

Abstract
Effective systemic treatment for hepatocellular carcinoma (HCC) remains urgently needed. Sorafenib is the first FDA-approved systemic treatment for HCC. However, individual HCC patents’ response to sorafenib varies greatly. How to enhance the anti-HCC effect of sorafenib is still a significant challenge. T cell immunoglobulin mucin-3 (Tim-3) is a newly identified immune checkpoint molecule and a promising target for HCC treatment. Herein, we developed a novel pH-triggered drug-eluting nanoparticle (CC@SR&SF@PP) for simultaneously delivery of Tim-3 siRNA and sorafenib to HCC in situ. By a single emulsification method, a representative HCC targeted-therapeutic drug sorafenib (SF) was encapsulated into the pH-triggered positive-charged mPEG5K-PAE10K (PP) nanoparticles, followed by condensing of negative-charged Tim-3 siRNA. Then, carboxymethyl chitosan (CMCS), an amphoteric polysaccharide with negative charge in the physiological pH and positive charge in the acidic environment of the tumor, was eventually adsorbed onto the surface of nanoparticles. This co-delivery nanoparticle rapidly and specifically accumulated in the tumor site of the liver and enhanced the targeted, specific and multiple release of siRNA and sorafenib. Enhanced Tim-3 siRNA transfected into tumor cells can not only directly inhibit the growth of tumor cells by knock down the expression Tim-3, but also induce the immune response and enhance the recruitment of cytotoxic T cells to kill tumor cells. The following pH-triggered sorafenib release from SF@PP NPs greatly inhibited the tumor proliferation and angiogenesis, resulting in remarkable tumor growth inhibition in a mouse hepatoma 22 (H22) orthotopic tumor model. Thus, co-delivery of Tim-3 siRNA and sorafenib via this novel pH triggered drug-eluting nanoparticle enhances their anti-tumor efficacy. We expect that such combination treatment strategy will have great potential in future clinical applications.
Keywords: Hepatocellular carcinoma, Tim-3 siRNA, Sorafenib, pH-triggered nanoparticle, Co-delivery
Abbreviations: HCC, Hepatocellular Carcinoma; SF, Sorafenib; Tim-3, T cell immunoglobulin and mucin domain 3; CMCS, carboxymethyl chitosan; CC@SR&SF@PP NPs, CMCS@Tim-3 siRNA&Sorfeinb@mPEG5K-PAE10K nanoparticles
Graphical abstract
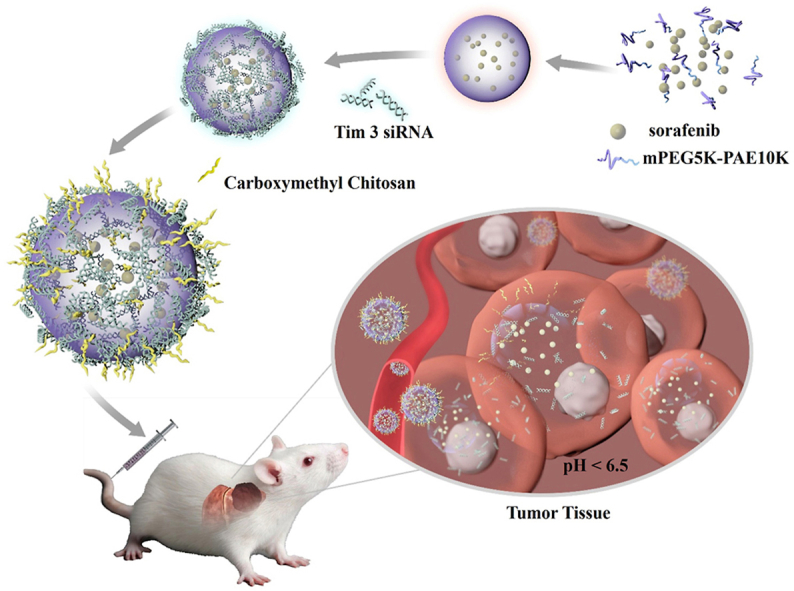
1. Introduction
Hepatocellular Carcinoma (HCC) is one of the most common malignant cancers in clinical practice [1,2]. Due to the insidious onset of HCC, most patients are in the middle or late stage of the disease when they are diagnosed, thus missing the optimal period of surgical resection [[3], [4], [5], [6]]. In addition, the remission rates of radiotherapy and chemotherapy for intermediate and advanced HCC are also very low [7]. The overall therapeutic effect of HCC around the world is not satisfactory [8]. Sorafenib (SF), the first drug approved by the U.S. Food and Drug Administration (FDA) and the most commonly used drug for the systemic treatment of HCC, is an effective targeted chemotherapeutic agent against tumors by inhibiting angiogenesis, proliferation and invasion [9,10]. However, due to poor aqueous solubility and various adverse effects such as elevated blood pressure, diarrhea, hand-foot syndrome and skin rash/desquamation, monotherapy with sorafenib is quite limited in clinical applications [[11], [12], [13], [14], [15]]. Therefore, there is an urgent need to develop novel therapeutic strategies and/or modality options of sorafenib for HCC [16].
The generation and metastasis of HCC is a multigene-related, multifactorial and multistep response process [17]. Gene therapy might be potential to achieve good therapeutic effects of HCC. Meanwhile, more and more researchers have proved that the combination therapy of gene therapy and chemotherapy shows a synergistic therapeutic effect for HCC [[18], [19], [20], [21], [22]]. PD-1 (programmed cell death protein 1) or PDL-1 (programmed cell death 1 ligand 1) combined with sorafenib has been used for the treatment of HCC and achieved a good synergistic therapeutic effect [[23], [24], [25], [26]]. It is expected to be a powerful therapeutic method in clinical application. What's more, other strategies for combining gene therapy with chemotherapy to treat HCC are also being investigated [27].
T cell immunoglobulin and mucin domain 3 (Tim-3), a novel immune checkpoint molecule [28], plays a vital role in the development of HCC. Studies have shown that, comparing with para-cancerous tissues and normal liver cells, Tim-3 is overexpressed in both HCC tumor tissues and cell lines, indicating that Tim-3 is associated with the occurrence of HCC [[29], [30], [31]]. In preclinical studies, targeting Tim-3 has also been proved to have anti-tumor efficiency. Meanwhile, clinical studies have also shown that the high expression of Tim-3 in the tumor tissue of patients with HCC often indicates poor prognosis [[32], [33], [34]]. All these suggest that inhibiting the expression of Tim-3 might be a novel therapeutic strategy for HCC. However, gene therapy is limited because of the poor targeting and specificity, low transfection efficiency, lacking of therapeutic effect, etc. [[35], [36], [37], [38], [39]]. More importantly, the potential virulence of commonly used adenovirus and lentivirus gene vectors may trigger the host's immune response and accelerate the host's clearance of vectors, which is a crucial obstacle for its clinical application [40]. Therefore, exploring new gene and drug delivery vectors to ensure the safety and improve the targeting and delivery efficiency is particularly important [41].
In this study, we reported a tumor micro-environmental triggered drug-eluting nanoparticle for simultaneous gene therapy and tumor vascular-targeted therapy in a mouse hepatoma 22 (H22) orthotopic tumor model. As illustrated in Scheme 1, a representative HCC targeted-therapeutic drug sorafenib (SF) was encapsulated into the pH-triggered mPEG5K-PAE10K (PP) nanoparticle [42,43] by a simple single emulsification method. The formed sorafenib@mPEG5K-PAE10K nanoparticle (SF@PP NP) was positively charged, which provided an excellent template for negatively charged Tim-3 siRNA (SR) to condense on the surface of the drug-carrying nanoparticles through electrostatic interaction. To protect the condensed Tim-3 siRNA and reduce the toxicity of cations, pH sensitive and negatively charged carboxymethyl chitosan (CMCS) [[44], [45], [46]] was eventually adsorbed onto the surface of the nanoparticle. This siRNA and sorafenib co-delivery system CMCS@Tim-3 siRNA&Sorfeinb@mPEG5K-PAE10K nanoparticles (CC@SR&SF@PP NPs) can rapidly accumulate in the tumor site of liver and enhance the targeted, specific and multiple release of siRNA and sorafenib in tumor tissues due to its acidic environment. Enhanced Tim-3 siRNA transfection greatly inhibited the expression of the target gene, induce the immune response and enhance the recruitment of cytotoxic T cells to kill tumor cells. Then, increased drug delivery of sorafenib into tumor tissues further induced extensive tumor apoptosis by inhibiting the tumor angiogenesis. In addition, the therapeutic effect of CC@SR&SF@PP NPs on tumor size was in situ monitored by taking advantage of the bioluminescence of luciferase labeled H22 cells. The results demonstrated that the as-proposed CC@SR&SF@PP NPs showed significant HCC inhibition effect in situ and promised greatly potential a novel platform for gene and drug co-delivery against other types of solid tumors.
Scheme 1.

Schematic representation of CC@SR&SF@PP NPs for co-delivery of Tim-3 siRNA and sorafenib in HCC treatment.
2. Experimental section
2.1. Materials
Analytical grade chloroform, methanol, paraformaldehyde, triethylamine (TEA) and acetonitrile (ACN, HPLC) were all obtained from Sinopharm Chemical Reagent Co. Ltd. (China). Biological materials and agents used in this study were listed as follows: sorafenib (SF, 99%, Aladdin), mPEG5K-PAE10K ((average MW 15000, Xi'an Ruixi Biological Technology Co, Ltd), carboxymethyl chitosan (CMCS, MW = 20,000–50000, carboxylation degrees: 87–90%, Solarbio Life Science, China). D-Luciferin potassium salt, diethyl pyrocarbonate (DEPC) water, Calcein-AM, and Hoechst 33,258 were purchased from Beyotime Biotechnology (China); Lipoamine-2000 was purchased from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). CCK-8 was purchased from Dojindo Co. LTD. All other reagents were of commercial special grade and used without further purification. Tim-3 antibody, CD31 antibody (Abcam, England) and CD8-α antibody (Santa Cruz). All the siRNAs used in this experiment were obtained from GenePharma Co, Ltd (Shanghai, China). PCR oligo primers were synthesized by Takara Biomedical Technology Co, Ltd (Beijing, China).
2.2. Mouse siRNA oligo sequence
Mouse-Tim-3 siRNA 307: sense: 5′-CCAGCAGAUACCAGCUAAATT-3’; antisense: 5′- UUUAGCUGGUAUCUGCUGGTT-3’.
Mouse-Tim-3 siRNA 538: sense: 5′-GAGAAAUGGUUCAGAGACATT-3’; antisense: 5′-UGUCUCUGAACCAUUUCUCTT-3’.
Negative control siRNA: sense: 5′- UUCUCCGAACGUGUCACGUTT -3’; antisense: 5′- ACGUGACACGUUCGGAGAATT -3’.
2.3. Preparation of sorafenib-loaded mPEG5K-PAE10K nanoparticles (SF@PP NPs)
Sorafenib-loaded mPEG5K-PAE10K NPs were prepared by a single emulsification method. Briefly, 1 mg of sorafenib was dissolved in 1 mL of chloroform/methanol (1:1, V/V), and the solution was added dropwise into the stirred solution containing 10 mg of mPEG5K-PAE10K in 10 mL of chloroform/methanol (1:1, V/V). Then, the mixture was gently stirred at room temperature for 1 h, followed by adding dropwise into 10 mL of stirred DEPC water and sonication using a probe-type sonifier (Ningbo Xinzhi Biotechnology Co. LTD, Scientz-950 E) at 275 W for 4 min, with the pulse turned off for 1.5 s after 8 s sonication to prevent heat build-up. After evaporating the organic solvent using a rotary evaporator, the sorafenib-loaded NPs (sorafenib@ mPEG5K-PAE10K, SF@PP) dispersed in DEPC water were centrifuged through ultrafiltration using an Amicon Ultra-15 Centrifugal Filter Unit with 100 kDa MW cut-off (Millipore) at 4000 rpm for 20 min to remove excess sorafenib. The filtrate was collected and excess sorafenib was determined through high performance liquid chromatograph (HPLC). The measurement was performed on a Phenomenex luna C18 4.6 × 250 mm column, using a mobile phase of water-ACN (37:63, v/v) + 0.03% TEA. The flow rate was 1.0 mL/min and the detection wavelength was 265 nm. The drug loading efficiencies (DLE) of sorafenib in the PP NPs were calculated by the following formula:
DLE (wt. %) = (weight of initial drug-weight of residual drug)/weight of mPEG5K-PAE10K.
At last, the suspension was passed through syringe filters (0.45 μm, Millipore) and the as-synthesized SF@PP NPs stock solution was stored in DEPC water at 4 °C for further use.
2.4. Preparation of Tim-3 siRNA and sorafenib co-loaded mPEG5K-PAE10K NPs (SR&SF@PP NPs)
5 μL of 20 μM Tim-3 siRNA (the weight ratio of Tim-3 siRNA 307:538 = 1:1) dissolved in DEPC water was mixed with different volume of 1 mg/mL (concentration of sorafenib) SF@PP NPs. Excess DEPC water was added to make sure the reaction volume was 100 μL. The mixture was vortexed vigorously for 15 s and incubated at room temperature for 30 min. Agarose gel electrophoresis was used to determine the optimal mass ratio of siRNA and SF@PP NPs. The SR&SF@PP NPs stock solution was stored in DEPC water at 4 °C for further use.
2.5. Preparation of CMCS encapsulated SR&SF@PP NPs (CC@SR&SF@PP NPs)
For the preparation of CC@SR&SF@PP NPs, the resulting SR&SF@PP NPs were simply added dropwise into stirred CMCS solution of different concentrations and kept at room temperature for 30 min. Agarose gel electrophoresis was used again to determine the optimal mass ratio of siRNA and CMCS. The CC@SR&SF@PP NPs solution was stored in DEPC water at 4 °C for further use.
2.6. Characterizations
Transmission electron microscopy (TEM) was conducted on an H-7650 TEM (Hitachi, Japan) at 80 kV, after negatively staining the nanoparticles with 2% (wt./vol.) uranyl acetate. UV–Visible (UV–Vis) absorption spectra were recorded on a UV-1900 Spectrophotometer (Shimadzu). Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) was used to measure particle size and zeta potential by dynamic light scattering. High performance liquid chromatography (HPLC) analysis was conducted on a Shimadzu HPLC system, which consisted of a 20 A UV detector, a 20 A pump and a temperature control column oven. Agarose gel was scanned on a BIO-RAD gel imaging system.
2.7. In vitro drug releasing
The release behavior of sorafenib from CC@SR&SF@PP NPs was studied at 37 °C in PBS with pH 7.4 and 6.5. In a typical experiment, 1 mL of CC@SR&SF@PP NPs solution (sorafenib concentration: 0.1 mg/mL) was transferred into a dialysis bag with a MW cut-off of 8–14 kDa. The dialysis bag was tied up with cotton thread and immersed into 13 mL of PBS with pH 7.4 or 6.5 (To increase the dispersion of sorafenib, 1% (v/v) Tween-80 was added into PBS solutions [15,47]) at 37 °C under constant shaking (150 rpm). 500 μL of released medium was taken out at desired time intervals (i.e., 0, 0.5, 1, 1.5, 2, 3, 5, 7, 17, 24, 41, 48, 65 and 72 h) for HPLC measurement in the same way as mentioned above. An equal volume of corresponding fresh medium was replenished. The release experiments were conducted in triplicate. The results presented were the average data.
2.8. Cell culture
Murine hepatic cancer cell line Hepa 1–6 and H22 were purchased from Procell Life Science & Technology Co, Ltd (China). Human umbilical vein endothelial cells (HUVECs) were purchased from BeNa Culture Collection (China). Luciferase or green fluorescent protein (GFP) labeled H22 cells (Luc-H22 or GFP-H22) were constructed by gene transfection with LV-LUC-PURO lentiviruses following the manufacturer's guidelines (HANBIO, China). H22 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin Solution (P/S). HUVECs were cultured in MCDB131 medium supplemented with 20% FBS, growth factor additives and 1% P/S. Hepa 1–6 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS, 1 mM of Sodium Pyruvate and 1% P/S. All the cell lines were cultured at 37 °C with 5% CO2.
2.9. In vitro cellular uptake assay
Hepa 1–6 cells were seeded into 6-well cell culture plates at 1 × 106 cells/well and incubated with fresh culture medium overnight. Cells were washed with PBS buffer for three times and dispersed in 1 mL of opti-MEM culture medium. 10 μL of Cy3-Tim-3-siRNA (20 μM, using Lipo2000 following the manufacturer's guidelines), Cy3-Tim-3-siRNA (20 μM) or CC@SR&SF@PP NPs (siRNA equivalents, incubation at pH 6.5 or 7.4) were added respectively and repeat three times for each group. The culture plates were incubated at 4 °C to avoid nonspecific adsorption for 6 h. Cells were washed three times with PBS before fluorescence imaging (Nikon Ti–S inverted fluorescence microscope, magnification: 100×).
2.10. Tubule formation assay
Tubule formation assay was performed referring to the literature [48]. Briefly, 50 μL per well of thawed Matrigel Matrix was added into precooled 96-well culture plates. The plates were then incubated at 37 °C for 30 min to gel. 2 × 104 cells/well of HUVECs cultured in MCDB131 medium (pH 6.5) containing 5 μg/mL of sorafenib, CC@SR&SF@PP NPs or equivalent CC@PP NPs were added to the matrigel-coated 96-well plates. The plates were incubated at 37 °C, 5% CO2 for 20 h. Then, the culture medium was removed, and the cells were washed for three times with PBS. 100 μL of 4 μg/mL Calcein-AM solution was added, followed by incubating for another 20 min. Then, dye solution was removed and the cells were washed for three times with PBS again. The cells were fixed with 4% paraformaldehyde at 4 °C. Micro-tubes formed were photographed using the Cytation 5 of BioTek.
2.11. In vitro Tim-3 interference assay
H22 cells were seeded into 6-well cell culture plates at 5 × 105 cells/well and incubated with fresh culture medium for 24 h. Culture medium was removed trough centrifuge. Cells were washed with PBS and dispersed in 1 mL of opti-MEM culture medium. 5 μL of Cy3-Tim-3-siRNA (20 μM, using Lipo2000 following the manufacturer's guidelines), CC@PP NPs or CC@SR&SF@PP NPs (siRNA equivalents, incubation at pH 6.5) were added respectively. After 6 h, 1.5 mL of fresh culture medium was replenished. Cells were collected at 72 h, and protein was extracted respectively for western blot analysis and semiquantitative analysis (compared with NC group) of protein band was conducted with Image J software.
2.12. In vitro cytotoxicity assay
The cytotoxicity of different groups of nanoparticles was measured through cell counting kit-8 (CCK-8) assay. Briefly, H22 cells were seeded into 96-well cell culture plates at 5 × 103 cells/well and incubated with fresh culture medium containing various PP NPs or siRNA & sorafenib, respectively, at siRNA concentrations at 0, 0.125, 0.25, 0.5, 0.75, 1.25 and 2.5 μg/mL (n = 5), at 37 °C under 5% CO2 for 72 h 10 μL of CCK-8 reagent was added, followed by another 2 h’ incubation. Absorbance at 450 nm of each well was measured using the Multilable Reader VARIOSKAN FLASH (Thermo Fisher). The cell growth viability was calculated through the formula shown below: Viability (%) = (mean of absorbance value of treatment group/mean absorbance value of control) × 100. In addition, cell growth viability-sorafenib concentration curves of every group were plotted respectively. IC50 of each group was calculated by the fitted curves and the final IC50s were presented as mean ± SD.
2.13. The orthotopic tumor model of hepatocellular carcinoma
Normal Balb/c mice were supplied by the Medical Animal Test Center of the Xi'an Jiaotong University and all the experiments were performed with the Guidelines for the Care and Use of Research Animals. The orthotopic tumor model of hepatocellular carcinoma was established as follows: Male Balb/c mice (5–6 week) were anesthetized by inhaling isoflurane using a small animal anesthesia machine. After skin preparation and disinfection, the abdomen was opened in the middle, and the middle lobe of the liver was pulled out with a cotton swab. 1 × 106 of Luc-H22 cells dissolved in 50 μL of PBS were injected into the liver using a 1 mL syringe. The middle lobe of the liver was inserted gently into the abdominal cavity, the abdomen was sutured layer by layer, and the incision was disinfected by iodophor. The mice were put back to the original feeding environment for ∼1 week after anesthesia and consciousness.
2.14. In vivo tumor uptake assay
For in vivo fluorescence imaging, tumor-bearing BALB/c mice were injected intravenously with a portion of 100 μL of Cy5-Tim-3 siRNA or CC@SR&SF@PP NPs (siRNA concentration of ∼0.5 mg/mL) respectively. Fluorescence Images were obtained 6 h post injection using the Xenogen IVIS SPECTRUM small animal imaging system. The mice were anesthetized by 5% chloral hydrate and mice's organs were fully exposed before imaging. After imaging, major organs including lung, heart, kidneys, liver and spleen were harvested and imaged on the same imaging system.
To analyze the distribution of drugs in mice tumor, tumor tissues were embedded with optimal cutting temperature compound (OCT). Tumor tissue slices were obtained on the RWD Minux FS800 and adhered to the slides. Then, the slices were fixed in 4% paraformaldehyde for 15 min. Next, 10 μg/mL Hochest 33,258 solution was used for nuclear staining for 10 min. The slices were washed again with PBS, protected by anti-fade PVP mounting medium. Fluorescence images were acquired on the multiphoton confocal microscopy Leica TCS SP8 DIVE system.
2.15. In vivo anticancer assay
The tumor-bearing mice were divided randomly into six groups (n = 5 for each group), including PBS, CC@PP, SR&SF, CC@SR@PP, CC@NSR&SF@PP and CC@SR&SF@PP NPs (Tim-3 siRNA 0.5 mg/mL, sorafenib: 2.0 mg/mL, PP: 20 mg/mL, CMCS: 0.5 mg/mL). 100 μL of different materials were injected intravenously at day 1, 4, 7, 10 and 13 with dosage of 2.5 mg/kg siRNA and 10 mg/kg sorafenib. One day after drug injection, after intraperitoneal injection with 150 μL of 15 mg/mL D-Luciferin potassium salt solution, tumor sizes were measured through the intensity of bioluminescence by the Xenogen IVIS SPECTRUM small animal imaging system (USA). Mice were sacrificed three days after the final drug injection. The whole livers were sampled and weighted. 20 mg of fresh tumor tissues were collected for western blot analysis and remaining livers were fixed in 4% paraformaldehyde for further use. Semiquantitative analysis (compared with PBS group) of protein band in western blot analysis was conducted with Image J software.
2.16. Histology and immunohistochemistry
The immunohistochemistry analysis of tumor vascular density, Tim-3 and CD8 were carried out by Servicebio (Wuhan, China) after obtaining the tumor sample. TUNEL assay was done completely based on the manufacturer's protocol of the Cell Death Detection Kit (Roche, Penzberg, Germany) by Freethink (Nanjing, China). The positive area and tumor vascularity were determined in 10 high-power fields per slide ( × 400) using Image-Pro Plus analytical software.
2.17. Statistical analysis
Data were expressed as means ± standard deviation. The statistical difference between two groups was determined using a one-way ANOVA (Origin 2022). p-values less than 0.05 were considered statistically significant. (∗: p < 0.05, ∗∗: p < 0.001, ∗∗∗: p < 0.001.)
3. Results and discussion
3.1. Preparation, characterization and drug release of the CC@SR&SF@PP NPs
To verify the successful preparation, the above-obtained CC@SR&SF@PP NPs were systematically characterized. Transmission electronic microscopy (TEM) image in Fig. 1(a) illustrated the good dispersibility and uniform spherical appearance of the nanoparticles with a diameter of 50.49 ± 5.34 nm. By monitoring the entire synthetic process using the dynamic light scattering (DLS) measurement (Fig. S3), we further demonstrated the successful formation of CC@SR&SF@PP with a hydrodynamic diameter of ∼132 nm. The negative charge (−3.39 ± 0.13 mV) was due to the presence of CMCS, an amphoteric polysaccharide which is negatively charged in the physiological pH and positively charged in the acidic environment of the tumor. Before entering the tumor tissue, the absorbed CMCS on the surface of CC@SR&SF@PP NPs protected the condensed siRNA and reduced the cationic toxicity of NPs. The optimal mass ratio of mPEG5K-PAE10K/sorafenib/Tim-3 siRNA/CMCS was determined to be 40:4:1:1 by using agarose gel electrophoresis and high performance liquid chromatography (HPLC) in Fig. S2. We also investigated the photo-physical properties of CC@SR&SF@PP NPs. Compared with absorbance spectra of equivalent of SF@PP NPs, free Tim-3 siRNA and PP NPs, the peak height, peak area and peak shape of CC@SR&SF@PP NPs showed the characteristics of superposition peak (Fig. 1(b)). Therefore, we concluded that both sorafenib and siRNA were loaded into PP NPs. The typical superposition absorbance peak of CC@SR&SF@PP NPs at ∼269 nm. We also demonstrated that the encapsulated drugs could be continuously released under acidic environment of tumor tissue. As shown in Fig. 1(d), the pH-dependent drug release profile from the CC@SR&SF@PP NPs was investigated by dialysis and HPLC. Under acidic physiological conditions (PBS, pH = 6.5), the release of sorafenib from CC@SR&SF@PP NPs became fast and maintained a steady release rate with increasing incubation time and more than 90% of sorafenib was released after incubation for 72 h. However, when incubated under normal physiological conditions (PBS, pH = 7.4), CC@SR&SF@PP NPs showed a relatively low release rate and even ceased releasing after 48 h, resulting in the maximum drug release rate of less than 40%. These results indicated that CC@SR&SF@PP NPs have great potential to be an ideal tumor microenvironment-triggered drug delivery system for tumor treatment. What's more, the stability analysis results showed that the CC@SR&SF@PP NPs maintained high colloidal stability even after 30 days of storage in DNA/RNA free sterile water (Fig. 1(c)).
Fig. 1.

(a) TEM image of CC@SR&SF@PP NPs (50.49 ± 5.34 nm). (b) UV–Vis absorbance spectra of CC@SR&SF@PP NPs and equivalent of SF@PP NPs, Tim-3 siRNA, CMCS, PP NPs and sorafenib. The mass ratio of mPEG5K-PAE10 K/sorafenib/Tim-3 siRNA/CMCS in CC@SR&SF@PP NPs was 40:4:1:1 and the concentration of sorafenib was 1.0 μg/mL for UV–Vis absorbance analysis. (c) The hydrodynamic diameters of CC@SR&SF@PP NPs at day 0 (∼132 nm) and day 30 (∼153 nm), stored in DNA/RNA free sterile water at 4 °C. (d) In vitro sorafenib release curves from CC@SR&SF@PP NPs over time in PBS buffer at pH 7.4 and pH 6.5.
3.2. Evaluation of in vitro efficacy and safety of CC@SR&SF@PP NPs
3.2.1. CC@SR&SF@PP NPs enhanced the cellular uptake of Tim-3 siRNA in acidic environment
To investigate the bioactivities of various components of CC@SR&SF@PP NPs, we then performed a series of in vitro experiments. To evaluate the influence of CMCS charge reversal on cellular uptake of CC@SR&SF@PP NPs, Hepa 1–6 cells were incubated for 6 h in a culture medium containing Cy3-Tim-3 siRNA or CC@SR&SF@PP NPs, at pH 7.4 or 6.5. As shown in Fig. 2(a), the Cy3-Tim-3 siRNA uptake intensity of Hepa 1–6 cells treated by CC@SR&SF@PP NPs at pH 6.5 was comparable to that treated by siRNA&Lipo2000 complex, whereas the cells treated by Cy3-Tim-3 siRNA alone or CC@SR&SF@PP NPs at pH 7.4 showed faint fluorescence. These results were mainly due to the charge reversal of CMCS in the acidic environment. The pH sensitive CMCS shell became positively charged at pH 6.5, and fell off from the surface of CC@SR&SF@PP NPs, followed by the exposure of SR&SF@PP NPs to cells and the cellular uptake. Encouraged by good results, we further investigated that whether the system could knock down the expression of Tim-3 at the cellular level. siRNA&Lipo2000 complex, CC@SR&SF@PP NPs or CC@PP NPs were used to carry out siRNA transfection experiment. Western blot analysis in Fig. 2(d and e) illustrated that the knockdown rate of Tim-3 by CC@SR&SF@PP NPs (Relative Tim-3 expression compared with NC groups: 0.191 ± 0.010) was comparable to that of siRNA&Lipo2000 complex (Relative Tim-3 expression compared with NC groups: 0.139 ± 0.013), whereas CC@PP NPs (Relative Tim-3 expression compared with NC groups: 0.913 ± 0.047) showed weak effects. These results indicated that pH-sensitive CC@SR&SF@PP NPs showed great potential as an ideal platform for enhancing the delivery of siRNA into tumor cells in the acidic environment of the tumor.
Fig. 2.

(a) In vitro cellular uptake assay of Hepa 1–6 cells treated with siRNA or CC@SR&SF@PP NPs in different pH conditions. (b) The tube images of HUVECs cells induced under 37 °C for 20 h with 5 μg/mL of PBS, sorafenib, CC@SR&SF@PP NPs or equivalent CC@PP NPs. (c) Concentration-dependent cell viability of H22 cells with different treatments for 72 h (n = 5) (d, e) In vitro Tim-3 Interference Assay. Western blot and quantitative analysis (compared with NC group) of Tim-3 extracted form H22 cells treated with different nanoparticles or siRNA. (∗: p < 0.05, ∗∗: p < 0.001, ∗∗∗: p < 0.001.)
3.2.2. CC@SR&SF@PP NPs enhanced the cellular uptake of sorafenib
Sorafenib is a representative tumor vascular-targeted therapeutic drug for HCC and displays the broad anticancer spectrum in clinical practice. To assess the anti-angiogenic effect of sorafenib loaded onto the surface of PP NPs, we performed a tubule formation assay by culturing HUVECs with different treatments. As shown in Fig. 2(b), under the stimulation of serum, HUVEC cells formed a complete network structure. At sorafenib concentration of 5 μg/mL, CC@SR&SF@PP NPs showed a little weak effect to inhibit tubule formation, compared with free sorafenib. This result attributed to that, the incubation time of this experiment was 20 h and only ∼40% of sorafenib was released from the nanoparticles. Moreover, compared with negative control, ∼80% of tubules were still suppressed even with less amount of released sorafenib, whereas CC@PP NPs showed a negligible or weak influence.
3.2.3. CC@SR&SF@PP NPs inhibited the proliferation of H22 cells in vitro
To evaluate the cytotoxicity of CC@SR&SF@PP NPs, a standard CCK-8 assay was executed in vitro (Fig. 2(c)). CC@SR&SF@PP NPs showed good cancer-killing effect with much lower half-maximal inhibitory concentration (IC50: 0.50 ± 0.07 μg/mL) in comparison to that of SR&SF (1.12 ± 0.28 μg/mL), CC@NSR&SF@PP NPs (2.91 ± 0.39 μg/mL) and CC@SR@PP NPs (7.59 ± 1.24 μg/mL). These results suggested the intact retention of the anti-Tim-3 expression, anti-angiogenesis ability and cancer cell lethality of CC@SR&SF@PP NPs, which showed great potential for tumor growth inhibition in vivo.
3.3. In vivo accurate delivery and enhanced siRNA uptake of CC@SR&SF@PP NPs
Next, we further investigated the capability of CC@SR&SF@PP NPs to enhance the delivery of loaded Cy5-Tim-3 siRNA and sorafenib to tumor tissues. As shown in Fig. 3(a), there was strong fluorescence intensity throughout the abdominal cavity treated with Cy5-Tim-3 siRNA for 6 h, indicating the rapid clearance of naked siRNA in vivo. In contrast, CC@SR&SF@PP NPs containing the same amount of Cy5-Tim-3 siRNA showed clear fluorescence solely on the liver and even more clusters were observed in the tumor sites of liver, demonstrating the in vivo stability and successfully enhanced delivery of siRNA to tumor regions. The ex vivo images of various tissues and organs in Fig. 3(b) further confirmed the higher accumulation of nanoparticles in tumors tissues on the liver, whereas naked siRNA was metabolized rapidly through the liver and kidneys. To prove the ability of accurate cellular delivery and release of CC@SR&SF@PP NPs at tumor regions, tumor tissue slices were obtained following the white dotted line in Fig. 3(b). As shown in Fig. 3(c), an enhanced Cy5 fluorescence signal was observed in the tumor site of the slices treated by CC@SR&SF@PP NPs, mainly distributed at the periphery of the liver lobe, corresponding to the location of tumor growth in Fig. 3(b). This result attributed to the excellent ability of CC@SR&SF@PP NPs releasing drugs accurately in the slightly acidic microenvironment of tumor tissues. In contrast, due to the non-specificity of naked siRNA, tumor slices treated by naked siRNA showed uniform fluorescence distribution. Both the live images and tumor slices images illustrated that CC@SR&SF@PP NPs could be accurately delivered and triggered by the acidic microenvironment of tumor tissues to release loaded siRNA to tumor cells. Meanwhile, as shown in Fig. S4, the in vivo cytotoxicity assay indicated that no observable tissue damage, necrosis, or inflammation was observed in slices treated by CC@SR&SF@PP NPs, either at the short term of 1 day or the long term of 7 days, which fully demonstrated that the CC@SR&SF@PP NPs were relatively safe for use in vivo.
Fig. 3.

In vivo tumor uptake assay. (a) Bioluminescence, brightfield and Cy5 fluorescence images of the tumors recruited obtained 6 h after the mice were injected with CC@SR&SF@PP NPs (up) or free Tim-3 siRNA (down). (b) Ex vivo merge images of tissues and organs obtained 6 h after the mice were injected with CC@SR&SF@PP (up) or free Tim-3 siRNA (down). The white dotted line shows the direction and approximate position of the slice. (c) Tumor slices images at 6 h after intravenous injection of different formulas.
3.4. In vivo anti-tumor efficacy of CC@SR&SF@PP NPs in a H22 orthotopic tumor model
3.4.1. CC@SR&SF@PP NPs inhibited the growth of tumors in vivo
Encouraged by the promising in vitro and in vivo results obtained above, we assessed the in vivo antitumor efficacy of CC@SR&SF@PP NPs in a mouse hepatoma 22 (H22) orthotopic tumor model. On the basis of in vitro experiment and in vivo tissue uptake property, CC@SR&SF@PP NPs were administered to the mice for five times every three days through tail vein injection, with PBS, CC@PP NPs, CC@NSR&SF@PP NPs, CC@SR@PP NPs and SR&SF groups as control. The administration dosage was 2.5 mg/kg for Tim-3 siRNA and 10 mg/kg for sorafenib. Due to the high sensitivity resolution of the bioluminescence, we monitored the tumor growth throughout the experimental process. As shown in Fig. 4(a), both CC@SR@PP NPs and CC@NSR&SF@PP NPs showed an unsatisfying effect on inhibiting tumor growth. Combination of therapy with Tim-3 siRNA and sorafenib based on free diffusion (SR&SF) could partially inhibit tumor growth. However, a nearly stagnant growth of tumors was observed after continuous injection of CC@SR&SF@PP NPs, suggesting the synergistically enhanced therapeutic effect of combing the Tim-3 siRNA for gene-therapy and sorafenib for tumor vascular-targeted therapy in vivo. In contrast, gradual increase of tumor volume was observed in the control groups and CC@PP NPs groups. By measuring the bioluminescence intensity of tumor tissues, the tumor growth curves were also achieved (Fig. 4(b)). The results further proved that only by combining the synergistically enhanced therapeutic effect of gene therapy and tumor vascular-targeted therapy could we achieve the remarkable tumor growth inhibition. Finally, the mice were sacrificed at day 16 after first injection and the whole livers were sampled. As expected, the group treated with CC@SR&SF@PP NPs exhibited the lowest level of tumor growth (Fig. 4(c)).
Fig. 4.

(a) In vivo real-time visualization of tumors in the H22 orthotopic tumor-bearing mouse. Bioluminescence images were acquired by intraperitoneal injection with 150 μL of 15 mg/mL D-Luciferin potassium salt solution on day 2, 5, 8, 11 and 14 (one day after drug injection) respectively. (b) Tumor volume growth curves of mice after various treatments (n = 5). Tumor sizes were measured through the intensity of bioluminescence by the Xenogen IVIS SPECTRUM small animal imaging system. (c) Representative photos of livers collected from mice at day 16 after different treatments.
3.4.2. CC@SR&SF@PP NPs enhanced the tumor cellular uptake of Tim-3 siRNA in acidic environment in vivo and the knock down of Tim-3
Next, a series of immunohistochemical examinations of the dissociated tumor tissues after continuous administration of CC@SR&SF@PP NPs and the controls were performed. As shown in Fig. 5(a, b), benefiting from the protective effect of CMCS shell and the pH-trigged delivery system, both CC@SR&SF@PP NPs and CC@SR@PP NPs could promote the delivery of the Tim-3 siRNA into tumor tissues and enhance the transfer of Tim-3 siRNA into tumor cells. The expression of Tim-3 in tumor tissues were reduced significantly (Relative Tim-3 expression compared with PBS group: CC@SR&SF@PP NPs: 0.083 ± 0.002; CC@SR@PP NPs: 0.1413 ± 0.021). Due to the instability, non-specific adsorption and rapid clearance by liver and kidney of the naked siRNA in vivo, Tim-3 siRNA and sorafenib based on free diffusion (Relative Tim-3 expression compared with PBS group: SR&SF: 0.461 ± 0.061) could only partially inhibit the expression of Tim-3 in vivo. In contrast, CC@PP (Relative Tim-3 expression compared with PBS group: 0.862 ± 0.043) and CC@NSR&SF@PP (Relative Tim-3 expression compared with PBS group: 0.950 ± 0.114) showed negligible effects. This effect was verified by immunohistochemical examination of the dissociated tumor tissue slices. As shown in Fig. 5 (c, d), the brown areas indicated the expression of Tim-3 in tumor tissues after treatment by different groups. By quantitative analysis of the brown area, we found that, compared with the negative control of PBS and CC@PP groups, ∼80% of the expression of Tim-3 was knocked down after being treated by free Tim-3 siRNA and sorafenib groups. However, the percentage of inhibition raised to more than 95% when it came to CC@SR&SF@PP NPs or CC@SR@PP NPs groups. This result verified effectively the significantly enhanced siRNA delivery and transfection ability of CC@SR&SF@PP NPs, indicating that this pH-sensitive CC@SR&SF@PP NPs could be an ideal platform for enhancing the delivery of siRNA into tumor cells in the acidic environment of the tumor in vivo and then enhanced the tumor inhibition.
Fig. 5.

(a, b) Western blot and quantitative analysis of Tim-3 extracted form tumor tissues treated with different nanoparticles or SR&SF. (c, d) Immunohistochemical Tim-3 staining and quantitative analysis (compared with PBS group) of the dissociated tumor tissues after different treatments. (∗: p < 0.05, ∗∗: p < 0.001, ∗∗∗: p < 0.001.) (e) Immunohistochemical CD8 staining of the dissociated tumor tissues after treated by PBS, SR&SF or CC@SR&SF@PP NPs.
3.4.3. CC@SR&SF@PP NPs induced the immune response and enhanced the recruitment of cytotoxic T cells to kill tumor cells in vivo
Considering that Tim-3 was also a novel immune checkpoint molecule, we also tested the expression of CD8 in tumor tissues to prove that whether knockdown of Tim-3 could also enhance the immune response of mice. As shown in Fig. 5(e), as expected, much more CD8 were detected in tumor tissues treated by CC@SR&SF@PP NPs with red fluorescence densely distributed throughout the tumor tissue, indicating that a large number of cytotoxic T cells were recruited to kill the tumor cells. There was also a small amount of CD8 detected in SR&SF treated groups. In contrast, CD8 expression was rare in PBS treated tumor tissues. These results indicated that enhanced Tim-3 siRNA transfection by CC@SR&SF@PP NPs could greatly inhibit the expression of the target gene, induce the immune response and enhance the recruitment of cytotoxic T cells to kill tumor cells.
3.4.4. CC@SR&SF@PP NPs promoted the delivery of sorafenib and induced the enhanced inhibition of tumor vasculature
What’ more, we also measured the density of blood vessels in tumor tissue slices. As demonstrated in CD31 immune staining in Fig. 6(a), there was a net organization of small and large vessels with an apparent lumen structure by CC@SR@PP NPs, CC@PP or PBS groups. Some middle-sized and small vessels were still observed in tumor slices treated by SR&SF group. In contrast, both CC@SR&SF@PP NPs and CC@NSR&SF@PP NPs treated groups formed very small undeveloped vessels, due to the encapsulation of sorafenib into PP NPs, confirming again the importance of sorafenib delivery and pH-triggered release in tumor therapy. Quantitative analysis of pathologic vascularity revealed a 95% reduction in tumor vascular density after treated with CC@SR&SF@PP NPs. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining further confirmed more regions of apoptotic cells in the CC@SR&SF@PP NPs group than in the control groups (Fig. 6(b)). These result indicated the synergistically enhanced in vivo tumor growth suppression of this combined gene therapy and tumor vascular-targeted therapy strategy.
Fig. 6.

(a) Immunohistochemical blood vessel staining and quantitative analysis of tumor tissue after different treatments. Red arrows indicate capillaries. (b) TUNEL staining of the deep region of the tumor after treatment of PBS or CC@SR&SF@PP NPs, respectively. Green region indicates apoptotic tumor cells.
Inspired by the good therapeutic effect of PD-1 or PDL-1 combined with sorafenib in treatment of HCC reported in literature, we focused on the novel immune checkpoint molecule Tim-3 [23,24]. Tim-3 had been proved overexpression in both HCC tumor tissues and cell lines and knocking down the expression of Tim-3 had been proved to have anti-tumor efficiency. However, limited by the poor targeting, low specificity, low transfection efficiency and the potential virulence of commonly used adenovirus and lentivirus gene vectors [36,39], the clinical application of Tim-3 remains challenging. In this paper, positively charged SF@PP NPs provided an excellent template for negatively charged Tim-3 siRNA to condense on its surface through electrostatic interaction. CMCS, being an amphoteric polysaccharide, possesses pH-sensitive property, since it bears both acidic and basic groups. CMCS shows negative charge in the physiological pH and positive charge in the acidic environment of the tumor. Therefore, the following absorbed CMCS protected the condensed Tim-3 siRNA in physiological environment and reduced the toxicity of cations. In acid environment of tumor tissues, CMCS underwent charge reversal process, released the protected Tim-3 siRNA to achieve the accurate release and enhanced enrichment of siRNA in tumor tissues. In general, nanoparticles with high surface charge density are more easily to be recognized and quickly cleared by the reticuloendothelial system (RES) in vivo. Negative or nearly neutral charged nanoparticles with particle size less than 300 nm can avoid the nonspecific capture of RES [37]. Polysaccharide modification on the surface of nanoparticles can give the carrier the ability of “invisibility” and prolong the blood circulation time of the carrier, so that the carrier has more chances to enter the tumor tissues through enhanced permeability and retention (EPR) effect [44]. Compared with free siRNA and free sorafenib, the final obtained pH-sensitive CC@SR&SF@PP NPs, with the hydrodynamic diameter of ∼132 nm and negative charge of ∼ -3.39 mV, enhanced the delivery of Tim-3 siRNA into tumor tissues and improved gene transfection greatly inhibited the expression of the target gene. Meanwhile, CC@SR&SF@PP induced the immune response and enhanced the recruitment of cytotoxic T cells to kill tumor cells. Then, increased delivery of sorafenib into tumor tissues further induced extensive tumor apoptosis by inhibiting the tumor angiogenesis and the enhanced recruitment of cytotoxic T cells by knocking down of Tim-3 expression could promote the anti-HCC efficiency of sorafenib.
4. Conclusion
In summary, a novel tumor micro-environmental triggered drug-eluting nanoparticle (CC@SR&SF@PP) for simultaneous delivery of Tim-3 siRNA and sorafenib was developed by a single emulsification method. In this nanoparticle, being a pH-sensitive amphoteric polysaccharide absorbed on the surface of the nanoparticle, CMCS protected the condensed siRNA and reduced cationic toxicity of CC@SR&SF@PP NPs. After entering the tumor tissues, CMCS underwent charge reversal and then released the loaded siRNA to realize the precise enhanced release of siRNA in tumor tissues. Enhanced therapeutic siRNA delivery into solid tumors directly inhibited tumor cells and more cytotoxic T cells were recruited to further kill tumor cells. The following tumor microenvironment triggered sorafenib release from SF@PP NPs greatly inhibited the tumor angiogenesis and further enhanced the inhibitory effect on tumor growth. We expect that such simultaneous gene therapy and tumor vascular-targeted therapy strategy may also be applicable for other types of tumors, and will have great potential in future clinical applications.
Credit author statement
Rongqian Wu and Chenghua Song contributed to the design of the study, data analysis and the writing of the manuscript. Chenghua Song, Jia Zhang, Ruichao Wen, Qingshan Li and Jiaxuan Zhou made substantial contributions to data acquisition. Zheng Wu, Xiaoli Liu and Yi Lv provided technical guidance.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Acknowledges
This work was financially supported by The National Natural Science Foundation of China (Grant No. 81701814), Fundamental Research Funds for the Central Universities of Xi'an Jiaotong University (Grant No. xjj2018270) and The Innovation Capacity Support Plan of Shaanxi Province (Grant No. 2020TD-040).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mtbio.2022.100350.
Contributor Information
Chenghua Song, Email: sch871224@xjtu.edu.cn.
Rongqian Wu, Email: rwu001@mail.xjtu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Kelley R.K., Greten T.F. Hepatocellular carcinoma - origins and outcomes. N. Engl. J. Med. 2021;385:280–282. doi: 10.1056/NEJMcibr2106594. [DOI] [PubMed] [Google Scholar]
- 2.Nhlane R., Kreuels B., Mallewa J., Chetcuti K., Gordon M.A., Stockdale A.J. Late presentation of hepatocellular carcinoma highlights the need for a public health programme to eliminate hepatitis B. Lancet. 2021;398 doi: 10.1016/S0140-6736(21)02138-3. 2288-2288. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X.Y., Wang Z., Tang W.X.F., Wang X.Y., Liu R., Bao H., Chen X., Wei Y.L., Wu S.Y., Bao H.R., Wu X., Shao Y., Fan J., Zhou J. Ultrasensitive and affordable assay for early detection of primary liver cancer using plasma cell-free DNA fragmentomics. Hepatology. 2022 doi: 10.1002/hep.32308. [DOI] [PubMed] [Google Scholar]
- 4.Gunasekaran G., Bekki Y., Lourdusamy V., Schwartz M. Surgical treatments of hepatobiliary cancers. Hepatology. 2021;73:128–136. doi: 10.1002/hep.31325. [DOI] [PubMed] [Google Scholar]
- 5.Heimbach J.K., Kulik L.M., Finn R.S., Sirlin C.B., Abecassis M.M., Roberts L.R., Zhu A.X., Murad M.H., Marrero J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67:358–380. doi: 10.1002/hep.29086. [DOI] [PubMed] [Google Scholar]
- 6.Forner A., Reig M., Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 7.Marrero J.A., Kulik L.M., Sirlin C.B., Zhu A.X., Finn R.S., Abecassis M.M., Roberts L.R., Heimbach J.K. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American association for the study of liver diseases. Hepatology. 2018;68:723–750. doi: 10.1002/hep.29913. [DOI] [PubMed] [Google Scholar]
- 8.Saffo S., Taddei T.H. Systemic management for advanced hepatocellular carcinoma: a review of the molecular pathways of carcinogenesis, current and emerging therapies, and novel treatment strategies. Dig. Dis. Sci. 2019;64:1016–1029. doi: 10.1007/s10620-019-05582-x. [DOI] [PubMed] [Google Scholar]
- 9.Pinyol R., Montal R., Bassaganyas L., Sia D., Takayama T., Chau G.Y., Mazzaferro V., Roayaie S., Lee H.C., Kokudo N., Zhang Z., Torrecilla S., Moeini A., Rodriguez-Carunchio L., Gane E., Verslype C., Croitoru A.E., Cillo U., de la Mata M., Lupo L., Strasser S., Park J.W., Camps J., Sole M., Thung S.N., Villanueva A., Pena C., Meinhardt G., Bruix J., Llovet J.M. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut. 2019;68:1065–1075. doi: 10.1136/gutjnl-2018-316408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koeberle D., Dufour J.F., Demeter G., Li Q., Ribi K., Samaras P., Saletti P., Roth A.D., Horber D., Buehlmann M., Wagner A.D., Montemurro M., Lakatos G., Feilchenfeldt J., Peck-Radosavljevic M., Rauch D., Tschanz B., Bodoky G., Swiss Group for Clinical Cancer R. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29) Ann. Oncol. 2016;27:856–861. doi: 10.1093/annonc/mdw054. [DOI] [PubMed] [Google Scholar]
- 11.Bondi M.L., Scala A., Sortino G., Amore E., Botto C., Azzolina A., Balasus D., Cervello M., Mazzaglia A. Nanoassemblies based on supramolecular complexes of nonionic amphiphilic cyclodextrin and sorafenib as effective weapons to kill human HCC cells. Biomacromolecules. 2015;16:3784–3791. doi: 10.1021/acs.biomac.5b01082. [DOI] [PubMed] [Google Scholar]
- 12.Calvisi D., Eferl R. CDK4/6 inhibition and sorafenib: a menage a deux in HCC therapy? Gut. 2017;66:1179–1180. doi: 10.1136/gutjnl-2016-313547. [DOI] [PubMed] [Google Scholar]
- 13.Younis M.A., Khalil I.A., Elewa Y.H.A., Kon Y., Harashima H. Ultra-small lipid nanoparticles encapsulating sorafenib and midkine-siRNA selectively-eradicate sorafenib-resistant hepatocellular carcinoma in vivo. J. Contr. Release. 2021;331:335–349. doi: 10.1016/j.jconrel.2021.01.021. [DOI] [PubMed] [Google Scholar]
- 14.So Y.J., Frentzas S., Segelov E. Is there a place for Ramucirumab after Sorafenib in patients with advanced HCC? Hepatobiliary Surg. Nutr. 2019;8:546–548. doi: 10.21037/hbsn.2019.04.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thapa R.K., Choi J.Y., Poudel B.K., Hiep T.T., Pathak S., Gupta B., Choi H.G., Yong C.S., Kim J.O. Multilayer-coated liquid crystalline nanoparticles for effective sorafenib delivery to hepatocellular carcinoma. ACS Appl. Mater. Interfaces. 2015;7:20360–20368. doi: 10.1021/acsami.5b06203. [DOI] [PubMed] [Google Scholar]
- 16.Kong F.H., Ye Q.F., Miao X.Y., Liu X., Huang S.Q., Xiong L., Wen Y., Zhang Z.J. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics. 2021;11:5464–5490. doi: 10.7150/thno.54822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Jong Y.P., Herzog R.W. Liver gene therapy and hepatocellular carcinoma: a complex web. Mol. Ther. : the journal of the American Society of Gene Therapy. 2021;29:1353–1354. doi: 10.1016/j.ymthe.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogorad R.L., Yin H., Zeigerer A., Nonaka H., Ruda V.M., Zerial M., Anderson D.G., Koteliansky V. Nanoparticle-formulated siRNA targeting integrins inhibits hepatocellular carcinoma progression in mice. Nat. Commun. 2014;5:3869. doi: 10.1038/ncomms4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelici B., Shen L., Schreiber J., Abraham A., Benenson Y. An AAV gene therapy computes over multiple cellular inputs to enable precise targeting of multifocal hepatocellular carcinoma in mice. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abh4456. [DOI] [PubMed] [Google Scholar]
- 20.Graepler F., Verbeek B., Graeter T., Smirnow I., Kong H.L., Schuppan D., Bauer M., Vonthein R., Gregor M., Lauer U.M. Combined endostatin/sFlt-1 antiangiogenic gene therapy is highly effective in a rat model of HCC. Hepatology. 2005;41:879–886. doi: 10.1002/hep.20613. [DOI] [PubMed] [Google Scholar]
- 21.Guo Y., Wu Z., Shen S., Guo R., Wang J., Wang W., Zhao K., Kuang M., Shuai X. Nanomedicines reveal how PBOV1 promotes hepatocellular carcinoma for effective gene therapy. Nat. Commun. 2018;9:3430. doi: 10.1038/s41467-018-05764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrone F., Craparo E.F., Cemazar M., Kamensek U., Drago S.E., Dapas B., Scaggiante B., Zanconati F., Bonazza D., Grassi M., Truong N., Pozzato G., Farra R., Cavallaro G., Grassi G. Targeted delivery of siRNAs against hepatocellular carcinoma-related genes by a galactosylated polyaspartamide copolymer. J. Contr. Release. 2021;330:1132–1151. doi: 10.1016/j.jconrel.2020.11.020. [DOI] [PubMed] [Google Scholar]
- 23.Tan Y., Xu Q., Wu Z., Zhang W., Li B., Zhang B., Xu X., Zhang B., Yan K., Song J., Lv T., Yang J., Jiang L., Shi Y., Yang J., Yan L. Overexpression of PD-L1 is an independent predictor for recurrence in HCC patients who receive sorafenib treatment after surgical resection. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.783335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudo M. Scientific rationale for combined immunotherapy with PD-1/PD-L1 antibodies and VEGF inhibitors in advanced hepatocellular carcinoma. Cancers. 2020;12 doi: 10.3390/cancers12051089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu P.Y., Chan S.H. Cure the incurable? Recent breakthroughs in immune checkpoint blockade for hepatocellular carcinoma. Cancers. 2021;13 doi: 10.3390/cancers13215295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyhl-Polk A., Mikkelsen M.K., Ladekarl M., Nielsen D.L. Clinical trials of immune checkpoint inhibitors in hepatocellular carcinoma. J. Clin. Med. 2021;10 doi: 10.3390/jcm10122662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eugen K. Current treatment options for hepatocellular carcinoma. Klin. Onkol. 2020;33:20–25. doi: 10.14735/amko20203S20. [DOI] [PubMed] [Google Scholar]
- 28.Romero D. Immunotherapy: PD-1 says goodbye, TIM-3 says hello. Nat. Rev. Clin. Oncol. 2016;13:202–203. doi: 10.1038/nrclinonc.2016.40. [DOI] [PubMed] [Google Scholar]
- 29.Liu F.F., Liu Y.N., Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 2018;11 doi: 10.1186/s13045-018-0667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakemi M.G., Jafarinia M., Azizi M., Rezaeepoor M., Isayev O., Bazhin A.V. The role of TIM-3 in hepatocellular carcinoma: a promising target for immunotherapy? Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H., Song Y., Yang H., Liu Z., Gao L., Liang X., Ma C. Tumor cell-intrinsic Tim-3 promotes liver cancer via NF-kappaB/IL-6/STAT3 axis. Oncogene. 2018;37:2456–2468. doi: 10.1038/s41388-018-0140-4. [DOI] [PubMed] [Google Scholar]
- 32.Li H., Wu K., Tao K.X., Chen L.B., Zheng Q.C., Lu X.M., Liu J., Shi L., Liu C.Q., Wang G.B., Zou W.P. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56:1342–1351. doi: 10.1002/hep.25777. [DOI] [PubMed] [Google Scholar]
- 33.Yan W.J., Liu X., Ma H.X., Zhang H.L., Song X.J., Gao L.F., Liang X.H., Ma C.H. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64:1593–1604. doi: 10.1136/gutjnl-2014-307671. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H.L., Song Y., Yang H.M., Liu Z.Y., Gao L.F., Liang X.H., Ma C.H. Tumor cell-intrinsic Tim-3 promotes liver cancer via NF-kappa B/IL-6/STAT3 axis. Oncogene. 2018;37:2456–2468. doi: 10.1038/s41388-018-0140-4. [DOI] [PubMed] [Google Scholar]
- 35.Liu C.H., Chern G.J., Hsu F.F., Huang K.W., Sung Y.C., Huang H.C., Qiu J.T., Wang S.K., Lin C.C., Wu C.H., Wu H.C., Liu J.Y., Chen Y. A multifunctional nanocarrier for efficient TRAIL-based gene therapy against hepatocellular carcinoma with desmoplasia in mice. Hepatology. 2018;67:899–913. doi: 10.1002/hep.29513. [DOI] [PubMed] [Google Scholar]
- 36.Rajasekaran D., Srivastava J., Ebeid K., Gredler R., Akiel M., Jariwala N., Robertson C.L., Shen X.N., Siddiq A., Fisher P.B., Salem A.K., Sarkar D. Combination of nanoparticle-delivered siRNA for astrocyte elevated gene-1 (AEG-1) and all-trans retinoic acid (ATRA): an effective therapeutic strategy for hepatocellular carcinoma (HCC) Bioconjugate Chem. 2015;26:1651–1661. doi: 10.1021/acs.bioconjchem.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu N., Zhang X., Li J., Gan Y. Targeting exosomal miRNA with pH-sensitive liposome coated chitosan-siRNA nanoparticles for inhibition of hepatocellular carcinoma metastasis. J. Contr. Release. 2015;213:e82. doi: 10.1016/j.jconrel.2015.05.136. [DOI] [PubMed] [Google Scholar]
- 38.Xiao S., Liu Z., Deng R., Li C., Fu S., Chen G., Zhang X., Ke F., Ke S., Yu X., Wang S., Zhong Z. Aptamer-mediated gene therapy enhanced antitumor activity against human hepatocellular carcinoma in vitro and in vivo. J. Contr. Release. 2017;258:130–145. doi: 10.1016/j.jconrel.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 39.Perrone F., Craparo E.F., Cemazar M., Kamensek U., Drago S.E., Dapas B., Scaggiante B., Zanconati F., Bonazza D., Grassi M., Truong N., Pozzato G., Farra R., Cavallaro G., Grassi G. Targeted delivery of siRNAs against hepatocellular carcinoma-related genes by a galactosylated polyaspartamide copolymer. J. Contr. Release. 2021;330:1132–1151. doi: 10.1016/j.jconrel.2020.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Dalwadi D.A., Torrens L., Abril-Fornaguera J., Pinyol R., Willoughby C., Posey J., Llovet J.M., Lanciault C., Russell D.W., Grompe M., Naugler W.E. Liver injury increases the incidence of HCC following AAV gene therapy in mice. Mol. Ther. : the journal of the American Society of Gene Therapy. 2021;29:680–690. doi: 10.1016/j.ymthe.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flynn M.J., Sayed A.A., Sharma R., Siddique A., Pinato D.J. Challenges and opportunities in the clinical development of immune checkpoint inhibitors for hepatocellular carcinoma. Hepatology. 2019;69:2258–2270. doi: 10.1002/hep.30337. [DOI] [PubMed] [Google Scholar]
- 42.Luo Z.L., Jiang J.W. pH-sensitive drug loading/releasing in amphiphilic copolymer PAE-PEG: integrating molecular dynamics and dissipative particle dynamics simulations. J. Contr. Release. 2012;162:185–193. doi: 10.1016/j.jconrel.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 43.Ko J., Park K., Kim Y.S., Kim M.S., Han J.K., Kim K., Park R.W., Kim I.S., Song H.K., Lee D.S., Kwon I.C. Tumoral acidic extracellular pH targeting of pH-responsive MPEG-poly (beta-amino ester) block copolymer micelles for cancer therapy. J. Contr. Release. 2007;123:109–115. doi: 10.1016/j.jconrel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Liu F., Li M., Liu C., Liu Y., Zhang N. PH-sensitive self-assembled carboxymethyl chitosan-modified DNA/polyethylenimine complexes for efficient gene delivery. J. Biomed. Nanotechnol. 2014;10:3397–3406. doi: 10.1166/jbn.2014.1901. [DOI] [PubMed] [Google Scholar]
- 45.Qi X., Qin J., Fan Y., Qin X., Jiang Y., Wu Z. Carboxymethyl chitosan-modified polyamidoamine dendrimer enables progressive drug targeting of tumors via pH-sensitive charge inversion. J. Biomed. Nanotechnol. 2016;12:667–678. doi: 10.1166/jbn.2016.2206. [DOI] [PubMed] [Google Scholar]
- 46.Yao Y., Su Z., Liang Y., Zhang N. pH-Sensitive carboxymethyl chitosan-modified cationic liposomes for sorafenib and siRNA co-delivery. Int. J. Nanomed. 2015;10:6185–6197. doi: 10.2147/IJN.S90524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Q., Yu M.Z., Wang J.C., Hou W.J., Gao L.Y., Ma X.F., Pei X.W., Niu Y.J., Liu X.Y., Qiu C., Pang W.H., Du L.L., Zhang Q. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials. 2014;35:5028–5038. doi: 10.1016/j.biomaterials.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Song C.H., Zhang Y.J., Li C.Y., Chen G.C., Kang X.F., Wang Q.B. Enhanced nanodrug delivery to solid tumors based on a tumor vasculature-targeted strategy. Adv. Funct. Mater. 2016;26:4192–4200. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.