

Abstract
Epidermal growth factor receptor (EGFR), the receptor for members of the epidermal growth factor family, regulates cell proliferation and signal transduction; moreover, EGFR is related to the inhibition of tumor cell proliferation, angiogenesis, invasion, metastasis, and apoptosis. Therefore, EGFR has become an important target for the treatment of cancer, including non-small cell lung cancer, head and neck cancer, breast cancer, glioma, cervical cancer, and bladder cancer. First- to third-generation EGFR inhibitors have shown considerable efficacy and have significantly improved disease prognosis. However, most patients develop drug resistance after treatment. The challenge of overcoming intrinsic and acquired resistance in primary and recurrent cancer mediated by EGFR mutations is thus driving the search for alternative strategies in the design of new therapeutic agents. In view of resistance to third-generation inhibitors, understanding the intricate mechanisms of resistance will offer insight for the development of more advanced targeted therapies. In this review, we discuss the molecular mechanisms of resistance to third-generation EGFR inhibitors and review recent strategies for overcoming resistance, new challenges, and future development directions.
Keywords: Epidermal growth factor receptor (EGFR), Drug resistance, Inhibitors, Structure–activity relationship, Tyrosine kinase, Cancer
Introduction
Epidermal growth factor receptor (EGFR) is a member of the receptor tyrosine kinase (RTK) superfamily that consists of exon boundaries and associated extracellular, transmembrane, and intracellular protein domains. EGFR is involved in multiple signaling pathways and regulates numerous cell functions (Fig. 1A). This transmembrane glycoprotein is composed of a cysteine-rich extracellular ligand binding domain, hydrophobic transmembrane domain, cytoplasmic RTK domain, and C-terminal domain. The RTK domain contains an N-lobe consisting of five β-sheet strands and one αC helix and a C-lobe containing the main helices of a highly flexible activation loop (A-loop) [1]. The deep cleft at the junction of these two lobes forms the binding pocket for the adenine ring of ATP. The conformation of three conserved structural elements, namely the Asp-Phe-Gly (DFG) motif, αC helix, and A-loop, critically regulates the activation or inactivation of the catalytic domain. When EGFR is in the active state, the important catalytic residue D855 is located in the ATP binding site, stabilizing the ATP-loaded complex (DFG-in) and αC helix (αC-in). In the inactive state, EGFR forms a Src-like structure, including a closed A-loop, αC-out, and DFG-in [2]. (Fig. 1B). EGFR can dimerize upon binding by ligands, such as amphiregulin, β-cytokines, epidermal growth factor (EGF), heparin-binding EGF-like growth factor (HB-EGF), and transforming growth factor (TGF). The activation of the intracellular tyrosine kinase domain and autophosphorylation, which initiates the Ras/RAF/MEK, signal transducer and activator of transcription (STAT), PI3K/AKT/mTOR and other downstream signaling pathways, are closely related to embryonic development and stem cell division [2–4]. Overexpression of wild-type (WT) EGFR protein with or without EGFR gene amplification or a kinase-activating mutation further enhances cell proliferation, migration, survival, and antiapoptotic responses through signaling cascades, and these processes are closely related to the occurrence and development of many types of epithelial-derived cancer, such as non-small cell lung cancer (NSCLC), breast cancer, glioma, head and neck cancer, cervical cancer, and bladder cancer. Among these cancers, lung cancer appears to be the most common and has the characteristics of aberrant proliferation, metastasis, and drug resistance [5–8]. Thus, EGFR has become a promising target for anticancer drug design and development. EGFR tyrosine kinase inhibitors (EGFR-TKIs) have achieved remarkable results in the clinic [9]. However, most patients develop acquired drug resistance to first- and second-generation EGFR-TKIs after 1–2 years. The mechanism of drug resistance for nearly half of cases relates to the T790M mutation. Third-generation EGFR-TKIs that target EGFR-TKI-sensitive mutations and the T790M mutation have been developed [10].
Fig. 1.

Structure and functions of EGFR. A EGFR exon boundaries and associated extracellular, transmembrane, and intracellular protein domains. EGFR is involved in multiple signaling pathways and regulates numerous cell functions. B The tyrosine kinase domain of EGFR and the activation or inactivation of the catalytic domain. C EGFR domains and the molecular mechanisms of acquired resistance. The intracellular domain contains a juxtamembrane domain, tyrosine kinase domain, and multiple C-terminal tyrosine residues. Multiple mutations within the tyrosine kinase domain are associated with resistance and sensitivity to EGFR-TKIs
Unfortunately, drug resistance caused by less-common mutations in the EGFR gene and components of signal transduction pathways continues to emerge. In addition to common secondary (T790M) and tertiary (C797S) mutations, other EGFR mutations (such as the L718Q, L796S, and L792H mutations and the exon 20 insertion), MET amplification, phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutations, HER2 amplification, oncogene fusions, and alterations in cell cycle-related genes have been observed [11] (Fig. 1C). There is an urgent need for better strategies to combat the inevitable molecular-targeted drug resistance associated with third-generation inhibitors. This review aims to provide a comprehensive overview of the mechanisms of resistance to third-generation EGFR-TKIs and to explore new insights and strategies for overcoming acquired resistance.
Third-generation EGFR-TKIs and drug resistance mechanisms
The development of third-generation EGFR-TKIs
The first-generation EGFR-TKIs form hydrogen bonds with Met793 in the ATP binding pocket of EGFR and reversibly compete with ATP for binding. Drug resistance occurs due to the EGFR T790M mutation (Thr790 in the hydrophobic ATP binding site encoded on exon 20 is replaced by methionine), subclonal selection (of a genetically resistant clone), and rare EGFR mutations (such as G719X, S768I, and L861Q). Thereafter, the development of second-generation EGFR-TKIs was reported; these inhibitors have the same quinazoline scaffold as first-generation EGFR-TKIs, but the side chain can irreversibly bind to Cys797 to inhibit the tyrosine kinase activity of EGFR. For example, the anilinoquinazoline derivative forms hydrogen bonds with the backbone of Met793 in the hinge region and interacts with the hydrophobic region. The acrylamide group binds covalently to Cys797 in the active conformation of EGFR, the furanyl group is exposed to solvent, and the 3-chloro-4-fluorophenyl group is situated next to the gatekeeper residue [12–14]. However, mutations such as T790M still emerge upon treatment with second-generation EGFR-TKIs, which have limited selectivity against WT-EGFR, resulting in serious side effects [15]. Fortunately, third-generation covalent inhibitors that bind irreversibly to the target and are mutation-selective have been developed. These compounds were designed based on a new aminopyrimidine scaffold and show preferable biological activities [16]. Early clinical trials have proven that these third-generation EGFR-TKIs are effective in patients with double-mutated tumors (EGFR L858R/T790M or ex19del/T790M) and have high selectivity for mutant EGFR, thereby eliminating the side effects in the skin and gastrointestinal system associated with the nonselective inhibition of WT-EGFR [17]. For example, the crystal structures of rociletinib (CO-1686) in complex with EGFR T790M and EGFR L858R have been published; in EGFR T790M, the anilinopyrimidine group of rociletinib forms hydrogen bonds with the Met793 amide and the carbonyl backbone, whereas in EGFR L858R, hydrophobic interactions between rociletinib and the protein were due to hydrogen bonds between nitrogens in the pyrimidine group and between the fluoromethyl and Thr790. In addition, the acrylamide group in rociletinib covalently binds to Cys797 in the DFG-in/αC-in active conformations [18]. The specificity for EGFR T790M may stem from hydrophobic interactions between the large methionine in mutant EGFR and pyrimidines. Drugs that have been approved for marketing include osimertinib (US), almonertinib (China), lazertinib (South Korea), and alflutinib (China) (Fig. 2).
Fig. 2.

Development of third-generation EGFR-TKIs
Mechanisms of resistance to third-generation EGFR-TKIs
Due to the covalent bond between the acrylamide (Michael acceptor) of third-generation EGFR-TKIs and the active thiol in the EGFR kinase domain, highly selective inhibitory activity has been achieved by targeting Cys797 and irreversible binding EGFR; thus, these compounds show excellent antitumor activity. Targeted therapy for patients with EGFR T790M and EGFR-activating mutations showed good efficacy in both first- and second-line settings. In patients who developed resistance to third-generation EGFR-TKIs as first-line therapy, genetic changes such as MET amplification, EGFR C797X mutation, PIK3CA amplification and mutation, HER2 amplification and mutation, K-RAS mutation, and BRAF mutation, as well as changes in cell cycle-related genes and oncogene fusions, have been reported, but no T790M mutations have been detected. The mechanism of resistance to second-line therapy is more complicated. Acquisition or deletion of the T790M mutation has been detected in patients [19], and other EGFR mutations (such as L718Q, L796S, L792H, and exon 20 insertion) have also been observed (Fig. 1B). In addition, the mechanisms of acquired resistance to third-generation EGFR-TKIs include alternative pathway activation and histologic and phenotypic transformation (Fig. 3); the details will be discussed in the following sections.
Fig. 3.

Molecular mechanisms of acquired resistance. The mechanisms include target gene modification, parallel alternative pathway activation, downstream pathway activation, and histological/phenotypic transformation. Both amplification and mutation of receptor tyrosine kinases (RTKs) can induce downstream survival signaling pathways. Moreover, direct overexpression and/or mutation of components of downstream pathways can contribute to acquired resistance by promoting cancer cell survival
Primary/intrinsic resistance
The differential sensitivity of TKIs to different EGFR mutations is a cause of primary drug resistance. In NSCLC patients, the in-frame deletion of exon 19 (ex19del) and the L858R point mutation in exon 21 are the most common somatic mutations, occurring in approximately 80% of cases. During EGFR-TKI treatment, patients with longer median survival have presented with more than 20 unique deletions of exon 19. Intrinsic drug resistance can all be triggered by other nonclassical sensitizing mutations (mainly exon 20 insertion) and inherent secondary genetic changes. Drug-resistant clones (for example, T790M) may already exist within the cancer cell population, leading to drug resistance during treatment [20]. Some studies have found that in nearly 1% of lung cancer patients, 2–3 simultaneous driver mutations can be detected before treatment. Some molecular and genetic changes have been reported to relate to intrinsic drug resistance, such as the lack of K-RAS/phosphatase and tensin homolog (PTEN) expression. These preexisting molecular and genetic alterations can stimulate the Ras/Raf/MEK/ERK and PI3K/AKT downstream pathways to promote cancer progression [21].
BIM deletion polymorphism
BIM is a proapoptotic member of the B-cell lymphoma-2 (Bcl-2) family [22]. Recent studies showed that lung cancer cells with the BIM deletion polymorphism and EGFR mutation are resistant to third-generation EGFR-TKIs, suggesting that the BIM deletion polymorphism has potential as a biomarker to predict the efficacy of third-generation EGFR-TKIs in patients [22].
EGFR exon 20 insertion
The molecular mechanism of drug resistance caused by the exon 20 insertion is not fully understood. Eck et al. [23] hypothesized that this mutation prevents binding to EGFR-TKIs due to the addition of residues to the N-lobe of EGFR. The crystal structure of EGFR exon 20 with the D770_N771insNPG insertion shows an unchanged ATP binding pocket and a rigid active conformation, leading to steric hindrance of the drug binding pocket and resistance to EGFR-TKIs.
Acquired resistance
Acquired drug resistance refers to the process by which tumor cells with prior sensitivity to treatment circumvent the inhibitory effects of drugs by changing their metabolic pathways. The mechanisms of acquired resistance to third-generation EGFR-TKIs can be divided into EGFR-dependent resistance and EGFR-independent resistance [24].
EGFR-dependent drug resistance mechanisms
Reappearance of an EGFR mutation
C797S mutation
One point mutation of EGFR (C797S) involves the replacement of Cys797 within the ATP binding site (exon 20) with serine [25]. Osimertinib binds covalently and irreversibly to EGFR T790M by interacting with Cys797. When the C797S mutation occurs, the osimertinib binding efficiency decreases [10], resulting in tumor resistance to all third-generation EGFR-TKIs.
G796R/D mutation
The G796R mutation has been detected in cancer patients who received treatment with a third-generation EGFR-TKI. Molecular docking predictions revealed that G796R sterically hinders the covalent binding of osimertinib. Because the bulky side chain and hydrophilic group hinder the binding of osimertinib to the hydrophobic region, the change in binding energy renders binding unfavorable. Compared with samples containing the double-mutant EGFR L858R/T790M, those harboring the triple-mutant EGFR L858R/T790M/G796R are 110 times more resistant to osimertinib [26]. G796D was reported for the first time in osimertinib-resistant NSCLC patients. In vitro studies have shown that the G796D mutation causes a 50-fold increase in the growth inhibitory 50% (GI50) value of osimertinib. Structural modeling showed that the side chain of the mutated G796D residue collides with the surface of osimertinib, resulting in steric hindrance and energy repulsion and ultimately the loss of binding affinity [27].
L792 mutation
The mutations at Leu792 include L792F, L792Y, and L792H. Structural prediction revealed that these mutations introduce a benzene ring or imidazole ring to the side chain of the residue at 792, which spatially disrupts the orientation of osimertinib, thereby potentially affecting the binding of osimertinib to the EGFR ATP binding site [28].
M766Q mutation
The homology simulation with the T790M and M766Q double mutant showed that M766Q seems to position T790M in the inhibitor binding site, thereby weakening osimertinib binding [29].
Mutations in exon 18
EGFR L718Q/V
EGFR L718Q was reported for the first time in a cell model of resistance to third-generation EGFR-TKIs. Subsequent studies have shown that NSCLC with EGFR L858R/T790M/L718Q is resistant to all EGFR-TKIs, but that with only L858R/L718Q remains sensitive to afatinib [30]. The crystallographic model revealed that the L718Q mutation reduces the efficiency of the formation of covalent bonds between the acrylamide warhead and the Cys797 thiol group, thus interfering with the irreversible binding of osimertinib [31, 32]. In addition, L718V resistance mutations in the kinase domain of EGFR have been detected, and these may interfere with the binding of osimertinib to the kinase domain [33]. Of note, EGFR L718Q/V is still sensitive to afatinib [32].
EGFR G724S
The G724S mutation in the ATP binding loop enriches this loop in glycine, which can lead to the development of resistance to EGFR-TKIs by changing the protein structure, enhancing ATP affinity, and stabilizing activating mutations [34]. However, this mutation does not lead to resistance to second-generation EGFR inhibitors [34].
Compound mutations
A compound mutation refers to the simultaneous detection of two or more different types of EGFR mutations in patient cancer cells [35]. The impact of compound mutations on EGFR-TKI sensitivity is listed in descending order: double classic mutations, compound mutations involving classic mutations and rare mutations, and compound mutations of only rare mutations [36, 37]. These EGFR mutations caused by treatment with third-generation EGFR-TKIs confer resistance to irreversible pyrimidine TKIs but not to quinazoline EGFR inhibitors [38].
T790M reduction or deletion
Deletion of T790M may result from third-generation EGFR-TKI treatment or may be one of the reasons for drug resistance related to tumor heterogeneity. In patients with EGFR T790M, resistance mechanisms are often associated with the C797S mutation or aberrant activation of compensatory pathways, whereas patients with the deletion of T790M typically exhibit different resistance mechanisms, most of which are not associated with EGFR signaling pathways [39].
EGFR amplification
Piotrowska and colleagues reported EGFR T790M allele amplification in rociletinib-resistant clones [40]. Nukaga et al. found that amplification of the WT allele of EGFR is sufficient to mediate resistance to third-generation TKIs. The mechanism of drug resistance may be that EGFR gene amplification leads to a relatively low TKI concentration that is insufficient to exert inhibitory activity [41].
EGFR-independent resistance mechanisms
Not all patients develop resistance to TKIs through EGFR mutation; other pathways of acquiring resistance to third-generation EGFR-TKIs include the activation of alternative or downstream signaling pathways, epithelial interstitial resistance, epithelial–mesenchymal transition (EMT), histologic and phenotypic transformation, oncogene fusion, and cell cycle-related gene abnormalities.
Bypass signal pathway activation
Abnormal activation of MET
There are two main drug resistance mechanisms caused by the abnormal activation of MET: the MET exon 14 skipping mutation (METex14) and MET amplification. METex14 leads to the loss of ubiquitin ligase binding sites, a reduction in receptor ubiquitination, and persistent MET activation, resulting in tumor cell survival and acquired resistance [42]. After treatment with third-generation EGFR-TKIs, MET gene amplification can promote drug resistance by activating MAPK/ERK, which is independent of EGFR [43].
HER2 amplification
Hus et al. found that H1975 cells expressing HER2D16 were resistant to osimertinib in vitro. HER2D16 can form a heterodimer with EGFR or a disulfide homodimer, which activates downstream signaling to achieve resistance to osimertinib [44]. HER2D16-driven drug resistance occurs in a manner unrelated to the kinase Src. In addition, other mutations in exon 20 of HER2 have been reported, including point mutations (such as G776C and L755S) and insertions that cause downstream activation [45, 46]. HER2 mutation occurs in approximately 2–4% of NSCLC cases, mostly in lung adenocarcinoma (LUAD) [47]. In NSCLC, HER2 oncogenic amplification occurs in approximately 3% of cases without EGFR-TKI treatment and accounts for approximately 10% of cases with EGFR-TKI resistance [48].
AXL activation
AXL is an RTK that regulates cell survival, proliferation, metastasis, and other cellular functions. Abnormalities in the AXL gene can generate acquired resistance to TKIs by activating relevant downstream signaling pathways. Osimertinib was found to trigger AXL activation by closing the negative feedback loop with SPRY4, thus triggering inherent osimertinib resistance [49].
Overexpression of HGF
Hepatocyte growth factor (HGF) is the ligand of the proto-oncogene c-Met; it can trigger MET activation through EGFR bypass signaling and induce lung cancer resistance to EGFR-TKIs. Yano et al. [50] found that high expression of HGF was related to the acquired and intrinsic drug resistance to EGFR-TKIs in patients with lung cancer. Tumor specimens from patients with acquired drug resistance showed high expression of HGF in the context of MET amplification and the T790M mutation.
Fibroblast growth factor receptor (FGFR) signaling
FGFR is a transmembrane RTK. Studies have shown that FGFR1 is amplified and fibroblast growth factor 2 (FGF2) mRNA levels are increased in patients with osimertinib resistance, suggesting that the FGFR2-FGFR1 autocrine loop may be related to drug resistance [51]. Patients with the T790M mutation have been reported to show disease progression after treatment with osimertinib and nilotinib. The FGFR3-TACC3 fusion was detected in ctDNA [52, 53]. These findings suggest that abnormalities in the FGFR signaling pathway may underlie the mechanism of acquired resistance to third-generation EGFR-TKIs.
Insulin-like growth factor receptor 1 (IGF1R)
IGF1R, a transmembrane heterotetrameric protein encoded by the gene located on chromosome 15q26.3, is involved in promoting the growth of tumor cells. Abnormal activation of IGF1R leads to EGFR-TKI resistance [54].
Aurora kinases (AURKs)
AURKs are an important category of enzymes within the serine/threonine kinase family consisting of three mammalian isoforms: Aurora kinase A (AURK A), AURK B, and AURK C [55, 56]. AURK A and AURK B are highly expressed in dividing cells and play important roles in mitotic progression. Mammalian AURK A and AURK B share approximately 71% similarity in the carboxy-terminal catalytic domain [57]. Aberrant expression of AURK A and AURK B is involved in a broad range of solid cancers and is associated with adverse prognosis and drug resistance [58, 59]. In addition, Tanaka et al. [60] reported that targeting AURK B can prevent and overcome resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis.
Downstream signaling pathway activation
The activation of signaling pathways downstream of oncogenic receptors can regulate cell proliferation, cell cycle progression, and cell survival. Therefore, the direct regulation of downstream signaling pathway-related factors can lead to acquired resistance.
K-RAS mutation
An epidemiological meta-analysis found that K-RAS mutations are present in NSCLC patients, and all patients with K-RAS mutations were resistant to EGFR-TKIs [61]. K-RAS mutation is related to activation of the RAS-MAPK pathway. The common K-RAS mutations include G12S, G12D, G12A, Q61H, and A146T. Studies have found that inhibiting mutant K-RAS can reduce tumor growth and render NSCLC patients sensitive to EGFR inhibitors [62].
BRAF (v-RAF murine sarcoma viral oncogene homologue B1) mutation
BRAF is a serine/threonine protein kinase that plays a key role in the MAPK/ERK pathway, including in EGFR/RAS/RAF signal transduction. BRAF can regulate cell survival, proliferation, differentiation, and apoptosis, as well as tumor induction. Many BRAF mutations (G469A, V600E, and V599E) have been found in cancer, including lung cancer [63]. Ohashi et al. [64] reported that in patients with lung cancer, BRAF mutations can induce acquired resistance to EGFR-TKIs. Preclinical data showed that the BRAF V600E mutation has a strong association with resistance to the third-generation EGFR-TKI osimertinib in patients with T790M-mutated LUAD.
PI3K/AKT/mTOR
PIK3CA is a driver gene of LUAD. Mutation of PIK3CA can promote tumor cell invasion and increase the activity of downstream PI3Ks. Studies have shown that PIK3CA amplification or mutation (including E453K, E545K, and H1047R) may occur in patients with osimertinib resistance [52, 65]. Increased PI3K activity leads to the activation of various downstream kinases, thereby increasing PI3K/AKT/mTOR pathway activity in the absence of coupling to upstream EGFR phosphorylation.
STAT3 activation
STAT proteins, especially STAT3, are key downstream signal sensors of EGFR activation. In studies on NSCLC, Zhao et al. [66] discovered the clinical significance of JAK2/STAT3 in angiogenesis. Chaib et al. [67] found that osimertinib treatment activates not only STAT3 but also SrcYAP1 signaling, which may act downstream of IL-6 to promote disease progression.
Loss of PTEN
PTEN is a tumor suppressor gene that encodes a protein with lipid phosphatase activity and thus regulates cellular protein phosphatase activity. PTEN has dual antitumor effects and is a key component of many signaling pathways in the body. If mutation or deletion of the PTEN gene or downregulation of PTEN expression can reduce or eliminate its antitumor activity [68], loss of PTEN leads to hyperactivation of the PI3K/AKT signaling pathway and resistance to EGFR-TKIs, including osimertinib.
Hyperactivation of activated Cdc42-associated kinase 1 (ACK1)
Hyperphosphorylation of ACK1 and the subsequent activation of antiapoptotic signaling through the AKT pathway are associated with resistance to third-generation EGFR-TKIs [69].
c-Myc gene
The c-Myc gene is an important member of the MYC gene family. The c-Myc gene can induce cells to proliferate indefinitely and can promote cell division; these activities are related to the occurrence and development of various types of cancer. Studies have shown that c-Myc levels are substantially elevated in different EGFR-mutant NSCLC cell lines with acquired resistance to the third-generation EGFR-TKI osimertinib compared with the corresponding parental cell lines; moreover, these increased levels cannot be reduced by osimertinib. Consistently, c-Myc levels are elevated in the majority of EGFR-mutant NSCLC tissues from patients who relapsed on EGFR-TKI treatment compared with the corresponding baseline c-Myc levels prior to treatment [70]. These findings indicate that c-Myc mediates the therapeutic efficacy of third-generation EGFR-TKIs and the development of acquired resistance to these TKIs.
Other mechanisms
Epithelial–mesenchymal transition (EMT)
In EMT, cancer cells lose their epithelial properties through the loss of E-cadherin, leading to increased vimentin expression and transformation into a mesenchymal phenotype. A previous study found that osimertinib-resistant H1975 cells have EMT characteristics in the absence of other EGFR mutations [71]. EMT is a coordinated process involving multiple regulatory factors, such as EMT-induced transcription factors (EMT-TFs), noncoding RNAs (ncRNAs), and various extracellular signals. EMT-TFs play an important role in all stages of EMT; the most well-known EMT-TFs are members of the SNAIL, ZEB, and TWIST families. Many studies have shown that SLUG and SNAIL overexpression can induce drug resistance [72].
miRNAs and EMT
Long noncoding RNAs (lncRNAs) and microRNAs (miRNAs) play important roles in regulating EMT and TKI resistance. Although most miRNAs have been found to inhibit EMT, some have activity that promotes EMT, including miR-21 and miR-155 [73, 74]. Some miRNAs can promote TKI resistance by activating the PI3K/AKT/mTOR signaling pathway; for example, miR-21 and miR-23a can target PTEN and activate AKT, leading to resistance to EGFR-TKIs [75, 76].
Epigenetic alterations
Epigenetic modifications involved in cancer initiation and progression include changes in DNA methylation patterns and histone modifications. Epigenetic changes are common in the development and progression of lung cancer [77]. Studies have shown that epigenetic disorders can make cancer patients susceptible to acquired resistance to EGFR-TKIs [78].
Oncogene fusion
The AURA-3 and FLAURA trials showed that oncogene fusion might be one mechanism of osimertinib resistance; the identified fusions included transforming growth factor receptor (TGFR)-transforming acidic coiled-coil protein 3 (TACC3), neurotrophic receptor tyrosine kinase 1 (NTRK1)-thrombopoietin mimetic peptide 3 (TMP3), ERC1-RET, SPTBN1-ALK, coiled-coil domain-containing protein 6 (CCDC6)-RET, GOPC-ROS1, AGK-BRAF, NCOA4-RET, ESYT2-BRAF, and echinoderm microtubule-associated protein-like 4 (EML4)-ALK. Oncogene fusions can coexist with the EGFR C797S mutation, MET amplification, and BRAF mutation [79].
Cell cycle-related gene abnormalities
Recent studies have shown that changes in cell cycle-related genes, including the CDKN2A E27fs mutation, cyclin D (CCND) amplification, cyclin-dependent kinase 4/6 (CDK4/6) amplification, and cyclin E1 (CCNE1) amplification, can cause resistance to third-generation EGFR-TKIs [65].
Histologic and phenotypic transformation
Histopathological transformation to small cell lung cancer (SCLC) from NSCLC has been reported as a mechanism of acquired resistance to EGFR-TKIs in 3–15% of patients [80–83]. Transformed SCLC mainly occurs in Asian patients with adenocarcinoma harboring EGFR-TKI-sensitive mutations (such as the EGFR ex19del/T790M mutation) who are nonsmokers. The widely accepted hypothesis for this transformation posits that adenocarcinoma and SCLC originate from type II alveolar cells. RB1 and TP53 mutations might be involved in SCLC transformation but are not sufficient for the induction of complete transformation. Additional genomic alterations, including those that activate the PI3K/AKT family and downregulate NOTCH signaling and those affecting the MYC and SOX families, AKT pathway activation and other molecules, also participate in the transformation from EGFR-mutant NSCLC. However, the precise mechanisms in other cases are unclear [84]. In addition, squamous cell transformation was recently identified as a mechanism of acquired EGFR-TKI resistance that occurs in approximately 15% of patients who received osimertinib as both first- and second-line therapy. Similar to the case in SCLC transformation, the primary EGFR mutation is preserved in squamous cell transformation [85].
Immune escape
EGFR is expressed in different hematopoietic cell types, including macrophages, monocytes, and certain T-cell subsets. Therefore, it is likely that EGFR inhibitors can interfere with the function of these leukocytes. Immune checkpoint inhibitors (ICIs) have adverse effects and poor efficacy in patients with an EGFR mutation or a secondary T790M mutation, largely because of low tumor mutational burden and a noninflamed tumor microenvironment [86–88]. A previous study showed that secreted phosphoprotein 1 (SPP1) promotes macrophage M2 polarization and PD-L1 expression in LUAD, which may influence the response to immunotherapy. SPP1 levels might be a useful marker of immunosuppression in patients with an EGFR mutation and could provide therapeutic insight [89]. In addition, HGF, MET amplification, and EGFR T790M lead to the upregulation of PD-L1 expression in NSCLC and promote immune escape by tumor cells through different mechanisms mediated by the PI3K-Akt, MAPK, and NF-κB pathways [90].
Strategies for overcoming third-generation EGFR-TKI resistance
Fourth-generation EGFR-TKIs: overcoming the L858R/T790M and C797S resistance mutations
Third-generation EGFR-TKIs had the potential for remarkable achievements, if not for the numerous mutations. The C797S mutation, which is a covalent anchor mutation, is located in the ATP binding site of the EGFR tyrosine kinase domain. This missense mutation in exon 20 at position Cys797 blocks the ability of third-generation EGFR-TKIs to form a covalent bond in the ATP binding region, with a consequent decrease in the binding affinity between EGFR and an EGFR-TKI [91]. The combination of the C797S mutation with exon 19 deletion, L858R mutation, or T790M mutation was reported both in vitro and in vivo [91]. Studies have shown that drug-resistant lung cancer cells with two mutations (EGFR-activating mutation/C797S) are sensitive to first- and second-generation EGFR-TKIs. However, lung cancer cells with three mutations (EGFR-activating mutation/T790M/C797S) show resistant to third-generation EGFR-TKIs if the C797S and T790M mutations are both in the trans conformation. Nonetheless, these cells are still sensitive to the combination of first- and third-generation EGFR-TKIs [92]. Of note, if C797S and T790M are mutated in the cis conformation, the cells show resistance to all existing EGFR-TKIs (either alone or in combination) [93]. The resistance to third-generation EGFR-TKIs caused by the trans-C797S mutation can be overcome by drugs targeting different kinase binding sites, including allosteric inhibitors, ATP-competitive inhibitors, and “dual-site” inhibitors that occupy both the ATP binding site and an allosteric site.
Allosteric inhibitors
EGFR has three binding sites: an inactive site, a competitive ATP binding site, and an allosteric site. Ligands and drugs cannot bind the inactive site. Recent studies have mostly focused on either ATP-competitive inhibitors targeting the ATP binding site or molecules that bind the allosteric site, which causes a conformational change in the protein that inhibits the signaling cascade [94]. To overcome the resistance of EGFR-TKIs mediated by the T790M and C797S mutations and to further identify and explore compounds that bind outside the ATP binding domain of EGFR, researchers have pursued the development of allosteric inhibitors, and this appears to be a promising strategy. The newly developed fourth-generation mutant-selective allosteric inhibitors can overcome the T790M and C797S mutations that develop in response to third-generation EGFR-TKIs by binding to sites outside the ATP binding pocket of EGFR.
Through molecular phenotypic screening, Engel et al. obtained quinazoline compound 1a (1), which specifically inhibits the drug-resistant H1975 cell line (L858R/T790M); further modification addressed the problem of off-target activity (nonspecific inhibition). X-ray crystallography verified that compound 1a (1) fits well in the tyrosine kinase domain of c-Src [95] (Fig. 4).
Fig. 4.
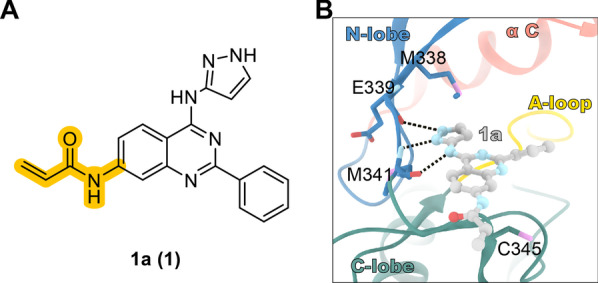
Screening hit 1a (1) from a phenotypic screen of NSCLC cell lines. A Structure of 1a (1). B X-ray crystal structure of 1a (1) in complex with c-Src-DM (PDB code: 5D12)
Jia et al. conducted counter-screening of active compounds against WT-EGFR and discovered the first non-ATP-competitive allosteric EGFR L858R/T790M/C797S inhibitor based on the thiazolamide scaffold (EAI001, 2) [96]. The X-ray crystal structure of EAI001 (2) in complex with EGFR T790M shows that EAI001 (2) can bind to the allosteric site of this receptor in the form of a “three-bladed propeller,” partly due to the outward displacement of the C-helix in the inactive conformation of the kinase. The hydrophilic side chain of the WT gatekeeper residue (Thr) cannot adapt to the thiazole of EAI001; therefore, there is no favorable interaction. The thiazole of EAI001 closely interacts with the hydrophobic side chain of Met790; specifically, aminothiazole group of EAI001 directly binds to Met790. The carbonyl oxygen of the isoindoline-1-one moiety is inserted between the mutant gatekeeper residue (Met) and the active site residue Lys745, forming another hydrogen bond with the ε-amine of the Lys745 side chain. The NH group of formamide acts as a hydrogen bond donor for Asp855 in the DFG motif. The cationic phenyl group occupies the hydrophobic pocket formed by Met766, Leu777, and Phe856. The 1-oxindolinyl group is exposed along the C-helix and extends to the solvent-accessible area. The ATP analog adenylyl imidodiphosphate (AMP-PNP) binds the active site cavity in an expected manner. The half maximal inhibitory concentration (IC50) of EAI001 (2) for EGFR L858R/T790M is 24 nmol/L, which is lower than that for WT-EGFR (IC50 > 50 μmol/L). The IC50s of EAI001 (2) for EGFR L858R and EGFR T790M is 0.75 μmol/L and 1.7 μmol/L, respectively. By introducing ortho-hydroxyl and meta-fluorine atoms on the benzene ring of EAI001 (2), the researchers synthesized another compound, EAI045 (3), that binds more tightly than EAI001 (2) to EGFR [96]. However, EAI045 has a major drawback: it must be used in combination with cetuximab to preserve its efficacy. While EAI045 (3) has good selectivity for WT-EGFR, cetuximab is expected to have off-target effects in clinical use. Lee et al. [97] designed the EGFR allosteric inhibitor TREA-0236 (4) based on the structure–activity relationships of EAI045 (3). The structure of EAI045 (3) was modified by cyclization, wherein the 2-aminothiazole amide was converted to quinazoline-4-one. To minimize hematological and methemoglobinemia toxicity and to obtain better safety and pharmacokinetic parameters, To et al. linked the 5-indole substituent to the isoindolinone of EAI001 (2) and obtained a new EGFR allosteric compound, JBJ-02-112-05 (5), with an IC50 of 15 nmol/L for EGFR L858R/T790M [98]. Additionally, EAI045 (3) was further optimized to generate another EGFR allosteric inhibitor, JBJ-04-125-02 (6), in which the 2-hydroxy-5-fluorophenyl of EAI045 (3) was combined with the phenylpiperazine on isoindolinone. This compound showed a significantly increased ability to inhibit EGFR L858R/T790M, with an IC50 of 0.26 nmol/L. Interestingly, combination with osimertinib enhanced the efficacy of JBJ-04-125-02 (6) and improved the targeting of JBJ-04-125-02 (6) to cancer cells [98], indicating that the combined use of covalent mutant-selective ATP-competitive inhibitors and EGFR allosteric inhibitors may be an effective treatment strategy for patients with EGFR-mutant disease (Fig. 5). Encouraged by the advantages of inhibiting allosteric sites in the EGFR tyrosine kinase domain, researchers have extensively designed and optimized allosteric inhibitors for EGFR [98–101], as shown in Table 1.
Fig. 5.

Chemical structures and structure–activity relationships of allosteric inhibitors. A The rational design of TERA-0236 (4), JBJ-02-112-05 (5), and JBJ-04-125-02 (6) and their inhibitory activities against EGFR. B X-ray cocrystal structure of EGFR with EAI001 (2) (PDB code: 5D41). C X-ray cocrystal structure of EGFR with EAI045 (3). D X-ray cocrystal structure of EGFR with EAI045 (in the active and inactive states) (PDB code: 5ZWJ). E X-ray cocrystal structure of EGFR with JBJ-04-125-02 (6) (PDB code: 6DUK)
Table 1.
EGFR allosteric inhibitors
| Compound (reference) | Structure | Activity | Interaction with EGFR allosteric site |
|---|---|---|---|
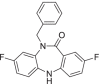
| 7, DDC4002 [100] |
|
EGFRL858R/T790M IC50 = 10 nmol/L EGFRL858R/T790M/C797S IC50 = 59 nmol/L |
Phe856 |
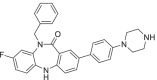
| 8 [100] |
|
EGFRL858R/T790M/C797S IC50 = 19 nmol/L |
Phe856 |
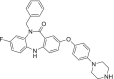
| 9 [100] |
|
EGFRL858R/T790M/C797S IC50 = 23 nmol/L |
Phe856 |
| 10 [100] |
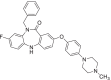
|
EGFRL858R/T790M/C797S IC50 = 13 nmol/L |
Phe856 |
| 11 [100, 101] |
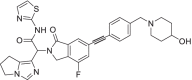
|
EGFRL858R/T790M IC50 = 1 nmol/L EGFRL858R/T790M/C797S IC50 = 5 nmol/L |
Phe856 |
| 12 [100, 101] |
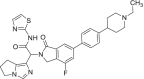
|
EGFRL858R/T790M IC50 = 3 nmol/L EGFRL858R/T790M/C797S IC50 = 4 nmol/L |
Phe856 |
The novelty of allosteric sites has attracted the attention of researchers, and these sites have become the most promising targets for the development of drugs for NSCLC and other diseases. Fourth-generation EGFR-TKIs require further investigation and development so that they are suitable as single-agent drugs targeting EGFR ex19del/T790M/C797S [98]. Allosteric inhibitors have now entered the stage of rapid development and are expected to enter clinical trials soon, with the goal of benefitting more patients.
ATP-competitive inhibitors
ATP-competitive inhibitors form one to three hydrogen bonds with amino acids in the hinge region of the target kinase, thereby mimicking the characteristic hydrogen bonds formed by the adenine ring of ATP. This type of inhibitor usually consists of a heterocyclic ring system that occupies the purine binding site, where it acts as a side chain scaffold that occupies the adjacent hydrophobic regions I and II. A high physiological or intracellular concentration of ATP may block the phosphotransferase activity of the target. The size of the amino acid side chain at the gatekeeper residue determines the relative accessibility of the hydrophobic pocket near the ATP binding site. To overcome drug resistance related to triple-mutant EGFR, it is particularly crucial to develop new ATP-competitive inhibitors based on structural design and optimization. Many ATP-competitive inhibitors have been reported; below, we summarize recent ATP-competitive inhibitors that can overcome the resistance to third-generation EGFR inhibitors (Table 2) [102–119].
Table 2.
ATP-competitive EGFR inhibitors
| Compound (reference) | Structure | Enzymatic activity | Biological activity | DMPK profile |
|---|---|---|---|---|
| 13, JND3229 [102] |

|
EGFRL858R/T790M/C797S IC50 = 5.8 nmol/L |
BaF3-EGFRL858R/T790M/C797S IC50 = 510 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 320 nmol/L |
N/A* |
| 14 [103] |
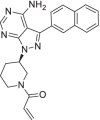
|
EGFRWT IC50 = 16 nmol/L EGFRL858R/T790M/C797S IC50 = 88 nmol/L |
A431 GI50 = 3600 nmol/L H1975 GI50 = 140 nmol/L |
N/A |
| 15 [104] |
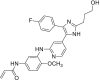
|
EGFRL858R/T790M/C797S IC50 = 8 nmol/L |
A431 EC50 = 4000 nmol/L H1975 EC50 = 400 nmol/L |
N/A |
| 16 [105, 106] |
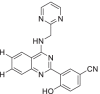
|
EGFRL858R/T790M/C797S IC50 = 630 nmol/L |
H1975 IC50 = 1200 nmol/L |
Liver microsomes (Human): t1/2 (min) = 66.6, CLint (mL/min/kg) = 20.8 |
| 17 [107] |
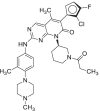
|
EGFRWT IC50 > 1000 nmol/L EGFRL858R/T790M/C797S IC50 = 27.5 nmol/L |
BaF3-EGFRL858R/T790M/C797S IC50 = 662 nmol/L |
T1/2 (rat, minutes) = 8.36, CLint (mL/min/kg) = 297.12 |
| 18 [108] |

|
EGFRL858R/T790M/C797S IC50 = 7.2 nmol/L |
HCC827 IC50 = 44 nmol/L H1975 IC50 = 400 nmol/L |
N/A |
| 19 [109, 110] |
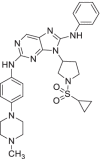
|
EGFRL858R/T790M/C797S IC50 = 18 nmol/L |
HCC827 IC50 = 0.88 nmol/L H1975 IC50 = 200 nmol/L A549 IC50 = 2910 nmol/L A431 IC50 > 10,000 nmol/L |
N/A |
| 20 [111] |
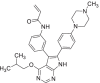
|
EGFRL858R/T790M/C797S IC50 = 8.5 nmol/L |
EGFR CTG EC50: HCC827 IC50 < 14 nmol/L H1975 IC50 = 51 ± 19 nmol/L A431 IC50 = 1675 ± 402 nmol/L A549 IC50 = 3700 nmol/L H358 IC50 = 3700 nmol/L |
In vivo PK (mice, IP, 20 mg/kg): AUCfree (h∙ng/mL) = 8.6 t1/2 (h) = 1.2 Cmax,free (μmol/L) = 0.012 |
| 21 [112] |

|
EGFRL858R/T790M/C797S IC50 = 3.1 nmol/L |
H1975 IC50 = 0.12 ± 0.09 μmol/L BaF3-EGFRL858R/T790M/C797S IC50 = 0.29 μmol/L BaF3-EGFR19D/T790M/C797S IC50 = 0.31 μmol/L |
N/A |
| 22 [113] |
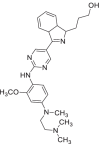
|
EGFRWT IC50 > 1000 nmol/L EGFRL858R/T790M/C797S IC50 = 218.3 nmol/L EGFRDel19/T790M/C797S IC50 = 15.3 nmol/L |
H1975 IC50 = 16,180 nmol/L A431 IC50 = 20,480 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 8510 nmol/L |
N/A |
| 23 [114] |
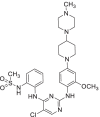
|
EGFRWT IC50 = 430 nmol/L EGFRDel19/T790M/C797S IC50 = 0.2 nmol/L |
BaF3-EGFRWT IC50 = 1000 nmol/L BaF3-EGFRDel19 IC50 = 180 nmol/L BaF3-EGFRDel19/T790M IC50 = 99 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 63 nmol/L |
N/A |
| 24 [115] |
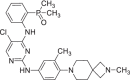
|
N/A |
Biochemical potency: BaF3 cells IC50 < 100 nmol/L Antiproliferative activity: BaF3 cells IC50 < 100 nmol/L |
N/A |
| 25 [116] |
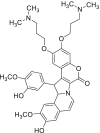
|
N/A |
Antiproliferative activity: PC-9-EGFRL858R/T790M/C797S IC50 = 595.7 nmol/L PC-9-EGFRDel19/T790M/C797S IC50 = 739.9 nmol/L A549 IC50 = 2861.7 nmol/L BaF3-EGFRWT IC50 = 519.82 nmol/L BaF3-EGFRL858R/T790M/C797S IC50 = 0.16 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 0.23 nmol/L |
N/A |
| 26 [117] |
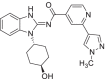
|
N/A |
BaF3-EGFRWT IC50 = 540 nmol/L BaF3-EGFRL858R/T790M/C797S IC50 = 48.2 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 12 nmol/L |
N/A |
| 27 [115] |
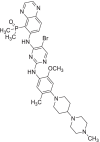
|
EGFRWT IC50 = 7.92 nmol/L EGFRL858R/T790M/C797S IC50 = 0.218 nmol/L EGFRDel19/T790M/C797S IC50 = 0.16 nmol/L |
Antiproliferative Activity: A431 IC50 = 154 nmol/L BaF3-EGFRDel19/T790M/C797S IC50 = 22 nmol/L |
In vivo PK (mice, per os, 15 mg/kg): AUC0-last = 57,037 (nmol/L∙h) t1/2 (h) = 10.0 Plasma (nmol/L), 2 h = 3553 Tumor (nmol/kg), 2 h = 16,667 |
| 28 [118] |
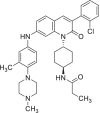
|
EGFRWT IC50 = 3.8 nmol/L EGFRL858R/T790M/C797S IC50 = 38.1 nmol/L |
BaF3-EGFRL858R/T790M/C797S IC50 < 1000 nmol/L |
N/A |
| 29, UPR1444 [119] |

|
EGFRWT IC50 = 30 ± 4.8 nmol/L EGFRL858R/T790M/C797S IC50 = 110 ± 33 nmol/L |
N/A | N/A |
*N/A not available
“Dual-site” inhibitors: occupying both the ATP binding site and the allosteric site
Based on the non-ATP-competitive EGFR L858R/T790M/C797S inhibitor EAI001 reported by Jia et al., the more potent compound EAI045 (3) was obtained through structural optimization [96]. EAI045 (3) binds to the allosteric site created by the outward displacement of the αC helix of EGFR, located next to the ATP binding pocket. Facilitated by molecular docking, researchers developed a series of new compounds that noncovalently occupy both the EGFR ATP binding site and the allosteric site; these fourth-generation reversible EGFR inhibitors have improved binding affinity for EGFR L858R/T790M/C797S, effectively compete with ATP, and further overcome resistance to third-generation EGFR inhibitors.
The compound vandetanib (30) [120] is a known EGFR inhibitor that shows moderate efficacy against EGFR L858R/T790M/C797S, with an IC50 value of 369.2 nmol/L. Via molecular docking simulation, Li et al. found that vandetanib can extend to the EGFR ATP binding pocket (gscore = − 8.2 kcal/mol). The docking model of vandetanib with EGFR T790M/V948R shows that the phenyl group of vandetanib binds the ATP binding site of EGFR, occupying a position such that it resembles the thiazole moiety of EAI001 (2). EAI001 (2) binds as a Y-shaped constellation in the allosteric site [121]. Modifying vandetanib to occupy both the ATP binding site and the allosteric site may be an effective way to improve its biological activity against EGFR L858R/T790M/C797S. To promote occupation of the allosteric site of EGFR, the structure of EAI045 (3) was modified such that the hydrophobic group oxyisoindole-2 phenylacetamide was introduced with an amide bond as the linker, generating compound 31. With this compound as a new lead, three moieties, namely the allosteric targeting region, the hinge targeting region, and the solvent exposure region, were studied and optimized. Finally, the EGFR L858R/T790M/C797S reversible inhibitor compound 32 (Fig. 6) was obtained, with an IC50 value of 2.2 nmol/L. The docking simulation showed that compound 32 occupies both the ATP binding region and the allosteric region. In addition, it extensively interacts with residues in the allosteric region, the solvent-accessible region, and the hinge region. The phenyl of the Y-shaped group (oxoisoindolin-2-phenylacetamide) and Phe856 of the allosteric cavity form π–π stacking interactions. Inside the ATP binding region, hydrogen bonds are formed between the quinazoline ring and the hinge residue Met793. In addition, the piperidine tail is surrounded by the solvent-exposed region. At a concentration of 0.1 μmol/L, compound 32 almost completely inhibited the phosphorylation of EGFR, showing comparable potency to that of EAI045 (3).
Fig. 6.

Chemical structures and structure–activity relationships of dual-site inhibitors: the rational design of compound 32 and its inhibitory activity against EGFR
To further design more potent inhibitors spanning both binding sites, considering the proximity of the ortho and allosteric positions, Wittlinger F et al. compared the binding of the EGFR ATP site inhibitor LN2057 (33) with the allosteric inhibitor EAI045 (3) and found that the 4-fluorophenyl of LN2057 (33) and the thiazole of EAI045 (3) had the same binding position [122]. Based on this, researchers designed and synthesized a series of compounds that combined a large portion of the isomerization inhibitor EAI045 (3) with the pyridyl-imidazole skeleton. For compound 34, the pyridinylimidazole scaffold partly binds the 2-fluoro-5-hydroxyphenyl moiety of EAI045 (3); 1-oxoisoindoline-2-yl was introduced into compound 35; and 1,3-dioxoisoindoline-2-yl was added to generate compound 36 to further explore the structure–activity relationship of the allosteric site. In addition, an N-(4-methoxyphenyl)acrylamide warhead was introduced to produce compound 37, and the influence of the C797-targeting capacity of these chimeric compounds, which are expected to form a covalent bond with C797, was assessed. The X-ray cocrystal analysis of the binding mode with EGFR T790M/V948R (Fig. 7) revealed that compounds 34 and 36 bind in the same way. Taking compound 36 as an example, the aminopyridine moiety forms a hydrogen bond with the M793 residue in the hinge region. The inhibitor is anchored at the ATP binding site, and the N atom of the imidazole moiety forms a hydrogen bond with K745, which is essential for the strong reversible binding of the imidazole skeleton. The phenylamide bond extending into the allosteric pocket is directed toward the T790M mutation, and the N atom on the amide forms hydrogen bonds with the T854 and D855 residues. Despite considerable efforts, the X-ray crystal structure of compound 37 in complex with EGFR was not obtained. Compound 37 was computationally docked to the EGFR T790M/V948R kinase domain, and the result was the same as that for compound 36. The methoxyphenyl acrylamide formed a covalent bond with C797. Importantly, no covalent binding of compound 37 to the EGFR L858R/T790M/C797S kinase domain was observed, confirming that this compound is a noncovalent inhibitor.
Fig. 7.

Chemical structures and structure–activity relationships of dual-site inhibitors. A Structure-guided design and synthesis of mutant-selective lead compounds and their inhibitory activities against EGFR. B Structural superposition of the ATP site binding inhibitor LN2057 (PDB code: 6V6K) and the allosteric inhibitor EAI045 (PDB code: 6P1L); C X-ray cocrystal structure of EGFR T790M/V948R with compound 34 (PDB code: 6WA2); D X-ray cocrystal structure of EGFR T790M/V948R with compound 36 (PDB code: 6WXN)
The inhibitory activity of the above compounds was tested, and the results showed that compound 34 exhibited strong inhibitory activity against all mutants, with IC50 values of 5–32 nmol/L, indicating that the introduction of 2-fluoro-5-hydroxyphenyl alone did not increase selectivity. With the introduction of oxyisoindolin-2-yl, the inhibitory activity of compound 35 decreased, but a certain inhibitory effect against EGFR L858R was observed. Furthermore, compound 1 inhibited all three EGFR mutants at the low nanomolar range. Compound 37 showed a moderate degree of mutation selectivity for WT-EGFR, possibly due to the methoxyphenyl acrylamide group. To assess the kinase selectivity of compound 37, a kinome screen including 335 WT kinases was performed; compound 37 exhibited high selectivity, with a selectivity score of 0.006 at an inhibitor concentration of 1 μmol/L. Next, the antiproliferative activity of these compounds was evaluated in Ba/F3 cells stably transfected with WT-EGFR, EGFR L858R, EGFR L858R/T790M, or EGFR L858R/T790M/C797S. Among the compounds, compound 37 showed an antiproliferative effect in the EGFR L858R and EGFR L858R/T790M cell lines, with IC50 values in the micromolar range in the presence and absence of cetuximab. The IC50 value of compound 37 in EGFR L858R Ba/F3 cells (1.2 ± 0.07 μmol/L) was comparable to that of EAI045 (3) combined with cetuximab (840 ± 700 nmol/L). Although compound 37 is potent and selective for kinases, its cellular activity is suboptimal. Kinase selectivity was achieved by increasing the molecular weight of the lead compound and increasing the number of hydrogen bond donors and acceptors, but these changes may have produced limited cell permeability and effects on cell viability; thus, this compound lacked sufficient activity in cells expressing EGFR L858R/T790M/C797S.
The selective EGFR inhibitor (compound 37) designed and developed in this study can bind to both the ATP site and the allosteric site of the EGFR kinase domain. Adding allosteric inhibitor elements to the compound skeleton at the ATP binding site contributes to the mutation selectivity of these compounds. The designed compound 37 has good kinase activity but nonideal cell activity. Future research and development could optimize the structure of this lead compound to further enhance its cellular activity.
PROTAC technology
Allosteric EGFR degrader
Resistance to third-generation EGFR-TKIs is a major obstacle to clinical targeted therapy. Due to changes in the EGFR protein [123], some kinase inhibitors are restricted to the catalytic pocket [124]. A proteolysis-targeting chimera (PROTAC) induces the proteasomal degradation of the target by recruiting it to a specific E3 ligase. The eradication of EGFR protein from cancer cells provides a promising strategy for overcoming drug resistance. The allosteric EGFR degrader is a heterobifunctional compound based on allosteric EGFR inhibitors. It includes a small molecule (protein-of-interest (POI) ligand) that binds the target protein and a small-molecule E3 ligase ligand that recruits cereblon (CRBN), von Hippel–Lindau (VHL), cellular inhibitor of apoptosis protein 1 (cIAP1) or murine double minute 2 (MDM2). After the addition of a linker connecting the two parts [125, 126], these chimeras can degrade mutant EGFR without affecting WT-EGFR.
Compared with classic “occupying” inhibitors, allosteric EGFR degraders can completely eliminate the function of the target protein, thereby improving the phenotypic potency. Moreover, since PROTAC molecules usually do not require strong binding to targets or long-term retention to achieve protein degradation, the development of drug-induced resistance mutations may be prevented. Compared with kinase inhibitors, PROTACs have the advantages of activity at lower concentrations, limited dose-dependent toxicity, and the potential to overcome drug resistance and target drug refractory disease [127–132]. These molecules have attracted considerable attention from academia and industry and have become an attractive therapeutic strategy in drug discovery.
Based on EAI001 (2), a compound that buries deeply in the allosteric pocket [96], Jang et al. introduced 1-(pyridin-2-yl)piperazine at the 6 position of isoindolinone and synthesized JBJ-07-149 (38), which has an IC50 value of 1.1 nmol/L for EGFR L858R/T790M. In combination with cetuximab, JBJ-07-149 has a half maximal effective concentration (EC50) of 0.148 nmol/L for EGFR L858R/T790M. However, this compound was less potent in the proliferation assay (EC50 = 4.9 nmol/L) [133].
Based on JBJ-07-149 (38), different linkers that bind the piperazine group and connect the CRBN ligand were evaluated. The compound with 3-PEG as the linker (DDC-01-163, 39) showed the strongest antiproliferative activity for EGFR L858R/T790M (Fig. 8). DDC-01-163 (39) induced the selective degradation mutant EGFR and inhibited the proliferation of cells expressing mutant EGFR in a dose- and time-dependent manner. DDC-01-163 (39) showed no activity in WT-EGFR Ba/F3 cells (EC50 > 10 μmol/L) but inhibited the proliferation of EGFR L858R/T790M Ba/F3 cells, including those expressing EGFR L858R/T790M (EC50 = 0.096 μmol/L), EGFR L858R/T790M/C797S (EC50 = 0.041 μmol/L) and EGFR L858R/T790M/L718Q (EC50 = 0.028 μmol/L). The results in H1975 cells were consistent with those in Ba/F3 cells. Osimertinib-resistant cell lines treated with 0.1 μmol/L DDC-01-163 (39) showed EGFR L858R/T790M/C797S and EGFR L858R/T790M/L718Q degradation rates of 74% and 71%, respectively.
Fig. 8.

Chemical structures and structure–activity relationships of an EGFR-targeted PROTAC: the rational design of DDC-01-163 (39)
Jang et al. also identified the 2-hydroxy-5-fluorophenyl allosteric inhibitor JBJ-04-125-02 (6), which can be used as a single agent to inhibit the proliferation of Ba/F3 cells. Following the same strategy as that used to develop DDC-01-163 (39), this group designed JBJ-04-125-02 (6) as a PROTAC molecule and synthesized the allosteric EGFR degrader JBJ-07–038 (40) (EC50 = 0.48 μmol/L). In addition, JBJ-07-200 (41) (EC50 = 0.15 μmol/L) was obtained by replacing the hydroxyl group of JBJ-04-125-02 (6) with fluorine (Fig. 9), which could potentially improve membrane permeability [133]. It is highly anticipated that the further characteristic optimization and development of allosteric EGFR PROTACs will produce a valuable therapeutic strategy that will benefit more patients with EGFR-mutant disease.
Fig. 9.

Chemical structures and EGFR inhibitory activities of EGFR-targeted PROTACs: the rational design of JBJ-07-200 (41)
According to the first report by Zhao et al., EGFR degradation induced by PROTACs may be related to the autophagy pathway [134]. Qu et al. [135] demonstrated for the first time that in addition to the well-known ubiquitin/proteasome pathway, the ubiquitin/autophagy/lysosomal pathway participates in PROTAC-induced EGFR degradation. Based on the EGFR inhibitor canertinib (41) and the CRBN ligand pomalidomide (an E3 ubiquitin ligase ligand), researchers generated two novel EGFR PROTACs (Fig. 10), namely SIAIS125 (42) and SIAIS126 (43). These two EGFR degraders showed effective and selective antitumor activity in EGFR-TKI-resistant lung cancer cells.
Fig. 10.

Chemical structures and EGFR inhibitory activities of novel EGFR PROTACs (through the ubiquitin/autophagy/lysosomal degradation system): the rational design of SIAIS125 (42) and SIAIS126 (43)
Dual PROTACs
The basic goal of modern drug discovery is to develop efficient and selective drugs for specific targets. However, complex diseases such as cancer usually result from interactions among multiple factors, synergistic effects of multiple disease-modifying factors, the upregulation of multiple receptors, and crosstalk between signaling networks. Tumor cells readily gain drug resistance by upregulating an alternative factor or transforming the signaling pathway that promotes proliferation; therefore, treatment focused on only a single target has limitations. In addition to its issues related to drug resistance, single-target drugs also show reduced efficacy and can decrease the quality of life of patients due to side effects and tissue toxicity.
To overcome the deficiencies of single-target drugs, single hybrid molecules fused to two or more pharmacophores have been designed to simultaneously target two or more antitumor epitopes or targets. These hybrid molecules can simultaneously modulate multiple targets or pathways and thus generally have better efficacy with fewer side effects. Based on this information and inspired by the great success of dual-targeted drugs, especially dual-specific antibodies, Professor Li et al. combined the concepts of PROTACs and dual targeting; this group used trifunctional natural amino acids as starlike core linkers to connect two independent inhibitors, gefitinib (44) and olaparib (45), that are linked to CRBN or VHL E3 ligands. The synthesized novel dual PROTACs can successfully and simultaneously degrade EGFR and poly(ADP-ribose) polymerase (PARP) in cancer cells [136]. Among the developed compounds, compound DP-V-4 (46) exhibited the best ability to degrade EGFR and PARP in a dose- and time-dependent manner in H1299 cells and human epidermal carcinoma A431 cells (Fig. 11). As the first successful example of a dual PROTAC, this research will inject new vitality into the field of combination therapy for cancer. Moreover, these findings will broaden the potential applications of the PROTAC method, open new fields of drug discovery, and overcome the limitations of single-target therapy against EGFR.
Fig. 11.

Chemical structure and antiproliferative activity of the dual PROTAC DP-V-4 (46) (through the ubiquitin–proteasome system)
Another new technology is the autophagy-targeting chimera (AUTAC), a small molecule that targets protein degradation through autophagy and contains both a degradation tag (guanine derivative) and a warhead to provide target specificity; AUTACs have a wider substrate panel than the ubiquitin–proteasome system [137–139]. Therefore, there is considerable potential for the design and development of AUTAC molecules to degrade EGFR.
Monoclonal antibodies and ADCs
For patients with EGFR-mutant disease, there are targeted therapies for tumors harboring EGFR-TKI-sensitizing mutations [140]. The EGFR monoclonal antibody can bind to the extracellular domain of EGFR to compete with EGF binding, thereby blocking downstream signaling. The variable fragment (Fv) is composed of parts of the light chain and heavy chain of the antibody and has unique antigen recognition function. The constant region (Fc) mediates innate immunity related to monoclonal antibodies, mainly by binding immune factors or cells to exert antitumor effects. These properties make antibodies a favorable approach in targeted therapy, especially in combination with other strategies. In addition, the internalization and degradation of EGFR monoclonal antibody and receptor complexes can downregulate EGFR on the surface of cancer cells. EGFR monoclonal antibodies are now standard-of-care therapies for head and neck cancer and colorectal cancer. Common EGFR monoclonal antibodies include cetuximab, necitumumab, panitumumab, matuzumab, and nimotuzumab. Antibody–drug conjugates (ADCs) are composed of three moieties: the antibody, linker, and drug (especially those with potential cytotoxicity) (Fig. 12). Antibodies are equivalent to precise arrows, and highly active cytotoxic drugs (the payload) correspond to the gunpowder on the arrows; these drugs mainly include tubulin inhibitors (monomethyl auristatin E, monomethyl auristatin F, mertansine, and ravtansine) and DNA-damaging agents (those that cause DNA double-strand breaks, DNA alkylation, DNA intercalation, and DNA cross-linking). It is difficult to effectively kill tumor cells with only cytotoxic drugs, but monoclonal antibodies alone are too inefficient. ADCs composed of both the cytotoxic drug and a monoclonal antibody represent a more powerful combination. ADCs can precisely target tumor cells by combining highly specific monoclonal antibodies with highly toxic cytotoxic drugs, thereby achieving a precise attack on EGFR-TKI-resistant cancer cells and filling the gap between antibody drugs and traditional chemotherapy drugs. The ADC approach can improve both the drug specificity and the treatment window. Being precise and efficient, ADCs have therapeutic potential across cancer types and can also induce tumor cell death via the bystander effect [141].
Fig. 12.

The mechanism of ADCs
He et al. developed a new ADC targeting EGFR, namely SHR-A1307 (47) (Fig. 13), for the treatment of solid tumors resistant or refractory to EGFR-targeted therapy [142]. SHR-A1307 (47) has intermediate ability to block EGFR affinity for hR3 and selectively binds to cancer cells expressing EGFR while avoiding inhibitory effects on normal cells. In addition to increasing stability and reducing systemic toxicity, Fc domain engineering improved the pharmacokinetics. Although less frequent drug administration may reduce toxin accumulation, effective tumor cell killing with minimal toxicity were observed. In addition, SHR-A1307 (47) can effectively kill cancer cells that do not respond to current EGFR inhibitors and shows low nanomolar in vitro cytotoxicity in a broad spectrum of cancer cells with different drug resistance mutations, thus providing an attractive treatment opportunity to overcome the drug resistance of patients with EGFR-overexpressing tumors.
Fig. 13.

Chemical structure of ADCs targeting EGFR: SHR-A1307 (47) and MRG003 (48) (*Specific research data not disclosed)
MRG003 (48) [143, 144], the first EGFR ADC to enter the clinical trials in China, is composed of a humanized anti-EGFR monoclonal antibody and the tubulin inhibitor MMAE coupled through a degradable VC (Val-Cit) linker (Fig. 13). The phase I dose escalation and expansion study for patients with refractory solid tumors has been completed. Based on the results of the phase Ia and Ib clinical trials, Lepu Biosciences is currently conducting phase II clinical trials of MRG003 monotherapy in China for recurrent or metastatic advanced head and neck squamous cell carcinoma, advanced NSCLC, biliary tract cancer, and nasopharyngeal carcinoma.
Combination therapy strategy
Resistance to third-generation EGFR inhibitors mediated by EGFR-independent mechanisms can develop through the activation of alternative bypass pathways and abnormal downstream signal transduction closely related to tumor growth, invasion and metastasis. In the clinic, HER2 mutation, high HGF expression, and abnormal activation of MET, AXL, IGF1R and the FGFR pathway were found in patients with acquired resistance to third-generation EGFR-TKIs. Mutation or abnormal expression of EGFR signaling pathway-related genes involved in the Ras/Raf/MEK/ERK/MARK, PI3K/PDK1/Akt, PLC-γ and JAK/STAT pathways was also found. Importantly, these aberrations can coexist in the same tumor and with EGFR-TKI tertiary mutations, which are the basis for the complexity and heterogeneity of cancer evolution in response to EGFR-TKI treatment. Therefore, in combination with third-generation EGFR-TKIs, targeting important components of alternative bypass pathways (Table 3) [145–154] and downstream signal transduction pathways (Table 4) [155–164] appears to be a promising treatment strategy.
Table 3.
Combination therapy with the bypass pathway target
| Target | Representative compound* | Structure | Reference |
|---|---|---|---|
| MET | Cabozantinib |
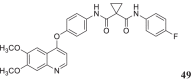
|
[145, 146] |
| MET | Crizotinib |
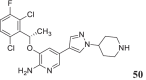
|
[147] |
| MET | Savolitinib |
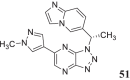
|
[148] |
| FGFR | AZD4547 |
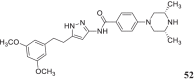
|
[149] |
| ALK | Lorlatinib |
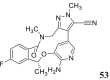
|
[150] |
| ALK | Brigatinib |
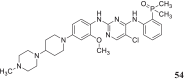
|
[151] |
| HER2 | JQ1 |

|
[152] |
| HER2 | Trastuzumab-DM1 |
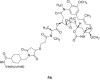
|
[153] |
| BRAF V600E | Encorafenib (LGX818) |
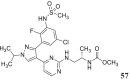
|
[64] |
| AURK B | PF-03814735 |
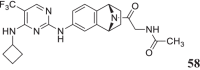
|
[60, 154] |
*Osimertinib is a representative third-generation EGFR-TKI
Table 4.
Combination therapy with targets in downstream signaling pathways
| Target | Representative compound* | Structure | Reference |
|---|---|---|---|
| MEK | Trametinib |

|
[155, 156] |
| MEK | Selumetinib |
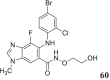
|
[155–157] |
| MEK | PD0325901 |
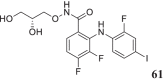
|
[155] |
| AKT | Uprosertib (GSK2141795) |
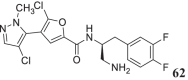
|
[158] |
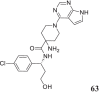
| AKT | Capivasertib (AZD5363) |
|
[158] |
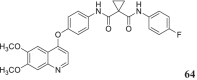
| AXL | Cabozantinib |
|
[159] |
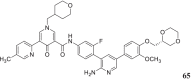
| AXL | DS-1205b |
|
[160] |
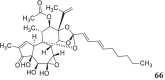
| AXL | Yuanhuadine (YD) |
|
[161] |
| AXL | Bemcentinib (R428) |
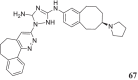
|
[162] |
| ACK1 | (R)-9b |
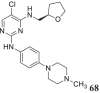
|
[163, 164] |
*Osimertinib is a representative third-generation EGFR-TKI
Multitarget inhibitors
Cancer is a multifactorial disease, and single-target treatments may have poor efficacy. As clinical targeted therapy, EGFR kinase inhibitors are effective only when the cancer cells contain specific EGFR-activating mutations that alter downstream signaling [165]. Moreover, only a small proportion of patients benefit from EGFR inhibitors [2]. In addition to activating mutations at the EGFR locus that lead to drug resistance, a large number of genetic and epigenetic abnormalities may also lead to resistance to third-generation EGFR-TKIs. The emergence of intrinsic and acquired resistance requires appropriate strategies to prevent serious side effects. Combination therapy has additive or even synergistic effects, but due to various dose-limiting toxicities and drug–drug interactions caused by changes in pharmacokinetics, the simultaneous use of two or more drugs in the clinic is challenging. Therefore, as an alternative to combination therapy, drugs targeting two or more objects have a lower risk of drug–drug interactions and better pharmacokinetic and safety profiles, which helps mitigate poor patient compliance, off-target effects, and high development costs. Such treatment regimens are more flexible and can represent an effective strategy for cancer therapy [166, 167]. The effectiveness of multitarget kinase inhibitors of WT and/or mutant EGFR has been extensively studied (Table 5) [59, 168–197]. Some EGFR-mutant cell lines are sensitive to multitarget inhibition and maintain certain levels of activity, highlighting the selectivity of multitarget compounds and suggesting that multitarget inhibition can be used to circumvent acquired multidrug resistance to EGFR-targeted therapy without serious side effects.
Table 5.
Multitarget inhibitors
| Number | Target | Pharmacophores | Structure | Activity |
|---|---|---|---|---|
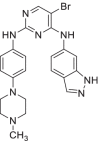
| 69 [168] | EGFR/FGFR1 | Pyrimidine-2,4-diamines |
|
EGFRL858R/T790M IC50 = 43.1 nmol/L EGFRWT IC50 = 1138.7 nmol/L FGFR1WT IC50 = 17.6 nmol/L H1975 cells IC50 = 336.3 nmol/L |
| 70 [169] | EGFR/Src | Pyrimidine-4-amines |
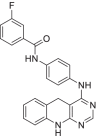
|
K562 cells IC50 = 220 nmol/L A549 cells IC50 = 250 nmol/L EGFR inhibition rate = 33.15% (10 µmol/L) Src inhibition rate = 72.12% (1 µmol/L) |
| 71 [170] | EGFR/HER4 | 3-Cyanoquizolines |
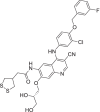
|
EGFRL858R IC50 = 419 nmol/L EGFRWT IC50 = 2.4 nmol/L HER4 IC50 = 0.03 nmol/L |
| 72 [171] | EGFR/COX2 | 1,3,4-Oxadiazole scaffold |
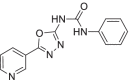
|
EGFR IC50 = 280 nmol/L COX2 IC50 = 170 nmol/L UO-31 cells IC50 = 5800 nmol/L |
| 73 [172] | EGFR/BRAF | Spirobenzo[h]chromene derivatives |
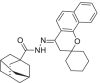
|
EGFR IC50 = 1200 nmol/L BRAF IC50 = 2600 nmol/L A549 cells IC50 = 1780 nmol/L MCF-7 cells IC50 = 4090 nmol/L HT-29 cells IC50 = 4450 nmol/L |
| 74 [173] | EGFRT790M/ALK | Pyrimidine-2,4-diamines |
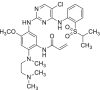
|
ALK IC50 = 18 nmol/L EGFRWT IC50 = 151 nmol/L EGFRT790M IC50 = 2 nmol/L EGFRL858R/T790M IC50 = 4 nmol/L DFCI032 cells IC50 = 170 nmol/L DFCI076 cells IC50 = 820 nmol/L |
| 75 [174] | EGFRWT and mutant EGFR/ALK | Pyrimidine-2,4-diamines |
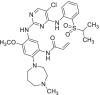
|
EGFRWT IC50 = 108 nmol/L EGFRT790M IC50 = 3.9 nmol/L EGFRL858R/T790M IC50 = 3.6 nmol/L ALKWT IC50 = 9.8 nmol/L ALKR1275Q IC50 = 0.82 nmol/L ALKL1196M IC50 = 0.59 nmol/L ALKF1174L IC50 = 0.92 nmol/L ALKC1156Y IC50 = 1.0 nmol/L H1975 cells GI50 = 15 nmol/L H3112 cells GI50 < 0.3 nmol/L |
| 76 [175] | EGFR/ATX | Pyrimidine-4-amines |
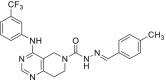
|
EGFR IC50 = 24.2 nmol/L ATX IC50 = 29.1 nmol/L A549 cells IC50 = 4960 nmol/L MKN-45 cells IC50 = 3430 nmol/L SGC cells IC50 = 2910 nmol/L CFs cells IC50 = 1490 nmol/L |
| 77 [176] | EGFR/AURK A | Pyrimidine-4-amines |
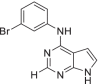
|
AURK A IC50 = 1990 nmol/L EGFR IC50 = 3.76 nmol/L |
| 78 [177] | EGFR/IGF1R | Pyrimidine-2-amines |
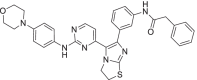
|
EGFR IC50 = 35.5 nmol/L EGFRT790M IC50 = 66.0 nmol/L IGF1R IC50 = 52.0 nmol/L |
| 79 [178] | EGFR/tubulin | Pyrimidine-4-amines |
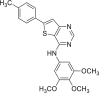
|
EGFR IC50 = 30 nmol/L Tubulin assembly IC50 = 710 nmol/L HeLa cells IC50 = 1 nmol/L HT-29 cells IC50 = 20 nmol/L Jurkat cells IC50 = 1 nmol/L RS4;11 cells IC50 = 1 nmol/L |
| 80 [179] | EGFR/tubulin | Chalcones |
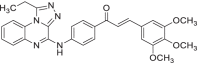
|
EGFR IC50 = 39 nmol/L Tubulin polymerization IC50 = 8840 nmol/L MCF-7 cells IC50 = 1650 nmol/L HCT-116 cells IC50 = 3610 nmol/L |
| 81 [180] | EGFR/AKT | Chalcones |
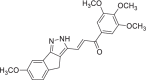
|
A549 cells IC50 = 3820 nmol/L MDA-MB-231 cells IC50 = 5890 nmol/L SKBR3 cells IC50 = 4790 nmol/L |
| 82 [181] | EGFR/HDACs | Pyrimidine-2-amines |
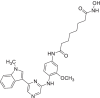
|
EGFRWT IC50 = 5700 nmol/L EGFRT790M IC50 = 5000 nmol/L HDACs IC50 = 85 nmol/L A549 cells IC50 = 2190 nmol/L HeLa cells IC50 = 1850 nmol/L MDA-MB-231 cells IC50 = 600 nmol/L MDA-MB-468 cells IC50 = 230 nmol/L |
| 83 [182] | EGFR/PDGFR-β | Pyrimidine-2,4-diamines |
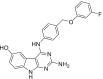
|
EGFR Ki IC50 = 170 nmol/L PDGFR-β IC50 = 81 nmol/L |
| 84 [183] | EGFR/NF-κB | Quinazolines |
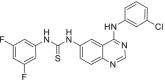
|
EGFR IC50 = 60.1 nmol/L NF-κB IC50 = 300 nmol/L |
| 85 [184] | EGFR/c-Met | 1,2,4-Oxadiazole derivate |
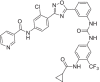
|
MDA-MB-231 cells IC50 = 200 nmol/L A459 cells IC50 = 200 nmol/L PC9 cells IC50 = 500 nmol/L H1975 cells IC50 = 300 nmol/L CL68 cells IC50 = 400 nmol/L CL97 cells IC50 = 500 nmol/L |
| 86 [185] | EGFRT790M/c-Met | Pyrimidine-2-amines |

|
EGFRT790M IC50 = 97 nmol/L c-Met IC50 = 518 nmol/L |
| 87 [186] | EGFR/VEGFR-2 | Pyrimidine-2,4-diamines |
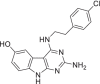
|
EGFR Ki IC50 = 80 nmol/L VEGFR-2 Ki IC50 = 3240 nmol/L NCI-H460 cells GI = 25% (10 µmol/L) |
| 88 [187] | EGFR/VEGFR-2 | Quinazolines |
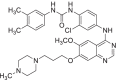
|
EGFR IC50 = 1.0 nmol/L VEGFR-2 IC50 = 79.0 nmol/L HT-29 cells IC50 = 1760 nmol/L MCF7 cells IC50 = 7280 nmol/L |
| 89 [188] | EGFR/VEGFR-2 | Quinazolines |
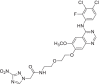
|
EGFR IC50 = 0.69 nmol/L VEGFR-2 IC50 = 67.84 nmol/L |
| 90 [59] | EGFR/VEGFR-2 | Quinazolines |
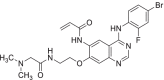
|
EGFR IC50 = 2.0 nmol/L VEGFR-2 IC50 = 103.0 nmol/L A431 cells IC50 = 14.0 nmol/L H1975 cells IC50 = 130.0 nmol/L |
| 91 [189] | EGFR/VEGFR-2 | Quinazolines |
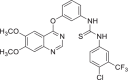
|
EGFR IC50 = 20 nmol/L VEGFR-2 IC50 = 50 nmol/L |
| 92 [190] | EGFR/HER2 | Quinazolines |
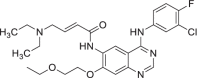
|
EGFR IC50 = 0.69 nmol/L HER2 IC50 = 42.1 nmol/L NCI-H1975 cells IC50 = 12.20 nmol/L HCC827 cells IC50 = 0.31 nmol/L A431 cells IC50 = 1.52 nmol/L MDA-MB-453 cells IC50 = 0.62 nmol/L |
| 93 [191] | EGFR/HER2 | Pyrimidine-4-amines |
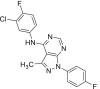
|
EGFR IC50 = 186 nmol/L VEGFR-2 IC50 = 254 nmol/L |
| 94 [192] | EGFR/HER2 | 3-Cyanoquizolines |
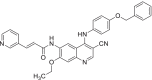
|
EGFR IC50 = 597 nmol/L IGF1R IC50 = 908 nmol/L A431 cells IC50 = 1890 nmol/L SKBR3 cells IC50 = 1930 nmol/L |
| 95 [193–195] | EGFR/HER2 | Pyrimidinones |
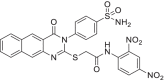
|
EGFR IC50 = 60 nmol/L HER2 IC50 = 300 nmol/L A549 cells IC50 = 280 nmol/L |
| 96 [196] | EGFR/CSK | Chalcones |
|
EGFR IC50 = 11,120 nmol/L CSK IC50 = 5160 nmol/L |
| 97 [197] | EGFR/CAIX | Quinazolines |
|
EGFRWT IC50 = 27.0 nmol/L EGFRT790M IC50 = 9.2 nmol/L hCAII IC50 = 278.2 nmol/L hCAIX IC50 = 115.0 nmol/L A549 cells (hypoxia) IC50 = 2210 nmol/L A549 cells (normoxia) IC50 = 6450 nmol/L HeLa cells IC50 = 1850 nmol/L H1975 cells (hypoxia) IC50 = 1050 nmol/L H1975 cells (normoxia) IC50 = 1940 nmol/L |
Natural products
The discovery of natural products offers new scaffolds for drug development. Natural products are an important source of compounds to overcome resistance to third-generation TKIs and provide ample possibilities for new drug discovery. Honokiol (HNK) (98) is a natural product purified from Magnolia used as a human nutritional supplement, with good tolerance and safety profiles. Many preclinical studies have shown that HNK (98) has potential antitumor activity against different types of cancer. Zang et al. proved that the decrease in Mcl-1 and the increase in BIM are the key mechanisms by which osimertinib induces the apoptosis of NSCLC cells with EGFR-TKI-sensitive mutations. HNK (98) and its derivative CAz-p (99) in combination with osimertinib effectively reduced the survival and induced the apoptosis of EGFR ex19del/C797S (trans) double-mutant PC-9/2 M cells and EGFR ex19del/T790M/C797S (cis) triple-mutant PC-9/3 M cells [198]. It is highly encouraging that HNK (98) and its derivatives may overcome clinical resistance to third-generation TKIs.
Overexpression of MCL-1 induces acquired resistance to osimertinib. Combination therapy with MCL-1 inhibitors and osimertinib is a potential strategy to overcome resistance. Bufalin (100) is a natural product that belongs to the class of bufadienolide analogs. A recent study found that bufalin (100) can reverse acquired resistance to osimertinib by inducing Ku70-mediated Mcl-1 degradation. Moreover, combined treatment with bufalin (100) and osimertinib triggered significant cell apoptosis and increased the levels of cleaved caspase-3 and PARP [199].
Wighteone (101) is a natural flavonoid compound widely found in plants. Sun et al. reported that wighteone (101) docks at the ATP binding site of EGFR L858R/T790M and forms two hydrogen bonds with the carbonyl group of Gln791 and the amino group of Met793, indicating that it may directly bind to EGFR L858R/T790M. Wighteone has a significant inhibitory effect on Ba/F3 and NCI-H1975 cells expressing EGFR L858R/T790M, with IC50 values of 1.88 μmol/L and 5.70 μmol/L, respectively [200] (Fig. 14).
Fig. 14.

Chemical structures of natural products with their synthetic analogs and other inhibitors (no specific IC50 values are shown due to variations in EGFR-mutant cell line survival and apoptosis assays)
Other strategies
EGFR degradation based on the FBXL2-Grp94-EGFR axis
Xiao’s research group found that the F-box protein Fbxl2 (an E3 ubiquitin ligase) can target EGFR and EGFR-TKI-resistant mutants for proteasome-mediated degradation independent of EGF stimulation. They also discovered that glucose regulatory protein 94 (Grp94) protects EGFR from degradation by blocking the binding of Fbxl2 to EGFR. Through virtual screening of the DrugBank database, small compounds that can bind to the Fbxo3-apag domain were scored. Nebivolol (102) can be placed in the dumbbell-shaped cavity of the APAG region. There are 5 amino acid residues in the center of this cavity (I331, E341, T367, T368 and F369); T367 and T368 project into the cavities of complementary shapes, forming hydrophobic interactions with the ligand. The binding affinity of the Fbxo3 protein for endogenous Fbxl2 is greatly reduced when these five amino acids are mutated individually or in combination. Data suggest the potential of nebivolol (102) as a small molecule that can disrupt the Fbxo3–Fbxl2 interaction. Increasing Fbxl2 levels with nebivolol (102) (Fig. 14) in combination with osimertinib or a Grp94 inhibitor (ganetespib) to target the FBXL2-Grp94-EGFR axis and thus destabilize EGFR is a possible therapeutic strategy to overcome resistance to third-generation EGFR-TKIs [201].
AKR1B1 inhibitors
Zhang et al. discovered that aldehyde ketone reductase family 1 member B1 (AKR1B1) interacts with STAT3 and activates the cystine transporter solute carrier family 7 member 11 (SLC7A11), which in turn leads to enhanced cystine uptake, glutathione synthesis flux, clearance of reactive oxygen species (ROS), protection against cell death, and EGFR-TKI resistance. The use of selective inhibitors (including the clinically approved anti-diabetic drug epalrestat) to inhibit AKR1B1 can restore the sensitivity of drug-resistant cell lines to EGFR-TKIs and delay drug resistance in mice harboring xenografted tumors derived from lung cancer patients [202].
PGAM1 inhibitors
Phosphoglycerate mutase 1 (PGAM1) is an important enzyme in the glycolysis pathway and is related to tumor cell metastasis [203]. HKB99 (103) (Fig. 14) is an allosteric inhibitor of PGAM1 that significantly inhibits the growth and metastasis of NSCLC by affecting the metabolic activity and nonmetabolic functions of PGAM1 [204]. The docking model of the PGAM1-HKB99 complex shows that HKB99 (103) binds to the allosteric site of the adjacent substrate-binding pocket of PGAM1, thereby inhibiting the conversion of 3-PG to 2-PG and significantly reducing the metabolic activity of PGAM1. In addition, HKB99 (103) can allosterically bind to PGAM1, weaken the interaction between PGAM1 and ACTA2, and inhibit the growth and metastasis of erlotinib-resistant lung cancer cells [205, 206]. Therefore, PGAM1 is a metabolic enzyme that may overcome EGFR-TKI resistance.
Nonoverlapping allosteric pockets—the X-Pocket
Qiu et al. revealed the underlying mechanism of reverse allosteric communication in dual-targeted therapy. Allosteric sites can be affected by orthomorphic drugs. The nonoverlapping allosteric pocket X-Pocket was discovered in EGFR mutants; this pocket is mainly composed of nonconserved residues, including the hot spots K867, S895, and K960, that can cooperate with traditional TKIs [207]. It is a promising target for the design of selective conformationally restricted drugs, with great potential in terms of affinity, efficacy, and selectivity.
DZ-SIM inhibitors
In addition, researchers found that a group of near-infrared heptamethine carbocyanine (DZ) fluorescent dyes, the prototype of which is heptamethylamine carbocyanine dye (IR-783) (104) (Fig. 14), have tumor-targeting activity through differentially expressed organic anion transport peptides on cancer cells [208]. This group of organic dyes can specifically deliver therapeutic payloads to tumor cells in the form of chemical conjugates. DZ-SIM was preliminarily synthesized; SIM specifically targets 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) in the endoplasmic reticulum. After specific uptake by tumor cells, DZ-SIM was enriched in subcellular organelles (including mitochondria and lysosomes). NSCLC cells were killed by mitochondrial damage, which mainly led to cytochrome C release into the cytoplasm, thereby activating the caspase-3-dependent apoptosis cascade. DZ-SIM inhibited the formation of cancer cell colonies resistant to first-generation (H1650 and H1975) and third-generation EGFR-TKIs (PC9AR), and most IC50 values were lower than 10 μmol/L. DZ-SIM represents a promising new therapy to overcome drug resistance in patients with EGFR-mutant disease.
Selection of individualized combination therapy
For patients who experience SCLC transformation, chemotherapy after the development of osimertinib resistance is an option. Research has shown that patients with transformation to SCLC have higher response rates to etoposide, cisplatin, and paclitaxel. For patients with unclear resistance mechanisms, chemotherapy is still a treatment option. If the patient is asymptomatic or has symptomatic local progression, osimertinib can be combined with local treatment according to National Comprehensive Cancer Network (NCCN) guidelines. Carboplatin, paclitaxel, bevacizumab, and atezolizumab (anti-PD-L1 antibody) are also options for patients who experience systemic progression after osimertinib treatment [209]. Whether chemotherapy can delay the development of resistance to third-generation EGFR-TKIs remains unknown. A study on osimertinib with or without chemotherapy as first-line therapy for patients with EGFR-mutant NSCLC is currently recruiting (NCT04035486) [210].
For most patients, the PD-1/PD-L1 pathway is not the sole rate-limiting factor for antitumor immunity, and blocking the PD-1/PD-L1 axis is insufficient to activate an effective antitumor immune response [211]. Strategies that lead to acquired EGFR-TKI resistance, such as HGF, MET amplification, and EGFR T790M, also promote immune escape in lung cancer by upregulating the expression of PD-L1. Many combination strategies, including α-PD-1/PD-L1 plus chemotherapy, radiotherapy, angiogenesis inhibitors, targeted therapy, other ICIs, agonists of the costimulatory molecule, stimulator of interferon gene agonists, epigenetic modulators, or metabolic modulators, have been confirmed to have superior antitumor efficacy and a higher response rate. The immunomodulatory effect of chemotherapy suggests that it might be a suitable partner for combination with α-PD-1/PD-L1 to achieve rapid and long-term cancer control. During the KEYNOTE series of clinical trials (such as KEYNOTE-021, KEYNOTE-189, and KEYNOTE-407), pembrolizumab combined with standard chemotherapy led to better overall survival (OS) and progression-free survival (PFS) in NSCLC patients and has been approved by the FDA as first-line treatment for advanced nonsquamous NSCLC [212, 213]. In addition, the National Medical Products Administration (NMPA) approved sintilimab plus gemcitabine and platinum as first-line treatment for advanced squamous NSCLC based on the results of ORIENT-12 [214]. In addition to α-PD-1-based approaches, α-PD-L1-based chemoimmunotherapy has also attracted intense attention. The IMpower150 trial was the pioneer of this series of studies, and the FDA-approved atezolizumab plus bevacizumab, paclitaxel, and carboplatin as first-line treatment for advanced nonsquamous NSCLC [215]. Subsequently, the FDA-approved atezolizumab plus nab-paclitaxel and carboplatin for nonsquamous NSCLC (based on the results of IMpower130). Radiotherapy can also induce immunogenic cell death and enhance the antitumor immune response. The results of a phase 1 study showed that α-PD-1/PD-L1 plus chemoradiotherapy was tolerable in advanced NSCLC (NCT02621398), with promising clinical outcomes. In multiple clinical studies, such as IMpower150, angiogenesis inhibitors enhanced the efficacy of α-PD-1/PD-L1 [216]. Moreover, dual immune checkpoint blockade or costimulatory molecule agonists plus α-PD-1/PD-L1 are also promising strategies. To date, the FDA has approved ipilimumab plus nivolumab for NSCLC and melanoma, among others. Agonists targeting costimulatory pathways such as CD27/CD70, CD40/CD40L, and 4-1BB/4-1BBL could also enhance T-cell activity and restore the antitumor immune response. However, bispecific/bifunctional antibodies simultaneously block two molecules and thus have a strategic advantage over combination therapy. For example, in the phase 1 NCT03710265 trial, SHR-1701 (TGF-β × PD-L1 bifunctional antibody) showed encouraging antitumor activity [217].
Given the heterogeneity of mutations across patients, the selection of individualized combination treatment strategies could improve outcomes and mitigate treatment resistance.
Discussion and future perspectives
EGFR is an important target on tumor cells that promotes mitosis and transformation. It is overexpressed in many diseases and is particularly related to the occurrence and development of cancer [3, 7, 8]. Tumors often have prominent genomic and transcriptional heterogeneity that is closely related to EGFR-TKI resistance [40, 218]. Studies have shown that drug resistance can develop through EGFR-dependent and EGFR-independent mechanisms [24, 219]. The emergence of resistance to third-generation EGFR-TKIs limits the clinical benefits for patients, thus necessitating the further development of more effective strategies.
To date, fourth-generation EGFR-TKIs show prominent antitumor activity. Recent research has shown that fourth-generation inhibitors targeting allosteric sites and ATP-competitive sites of EGFR can achieve remarkable results against EGFR L858R/T790M and C797S. In addition to fourth-generation EGFR-TKIs, combination treatments, monoclonal antibodies, and bispecific antibodies are significantly contributing to the treatment of tumors harboring the C797S mutation. While the C797S mutation is only one of the numerous drug resistance mechanisms, it is necessary to overcome other mutations by designing and developing new noncovalent ATP-competitive inhibitors that form hydrogen bonds with mutated residues in the EGFR ATP pocket (such as Lys745 and Asn842). The rational design of selective EGFR inhibitors that bind to both the ATP and allosteric sites of the EGFR kinase domain, that is, adding allosteric inhibitor elements to the compound skeleton at the ATP binding site, will help optimize and improve the mutation selectivity of compounds and lead to the identification of small molecules with good kinase inhibitory activity. However, the cellular activity of such compounds needs to be further improved, and future research directions should focus on the structural optimization of current lead compounds to obtain EGFR inhibitors with better mutation selectivity. Targeted protein degradation technology provides a new research direction for overcoming resistance to third-generation EGFR inhibitors. Considering the significance of overcoming allosteric hindrance by triple-mutant EGFR, allosteric EGFR degraders were developed. In addition, dual PROTACs have emerged in the field of cancer combination therapy; dual PROTACs can be designed with two targets, such as tumor immune targets plus adjuvant immune targets or energy metabolism targets and epigenetic targets plus antiapoptotic targets, to further overcome resistance of third-generation EGFR inhibitors and provide a better curative effect. Of course, the larger molecular weight of dual PROTACs will affect their druggability and pharmacokinetics, but perhaps nanodrug delivery systems can be utilized to improve drug absorption or optimized by simplifying the inhibitor moiety and maintaining the minimum pharmacophore. In addition, ADCs containing a small-molecule cytotoxic compound and a monoclonal antibody targeting a cancer target have attracted attention. The ADC MRG003 has entered clinical trials with great development and application prospects. The activation of alternative pathways and histological transformation are important mechanisms of resistance to third-generation EGFR inhibitors. The combined use of third-generation inhibitors and related pathway blockers is another important approach. To prevent the toxicity and side effects of multidrug combinations, drugs with multiple pharmacological activities were developed and proven to have more advantages than combination therapy. Multitarget kinase drugs have become a favorable choice due to their attractive pharmacokinetic characteristics and safety profiles. Natural compounds have received much research attention due to their potential antitumor effects. Based on the molecular mechanism of inhibition, natural compounds can be modified to provide new insights for effectively overcoming resistance to third-generation EGFR-TKIs. Last but not least, the discovery of EGFR degraders based on the FBXL2-Grp94-EGFR axis, AKR1B1 and PGAM1 inhibitors, DZ-SIM, and the nonoverlapping allosteric pocket X-Pocket provides promising support for the further development of strategies to overcome resistance to third-generation EGFR inhibitors. The mechanism of resistance to third-generation EGFR-TKIs is very complex, is impacted by EGFR mutations, and differs among patients and tumor sites. Thus, next-generation sequencing (NGS) of blood-based circulating tumor DNA (ctDNA) or tissue samples to elucidate the resistance mechanism will be valuable for guiding future therapeutic approaches and for clinical research on novel combination therapies to overcome drug resistance. Moreover, individualized combination treatment strategies could also improve treatment efficacy and mitigate treatment resistance.
EGFR is a verified target for antitumor therapy in a broad spectrum of cancers. Facilitated by versatile strategies in the field of medicinal chemistry, better approaches are anticipated for overcoming the hurdle of drug resistance to provide new hope for patients.
Conclusion
As a crucial “controller” that is related to the inhibition of tumor cell proliferation, angiogenesis, invasion, metastasis, and apoptosis, EGFR actively participates in malignant disease progression. However, the intrinsic and acquired resistance in primary and recurrent cancer which is mediated by EGFR mutations after target treatment leads to difficult therapeutic. Understanding the complex resistance mechanisms of EGFR-TKIs and developing potential strategies to combat it could be of potential interest for improving the individual therapeutic strategies for cancer.
Acknowledgements
Not applicable.
Abbreviations
- ACK1
Activated Cdc42-associated kinase 1
- ADC
Antibody drug conjugates
- AKR1B1
Aldehyde–ketone reductase family 1 member B1
- A-loop
Activation loop
- ATP
Adenosine triphosphate
- AURK
Aurora kinases
- AUTAC
Autophagy-targeting chimera
- Bcl-2
B cell lymphoma-2
- BRAF
V-RAF murine sarcoma viral oncogene homolog B1
- CCND
Cyclin D
- CCNE1
Cyclin E1
- CDK4/6
Cyclin-dependent kinase 4/6
- cIAP1
Cellular inhibitor of apoptosis protein 1
- CRBN
Cereblon
- DFG
Asp-Phe-Gly
- DZ-SIM
DZ-SIMvastatin
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- EGFR-TKIs
Epidermal growth factor receptor tyrosine kinase inhibitors
- EMT
Epithelial–mesenchymal transformation
- EMT-TFs
EMT-induced transcription factors
- ERC1
Excision repair cross-complementation 1
- FGF
Fibroblast growth factor
- FGFR
Fibroblast growth factor receptor
- Fv
Variable region fragment
- Grp94
Glucose regulatory protein 94
- HB-EGF
EGF-like growth factor
- HMGCR
3-Hydroxy-3-methylglutaryl-CoA reductase
- HNK
Honokiol
- ICIs
Immune checkpoint inhibitors
- IGF1R
Insulin-like growth factor receptor 1
- LUAD
Lung adenocarcinoma
- MDM2
Mouse double minute 2
- NCCN
National Comprehensive Cancer Network
- NSCLC
Non-small cell lung cancer
- NTRK1
Neurotrophic tyrosine receptor kinase 1
- PARP
Poly(ADP-ribose) polymerase
- PGAM1
Phosphoglycerate mutase 1
- PIK3CA
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit
- PROTAC
Proteolysis-targeting chimera
- PTEN
Phosphatase and tensin homolog
- ROS
Reactive oxygen species
- RTK
Receptor tyrosine kinase
- SCLC
Small cell lung cancer
- SLC7A11
Solute carrier family 7 member 11
- SPP1
Secreted phosphoprotein 1
- STAT
Signal sensor and transcription activator
- TACC3
Transforming acid helix protein 3
- TGF
Transforming growth factor
- TMP3
Thrombopoietin mimetic peptide 3
- VHL
Von Hippel–Lindau
- WT
Wild-type
- WT-EGFR
Wild-type EGFR
Author contributions
WL, YW, and LO conceived the project, supervised the project, and revised the manuscript. KS, GW, and JP summed up the literature, drafted the manuscript, and drew the figures. JP and JZ collected and organized the inhibitors. JW and GW proofread the structures and figures. All authors approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grants 82037718, 81922064, 22177083), the Fundamental Research Funds for the Central Universities (SCU2022D025), 1.3.5 project for disciplines of excellent, West China Hospital, Sichuan University (ZYJC18001), the Sichuan Science and Technology Program (grant number 2022NSFSC1290, 2019YFS0003), and West China Nursing Discipline Development Special Fund Project, Sichuan University (Grant HXHL21011).
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kunyu Shi, Guan Wang and Junping Pei contributed equally to this work
Contributor Information
Liang Ouyang, Email: ouyangliang@scu.edu.cn.
Yuxi Wang, Email: yuxiwang@scu.edu.cn.
Weimin Li, Email: weimi003@scu.edu.cn.
References
- 1.Amelia T, Kartasasmita RE, Ohwada T, Tjahjono DH. Structural insight and development of EGFR tyrosine kinase inhibitors. Molecules. 2022;27:819. doi: 10.3390/molecules27030819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.da Cunha SG, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69. doi: 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 3.Campbell ID, Bork P. Epidermal growth factor-like modules. Curr Opin Struct Biol. 1993;3:385–392. doi: 10.1016/S0959-440X(05)80111-3. [DOI] [Google Scholar]
- 4.Roskoski R. The ERBB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS, Langer CJ. Epidermal growth factor receptors as a target for cancer treatment: the emerging role of IMC-C225 in the treatment of lung and head and neck cancers. Semin Oncol. 2002;29:27–36. doi: 10.1053/sonc.2002.31525. [DOI] [PubMed] [Google Scholar]
- 7.Normanno N, Bianco C, De Luca A, Salomon DS. The role of EGF-related peptides in tumor growth. Front Biosci. 2001;6:D685–D707. doi: 10.2741/Normano. [DOI] [PubMed] [Google Scholar]
- 8.Thomas R, Weihua Z. Rethink of EGFR in cancer with its kinase independent function on board. Front Oncol. 2019;9:800. doi: 10.3389/fonc.2019.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabbah DA, Hajjo R, Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr Top Med Chem. 2020;20:815–834. doi: 10.2174/1568026620666200303123102. [DOI] [PubMed] [Google Scholar]
- 10.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol. 2018;29:i28–i37. doi: 10.1093/annonc/mdx705. [DOI] [PubMed] [Google Scholar]
- 12.Roskoski R., Jr Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48. doi: 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, et al. Phase iii study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 14.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H. Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer. Drug Des Devel Ther. 2016;10:3867–3872. doi: 10.2147/DDDT.S119162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang T, Zhou C. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients with EGFR inhibitor-resistant non-small cell lung cancer. Transl Lung Cancer Res. 2014;3:370–372. doi: 10.3978/j.issn.2218-6751.2014.08.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan XE, Zhu SJ, Liang L, Zhao P, Choi HG, Yun CH. Structural basis of mutant-selectivity and drug-resistance related to CO-1686. Oncotarget. 2017;8:53508–53517. doi: 10.18632/oncotarget.18588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–737. doi: 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guibert N, Barlesi F, Descourt R, Lena H, Besse B, Beau-Faller M, et al. Characteristics and outcomes of patients with lung cancer harboring multiple molecular alterations: results from the IFCT study biomarkers france. J Thorac Oncol. 2017;12:963–973. doi: 10.1016/j.jtho.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Wang S, Li B, Wang Z, Shang S, Shao Y, et al. Bim deletion polymorphism confers resistance to osimertinib in EGFR T790M lung cancer: a case report and literature review. Target Oncol. 2018;13:517–523. doi: 10.1007/s11523-018-0573-2. [DOI] [PubMed] [Google Scholar]
- 23.Eck MJ, Yun CH. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta. 2010;1804:559–566. doi: 10.1016/j.bbapap.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–i19. doi: 10.1093/annonc/mdx703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Zhang XC, Yang JJ, Yang ZF, Bai Y, Su J, et al. EGFR L792H and G796R: two novel mutations mediating resistance to the third-generation EGFR tyrosine kinase inhibitor osimertinib. J Thorac Oncol. 2018;13:1415–1421. doi: 10.1016/j.jtho.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 27.Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C, et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget. 2017;8:49671–49679. doi: 10.18632/oncotarget.17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ou SI, Cui J, Schrock AB, Goldberg ME, Zhu VW, Albacker L, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017;108:228–231. doi: 10.1016/j.lungcan.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Castellano GM, Aisner J, Burley SK, Vallat B, Yu HA, Pine SR, et al. A novel acquired exon 20 EGFR M766Q mutation in lung adenocarcinoma mediates osimertinib resistance but is sensitive to neratinib and poziotinib. J Thorac Oncol. 2019;14:1982–1988. doi: 10.1016/j.jtho.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Jin B, Su H, Qu X, Liu Y. Afatinib helped overcome subsequent resistance to osimertinib in a patient with NSCLC having leptomeningeal metastasis baring acquired EGFR L718Q mutation: a case report. BMC Cancer. 2019;19:702. doi: 10.1186/s12885-019-5915-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, et al. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR -mutated NSCLC. J Thorac Oncol. 2016;11:e121–e123. doi: 10.1016/j.jtho.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 32.Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, et al. L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chem Sci. 2018;9:2740–2749. doi: 10.1039/C7SC04761D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z, Yang J, Chen Y, Shao YW, Wang X. Acquired EGFR L718V mutation as the mechanism for osimertinib resistance in a T790M-negative non-small-cell lung cancer patient. Target Oncol. 2019;14:369–374. doi: 10.1007/s11523-019-00652-6. [DOI] [PubMed] [Google Scholar]
- 34.Fassunke J, Muller F, Keul M, Michels S, Dammert MA, Schmitt A, et al. Overcoming EGFR (G724S)-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat Commun. 2018;9:4655. doi: 10.1038/s41467-018-07078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu HY, Ke EE, Yang JJ, Sun YL, Yan HH, Zheng MY, et al. A comprehensive review of uncommon EGFR mutations in patients with non-small cell lung cancer. Lung Cancer. 2017;114:96–102. doi: 10.1016/j.lungcan.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Xu J, Jin B, Chu T, Dong X, Yang H, Zhang Y, et al. EGFR tyrosine kinase inhibitor (TKI) in patients with advanced non-small cell lung cancer (NSCLC) harboring uncommon EGFR mutations: a real-world study in china. Lung Cancer. 2016;96:87–92. doi: 10.1016/j.lungcan.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Shen YC, Tseng GC, Tu CY, Chen WC, Liao WC, Chen WC, et al. Comparing the effects of afatinib with gefitinib or erlotinib in patients with advanced-stage lung adenocarcinoma harboring non-classical epidermal growth factor receptor mutations. Lung Cancer. 2017;110:56–62. doi: 10.1016/j.lungcan.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21:3913–3923. doi: 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR -mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8:1529–1539. doi: 10.1158/2159-8290.CD-18-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5:713–722. doi: 10.1158/2159-8290.CD-15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017;77:2078–2089. doi: 10.1158/0008-5472.CAN-16-2359. [DOI] [PubMed] [Google Scholar]
- 42.Huang C, Zou Q, Liu H, Qiu B, Li Q, Lin Y, et al. Management of non-small cell lung cancer patients with MET exon 14 skipping mutations. Curr Treat Opt Oncol. 2020;21:33. doi: 10.1007/s11864-020-0723-5. [DOI] [PubMed] [Google Scholar]
- 43.Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K, Boerner JL. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of MET. Breast Cancer Res. 2012;14:R104. doi: 10.1186/bcr3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu CC, Liao BC, Liao WY, Markovets A, Stetson D, Thress K, et al. Exon 16-skipping HER2 as a novel mechanism of osimertinib resistance in EGFR L858R/T790M-positive non-small cell lung cancer. J Thorac Oncol. 2020;15:50–61. doi: 10.1016/j.jtho.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Ou S-HI, Madison R, Robichaux JP, Ross JS, Miller VA, Ali SM, et al. Characterization of 648 non-small cell lung cancer (NSCLC) cases with 28 unique HER2 exon 20 insertions. J Clin Oncol. 2019;37:9063–63. doi: 10.1200/JCO.2019.37.15_suppl.9063. [DOI] [Google Scholar]
- 46.Gao G, Li X, Wang Q, Zhang Y, Chen J, Shu Y, et al. Single-arm, phase ii study of pyrotinib in advanced non-small cell lung cancer (NSCLC) patients with HER2 exon 20 mutation. J Clin Oncol. 2019;37:9089–9189. doi: 10.1200/JCO.2019.37.15_suppl.9089. [DOI] [Google Scholar]
- 47.Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17:38. doi: 10.1186/s12943-018-0777-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J, Yang Q, Xu W. Iterative upgrading of small molecular tyrosine kinase inhibitors for EGFR mutation in NSCLC: necessity and perspective. Pharmaceutics. 2021;13:1500. doi: 10.3390/pharmaceutics13091500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10:259. doi: 10.1038/s41467-018-08074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 51.Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. Mechanisms of acquired resistance to AZD9291 a mutation-selective, irreversible EGFR inhibitor. J Thorac Oncol. 2015;10:1736–1744. doi: 10.1097/JTO.0000000000000688. [DOI] [PubMed] [Google Scholar]
- 52.Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:741–841. doi: 10.1093/annonc/mdy424.064. [DOI] [Google Scholar]
- 53.Tanaka H, Sakagami H, Kaneko N, Konagai S, Yamamoto H, Matsuya T, et al. Mutant-selective irreversible EGFR inhibitor, naquotinib, inhibits tumor growth in NSCLC models with EGFR-activating mutations, T790M mutation, and AXL overexpression. Mol Cancer Ther. 2019;18:1366–1373. doi: 10.1158/1535-7163.MCT-18-0976. [DOI] [PubMed] [Google Scholar]
- 54.Park JH, Choi YJ, Kim SY, Lee JE, Sung KJ, Park S, et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget. 2016;7:22005–22015. doi: 10.18632/oncotarget.8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 56.Cheetham GMT, Knegtel RMA, Coll JT, Renwick SB, Swenson L, Weber P, et al. Crystal structure of aurora-2, an oncogenic serine/threonine kinase*. J Biol Chem. 2002;277:42419–42422. doi: 10.1074/jbc.C200426200. [DOI] [PubMed] [Google Scholar]
- 57.Pradhan T, Gupta O, Singh G, Monga V. Aurora kinase inhibitors as potential anticancer agents: recent advances. Eur J Med Chem. 2021;221:113495. doi: 10.1016/j.ejmech.2021.113495. [DOI] [PubMed] [Google Scholar]
- 58.Falchook GS, Bastida CC, Kurzrock R. Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin Oncol. 2015;42:832–848. doi: 10.1053/j.seminoncol.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 59.Hu L, Fan M, Shi S, Song X, Wang F, He H, et al. Dual target inhibitors based on EGFR: promising anticancer agents for the treatment of cancers (2017-) Eur J Med Chem. 2022;227:113963. doi: 10.1016/j.ejmech.2021.113963. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka K, Yu HA, Yang S, Han S, Selcuklu SD, Kim K, et al. Targeting aurora b kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 2021;39(1245–61):e6. doi: 10.1016/j.ccell.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010;69:272–278. doi: 10.1016/j.lungcan.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10:336–346. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L, et al. Braf in non-small cell lung cancer (NSCLC): pickaxing another brick in the wall. Cancer Treat Rev. 2018;66:82–94. doi: 10.1016/j.ctrv.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 64.Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang JC. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12:567–572. doi: 10.1016/j.jtho.2016.11.2231. [DOI] [PubMed] [Google Scholar]
- 65.Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase iii flaura study. Ann Oncol. 2018;29:viii740. doi: 10.1093/annonc/mdy424.063. [DOI] [Google Scholar]
- 66.Zhao M, Gao FH, Wang JY, Liu F, Yuan HH, Zhang WY, et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73:366–374. doi: 10.1016/j.lungcan.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Chaib I, Karachaliou N, Pilotto S, Codony Servat J, Cai X, Li X, et al. Co-activation of STAT3 and YES-associated protein 1 (YAP1) pathway in EGFR-mutant NSCLC. J Natl Cancer Inst. 2017;109. [DOI] [PMC free article] [PubMed]
- 68.Soria J-C, Lee H-Y, Lee JI, Wang L, Issa J-P, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178–1184. [PubMed] [Google Scholar]
- 69.Zhang T, Qu R, Chan S, Lai M, Tong L, Feng F, et al. Discovery of a novel third-generation EGFR inhibitor and identification of a potential combination strategy to overcome resistance. Mol Cancer. 2020;19:90. doi: 10.1186/s12943-020-01202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu L, Chen Z, Zang H, Fan S, Gu J, Zhang G, et al. Targeting c-Myc to overcome acquired resistance of EGFR mutant NSCLC cells to the third-generation EGFR tyrosine kinase inhibitor, osimertinib. Cancer Res. 2021;81:4822–4834. doi: 10.1158/0008-5472.CAN-21-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weng CH, Chen LY, Lin YC, Shih JY, Lin YC, Tseng RY, et al. Epithelial-mesenchymal transition (emtEMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38:455–468. doi: 10.1038/s41388-018-0454-2. [DOI] [PubMed] [Google Scholar]
- 72.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–134. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 73.Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu CH, Huang Q, Jin ZY, Zhu CL, Liu Z, Wang C. miR-21 and KLF4 jointly augment epithelialmesenchymal transition via the Akt/ERK1/2 pathway. Int J Oncol. 2017;50:1109–1115. doi: 10.3892/ijo.2017.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han Z, Zhou X, Li S, Qin Y, Chen Y, Liu H. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol Rep. 2017;38:3064–3070. doi: 10.3892/or.2017.5938. [DOI] [PubMed] [Google Scholar]
- 76.Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE. 2014;9:e103305. doi: 10.1371/journal.pone.0103305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fardi M, Solali S, Farshdousti HM. Epigenetic mechanisms as a new approach in cancer treatment: an updated review. Genes Dis. 2018;5:304–311. doi: 10.1016/j.gendis.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Del Re M, Arrigoni E, Restante G, Passaro A, Rofi E, Crucitta S, et al. Concise review: resistance to tyrosine kinase inhibitors in non-small cell lung cancer: the role of cancer stem cells. Stem Cells. 2018;36:633–640. doi: 10.1002/stem.2787. [DOI] [PubMed] [Google Scholar]
- 79.Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:viii741. doi: 10.1093/annonc/mdy424.064. [DOI] [Google Scholar]
- 80.Dorantes-Heredia R, Ruiz-Morales JM, Cano-Garcia F. Histopathological transformation to small-cell lung carcinoma in non-small cell lung carcinoma tumors. Transl Lung Cancer Res. 2016;5:401–412. doi: 10.21037/tlcr.2016.07.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165–172. doi: 10.1016/S1470-2045(14)71180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Norkowski E, Ghigna MR, Lacroix L, Le Chevalier T, Fadel E, Dartevelle P, et al. Small-cell carcinoma in the setting of pulmonary adenocarcinoma: new insights in the era of molecular pathology. J Thorac Oncol. 2013;8:1265–1271. doi: 10.1097/JTO.0b013e3182a407fa. [DOI] [PubMed] [Google Scholar]
- 84.Yin X, Li Y, Wang H, Jia T, Wang E, Luo Y, et al. Small cell lung cancer transformation: from pathogenesis to treatment. Semin Cancer Biol. 2022 doi: 10.1016/j.semcancer.2022.03.006. [DOI] [PubMed] [Google Scholar]
- 85.Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;26:2654–2663. doi: 10.1158/1078-0432.CCR-19-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garassino MC, Cho BC, Kim JH, Mazieres J, Vansteenkiste J, Lena H, et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol. 2018;19:521–536. doi: 10.1016/S1470-2045(18)30144-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585–4593. doi: 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haratani K, Hayashi H, Tanaka T, Kaneda H, Togashi Y, Sakai K, et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann Oncol. 2017;28:1532–1539. doi: 10.1093/annonc/mdx183. [DOI] [PubMed] [Google Scholar]
- 89.Zheng Y, Hao S, Xiang C, Han Y, Shang Y, Zhen Q, et al. The correlation between SPP1 and immune escape of EGFR mutant lung adenocarcinoma was explored by bioinformatics analysis. Front Oncol. 2021;11:592854. doi: 10.3389/fonc.2021.592854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165. doi: 10.1186/s12943-019-1073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, et al. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015;1:982–984. doi: 10.1001/jamaoncol.2015.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J Thorac Oncol. 2017;12:1723–1727. doi: 10.1016/j.jtho.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 93.Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21:3924–3933. doi: 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsai CJ, Nussinov R. Emerging allosteric mechanism of EGFR activation in physiological and pathological contexts. Biophys J. 2019;117:5–13. doi: 10.1016/j.bpj.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Engel J, Richters A, Getlik M, Tomassi S, Keul M, Termathe M, et al. Targeting drug resistance in EGFR with covalent inhibitors: a structure-based design approach. J Med Chem. 2015;58:6844–6863. doi: 10.1021/acs.jmedchem.5b01082. [DOI] [PubMed] [Google Scholar]
- 96.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee S, Kim J, Duggirala KB, Go A, Shin I, Cho BC, et al. Allosteric inhibitor TREA-0236 containing non-hydrolysable quinazoline-4-one for EGFR T790M/C797S mutants inhibition. Bull Korean Chem Soc. 2018;39:895–898. doi: 10.1002/bkcs.11491. [DOI] [Google Scholar]
- 98.To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019;9:926–943. doi: 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maity S, Pai KSR, Nayak Y. Advances in targeting EGFR allosteric site as anti-NSCLC therapy to overcome the drug resistance. Pharmacol Rep. 2020;72:799–813. doi: 10.1007/s43440-020-00131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Clercq DJH, Heppner DE, To C, Jang J, Park E, Yun C-H, et al. Discovery and optimization of dibenzodiazepinones as allosteric mutant-selective EGFR inhibitors. ACS Med Chem Lett. 2019;10:1549–1553. doi: 10.1021/acsmedchemlett.9b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duplessis M, Goergler A, Jaeschke G, Kocer B, Kuhn B, Lazarski K, et al. COMPOUNDS. Publication number: 20210079005, March 18, 2021.
- 102.Lu X, Zhang T, Zhu SJ, Xun Q, Tong L, Hu X, et al. Discovery of JND3229 as a new EGFR(C797S) mutant inhibitor with in vivo monodrug efficacy. ACS Med Chem Lett. 2018;9:1123–1127. doi: 10.1021/acsmedchemlett.8b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Engel J, Becker C, Lategahn J, Keul M, Ketzer J, Muhlenberg T, et al. Insight into the inhibition of drug-resistant mutants of the receptor tyrosine kinase EGFR. Angew Chem Int Ed Engl. 2016;55:10909–10912. doi: 10.1002/anie.201605011. [DOI] [PubMed] [Google Scholar]
- 104.Gunther M, Lategahn J, Juchum M, Doring E, Keul M, Engel J, et al. Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. J Med Chem. 2017;60:5613–5637. doi: 10.1021/acs.jmedchem.7b00316. [DOI] [PubMed] [Google Scholar]
- 105.Park H, Jung HY, Mah S, Hong S. Discovery of EGF receptor inhibitors that are selective for the D746–750/T790M/C797S mutant through structure-based de novo design. Angew Chem Int Ed Engl. 2017;56:7634–7638. doi: 10.1002/anie.201703389. [DOI] [PubMed] [Google Scholar]
- 106.Zhang M, Wang Y, Wang J, Liu Z, Shi J, Li M, et al. Design, synthesis and biological evaluation of the quinazoline derivatives as L858R/T790M/C797S triple mutant epidermal growth factor receptor tyrosine kinase inhibitors. Chem Pharm Bull (Tokyo) 2020;68:971–980. doi: 10.1248/cpb.c20-00411. [DOI] [PubMed] [Google Scholar]
- 107.Shen J, Zhang T, Zhu SJ, Sun M, Tong L, Lai M, et al. Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFR(L858R/T790M/C797S) J Med Chem. 2019;62:7302–7308. doi: 10.1021/acs.jmedchem.9b00576. [DOI] [PubMed] [Google Scholar]
- 108.Zhang H, Wang J, Shen Y, Wang HY, Duan WM, Zhao HY, et al. Discovery of 2,4,6-trisubstitued pyrido[3,4-d]pyrimidine derivatives as new EGFR-TKIs. Eur J Med Chem. 2018;148:221–237. doi: 10.1016/j.ejmech.2018.02.051. [DOI] [PubMed] [Google Scholar]
- 109.Hei YY, Shen Y, Wang J, Zhang H, Zhao HY, Xin M, et al. Synthesis and evaluation of 2,9-disubstituted 8-phenylthio/phenylsulfinyl-9H-purine as new EGFR inhibitors. Bioorg Med Chem. 2018;26:2173–2185. doi: 10.1016/j.bmc.2018.03.025. [DOI] [PubMed] [Google Scholar]
- 110.Lei H, Fan S, Zhang H, Liu YJ, Hei YY, Zhang JJ, et al. Discovery of novel 9-heterocyclyl substituted 9H-purines as L858R/T790M/C797S mutant EGFR tyrosine kinase inhibitors. Eur J Med Chem. 2020;186:111888. doi: 10.1016/j.ejmech.2019.111888. [DOI] [PubMed] [Google Scholar]
- 111.Lategahn J, Keul M, Klovekorn P, Tumbrink HL, Niggenaber J, Muller MP, et al. Inhibition of osimertinib-resistant epidermal growth factor receptor EGFR-T790M/C797S. Chem Sci. 2019;10:10789–10801. doi: 10.1039/C9SC03445E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu X, Xun Q, Zhang T, Zhu S-J, Li Q, Tong L, et al. 2-Oxo-3,4-dihydropyrimido[4,5-d] pyrimidines as new reversible inhibitors of EGFR C797S (Cys797 to Ser797) mutant. Chin Chem Lett. 2020;31:1281–1287. doi: 10.1016/j.cclet.2019.09.044. [DOI] [Google Scholar]
- 113.Su Z, Yang T, Wang J, Lai M, Tong L, Wumaier G, et al. Design, synthesis and biological evaluation of potent EGFR kinase inhibitors against 19D/T790M/C797S mutation. Bioorg Med Chem Lett. 2020;30:127327. doi: 10.1016/j.bmcl.2020.127327. [DOI] [PubMed] [Google Scholar]
- 114.Lee Kwangho, SHIN Inji, CHOI Gildon, CHAE Chong Hak, Choe Hyeon Jeong, JUNG Myoung Eun, et al. N2,N4-diphenylpyrimidine-2,4-diamine derivative, method for preparing same, and pharmaceutical composition containing same as active ingredient for prevention or treatment of cancer. WO2018230934, 2018.
- 115.Wu L, Liu X, Ding CZ, Chen S, Hu L, Zhao L, et al. Spiro-aryl-phosphorus-oxygen compound as fourth generation of EGFR kinase inhibitor. WO 2018108064 A1, 2016.
- 116.Iwao M, Fukuda T, Ishibashi F, Uehara Y, Nishiya N, Oku Y, et al. Fourth-generation EGFR tyrosine kinase inhibitor. CN 110461850 A, 2019.
- 117.Boese D, Dahmann G, Engelhardt H, Petronczki M, Scharn D. New benzimidazole compounds and derivatives as EGFR inhibitors. WO 2019162323 A1, 2019.
- 118.Ding K, Ding J, Shen J, Geng M, Lu X, Xie H, et al. Pyrimidopyridone or pyridopyridone compound and use thereof. WO 2019015593 A1, 2019.
- 119.Ferlenghi F, Scalvini L, Vacondio F, Castelli R, Bozza N, Marseglia G, et al. A sulfonyl fluoride derivative inhibits EGFR(L858R/T790M/C797S) by covalent modification of the catalytic lysine. Eur J Med Chem. 2021;225:113786. doi: 10.1016/j.ejmech.2021.113786. [DOI] [PubMed] [Google Scholar]
- 120.Morabito A, Piccirillo MC, Falasconi F, De Feo G, Del Giudice A, Bryce J, et al. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (vEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: Current status and future directions. Oncologist. 2009;14:378–390. doi: 10.1634/theoncologist.2008-0261. [DOI] [PubMed] [Google Scholar]
- 121.Li Q, Zhang T, Li S, Tong L, Li J, Su Z, et al. Discovery of potent and noncovalent reversible EGFR kinase inhibitors of EGFR(L858R/T790M/C797S) ACS Med Chem Lett. 2019;10:869–873. doi: 10.1021/acsmedchemlett.8b00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wittlinger F, Heppner DE, To C, Gunther M, Shin BH, Rana JK, et al. Design of a “two-in-one” mutant-selective epidermal growth factor receptor inhibitor that spans the orthosteric and allosteric sites. J Med Chem. 2022;65:1370–1383. doi: 10.1021/acs.jmedchem.1c00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: Insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 125.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule protacs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Scheepstra M, Hekking KFW, van Hijfte L, Folmer RHA. Bivalent ligands for protein degradation in drug discovery. Comput Struct Biotechnol J. 2019;17:160–176. doi: 10.1016/j.csbj.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.An S, Fu L. Small-molecule protacs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–562. doi: 10.1016/j.ebiom.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat Chem Biol. 2019;15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 130.Churcher I. Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem. 2018;61:444–452. doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- 131.Cromm PM, Crews CM. Targeted protein degradation: from chemical biology to drug discovery. Cell Chem Biol. 2017;24:1181–1190. doi: 10.1016/j.chembiol.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Toure M, Crews CM. Small-molecule protacs: new approaches to protein degradation. Angew Chem Int Ed Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 133.Jang J, To C, De Clercq DJH, Park E, Ponthier CM, Shin BH, et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew Chem Int Ed Engl. 2020;59:14481–14489. doi: 10.1002/anie.202003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao HY, Yang XY, Lei H, Xi XX, Lu SM, Zhang JJ, et al. Discovery of potent small molecule protacs targeting mutant EGFR. Eur J Med Chem. 2020;208:112781. doi: 10.1016/j.ejmech.2020.112781. [DOI] [PubMed] [Google Scholar]
- 135.Qu X, Liu H, Song X, Sun N, Zhong H, Qiu X, et al. Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTAC s through both proteosome and autophagy/lysosome degradation systems. Eur J Med Chem. 2021;218:113328. doi: 10.1016/j.ejmech.2021.113328. [DOI] [PubMed] [Google Scholar]
- 136.Zheng M, Huo J, Gu X, Wang Y, Wu C, Zhang Q, et al. Rational design and synthesis of novel dual PROTACS for simultaneous degradation of EGFR and PARP. J Med Chem. 2021;64:7839–7852. doi: 10.1021/acs.jmedchem.1c00649. [DOI] [PubMed] [Google Scholar]
- 137.Kim JH, Nam B, Choi YJ, Kim SY, Lee JE, Sung KJ, et al. Enhanced glycolysis supports cell survival in EGFR-mutant lung adenocarcinoma by inhibiting autophagy-mediated EGFR degradation. Cancer Res. 2018;78:4482–4496. doi: 10.1158/0008-5472.CAN-18-0117. [DOI] [PubMed] [Google Scholar]
- 138.Takahashi D, Arimoto H. Targeting selective autophagy by AUTAC degraders. Autophagy. 2020;16:765–766. doi: 10.1080/15548627.2020.1718362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. Autacs: Cargo-specific degraders using selective autophagy. Mol Cell. 2019;76(797–810):e10. doi: 10.1016/j.molcel.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 140.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 141.Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019;394:793–804. doi: 10.1016/S0140-6736(19)31774-X. [DOI] [PubMed] [Google Scholar]
- 142.He K, Xu J, Liang J, Jiang J, Tang M, Ye X, et al. Discovery of a novel EGFR-targeting antibody-drug conjugate, SHR-A1307, for the treatment of solid tumors resistant or refractory to anti-EGFR therapies. Mol Cancer Ther. 2019;18:1104–1114. doi: 10.1158/1535-7163.MCT-18-0854. [DOI] [PubMed] [Google Scholar]
- 143.Xu R-h, Qiu M-Z, Zhang Y, Wei X-L, Hu C. First-in-human dose-escalation study of anti-EGFR adc MRG003 in patients with relapsed/refractory solid tumors. J Clin Oncol. 2020;38:3550–50. doi: 10.1200/JCO.2020.38.15_suppl.3550. [DOI] [Google Scholar]
- 144.Li Z, Wang M, Yao X, Luo W, Qu Y, Yu D, et al. Development of a novel EGFR-targeting antibody-drug conjugate for pancreatic cancer therapy. Target Oncol. 2019;14:93–105. doi: 10.1007/s11523-018-0616-8. [DOI] [PubMed] [Google Scholar]
- 145.Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016;380:494–504. doi: 10.1016/j.canlet.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 146.Giroux-Leprieur E, Dumenil C, Chinet T. Combination of crizotinib and osimertinib or erlotinib might overcome MET-mediated resistance to EGFR tyrosine kinase inhibitor in EGFR-mutated adenocarcinoma. J Thorac Oncol. 2018;13:e232–e234. doi: 10.1016/j.jtho.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 147.Kang J, Chen HJ, Wang Z, Liu J, Li B, Zhang T, et al. Osimertinib and cabozantinib combinatorial therapy in an EGFR-mutant lung adenocarcinoma patient with multiple MET secondary-site mutations after resistance to crizotinib. J Thorac Oncol. 2018;13:e49–e53. doi: 10.1016/j.jtho.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 148.Fujino T, Suda K, Mitsudomi T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin Emerg Drugs. 2020;25:229–249. doi: 10.1080/14728214.2020.1791821. [DOI] [PubMed] [Google Scholar]
- 149.Quintanal-Villalonga A, Molina-Pinelo S, Cirauqui C, Ojeda-Marquez L, Marrugal A, Suarez R, et al. FGFR1 cooperates with EGFR in lung cancer oncogenesis, and their combined inhibition shows improved efficacy. J Thorac Oncol. 2019;14:641–655. doi: 10.1016/j.jtho.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 150.Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18:1590–1599. doi: 10.1016/S1470-2045(17)30680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun. 2017;8:14768. doi: 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu S, Li S, Hai J, Wang X, Chen T, Quinn MM, et al. Targeting HER2 aberrations in non-small cell lung cancer with osimertinib. Clin Cancer Res. 2018;24:2594–2604. doi: 10.1158/1078-0432.CCR-17-1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.La Monica S, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, Digiacomo G, et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J Exp Clin Cancer Res. 2017;36:174. doi: 10.1186/s13046-017-0653-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jani JP, Arcari J, Bernardo V, Bhattacharya SK, Briere D, Cohen BD, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9:883–894. doi: 10.1158/1535-7163.MCT-09-0915. [DOI] [PubMed] [Google Scholar]
- 155.Kim C, Giaccone G. MEK inhibitors under development for treatment of non-small-cell lung cancer. Expert Opin Investig Drugs. 2018;27:17–30. doi: 10.1080/13543784.2018.1415324. [DOI] [PubMed] [Google Scholar]
- 156.Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22:4837–4847. doi: 10.1158/1078-0432.CCR-15-1915. [DOI] [PubMed] [Google Scholar]
- 157.Della Corte CM, Ciaramella V, Cardone C, La Monica S, Alfieri R, Petronini PG, et al. Antitumor efficacy of dual blockade of EGFR signaling by osimertinib in combination with selumetinib or cetuximab in activated EGFR human NCLC tumor models. J Thorac Oncol. 2018;13:810–820. doi: 10.1016/j.jtho.2018.02.025. [DOI] [PubMed] [Google Scholar]
- 158.Jacobsen K, Bertran-Alamillo J, Molina MA, Teixido C, Karachaliou N, Pedersen MH, et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat Commun. 2017;8:410. doi: 10.1038/s41467-017-00450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Namba K, Shien K, Takahashi Y, Torigoe H, Sato H, Yoshioka T, et al. Activation of AXL as a preclinical acquired resistance mechanism against osimertinib treatment in EGFR-mutant non-small cell lung cancer cells. Mol Cancer Res. 2019;17:499–507. doi: 10.1158/1541-7786.MCR-18-0628. [DOI] [PubMed] [Google Scholar]
- 160.Jimbo T, Hatanaka M, Komatsu T, Taira T, Kumazawa K, Maeda N, et al. DS-1205b, a novel selective inhibitor of AXL kinase, blocks resistance to EGFR-tyrosine kinase inhibitors in a non-small cell lung cancer xenograft model. Oncotarget. 2019;10:5152–5167. doi: 10.18632/oncotarget.27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim D, Bach DH, Fan YH, Luu TT, Hong JY, Park HJ, et al. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis. 2019;10:361. doi: 10.1038/s41419-019-1601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu YN, Tsai MF, Wu SG, Chang TH, Tsai TH, Gow CH, et al. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int J Cancer. 2019;145:1609–1624. doi: 10.1002/ijc.32487. [DOI] [PubMed] [Google Scholar]
- 163.Gu J, Qian L, Zhang G, Mahajan NP, Owonikoko TK, Ramalingam SS, et al. Inhibition of ACK1 delays and overcomes acquired resistance of EGFR mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib. Lung Cancer. 2020;150:26–35. doi: 10.1016/j.lungcan.2020.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lawrence HR, Mahajan K, Luo Y, Zhang D, Tindall N, Huseyin M, et al. Development of novel ACK1/TNK2 inhibitors using a fragment-based approach. J Med Chem. 2015;58:2746–2763. doi: 10.1021/jm501929n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sequist LV, Lynch TJ. EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med. 2008;59:429–442. doi: 10.1146/annurev.med.59.090506.202405. [DOI] [PubMed] [Google Scholar]
- 166.Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, et al. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9:843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 167.Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 168.Chen G, Bao Y, Weng Q, Zhao Y, Lu X, Fu L, et al. Compound 15c, a novel dual inhibitor of EGFR(L858R/T790M) and FGFR1, efficiently overcomes epidermal growth factor receptor-tyrosine kinase inhibitor resistance of non-small-cell lung cancers. Front Pharmacol. 2019;10:1533. doi: 10.3389/fphar.2019.01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cui Z, Chen S, Wang Y, Gao C, Chen Y, Tan C, et al. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur J Med Chem. 2017;136:372–381. doi: 10.1016/j.ejmech.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 170.Mansour TS, Pallepati RR, Basetti V. Potent dual EGFR/HER4 tyrosine kinase inhibitors containing novel (1,2-dithiolan-4-yl)acetamides. Bioorg Med Chem Lett. 2020;30:127288. doi: 10.1016/j.bmcl.2020.127288. [DOI] [PubMed] [Google Scholar]
- 171.El-Sayed NA, Nour MS, Salem MA, Arafa RK. New oxadiazoles with selective- COX-2 and EGFR dual inhibitory activity: design, synthesis, cytotoxicity evaluation and in silico studies. Eur J Med Chem. 2019;183:111693. doi: 10.1016/j.ejmech.2019.111693. [DOI] [PubMed] [Google Scholar]
- 172.Abdelatef SA, El-Saadi MT, Amin NH, Abdelazeem AH, Omar HA, Abdellatif KRA. Design, synthesis and anticancer evaluation of novel spirobenzo[h]chromene and spirochromane derivatives with dual EGFR and B-RAF inhibitory activities. Eur J Med Chem. 2018;150:567–578. doi: 10.1016/j.ejmech.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 173.Jang J, Son JB, To C, Bahcall M, Kim SY, Kang SY, et al. Discovery of a potent dual ALK and EGFR T790M inhibitor. Eur J Med Chem. 2017;136:497–510. doi: 10.1016/j.ejmech.2017.04.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen Y, Wu J, Wang A, Qi Z, Jiang T, Chen C, et al. Discovery of n-(5-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-4-met hoxy-2-(4-methyl-1,4-diazepan-1-yl)phenyl)acrylamide (chmfl-alk/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC. Eur J Med Chem. 2017;139:674–697. doi: 10.1016/j.ejmech.2017.08.035. [DOI] [PubMed] [Google Scholar]
- 175.Jing T, Miao X, Jiang F, Guo M, Xing L, Zhang J, et al. Discovery and optimization of tetrahydropyrido[4,3-d]pyrimidine derivatives as novel ATX and EGFR dual inhibitors. Bioorg Med Chem. 2018;26:1784–1796. doi: 10.1016/j.bmc.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 176.Kurup S, McAllister B, Liskova P, Mistry T, Fanizza A, Stanford D, et al. Design, synthesis and biological activity of n(4)-phenylsubstituted-7h-pyrrolo[2,3-d]pyrimidin-4-amines as dual inhibitors of aurora kinase a and epidermal growth factor receptor kinase. J Enzyme Inhib Med Chem. 2018;33:74–84. doi: 10.1080/14756366.2017.1376666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gadekar PK, Urunkar G, Roychowdhury A, Sharma R, Bose J, Khanna S, et al. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[2,1-b]thiazoles as dual EGFR and IGF1R inhibitors. Bioorg Chem. 2021;115:105151. doi: 10.1016/j.bioorg.2021.105151. [DOI] [PubMed] [Google Scholar]
- 178.Romagnoli R, Prencipe F, Oliva P, Baraldi S, Baraldi PG, Schiaffino Ortega S, et al. Design, synthesis, and biological evaluation of 6-substituted thieno[3,2-d]pyrimidine analogues as dual epidermal growth factor receptor kinase and microtubule inhibitors. J Med Chem. 2019;62:1274–1290. doi: 10.1021/acs.jmedchem.8b01391. [DOI] [PubMed] [Google Scholar]
- 179.Alswah M, Bayoumi AH, Elgamal K, Elmorsy A, Ihmaid S, Ahmed HEA. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo[4,3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules. 2017;23:48. doi: 10.3390/molecules23010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Khan I, Garikapati KR, Setti A, Shaik AB, Kanth Makani VK, Shareef MA, et al. Design, synthesis, in silico pharmacokinetics prediction and biological evaluation of 1,4-dihydroindeno[1,2-c]pyrazole chalcone as EGFR/AKT pathway inhibitors. Eur J Med Chem. 2019;163:636–648. doi: 10.1016/j.ejmech.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 181.Dong H, Yin H, Zhao C, Cao J, Xu W, Zhang Y. Design, synthesis and biological evaluation of novel osimertinib-based HDAC and EGFR dual inhibitors. Molecules. 2019;24:2407. doi: 10.3390/molecules24132407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fischer T, Najjar A, Totzke F, Schachtele C, Sippl W, Ritter C, et al. Discovery of novel dual inhibitors of receptor tyrosine kinases EGFR and PDGFR-β related to anticancer drug resistance. J Enzyme Inhib Med Chem. 2018;33:1–8. doi: 10.1080/14756366.2017.1370583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hamed MM, Darwish SS, Herrmann J, Abadi AH, Engel M. First bispecific inhibitors of the epidermal growth factor receptor kinase and the NF-κB activity as novel anticancer agents. J Med Chem. 2017;60:2853–2868. doi: 10.1021/acs.jmedchem.6b01774. [DOI] [PubMed] [Google Scholar]
- 184.Dokla EME, Fang CS, Abouzid KAM, Chen CS. 1,2,4-oxadiazole derivatives targeting EGFR and c-Met degradation in TKI resistant NSCLC. Eur J Med Chem. 2019;182:111607. doi: 10.1016/j.ejmech.2019.111607. [DOI] [PubMed] [Google Scholar]
- 185.Singh PK, Silakari O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-Met. Bioorg Chem. 2018;79:163–170. doi: 10.1016/j.bioorg.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 186.Fischer T, Kruger T, Najjar A, Totzke F, Schachtele C, Sippl W, et al. Discovery of novel substituted benzo-anellated 4-benzylamino pyrrolopyrimidines as dual EGFR and vEGFR2 inhibitors. Bioorg Med Chem Lett. 2017;27:2708–2712. doi: 10.1016/j.bmcl.2017.04.053. [DOI] [PubMed] [Google Scholar]
- 187.Zhang HQ, Gong FH, Ye JQ, Zhang C, Yue XH, Li CG, et al. Design and discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and vEGFR-2. Eur J Med Chem. 2017;125:245–254. doi: 10.1016/j.ejmech.2016.09.039. [DOI] [PubMed] [Google Scholar]
- 188.Wei H, Duan Y, Gou W, Cui J, Ning H, Li D, et al. Design, synthesis and biological evaluation of novel 4-anilinoquinazoline derivatives as hypoxia-selective EGFR and vEGFR-2 dual inhibitors. Eur J Med Chem. 2019;181:111552. doi: 10.1016/j.ejmech.2019.07.055. [DOI] [PubMed] [Google Scholar]
- 189.Sun S, Zhang J, Wang N, Kong X, Fu F, Wang H, et al. Design and discovery of quinazoline- and thiourea-containing sorafenib analogs as EGFR and vEGFR-2 dual TK inhibitors. Molecules. 2017;23:24. doi: 10.3390/molecules23010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Das D, Xie L, Wang J, Xu X, Zhang Z, Shi J, et al. Discovery of new quinazoline derivatives as irreversible dual EGFR/HER2 inhibitors and their anticancer activities: part 1. Bioorg Med Chem Lett. 2019;29:591–596. doi: 10.1016/j.bmcl.2018.12.056. [DOI] [PubMed] [Google Scholar]
- 191.Maher M, Kassab AE, Zaher AF, Mahmoud Z. Novel pyrazolo[3,4-d]pyrimidines: design, synthesis, anticancer activity, dual EGFR/ErbB2 receptor tyrosine kinases inhibitory activity, effects on cell cycle profile and caspase-3-mediated apoptosis. J Enzyme Inhib Med Chem. 2019;34:532–546. doi: 10.1080/14756366.2018.1564046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zou M, Li J, Jin B, Wang M, Chen H, Zhang Z, et al. Design, synthesis and anticancer evaluation of new 4-anilinoquinoline-3-carbonitrile derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg Chem. 2021;114:105200. doi: 10.1016/j.bioorg.2021.105200. [DOI] [PubMed] [Google Scholar]
- 193.Alsaid MS, Al-Mishari AA, Soliman AM, Ragab FA, Ghorab MM. Discovery of benzo[g]quinazolin benzenesulfonamide derivatives as dual EGFR/HER2 inhibitors. Eur J Med Chem. 2017;141:84–91. doi: 10.1016/j.ejmech.2017.09.061. [DOI] [PubMed] [Google Scholar]
- 194.Ghorab MM, Alsaid MS, Soliman AM. Dual EGFR/HER2 inhibitors and apoptosis inducers: new benzo[g]quinazoline derivatives bearing benzenesulfonamide as anticancer and radiosensitizers. Bioorg Chem. 2018;80:611–620. doi: 10.1016/j.bioorg.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 195.Soliman AM, Alqahtani AS, Ghorab M. Novel sulphonamide benzoquinazolinones as dual EGFR/HER2 inhibitors, apoptosis inducers and radiosensitizers. J Enzyme Inhib Med Chem. 2019;34:1030–1040. doi: 10.1080/14756366.2019.1609469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liu X, Du Q, Tian C, Tang M, Jiang Y, Wang Y, et al. Discovery of cape derivatives as dual EGFR and CSK inhibitors with anticancer activity in a murine model of hepatocellular carcinoma. Bioorg Chem. 2021;107:104536. doi: 10.1016/j.bioorg.2020.104536. [DOI] [PubMed] [Google Scholar]
- 197.Zhang B, Liu Z, Xia S, Liu Q, Gou S. Design, synthesis and biological evaluation of sulfamoylphenyl-quinazoline derivatives as potential EGFR/CAIX dual inhibitors. Eur J Med Chem. 2021;216:113300. doi: 10.1016/j.ejmech.2021.113300. [DOI] [PubMed] [Google Scholar]
- 198.Zang H, Qian G, Arbiser J, Owonikoko TK, Ramalingam SS, Fan S, et al. Overcoming acquired resistance of EGFR-mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib, with the natural product honokiol. Mol Oncol. 2020;14:882–895. doi: 10.1002/1878-0261.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cao F, Gong YB, Kang XH, Lu ZH, Wang Y, Zhao KL, et al. Degradation of MCL-1 by bufalin reverses acquired resistance to osimertinib in EGFR-mutant lung cancer. Toxicol Appl Pharmacol. 2019;379:114662. doi: 10.1016/j.taap.2019.114662. [DOI] [PubMed] [Google Scholar]
- 200.Sun P, Qu Y, Wang Y, Wang J, Wang X, Sheng J. Wighteone exhibits an antitumor effect against EGFR L858R/T790M mutation non-small cell lung cancer. J Cancer. 2021;12:3900–3908. doi: 10.7150/jca.54574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Niu M, Xu J, Liu Y, Li Y, He T, Ding L, et al. FBXL 2 counteracts Grp94 to destabilize EGFR and inhibit EGFR-driven NSCLC growth. Nat Commun. 2021;12:5919. doi: 10.1038/s41467-021-26222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang KR, Zhang YF, Lei HM, Tang YB, Ma CS, Lv QM, et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci Transl Med. 2021;13:eabg6428. doi: 10.1126/scitranslmed.abg6428. [DOI] [PubMed] [Google Scholar]
- 203.Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liang Q, Gu WM, Huang K, Luo MY, Zou JH, Zhuang GL, et al. HKB99, an allosteric inhibitor of phosphoglycerate mutase 1, suppresses invasive pseudopodia formation and upregulates plasminogen activator inhibitor-2 in erlotinib-resistant non-small cell lung cancer cells. Acta Pharmacol Sin. 2021;42:115–119. doi: 10.1038/s41401-020-0399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huang K, Liang Q, Zhou Y, Jiang LL, Gu WM, Luo MY, et al. A novel allosteric inhibitor of phosphoglycerate mutase 1 suppresses growth and metastasis of non-small-cell lung cancer. Cell Metab. 2021;33:223. doi: 10.1016/j.cmet.2020.12.013. [DOI] [PubMed] [Google Scholar]
- 206.Huang K, Liang Q, Zhou Y, Jiang L-l, Gu W-m, Luo M-y, et al. A novel allosteric inhibitor of phosphoglycerate mutase 1 suppresses growth and metastasis of non-small-cell lung cancer. Cell Metab. 2019;30:1107–19.e8. doi: 10.1016/j.cmet.2019.09.014. [DOI] [PubMed] [Google Scholar]
- 207.Qiu Y, Yin X, Li X, Wang Y, Fu Q, Huang R, et al. Untangling dual-targeting therapeutic mechanism of epidermal growth factor receptor (EGFR) based on reversed allosteric communication. Pharmaceutics. 2021;13:747. doi: 10.3390/pharmaceutics13050747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yin L, Zhang Y, Yin L, Ou Y, Lewis MS, Wang R, et al. Novel mitochondria-based targeting restores responsiveness in therapeutically resistant human lung cancer cells. Mol Cancer Ther. 2021;20(12):2527–2538. doi: 10.1158/1535-7163.MCT-20-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.He J, Huang Z, Han L, Gong Y, Xie C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review) Int J Oncol. 2021;59:90. doi: 10.3892/ijo.2021.5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Planchard D, Feng PH, Karaseva N, Kim SW, Kim TM, Lee CK, et al. Osimertinib plus platinum–pemetrexed in newly diagnosed epidermal growth factor receptor mutation-positive advanced/metastatic non-small-cell lung cancer: safety run-in results from the FLAURA2 study. ESMO Open. 2020;6:100271. doi: 10.1016/j.esmoop.2021.100271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28. doi: 10.1186/s12943-021-01489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 213.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379:2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 214.Zhou C, Wu L, Fan Y, Wang Z, Liu L, Chen G, et al. Sintilimab plus platinum and gemcitabine as first-line treatment for advanced or metastatic squamous nsclc: results from a randomized, double-blind, phase 3 trial (ORIENT-12) J Thorac Oncol. 2021;16:1501–1511. doi: 10.1016/j.jtho.2021.04.011. [DOI] [PubMed] [Google Scholar]
- 215.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–2301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 216.Jabbour SK, Berman AT, Decker RH, Lin Y, Feigenberg SJ, Gettinger SN, et al. Phase 1 trial of pembrolizumab administered concurrently with chemoradiotherapy for locally advanced non-small cell lung cancer: a nonrandomized controlled trial. JAMA Oncol. 2020;6:848–855. doi: 10.1001/jamaoncol.2019.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Liu D, Gong J, Liu T, Li K, Yin X, Liu Y, et al. Phase 1 study of SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with advanced solid tumors. J Clin Oncol. 2021;39:2503–2503. doi: 10.1200/JCO.2021.39.15_suppl.2503. [DOI] [Google Scholar]
- 218.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Trans Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wu L, Ke L, Zhang Z, Yu J, Meng X. Development of EGFR TKIs and options to manage resistance of third-generation EGFR TKI osimertinib: conventional ways and immune checkpoint inhibitors. Front Oncol. 2020;10:602762–602862. doi: 10.3389/fonc.2020.602762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The material supporting the conclusion of this review has been included within the article.