

Abstract
Increasing evidence indicates that a mitochondria-specific stress response referred to as the mitochondrial unfolded protein response (UPRmt) is activated to maintain mitochondrial integrity and support tumor growth. In this forum article, we discuss the recent advances and current challenges in therapeutically targeting UPRmt in cancer.
Keywords: Mitochondrial unfolded protein response, mitochondrial chaperonins, mitochondrial proteases, mitochondrial proteostasis, cancer
Mitochondrial Stress and Mitochondrial Unfolded Protein Response
Mitochondria are essential for tumor growth and progression. Mitochondria supply bioenergetic and biosynthetic demands during tumor growth through production of ATP by oxidative phosphorylation (OXPHOS); and synthesis of nucleotides, amino acids, and lipids [1]. Mitochondria are crucial for metabolic reprogramming to coordinate energy production during stressful conditions. However, an increased demand for mitochondrial activity by highly proliferating cells leads to excessive generation of reactive oxygen species (ROS), toxic byproducts of mitochondria. ROS induce oxidative damage and promote the unfolding/aggregation of proteins in mitochondria [2].
UPRmt, best studied in C. elegans, is activated during mitochondrial stress to maintain mitochondrial health [3]. UPRmt induces the expression of (1) chaperones that promote folding of mitochondrial proteins that have unfolded/aggregated due to excessive ROS, and (2) proteases that eliminate mitochondrial proteins that have been irreparably damaged by ROS (Figure 1). Lead chaperones include heat shock protein 60 (HSP60), heat shock protein 10 (HSP10), and mitochondrial heat shock protein 70 (mtHSP70). Key proteases include Lon peptidase 1 (LONP1) and Caseinolytic protease (ClpP).
Figure 1: The mitochondrial unfolded protein response (UPRmt) promotes tumor growth and progression.
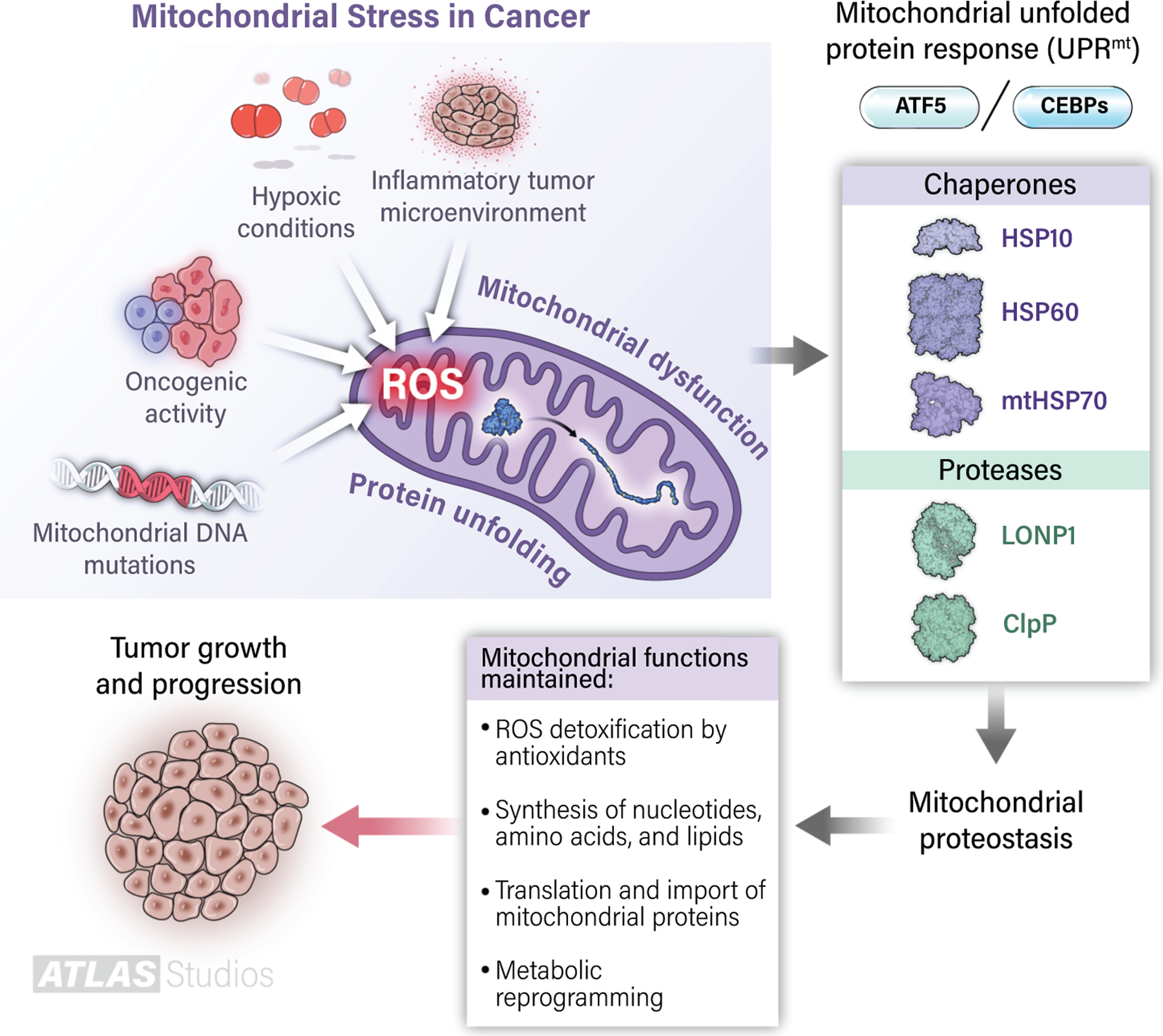
Various sources of endogenous stress, including genetic alterations, hypoxia, and the presence of inflammatory immune cells, alter mitochondria and enhance ROS production. ROS inflict damage and promote the unfolding/aggregation of mitochondrial proteins. Under these circumstances, the UPRmt activates transcription factors such as ATF5 and CEBPs to induce the upregulation of chaperones (HSP60, HSP10, mtHSP70) and proteases (LONP1, ClpP), which fold proteins into their proper conformation and dispose of excessively damaged proteins, respectively. Together, this stress-responsive system allows mitochondrial proteostasis and maintenance of multiple tumor-supporting activities by mitochondria.
Studies of UPRmt in cancer have mostly focused on the ability of individual UPRmt components to promote tumor growth. Emerging evidence highlights the ability of UPRmt as a whole to attenuate mitochondrial stress and support tumor growth in vivo [4, 5]. Given the tumor-supportive role of UPRmt, various groups have endeavored to target UPRmt proteins (Table 1). While many inhibitors of individual UPRmt components are known, we highlight only those inhibitors that have either reached the clinical stage or show promising preclinical data.
Table 1:
Key cancer therapeutics that target the mitochondrial unfolded protein response (UPRmt)
| UPRmt Target | Drug | Mechanism | Phase | References |
|---|---|---|---|---|
| HSP60 | Epolactaene and derivatives | Inhibits HSP60/HSP10 chaperone activity | Preclinical | [7] |
| HSP60 | Gold (III) porphyrins | Inhibits HSP60/HSP10 chaperone activity | Preclinical | [8] |
| HSP60 | BSP-SCA hybrid analogs | Inhibits HSP60/HSP10 chaperone activity | Preclinical | [9] |
| mtHSP70 | MKT 077 | Inhibits interactions between mtHSP70 and Bag co-chaperones | Phase 1 | [10] |
| mtHSP70 | JG-231 | Inhibits interactions between mtHSP70 and nucleotide exchange factors | Preclinical | [11] |
| LONP1 | CDDO-Me | Inhibits LONP1 protease activity | Phase 1 | [12,I*] |
| ClpP | ONC201 | Hyperactivates ClpP protease activity | Phase 1,2 | [IV*] |
| ClpP | A2-32-01 | Inhibits ClpP protease activity | Preclinical | [13] |
| ATF5, CEBPs | (d/n)-ATF5 peptides | Prevents dimerization of transcription factors | Preclinical | [14] |
Indicates references from ClinicalTrials.gov
Targeting UPRmt-specific Chaperones in Cancer
HSP60/HSP10
HSP60 forms a complex with HSP10 to function as a key chaperone in mitochondria. Majority of HSP60/HSP10 inhibitors target HSP60 of this complex. Mizoribine, derived from Eupenicillium brefeldianum, was the first natural molecule identified as an HSP60 inhibitor [6]. Traditionally used as an immunosuppressant after renal transplantation, mizoribine was later shown to bind HSP60 and inhibit the chaperonin activity of HSP60/HSP10 in the mM range. Randomized, double-blind, placebo-controlled phase 1 trials enrolled healthy males for administration of mizoribine to determine the mizoribine tolerability. Values were compared by one-way analysis of variance (ANOVA) and mizoribine displayed a favorable safety profile, except for transient elevations in serum uric acid at the highest dose (12 mg/kg). However, mizoribine was unable to reach concentrations required to inhibit HSP60/HSP10 and peaked at plasma concentrations of ~30 μM in the clinic [6].
Epolactaene, isolated from Penicillium sp. BM 1689-P, and its synthetic derivatives selectively target HSP60 in the 2–4 μM range and induce cell cycle arrest in cancer cells [7]. Other synthetic molecules, such as gold (III) porphyrin complexes and BSP-SCA (5-sulfonamido-2-phenylbenzoxazole and salicylanilide) hybrid analogs, directly target HSP60 and exert anti-cancer effects [8, 9]. However, the specificity and efficacy of such compounds in vivo remain to be established. Future studies must explore these properties before such inhibitors can be tested in the clinic.
mtHSP70
Another crucial mitochondrial chaperone is mtHSP70. MKT 077, a rhodacyanine dye, accumulates in cancer mitochondria due to the increased electrochemical gradient of the mitochondrial membrane. MKT 077 disrupts interactions between mtHSP70 and Bag co-chaperones to reduce the chaperone activity of mtHSP70.
MKT 077 is cytotoxic in many preclinical models of human cancer. MKT 077 was subjected to a phase 1 clinical trial to treat patients with advanced solid tumors refractory to standard chemotherapies [10]. Tumors with an increased mitochondrial membrane gradient, such as colon, were included for treatment. The primary aim was to determine MTK 077 toxicity to patients. Comparisons were evaluated using a Student’s t-test. Reversible renal toxicity was frequently observed. Therefore, recruitment for this study was discontinued. However, this trial did demonstrate that MKT 077 can safely inhibit mitochondrial functions and that rhodacyanine analogues with a higher therapeutic index could plausibly target mtHSP70 in cancer.
Structure-based design identified new rhodacyanine analogues, such as JG-231, that display increased selectivity and bioavailability, and are well-tolerated in vivo [11]. However, further challenges lie ahead as these rhodacyanine analogues are light sensitive, and difficult to synthesize and solubilize.
Targeting UPRmt-specific Proteases in Cancer
LONP1
Triterpenoids isolated from plants and synthetic derivatives, such as CDDO-Me, block LONP1-mediated proteolysis in cancer cells and induce apoptosis [12]. A non-randomized, phase 1 clinical study of CDDO-Me enrolled advanced solid tumors and lymphoma patients refractory to standard therapy (NCT00508807)I. The purpose was to determine the dose-limiting toxicities and maximum tolerated dose, in order to guide dosing for phase 2 studies. Dose escalation by accelerated titration within single-patient cohorts was performed until grade-2 adverse events were observed. Upon completion of this study, comparisons were evaluated using a Student’s t-test. Common adverse events included fatigue and nausea. The maximum tolerated dose was 900 mg/d and patients who received 1300 mg/d displayed reversible hepatotoxicity.
Although CDDO-Me targets LONP1, it also inhibits PPAR-γ, ubiquitin-specific-processing protease 7, and IκB kinase to exert its anti-cancer functions [12]. CDDO-Me was selected for clinical trials not simply based on its ability to inhibit LONP1 of the UPRmt response, but for its inhibition of a range of tumor-supportive pathways. This precludes the therapeutic use of triterpenoids as LONP1-specific inhibitors. Interestingly, CDDO-Me also increased glomerular filtration rate. Consequently, CDDO-Me is now enrolling patients for clinical trials to treat chronic kidney disease (NCT03749447, NCT04702997)II, III.
ClpP
The imipridone molecule ONC201 binds and hyperactivates ClpP, leading to excessive proteolysis, mitochondrial failure, and cell death in multiple cancer cell lines. Patients with advanced solid tumors and resistant to standard therapies (NCT02250781)IV were enrolled in an open label, single group assignment, phase 1 dose-escalation study for ONC201. The primary objective was to determine the safety and tolerability of ONC201 helping establish doses for phase 2 studies. Comparisons were evaluated upon completion of the study using descriptive statistics. Adverse events included cases of nausea and emesis. Overall, results indicated a favorable safety profile at the recommended phase 2 dose of 625 mg per week.
However, in addition to hyperactivating ClpP, ONC201 is also known to inactivate AKT/ERK signaling and inhibit dopamine receptor D2. In fact, the ability of ONC201 to hyperactivate ClpP was not known until after the clinical trial had commenced. The non-selective nature of ONC201 prevents its sole designation as a ClpP-targeting agent.
Notably, inhibition of ClpP activity by (3RS,4RS)-3-(non-8-en-1-yl)-4-(2-(pyridin-3-yl)ethyl)oxetan-2-one (A2-32-01) also shows beneficial effects in a preclinical model of leukemia [13]. However, the specificity and bioavailability of A2-32-01 in humans remain unknown and must be addressed before A-32-01 can be used for clinical trials.
Targeting UPRmt Transcription Factors
A family of leucine zipper proteins initiate the transcription of chaperones and proteases during the UPRmt response. Activating transcription factor 5 (ATF5) and CCAAT-enhancer-binding proteins (CEBPs) are key transcription factors, which exert their function either via homodimerization or heterodimerization. ATF5 is crucial for upregulating the transcription of UPRmt components during mitochondrial stress [3], maintains mitochondrial proteostasis, and allows mitochondria to promote tumor growth and progression.
ATF5 protein was modified so that the DNA binding domain was mutated to eliminate transcriptional activity [14]. However, with the leucine zipper left intact, this dominant-negative (d/n)-ATF5 could bind the DNA interaction domains of ATF5, CEBPB, and CEBPD, and prevent their dimerization. Later, d/n-ATF5 was fused to cell-penetrating domains to produce (d/n)-ATF5 peptides that can be administered via injection to selectively trigger apoptosis in a broad range of treatment-resistant tumors in mice [14]. Such potent and tumor-selective effects allow (d/n)-ATF5 peptides to be ideal candidates for clinical studies.
Final Remarks
Altough UPRmt is an attractive therapeutic target, many outstanding questions including the mechanism of UPRmt activation in cancer and how does UPRmt interact with oncogenes/tumor suppressors require further investigation.
Many challenges remain as the current inhibitors of UPRmt appear to have issues with potency, specificity, and toxicity. As a result, only a few of these inhibitors have entered clinical trials and none have yet been approved for cancer therapy. These issues could eventually be overcome by the use of in silico screening and X-ray crystallography to guide the development of agents, which selectively target UPRmt proteins. Drug uptake can be enhanced by the combined use of lipid nanoparticles for tumor cell delivery and attachment of cationic groups, which utilize high transmembrane potential of the mitochondrial membrane for mitochondrial targeting [15].
The success of inhibiting UPRmt as a form of therapy likely correlates with the dependency of individual tumors upon UPRmt for growth and survival. Genomic profiling of tumors for signs of mitochondrial stress and dysfunction could help predict the efficacy of targeting UPRmt. In addition, tumor cells that emerge after chemotherapy are highly dependent upon mitochondria for proliferation. Optimal timing and combinational use of UPRmt inhibitors with standard therapies could enhance current care [15]. Overall, cancer dependency on UPRmt provides the vast, and untapped potential of appropriately targeting this response for the treatment and management of cancer burden.
Acknowledgements
This work was supported by the NCI of the NIH under Award Number RO1-CA246437 to D. Chandra, and in part by the NCI Center Support Grant P30-CA016056 to the Roswell Park Comprehensive Cancer Center. We apologize to those colleagues whose publications inadvertently were not cited.
Footnotes
Declaration of interests: The authors declare no competing interests.
References
- 1.Spinelli JB and Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20 (7), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenny TC et al. (2017) The Mitochondrial Unfolded Protein Response as a Non-Oncogene Addiction to Support Adaptation to Stress during Transformation in Cancer and Beyond. Front Oncol 7, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiorese CJ et al. (2016) The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol 26 (15), 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng P and Haynes CM (2017) Mitochondrial dysfunction in cancer: Potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol 47, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karpel-Massler G et al. (2016) A Synthetic Cell-Penetrating Dominant-Negative ATF5 Peptide Exerts Anticancer Activity against a Broad Spectrum of Treatment-Resistant Cancers. Clin Cancer Res 22 (18), 4698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda M et al. (2006) Population pharmacokinetics of higher-dose mizoribine in healthy male volunteers. Biol Pharm Bull 29 (12), 2460–4. [DOI] [PubMed] [Google Scholar]
- 7.Nagumo Y et al. (2004) Structure-activity relationships of epolactaene derivatives: structural requirements for inhibition of Hsp60 chaperone activity. Bioorg Med Chem Lett 14 (17), 4425–9. [DOI] [PubMed] [Google Scholar]
- 8.Hu D et al. (2016) Anticancer Gold(III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew Chem Int Ed Engl 55 (4), 1387–91. [DOI] [PubMed] [Google Scholar]
- 9.Ray AM et al. (2021) Exploiting the HSP60/10 chaperonin system as a chemotherapeutic target for colorectal cancer. Bioorg Med Chem 40, 116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Propper DJ et al. (1999) Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol 10 (8), 923–7. [DOI] [PubMed] [Google Scholar]
- 11.Shao H et al. (2018) Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem 61 (14), 6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borella R et al. (2019) Synthesis and Anticancer Activity of CDDO and CDDO-Me, Two Derivatives of Natural Triterpenoids. Molecules 24 (22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole A et al. (2015) Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 27 (6), 864–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun X et al. (2020) Dominant-Negative ATF5 Compromises Cancer Cell Survival by Targeting CEBPB and CEBPD. Mol Cancer Res 18 (2), 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong L and Neuzil J (2019) Targeting mitochondria as an anticancer strategy. Cancer Commun (Lond) 39 (1), 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- I.This study is registered with ClinicalTrials.gov under the following link: https://clinicaltrials.gov/ct2/show/NCT00508807
- II.This study is registered with ClinicalTrials.gov under the following link: https://clinicaltrials.gov/ct2/show/NCT03749447
- III.This study is registered with ClinicalTrials.gov under the following link: https://clinicaltrials.gov/ct2/show/NCT04702997
- IV.This study is registered with ClinicalTrials.gov under the following link: https://clinicaltrials.gov/ct2/show/NCT02250781