

Summary
At the dawn of the personalized medicine era, the number of rare diseases has been estimated at 10,000. By considering the influence of environmental factors together with genetic variations and our improved diagnostic capabilities, an assessment suggests a considerably larger number. The majority would be extremely rare, and hence, we introduce the term “hyper-rare,” defined as affecting <1/108 individuals. Such disorders would potentially outnumber all currently known rare diseases. Because autosomal recessive disorders are likely concentrated in consanguineous populations, and rare toxicities in rural areas, establishing their existence necessitates a greater reach than is currently viable. Moreover, the randomness of X-linked and gain-of-function mutations greatly compound this challenge. However, whether concurrent diseases actually cause a distinct illness will depend on if their pathological mechanisms interact (phenotype conversion) or not (phenotype maintenance). The hyper-rare disease concept will be important in precision medicine with improved diagnosis and treatment of rare disease patients.
Subject areas: Health sciences, Clinical genetics, Human genetics, Biological sciences, Genetics Disease
Graphical abstract
Health sciences, Clinical genetics, Human genetics, Biological sciences, Genetics, Disease
Introduction
The total number of diseases is a topic that has been frequently discussed. Recently, in a commentary entitled “Why rare disease needs precision medicine—and why precision medicine needs rare disease” (Might and Crouse, 2022), the authors stated the figure of 10,000 rare diseases citing Haendel et al. (2020). A crucial aspect is the definition of the term disease, and because this is a complex issue itself, we refer to publications specifically deciphering this topic (Boorse, 1977, 2014; Schwartz, 2014; Scully, 2004; Tikkinen et al., 2012). Although the number of 10,000 is practical for many purposes (Haendel et al., 2020), we believe that it is a gross underestimate when considering the number of potential diseases. There are two primary reasons: First, diseases depend on our ability to define and distinguish them, which will continually increase with our understanding and capacity to diagnose them (Smedley et al., 2021; Chong et al., 2015; Degasperi et al., 2022; Ferreira 2019). Second, disease can be influenced or caused by a myriad of factors including infections, allergens, physical insults, toxins, environmental conditions, such as altitude or humidity, as well as both inherited and acquired genetic variants (Figure 1). Even if rare diseases are, by definition, scarce, together they have been estimated to constitute as much as 1/10 of all human illnesses (Haendel et al., 2020). The British National Health Service lists 322 common diseases, while the Human Phenotype Ontology derived the number 3,145 by manual selection from a total of 4,620 unique entries comprised of medical subject headings Category C (diseases). These estimates are considerably lower than the 10,000 estimated rare diseases (https://www.nhsinform.scot/illnesses-and-conditions/a-to-z; Groza et al., 2015; Tudor Groza, pers commun). Such a number is always dependent on the definition of “disease,” but we do not believe that the total number of common disorders exceeds the proposed figure for rare diseases.
Figure 1.

The interaction between external factors such as environment, infection, injury, toxicity, and genetic factors causes disease
Not included are allergens, which can be derived from various sources. The red cross marks a genetic disease variant, inherited or acquired.
Rare diseases have different definitions regarding their prevalence: around <1/1650 affected individuals in the US (<200,000 Americans) (Herder, 2017) and <1/2000 in Europe (Ferreira 2019). Extremely rare diseases are sometimes referred to as ultra-rare with a prevalence of <1/50,000 (Hughes et al., 2005). Although the ultra-rare designation is considerably less frequently used as compared to rare, there are >500 citations in PubMed. The Orphanet organization carries out systematic literature surveys to assess the prevalence and incidence of rare disorders (https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf). In their most recent listing of diseases, they include osteoclastic giant cell tumor of pancreas for which the incidence based on European data is 1/108.
Along these lines, we propose that there are virtually innumerable diseases that are extremely infrequent. The incidence for many of them is so low that chances are there is not even a single affected individual currently living on our planet, with the current approximation of the world population being ∼8 × 109. Despite this, there are benefits to considering such disorders, which is why we introduce the term hyper-rare; here defined as affecting <1/108 individuals. This is because hyper-rare diseases likely represent a very large group of disorders of unknown future prevalence, as discussed later in discussion, and this has implications for the concept of precision medicine.
Hyper-rare diseases are likely to exist among the group of undiagnosed rare disease patients. The major challenges will be to define and study such disease phenotypes, given that for most of these disorders only single patients will be diagnosed (Landrum et al., 2020; Vihinen, 2021). However, it is important to consider that genomic efforts over recent years have revolutionized the identification of rare diseases (Green et al., 2020). As an example, DECIPHER, established in 2004 is a web-based platform for secure deposition, analysis, and sharing of plausibly pathogenic genomic variants from well-phenotyped patients suffering from rare genetic disorders (Foreman et al., 2022). This work also includes the formation of the Undiagnosed Disease Program by NIH in 2008 (Gahl and Tifft, 2011), followed by the International Rare Disease Research Consortium (IRDiRC) in 2011 (Boycott et al., 2017) and the Undiagnosed Disease Network in 2014 (Splinter et al., 2018; Wise et al., 2019). A major contributor in this field is the Human Phenotype Ontology (HPO), which was created in 2008 to capture symptoms and phenotypic findings using a logically constructed hierarchy of defined phenotypic terms (Köhler et al., 2021). In spite of these efforts, numerous patients remain undiagnosed, which negatively influences the life of the patient and their families (Kliegman et al., 2017; Lewis et al., 2010). Thus, this perspective aims to highlight the extensive diversity of disease in order to promote taking full advantage of future advances in diagnostics and personalized therapeutics.
Phenotype and diagnosis
Perhaps the most important consideration when discussing how many diseases could exist is how we define and distinguish them. The World Health Organization’s 1946 definition of health is “a state of complete physical, mental and social wellbeing and not merely the absence of disease or infirmity.” Although sickness is often framed as an absolute state, we subscribe to the dynamic definition of disease as a pathological condition, which relies on statistical abnormality relative to an individual’s peers as described by the Biostatistical Theory (Boorse, 2014; Schwartz, 2014). It is a type of internal state, which impairs health, by reducing one or more functional abilities below typical efficiency (Boorse, 1977). The subjective nature of this outlook can potentially give rise to many novel, distinct disease states, but more closely matches an individual’s experience of their wellbeing than what is possible via clinical diagnostics. However, it should be noted that many genetic variations result in synthetic lethal combinations, which are not tolerated and lead to fetal death, which is not generally referred to as disease. Although this practice can be questioned, the inclusion of mutations (here defined as acquired variations) causing miscarriage is not crucial for demonstrating that 10,000 rare diseases are a number that is too low owing to our increasing ability to distinguish diseases.
When discussing how to distinguish diseases, it is important to consider phenotype—when should different phenotypes caused by similar insults or mutations in the same gene be classified as different diseases? To define this, the objective of the International Consortium of Human Phenotype Terminologies and HPO is to provide the community with standards to allow the linking of phenotype and genotype databases for rare diseases (https://irdirc.org/activities/task-forces/international-consortium-of-human-phenotype-terminologies/; https://www.ebi.ac.uk/ols/ontologies/mondo; Groza et al., 2015). The HPO is considered to be a worldwide standard for phenotype exchange.
A classic example of variations causing different phenotypes is Becker dystrophy—an inherited recessive X-linked muscle disorder. Becker dystrophy presents as a milder form of Duchenne muscular dystrophy, is caused by internal in-frame deletions in the DMD gene and is classified as a separate disease entity (Hoffman et al., 1988). However, it is likely that many genes can give rise to distinct diseases dependent on the specific mutation concerned. Analogously, similar insults often lead to distinct syndromes in different individuals, some being infrequent, while others are more common, such as “long-COVID-19,” which is often referred to as a distinct disease (Sudre et al., 2021). Another example of the pandemic is the extreme susceptibility to severe COVID-19 in patients with hematological malignancies (Blixt et al., 2022). Because the increased risk may be secondary to tumor-induced reduction of plasmacytoid dendritic cells (Smith et al., 2022), would this qualify as a separate disease? Although semantic, this issue becomes expansive when considering various classes of illness, such as mental health. For instance, recent progress in genome-wide association studies of schizophrenia revealed hundreds of common genetic variants, whereas complementing exome sequencing identified, what was referred to as ultra-rare, coding alterations, including de novo mutations (Iyegbe and O’Reilly, 2022; Singh et al., 2022; Trubetskoy et al., 2022). We predict that hyper-rare variants will also exist, and a few may already appear within these cohorts, but a causative relationship cannot be established because of their extremely low frequency. This is a good example of how increased knowledge of disease mechanisms can lead to a single disease fragmenting into multiple related disorders.
In contrast, when phenotypes are known to be caused by distinct mechanisms, these will be defined as different diseases. However, this convention also reflects more on our knowledge of biology than the medical implication of diseases. Thus, it may also be appropriate to distinguish diseases with similar causes that require different treatments to maximize the medical utility of our definitions. This can be illustrated by antibiotic-resistant strains of Mycobacterium tuberculosis, where separating these would allow for improved first-line treatment. As with the examples above, defining these pathogens as separate entities would further add to the absolute number of known diseases, but would also increase the usefulness of those definitions (Abate et al., 1998).
At this point, it is relevant to consider the advantages and disadvantages of distinguishing related diseases and classifying them as unique. This is related to Darwin’s mentioning already 165 years ago of species “hair-splitters and lumpers” (Darwin, 1857). As in the examples above, splitting definitions has the benefits of descriptive accuracy, continued evolution, and potentially improved treatment. However, this can make discussions of disease needlessly pedantic and may lead clinicians to overlook important commonalities between syndromes. Moreover, this extends the challenges of working with rare diseases, such as cohort aggregation, clinical study design, and obtaining funding, to additional maladies. Despite these challenges, the overall direction of the medical field is toward more specific and accurate disease definitions. Fortunately, as we increase our ability to distinguish and understand the disease by more precisely measuring variation, we will be better able to provide prognosis and treatment to individual patients. Numerous examples of this come from the profound increase in nucleic acid sequencing data available to distinguish related cancers leading to improved specific therapeutics to treat them (Degasperi et al., 2022; Incerti et al., 2022). To this end, many fields are benefiting from our unprecedented ability to collect patient data and make sense of it in novel ways, as exemplified by surveillance of diseases by mobile device data collection (Wood et al., 2019). Despite introducing challenges in communication and research logistics, a fractal perspective of disease and improved diagnostics may lead to a better understanding of phenotypes and a more accurate prescription of therapies.
Exogenous agents – the effect of quantity
One crucial aspect for infections as well as toxin- or allergen-induced diseases is the quantity of the entities that someone is exposed to. Human salmonellosis is an infectious disease highly prevalent in certain geographic regions, but rare in others, which brings in another component when classifying diseases as rare or common. Apart from the dose of Salmonella, which is essential, stomach acidity is also of importance, making the very young and the elderly particularly susceptible (Blaser and Newman, 1982). This is an example of the interplay between a contagious agent and age-dependent physiological conditions. Various forms of this interaction are likely to apply also for rare infections. In the main, knowledge about the dosage effect for exogenous agents is scarce. Some exceptions include surveillance of allergens (Sheikh et al., 2007), measurement of radon (Al-Zoughool and Krewski, 2009), sampling of airborne particulate matter (Chen et al., 2022), and of pathogens in recreational water (Korajkic et al., 2018), as well as heat measurements (Fatima et al., 2021).
Upon exposure to an unknown toxin, allergen, or infectious agent in a rural area, it is furthermore highly unlikely that the origin is revealed because of its rarity and the limited available resources to perform proper diagnostic procedures. Given that the Earth is predicted to be home to upward of 1012 microbial species (Locey and Lennon, 2016), more than six million fungal strains (Větrovský et al., 2020), and 391,000 plant species (Royal Botanic Gardens Kew, 2016), there is no lack of potential new disease entities caused by infections, toxicities or hypersensitivity reactions. Albeit exposure to many microbial species and toxins may be highly infrequent among adults, the situation is different for toddlers who are prone to ingest foreign materials.
A prime example of a toxic compound is the drug thalidomide, which caused multiple birth defects in thousands of children from week three to eight of gestation, and whose target later was found to be the E3 ubiquitin ligase cereblon (Ito et al., 2010). A major reason for identifying this relationship was the magnitude of this severe adverse effect. However, many drug-induced gestational effects likely go unnoticed because they are highly infrequent, while even the relatively common thalidomide-induced birth defects belong to the group of rare diseases. Moreover, all these factors can interact in countless ways over time and organ systems. As an example, it was recently demonstrated that non-heritable immune perturbations influence the risk of developing multiple sclerosis in identical twins (Ingelfinger et al., 2022). Whereas multiple sclerosis is common in certain locations, similar influences are expected to occur in rare diseases and a disorder is always dependent on a combination of components as depicted in Figure 1.
Genetic disease – inherited variations
The most common polymorphisms in the genome are single nucleotide variations (SNVs) (polymorphism and variation are here used as synonyms), which are found throughout the genome, including within coding regions (Klein et al., 2022; Wainschtein et al., 2022). The total number of genes has been estimated to be at least 24,000 (International Human Genome Sequencing Consortium, 2004; Salzberg, 2018), each of which could harbor such variations.
In this context, we would like to mention an important parameter in genetic disease; namely gene size, as the shorter the gene, the lesser the likelihood that there exists a variation in the coding sequence. A case in point here is micro-RNAs (miRs) because their corresponding genes are tiny. As expected, only extremely rare, single-gene, variations have been reported for miRs (Mencía et al., 2009; Grigelioniene et al., 2019). For instance, gain-of-function mutations in the MIR140 gene cause autosomal dominant skeletal dysplasia and according to Giedre Grigelioniene (pers commun) only 3 cases have been identified worldwide. The low number of recognized patients suggests that this disease is hyper-rare, i.e. affects <1/108 individuals. Interestingly, both the variation in MIR96 and MIR140 genes show dominant inheritance. The phenotype caused by mutations in MIR140 represents a gain-of-new-function, whereas the mechanism underlying non-syndromal hearing loss in MIR96 remains elusive, although the processing of miR-96 seems to be impaired, suggesting haplotype insufficiency.
Moreover, the first miR gene ever reported (Lee et al., 1993; Wightman et al., 1993), LET-7, is a good example of redundancy, as it is encoded by eight loci in mice and humans (Gurtan and Sharp, 2013). Although there is some clustering of the LET-7 genes, where deletions could simultaneously remove more than a single copy, it still means that multiple, independent variations are needed to completely remove the expression of this miR. Although a phenotype caused by such a combination of infrequent events is expected to be extremely rare, it is anticipated to occur, although no human being may be affected at this time point.
Another important consideration is when a patient suffers simultaneously from two or more independent genetic variations that together produce a distinct phenotype, which can be difficult to diagnose for clinicians. Examples are diseases arising from polygenic inheritance, which make diagnosis highly complex, because to date only a fraction of the contributing genetic determinants have been identified (Sun et al., 2022). Confounding this issue is that polymorphisms can have various effects on gene activity. Although the majority of these are benign, there are both loss-of-function alterations and potential gain-of-function variations with diverse functional effects. Many of these variations may not be individually rare, but at the other end of the spectrum, there are numerous polymorphisms that are exceedingly rare. Polygenic diseases result from some poorly understood combination of genetic and environmental influences. Related to this is a recent report from the “100,000 genomes” study, in which diagnosis was much more robust for monogenic rare diseases as compared to those of complex origin, where the cause could only be determined in 11% of cases (Smedley et al., 2021). As such, Figure 2, panel A schematically depicts the genetics of polygenic disorders such as cardiovascular disease and diabetes.
Figure 2.

Genetic variations causing disease
(A) Inherited genomic polymorphisms, which individually do not cause human illness, but when combined result in polygenic disease.
(B) The combination of rare, inherited, disease-causing variants together with common or rare polymorphisms, which by themselves do not cause disease. Jointly they induce a qualitatively different phenotype (phenotype conversion) as compared to the individual disorders themselves. Because the combination (disease 4) would be extremely infrequent it would qualify as hyper-rare disease.
(C) Three acquired chromosomal deletions of different lengths yielding different diseases with most abnormalities being infrequent and with some being extremely rare and belonging to the hyper-rare category. Boxes indicate changes resulting in disease. Letters correspond to different chromosomes. Blue color marks a common variation; red indicates a rare variation; red cross marks genetic abnormality causing disease.
Furthermore, in contrast to the majority of polygenic diseases, for which the individual polymorphisms are not considered to lead to any overt symptoms, true disease-causing variations in two or more different genes can also occur simultaneously. This may result in synthetic dysregulation that could be lethal. Although such combinations are expected to be extremely infrequent in an outbred population, the situation is dramatically different in the case of consanguinity, or when individuals with rare diseases are coming together and this results in progeny. It is among individuals from this group that hyper-rare diseases caused by synthetic, non-lethal dysregulation may be found. This is because infrequent combinations of very rare biallelic variations are highly overrepresented in this context. It is common practice in a consanguineous situation to classify siblings affected by related symptoms as having the same disease. Although this represents a pragmatic approach, we suggest that it is not uncommon that sick siblings instead have different diseases, and we believe that such genetically determined phenotypic divergence among siblings is greatly underappreciated. Thus, even if there is a primary loss-of-function defect involved that affects a particular organ, the observed phenotype could be heavily influenced by distinct combinations of other biallelic variants. In contrast, in an outbred population the likelihood of other genetic variants influencing the phenotype in a similar way is considerably reduced. Contrasting the phenotypic outcomes resulting from in-versus outbreeding could help in the disease classification. However, this necessitates that a sufficient number of individuals are affected.
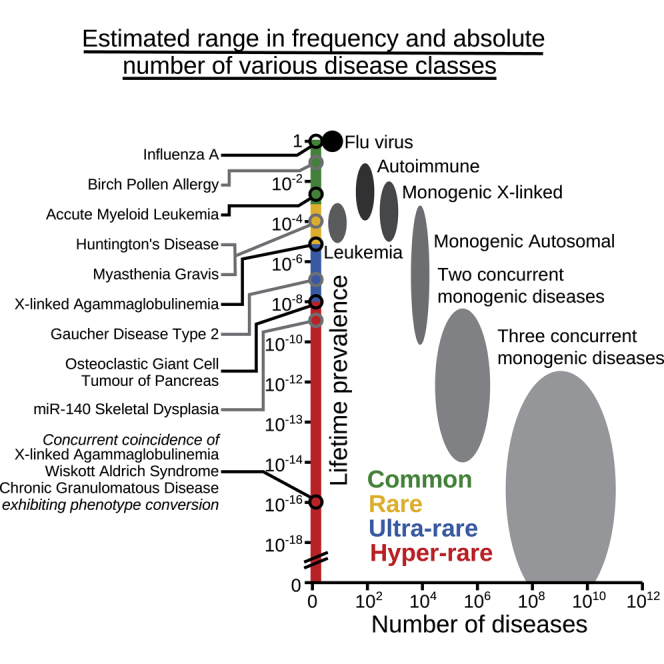
The resulting phenotype may correspond to the sum of the characteristics of the individual diseases, each with its specific pathology. However, variations can also influence each other mechanistically at the post-transcriptional level, with certain phenotypes being aggravated, reduced, or unique. Whereas aggravation or amelioration would not normally be regarded as a distinct disease, when unique phenotypes arise, the resulting illness could potentially qualify as a novel disorder. In Figure 3, we have tried to estimate both the frequency and potential number of diseases, whereby there are two or three concurrent monogenic diseases in the same individual. Figure 3 represents the situation in the Western world when there is no incidence of consanguinity. To compensate for the situation where the phenotypes do not affect each other, and hence the combination would not be recognized as a distinct disease (phenotype maintenance), we have introduced a hypothetical correction coefficient assuming that there will only be such an influence for 1/3 of genes (for two concurrent diseases the combined frequency is thus multiplied by 1/9). For the depicted concurrent coincidence of three primary immunodeficiency diseases, including X-linked agammaglobulinemia (XLA), at least two phenotypes are predicted to be unique (phenotype conversion), namely the cellular characteristics and the propensity for infections. To this end, we believe that “phenotype conversion” is a useful term when describing an alteration of the phenotype which is sufficiently significant, as measured by certain criteria, to warrant the assignment as a distinct disease.
Figure 3.

Estimated range in frequency and absolute number of various disease classes
Left, Distinct diseases, including the concurrent coincidence of three primary immunodeficiencies, where the prevalence for chronic granulomatous disease corresponds to mutations in the autosomal NCF1 gene, encoding a 47 kDa cytosolic subunit of neutrophil NADPH oxidase. Right, the oval-shaped areas correspond to various disease classes. To compensate for that phenotype conversion only appears in certain disease combinations a correction coefficient was introduced, which for three concurrent diseases amounts to [⅓]3 = ½7.
However, the most illustrative examples of unique phenotypes arising originate from combining mutations in experimental animals. Let us exemplify this by the human disease XLA, which is caused by mutations in the BTK gene resulting in an essential absence of B-lymphocytes (Vetrie et al., 1993; Smith 2017). In mice with mutations in the corresponding gene, the phenotype is mild (Thomas et al., 1993; Khan et al., 1995), but when coupled with the inactivation of the Tec gene, which by itself causes a very mild phenotype, the resulting combination generates a very severe B-cell lineage developmental defect (Ellmeier et al., 2000). Figure 2, panel B depicts an individual simultaneously affected by two rare genetic diseases, and who also carries a set of common and rare SNVs, which contribute to a novel disease phenotype. These SNVs may not cause any symptoms by themselves but will lead to synthetic dysregulation when combined with other genetic variants. Such an infrequent combination would be an example of a hyper-rare disorder.
The influence of polymorphisms is related to the concept of modifier genes, which are not normally expected to cause unique phenotypic changes. However, the classification of polymorphism as a modifier may not be definitive, as the same gene product could serve as a modifier under certain conditions but induce rare, unique changes in a different context. Such variations can be epigenetic, and though we will not specifically discuss epigenetic changes, suffice it to state that these are known to make a major contribution to disease. Moreover, individuals affected by genetic disease can also be exposed to infectious, physical, or toxic exogenous factors, or be influenced by age-induced senescence (Zhang et al., 2020). As an example, host genetics, including ultra-rare coding variants, are also of importance for infectious disease severity (Fallerini et al., 2022). The combination of these possibilities serves to amplify the number of potential unique disease phenotypes.
Genetic disease – acquired variations
While genomic and chromosomal abnormalities can be inherited, they most often occur de novo through acquired errors in egg, sperm, or tissue-resident stem cell populations. They are mainly classified into two groups: structural and numerical alterations. Structural rearrangements involve deletions, duplications, translocations between chromosomes, inversions, and gene amplifications, whereas numerical abnormalities result in aneuploidy or polyploidy. Certain aberrations are overrepresented because they occur owing to recombination processes that are facilitated by sequence homologies. However, this does not mean that other chromosomal errors do not exist; they are simply considerably less frequent, and an overwhelming majority of these have likely not yet been described. Figure 2, panel C depicts chromosomal deletions of varying lengths. Loss of large stretches of chromosomes is expected to cause complex phenotypes and for deletions, these are mainly in the form of haploinsufficiency. Another example is trisomy 21 causing Down syndrome, where instead extra genetic material is responsible for the phenotype.
Additionally, a large group of diseases result from variations acquired at a later stage, which are dominated by, but not limited to, tumors (Martincorena and Campbell, 2015). Neoplasms are mainly caused by acquired variations, also known as mutations (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020). However, for many cancers, certain rare inherited variations predispose the individual to the development of a tumor, as first hypothesized in retinoblastoma (Knudson, 1971). This report also indicates the importance of what was later named tumor suppressor genes. Malignancies can be caused by incrementally acquired SNVs, but also by a single catastrophic event leading to up to hundreds of rearrangements, first described in the form of chromothripsis (Stephens et al., 2011).
Both the number and the spectrum of mutations vary profoundly not only among different neoplasms but also within a tumor. Most acquired variants are passengers, whereas a selected few act as drivers providing a selective advantage to the mutated cell. So, when do these acquired alterations represent different diseases? This is a difficult question to answer. When the phenotype and treatment responses vary according to the specific type of mutation, this is normally considered a basis for subgrouping into different stages or even disease entities. As genome-wide analyses of tumors have increased profoundly over the last few years (Degasperi et al., 2022), they have already led to an improved appreciation of distinct malignant diseases (Calderaro et al., 2017). Additionally, liquid biopsy analysis has the potential to revolutionize our understanding of tumor evolution by providing information in real-time, though research into the biology of blood-borne tumor material is still in its infancy (Hagey et al., 2021). To this end, we believe that many more studies are needed before it is possible to reach any consensus on how many tumor subtypes exist and to what extent they represent different entities. However, there is no doubt that there are many forms of tumors, and that the characterization of subtypes will continue (Degasperi et al., 2022; https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga), likely resulting in numerous novel cancer subgroups being classified as different diseases.
The accumulation of somatic mutations causes what is known as genetic mosaicism (Biesecker and Spinner, 2013). Thus, the mutations occurring in cancer cells represent a form of mosaicism, though the term is mainly used in the context of mutations acquired during gestation. Such examples would relate to the large chromosomal deletions depicted in Figure 2, panel C. When an afflicted individual also carries, or acquires, a somatic loss-of-function mutation on the unaffected allele, both copies of this gene would be inactive with immediate phenotypic implications. As mutations occur at every cell division, essentially all cells within the same individual have a different genetic makeup. Because the acquired mutations are in essence random, somatic mosaicism is the underlying mechanism by which “identical twins” have distinct genomes. Such mosaicism rarely results in any illness because the majority of genetic alterations occur in non-coding chromosomal regions outside of control elements and are therefore not manifested as overt disease. However, certain acquired mutations will cause a disease phenotype. The phenotypic manifestation will depend on the cell type in which the acquisition occurs as well as when it happens during ontogeny. This creates a multitude of different potential phenotypes. Mutations occurring early during gestation have the greatest impact because more cells and organs are affected, but it is an open question to what extent various forms of mosaicism should be regarded as different diseases.
Theoretical calculations on the number of diseases
The average human genome is estimated to contain ∼100 loss-of-function variants with ∼20 genes completely inactivated (MacArthur et al., 2012). Notably, many of them never cause disease as exemplified by those related to olfactory reception (MacArthur et al., 2012). In order to put a true estimate on the number of possible diseases, we must first appreciate the limitations of our ability to define them, as well as the multitude of potential pathogenic mechanisms. From here, how often multiple disease combinations co-occur could be treated as a purely mathematical question. Thus, the number of disease-causing variants in all human genes can be combined with all the external insults, which together cause distinct phenotypes.
The number of inherited, purely genetic diseases resulting from this thought experiment would be unimaginably large. This could theoretically correspond to a factorial of 12,000, based on the assumption that single variations in just half of the human genes would cause disease. Although we believe that many are expected to cause fetal death, this number is without introducing any exogenous disease-causing factors. Hence, in the same way as different genetic variations can be combined, so could exogenous components, and a factorial of just 10 corresponds to over 3.6 million diseases. Moreover, gain-of-function mutations have the potential to produce orders of magnitude more disease phenotypes than loss-of-function mutations, but occur much more seldom.
Additional calculations were made for Figure 3. When estimating the number of disorders occurring when there are concurrent genetic diseases, we introduced the aforementioned correction coefficient. This should limit the amount to only those instances when there is phenotype conversion as opposed to phenotype maintenance. Taking into account a large number of hypothetical gain-of-function variations would presumably make the shape of the “concurrent genetic disease areas” in Figure 3 triangular, with rarer disorders being more numerous, but would also significantly increase the amount of potential monogenic diseases. Although this may seem outlandish, it is important to remember that only a small and healthy fraction of the world’s population is properly screened for the disease. Taken together, this illustrates that the possible number of rare diseases is enormously much larger than the estimate of 10,000 (Haendel et al., 2020) and compatible with the idea of hyper-rare disease.
Finally, related to the concept of precision medicine, we believe that as advances in diagnostics progress, the number of definable disorders may continue to increase to the point when sometimes it is instead the idea of common diseases that requires questioning. Fortunately, this will most likely occur in parallel with improvements in our ability to cure these disorders. A recent example is the “N-of-1 study,” in which a unique splice-site mutation was identified in a child with a very rare neurodegenerative disorder. Within one year the patient was treated with a newly designed oligonucleotide therapy (Kim et al., 2019).
Concluding remarks
There are numerous causes of illness, many of which are highly infrequent. We propose that together they correspond to a very large number of disorders, much greater than the frequently used estimate of 10,000 rare diseases. These include scarce inherited variations and acquired mutations, epigenetic modifications, physical insults, dosage-dependent effects of toxic and infectious agents, as well as environmental factors. In this perspective, we suggest that the number of diseases referred to as common, rare, or ultra-rare should be complemented with those that are hyper-rare. We also suggest that consanguinity profoundly increases the likelihood of finding genetically determined hyper-rare diseases, with disorders differing even among siblings, owing to the enhanced number of sibling-unique combinations of multiple biallelic variants resulting in phenotype conversion.
An understudied area is the influence of such factors during gestation and there is also the issue of whether lethality, including synthetic lethality, caused by various mechanisms during this period should be classified as different diseases. Even if the incidence of hyper-rare disorders is extremely low, the total number of diseases belonging to this group is likely very high. Thus, hyper-rare diseases, defined as affecting <1 individual per 108, are estimated to outnumber the other categories by orders of magnitude. Though we do not wish for this definition to be used to stratify established rare disease patient groups, this definition is reflected in the unknown number of patients who cannot be diagnosed and are lost from surveillance by the healthcare system. Defining these disorders would be valuable to patients by putting a renewed focus on diagnosis and stimulating research into the causes of disease. It would also lead to a better understanding of developmental pathways during and after embryogenesis. Because hyper-rare diseases occur so infrequently, a major challenge will be to define their phenotypes, as in many instances there may only be a single affected individual.
Acknowledgments
This work was supported by CIMED, the Center for Innovative Medicine, and the Swedish Cancer Society. We would like to thank Giedre Grigelioniene, Karin Lundin and Tea Umek, Karolinska Institutet, and Mauno Vihinen, University of Lund, for valuable comments. Images were produced using Biorender.
Author contributions
Development of the original concept CIES. Further concept development PB & DWH. Images were rendered by DWH. CIES & DWH wrote the article and all the authors edited and reviewed it.
Declaration of interests
The authors declare no competing interests.
References
- Abate G., Miörner H., Ahmed O., Hoffner S.E. Drug resistance in Mycobacterium tuberculosis strains isolated from re-treatment cases of pulmonary tuberculosis in Ethiopia: susceptibility to first-line and alternative drugs. Int. J. Tubercul. Lung Dis. 1998;2:580–584. [PubMed] [Google Scholar]
- Al-Zoughool M., Krewski D. Health effects of radon: a review of the literature. Int. J. Radiat. Biol. 2009;85:57–69. doi: 10.1080/09553000802635054. [DOI] [PubMed] [Google Scholar]
- Biesecker L.G., Spinner N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013;14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- Blaser M.J., Newman L.S. A review of human salmonellosis: I. Infective dose. Rev. Infect. Dis. 1982;4:1096–1106. doi: 10.1093/clinids/4.6.1096. [DOI] [PubMed] [Google Scholar]
- Blixt L., Bogdanovic G., Buggert M., Gao Y., Hober S., Healy K., Johansson H., Kjellander C., Mravinacova S., Muschiol S., et al. Covid-19 in patients with chronic lymphocytic leukemia: clinical outcome and B- and T-cell immunity during 13 monthsin consecutive patients. Leukemia. 2022;36:476–481. doi: 10.1038/s41375-021-01424-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorse C. Health as a theoretical concept. Philos. Rev. 1977;44:542–573. doi: 10.1086/288768. [DOI] [Google Scholar]
- Boorse C. A second rebuttal on health. J. Med. Philos. 2014;39:683–724. doi: 10.1093/jmp/jhu035. [DOI] [PubMed] [Google Scholar]
- Boycott K.M., Rath A., Chong J.X., Hartley T., Alkuraya F.S., Baynam G., Brookes A.J., Brudno M., Carracedo A., den Dunnen J.T., et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 2017;100:695–705. doi: 10.1016/j.ajhg.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderaro J., Couchy G., Imbeaud S., Amaddeo G., Letouzé E., Blanc J.F., Laurent C., Hajji Y., Azoulay D., Bioulac-Sage P., et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017;67:727–738. doi: 10.1016/j.jhep.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Chen H., Oliver B.G., Pant A., Olivera A., Poronnik P., Pollock C.A., Saad S. Effects of air pollution on human health - mechanistic evidence suggested by in vitro and in vivo modelling. Environ. Res. 2022;212:113378. doi: 10.1016/j.envres.2022.113378. [DOI] [PubMed] [Google Scholar]
- Chong J., Buckingham K., Jhangiani S., Boehm C., Sobreira N., Smith J., Harrell T., McMillin M., Wiszniewski W., Gambin T., et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. Darwin Correspondence Project. University of Cambridge; 1857. Letter no. 2130. [Google Scholar]
- Degasperi A., Zou X., Dias Amarante T., Martinez-Martinez A., Koh G.C.C., Dias J.M.L., Heskin L., Chmelova L., Rinaldi G., Wang V.Y.W., et al. Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science. 2022;376:368. doi: 10.1126/science.abl9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmeier W., Jung S., Sunshine M.J., Hatam F., Xu Y., Baltimore D., Mano H., Littman D.R. Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk. J. Exp. Med. 2000;192:1611–1624. doi: 10.1084/jem.192.11.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallerini C., Picchiotti N., Baldassarri M., Zguro K., Daga S., Fava F., Benetti E., Amitrano S., Bruttini M., Palmieri M., et al. Common, low-frequency, rare, and ultra-rare coding variants contribute to COVID-19 severity. Hum. Genet. 2022;141:147–173. doi: 10.1007/s00439-021-02397-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima S.H., Rothmore P., Giles L.C., Varghese B.M., Bi P. Extreme heat and occupational injuries in different climate zones: a systematic review and meta-analysis of epidemiological evidence. Environ. Int. 2021;148:106384. doi: 10.1016/j.envint.2021.106384. [DOI] [PubMed] [Google Scholar]
- Ferreira C.R. The burden of rare diseases. Am. J. Med. Genet. 2019;179:885–892. doi: 10.1002/ajmg.a.61124. [DOI] [PubMed] [Google Scholar]
- Foreman J., Brent S., Perrett D., Bevan A.P., Hunt S.E., Cunningham F., Hurles M.E., Firth H.V. DECIPHER: Supporting the interpretation and sharing of rare disease phenotype-linked variant data to advance diagnosis and research. Hum. Mutat. 2022;43:682–697. doi: 10.1002/humu.24340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl W.A., Tifft C.J. The NIH undiagnosed diseases program: lessons learned. JAMA. 2011;305:1904. doi: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- Green E.D., Gunter C., Biesecker L.G., Di Francesco V., Easter C.L., Feingold E.A., Felsenfeld A.L., Kaufman D.J., Ostrander E.A., Pavan W.J., et al. Strategic vision for improving human health at the forefront of genomics. Nature. 2020;586:683–692. doi: 10.1038/s41586-020-2817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigelioniene G., Suzuki H.I., Taylan F., Mirzamohammadi F., Borochowitz Z.U., Ayturk U.M., Tzur S., Horemuzova E., Lindstrand A., Weis M.A., et al. Gain-of-function mutation of microRNA-140 in human skeletal dysplasia. Nat. Med. 2019;25:583–590. doi: 10.1038/s41591-019-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groza T., Köhler S., Moldenhauer D., Vasilevsky N., Baynam G., Zemojtel T., Schriml L., Kibbe W., Schofield P., Beck T., et al. The Human Phenotype Ontology: semantic unification of common and rare disease. Am. J. Hum. Genet. 2015;97:111–124. doi: 10.1016/j.ajhg.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtan A.M., Sharp P.A. The role of miRNAs in regulating gene expression networks. J. Mol. Biol. 2013;425:3582–3600. doi: 10.1016/j.jmb.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendel M., Vasilevsky N., Unni D., Bologa C., Harris N., Rehm H., Hamosh A., Baynam G., Groza T., McMurry J., et al. How many rare diseases are there? Nat. Rev. Drug Discov. 2020;19:77–78. doi: 10.1038/d41573-019-00180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagey D.W., Kordes M., Görgens A., Mowoe M.O., Nordin J.Z., Moro C.F., Löhr J.M., El Andaloussi S. Extracellular vesicles are the primary source of blood-borne tumour-derived mutant KRAS DNA early in pancreatic cancer. J. Extracell. Vesicles. 2021;10:e12142. doi: 10.1002/jev2.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herder M. What is the purpose of the orphan drug act? PLoS Med. 2017;14:e1002191. doi: 10.1371/journal.pmed.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman E.P., Fischbeck K.H., Brown R.H., Johnson M., Medori R., Loire J.D., Harris J.B., Waterston R., Brooke M., Specht L., et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N. Engl. J. Med. 1988;318:1363–1368. doi: 10.1056/nejm198805263182104. [DOI] [PubMed] [Google Scholar]
- Hughes D.A., Tunnage B., Yeo S.T. Drugs for exceptionally rare diseases: do they deserve special status for funding? QJM. 2005;98:829–836. doi: 10.1093/qjmed/hci128. [DOI] [PubMed] [Google Scholar]
- Incerti D., Xu X.M., Chou J.W., Gonzaludo N., Belmont J.W., Schroeder B.E. Cost-effectiveness of genome sequencing for diagnosing patients with undiagnosed rare genetic diseases. Genet. Med. 2022;24:109–118. doi: 10.1016/j.gim.2021.08.015. [DOI] [PubMed] [Google Scholar]
- Ingelfinger F., Gerdes L.A., Kavaka V., Krishnarajah S., Friebel E., Galli E., Zwicky P., Furrer R., Peukert C., Dutertre C.A., et al. Twin study reveals non-heritable immune perturbations in multiple sclerosis. Nature. 2022;603:152–158. doi: 10.1038/s41586-022-04419-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Iyegbe C.O., O’Reilly P.F. Genetic origins of schizophrenia find common ground. Nature. 2022;604:433–435. doi: 10.1038/d41586-022-00773-5. [DOI] [PubMed] [Google Scholar]
- Khan W.N., Alt F.W., Gerstein R.M., Malynn B.A., Larsson I., Rathbun G., Davidson L., Müller S., Kantor A.B., Herzenberg L.A., et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Kim J., Hu C., Moufawad El Achkar C., Black L.E., Douville J., Larson A., Pendergast M.K., Goldkind S.F., Lee E.A., Kuniholm A., et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019;381:1644–1652. doi: 10.1056/nejmoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L., D'Urso S., Eapen V., Hwang L.-D., Lin P.-I. Exploring polygenic contributors to subgroups of comorbid conditions in autism spectrum disorder. Sci. Rep. 2022;12:3416. doi: 10.1038/s41598-022-07399-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliegman R.M., Bordini B.J., Basel D., Nocton J.J. How doctors think: common diagnostic errors in clinical judgment-lessons from an undiagnosed and rare disease program. Pediatr. Clin. 2017;64:1–15. doi: 10.1016/j.pcl.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Knudson A.G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S., Gargano M., Matentzoglu N., Carmody L.C., Lewis-Smith D., Vasilevsky N.A., Danis D., Balagura G., Baynam G., Brower A.M., et al. The human phenotype Ontology in 2021. Nucleic Acids Res. 2021;49:D1207–D1217. doi: 10.1093/nar/gkaa1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korajkic A., McMinn B., Harwood V. Relationships between microbial indicators and pathogens in recreational water settings. Int. J. Environ. Res. Publ. Health. 2018;15:2842. doi: 10.3390/ijerph15122842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C., et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–D844. doi: 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lewis C., Skirton H., Jones R. Living without a diagnosis: the parental experience. Genet. Test. Mol. Biomarkers. 2010;14:807–815. doi: 10.1089/gtmb.2010.0061. [DOI] [PubMed] [Google Scholar]
- Locey K.J., Lennon J.T. Scaling laws predict global microbial diversity. Proc. Natl. Acad. Sci. USA. 2016;113:5970–5975. doi: 10.1073/pnas.1521291113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I., Campbell P.J. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- Mencía Á., Modamio-Høybjør S., Redshaw N., Morín M., Mayo-Merino F., Olavarrieta L., Aguirre L.A., del Castillo I., Steel K.P., Dalmay T., et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat. Genet. 2009;41:609–613. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- Might M., Crouse A.B. Why rare disease needs precision medicine—and precision medicine needs rare disease. Cell Rep. Med. 2022;3:100530. doi: 10.1016/j.xcrm.2022.100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal Botanic Gardens Kew . 2016. State of the World’s Plants 2016. https://stateoftheworldsplants.org/2016/report/sotwp_2016.pdf. [Google Scholar]
- Salzberg S.L. Open questions: how many genes do we have? BMC Biol. 2018;16:94. doi: 10.1186/s12915-018-0564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully J.L. What is a disease? EMBO Rep. 2004;5:650–653. doi: 10.1038/sj.embor.7400195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P.H. Reframing the disease debate and defending the biostatistical theory. J. Med. Philos. 2014;39:572–589. doi: 10.1093/jmp/jhu039. [DOI] [PubMed] [Google Scholar]
- Sheikh A., Singh Panesar S., Dhami S., Salvilla S. Seasonal allergic rhinitis in adolescents and adults. Clin. Evid. 2007;2007:0509. [PubMed] [Google Scholar]
- Singh T., Poterba T., Curtis D., Akil H., Al Eissa M., Barchas J.D., Bass N., Bigdeli T.B., Breen G., Bromet E.J., et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature. 2022;604:509–516. doi: 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100,000 Genomes Project Pilot Investigators. Smedley D., Smith K.R., Martin A., Thomas E.A., McDonagh E.M., Cipriani V., Ellingford J.M., Arno G., Tucci A., et al. 100,000 genomes pilot on rare-disease diagnosis in health care - preliminary report. N. Engl. J. Med. 2021;385:1868–1880. doi: 10.1056/NEJMoa2035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.I.E. From identification of the BTK kinase to effective management of leukemia. Oncogene. 2017;36:2045–2053. doi: 10.1038/onc.2016.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.I.E., Zain R., Österborg A., Palma M., Buggert M., Bergman P., Bryceson Y. Do reduced numbers of plasmacytoid dendritic cells contribute to the aggressive clinical course of COVID-19 in chronic lymphocytic leukemia (CLL)? Scand. J. Immunol. 2022;95:e13153. doi: 10.1111/sji.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter K., Adams D.R., Bacino C.A., Bellen H.J., Bernstein J.A., Cheatle-Jarvela A.M., Eng C.M., Esteves C., Gahl W.A., Hamid R., et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 2018;379:2131–2139. doi: 10.1056/nejmoa1714458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P.J., Greenman C.D., Fu B., Yang F., Bignell G.R., Mudie L.J., Pleasance E.D., Lau K.W., Beare D., Stebbings L.A., et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudre C.H., Murray B., Varsavsky T., Graham M.S., Penfold R.S., Bowyer R.C., Pujol J.C., Klaser K., Antonelli M., Canas L.S., et al. Attributes and predictors of long COVID. Nat. Med. 2021;27:626–631. doi: 10.1038/s41591-021-01292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B.B., Kurki M.I., Foley C.N., Mechakra A., Chen C.Y., Marshall E., Wilk J.B., Sun B.B., Ghen C.Y., Marshall E., et al. Genetic associations of protein-coding variants in human disease. Nature. 2022;603:95–102. doi: 10.1038/s41586-022-04394-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.D., Sideras P., Smith C.I.E., Vořechovský I., Chapman V., Paul W.E. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- Tikkinen K.A.O., Leinonen J.S., Guyatt G.H., Ebrahim S., Järvinen T.L.N. What is a disease? Perspectives of the public, health professionals and legislators. BMJ Open. 2012;2:e001632. doi: 10.1136/bmjopen-2012-001632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trubetskoy V., Pardiñas A.F., Qi T., Panagiotaropoulou G., Awasthi S., Bigdeli T.B., Bryois J., Chen C.-Y., Dennison C.A., Hall L.S., et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–508. doi: 10.1038/s41586-022-04434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrie D., Vořechovský I., Sideras P., Holland J., Davies A., Flinter F., Hammarström L., Kinnon C., Levinsky R., Bobrow M., et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- Větrovský T., Morais D., Kohout P., Lepinay C., Algora C., Awokunle Hollá S., Bahnmann B.D., Bílohnědá K., Brabcová V., D’Alò F., et al. GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci. Data. 2020;7:228. doi: 10.1038/s41597-020-0567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. Measuring and interpreting pervasive heterogeneity, poikilosis. FASEB Bioadv. 2021;3:611–625. doi: 10.1096/fba.2021-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainschtein P., Jain D., Zheng Z., TOPMed Anthropometry Working Group. Psaty B.M., Kooperberg C., Liu C.T., Albert C.M., Roden D., Chasman D.I., Darbar D., et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet. 2022;54:263–273. doi: 10.1038/s41588-021-00997-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Wise A.L., Manolio T.A., Mensah G.A., Peterson J.F., Roden D.M., Tamburro C., Williams M.S., Green E.D. Genomic medicine for undiagnosed diseases. Lancet. 2019;394:533–540. doi: 10.1016/s0140-6736(19)31274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C.S., Thomas M.R., Budd J., Mashamba-Thompson T.P., Herbst K., Pillay D., Peeling R.W., Johnson A.M., McKendry R.A., Stevens M.M. Taking connected mobile-health diagnostics of infectious diseases to the field. Nature. 2019;566:467–474. doi: 10.1038/s41586-019-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Qu J., Liu G.-H., Belmonte J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020;21:137–150. doi: 10.1038/s41580-019-0204-5. [DOI] [PubMed] [Google Scholar]