

Abstract
We have developed DNA microarray techniques for studying Corynebacterium glutamicum. A set of 52 C. glutamicum genes encoding enzymes from primary metabolism was amplified by PCR and printed in triplicate onto glass slides. Total RNA was extracted from cells harvested during the exponential-growth and lysine production phases of a C. glutamicum fermentation. Fluorescently labeled cDNAs were prepared by reverse transcription using random hexamer primers and hybridized to the microarrays. To establish a set of benchmark metrics for this technique, we compared the variability between replicate spots on the same slide, between slides hybridized with cDNAs from the same labeling reaction, and between slides hybridized with cDNAs prepared in separate labeling reactions. We found that the results were both robust and statistically reproducible. Spot-to-spot variability was 3.8% between replicate spots on a given slide, 5.0% between spots on separate slides (though hybridized with identical, labeled cDNA), and 8.1% between spots from separate slides hybridized with samples from separate reverse transcription reactions yielding an average spot to spot variability of 7.1% across all conditions. Furthermore, when we examined the changes in gene expression that occurred between the two phases of the fermentation, we found that results for the majority of the genes agreed with observations made using other methods. These procedures will be a valuable addition to the metabolic engineering toolbox for the improvement of C. glutamicum amino acid-producing strains.
Corynebacterium glutamicum is used in the commercial production of lysine and glutamic acid (reviewed in reference 9). High-level amino acid biosynthesis draws resources from many different biosynthetic pathways, and it places physiological stress upon the cell (e.g., reference 6). To understand how Corynebacterium accommodates these demands, research in recent years has sought to divine the overall regulatory processes that coordinate the cell's physiology during amino acid production.
During the last 5 years, Brown and coworkers (2, 11) have described methods for studying gene regulation on a global scale using DNA microarrays. These techniques should be useful for studying how gene expression changes during amino acid biosynthesis and secretion. However, the literature offers few examples of the successful application of DNA microarray technology in gram-positive bacteria (e.g., references 3, 19, and 21). In addition, we were interested in evaluating the reproducibility of microarray technologies. To address these issues, we have adapted the methods developed by the Brown lab (herein called the Stanford protocol [http://cmgm.stanford.edu/pbrown/]). We have printed and tested pilot microarrays of Corynebacterium genes and used them to evaluate the robustness and reproducibility of the technique for studying gene expression in Corynebacterium.
MATERIALS AND METHODS
Preparation of DNA microarrays.
Oligonucleotide primers (Life Technologies, Inc., Rockville, Md.) were designed to amplify 52 specific open reading frames (ORFs) or portions of the ORFs up to ∼1 kb in length from Corynebacterium (Table 1). One of the genes was amplified by two different sets of primers resulting in a total of 53 PCR products representing 52 genes. In cases when the PCR product did not span the entire length of the ORF, the 3′-most portion of the ORF was amplified. Primers contained 18 to 20 nucleotides (nt) specific to each gene and an additional 18-nt adaptor at their 5′ ends for easy reamplification. Genomic DNA was prepared from C. glutamicum ATCC 21253 by the method described by Treadway et al. (15). PCR was carried out with HotStarTaq DNA polymerase (Qiagen, Valencia, Calif.) in a Robocycler thermocycler (Stratagene, La Jolla, Calif.).
TABLE 1.
Corynebacterium ORFs printed onto microarrays and significant changes in gene expression during lysine production
| Gene | Assigned function of gene product | PCR product length (bp) | Accession no. or source | Fold inductiona |
|---|---|---|---|---|
| aat | aspartate aminotransferase | 1,050 | E16763 | ns |
| accBC | acyl-CoA carboxylase (biotinylated subunit) | 1,050 | U35023 | ns |
| accD | acetyl-CoA carboxylase (transcarboxylase subunit) | 1,100 | L. B. Willis, unpublished data | ns |
| aceA | isocitrate lyase | 1,040 | L28760 | ns |
| aceB | malate synthase | 1,090 | L27123 | ns |
| amt | ammonium transport system | 1,340 | X93513 | ns |
| amtR | repressor of (methyl) ammonium uptake system | 590 | AJ133719 | ns |
| argS | arginyl-tRNA synthase | 1,100 | E16355 | ns |
| asd | aspartate-semialdehyde dehydrogenase | 900 | L16848 | ns |
| ask | aspartokinase | 1,060 | L16848 | ns |
| bioB | biotin synthase | 890 | U31281 | ns |
| dapA | dihydrodipicolinate synthase | 850 | Z21502 | ns |
| dapB | dihydrodipicolinate reductase | 700 | Z21502 | ns |
| dapD | tetrahydrodipicolinate succinylase | 540 | AJ004934 | ns |
| dapE | succinyl-diaminopimelate desuccinylase | 1,050 | X81379 | ns |
| ddc | diaminopimelic acid decarboxylase | 1,070 | E16355 | ns |
| ddh | m̄eso-diaminopimelate d-dehydrogenase | 950 | Y00151 | ns |
| dtsR1 | acyl-CoA carboxylase (transcarboxylase subunit) | 1,080 | AB018531 | +2.8 |
| dtsR2 | acyl-CoA carboxylase (transcarboxylase subunit) | 1,080 | AB018531 | ns |
| ectP | transport of ectoine, glycine betaine, and proline | 1,090 | AJ001436 | ns |
| fda | fructose-bisphosphate aldolase | 1,000 | X17313 | ns |
| gap | glyceraldehyde-3-phosphate dehydrogenase | 970 | X59403 | ns |
| gdh | glutamate dehydrogenase | 1,020 | X59404 | ns |
| glnA | glutamine synthetase | 1,130 | AF005635 | +1.8 |
| gltA | citrate synthase | 1,050 | X66112 | −2.6 |
| gltB | glutamine 2-oxoglutarate aminotransferase (large subunit) | 1,080 | AB024708 | ns |
| glyA | serine hydroxymethyltransferase | 1,040 | E12594 | ns |
| gpd | 6-phosphogluconate dehydrogenase | 990 | E13660 | ns |
| hom | homoserine dehydrogenase | 1,040 | Y00546 | ns |
| icd | monomeric isocitrate dehydrogenase | 1,060 | X71489 | ns |
| ilvA | threonine dehydratase | 1,030 | L01508 | ns |
| ilvB | AHAS (large subunit) | 1,020 | L09232 | +1.8 |
| ilvC | acetohydroxy acid isomeroreductase | 930 | L09232 | ns |
| ilvN | AHAS (small subunit) | 470 | L09232 | ns |
| leuB | 3-isopropylmalate dehydrogenase | 990 | Y09578 | ns |
| lysE | lysine export | 670 | X96471 | +1.8 |
| lysG | lysine export regulator | 810 | X96471 | ns |
| metA | homoserine O-acetyltransferase | 1,090 | AF052652 | ns |
| ndh | NADH dehydrogenase | 1,050 | AJ238250 | ns |
| odhA | 2-oxoglutarate dehydrogenase | 1,250 1,050b | D84102 | −3.7, −3.2b |
| panC | pantothenate β-alanine ligase | 780 | X96580 | ns |
| pgk | phosphoglycerate kinase | 1,060 | X59403 | ns |
| ppc | phosphoenolpyruvate carboxylase | 1,040 | E16358 | ns |
| proP | proline/ectoine uptake | 1,050 | Y12537 | ns |
| pta | phosphate acetyltransferase | 930 | X89084 | −2.2 |
| ptsM | phosphoenolpyruvate sugar phosphotransferase | 1,060 | L18874 | ns |
| pyc | pyruvate carboxylase | 1,190 | AF038548 | ns |
| pyk | pyruvate kinase | 1,060 | L27126 | +1.5 |
| thrB | homoserine kinase | 870 | Y00546 | ns |
| thrC | threonine synthase | 1,140 | X56037 | ns |
| tpi | triosephosphate isomerase | 750 | X59403 | ns |
| zwf | glucose-6-phosphate dehydrogenase | 1,040 | E13655 | ns |
Relative to the mean and based upon analysis of the AR data set for each gene; ns, no significant change in expression.
Two different PCR products were prepared from odhA and printed separately on the array.
All PCR products were verified by agarose gel electrophoresis to ensure that only a single product of the expected length had been amplified with each primer pair. Lengths of PCR products ranged from 470 to 1,250 bp. PCR products were purified using a PCR cleanup kit (Qiagen), precipitated with ethanol, resuspended in 3× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (10) to concentrations of 0.2 mg/ml, and distributed into a 96-well microtiter dish. Using a GMS model 417 pin-and-ring arraying robot (Affymetrix, Santa Clara, Calif.), these PCR products were printed onto poly-l-lysine-coated microscope slides (Cel Associates, Houston, Tex.), with the pin touching the slides three times per spot and printing three spots per gene. PCR products representing two ORFs from Saccharomyces cerevisiae (YAL058C-A and YAL047C) were also printed onto the arrays to serve as internal controls. Slides were processed as described in the Stanford protocol and stored covered at room temperature for up to two months.
Growth of C. glutamicum.
Fermentations and determinations of biomass and lysine were carried out as described by Guillouet et al. (6). C. glutamicum ATCC 21253 (American Type Culture Collection, Manassas, Va.) was fermented in a defined medium (FM4 [16]) containing excess threonine (Fig. 1). Samples were harvested during exponential growth with no lysine production (phase I) and later when threonine had been depleted and lysine was being secreted into the medium (phase II). Immediately after the harvesting, defined portions of C. glutamicum cultures (before centrifugation) were flash frozen by pouring the cultures slowly into a flask containing liquid nitrogen.
FIG. 1.

Sampling of Corynebacterium culture before and during lysine production. Changes in biomass (●), external glucose (▵), and external lysine (○) were monitored during the course of fermentation of C. glutamicum ATCC 21253 in medium FM4. Samples were withdrawn at time points I (growth phase) and II (growth-and-lysine-production phase) and used for the preparation of total RNA.
Purification of RNA.
Frozen bacterial cells were thawed on ice, centrifuged for 2 min at 5,000 × g at 4°C, and resuspended in cold RLT buffer containing β-mercaptoethanol (RNeasy Mini kit; Qiagen) to 25 g (dry cell weight [DCW])/liter. Then, 3 ml of cells and 3 ml of 106-μm acid-washed glass beads (catalog number G-4649; Sigma, St. Louis, Mo.) were lysed at 4°C in a Spex Certiprep (Metuchen, N.J.) bead mill in six 1-min pulses, separated by 1-min rests on ice. Subsequently, the RNA was purified with the RNeasy Mini kit (Qiagen). RNA samples were incubated at 37°C for 30 min with 30 U of DNase and 300 U of RNase Inhibitor (Roche, Indianapolis, Ind.) and then repurified with RNeasy Mini kit spin columns (Qiagen). Typically, 50 to 200 μg of RNA are recovered from 3 ml of cells resuspended to 25 g (DCW)/liter with this method. Each RNA sample was examined by gel electrophoresis, which revealed distinct rRNA bands and little evidence of degradation. These samples reported A260/A280 ratios in the range of 1.7, as measured in unbuffered water.
Preparation of labeled cDNA.
Following the Stanford protocol for reverse transcription of E. coli total RNA, labeled cDNA from Corynebacterium was prepared with random hexamer primers (Roche) in the presence of either Cy3-dUTP or Cy5-dUTP (NEN, Boston, Mass.). Each reaction contained 50 μg of total Corynebacterium RNA (which is sufficient for two slides) and 1 μg of in vitro-transcribed RNA corresponding to the yeast genes that had been arrayed on the microscope slides. cDNA synthesis was carried out essentially as described in the Stanford protocol, except that RNA was destroyed by treating the cDNA with RNase H and RNase (Roche) for 1 h at 37°C and the ethanol precipitation step was omitted. The cDNA samples were then combined, washed four times with 4× SSC in a Centricon YM-30 (Millipore Corporation, Bedford, Mass.) to remove unincorporated dye, concentrated to ∼25 μl, recovered in a 1.5-ml tube, and centrifuged briefly to remove aggregated dyes. The supernatant of the ∼25-μl sample was used directly for hybridization of two replicate slides. With each of the two RNA samples, we performed two separate reverse transcription reactions. cDNAs from one set of reverse transcriptions were hybridized to slides 15 and 21, whereas cDNAs from the second set of reverse transcriptions were hybridized to slides 35 and 45. Slide numbers represent the order of the slides during printing, showing that 30 slides were printed between the first and the last slide we used in the hybridization experiments.
Hybridization and analysis.
Hybridization and postprocessing were carried out according to the Stanford protocol in an ArrayIt hybridization cassette (TeleChem International, Sunnyvale, Calif.) using 20 μg of sheared herring sperm competitor DNA. The entire hybridization chamber was submerged in a 65°C water bath for 15 h. Following the wash steps, microarrays were analyzed using an ArrayWoRx CCD scanner (Applied Precision, Issaquah, Wash.). The ArrayWoRx scanner was used to measure fluorescence of the Cy3- and Cy5-labeled cDNAs bound to the DNA microarray. The signal of the Cy5 channel was normalized with respect to the Cy3 channel, using total signal intensity. Raw data from the scans was collected into a Microsoft Excel spreadsheet and visualized using SpotFire Pro (SpotFire, Inc., Cambridge, Mass.).
Statistical analysis. (i) Normalized weighted average log (AL) data set: AL-Cy3 and AL-Cy5.
The spot intensity of each channel (Cy3 or Cy5) and its associated standard deviation were normalized with respect to total intensity from each channel for the entire array (hereafter called the scanner normalization factor). The log of the normalized value (LNV) of each channel for each spot was taken, and its standard deviation was determined using the standard method for propagating errors through the log transformation:
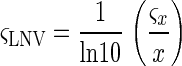 |
For a given slide, each gene was assigned a single value, with associated standard deviation, for each channel by a weighted average of the three spots per chip corresponding to that gene. The gene that was represented by two different sets of triplicate spots was treated as two separate genes for the purpose of analysis. Using the method of maximum likelihood, the LNV for each gene was calculated for each channel as the weighted average of the three spots for each gene as follows:
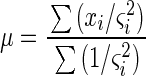 |
where each data point xi in the sum is weighted inversely by its own variance ςi2 (1). The standard deviation of the weighted average is then expressed as follows:
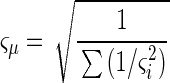 |
This is sufficient for relative comparisons on a single chip. To transform the data into a form that is comparable across chips, the weighted average LNV and its associated standard deviations were expressed as a fraction of the sum of the LNV of each channel. The resultant data sets (AL-Cy3 and AL-Cy5) are presented as a scatter plot in Fig. 2A and used in later statistical analysis.
FIG. 2.

Scatter plot of log Cy3- and log Cy5-derived fluorescence. (A) Compiled normalized weighted average log (AL-Cy3 and AL-Cy5) data from four microarray slides. Each data point corresponds to the weighted average of the three spots on a single microarray representing an individual Corynebacterium or S. cerevisiae gene. The four data points corresponding to the same gene from each of the four slides are represented in the same color. Dashed lines demarcate threshold values for significant difference in gene expression relative to the normal line (solid). (B) Genes with a significant difference in gene expression relative to the mean are plotted with two-dimensional error bars. Data points corresponding to the same gene in each plot are represented in the same color. Dashed lines demarcate threshold values for significant difference in gene expression relative to the normal line (solid).
(ii) Normalized weighted average ratio (AR) data set.
The average pixel-by-pixel ratio (Cy5/Cy3) within a spot and its associated standard deviation was normalized using the scanner normalization factor. For a given slide, each gene was assigned a single ratio, with associated standard deviation, by a weighted average of the three spots per chip corresponding to that gene using the method described in calculation of the AL data set. The resultant data are presented as a histogram in Fig. 3 and used in later statistical analysis.
FIG. 3.

Histogram of normalized weighted AR data. Compiled normalized weighted AR data from four microarray slides. The fold induction relative to the mean is plotted versus the frequency. Significant differences in gene expression relative to the mean are indicated on the histogram as darkened bars. Dashed lines demarcate threshold values for significant differences in gene expression relative to the normal line (solid line).
(iii) Analysis of means.
To analyze the significance of differences in the data sets, a two-tailed Student's t test of H0: μ1 − μ2 = 0 was calculated between the AL-Cy5, AL-Cy3, and the AR data sets of each chip using the following formula:
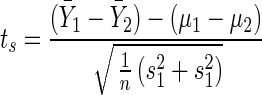 |
where the degrees of freedom value is equal to 108 [df = 2(n − 1); see reference 12 for definitions and further explanation of the statistical methods]. The critical values of t0.05[108] = 1.982 and t0.9[108] = 0.126 are as previously described (12). These values are tabulated in Table 2.
TABLE 2.
t test for the comparison of means
| Data set 1a | Data set 2a | t statisticb | P(μ1 = μ2)c (%) |
|---|---|---|---|
| AL-Cy5 slide 15 | AL-Cy5 slide 21 | −0.0217 | 98.3 |
| AL-Cy5 slide 15 | AL-Cy5 slide 35 | −0.0039 | 99.7 |
| AL-Cy5 slide 15 | AL-Cy5 slide 45 | −0.0539 | 95.7 |
| AL-Cy5 slide 21 | AL-Cy5 slide 35 | 0.0152 | 98.8 |
| AL-Cy5 slide 21 | AL-Cy5 slide 45 | −0.0341 | 97.3 |
| AL-Cy5 slide 35 | AL-Cy5 slide 45 | −0.0448 | 96.5 |
| AL-Cy3 slide 15 | AL-Cy3 slide 21 | −0.0705 | 94.4 |
| AL-Cy3 slide 15 | AL-Cy3 slide 35 | −0.0033 | 99.7 |
| AL-Cy3 slide 15 | AL-Cy3 slide 45 | −0.0537 | 95.8 |
| AL-Cy3 slide 21 | AL-Cy3 slide 35 | 0.0623 | 95.1 |
| AL-Cy3 slide 21 | AL-Cy3 slide 45 | 0.0149 | 98.8 |
| AL-Cy3 slide 35 | AL-Cy3 slide 45 | −0.0469 | 96.3 |
| AR slide 15 | AR slide 21 | −0.0304 | 97.6 |
| AR slide 15 | AR slide 35 | −0.0075 | 99.4 |
| AR slide 15 | AR slide 45 | −0.0075 | 99.4 |
| AR slide 21 | AR slide 35 | 0.0237 | 98.1 |
| AR slide 21 | AR slide 45 | 0.0237 | 98.1 |
| AR slide 35 | AR slide 45 | 0.0001 | 99.9 |
Slides 15 and 21 were hybridized with cDNAs from one set of reverse transcriptions; slides 35 and 45 were hybridized with cDNAs from the second set of reverse transcriptions.
Two-tailed t test of H0: μ1 − μ2 = 0; df = 2(n − 1) = 108.
The critical value of t0.05[108] = 1.982 and t0.9[108] = 0.126.
(iv) Correlation analysis and assignment of confidence intervals.
To analyze data correlation, weighted Pearson product-moment correlations, rxy, were calculated between the AL-Cy5, AL-Cy3, and AR data sets of each chip with each data point weighted inversely by its own variance in the following calculation (12):
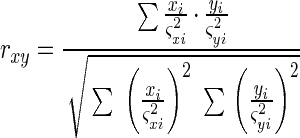 |
For the product-moment correlation coefficients, the critical value for significance was 0.354 at the 1% level with 53 degrees of freedom (12). The 99% confidence limits (t0.01[∞] = 2.275) were set to each rxy using the z transformation (12). These values are tabulated in Table 3.
TABLE 3.
Weighted Pearson product-moment correlation coefficientsa
| Data set 1b | Data set 2b | r1.2c | 99% Confidence intervalc |
|---|---|---|---|
| AL-Cy5 slide 15 | AL-Cy5 slide 21 | 0.8158 | 0.6567–0.9054 |
| AL-Cy5 slide 15 | AL-Cy5 slide 35 | 0.7683 | 0.5777–0.8795 |
| AL-Cy5 slide 15 | AL-Cy5 slide 45 | 0.7874 | 0.6090–0.8900 |
| AL-Cy5 slide 21 | AL-Cy5 slide 35 | 0.8755 | 0.7612–0.9371 |
| AL-Cy5 slide 21 | AL-Cy5 slide 45 | 0.8816 | 0.7722–0.9403 |
| AL-Cy5 slide 35 | AL-Cy5 slide 45 | 0.9729 | 0.9454–0.9867 |
| AL-Cy3 slide 15 | AL-Cy3 slide 21 | 0.6911 | 0.4565–0.8359 |
| AL-Cy3 slide 15 | AL-Cy3 slide 35 | 0.6799 | 0.4400–0.8294 |
| AL-Cy3 slide 15 | AL-Cy3 slide 45 | 0.6653 | 0.4177–0.8208 |
| AL-Cy3 slide 21 | AL-Cy3 slide 35 | 0.7302 | 0.5168–0.8582 |
| AL-Cy3 slide 21 | AL-Cy3 slide 45 | 0.7738 | 0.5866–0.8825 |
| AL-Cy3 slide 35 | AL-Cy3 slide 45 | 0.9881 | 0.9759–0.9942 |
| AR slide 15 | AR slide 21 | 0.8038 | 0.6363–0.8989 |
| AR slide 15 | AR slide 35 | 0.7383 | 0.5396–0.8627 |
| AR slide 15 | AR slide 45 | 0.6627 | 0.4140–0.8194 |
| AR slide 21 | AR slide 35 | 0.7312 | 0.5183–0.8587 |
| AR slide 21 | AR slide 45 | 0.6394 | 0.3800–0.8056 |
| AR slide 35 | AR slide 45 | 0.9495 | 0.8995–0.9750 |
df = n − 2 = 53; P0.01 = 0.354. The 99% confidence limits (t0.01[∞] = 2.275) were set to each rxy using the z transformation.
Slides 15 and 21 were hybridized with cDNAs from one set of reverse transcriptions; slides 35 and 45 were hybridized with cDNAs from the second set of reverse transcriptions.
P < 0.01.
(v) Outlier analysis of AL data set.
To determine which genes differed statistically from the mean, an iterative method of outlier determination was utilized. The data sets from each of the four slides were combined to yield a single AL-Cy3 and a single AL-Cy5 data set. These two data sets were then used to calculate the standard deviation about the mean, and the threshold value of significance was set at two standard deviations from the mean. Residuals were then calculated for each gene. Genes with residual values greater than the threshold level were considered significant and removed from the core data set. A new standard deviation was calculated based on the revised data set, and the process was repeated for a total of three iterations. The threshold value set in the third iteration is indicated in the scatter plots of Fig. 2. Genes determined to differ statistically from the mean are plotted as a scatter plot with two-dimensional error bars in Fig. 2B.
(vi) Outlier analysis of AR data set.
To determine which genes differed statistically from the mean in this average ratio data set, an iterative method of outlier determination was also employed. The data set from each slide was combined to yield a single AR data set. The standard deviation was calculated, and the threshold value of significance was set at two standard deviations from the mean. Genes with ratios falling outside the threshold value were considered significant and removed from the core data set. A new standard deviation was calculated based on the revised data set, and the process was repeated for a total of three iterations. The threshold value set in the third iteration is indicated in the histogram of Fig. 3. Each gene that was considered significant was assigned a single ratio, with an associated standard deviation, by a weighted average of the ratios calculated in each of the four chips corresponding to that gene using the method described in the calculation of the AL data set. Error bars were calculated, and a gene was considered significantly different from the mean if the weighted ratio and the associated error bars were outside the threshold value set in the third iteration. Genes that differed significantly from the mean under this criterion are indicated in Table 1.
(viii) Spot-to-spot variation.
The spot-to-spot variability was calculated from the average pixel-by-pixel Cy5/Cy3 ratio within a spot normalized using the scanner normalization factor. An average ratio and standard deviation for a gene was calculated as an average of the normalized ratios. The percent error, which represents the spot-to-spot variation, was calculated by dividing the standard deviation by the average ratio for each gene. Spot-to-spot variation was calculated both by slide (the average of three spots from the same slide), by pairwise slide comparisons (the average of six spots from two slides), and across all slides (the average of 12 spots from two slides each for two separate reverse transcriptions). Average spot-to-spot variation by slide was calculated as an average of the gene spot-to-spot variations on that slide. Average spot-to-spot variation of pairwise comparisons was calculated as an average of the gene spot-to-spot variation based on the six spots of the two slides being compared. Average spot-to-spot variation between reverse transcriptions was calculated as an average of the average spot-to-spot variations of all the pairwise comparisons that used probes generated through different reverse transcription reactions. Thus, calculations of the variation between different cDNA synthesis reactions combine the variations due to spot-to-spot, slide-to-slide, and cDNA-to-cDNA variability. Total average spot-to-spot variation was calculated as an average of the gene spot-to-spot variations based on the 12 spots of the data set.
RESULTS
Preparation of cDNA microarrays and RNA samples.
To begin our development of Corynebacterium microarrays, we first identified 52 Corynebacterium gene sequences from public databases (Table 1). The selected genes encode enzymes involved in many different aspects of primary metabolism, most notably amino acid biosynthesis and central carbon metabolism. Portions of these ORFs were amplified from C. glutamicum ATCC 21253 genomic DNA by PCR, purified, and printed in triplicate on poly-l-lysine-coated microscope slides. As internal controls, we also printed ORFs from S. cerevisiae (YAL058C-A and YAL047C) on the arrays.
To examine whether the expression of these genes changes under different physiological conditions, we carried out a fermentation of C. glutamicum ATCC 21253 in a defined medium containing threonine and excess methionine. C. glutamicum ATCC 21253 is auxotrophic for homoserine and therefore requires supplementation of minimal medium with either homoserine or its derivatives methionine and threonine. When the exogenously supplied threonine is exhausted by the culture, feedback repression of enzymes involved in the production of aspartate semialdehyde, a precursor of both homoserine and lysine, is relieved. Since the cell carries a mutation blocking homoserine production, carbon is diverted toward the production of lysine. We harvested cells during the exponential growth phase of the fermentation (specific growth rate of 0.35 h−1) when no lysine is detectable in the medium (phase I) and later in the fermentation after threonine was depleted, the growth rate slowed to 0.04 h−1 and lysine was being secreted into the medium (phase II) (Fig. 1).
These samples were used for the isolation of total RNA. The quality of RNA directly affects the outcome of cDNA synthesis and all downstream steps in analysis of gene expression. Preparation of high-quality RNA from gram-positive Corynebacterium requires both efficient cell lysis and effective RNA purification protocols. We found that lysing the cells with a bead mill followed by purification of RNA with a Qiagen RNeasy kit results in the isolation of high-quality RNA.
Corynebacterium mRNAs do not contain poly(A) tails. Rather than risk the loss of some messages during removal of ribosomal and transfer RNAs, we synthesized cDNA from total RNA using random hexamer primers. RNA isolated from the phase I sample was labeled with the fluorescent dye Cy3. RNA isolated from the phase II sample was labeled with the fluorescent dye Cy5. We included, as internal controls for assessing the effectiveness of the reverse transcription reaction, 1 μg of in vitro-transcribed RNA that corresponded to the two yeast control genes that had been arrayed on the microscope slides. We found that after cDNA synthesis it is important to remove aggregated unincorporated dyes by centrifuging the samples briefly. If this step is omitted, the aggregated dyes cause high background in the microarray images.
The Cy3- and Cy5-labeled cDNAs were mixed and hybridized to identical microarray slides. Following hybridization and washing, we normalized the total Cy3- and Cy5-derived signals using the scanner normalization factor. Following this normalization of the population as a whole, we found that the internal yeast control spots each reported a Cy5/Cy3 ratio of 1, as expected.
Reproducibility of data.
To gauge the reproducibility of the technique, we carried out statistical analyses of data from each of the four slides described in Materials and Methods, representing separate hybridizations as well as separate reverse transcription reactions. The robustness of our microarray analysis was tested at both the spot level and the level of the entire population of data.
We examined whether results would differ from slide to slide, that is, whether separately printed microarrays would give the same results if used with the same labeled cDNA in replicate experiments. Using a standard, two-tailed t test between the AL-Cy5, AL-Cy3, and AR data sets (see Materials and Methods) of two microarrays that had been hybridized to labeled cDNA from a common Cy3- and Cy5-labeling reaction, the average probability that the data sets share the same mean was calculated to be 97.2% (range, 94.4 to 99.9%; Table 2). As an additional validation of these data, weighted Pearson product-moment correlations, rxy, were calculated between the AL-Cy5, AL-Cy3, and AR data sets of each slide, with each data point weighted inversely by its own variance in the calculation (see Table 3). The 99% confidence limits (t0.01[∞] = 2.275) were set to each rxy using the z transformation. The significance of the correlations was calculated to be greater than 99% across 99% confidence limits.
As an additional measure of the robustness of this technique, we tested the reproducibility of the cDNA synthesis step. Using the same source of RNA as before, we repeated the reverse transcription and labeling steps. Following hybridization to two additional slides from the same printing as the original two, we performed mean and correlation analysis. Again, we found a low level of variation between the two slides; the average probability that the data sets share the same mean was calculated to be 98% (range, 95.1 to 99.7%; Table 2). The significance of the correlations was calculated to be greater than 99% across 99% confidence limits (see Table 3). On these slides, we observed very similar intensities among the yeast control spots, indicating the uniformity of the reverse transcription reaction. The yeast controls are also useful for gauging the quality of the RNA used in the labeling reaction. In other experiments (not shown) we have found that poor quality RNA will generate a low fluorescent signal relative to other experiments, while the yeast control RNAs will perform similarly across experiments. Furthermore, the RNA recovered from very early stage cultures (optical density at 600 nm of <1.0) was of poorer quality (not shown).
Across all conditions, the average probability that the data sets share the same mean was calculated to be 97.7% (range, 94.4 to 99.9%; Table 2). The significance of the correlations was calculated to be greater than 99% across 99% confidence limits. We found that spot-to-spot variability was 3.8% (range, 3.0 to 5.4% by slide) between replicate spots on a given slide, 5.0% (range, 3.7 to 6.3% by slide) between spots on separate slides (though hybridized with identical, labeled cDNA), and 8.1% (range, 7.2 to 9.0% by slide) between spots from separate slides hybridized with samples from separate reverse transcription reactions, yielding an average spot to spot variability of 7.1% (range, 3.7 to 9.0% by slide) across all conditions by pairwise analysis. Additionally, gene spot-to-spot variation (based on the average of 12 spots from four slides) provides an additional measure of variability across all conditions and was calculated to be an average of 8.5% (range, 2.5 to 22.0% by gene; low-intensity spots inherently have a higher percent error assuming equivalent measurement precision). This estimates the variation inherent to our methods to be 7.1 to 8.5%. These tests established the reproducibility of our technique, meaning that a common RNA sample will yield equivalent microarray hybridization data. Biological variation of transcription in equivalent culture conditions is not included in these error estimates. The combined biological variation and methodological variation is necessarily higher and reflects the higher variation encountered in current microarray techniques.
We also determined that signal intensity is independent of either the probe size or the actual length of the gene across the samples tested. The smallest probe that we used was 470 bp, and the largest was 1,250 bp (Table 1). We calculated the average total signal intensity for each gene as an average of the total intensity (measured as the sum of the intensity from the Cy3 and Cy5 channels) of the three spots representing that gene. We then plotted the average signal intensity for each of the 52 genes against either the length of the probe or the actual length of the gene and found no visible correlation between these parameters (data not shown).
Identification of differentially expressed genes.
Having shown statistically that our microarray preparation and scanning methods are robust, we used an iterative outlier analysis method to distinguish genes whose expression differed significantly from the mean (Table 1 and Fig. 2B). Under the growth conditions tested, we found that mRNAs from a small subset of the total number of genes became more abundant during the shift to lysine production conditions (Fig. 2A), as indicated by higher Cy5/Cy3 ratio in the ArrayWoRx scan. Similarly, mRNAs from a small number of genes were less abundant in the lysine-producing cells. However, the majority of the messages showed similar abundance in both samples, indicating that the relative concentrations of these mRNAs did not change significantly under these growth conditions.
DISCUSSION
Validating experimental and analytical techniques is an important first step in developing DNA microarrays to study a given organism. It is essential to establish that equivalent results can be obtained with identical starting materials before attempting to quantitate any biological variability. Investigating biological variation before benchmarking the technique creates a situation where it is impossible to determine how much data variation is due to biological effects and how much is inherent to the method itself.
In an analysis of data from microarrays, data points should not be removed from a set based on an arbitrary judgement of spot quality. Besides being in poor statistical form, the method of manually eliminating data points from a set becomes unwieldy, if not impossible, as the size of the data set increases. In addition, maintaining the integrity of data sets is essential if large volumes of data are to be shared among researchers in a centralized database system. Conventional analysis of array data has not been very rigorous in the propagation of error and has relied upon the calculation of averages and standard correlations that carry the implicit assumption of standard deviation on all data in a set. Microarray data does not meet this assumption. Every piece of microarray data has its own associated deviation that is reflective not only of standard measurement error but also of spot quality. To address both of these issues, we have employed weighted averages and correlations in our data analysis, where each data point is weighted inversely by its own variance. This method allows for the rigorous propagation of error across data transformations while minimizing the contribution of “low quality” spots.
Parallel analysis was performed on two data sets, one based on channel intensity data per spot and the other based on average pixel-by-pixel ratio within a spot. Performing congruent analysis is essential because it provides an internal validation of analytical technique. We used two separate analyses of our data sets to test the reproducibility of the method, and statistical manipulations demonstrated the robustness of the technique. First, upon examining the channel intensity data per spot (that is the Cy3 or Cy5 signals, separately), we found that both mean and correlation analysis were unable to find significant differences in the data from the four microarray slides tested. Second, when weighted average pixel-by-pixel Cy5/Cy3 ratios were compared, the data were similarly indistinguishable across the four microarray slides tested. Additionally, parallel analysis carried out on both data sets independently to identify genes with expression ratios that differed significantly from the mean yielded the identical genes at the same levels of significance.
Our experiments with pilot C. glutamicum microarrays show that this methodology for cDNA labeling, slide preparation, hybridization, and detection are both reproducible and robust, making it possible to detect even subtle changes in RNA abundance from an individual gene. Through an iterative outlier analysis of our small data set, we were able to distinguish genes with changes in expression as small as −2.2- or +1.5-fold from the rest of the population with statistical confidence. In our experiments with C. glutamicum ATCC 21253, we have observed apparent changes in gene expression between the “growth only” phase and the “growth and lysine production” phase. Upon reviewing the list of genes identified in this manner, we found that our experimental data are consistent with observations that have been reported in the literature.
The expression of the majority of the genes on the array changed less than 30% upon shifting to lysine production (Fig. 3). In contrast, transcription of lysE, which encodes a lysine transporter (17), was induced under lysine production conditions (1.8-fold). Our observation of an increase in the proportion of lysE transcript during lysine production is consistent with the hypothesis (17) that transcription of lysE is positively regulated in response to increasing intracellular lysine.
dtsR1, which encodes a putative transcarboxylase subunit of propionyl-coenzyme A (CoA) carboxylase (7), was strongly upregulated (2.8-fold) during lysine production. Previous studies have shown an inverse relationship between glutamate production and lysine production. Also, an inverse relationship between glutamate production and DtsR1 protein levels has been reported (8). Here we found a direct relationship between transcription of dtsR1 and lysine production. We can speculate that this is due to biotin levels. DtsR1 is known to be associated with a biotin-containing protein (8). Our fermentations were carried out under conditions of excess biotin. Biotin is a key switch as the cells shift their carbon flux from glutamate accumulation under biotin limitation (13) to lysine accumulation when biotin is in excess (14). Other genes believed to encode subunits of acyl-CoA carboxylases (accBC, accD, and dtsR2) were not significantly induced.
ilvB, which encodes the large subunit of acetohydroxy acid synthase (AHAS), an enzyme required to convert threonine into isoleucine, was also upregulated (1.8-fold) as lysine production increased. Lysine production was induced in the fermentation by the depletion of threonine from the medium. As threonine levels decrease so will the cells' supply of isoleucine. The observation that ilvB expression increases during the transition to lysine production is thus consistent with the findings of Eggeling et al. (4), who have shown that isoleucine limitation derepresses AHAS expression.
Genes encoding enzymes in the tricarboxylic acid (TCA) cycle (gltA and odhA) were downregulated in the growth-and-lysine-production phase. This may be in response to the fact that growth rate slows (from 0.35 to 0.04 h−1), and glucose uptake rate slows as well [from 0.35 to 0.06 g of Glc/g (DCW)/h] during this phase of the fermentation, as the cells react to threonine starvation and begin accumulating lysine. This change in transcription may also reflect the changes in carbon flux during lysine production when oxaloacetate is withdrawn from the TCA cycle and converted into aspartate and downstream compounds.
The gene encoding pyruvate kinase, pyk, was upregulated in the growth-and-lysine-production phase, although the level of induction was barely above the threshold in the analysis of statistically significant changes in gene expression. Although it is known that pyruvate kinase is important for lysine biosynthesis (5), we know of no studies that have examined the transcriptional regulation of the pyk gene under lysine production conditions.
Our microarray data also indicate that transcriptional changes are occurring in genes involved in nitrogen metabolism and acetate metabolism. We found that the gene encoding glutamine synthetase (glnA), which interconverts glutamate and glutamine, was induced. This is in line with expectations. During growth, lysine biosynthesis depends primarily on glutamine as a nitrogen source (18). However, as cells begin overproducing lysine, the preferred nitrogen source shifts to ammonium. Thus, an increase in glutamine synthetase may reflect the cells' changing requirements for nitrogen. In contrast, the acetate-activating phosphoacetyltransferase gene (pta) was downregulated in the lysine production phase.
In this study, we have described a method for the efficient extraction of total RNA from C. glutamicum and the conditions for labeling and hybridizing this material for the transcriptional profiling of this industrially important strain. We have shown that this method is robust and statistically reproducible. The changes in gene expression results we observed are consistent with reported data that were gathered by other methods.
Although the data presented here support the reproducibility of Corynebacterium DNA microarray techniques, separate experimentation must be done to address the issue of whether biological significance can be inferred from microarray data. The low variability that we have seen from spot-to-spot, from slide-to-slide, and from cDNA synthesis-to-cDNA synthesis makes us confident that we can distinguish small changes in gene expression between any two experimental samples. However, as others (20) have pointed out, experiments such as those described above do not assess the biological significance of variability in expression for any of the genes on the array. Under seemingly identical culture conditions, we may find that the level of expression for some genes varies greatly, with little physiological impact. In contrast, minor changes in the transcription of other genes may reflect (or cause) significant physiological changes, because these genes are more exquisitely regulated. From this argument, it follows that variability of gene expression must be assessed for each gene on the array through multiple replications of an experiment. Once the normal range of variability can be defined for each element, it may be possible to define threshold values above which a certain “fold change” in expression for each particular gene may be called relevant or significant. To address this, Wittes and Friedman (20) have proposed using scale invariant statistical methods of analysis for identifying genes whose changes in expression are worthy of further study.
The techniques we have described should permit further experimentation with large numbers of genes from Corynebacterium or related bacteria. Since the genes we used were all available in public databases, our observations about changes in gene expression suggest that useful experiments can be done even though the entire genomic sequence is not known. Small, inexpensive microarrays can provide statistically significant data and are valuable for understanding the interplay of known genes in controlling metabolism.
ACKNOWLEDGMENTS
This work was funded by a grant from the Archer Daniels Midland Corporation.
We thank Rick Damren for computing assistance and Danimal O'Brien for thoughtful discussions on statistical analysis and error propagation.
REFERENCES
- 1.Bevington P R, Robinson D K. Data reduction and error analysis for the physical sciences. 2nd ed. New York, N.Y: McGraw-Hill; 1992. [Google Scholar]
- 2.Brown P O, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21(Suppl. 1):33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 3.de Saizieu A, Gardes C, Flint N, Wagner C, Kamber M, Mitchell T J, Keck W, Amrein K E, Lange R. Microarray-based identification of a novel Streptococcus pneumoniae regulon controlled by an autoinduced peptide. J Bacteriol. 2000;182:4696–4703. doi: 10.1128/jb.182.17.4696-4703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eggeling I, Cordes C, Eggeling L, Sahm H. Regulation of acetohydroxy acid synthase in Corynebacterium glutamicum during fermentation of α-ketobutyrate to l-isoleucine. Appl Microbiol Biotechnol. 1987;25:346–351. [Google Scholar]
- 5.Gubler M, Jetten M, Lee S H, Sinskey A J. Cloning of the pyruvate kinase gene (pyk) of Corynebacterium glutamicum and site-specific inactivation of pyk in a lysine-producing Corynebacterium lactofermentum strain. Appl Environ Microbiol. 1994;60:2494–2500. doi: 10.1128/aem.60.7.2494-2500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guillouet S, Rodal A A, An G, Lessard P A, Sinskey A J. Expression of the Escherichia coli catabolic threonine dehydratase in Corynebacterium glutamicum and its effect on isoleucine production. Appl Environ Microbiol. 1999;65:3100–3107. doi: 10.1128/aem.65.7.3100-3107.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimura E, Abe C, Kawahara Y, Nakamatsu T. Molecular cloning of a novel gene, dtsR, which rescues the detergent sensitivity of a mutant derived from Brevibacterium lactofermentum. Biosci Biotechnol Biochem. 1996;60:1565–1570. doi: 10.1271/bbb.60.1565. [DOI] [PubMed] [Google Scholar]
- 8.Kimura E, Yagoshi C, Kawahara Y, Ohsumi T, Nakamatsu T, Tokuda H. Glutamate overproduction in Corynebacterium glutamicum triggered by a decrease in the level of a complex comprising DtsR and a biotin-containing subunit. Biosci Biotechnol Biochem. 1999;63:1274–1278. doi: 10.1271/bbb.63.1274. [DOI] [PubMed] [Google Scholar]
- 9.Lessard P A, Guillouet S, Willis L B, Sinskey A J. Corynebacteria, Brevibacteria. In: Flickinger M C, Drew S W, editors. Encyclopedia of bioprocess technology: fermentation, biocatalysis and bioseparation. New York, N.Y: John Wiley & Sons, Inc.; 1999. pp. 729–740. [Google Scholar]
- 10.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 11.Schena M, Shalon D, Davis R W, Brown P O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 12.Sokal R R, Rohlf F J. Introduction to biostatistics. 2nd ed. New York, N.Y: Freeman; 1987. [Google Scholar]
- 13.Takinami K, Yamada Y, Okada H. Biochemical effects of fatty acid and its derivatives on l-glutamic acid fermentation. Agric Biol Chem. 1966;30:674. [Google Scholar]
- 14.Tosaka O, Hirakawa H, Takinami K. Effects of biotin levels on L-lysine formation in Brevibacterium lactofermentum. Agric Biol Chem. 1979;43:491–495. [Google Scholar]
- 15.Treadway S L, Yanagimachi K S, Lankenau E, Lessard P A, Stephanopoulos G, Sinskey A J. Isolation and characterization of indene bioconversion genes from Rhodococcus strain I24. Appl Microbiol Biotechnol. 1999;51:786–793. doi: 10.1007/s002530051463. [DOI] [PubMed] [Google Scholar]
- 16.Vallino J J, Stephanopoulos G. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol Bioeng. 1993;41:633–646. doi: 10.1002/bit.260410606. [DOI] [PubMed] [Google Scholar]
- 17.Vrljic M, Sahm H, Eggeling L. A new type of transporter with a new type of cellular function: l-lysine export from Corynebacterium glutamicum. Mol Microbiol. 1996;22:815–826. doi: 10.1046/j.1365-2958.1996.01527.x. [DOI] [PubMed] [Google Scholar]
- 18.Wehrmann A, Phillipp B, Sahm H, Eggeling L. Different modes of diaminopimelate synthesis and their role in cell wall integrity: a study with Corynebacterium glutamicum. J Bacteriol. 1998;180:3159–3165. doi: 10.1128/jb.180.12.3159-3165.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson M, DeRisi J, Kristensen H H, Imboden P, Rane S, Brown P O, Schoolnik G K. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci USA. 1999;96:12833–12838. doi: 10.1073/pnas.96.22.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wittes J, Friedman H P. Searching for evidence of altered gene expression: a comment on statistical analysis of microarray data. J Natl Cancer Inst. 1999;91:400–401. doi: 10.1093/jnci/91.5.400. [DOI] [PubMed] [Google Scholar]
- 21.Ye R W, Tao W, Bedzyk L, Young T, Chen M, Li L. Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions. J Bacteriol. 2000;182:4458–4465. doi: 10.1128/jb.182.16.4458-4465.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]