

Abstract
Purpose of Review
Neutrophilic dermatoses are a heterogeneous group of disorders with significant overlap in associated conditions, clinical presentation, and histopathologic features. This review provides a structural overview of neutrophilic dermatoses that may present in the inpatient setting along with diagnostic work-up and management strategies.
Recent Findings
Sweet’s syndrome has been found in patients with coronavirus disease 2019 (COVID-19). Pyoderma gangrenosum (PG) has been shown to be equally associated with ulcerative colitis and Crohn’s disease. A clinical trial shows that cyclosporine is equally effective as prednisone in treating PG. Neutrophilic eccrine hidradenitis has been found in the setting of newer antineoplastic medications, such as BRAF inhibitors, as well as in the setting of malignancy without chemotherapy exposure.
Summary
Neutrophilic dermatoses are a rare and complex group of dermatoses with varying and overlapping clinical presentations. Physicians should be aware of the growing list of associated diseases in order to build a better differential diagnosis or to potentially investigate for co-existing disease.
Keywords: Neutrophilic dermatosis, Systemic disease, Autoinflammatory disease, Hospital medicine, Complex medical dermatology
Introduction
Neutrophils are an essential inflammatory cell, comprising 40–70% of total white blood cells [1–3]. They accumulate rapidly at sites of infection or tissue injury, where they ingest, kill, and digest microbial pathogens through the use of granule-packed proteases and reactive oxygen intermediates [3]. However, neutrophil accumulation in the absence of an identifiable cause leads to injury of normal tissue [4, 5•]. Neutrophilic dermatoses (ND) are a heterogeneous group of dermatologic disorders characterized by the shared histological feature of cutaneous infiltration of mature neutrophils without an identifiable infectious agent. Clinical presentation can range from vesicles, papules, nodules, or ulcerations, along with extracutaneous involvement [4]. Given the substantial overlap between history, clinical, and pathologic features, diagnosis between subtypes of neutrophilic dermatoses can be challenging, especially in hospitalized patients with underlying inflammatory or paraneoplastic syndromes. This article provides an overview of the known pathophysiology, clinical presentation, approach to diagnostic work-up, and management of a subset of NDs, such as Sweet’s syndrome, pyoderma gangrenosum, neutrophilic eccrine hidradenitis, and Behçet disease, that may present in the inpatient setting.
Overview of Pathophysiology
The etiopathogenesis of ND has continuously evolved with scientific discovery, yet it is incompletely understood. Neutrophils play a large role in the innate immune response through the release of various chemo-attractants causing activation and migration of additional neutrophils, monocytes/macrophages, dendritic cells, natural killer cells, T helper type 1 (Th1), and Th17 cells into damaged tissues [5•]. While they are an important function of the immune response, the proteolytic granules housed by neutrophils carry the potential to cause damage in infiltrated tissues. For example, it has been shown that neutrophils release serine and matrix metalloproteinases that cleave extracellular matrix components, in turn, destroying tissue architecture [6, 7]. Prior exposure to certain pathogens and immune response to the bacterial, viral, tumor, or other antigens is thought to influence the development of NDs through the dysregulation of cytokines promoting neutrophil infiltration, although the exact mechanisms are incompletely understood [8]. Granulocyte colony-stimulating factor (G-CSF) is a known major regulator of neutrophil differentiation, function, and therefore an important factor in maintaining the balance between sustained immune response and resolution of inflammation [9]. G-CSF not only has been found to be a cause of drug-induced NDs, such as Sweet’s syndrome, but has also been found to be elevated in the serum of patients with active disease [10, 11]. Increased production of G-CSF by tumor cells has also been proposed as a potential factor in malignancy-associated disease [12].
Concept of Pathergy
All NDs exhibit a hypersensitivity reaction called pathergy [13]. Although the pathophysiology is unclear, cutaneous trauma is thought to trigger a cytokine-mediated inflammatory response leading to new lesions or aggravation of existing lesions [14]. This is especially important for the inpatient physician as the development of multiple skin reactions secondary to biopsies, intravenous catheter placement, vaccination, and/or venipuncture have been reported to occur in the setting of inpatient work-up and management [15].
General Work-up
Clinical assessment should include timing of disease onset as well as a thorough review of systems to initially evaluate for conditions such as recent infection, malignancy, pregnancy, and autoimmune disease, due to their known associations with ND. Surveillance of the entire skin should be performed as the distribution of lesions of the same disease can vary between patients (e.g., cases of both proximal and distal distributions of neutrophilic eccrine hidradenitis have been described), as well as the overlap between lesions of different NDs have also been reported [16]. While the appropriate description of eruption morphology is crucial to initial differential diagnoses, clinico-pathologic correlation is essential for diagnosis and is often a major diagnostic criterion for NDs [4]. In patients with inflammatory papules or plaques, a 4-mm punch biopsy is usually sufficient for histological examination. Microbial pathology stains should be performed along with an additional second punch biopsy for bacterial, fungal, and mycobacterial cultures, as infectious etiologies are often included in the initial differential diagnosis [4].
Sweet’s Syndrome
Sweet’s syndrome (SS), also known as acute febrile neutrophilic dermatosis, was first recognized in 1964. It was initially called Gomm-Button disease, and careful documentation over the years has led to the classification of three distinct subtypes based on etiology: classical SS (idiopathic Sweet’s syndrome), malignancy-associated SS, and drug-induced SS [17–19]. Given the rarity of the disease, exact epidemiological data is unclear, although known data will be summarized within SS subtypes.
Classical Sweet’s Syndrome
Classical, or idiopathic, Sweet’s syndrome constitutes the majority of cases and remains a diagnosis of exclusion. This subtype predominantly affects women over men at a 4:1 ratio and most commonly presents between 30 and 60 years of age. Patients with idiopathic SS often present in the setting of inflammatory states (e.g., post-infection, in the setting of autoimmune disorders) or pregnancy. The most common preceding infections include gastrointestinal and upper respiratory infections [19]. More recently, three cases of SS have been noted in post-COVID-19 patients [20, 21, 22•].
Drug-induced Sweet’s Syndrome
Drug-induced SS was first reported in 1986, attributed to trimethoprim-sulfamethoxazole [23]. Although several hundred cases of SS have been reported in the literature, reviews report that drug-induced SS can make up anywhere from 1 to 27% of cases [15, 24]. Currently, the most common instigating drug is G-CSF, although cases secondary to non-steroidal anti-inflammatory drugs, antivirals, antibacterials, and several other drug groups have also been reported [13]. Given the rarity of occurrence, patients with suspected SS should be asked about new medication use.
Malignancy-associated Sweet’s Syndrome
Malignancy-associated SS was initially categorized under the same diagnostic criteria as SS; however, as cancer-related cases became increasingly prevalent, a separated subtype was created. Approximately 85% of reported cases of malignancy-associated SS are associated with underlying hematological neoplasia, most commonly acute myeloblastic leukemia (AML). Reported solid malignancies, although rare, include carcinomas of the genitourinary organs, breast, and gastrointestinal tract [19, 25–27]. Recognizing malignancy-associated SS is critical for the inpatient physician as it can lead to early discovery and management of new or recurrent malignancy [19].
Clinical Presentation
SS classically presents as a constellation of both physical and pathologic findings which include fever, leukocytosis, and tender erythematous skin lesions [19]. Due to the nature of the illness and its association with other disease states, patients with SS often present with significant comorbidities.
Timing
Initial symptoms of SS typically present several days after upper respiratory tract infection of acute gastroenteritis [28]. Case reports of drug-related SS demonstrate disease onset anywhere from 5 to 10 days of therapy onset with a general consensus of SS onset within 2 weeks of inciting drug exposure [19, 24]. Cutaneous manifestations can be preceded by several days to weeks of isolated fever and can occur concurrently during the entire course of dermatosis. In cases of malignancy-associated SS, pyrexia can be absent and/or replaced with other co-occurring symptoms such as arthralgias, general malaise, headache, and myalgias [19].
Cutaneous Lesions
Skin lesions often appear abruptly as tender, non-pruritic, red or purple-red papules, or nodules (Fig. 1). Eruptions are often bilateral, but asymmetrical, with favoring of the head, neck, and upper extremities [29]. Cohen and colleagues note that some lesions may appear similar to bullae with a transparent, pseudovesicular appearance due to edema in the upper dermis [19]. In patients with malignancy, lesions may appear more pseudopustular or pseudovesicular, develop ulceration with central clearing, and potentially mimic the features of pyoderma gangrenosum [30]. Mucosal involvement, typically presenting as ulceration, has also been reported, although more commonly in patients with hematologic malignancies [19, 28].
Fig. 1.

Sweet’s syndrome. A Classical Distribution of lesions on upper extremities and trunk. B Pseudopustular appearance of lesions seen in SS, most commonly seen in cases associated with malignancy. Taken from DermnetNZ.org, “Acute Febrile Neutrophilic Dermatosis”
Necrotizing Sweet’s Syndrome
A clinical variant of SS, necrotizing Sweet’s syndrome (N-SS), was first described by Kroshinsky et al. in 2012. Patients presented with rapidly progressive erythematous, edematous cutaneous lesions with necrotic involvement of the underlying soft tissues, including myonecrosis. Although the acute clinical presentation led to high clinical suspicion of necrotizing fasciitis, a rapidly progressive and often fatal microbial soft tissue infection that tracks along fascial planes, negative cultures and histopathologic findings consistent with SS led to favoring of N-SS over necrotizing fasciitis. Although initial discrimination between necrotizing fasciitis and N-SS can be difficult, pathergy is a key distinguishing finding. Early recognition of this clinical variant of SS is vital as debridement, the treatment of choice for necrotizing fasciitis, may induce further pathergy and clinical decline [31].
Work-up and Diagnosis
Laboratory Studies/Serological Tests
Laboratory investigation should include a complete blood count with differential. Although non-specific, peripheral leukocytosis with neutrophilia and an elevated ESR/C-reactive protein (CRP) is frequently seen [19]. Elevated serum levels of antineutrophil cytoplasmic antibody (ANCA) have also been reported, though it has limited evidence as a marker of disease [32]. Evaluation for pregnancy should be done in women of childbearing age.
Biopsy
Two 4-mm punch biopsies are sufficient for histological evaluation and tissue culture in order to exclude infection. When patients present with primarily subcutaneous nodules, excisional biopsy may be preferred.
Evaluation for Malignancy or Other Associated Conditions
In the absence of other explanations and presence of concerning constitutional symptoms, malignancy work-up can be considered in the inpatient or outpatient setting. Of note, any abnormalities in the CBC count should prompt consideration of bone marrow biopsy. Beginning with age-appropriate screenings followed by work-up for the most commonly associated malignancies is recommended. Evaluation for autoimmune disorders or inflammatory bowel disease (IBD) can be considered based on clinical suspicion, though it may be more appropriate in the outpatient setting.
Diagnostic Criteria
In 1986, Su and Liu proposed two major and four minor criteria for the diagnosis of either classical or malignancy-associated SS which have been widely accepted and are outlined in Table 1 [33]. Patients suspected of either SS subtype must meet both major criteria which include (1) sudden onset of characteristic plaques or nodules and (2) neutrophilic infiltrate in the dermis without vasculitis. Additionally, confirmation of SS requires the presence of at least two minor clinical features, as listed in Table 1. For drug-induced SS, a different set of diagnostic criteria was set in 1996 (Table 1), for which all five diagnostic criteria must be met for diagnosis.
Table 1.
Criteria for diagnosis of Sweet’s syndrome
| Criteria for diagnosis of classic and drug-induced Sweet’s syndrome | |
|---|---|
| Major criteria | |
|
1. Abrupt onset of typical cutaneous lesions (tender erythematous plaques or nodules, occasionally with vesicles, pustules, or blisters) 2. Histopathology consistent with Sweet’s syndrome (predominantly neutrophilic dermal infiltrate without leukoclastic vasculitis) | |
| Minor criteria | |
|
1. Preceded by an associated infection (gastrointestinal or respiratory) or vaccination or associated with - Inflammatory diseases such as chronic autoimmune disorders, infections - Hemoproliferative disorders or solid malignant tumor - Pregnancy 2. Presence of fever (> 38 °C) 3. Abnormal laboratory values - Erythrocyte sediment rate > 20 mm/h - Elevated C-reactive protein levels - Leukocytosis > 8000 - Neutrophilic > 70% 4. Excellent response to systemic corticosteroids |
| Criteria for diagnosis of drug-induced Sweet’s syndrome | |
|---|---|
|
1. Abrupt onset of painful erythematous plaques or nodules 2. Histopathological evidence of a dense neutrophilic infiltrate without evidence of leukoclastic vasculitis 3. Presence of fever (38 °C) 4. Temporal relationship between drug ingestion and clinical presentation or temporally related recurrence after oral challenge 5. Temporally related resolution of lesions after drug withdrawal or treatment with systemic corticosteroids |
In order for the diagnosis of SS, including classic SS and/or malignancy-associated SS, both major criteria along with at least two minor criteria must be met. For drug-induced SS, all five criteria must be met for diagnosis
Histopathology
The characteristic histologic presentation is a dense neutrophilic infiltrate within the reticular dermis that may appear diffusely perivascular, with significant papillary dermal edema. Although the findings of frank vasculitis are not expected, some features such as leukocytoclasis and red cell extravasion may be seen and are thought to be secondary to noxious products released from neutrophils [34]. In general, there are no significant epidermal changes [29]. First described by Requena et al. in 2005, histiocytoid Sweet’s syndrome (H-SS) is a histological variant of Sweet’s syndrome characterized by a dermal infiltrate mainly composed of lymphocytes and histiocytoid myeloperoxidase-positive cells and appears to be associated with hematologic malignancy at higher frequencies than classic neutrophilic SS [35].
Management
When limited to the skin, SS is a benign condition that can resolve spontaneously without scaring, within weeks to months, especially after discontinuation of causative medications or treatment of underlying disease [13, 28, 36]. Although no standardized treatment guidelines exist for SS, the most effective therapy for Sweet’s syndrome is oral prednisone (0.5–1.0 mg/kg/day) for 2–6 weeks which can be started in the inpatient setting and then managed by outpatient dermatology [19]. Clinical response to systemic corticosteroids remains a minor diagnostic criterion [33]. When there are only a few localized lesions, topical corticosteroids (typically super potent or intralesional) may be trialed first. Evidence for alternative drugs is limited by case reports or small case series; however, steroid-sparing medications that have some evidence as potential treatment modalities include potassium iodide, dapsone, and colchicine [19]. Recurrences develop in approximately 30% of patients and occur more often in those with hematologic disorders (50%), which makes dermatology follow-up an important component of the post-discharge care plan [19].
Pyoderma Gangrenosum
Pyoderma gangrenosum (PG) is a rare, chronic, and recurrent ulcerative disease with a distinct morphological presentation. Investigation of US inpatient data demonstrated an increase in hospitalizations secondary to PG from 2002 to 2012, highlighting the need for an improved understanding of disease management [37•]. In this section, we will focus mostly on classic ulcerative PG, though the classification of PG into other major clinical subtypes (pustular, bullous, and vegetative) has also been proposed [38•]. In addition to morphological subtypes, classification of cases based on location or etiology has been described (e.g., peristomal PG, genital PG, and post-operative PG) [39].
Associated Conditions
Literature suggests that 25–50% of PG cases are associated with an underlying co-morbidity. In the largest case series including data from 356 validated cases, 66.9% of cases were associated with another condition. These included inflammatory bowel disease in 146 (41%) of patients, inflammatory arthritis in 73 (20.5%) of patients, solid organ malignancy in 23 (6.5%) of patients, and hematologic malignancy in 21 (5.9%) of patients [40]. Previously thought to be more frequently associated with ulcerative colitis (UC), recent research recognizes that regional enteritis and Crohn’s disease are associated with PG at similar rates as UC. Additionally, PG activity does not appear to be related to IBD disease course, as shown by cases during IBD clinical remission [41, 42]. Other clinical subtypes of PG may be associated with underlying systemic diseases at different rates than classical PG. For example, it has been reported that 70% of Bullous PG cases develop alongside hematologic diseases, such as myelogenous leukemia, lymphoma, monoclonal gammopathy, and myelodysplastic syndromes [43, 44].
Clinical Presentation
Cutaneous Lesions
PG ulcers form rapidly and frequently appear well developed at initial clinical evaluation, often following minor trauma, similar to SS. The classic presentation involves an initial inflammatory papulopustule, nodule, or bulla that quickly breaks down over the course of days leading to painful necrotic ulceration (Fig. 2). Central necrosis and loss of tissue may expose underlying muscles or tendons. A violaceous or “gun-metal” gray undermined border with areas of peripheral macular erythema is a potential sign of active lesions, and lesions often spread with the centrifugal spreading of lesions. Re-epithelialization occurs from the margin and often heals with atrophic cribriform pigmented scars [41, 45–49].
Fig. 2.

Pyoderma gangrenosum. A A violaceous plaque with an erythematous border and slight central ulceration in the pretibial region. B Close-up of a large ulcer on the medial malleolus with an overlying yellow-ish slough and a hemorrhagic border that appears undermined in some areas. Taken from DermnetNZ.org, “Pyoderma Gangrenosum”
Distribution
Classical cutaneous lesions most frequently appear on lower extremities, especially the pretibial area, but can occur anywhere on the body, including mucosal and peristomal sites. The number of ulcers can vary from isolated to > 12, and occasional cases of coalescing lesions have been reported [45–48]. In patients with hematologic disorders, initial lesions may appear acutely as hemorrhagic or purulent bullous lesions with a widespread distribution [40]. In patients with IBD, a chronic, slowly enlarging ulcer with excessive granulation tissue is more characteristic [41].
Work-up and Diagnosis
Similar to other neutrophilic dermatoses, laboratory and histopathological findings can vary and clinico-pathologic correlation is required for diagnosis. Evaluation should begin with a thorough medical history to the assessment of risk factors and associated diseases including the history of IBD, autoimmune arthritis, and/or malignancy. Temporal evaluation relating to underlying and triggering events should be considered, although initial PG diagnosis has been shown to both precede, coincide with, and/or follow underlying systemic disease activity [44].
Laboratory Evaluation
Studies may be useful to rule out other potential causes and/or look for comorbid disease states. The initial evaluation should include a complete blood count and a comprehensive metabolic panel to obtain baseline studies prior to beginning immunosuppressive therapies. Further evaluation in the outpatient setting can include antinuclear antibody titers, antineutrophilic cytoplasmic antibodies, hepatitis panel, rheumatoid factor, and/or endoscopy/colonoscopy based on clinical suspicion.
Biopsy
Biopsies are indicated in patients without a known history of PG and in patients with established PG who present with atypical features or lesions that fail to respond to therapy. Although there remains a concern for biopsy-induced pathergy, the need to evaluate for other disorders and to ensure proper treatment initiation supersedes risk. In contrast to SS, an elliptical incisional biopsy that incorporates both the inflamed lesion border and ulcer edge while extending vertically into the subcutaneous fat can be considered for PG [43]. Culture for bacterial (including atypical mycobacteria) and fungal infection should be performed. It is important to mention that superinfected PG ulcers may lead to positive bacterial cultures, although not the primary cause of the ulcers [50].
Diagnosis
There are no definitive tests for PG, contributing to the difficulty of diagnosis. Clinical history, biopsy, and directed laboratory evaluation can aid in distinguishing PG from other disease states. Proposed diagnostic criteria for classic ulcerative PG include meeting both major criteria of (1) rapid progression of a painful, necrolytic cutaneous ulcer, and (2) other causes of cutaneous ulceration have been excluded [43]; however, a high rate of misdiagnosis of PG still exists [51]. Reassessment of a PG diagnosis is indicated if the disease fails to respond to therapy as expected.
Histopathology
Histopathologic findings of PG are non-specific and evolve depending on the stage of the lesion. Biopsies are therefore often more useful to exclude other dermatologic disorders with similar findings. Early lesions may reveal dermal neutrophilia centered on follicles, while severe skin lesions may show tissue necrosis with a surrounding mononuclear cell infiltrate [52•].
Management
PG remains a difficult dermatological disease to manage. Only two published randomized clinical trials examining treatment for PG exist, and current treatments are largely based on anecdotal data and case studies. Additionally, a lack of defined and validated PG outcome measures presents a challenge when attempting to investigate and describe treatment response. Current treatment modalities focus on long-term immunosuppression and wound care along with treatment of concurrent illnesses. Initial treatment often involves the use of a fast-acting immunosuppressive agent, especially in the inpatient setting [42, 47, 53]. Systemic corticosteroids have historically been the therapy of choice, though recent results from one randomized clinical trial revealed that cyclosporine may be just as effective and is associated with fewer adverse events [54]. Depending on the response, which can be defined as no new ulcers and/or cessation of progression of existing ulcers, outpatient clinicians can consider the addition of a steroid-sparing agent, such as mycophenolate mofetil or biologics, which may exhibit maximum effectiveness between 1 and 4 months of use [53]. Additionally, non-immunosuppressive options that target neutrophils, such as colchicine or dapsone, can be considered [55]. To avoid polypharmacy, associated diseases should be considered when developing long-term treatment plans. Treating patients with PG and concurrent malignancies may pose a particular challenge given direct interactions of anti-cancer treatments or contraindications in hematologic malignancies.
From a wound care standpoint, pathergy makes the management of PG ulcers difficult. PG ulcers on the lower extremities should be gently cleansed and monitored for edema, and wound care teams should be instructed to minimize excessive compression until appropriate treatment is started to avoid further lesions. Additionally, wound debridement and surgical intervention are contraindicated in active PG ulcers, though presenting post-debridement due to misdiagnosis is not uncommon [56].
Neutrophilic Eccrine Hidradenitis
Neutrophilic eccrine hidradenitis (NEH) is a reactive disorder characterized by the deposition of neutrophilic infiltrate around eccrine coils and glands [57].
Associated Diseases/Medications
First described in 1982, NEH was thought to result from chemotherapy; however, case reports have since documented occurrence due to malignancy (in the absence of chemotherapy), infections, and medication use [57]. In a review of 51 patients by Bachmeyer and Aractingi, acute myeloid leukemia (AML) was the most frequently associated malignancy. Most patients (n = 43/51, 84%) were exposed to cytarabine and/or an anthracycline about 9.7 days prior to rash onset. Although hematologic malignancies represent the most commonly associated disease, cases due to solid tumors and infection including human immunodeficiency virus (HIV) and bacterial pathogens (Serratia, Staphylococcus, Enterobacter, and Nocardia) have been reported [57–59]. Many antineoplastic regiments that have been listed as potential risk factors for NEH include epidermal growth factor receptor inhibitors (EGFR), tyrosine kinase inhibitors, and BRAF inhibitors [60–63]. Interestingly, NEH due to BRAF inhibitors, such as dabrafenib or vemurafenib, has been reported to present earlier (3–4 days after drug exposure) compared to other anti-cancer medications [60]. Due to the frequent association of NEH with chemotherapy, some clinicians believe NEH to be within the spectrum of toxic erythema of chemotherapy, a broad range of nonallergic cutaneous eruptions secondary to the use of antineoplastic drugs [56].
Clinical Presentation
Symptoms
Fever is frequently observed, though the link between NEH and fever is controversial given confounders such as co-occurring chemotherapy-induced neutropenia [57, 64].
Cutaneous Lesions
NEH most commonly presents as tender erythematous or purpuric macules. Other presentations include asymptomatic or painful erythematous papules, pustules, nodules, or plaques. Epidermal changes are atypical, though isolated pustules within the erythematous plaques have been reported in prior cases (Fig. 3) [57].
Fig. 3.
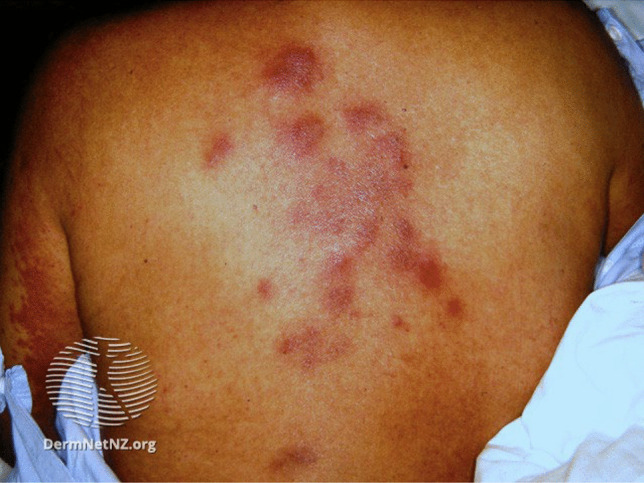
Neutrophilic eccrine hidradenitis. Erythematous/violaceous and edematous papules and plaques on the back of a patient receiving chemotherapy. Taken from DermnetNZ.org, “Urticaria Like Conditions: Neutrophilic Eccrine Hidradenits”
Distribution
Lesions tend to affect the trunk, upper extremities, and face, particularly around the periorbital areas. The groin and axilla are usually spared.
Work-up and Diagnosis
Laboratory Evaluation
Laboratory investigation should include a complete blood count with differential to evaluate for chemotherapy-induced neutropenia in patients undergoing treatment. Neutropenia is an important perspective through which to view histopathology as results may appear atypical, as discussed later. If a patient presents with NEH without known malignancy, age-appropriate evaluation for underlying neoplasm should be performed.
Biopsy
4-mm punch biopsies of new inflammatory lesions are recommended to rule out infection, other NDs (such as SS), or alternative paraneoplastic phenomena.
Histopathology
Although a dense neutrophilic infiltrate around the eccrine apparatus is considered “classical,” this finding is often absent in patients with underlying neutropenia. Given the highly polymorphic clinical presentation, histopathological findings are, nonetheless, important for diagnosis. Other findings consistent with NEH include vacuolar degeneration and/or necrosis of the eccrine epithelium and eccrine squamous syringometaplasia [57, 64–66].
Management
NEH resolves spontaneously without scarring within days to weeks in almost all patients, even in the absence of treatment [67]. Similar to other rare NDs, treatment modalities are largely anecdotal based on case reports without any existing clinical trials. Reported management strategies include antibiotics (secondary to febrile neutropenia versus management for NEH specifically), systemic corticosteroids, and non-steroidal anti-inflammatory drugs for pain and fever [65, 68, 69]. Relapse has been reported, most commonly occurring in patients receiving the same chemotherapy medication responsible for the initial presentation, although the majority of patients are able to continue chemotherapy regimens without further reaction. In one patient with Hodgkin’s lymphoma with recurring episodes, relapse was prevented by prophylactic treatment with 100 mg/day of dapsone [68].
Behçet Disease
Behçet disease (BD) is a multisystem inflammatory disease with an unpredictable disease course, characteristic of oral ulcers, genital ulcers, and uveitis [70]. Equally distributed between sexes, BD typically begins between 30 and 40 years of age and is thought to be triggered by a combination of environmental factors, such as microbial infection, with genetic predisposition [Fietta 2005]. BD has a distinct geographical distribution along the “Silk Route,” a region stretching from Japan to Middle Eastern and Mediterranean countries [67]. Prevalence correlates to the genetic distribution of human leukocyte antigen (HLA)-B51, with an estimated frequency of 20–25% among the Silk Route population compared to an HLA-B51 frequency of 2–8% among the general Northern Europe/USA populations [71].
Clinical Presentation
Mucocutaneous Lesions
Recurrent mucocutaneous lesions are the hallmark of BD. Oral ulcers are a major criterion (as described in the diagnostic section) and are frequently the first presenting symptom (65–70% of patients) [70]. Oral ulcers begin as an erythematous papule or vesiculopustular lesion that develops a gray to yellow pseudomembrane, eventually evolving into a non-scarring round ulcer that heals within 1–4 weeks. The individual oral ulcers are similar to those occurring in recurrent oral aphthosis; however, differentiating factors include the increased number of ulcers (> 6) and presence of mucosal pathergy, and soft palate and oropharyngeal involvement [72, 73]. The genital ulcers primarily involve the scrotum and penis in males and the labia majora in females, although they can present widely in the anogenital area including the labia minora, vaginal mucosa, cervix, glans penis, and perineum [70]. Genital ulcers appear similar to oral ulcers, although they tend to be larger and more painful with increased potential for scarring [70, 73, 74].
Cutaneous Lesions
Various cutaneous lesions of BD have been described including erythema nodosum-like lesions (favoring women and typically on the lower extremities), sterile vesiculopustular and papules (occurring on the face and mimicking acneiform lesions), and superficial thrombophlebitis (occurring in up to 30% of patients with BD) (Table 2) [70].
Table 2.
| Prevalence within BD | Clinical appearance | |
|---|---|---|
| Papulopustular lesions | Unknown prevalence; increased frequency in patients with a positive SPT arthritis | Acral and facial vesiculopustular and/or purpuric papules lesions on an erythematous base; described as follicular or acneiform |
| Erythema nodosum lesions | Females > males; 33% of BD patients | Bilateral, pretibial, painful erythematous nodules. It can also localize to the face, neck, forearms, and buttocks |
| Superficial thrombophlebitis | Males > females, 30% of BD patients | Erythematous, tender, subcutaneous nodules arranged in a linear fashion. “String-like” palpation of veins and erythema of overlying skin |
SPT skin pathergy test
Extracutaneous Manifestations
Ocular involvement is another hallmark of BD, with panuveitis being the most common ocular lesion, although isolated anterior and posterior uveitis, retinal vasculitis, papillitis, and chorioretinitis have all been reported [75]. BD is considered a multisystem disease and has well-documented neurological, vascular, articular, and gastrointestinal manifestations that will not be discussed in the scope of this review [72].
Work-up and Diagnosis
Laboratory Evaluation
There are no pathognomonic laboratory tests in BD. Laboratory markers of inflammation, such as erythrocyte sedimentation rate (ESR) and CRP, may be elevated in association with increased disease activity, although this remains a non-specific finding [76]. Exclusion of other causes of oral and genital ulcerations, such as sexually transmitted infections, should be included in the initial work-up based on patient history and clinical suspicion [77].
Diagnosis
BD, similar to many of the discussed NDs, primarily remains a diagnosis based on clinical findings and exclusion of alternative diagnosis. The International Study Group for Behçet’s Disease Criteria are the most commonly accepted diagnostic criteria which require recurrent oral aphthae (at least three times in one year) plus two of the following clinical features: recurrent genital aphthae (aphthous ulceration or scarring}; eye lesions (including anterior or posterior uveitis, cells in vitreous on slit lamp examination or retinal vasculitis observed by an ophthalmologist); skin lesions (including erythema nodosum, pseudofolliculitis, papulopustular lesions, or acneiform nodules); and positive pathergy test [78].
Management
Evidence on the management of BD largely comes from randomized controlled trials. The European League Against Rheumatism (EULAR) published recommendations for the treatment of BD in 2008, from which treatment of mucocutaneous involvement will be focused on here [79]. In general, treatment should be tailored to each patient’s impact on quality of life. Topical measures, such as corticosteroids, should be the first line for isolated oral and genital lesions along with lidocaine gel, chlorhexidine, and sucralfate suspension. Systemic treatments should be reserved for patients with resistant cases. Colchicine, although widely used, has not been proven efficacious other than in erythema nodosum-like lesions, for which it is the first line. Other systemic agents for mucocutaneous lesions include azathioprine, thalidomide, interferon-alpha, and TNF-alpha antagonists [80, 81]. Reports since the EULAR publication show efficacy of newer agents including alemtuzumab, anakinra, apremilast, azithromycin, canakinumab, isotretinoin, and ustekinumab [82–88].
Conclusions
Neutrophilic dermatoses include a rare, but important, pattern of diseases characterized by a neutrophil-rich cutaneous infiltrate. Although outpatient dermatologic follow-up is frequently necessary, prompt evaluation and diagnosis by the inpatient dermatologist may lead to improved outcomes, especially in patients with multiple comorbidities [76]. Clinical suspicion of such associated diseases (e.g., malignancy or autoimmune disease) should prompt further work-up in either the inpatient or outpatient setting.
Compliance with Ethical Standards
Conflict of Interest
STC has received honoraria from Pfizer and Novartis for serving on an advisory board for digital media.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
Nishi Shah and Maria S. Asdourian contributed equally to this work.
This article is part of the Topical Collection on Hospital-Based Dermatology
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Phillipson M, Kubes P. The healing power of neutrophils. Trends Immunol. 2019;40(7):635–647. doi: 10.1016/j.it.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17(11):1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi SD, Malachowa N, DeLeo FR. Neutrophils and bacterial immune evasion. J Innate Immun. 2018;10(5–6):432–441. doi: 10.1159/000487756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callen JP. Neutrophilic dermatoses. Dermatol Clin. 2002;20(3):409–419. doi: 10.1016/s0733-8635(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 5.• Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503. 10.1038/s41568-020-0281-y. Review discussing the key role of neutrophils as part of the innate immune system as well as the discussing neutrophils as part of the tumor microenvironment as tumor-associated neutrophils (TAN). [DOI] [PubMed]
- 6.Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162(1):100–107. doi: 10.1111/j.1365-2249.2010.04201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11(3):e1004651. doi: 10.1371/journal.ppat.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schröder J-M, Szperalski B, Koh C-J, Christophers E. IgA-associated inhibition of polymorphonuclear leukocyte chemotaxis in neutrophilic dermatoses. J Investig Dermatol. 1981;77(6):464–468. doi: 10.1111/1523-1747.ep12497599. [DOI] [PubMed] [Google Scholar]
- 9.Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils – forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol. 2006;2(9):500–510. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- 10.Park JW, Mehrotra B, Barnett BO, Baron AD, Venook AP. The Sweet syndrome during therapy with granulocyte colony-stimulating factor. Ann Intern Med. 1992;116(12 Pt 1):996–998. doi: 10.7326/0003-4819-116-12-996. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami T, Ohashi S, Kawa Y, Takahama H, Ito M, Soma Y, et al. Elevated serum granulocyte colony-stimulating factor levels in patients with active phase of Sweet syndrome and patients with active Behcet disease: implication in neutrophil apoptosis dysfunction. Arch Dermatol. 2004;140(5):570–574. doi: 10.1001/archderm.140.5.570. [DOI] [PubMed] [Google Scholar]
- 12.Uhara H, Saida T, Nakazawa H, Ito T. Neutrophilic dermatoses with acute myeloid leukemia associated with an increase of serum colony-stimulating factor. J Am Acad Dermatol. 2008;59(2):S10–S12. doi: 10.1016/j.jaad.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Nelson CA, Noe MH, McMahon CM, Gowda A, Wu B, Ashchyan HJ, et al. Sweet syndrome in patients with and without malignancy: a retrospective analysis of 83 patients from a tertiary academic referral center. J Am Acad Dermatol. 2018;78(2):303–9.e4. doi: 10.1016/j.jaad.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Rahman S, Daveluy S. Pathergy test. Treasure Island (FL): StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- 15.Nelson CA, Stephen S, Ashchyan HJ, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79(6):987–1006. doi: 10.1016/j.jaad.2017.11.064. [DOI] [PubMed] [Google Scholar]
- 16.Ajili F, Souissi A, Bougrine F, Boussetta N, Abdelhafidh NB, Sayhi S, et al. Coexistence of pyoderma gangrenosum and Sweet’s syndrome in a patient with ulcerative colitis. Pan Afr Med J. 2015;21:151. doi: 10.11604/pamj.2015.21.151.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweet RD. Acute febrile neutrophilic dermatosis–1978. Br J Dermatol. 1979;100(1):93–99. doi: 10.1111/j.1365-2133.1979.tb03573.x. [DOI] [PubMed] [Google Scholar]
- 18.Sweet RB. An acute febrile neutrophtlic dermatosts. Br J Dermatol. 1964;76(8–9):349–356. doi: 10.1111/j.1365-2133.1964.tb14541.x. [DOI] [PubMed] [Google Scholar]
- 19.Cohen PR. Sweet’s syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agharbi F-Z, Oqbani K, Allaoui A, Chikhaoui I, Chiheb S. Sweet syndrome in post-COVID-19 infection: a case report. Travel Med Infect Dis. 2021;44:102188. doi: 10.1016/j.tmaid.2021.102188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taşkın B, Vural S, Altuğ E, Demirkesen C, Kocatürk E, Çelebi İ, et al. Coronavirus 19 presenting with atypical Sweet’s syndrome. J Eur Acad Dermatol Venereol. 2020;34(10):e534–e535. doi: 10.1111/jdv.16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.• Berro S, Calas A, Sohier P, Darbord D, Dupin N. Sweet’s syndrome three weeks after a severe COVID-19 infection: a case report. Acta Derm Venereol. 2021;101(6):adv00486. 10.2340/00015555-3850. Explanation of importance relating to references 20, 21, and 22: the novel coranavirus (SARS-CoV-2) has caused many different infection-related cutaneous manifestations. These case studies continue to provide evidence that Sweet’s syndrome is related to a hypersensitivity reaction triggered by the release of cytokines/activation of neutrophils in the setting of an inflammatory process. [DOI] [PMC free article] [PubMed]
- 23.Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5):918–923. doi: 10.1016/S0190-9622(96)90080-8. [DOI] [PubMed] [Google Scholar]
- 24.Tien V, Jones AD, Aronowitz PB. Drug-induced Sweet’s syndrome. J Gen Intern Med. 2017;32(8):953–954. doi: 10.1007/s11606-017-4022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hensley CD, Caughman SW. Neutrophilic dermatoses associated with hematologic disorders. Clin Dermatol. 2000;18(3):355–367. doi: 10.1016/S0738-081X(99)00127-3. [DOI] [PubMed] [Google Scholar]
- 26.Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82(6):1220–1226. doi: 10.1016/0002-9343(87)90229-4. [DOI] [PubMed] [Google Scholar]
- 27.Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46(1):106–111. doi: 10.1111/j.1365-4632.2006.02605.x. [DOI] [PubMed] [Google Scholar]
- 28.Rochet NM, Chavan RN, Cappel MA, Wada DA, Gibson LE. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69(4):557–564. doi: 10.1016/j.jaad.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 29.von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis) J Am Acad Dermatol. 1994;31(4):535–556. doi: 10.1016/s0190-9622(94)70215-2. [DOI] [PubMed] [Google Scholar]
- 30.Tacke J, Diepgen TL, Albrecht HP, von den Driesch P. Idiopathic pustular and bullous variants of Sweet syndrome. Hautarzt. 1994;45(3):184–187. doi: 10.1007/s001050050061. [DOI] [PubMed] [Google Scholar]
- 31.Kroshinsky D, Alloo A, Rothschild B, Cummins J, Tan J, Montecino R, et al. Necrotizing Sweet syndrome: a new variant of neutrophilic dermatosis mimicking necrotizing fasciitis. J Am Acad Dermatol. 2012;67(5):945–954. doi: 10.1016/j.jaad.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 32.Arun Kumar AU, Elsayed ME, Alghali A, Ali AA, Mohamed H, Hussein W, et al. Sweet syndrome: a rare feature of ANCA-associated vasculitis or unusual consequence of azathioprine-induced treatment. Allergy Asthma Clin Immunol. 2018;14:46. doi: 10.1186/s13223-018-0265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37(3):167–174. [PubMed] [Google Scholar]
- 34.Malone JC, Slone SP, Wills-Frank LA, Fearneyhough PK, Lear SC, Goldsmith LJ, et al. Vascular inflammation (Vasculitis) in Sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol. 2002;138(3):345–349. doi: 10.1001/archderm.138.3.345. [DOI] [PubMed] [Google Scholar]
- 35.Requena L, Kutzner H, Palmedo G, Pascual M, Fernández-Herrera J, Fraga J, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141(7):834–842. doi: 10.1001/archderm.141.7.834. [DOI] [PubMed] [Google Scholar]
- 36.Amouri M, Masmoudi A, Ammar M, Boudaya S, Khabir A, Boudawara T, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55(9):1033–1039. doi: 10.1111/ijd.13232. [DOI] [PubMed] [Google Scholar]
- 37.• Narla S, Silverberg JI. The inpatient burden and comorbidities of pyoderma gangrenosum in adults in the United States. Arch Dermatol Res. 2021;313(4):245–53. 10.1007/s00403-020-02098-7. An analysis of hospital admission relating to pyoderma gangrenosum data within the United States over a ten-year period. This particular article highlights the importance of accurate diagnosis within the inpatient setting, given the rise of cases and the burden on the healthcare system. [DOI] [PubMed]
- 38.• Maverakis E, Marzano AV, Le ST, Callen JP, Brüggen MC, Guenova E, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020;6(1):81. 10.1038/s41572-020-0213-x. Pyoderma gangrenosum is a difficult-to-diagnose disorder, and this review highlighted ongoing management strategies as well as areas of future research regarding outcome measures and quality of life studies. [DOI] [PubMed]
- 39.Ruocco E, Sangiuliano S, Gravina AG, Miranda A, Nicoletti G. Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol. 2009;23(9):1008–1017. doi: 10.1111/j.1468-3083.2009.03199.x. [DOI] [PubMed] [Google Scholar]
- 40.Ashchyan HJ, Butler DC, Nelson CA, Noe MH, Tsiaras WG, Lockwood SJ, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154(4):409–413. doi: 10.1001/jamadermatol.2017.5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callen JP, Jackson JM. Pyoderma gangrenosum: an update. Rheum Dis Clin North Am. 2007;33(4):787–802. doi: 10.1016/j.rdc.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 42.Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012;13(3):191–211. doi: 10.2165/11595240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 43.Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800. doi: 10.1111/j.1365-4632.2004.02128.x. [DOI] [PubMed] [Google Scholar]
- 44.Vacas AS, Bollea-Garlatti ML, Torre AC, Galimberti RL. Bullous pyoderma gangrenosum as a predictor of hematological malignancies. An Bras Dermatol. 2018;93(1):133–134. doi: 10.1590/abd1806-4841.20187031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18(3):355–372. doi: 10.1007/s40257-017-0251-7. [DOI] [PubMed] [Google Scholar]
- 46.Ye MJ, Ye JM. Pyoderma gangrenosum: a review of clinical features and outcomes of 23 cases requiring inpatient management. Dermatol Res Pract. 2014;2014:461467. doi: 10.1155/2014/461467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165(6):1244–1250. doi: 10.1111/j.1365-2133.2011.10565.x. [DOI] [PubMed] [Google Scholar]
- 48.Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB, Jr, White WL, Callen JP. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine. 2000;79(1):37–46. doi: 10.1097/00005792-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 49.von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol. 1997;137(6):1000–1005. doi: 10.1111/j.1365-2133.1997.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 50.Gavioli EM, Casias M, Ngo L. Pyoderma gangrenosum and superimposed infection: a case report. Adv Skin Wound Care. 2020;33(6):1–3. doi: 10.1097/01.ASW.0000661796.90753.f3. [DOI] [PubMed] [Google Scholar]
- 51.Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347(18):1412–1418. doi: 10.1056/NEJMoa013383. [DOI] [PubMed] [Google Scholar]
- 52.• George C, Deroide F, Rustin M. Pyoderma gangrenosum – a guide to diagnosis and management Clin Med (Lond). 2019;19(3):224–8. 10.7861/clinmedicine.19-3-224. This review focuses on the differential diagnosis of pyoderma gangrenosum, given the high misdiagnose rate. They also include a summary of the largely anecdotal evidence regarding management options for the treatment of PG.
- 53.Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53(2):273–283. doi: 10.1016/j.jaad.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Ormerod AD, Thomas KS, Craig FE, Mitchell E, Greenlaw N, Norrie J, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ Br Med J. 2015;350:h2958. doi: 10.1136/bmj.h2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kontochristopoulos GJ, Stavropoulos PG, Gregoriou S, Zakopoulou N. Treatment of Pyoderma gangrenosum with low-dose colchicine. Dermatology. 2004;209(3):233–236. doi: 10.1159/000079897. [DOI] [PubMed] [Google Scholar]
- 56.Patel DK, Locke M, Jarrett P. Pyoderma gangrenosum with pathergy: a potentially significant complication following breast reconstruction. J Plast Reconstr Aesthet Surg. 2017;70(7):884–892. doi: 10.1016/j.bjps.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 57.Bachmeyer C, Aractingi S. Neutrophilic eccrine hidradenitis. Clin Dermatol. 2000;18(3):319–330. doi: 10.1016/s0738-081x(99)00123-6. [DOI] [PubMed] [Google Scholar]
- 58.Ruiz-López P, Martínez-Luna E, Toussaint-Caire S, Fuentes-Suárez A, Vega-Memije E. Neutrophilic eccrine hidradenitis in an HIV-infected patient. Skinmed. 2017;15(4):297–299. [PubMed] [Google Scholar]
- 59.Rouanet I, Jantac M, Lechiche C, Hope-Rapp E, Sotto A. Neutrophilic eccrine hidradenitis in an HIV-1-infected patient. AIDS. 2012;26(6):775–776. doi: 10.1097/QAD.0b013e328351f64b. [DOI] [PubMed] [Google Scholar]
- 60.Herms F, Franck N, Kramkimel N, Fichel F, Delaval L, Laurent-Roussel S, et al. Neutrophilic eccrine hidradenitis in two patients treated with BRAF inhibitors: a new cutaneous adverse event. Br J Dermatol. 2017;176(6):1645–1648. doi: 10.1111/bjd.15259. [DOI] [PubMed] [Google Scholar]
- 61.Bishnoi A, Daroach M, Aggarwal D, Radotra BD, Panda P, Parsad D. Ticagrelor induced neutrophilic eccrine hidradenitis: a unique adverse effect of a new antiplatelet drug. Postgrad Med J. 2019;95(1123):279–280. doi: 10.1136/postgradmedj-2019-136426. [DOI] [PubMed] [Google Scholar]
- 62.Wang F-Y, Chen W-T, Shen M-H, Chung W-H, Chen C-B. Pazopanib as a new culprit in neutrophilic eccrine hidradenitis. Dermatol Sin. 2019;37(4):222–225. doi: 10.4103/ds.ds_6_19. [DOI] [Google Scholar]
- 63.Dib EG, Ifthikharuddin JJ, Scott GA, Partilo SR. Neutrophilic eccrine hidradenitis induced by imatinib mesylate (Gleevec) therapy. Leuk Res. 2005;29(2):233–234. doi: 10.1016/j.leukres.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 64.Prat L, Bouaziz JD, Wallach D, Vignon-Pennamen MD, Bagot M. Neutrophilic dermatoses as systemic diseases. Clin Dermatol. 2014;32(3):376–388. doi: 10.1016/j.clindermatol.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 65.Bernstein EF, Spielvogel RL, Topolsky DL. Recurrent neutrophilic eccrine hidradenitis. Br J Dermatol. 1992;127(5):529–533. doi: 10.1111/j.1365-2133.1992.tb14854.x. [DOI] [PubMed] [Google Scholar]
- 66.Bhawan J, Petry J, Rybak ME. Histologic changes induced in skin by extravasation of doxorubicin (adriamycin) J Cutan Pathol. 1989;16(3):158–163. doi: 10.1111/j.1600-0560.1989.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 67.Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10(5):301–312. doi: 10.2165/11310730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 68.Shear NH, Knowles SR, Shapiro L, Poldre P. Dapsone in prevention of recurrent neutrophilic eccrine hidradenitis. J Am Acad Dermatol. 1996;35(5):819–822. doi: 10.1016/S0190-9622(96)90092-4. [DOI] [PubMed] [Google Scholar]
- 69.Bassas-Vila J, Fernández-Figueras MT, Romaní J, Ferrándiz C. Infectious eccrine hidradenitis: a report of 3 cases and a review of the literature. Actas Dermosifiliogr. 2014;105(2):e7–e12. doi: 10.1016/j.adengl.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 70.Alpsoy E. Behçet’s disease: a comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol. 2016;43(6):620–632. doi: 10.1111/1346-8138.13381. [DOI] [PubMed] [Google Scholar]
- 71.Dalvi SR, Yildirim R, Yazici Y. Behcet’s syndrome. Drugs. 2012;72(17):2223–2241. doi: 10.2165/11641370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 72.Scherrer MAR, Rocha VB, Garcia LC. Behçet’s disease: review with emphasis on dermatological aspects. An Bras Dermatol. 2017;92(4):452–464. doi: 10.1590/abd1806-4841.20177359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alpsoy E, Zouboulis CC, Ehrlich GE. Mucocutaneous lesions of Behçet’s disease. Yonsei Med J. 2007;48(4):573–585. doi: 10.3349/ymj.2007.48.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarica-Kucukoglu R, Akdag-Kose A, Kayabalı M, Yazganoglu KD, Disci R, Erzengin D, et al. Vascular involvement in Behçet’s disease: a retrospective analysis of 2319 cases. Int J Dermatol. 2006;45(8):919–921. doi: 10.1111/j.1365-4632.2006.02832.x. [DOI] [PubMed] [Google Scholar]
- 75.Hussein MA, Eissa IM, Dahab AA. Vision-threatening Behcet’s disease: severity of ocular involvement predictors. J Ophthalmol. 2018;2018:9518065. doi: 10.1155/2018/9518065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Melikoglu M, Topkarci Z. Is there a relation between clinical disease activity and acute phase response in Behcet’s disease? Int J Dermatol. 2014;53(2):250–254. doi: 10.1111/ijd.12224. [DOI] [PubMed] [Google Scholar]
- 77.Nair JR, Moots RJ. Behcet's disease. Clin Med. 2017;17(1):71–77. doi: 10.7861/clinmedicine.17-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.International Study Group for Behçet's D Criteria for diagnosis of Behcet’s disease. Lancet. 1990;335(8697):1078–1080. doi: 10.1016/0140-6736(90)92643-V. [DOI] [PubMed] [Google Scholar]
- 79.Hatemi G, Silman A, Bang D, Bodaghi B, Chamberlain AM, Gul A, et al. EULAR recommendations for the management of Behçet disease. Ann Rheum Dis. 2008;67(12):1656. doi: 10.1136/ard.2007.080432. [DOI] [PubMed] [Google Scholar]
- 80.Cheng S, Murphy R. Refractory aphthous ulceration treated with thalidomide: a report of 10 years’ clinical experience. Clin Exp Dermatol. 2012;37(2):132–135. doi: 10.1111/j.1365-2230.2011.04169.x. [DOI] [PubMed] [Google Scholar]
- 81.Ambrose NL, Haskard DO. Differential diagnosis and management of Behçet syndrome. Nat Rev Rheumatol. 2013;9(2):79–89. doi: 10.1038/nrrheum.2012.156. [DOI] [PubMed] [Google Scholar]
- 82.Baerveldt EM, Kappen JH, Thio HB, van Laar JAM, van Hagen PM, Prens EP. Successful long-term triple disease control by ustekinumab in a patient with Behçet's disease, psoriasis and hidradenitis suppurativa. Ann Rheum Dis. 2013;72(4):626. doi: 10.1136/annrheumdis-2012-202392. [DOI] [PubMed] [Google Scholar]
- 83.Ryu HJ, Seo MR, Choi HJ, Baek HJ. Infliximab for refractory oral ulcers. Am J Otolaryngol. 2014;35(5):664–668. doi: 10.1016/j.amjoto.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 84.Mohammad AJ, Smith RM, Chow YW, Chaudhry AN, Jayne DRW. Alemtuzumab as remission induction therapy in Behçet disease: a 20-year experience. J Rheumatol. 2015;42(10):1906. doi: 10.3899/jrheum.141344. [DOI] [PubMed] [Google Scholar]
- 85.Cantarini L, Tinazzi I, Caramaschi P, Bellisai F, Brogna A, Galeazzi M. Safety and efficacy of etanercept in children with juvenile-onset Behcet’s disease. Int J Immunopathol Pharmacol. 2009;22(2):551–555. doi: 10.1177/039463200902200235. [DOI] [PubMed] [Google Scholar]
- 86.Cantarini L, Vitale A, Scalini P, Dinarello CA, Rigante D, Franceschini R, et al. Anakinra treatment in drug-resistant Behcet’s disease: a case series. Clin Rheumatol. 2015;34(7):1293–1301. doi: 10.1007/s10067-013-2443-8. [DOI] [PubMed] [Google Scholar]
- 87.Hatemi G, Melikoglu M, Tunc R, Korkmaz C, Turgut Ozturk B, Mat C, et al. Apremilast for Behçet’s syndrome – a phase 2, placebo-controlled study. N Engl J Med. 2015;372(16):1510–1518. doi: 10.1056/NEJMoa1408684. [DOI] [PubMed] [Google Scholar]
- 88.Vitale A, Rigante D, Caso F, Brizi MG, Galeazzi M, Costa L, et al. Inhibition of interleukin-1 by canakinumab as a successful mono-drug strategy for the treatment of refractory Behçet’s disease: a case series. Dermatology. 2014;228(3):211–214. doi: 10.1159/000358125. [DOI] [PubMed] [Google Scholar]