additional evaluation of functional rescue, preliminary investigation of the interacting partners of TECPR2 and TECPR2ΔEx8 in HEK293 cells using pull-down experiments followed by mass spectrometry-based proteomics suggests that the percentage of retained interactions in these non-neuronal cells ranges from 33% to 72%, depending on the statistical thresholds applied (Figure S9). The evidence that a significant percentage of TECPR2 protein interactions are retained by TECPR2ΔEx8 suggests that TECPR2ΔEx8 may at least partially restore TECPR2 function.
Single-dose tolerability study of lead ASO candidates in cynomolgus monkeys
To determine if the lead ASO candidate ASO-005-02 and two back-up candidates (ASO-059 and ASO-066) have TECPR2 exon 8-skipping activity in vivo and are tolerated in non-human primates, a single-dose (intrathecal; 20 mg) 14-day tolerability study was performed in cynomolgus monkeys (n = 2 per ASO) (Figure 6A). Prior to the in vivo study, the efficacy of the ASO candidates was confirmed in vitro in cultured cynomolgus monkey fibroblasts using an RT-PCR assay analogous to that used for human fibroblasts (Figure S10). Clinical observations of animals treated with the ASOs indicated that all three ASO candidates had an acceptable tolerability profile with similar non-adverse transient clinical signs and neurological changes (Figure S11). For ASO-005-02, transient behavioral changes included tremors, postural changes, as well as effects on postural reactions and flexor reflexes, which generally resolved by 24 h post dose.
Figure 6.

Lead ASO candidate ASO-005-02 is effective in CNS tissues and well tolerated in non-human primates
(A) In vivo ASO tolerability study design. A single 20-mg intrathecal dose of each of the three lead ASOs was delivered to cynomolgus monkeys (n = 2 per ASO). Elliotts B solution was used as vehicle control to establish baseline conditions for the different assays. Fourteen days after ASO treatment, animals were necropsied, and different CNS tissues were obtained for pharmacokinetic (PK), pharmacodynamic (PD), and postmortem histopathology analyses. ASO concentrations (ng/g) in the different CNS tissues were determined using a hybridization ELISA assay and percentage of cynomolgus TECPR2 exon 8 skipping was estimated using a modified version of the patient RT-PCR assay with cynomolgus monkey-specific PCR primers. (B) Diagram of RT-PCR assay for detection of cynomolgus monkey TECPR2 exon 8 skipping, which corresponds to enrichment of a lower PCR amplicon (∼386 bp). (C) DNA gel inverted images showing PCR results for TECPR2 exon 8-skipping assay for different CNS tissues (spinal cord cervical, lumbar, thoracic, pons, medulla, and frontal cortex) obtained from monkeys treated with each of three ASO leads (ASO-059, ASO-005-02, ASO-066). cDNA samples from cultured monkey fibroblasts transfected with the same three ASOs were used as positive controls for the assay, and molecular biology-grade water (−) was used as negative control for the PCR assay. Significant enrichment of lower PCR amplicon (∼386 bp, indicated by red asterisks [∗∗]) can be easily detected for the two animals treated with ASO-005-02, which was selected as the therapeutic candidate based on this superior in vivo TECPR2 exon 8-skipping activity profile. (D) PK/PD scatterplot showing good correlation between the concentration (ng/g) of ASO-005-02 estimated in different CNS tissues (using hybridization ELISA) and the percentage of TECPR2 exon 8 skipping (as measured by RT-PCR) detected in those tissues for the two animals (animals 3001 and 3501) treated with this ASO candidate. SP Lumbar, lumbar spinal cord tissue; SP Thoracic, thoracic spinal cord thoracic tissue; Fr Cortex, frontal cortex tissue.
Histopathology analyses of brain, spinal cord (lumbar, thoracic, and cervical), and dorsal root ganglia tissues indicated that the single 20-mg dose of ASO-005-02, ASO-059, or ASO-066 to cynomolgus monkeys via intrathecal injection did not result in any test item-related gross or microscopic changes at study termination (Figure S11). Intrathecal injection resulted in broad uptake of ASO-005-02 in most animals throughout the neuraxis, as measured by hybridization ELISA (Figure 6D). Pharmacodynamic (PD) analyses using RT-PCR in CNS tissues led to the selection of ASO-005-02 as the therapeutic candidate based on its superior in vivo TECPR2 exon 8-skipping activity (Figures 6B and 6C), correlating with ASO concentration in the CNS tissues analyzed (Figure 6D), confirming in vivo target engagement via this route of administration. In sum, the single-dose in vivo tolerability study indicated that ASO-005-02 is active and has an acceptable tolerability profile when administered by intrathecal injection in cynomolgus monkeys.
Discussion
Here, we report the identification of an ASO therapeutic strategy suitable for patients with an ultra-rare neurological disorder (SPG49) resulting from homozygous truncating mutations in exon 8 of the TECPR2 gene. The strategy could also benefit patients with the exon 8 mutation in compound heterozygous context. The therapeutic is designed to be administered by intrathecal injection to address the severe, life-threatening neurological symptoms of the disease. The mechanism of action is to induce exon 8 skipping to restore the TECPR2 mRNA reading frame and induce expression of a form of TECPR2 protein (TECPR2ΔEx8) that lacks 111 amino acids of an apparently unstructured central region of full-length TECPR2.
Following the identification of ASO sequences with TECPR2 exon 8-skipping activity, optimization studies led to identification of a lead candidate, ASO-005-02, with potent exon 8-skipping activity. Treatment of patient-derived fibroblasts and neurons with ASO-005-02 induced expression of TECPR2ΔEx8 in both patient cell types. TECPR2ΔEx8 protein was shown to retain the distinct, punctate neuronal expression pattern of full-length TECPR2, providing evidence that it is synthesized, folded, and transported to discrete subcellular domains similar to the full-length protein. In addition, preliminary protein interactome studies in HEK293 cells suggest that TECPR2ΔEx8 retains a significant portion of the protein binding interactions of full-length TECPR2. Future interactome studies in human iPSC-derived neurons will be informative to further evaluate TECPR2ΔEx8 in a neuronal context. Based on these observed features of TECPR2ΔEx8, it is anticipated that TECPR2 exon 8-skipping ASOs may at least partially rescue the function of full-length TECPR2.
Analysis of the effects of exon 8-skipping ASOs in patient-derived cells to confirm and characterize activity in disease-relevant cellular contexts was an important aspect of this study. For these experiments, we utilized dermal fibroblast cells and iPSC-derived neurons from a single SPG49 patient homozygous for the exon 8 TECPR2 mutation. We were not able to access cell lines from additional patients as a result of the rarity of the disease. However, since the ASO leads target wild-type sequences within the TECPR2 pre-mRNA and not the patient mutation, we predicted and confirmed exon 8-skipping activity of the candidate ASO in unaffected control cells from different genetic backgrounds (Figure S12). Therefore, we anticipate that the effects of the ASO candidate should generalize to the population of SPG49 patients harboring the exon 8 mutation, which may include up to a few hundred individuals globally, based on estimated mutation carrier frequencies.
Exon-skipping ASO therapeutics that address loss-of-function mutations have been developed for DMD. This strategy led to approval of eteplirsen22 and golodirsen,11,23,24 which induce skipping of DMD exons 51 and 53, respectively, rescuing expression of shortened forms of dystrophin lacking the exon-encoded sequences. In-frame deletions including exons 51 or 53 were known to result in Becker muscular dystrophy, a less severe form of muscular dystrophy, providing genetic validation for the ASO therapeutic concept.25,26 The lack of such genetic concept validation for TECPR2 exon 8 skipping, and the lack of understanding of TECPR2 function and the SPG49 disease mechanism, present challenges in applying an analogous approach to SPG49. We addressed this by using high-resolution subcellular localization and proteomics studies to identify surrogate measures of TECPR2 functionality. We propose that the full preservation by TECPR2ΔEx8 of the distinct neuronal subcellular distribution identified for full-length TECPR2 and the partial preservation of its protein interactome are valid surrogates for partial functional restoration.
The distinct neuronal subcellular distribution pattern identified for TECPR2 and its co-localization with VPS11, a core member of CORVET (class C core vacuole/endosome tethering) and HOPS (homotypic fusion and vacuole protein sorting) tethering complexes,27,28 may shed light on TECPR2 functions relevant to SPG49. Further characterization of the various TECPR2-positive puncta to define these subcellular compartments and investigate how their perturbation could lead to pathogenic manifestations of SPG49 is an important area for future work.
ASO-005-02 employs the same chemical modifications as in nusinersen and was inactive in human peripheral blood mononuclear cell (PBMC) cytokine release assays, suggesting minimal potential for immune stimulatory effects (Figure S13). Finally, a single 20-mg intrathecal dose of ASO-005-02 in cynomolgus monkeys had an acceptable tolerability profile, with some transient non-adverse behavioral effects with no correlating histopathology. The ASO distributed throughout the CNS and induced detectable exon 8 skipping in multiple CNS tissues, demonstrating activity in vivo and the applicability of the cynomolgus monkey for assessing toxicology of the candidate. To our knowledge, this is the first report of exon skipping induced in the CNS of non-human primates by an intrathecally administered ASO. Additional long-term toxicology studies in non-human primates across a range of doses with comprehensive toxicology evaluation are needed to gain a complete understanding of the safety profile of this therapeutic candidate and establish whether the candidate has a sufficient therapeutic window to support clinical development. In addition, profiling the effects of the ASO candidate on the transcriptome using RNA sequencing analysis in relevant cellular contexts will be important to examine whether the candidate induces off-target effects at a therapeutically relevant concentration, as potential for off-target effects on transcript expression has been reported for splice-switching ASOs.29,30 Given the grave prognosis for SPG49 patients, the potential of TECPR2 exon 8-skipping ASOs to restore TECPR2 expression and function, and the relatively safe profile of this drug class, we expect that this therapeutic approach could ultimately provide clinical benefit to SPG49 patients suffering from TECPR2 exon 8 mutation-induced disease.
Materials and methods
Derivation and culture of human dermal fibroblast cell lines
Under Institutional Review Board (IRB)-approved protocol and consent forms, we obtained skin punch biopsies from the forearm of an SPG49 patient (TECPR2-/-) and from healthy control relatives (TECPR2+/- and TECPR2+/+). Skin biopsies were cultured using DMEM basal medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Hyclone FBS Defined (US)-Heat Inactivated) and 1× penicillin:streptomycin solution (Corning), until dermal fibroblast cells expanded and reached 80%–90% confluency in a surface area of ∼3.8 cm2. Fibroblasts were then dissociated with 0.05% Trypsin EDTA (Corning) and passaged by seeding at 1:3 to 1:5 dilution densities. This process was repeated until establishing cryostocks of fibroblasts at passage (P) 3 to 6. All experiments presented here were carried out using fibroblast cells at P6 to P10. Cells were routinely cultured in 96-well Ibidi cell culture clear plates for RT-PCR and immunocytochemistry assays.
Derivation and culture of human iPSC lines and neuronal production
Under IRB-approved protocol and consent forms, we obtained peripheral blood samples (2–4 mL) from an SPG49 patient (TECPR2-/-) and from healthy control relatives (TECPR2+/- and TECPR2+/+). PBMCs were then isolated by density gradient centrifugation with Ficoll. PBMCs were reprogrammed into iPSCs using an established non-integrating method whereby pluripotency-associated factors (OCT4, KLF4, SOX2, and c-Myc) are transiently expressed via Sendai virus particles (Cytotune 2.0 kit, Life Technologies). At least three to five clones per donor were established into iPSC lines, and these cells were routinely expanded in culture and maintained using mTeSR1 medium (STEMCELL Technologies) and cell culture surfaces pre-coated with Matrigel (Corning) according to manufacturers’ recommendations. TECPR2+/+, TECPR2+/-, and TECPR2-/- iPSC lines generated as part of this study are available upon reasonable request to the Luke Heller TECPR2 Foundation and Q-State Biosciences. iPSC lines were differentiated into cortical excitatory NGN2 neurons using a transcriptional programming approach, as previously described.17,18 Briefly, iPSC lines were initially transduced with lentiviral particles expressing the reverse tetracycline transactivator (rtTA) and a tetracycline-responsive (TetOp) construct driving the expression of the proneuronal transcription factor Neurogenin-2 (NGN2) and a puromycin antibiotic resistance enzyme. These genetically modified iPSC lines were then expanded in mTeSR1 medium for three to six passages prior to induction of NGN2 expression via doxycycline. For neuronal production, iPSCs were dissociated with Accutase (STEMCELL Technologies) according to manufacturer’s recommendations and plated at a density of 300,000 cells/cm2 using mTeSR1 medium supplemented with 10 μM Rock Inhibitor (Sigma) and 2 μg/mL doxycycline (Sigma) to initiate NGN2 overexpression. A day later, the medium was switched to a 1:1 DMEM/F-12:Neurobasal Medium (Thermo Fisher Scientific) supplemented with 1× GlutaMAX (Life Technologies), 1× non-essential amino acids (Life Technologies), 1× N2 (Gibco), 1× B-27 (Gibco), 2 μg/mL doxycycline (Sigma), and 2 μg/mL puromycin (Sigma). Medium was exchanged daily with fresh medium for three additional days, which then resulted in a highly homogenous population of post-mitotic neurons, which were then dissociated with Accutase and plated at a density of 80,000–100,000 cells/cm2 onto poly-D-lysine/laminin pre-coated single-well dishes (MatTek) for ASO treatments and immunofluorescence and RT-PCR assays. Human neuronal cultures were seeded and maintained for 30 to 45 days in Neurobasal A Medium supplemented with 1× GlutaMAX (Life Technologies), 1× non-essential amino acids (Life Technologies), 1× N2 (Gibco), 1× B-27, 10 ng/mL BDNF (R&D), and 10 ng/mL GDNF (R&D). In order to allow for neuronal maturation of the differentiated cells, the cultures were supplemented 3 days after the initial seeding with 40,000 cells/cm2 of primary mouse cortical glial cells, prepared as previously described.31
ASO synthesis and delivery into cultured cells
All ASO molecules used for in vitro studies were synthesized (100–250 nmol scale) at Integrated DNA Technologies (IDT) using either 2′-O-methyl or 2′-MOE modified ribonucleosides and phosphorothioate internucleotide linkages. For ASO delivery into human and non-human primate (cynomolgus) fibroblast cells, we used a lipid-based transfection system (Lipofectin, Thermo Fisher Scientific) in which the ASO was incubated with the transfection reagent in Opti-MEM (Themo Fisher Scientific) for 15–20 min prior to the addition of this solution to the cultured cells. For these experiments, we routinely used 0.5 μL of Lipofectin for every 200 μL of cell culture medium, and serum concentration in the medium was reduced from 10% to 2% for the duration of the ASO treatment (typically 3–5 days). For ASO delivery into human iPSC-neuronal cultures, we used gymnotic delivery (no transfection agent) as we observed significant cellular toxicity with lipid-based transfection methods. For gymnotic delivery, the ASO was added to the medium at the desired concentration (typically 2.5 to 5 μM) with a single treatment at the moment of neuronal plating. Medium was exchanged once a week starting 3 days after plating, and ASO was not re-plenished with fresh medium additions.
ASO synthesis and intrathecal delivery into non-human primates
For in vivo studies, the top three lead ASOs: ASO-059, ASO-005-02, and ASO-066, were re-synthesized at the 1.1 g scale at Axolabs (Germany) and additional quality control was carried out on these production batches with acceptable endotoxin levels (<0.10 EU/mg). The new corresponding production identity numbers (IDs) for these three new batches of ASOs were QS0321489-05 (same sequence as ASO-059 and also designated in some figures as 89-05), QS0321490-05 (same sequence as ASO-005-02 and also designated in some figures as 90-05), and QS0321491-05 (same sequence as ASO-066 and also designated in some figures as 91-05). ASO in vivo work was carried out at Charles River Laboratories (Charles River Laboratories Montreal ULC; Senneville Site) under their Institutional Animal Care and Use Committee (IACUC)-approved protocols. Briefly, ASOs as lyophilized powders were dissolved in Elliotts B solution at 20 mg/mL and sterile filtered using a 0.22-μm PVDF membrane. Animals were dosed under general anesthesia in lateral recumbency position by direct intrathecal puncture at the lumbar level using a spinal needle. Dosing volume was 1 mL, followed by a 0.25-mL flush of Elliotts B solution. The term of the study was 2 weeks, at which point animals were euthanized, and necropsy and terminal analyses performed. Endpoints evaluated in the study included mortality, clinical observations, body weights, neurological examinations, cerebrospinal fluid (CSF) and tissue bioanalysis for ASO concentration by hybridization ELISA (samples collected at necropsy), tissue TECPR2 mRNA splicing analysis by RT-PCR, gross necropsy findings, and histopathological examinations. At study termination, CNS tissues, CSF, and liver and kidney tissues were collected for the pharmacokinetic (tissue bioanalysis) and pharmacodynamic (TECPR2 exon 8-skipping RT-PCR assay) analyses. ASO concentrations in tissues were determined by hybridization ELISA (HELISA) using a dual probe capture/detection method as previously reported.32 The HELISA quantification method for ASO-005-02, ASO-059, and ASO-066 used a capture probe that consisted of sequence GTCTGGATG linked to biotin at the 3′ end and a detection probe that consisted of sequence TCTGGAGAT linked to digoxigenin at the 5′ end, and the method was further optimized and validated as a qualified analytical procedure at Charles River Laboratories (Montreal). The TECPR2 exon 8-skipping RT-PCR assay for PD analyses is described next.
RT-PCR (TECPR2 exon 8 skipping) assays
For RT-PCR exon 8-skipping assay, two different methods were used for RNA and cDNA preparations, depending on the initial source (cultured cells versus in vivo tissues). For cultured fibroblasts, total RNA was initially prepared 3–5 days after ASO treatment using the RNA lysis reagents from the Cell-to-CT kit (Thermo Fisher Scientific), following manufacturer’s recommendations, and a total of 50 μL of lysis solution (containing DNase I) per cell culture well. cDNA for these RNA samples was prepared in a 50-μL volume using the RT reagents of the same kit, 10 μL of lysed sample, and following manufacturer’s RT protocol (37°C 1 h, 95°C 5min). cDNA was then diluted 1:2 to 1:10 in molecular biology-grade nuclease-free water prior to setting up PCR assays. For tissue samples from the in vivo study, tissue was homogenized, and total RNA was obtained using the RiboPure RNA Purification kit (Thermo Fisher Scientific). For this particular set of samples, RNA was quantified using a Nanodrop and was diluted to 10 ng/μL in nuclease-free water prior to cDNA synthesis. This purified total RNA was reverse transcribed to cDNA using iScript Reverse Transcription Supermix (Bio-Rad Laboratories) with 100 ng (10 μL) of the RNA template and combined oligo(dT) and random primers according to the manufacturer’s protocol: priming at 25°C for 5 min, RT at 46°C for 20 min, and RT inactivation at 95°C for 1 min. The resulting cDNA was then diluted 1:2 to 1:10 in nuclease-free water. All PCR assays were carried out in 96-well plates using a Mastercycler Pro S Thermocycler 6325 (Eppendorf). PCR-based TECPR2 exon 8-skipping assays used a 25-μL reaction containing KOD Hot Start Master Mix (Sigma), 400 nM solutions of each of two primers, and 10 μL of diluted cDNA sample. The following primer pairs were used on human and non-human primate samples, respectively: human TECPR2 forward primer #25 (GGGACTGTTCAAGCCACGTTTATC) and human TECPR2 reverse primer #30 (AAATTCGGAGCAGGGTGTCGATTC); cynomolgus TECPR2 forward primer #96 (AGATGCTTTTGCCGGGGGAGTC) and cynomolgus TECPR2 reverse primer #97 (GGGGACTGTCCCCAGGAAAT). PCR amplification protocol consisted of the following steps: 95°C for 2 min, 35× (95°C for 20 s, 58°C for 10 s, 70°C for 30 s), and 70°C for 5 min. RT-PCR products were resolved using electrophoresis with 1% agarose gel stained with SYBR Safe DNA Gel Stain (Thermo Fisher Scientific) and visualized with a Chemidoc Touch Imaging System (Bio-Rad Laboratories). A 1-kb DNA ladder (New England Biolabs #N0550S) was loaded into each gel row to serve as a marker for the size of the TECPR2 PCR products. A PCR sample showing successful TECPR2 exon 8 skipping appears as enrichment of a lower-molecular-weight PCR product (reduced by 333 bp) in comparison with the upper PCR band, which includes exon 8 (wild type or full-length TECPR2 product). In order to quantify the percentage of TECPR2 exon8-skipped product and wild-type TECPR2 product, the intensities of the PCR bands were evaluated using Image Lab software (Bio-Rad Laboratories). The output of this algorithm is designated as “adj volume” and represents the adjusted volume of each band after background subtraction. After adjusted volumes are calculated for the PCR bands present on a gel, the sum of the total adj volume for all PCR bands in a sample is designated as 1.00 and the contribution of the two PCR bands (full-length TECPR2 versus TECPR2ΔExon8) to this sum is expressed as percentages, by dividing the adj volume for each PCR band by the sum and then multiplying by 100.
Immunocytochemistry assays
Immunocytochemistry (ICC) assays were carried out as previously reported.18 Briefly, fibroblast and neuronal cultures were fixed using 4% paraformaldehyde diluted in 1× PBS from a 16% aqueous solution (Electron Microscopy Sciences) for 20 min and then permeabilized with 0.2% Triton X-100 (Sigma). Cell culture dishes were blocked with 10% donkey serum in 0.1% PBS-Tween for 1 h and then incubated with primary antibodies overnight at 4°C. After five washes with 0.1% PBS-Tween, dishes were treated with secondary (Alexa -conjugated) antibodies for 1 h at room temperature. Cell nuclei (DNA) were stained with Hoechst 33342. Primary antibodies used in this study were the cytoskeletal pan-neuronal markers MAP2 (Abcam; ab5392, 1:3,000) and β-III TUBULIN (TUJ1, BioLegend; #801202, 1:1,000), custom rabbit polyclonal antibody against TECPR24 1:300 (produced in the laboratory of Dr. Christian Behrends), VPS11 (Santa Cruz; sc-515094, 1:300). For fibroblast cultures in 96-well Ibidi plates, images were acquired as maximum-intensity Z-projections using the automated confocal microscope GE Healthcare IN Cell Analyzer 6000 with 20× (0.75NA) objective. Single-cell TECPR2 expression in fibroblasts was estimated using a custom automated microscopy pipeline. Briefly, cell nuclei were first identified via histogram splitting on preprocessed images from the Hoechst33342 channel, with a second pass via a local watershed algorithm to split nuclei detected as possibly fused. Nuclear segmentations for each field were manually appraised and rejected in cases of gross proliferation or poor segmentations. Individual nuclei were then subjected to algorithmic quality criteria assessing size, eccentricity, snowiness, solidity, and average Hoechst33342 brightness (to catch fluorescent artifacts). As the TECPR2 staining pattern was expected to be punctate, fluorescence values from the TECPR2 channel were captured using a 20-pixel halo that was drawn around each annotated cell nucleus, with duplicate claims on specific pixels resolved via nearest neighbors. For statistical analysis, we carried out a one-way ANOVA followed by pairwise t tests to evaluate differences between the five groups. p values were false discovery rate (FDR) corrected to account for multiple hypothesis testing. Analysis was performed in R (version 3.6.3). For human neuronal cultures treated with ASOs, ICC images were acquired using the LSM880 confocal microscope with 60× objective.
Autophagy and immunoblotting assays
Autophagy assays with Bafilomycin A1 were carried out as previously described.33 For detection of protein levels using immunoblotting assays, protein concentrations were determined using BCA assay on a Nanodrop, and 10–30 μg of protein sample were loaded and separated using SDS-PAGE. After protein transfer onto membranes, these were incubated with primary antibodies overnight and corresponding HRP-conjugated secondary antibodies for 1–2 h prior to developing and measuring protein signal. Primary antibodies and dilution used were MAP1LC3B (Cell Signaling Technology 2775, 1:500 dilution), SQSTM1 (Cell Signaling Technology 5114S, 1:500 dilution), GAPDH (Abcam ab9484, 1:2,000 dilution), and custom rabbit polyclonal antibody against TECPR24 1:300 (produced in the laboratory of Dr. Christian Behrends).
Human PBMC assays
Human PBMC assays were carried out at Axolabs (Germany). Starting material for human PBMCs was buffy coat blood corresponding to 500 mL of full-blood transfusion units. Each unit was obtained from healthy volunteers, and glucose-citrate was used as anti-agglutinant. Initial buffy coat volume was 28–32 mL, and the sealed bag was opened about 40 h after blood donation, when isolation of human PBMCs was carried out via Ficoll gradient centrifugation. PBMCs were resuspended in 10 mL of RPMI1640 complete medium (1× L-glutamine, 1× penicillin-streptomycin (Pen/Strep), and 10% fetal calf serum). PBMCs were seeded at 100,000 cells/well of 96-well cell culture plates in 100 μL of complete medium and maintained inside a 37 °C/5% CO2 incubator. ASO molecules (ASO-005-02, ASO-059, and ASO-066) were delivered at three different concentrations (50 nM, 150 nM, 300 nM) using lipid-based transfection via OptiMem and Lipofectamine 2000 (0.5 mL/well). Twenty-four hours after transfection and incubation (37°C/5% CO2), multiple cytokines (interferon [IFN]-α2a, IFN-β, interleukin [IL]-1β, IL-6, tumor necrosis factor [TNF]-α) were measured and analyzed via the MSD platform. Multiple positive controls (inflammatory stimuli) were used, including a TLR7/8 agonist (XD-01024, a cholesterol-conjugated ApoB siRNA); a TLR9 agonist (ODN2216, a 20-mer CpG oligo containing unmethylated CpG); a TLR7/8 agonist (XD-00366, a 25-mer double-stranded, unmodified, blunt-ended LacZ RNA duplex); and a TLR3, RIG-I/MDA5, and PKR agonist (low-molecular-weight polyinosinic-polycytidylic acid [LMW poly(I:C)]). A chemically modified siRNA (XD-03999) against factor VII (FVII) was used as negative control.
TECPR2 interactome studies in HEK cell lines
HEK293 TECPR2-/- cells were generated by CRISPR-Cas9 gene editing followed by clonal selection. A stable TECPR2-/- HEK293 line was generated and shown to have no TECPR2 protein expression by immunocytochemistry with the anti-TECPR2 antibody. The TECPR2-/- HEK293 cell line was separately transfected with expression constructs for hemagglutinin (HA)-tagged TECPR2 and HA-tagged TECPR2ΔEx8, and stable clones selected and expanded. The following four resulting cell lines were used for the pull-down experiment: (1) TECPR2-/- HEK293-HA-TECPR2 (TECPR2/- stably expressing full-length HA-TECPR2), (2) TECPR2-/- HEK293-HA-TECPR2ΔEx8 (TECPR2-/- stably expressing HA-TECPR2ΔEx8), (3) TECPR2-/- HEK293-non-transfected (negative control for anti-HA pull-down), (4) TECPR2+/+ parental HEK293-non-transfected (negative control for anti-HA pull-down). Eight replicates of each of the four cell lines were used for pull-down experiments for a total of 32. The combination of the stable HA-tagged bait expression protocol with the large number of replicates provided an optimal design and well-powered experiment for detecting potential interactors of the two forms of TECPR2 in the HEK293 cellular background. Cells were grown to 75% confluency, and protein lysates were generated. Pull-downs were performed using beads conjugated to anti-HA antibody, followed by extensive washing of beads. Pulled down proteins were eluted by adding an excess of HA-tagged peptide to the beads and isolating the supernatant. The eluted protein samples were analyzed by mass-spectrometry-based proteomics analysis. Proteome informatics results and protein database FASTA files including spectral count data were provided to A2Idea (Ann Arbor, MI) for statistical analysis to define significant interactors. Prior to statistical analysis, proteins commonly observed in affinity-purification coupled to mass spectrometry (AP-MS) experimental blanks were identified in and removed from all comparisons. These proteins represent non-specific binding to the assay antibody used (anti-HA-tag) and must be removed from any label-free analysis as they do not represent a true interaction. The non-specific binding proteins were identified using a public database resource for AP-MS (crapome.org) that enables users to download lists of common contaminants in AP-MS experiments.34 Four replicate experiments using the HA-tag were downloaded from crapome. All proteins found in common among these four experiments were taken to be contaminants. Any occurrence of these proteins was removed from the spectral count data prior to running significance analysis. Significant interactors were identified using the significance analysis of interactome (SAINT) algorithm,35 a computational tool that assigns confidence scores to protein-protein interaction data generated using AP-MS. The method utilizes label-free quantitative data (i.e., spectral counts). Using spectral counts, SAINT constructs separate distributions for true and false interactions to derive the probability of a bona fide protein-protein interaction. A stringent Bayesian FDR (BFDR) of 0.01 statistical threshold was used to define interactors. Log of odds ratios (LORs) of 40, 30, and 20 were applied to the identified interactors to define likely significant interaction candidates over a range of statistical thresholds.
Data availability
The most relevant datasets generated and analyzed as part of this study are included in this published article (and its supplementary information files). Additional data analyzed in the study are available from the corresponding authors on reasonable request. Custom code used for the automated microscopy pipeline is restricted from outside use.
Acknowledgments
The project was funded by the TECPR2 Foundation. We would like to thank Dr. Charles Berde and Dr. Robert J. Graham at Boston Children’s Hospital for their insights on preclinical in vivo studies.
Author contributions
G.T.D, A.K., L.A.W., D.J.G., A.E., W.C.T., and D.M. designed ASO strategy. L.A.W. and W.C.T. designed all in vitro ASO experiments in fibroblasts and human iPSC neurons. W.C.T., V.J., S.H.R., H.S., and L.A.W. executed ASO experiments in fibroblasts. V.B. carried out immunocytochemistry (ICC) assays. N.D.B. developed qICC pipeline for fibroblast assays. K.H. and H.U. carried out iPSC-neuronal production and cultures. V.J., A.D., and J.M. carried out TECPR2 immunoblotting assays. S.J.R., C.L., and N.D.B. assisted with data analyses. L.B., J.D., and S.C. supervised and carried out behavioral studies in cynomolgus monkeys. H.L. carried out HELISA assays. C.A., G.M., H.S., and L.A.W. executed PD RT-PCR assays in non-human primate mRNA samples. A.S. and L.G. carried out autophagy assays. C.B. assisted with validation of TECPR2 immunoreagents. K.N. and C.B. carried out interactome experiments in HEK293 cells. T.W.Y. carried out initial genotyping of TECPR2 mutation in the SPG49 patient. C.L., D.B., and S.A. designed ASO sequences. L.W., D.J.G., A.E., A.K., and G.T.D. provided scientific oversight over interpretation of experimental results and wrote the manuscript.
Declaration of interests
We declare that one or more of the authors have a competing interest: L.A.W., D.J.G., A.E., V.B., N.D.B., C.A., G.M., V.J., H.S., K.H., H.U., S.H.R., A.D., J.M., S.J.R., C.L., S.A., D.M., and G.T.D. are employees or consultants of Q-State Biosciences and hold stock.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.06.015.
Contributor Information
Alan Kopin, Email: akopin55@gmail.com.
Graham T. Dempsey, Email: graham.dempsey@qstatebio.com.
Supplemental information
References
- 1.Oz-Levi D., Ben-Zeev B., Ruzzo E.K., Hitomi Y., Gelman A., Pelak K., Anikster Y., Reznik-Wolf H., Bar-Joseph I., Olender T., et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012;91:1065–1072. doi: 10.1016/j.ajhg.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heimer G., Oz-Levi D., Eyal E., Edvardson S., Nissenkorn A., Ruzzo E.K., Szeinberg A., Maayan C., Mai-Zahav M., Efrati O., et al. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur. J. Paediatr. Neurol. 2016;20:69–79. doi: 10.1016/j.ejpn.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Patwari P.P., Wolfe L.F., Sharma G.D., Berry-Kravis E. TECPR2 mutation-associated respiratory dysregulation: more than central apnea. J. Clin. Sleep Med. 2020;16:977–982. doi: 10.5664/jcsm.8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stadel D., Millarte V., Tillmann K.D., Huber J., Tamin-Yecheskel B.C., Akutsu M., Demishtein A., Ben-Zeev B., Anikster Y., Perez F., et al. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol. Cell. 2015;60:89–104. doi: 10.1016/j.molcel.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraiberg M., Tamim-Yecheskel B.C., Kokabi K., Subic N., Heimer G., Eck F., Nalbach K., Behrends C., Ben-Zeev B., Shatz O., et al. Lysosomal targeting of autophagosomes by the TECPR domain of TECPR2. Autophagy. 2021;17:3096–3108. doi: 10.1080/15548627.2020.1852727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn K., Rohdin C., Jagannathan V., Wohlsein P., Baumgärtner W., Seehusen F., Spitzbarth I., Grandon R., Drögemüller C., Jäderlund K.H. TECPR2 associated neuroaxonal dystrophy in Spanish water dogs. PLoS One. 2015;10:e0141824. doi: 10.1371/journal.pone.0141824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamim-Yecheskel B.-C., Fraiberg M., Kokabi K., Freud S., Shatz O., Marvaldi L., Subic N., Brenner O., Tsoory M., Eilam-Altstadter R., et al. A tecpr2 knockout mouse exhibits age-dependent neuroaxonal dystrophy associated with autophagosome accumulation. Autophagy. 2021;17:3082–3095. doi: 10.1080/15548627.2020.1852724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D., Mastaglia F.L., Fletcher S., Wilton S.D. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol. Sci. 2018;39:982–994. doi: 10.1016/j.tips.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Wilton S.D., Lloyd F., Carville K., Fletcher S., Honeyman K., Agrawal S., Kole R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999;9:330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 11.Charleston J.S., Schnell F.J., Dworzak J., Donoghue C., Lewis S., Chen L., Young G.D., Milici A.J., Voss J., DeAlwis U., et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90:e2146–e2154. doi: 10.1212/wnl.0000000000005680. [DOI] [PubMed] [Google Scholar]
- 12.Ottesen E.W. ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy. Transl. Neurosci. 2017;8:1–6. doi: 10.1515/tnsci-2017-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parente V., Corti S. Advances in spinal muscular atrophy therapeutics. Ther. Adv. Neurol. Disord. 2018;11 doi: 10.1177/1756285618754501. 175628561875450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett C.F., Kordasiewicz H.B., Cleveland D.W. Antisense drugs make sense for neurological diseases. Annu. Rev. Pharmacol. Toxicol. 2021;61:831–852. doi: 10.1146/annurev-pharmtox-010919-023738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuser S., Brechmann B., Heimer G., Brösse I., Schubert S., O’grady L., Zech M., Srivastava S., Sweetser D.A., Houlden H., et al. Clinical, neuroimaging, and molecular spectrum of TECPR2-associated hereditary sensory and autonomic neuropathy with intellectual disability. Hum. Mutat. 2021;42:762–776. doi: 10.1002/humu.24206. [DOI] [PubMed] [Google Scholar]
- 16.Roberts T.C., Langer R., Wood M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams L.A., Joshi V., Murphy M., Ferrante J., Werley C.A., Brookings T., McManus O., Grosse J., Davies C.H., Dempsey G.T. Scalable measurements of intrinsic excitability in human iPS cell-derived excitatory neurons using all-optical electrophysiology. Neurochem. Res. 2019;44:714–725. doi: 10.1007/s11064-018-2694-5. [DOI] [PubMed] [Google Scholar]
- 19.Jafar-Nejad P., Powers B., Soriano A., Zhao H., Norris D.A., Matson J., DeBrosse-Serra B., Watson J., Narayanan P., Chun S.J., et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021;49:657–673. doi: 10.1093/nar/gkaa1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hori S.I., Yamamoto T., Waki R., Wada S., Wada F., Noda M., Obika S. Ca2+ enrichment in culture medium potentiates effect of oligonucleotides. Nucleic Acids Res. 2015;43:e128. doi: 10.1093/nar/gkv626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen X., Beasley S., Putman J.N., Li Y., Prakash T.P., Rigo F., Napierala M., Corey D.R. Efficient electroporation of neuronal cells using synthetic oligonucleotides: identifying duplex RNA and antisense oligonucleotide activators of human frataxin expression. RNA. 2019;25:1118–1129. doi: 10.1261/rna.071290.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim K.R.Q., Maruyama R., Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017;11:533–545. doi: 10.2147/dddt.s97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heo Y.A. Golodirsen: first approval. Drugs. 2020;80:329–333. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- 24.Frank D.E., Schnell F.J., Akana C., El-Husayni S.H., Desjardins C.A., Morgan J., Charleston J.S., Sardone V., Domingos J., Dickson G., et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270–e2282. doi: 10.1212/wnl.0000000000009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anthony K., Cirak S., Torelli S., Tasca G., Feng L., Arechavala-Gomeza V., Armaroli A., Guglieri M., Straathof C.S., Verschuuren J.J., et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: implications for clinical trials. Brain. 2011;134:3547–3559. doi: 10.1093/brain/awr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldrop M.A., Yaou R.B., Lucas K.K., Martin A.S., O’Rourke E., Ferlini A., Ferlini A., Muntoni F., Leturcq F., Weiss R.B., et al. Clinical phenotypes of DMD exon 51 skip equivalent deletions: a systematic review. J. Neuromuscul. Dis. 2020;7:217–229. doi: 10.3233/jnd-200483. [DOI] [PubMed] [Google Scholar]
- 27.Van Der Kant R., Jonker C.T.H., Wijdeven R.H., Bakker J., Janssen L., Klumperman J., Neefjes J. Characterization of the mammalian CORVET and HOPS complexes and their modular restructuring for endosome specificity. J. Biol. Chem. 2015;290:30280–30290. doi: 10.1074/jbc.m115.688440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balderhaar H.J. klein., Ungermann C. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J. Cell Sci. 2013;126:1307–1316. doi: 10.1242/jcs.107805. [DOI] [PubMed] [Google Scholar]
- 29.Scharner J., Ma W.K., Zhang Q., Lin K.-T., Rigo F., Bennett C.F., Krainer A.R. Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides. Nucleic Acids Res. 2020;48:802–816. doi: 10.1093/nar/gkz1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ottesen E.W., Luo D., Singh N.N., Singh R.N. High concentration of an iss-n1-targeting antisense oligonucleotide causes massive perturbation of the transcriptome. Int. J. Mol. Sci. 2021;22:8378. doi: 10.3390/ijms22168378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Giorgio F.P., Boulting G.L., Bobrowicz S., Eggan K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Efler S.M., Zhang L., Noll B.O., Uhlmann E., Davis H.L. Quantification of oligodeoxynucleotides in human plasma with a novel hybridization assay offers greatly enhanced sensitivity over capillary gel electrophoresis. Oligonucleotides. 2005;15:119–131. doi: 10.1089/oli.2005.15.119. [DOI] [PubMed] [Google Scholar]
- 33.Yamazaki T., Kirchmair A., Sato A., Buqué A., Rybstein M., Petroni G., Bloy N., Finotello F., Stafford L., Navarro Manzano E., et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020;21:1160–1171. doi: 10.1038/s41590-020-0751-0. [DOI] [PubMed] [Google Scholar]
- 34.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi H., Larsen B., Lin Z.Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The most relevant datasets generated and analyzed as part of this study are included in this published article (and its supplementary information files). Additional data analyzed in the study are available from the corresponding authors on reasonable request. Custom code used for the automated microscopy pipeline is restricted from outside use.