

Abstract
Obtaining high-resolution structures of Receptor Tyrosine Kinases that visualize extracellular, transmembrane and intracellular kinase regions simultaneously is an eagerly pursued but still unmet challenge of structural biology. The Human Epidermal Growth Factor Receptor 3 (HER3) that has a catalytically inactive kinase domain (pseudokinase) forms a potent signaling complex upon binding of growth factor neuregulin 1β (NRG1β) and dimerization with a close homolog, the HER2 receptor. The HER2/HER3/NRG1β complex is often referred to as an oncogenic driver in breast cancer and is an attractive target for anti-cancer therapies. After overcoming significant hurdles in isolating sufficient amounts of the HER2/HER3/NRG1β complex for structural studies by cryo-electron microscopy (cryo-EM), recently we obtained the first high-resolution structures of the extracellular portion of this complex. Here we describe a step-by-step protocol for obtaining a stable and homogenous HER2/HER3/NRG1β complex for structural studies and our recommendation for collecting and processing cryo-EM data collected for this sample. We also show improved EM density for the transmembrane and kinase domains of the receptors, which continue to evade structural determination at high resolution. The discussed strategies are tunable and applicable to other membrane receptor complexes.
1. Introduction
Receptor Tyrosine Kinases (RTKs) are key mediators of transmembrane (TM) signaling and are one of the most often deregulated receptor classes in disease (Arteaga & Engelman, 2014; Du & Lovly, 2018; Lemmon & Schlessinger, 2010). RTKs couple binding of the extracellular ligands to their ectodomains with activation of the intracellular tyrosine kinase domains, resulting in phosphorylation of the flexible C-terminal receptor tails that then recruit downstream signaling molecules (Lemmon & Schlessinger, 2010). Activation occurs via receptor dimerization, or sometimes higher-order oligomerization, which is initiated by binding of the extracellular ligands (Garrett et al., 2002; Himanen & Nikolov, 2003; Lemmon & Schlessinger, 2010; H. Liu, Chen, Focia, & He, 2007; Trenker & Jura, 2020; Ullrich & Schlessinger, 1990; van Lengerich, Agnew, Puchner, Huang, & Jura, 2017; Wiesmann et al., 1997). Disease mutations have been shown to map across RTK domains and perturb normal receptor regulation resulting in oncogenic signaling (Du & Lovly, 2018). Understanding the detailed mechanisms by which ligand binding is transduced into kinase activation is of importance for understanding the pathology of disease mutations, and for enabling the development of therapeutics that target this important family of receptors.
Most of what is known about the structural changes that the receptors undergo during ligand-induced activation is based on structures of isolated extracellular, transmembrane and intracellular domains (Kovacs, Zorn, Huang, Barros, & Kuriyan, 2015). While such studies had brought fundamental insights into modes of ligand/receptor binding and architecture of the individual receptor domains, the information about allosteric coupling between receptor domains across the plasma membrane is inevitably lost in such investigations. Several recent structural studies by single particle negative-stain (NS) and cryo-electron microscopy (cryo-EM) have focused on full-length RTK preparations - signaling the beginning of the exciting new era in structural studies of RTKs (Chen, Unger, & He, 2015; Diwanji, Thaker, & Jura, 2019; Gutmann, Kim, Grzybek, Walz, & Coskun, 2018; Huang et al., 2021; Li, Choi, Yu, & Bai, 2019; Mi et al., 2008; Mi et al., 2011; Opatowsky et al., 2014; Uchikawa, Choi, Shang, Yu, & Bai, 2019). Despite the efforts, all reported studies fell short of reconstructing high-resolution structures of receptor complexes that visualize all functional domains simultaneously. Thus, such structures remain an unsolved problem in RTK structural biology and are being eagerly pursued.
Human epidermal growth factor receptors (HERs) are a subfamily of RTKs that encompass four closely related receptors, EGFR, HER2, HER3 and HER4 (Lemmon & Schlessinger, 2010). Activation of HERs proceeds through homo- or heterodimerization induced by ligand binding (Alroy & Yarden, 1997; Freed et al., 2017; Rush, Peterson, & Ceresa, 2018; van Lengerich et al., 2017). Depending on a type of bound ligand and receptors in a dimer, the active HER dimer will elicit unique downstream signaling responses (Freed et al., 2017; Huang et al., 2021; Sweeney & Carraway, 2000; Trenker & Jura, 2020). HER3 is a unique family member because it has a catalytically-inactive domain - a pseudokinase domain (Guy, Platko, Cantley, Cerione, & Carraway, 1994; Jura, Shan, Cao, Shaw, & Kuriyan, 2009; Sierke, Cheng, Kim, & Koland, 1997). Despite having negligible catalytic activity, HER3 can form signaling-competent complexes through heterodimerization with other HER receptors. This is induced by binding of the HER3 extracellular domain (ECD) to growth factor ligands, which belong to the neuregulin (NRG) family of growth factors. In the heterodimeric complexes, the HER3 pseudokinase domain serves as an allosteric activator of the kinase domain of the catalytically-active HER dimerization partner (Littlefield et al., 2014; Zhang, Gureasko, Shen, Cole, & Kuriyan, 2006). HER3 preferentially dimerizes with HER2, which also depends on heterodimerization with other HER receptors for signaling (Sliwkowski et al., 1994; Wallasch et al., 1995). This is because HER2 is an orphan receptor for which no known activating ligands have been identified. Thus, in the active HER2/HER3/NRG1β signaling complex HER3 is the ligand-sensing and HER2 is the kinase-active component.
The functionally asymmetric HER2/HER3 heterodimer is one of the most potent signaling HER receptor complexes. Its potency is in large part determined by the ability of phosphorylated HER3 to recruit and activate multiple copies of PI3K (Wallasch et al., 1995). HER2 does not bind PI3K directly and on its own it cannot efficiently activate it. Thus, heterodimerization with HER3 is an essential component of HER2-dependent transformation and is considered to be a driving force of cancer progression in several tissues, most predominantly - in the breast (Moasser, 2007). Despite its importance, no structures of the HER2/HER3 complexes have been solved until our recent study in which we report cryo-EM reconstructions of the extracellular domain module of the NRG1β-bound HER3 with the wild type HER2 or an oncogenic cancer mutant HER2-S310F (Diwanji et al., 2021). These structures have revealed unique features of the functionally asymmetric HER2/HER3 complex, including an unusual mode of engagement of the dimerization arms, which are key structural elements contributing to the dimerization of the HER receptor extracellular domains. We found that HER2 is unable to engage a dimerization arm from HER3, making dimerization reliant on HER3’s ability to engage the HER2 arm. We hypothesized that this is why HER2 cannot form homodimers itself, which had been a curious puzzle in the field. By solving the structure of HER3 in complex with an oncogenic HER2 mutant (S310F) that mutation restores HER3 dimerization arm engagement, which provided a mechanistic explanation for its activating effect on HER2/HER3 signaling (Diwanji et al., 2021).
The structural characterization of the HER2/HER3/NRG1β was enabled by the development of the experimental protocols of purification and reconstitution of a stable dimeric complex. Despite the fact that we have only resolved the extracellular domain module of the HER2/HER3 heterodimer, our studies had taught us that the inclusion of the intracellular domains in our constructs was necessary for purification of the stable dimers. While, we have not yet been able to obtain a high-resolution reconstruction of the intracellular domain module of the HER2/HER3 dimer, we made progress in data collection and analysis that focus on resolution of these domains. Here we describe a detailed protocol for assembling the stable HER2/HER3/NRG1β and HER2-S310F/HER3/NRG1β complexes and for collecting and processing cryo-EM data with improved EM density for transmembrane and kinase domains (Diwanji et al., 2021). These strategies are tunable and applicable to other membrane receptor complexes.
2. Expression and purification of NRG1β
All ligands known to bind to HER receptors feature an EGF-like domain, which is required and sufficient for receptor binding. To reconstitute the HER2/HER3/NRG1β complex, we first established a protocol for expression and purification of the EGF-like domain of the HER3-binding growth factor ligand, NRG1β. The EGF-like domain is held together by three conserved disulfide bonds, which poses a major challenge for recombinant expression of the correctly folded HER receptor ligands. Based on a previous protocol, we express the NRG1β EGF-like domain (residues 177–236) as a fusion protein to thioredoxin (TrxA) and C-terminal FLAG (DYKDDDDK)- and 6x polyhistidine (His)-tag in E. coli Origami B (DE3) pLysS cells (Diwanji et al., 2021; P. Liu et al., 2012). This E. coli strain features several mutations that lower the reducing capability of the E. coli cytoplasm which aids the formation of disulfide bonds (Figure 1a–b). The TrxA may also assist in disulfide bond formation and may support solubility of the fusion protein (LaVallie et al., 1993). The TrxA-NRG1β-FLAG-His expression construct features a 3C protease cleavage site between TrxA and NRG1β (Figure 1a). The His-tag in this construct is utilized for affinity purification of the protein from E. coli origami cells (see protocol below, section 2). As discussed in section 3, the additional FLAG-tag can be used to prepare a NRG1β-coated resin by incubating the purified NRG1β with anti-FLAG resin. This resin then allows for pulldown of the HER3 receptor from HER3-expressing cell lysates (protocol in section 3).
Figure 1: Expression and purification of NRG1β.
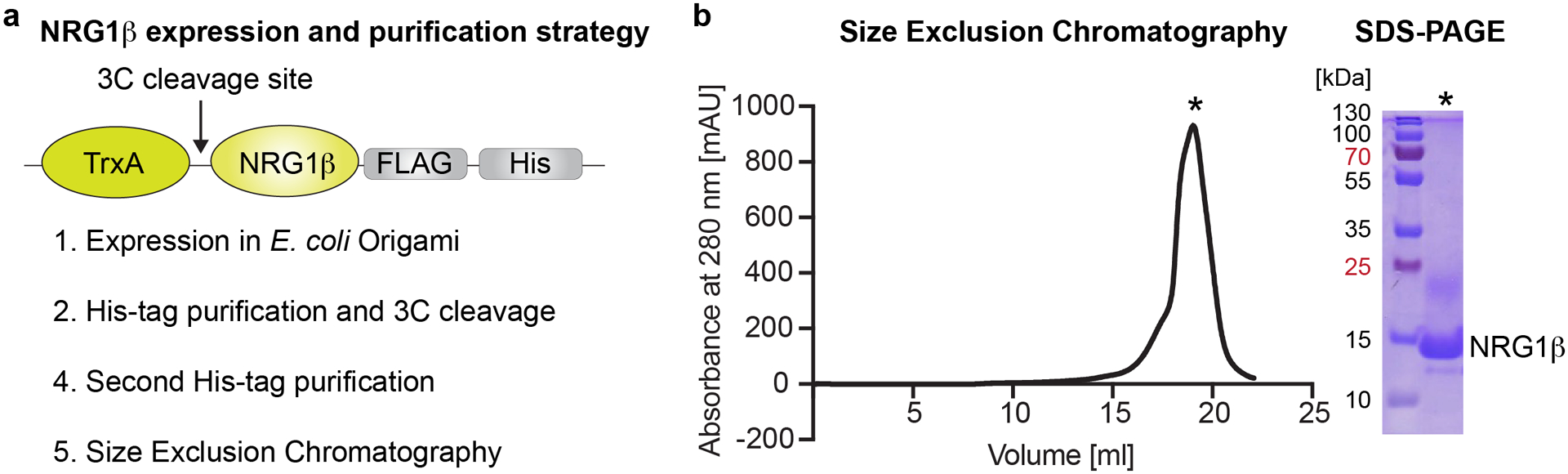
a, Overview of the expression and purification pipeline. Thioredoxin-NRG1β (TrxA-NRG1β-FLAG-His) fusion protein is expressed in E. coli origami B (DE3) pLysS cells and purified via a His-tag. The TrxA protein is cleaved off with the 3C protease and NRG1β is purified using another His-tag purification followed by Size Exclusion Chromatography (SEC). b, The SEC chromatogram of NRG1β loaded onto a Superdex 200 increase column shows a homogenous peak of NRG1β elution at 18–19 ml in high purity as shown on the Coomassie-stained SDS-PAGE gel with 10 μl of sample loaded.
2.1. Key Resources and equipment
Key resources and consumables
E. coli origami B (DE3) pLysS (made chemically competent in house and are stored at −80 °C at an OD600 of ~12.5. Can be purchased from Millipore Sigma, #70839)
pET32a expression plasmid with Ampicillin resistance (LaVallie et al., 1993). Empty plasmid available from Millipore Sigma, # 69015). Construct was cloned in house and encodes a Thioredoxin-NRG1β (177–236)-FLAG-His fusion protein with 3C cleavage site between thioredoxin and NRG1β (Figure 1).
Tryptone (Thermo Fisher Scientific, # BP9726-2)
Yeast Extract (Thermo Fisher Scientific, # BP97275)
Glycerol (Millipore Sigma, # GX0185),
Potassium phosphate, Monobasic, (KH2PO4 (Millipore Sigma, # PX1565)
Potassium phosphate, Dibasic (K2HPO4, Millipore Sigma, # PX1570))
LB Broth with agar (Millipore Sigma, # L3147)
LB-Agar plates (made in house using 40 g of LB Agar per 1 l ddH2O)
Chloramphenicol (AllStar Scientific, #480-045)
Kanamycin Sulfate (VWR, #97061-598)
Tetracycline (Tetracycline Hydrochloride, Gold Biotechnology, # T-101-25)
Ampicillin (Calbiochem, sold by Millipore Sigma, #171254)
Two-Sided Disposable Plastic Cuvettes, Semi-Micro 1.3- 3 ml (VWR, 97000-586)
TRIS (Trizma base Millipore Sigma, #T6066)
Sodium chloride (NaCl, Millipore Sigma, #SX0420)
Imidazole (Millipore Sigma, #5720)
Isopropyl β-D-1-thiogalactopyranoside (IPTG, Gold Biotechnology, #I2481C100)
Phenylmethylsulfonyl fluoride (PMSF, VWR, #0754)
DNase I, grade II, from bovine pancreas (Roche, sold by Millipore Sigma, #10104159001)
HisPur Ni-NTA Resin (Thermo Fisher Scientific, PI88222)
3C protease (prepared in house with non-cleavable His-tag, stored in 1 mg aliquots at −80 °C at ~1.5 mg/ml, adjusted from (Lee, Lolicato, Arrigoni, & Minor, 2021))
Liquid nitrogen
Amicon Ultra-15 Centrifugal Filter Units, 3,000 and 10,000 MWCO, (Millipore Sigma, #UFC900324 and #UFC901024, respectively)
Conical tubes, 15 and 50 ml, polypropylene (VWR, #21008)
Microcentrifuge tubes, 1.7 ml (VWR, #87003-294)
Econo-Pac(R) Chromatography Columns, 20 ml bed volume (Bio-Rad, #7321011)
Syringes and Millex-HV Syringe Filter Unit, 0.45 um, Millipore Sigma, #SLHVM33RS)
Liquid nitrogen
Buffers and media
TBS (50 mM TRIS pH 7.4, 150 mM NaCl
5 M imidazole stock pH 8.0
Terrific Broth (1 liter: 12 g Tryptone, 24 g Yeast Extract, 4 ml glycerol, dissolve in 900 ml ddH2O and autoclave. Before use, add 100 ml Phosphate buffer at 0.17 M KH2PO4 and 0.72 M K2HPO4)
L Broth (1 liter: 10 g Tryptone, 10 g NaCl, 5 g Yeast Extract, adjust to pH 7)
IPTG stock solution, 1 M (2.38 g IPTG for 10 ml, dissolve in ddH2O)
Equipment
37 °C static incubator (VWR)
37 °C shaking incubator (New Brunswick Innova 4300, Marshall Scientific, #NB-4300)
Sonicator for cell lysis (S-4000 Ultrasonic Liquid Processor, Misonix, probe diameter 1 cm)
Non-baffled 2 l sterile glass flasks
Non-baffled 500 ml sterile glass flasks
Glass beaker (50 ml volume per bacterial pellet)
Glass bottle (50 ml per 1 l bacterial pellet)
Centrifuge to pellet 1 l bacterial cultures (Avanti J-26 XP, Beckman Coulter, rotor JLA-8.1000)
Polypropylene Bottle Assembly, 1000 ml (Beckman Coulter, # C31597)
Ultracentrifuge (Optima L-90K, Beckman Coulter, rotor Type 45 Ti)
Polycarbonate Centrifuge Tubes, 70 ml with cap (Beckman Coulter, #355620)
Table-top centrifuge (for 15/50 ml conical tubes, Allegra X-15R, Beckman Coulter)
Magnetic stirrer plate
Nutating mixer (Fisher scientific)
Spectrophotometer optical density measurements of bacterial cultures (Nanodrop 2000c, Thermo Scientific)
Superdex 200 Increase 10/300 column (GE HEalthcare, now Cytiva #28990944)
Akta purification system (Akta Pure, Cytiva)
2.2. Protocol:
Note: E. coli Origami cells grow slowly thus allowing for longer expansion times as outlined below. The protocol below is optimized for 6 liter cultures and typically yields in ~15–20 mg of the pure NRG1β EGF-like domain.
2.2.1. Transformation
-
1
Transform 1 μg of DNA into 50 μl chemically competent E. coli Origami B (DE3) pLysS cells and plate onto LB-Agar plates containing Ampicillin (100 μg/ml), Kanamycin (30 μg/ml), Chloramphenicol (35 μg/ml) and Tetracycline (10 μg/ml) using any standard transformation protocol with a recovery time of one hour.
-
2
Incubate at 37 °C in a static incubator for 24–40 h. Cells grow very slowly and we advise transforming in the morning one day or in the evening two days before starter cultures are grown.
2.2.2. Expression
-
3
In the morning, streak across 5–10 colonies on the LB-Agar plate and inoculate a 10 ml starter culture in LB containing Ampicillin (100 μg/ml), Kanamycin (30 μg/ml), Chloramphenicol (35 μg/ml) and Tetracycline (10 μg/ml) throughout the day at 37 °C and 200 rpm.
-
4
Set up overnight culture. Transfer 10 ml starter culture into 100 ml LB supplemented with antibiotics (see step 3) and grow overnight at 37 °C and 200 rpm in a non-baffled 500 ml flask.
-
5
The next day, add 15 ml of the overnight culture to 1 liter Terrific Broth + antibiotics and grow to an OD600 of 1 at 37 °C and 200 rpm in a non-baffled 2 liter flask. This usually takes 3–4 hours. OD600 measurements are performed using a Nanodrop in Semi-Micro Plastic Cuvettes.
-
6
Induce with 1 mM IPTG (1 ml of a 1 M fresh stock solution) and reduce temperature of the incubator to 25 °C.
-
7
The next morning, pellet cells by centrifugation using 1000 ml Polypropylene Bottles at 4000 rpm (3397 × g, Avanti J-26 XP, Beckman Coulter JLA-8.1000 rotor) for 20 min at 25 °C and flash freeze the pellet in liquid nitrogen. Either store pellet at −80 °C or continue with the purification.
2.2.3. Purification
-
8
Thaw bacterial pellet(s) in a cold-water bath and resuspend in 40 ml TBS supplemented with 1 mM PMSF and 1 mg/ml DNase I per 1 liter pellet.
-
9
Sonicate in a glass beaker on ice for at least 10 min total at power 40, 4 sec on, 4 sec off, or until thoroughly lysed.
-
10
Transfer lysate into required numbers of 70 ml Polycarbonate Centrifuge Tubes and pellet cell debris by ultracentrifugation at 40,000 rpm (1865511 × g, Optima L-90K, Beckman Coulter, rotor Type 45 Ti) for 1 h at 4°C. If required, add TBS to fill tubes to the top.
-
11
Filter supernatant through a 0.45 μm syringe filter into a glass bottle.
-
12
Equilibrate ~1 ml Ni-NTA resin per 1 liter expression culture in TBS, add to the lysate and incubate overnight at 4 °C while stirring the beads on a magnetic stirrer plate.
-
13
Transfer the lysate + beads into Econo-Pac Chromatography Columns. The beads usually appear brown for well-expressing proteins.
-
14
Wash beads with at least 5 column volumes of TBS + 20 mM imidazole, followed by 5 column volumes of TBS + 50 mM imidazole.
-
15
Elute in 3–5 × column volumes of TBS + 300 mM imidazole, or until beads return to their initial blue color, into a fresh 50 ml conical tube.
-
16
To reduce the imidazole concentration to 30 mM or lower, concentrate the eluate to a volume of 4 ml or less using an Amicon-15 Centrifugal Filter Unit with a molecular weight cut-off (MWCO) of 10 kDa and spinning at 4000 × g for 20 min intervals in a table-top centrifuge at 4 °C. Transfer the sample to a 50 ml conical tube and add TBS to a total volume of 40 ml.
-
17
Add 3C protease to cleave off thioredoxin from the NRG1β-FLAG-His ligand, mix by inverting the tube 2–3 x and incubate overnight at 4°C without agitation. We use 1 mg of protease for 6 liter of expression culture.
-
18
The following day, add ~1 ml equilibrated Ni-NTA resin per 1 liter expression culture to the sample and incubate overnight at 4 °C while rocking the tube on a nutating mixer.
-
19
Transfer beads to an Econo-Pac Chromatography Column and wash with 5 column volumes of TBS.
-
20
Elute in 3–5 column volumes TBS + 300 mM imidazole into a fresh 50 ml conical tube.
-
21
Concentrate the eluate to a volume of 0.5 ml per liter of expression culture using an Amicon-15 Centrifugal Filter Unit with a MWCO of 3 kDa and spinning at 4000 × g for 20 min intervals in a table-top centrifuge at 4 °C. The eluate usually appears yellow/brown in color.
-
22
Transfer the sample into a microcentrifuge tube, flash freeze in liquid nitrogen and store at −80 °C or proceed directly to Size Exclusion Chromatography (SEC). For larger preparations of more than 1 liter, freeze 500 μl aliquots for each liter expression culture. If precipitation occurs, remove precipitate by centrifugation at ~21,000 × g for 3 min and 4 °C in a table-top microcentrifuge before freezing and/or SEC.
-
23
Equilibrate a Superdex 200 increase 10/300 GE column with TBS using an Akta Pure instrument, and load 500 μl of sample on the equilibrated column. Collect fractions of high-purity NRG1β around an elution volume of 18 ml (Figure 1a–b).
-
24
Alternatively, the second Ni-NTA step can be replaced by ion exchange chromatography to remove the protease and thioredoxin A as described in a similar protocol (P. Liu et al., 2012). We have also successfully performed on-column digests after step 14 prior to elution. Transfer the protein-bound Ni-NTA beads into a 50 ml conical tube and adjust to a total volume of 40 ml with TBS. Add 3C protease and incubate overnight at 4 °C while rocking the tube on a nutating mixer. After the on-column digest, transfer the resin into an Econo-Pac Chromatography Column, wash in 5 bed volumes of TBS + 50 mM imidazole and elute in TBS + 300 mM imidazole. Afterwards proceed with step 21.
-
25
Freeze protein as 1 mg aliquots in microcentrifuge tubes in liquid nitrogen and store at −80 °C. The extinction coefficient of the NRG1β-FLAG-His ligand is 6335 with a molecular weight of 9.73 kDa (Abs 0.1% =0.651 g/l, determined via the Expasy ProtParam online tool). Absorbance readings are performed using a nanodrop.
Expected yield: We typically obtain ~15–20 mg of the pure NRG1β EGF-like domain for 6 liters of culture.
3. Purification, crosslinking and preparation of the HER2/HER3/NRG1β sample for single particle electron microscopy imaging.
Previous efforts aimed at generating a stable HER2/HER3/NRG1β complex using isolated extracellular domains of the receptors were unsuccessful (Ferguson, Darling, Mohan, Macatee, & Lemmon, 2000). We set to assemble this signaling complex between nearly full-length receptors, which only had their flexible C-terminal tails truncated to facilitate expression, purification and structural analysis. HER3 (1–1022) was expressed with a C-terminal Twin-Strep (TS) tag, HER2 (1–1029) featured C-terminal TS and Maltose-Binding-Protein (MBP) tags (Figure 2a). Both receptors were expressed individually in Expi293F cells using ExpiFectamine-based transfection. Receptor pellets were lysed and the complex was purified from mixed cell lysates using tandem affinity purification to pull specifically on HER3 and then HER2. In the first step, HER3 was specifically pulled down via NRG1β-coated beads. In a second step HER2 was specifically pulled down via the MBP-tag followed by SEC. Below we describe a detailed procedure for purifying the HER2/HER3/NRG1β complex, discuss optimization of the constructs and the additional steps we took to obtain an optimal sample for structure determination by cryo-EM.
Figure 2: Expression and purification of the HER2/HER3/NRG1β complexes.
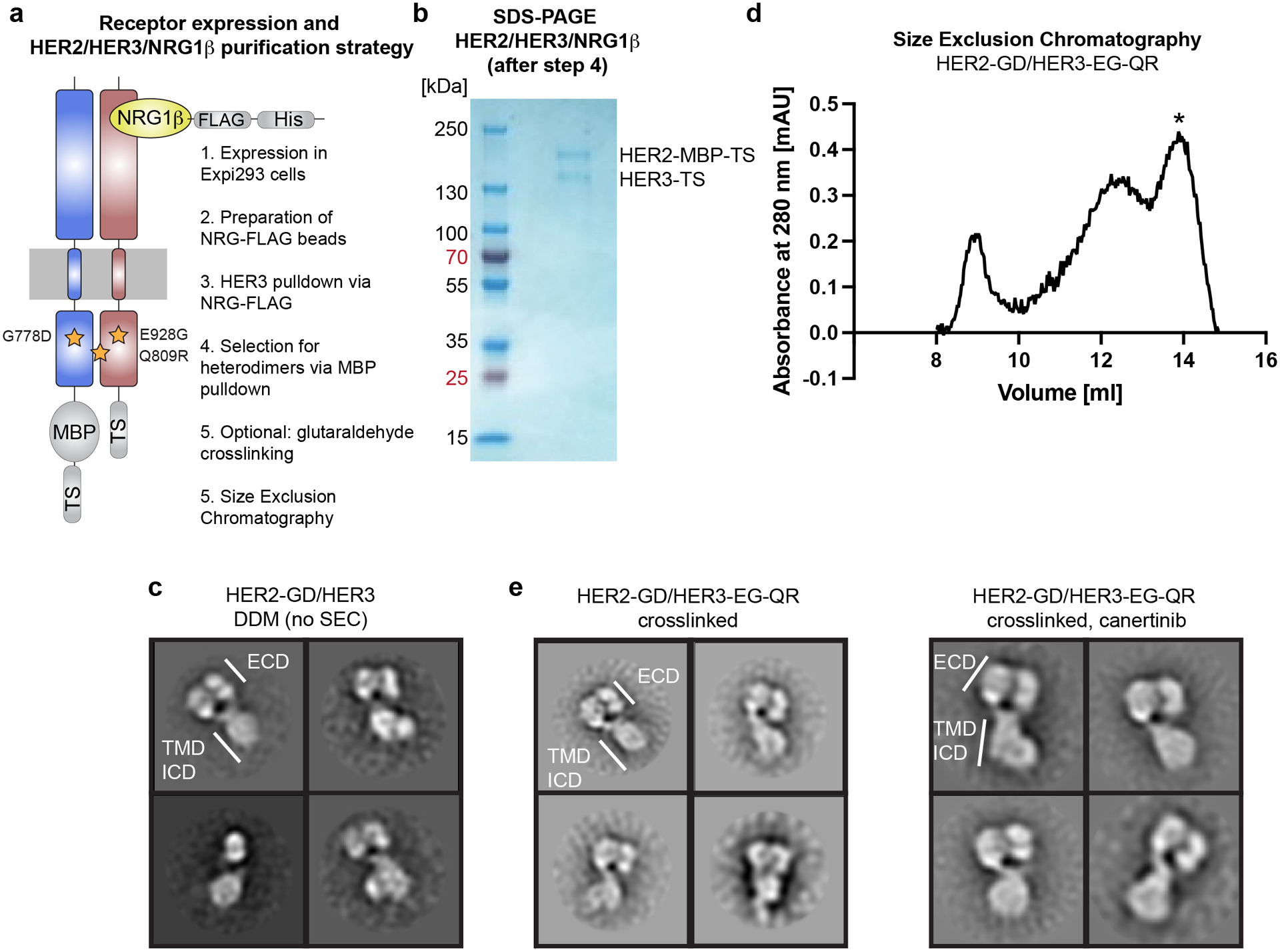
a, Cartoon of the HER2/HER3/NRG1β complex with overview of the expression/purification pipeline. The optimized HER2 construct has the G778D (GD) mutation, and HER3 has the E928G (EG) and Q809R (QR) mutations. Receptors are expressed in Expi293 suspension cells and purified via tandem affinity chromatography using NRG1β-coated FLAG beads and amylose resin, followed by SEC. b, Coomassie-stained SDS-PAGE gel with 10 μl of the ~4 ml elution volume loaded shows a stoichiometric ration of HER2 and HER3 receptors. NRG1β is less visible due to small molecular weight. c, NS-EM 2D class averages of the HER2-GD/HER3/NRG1β complex after tandem affinity purification with the expected heart-shaped ECD dimers in some classes. Images were collected on a FEI Tecnai T12 microscope at 52,000 × magnification. d, Representative SEC chromatogram shows a profile of the crosslinked HER2-GD/HER3-EG-QR/NRG1β complex with the heterodimer peak eluting at 14 ml (marked with *). e, NS-EM 2D class averages of the cross-linked HER2-GD/HER3-EG-QR/NRG1β complexes with and without canertinib. Images were collected on a FEI Tecnai T12 microscope at 52,000 × magnification.
3.1. Key resources and equipment
Key resources and consumables
Plasmid DNA for expression in mammalian cells encoding for HER2(1–1029)-MBP-TS and HER3(1–1022)-TS. Here pFastBac1 plasmids (Thermo Fisher Scientific, #10360014) were use in which the baculovirus promoter was replaced by a CMV promoter that is active in mammalian cells (Sung et al., 2014). We also commonly use pcDNA3.1 and pcDNA4/TO for this protocol (Thermo Fisher Scientific, # V79020 and # V102020).
ExpiFectamine 293 Transfection Kit (Thermo Fisher Scientific, #A14524)
Expi293 Expression Medium (Thermo Fisher Scientific, #A1435101)
Expi 293F human cells (Thermo Fisher Scientific, #A14527)
Opti-MEM (Thermo Fisher Scientific, #31985070)
Canertinib, 10 mM in DMSO (MedChem Express, #HY-10367)
NRG1β-FLAG (in house, see section 2)
n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace, #D310)
n-Dodecyl-β-D-Maltopyranoside (DDM, Inalco, #1758-1350, used only for mammalian
TRIS (Triszma base, Millipore Sigma, #T6066)
HEPES (Thermo Fisher Scientific, # FLBP3101)
Sodium chloride (NaCl, Millipore Sigma, #SX0420)
Ethylenediaminetetraacetic acid (EDTA, Millipore Sigma, #324503)
Sodium orthovanadate (Millipore Sigma, #S6508)
Sodium fluoride (NaF, Millipore Sigma, # SX0550)
cOmplete EDTA-free Protease Inhibitor Cocktail Mini Tablets (Roche, sold by Millipore Sigma, #11836170001)
DNase I, grade II, from bovine pancreas (Roche, sold by Millipore Sigma, #10104159001)
Anti-FLAG resin (Anti-DYKDDDDK G1 Affinity Resin, GenScript, #L00432-10)
FLAG peptide (DYKDDDDK synthetic peptide, Sino Biological, #PP101274)
Glutaraldehyde Aqueous 8%, EM Grade (Electron Microscopy Sciences, #16019)
Amylose resin (NEB, E8021L)
Maltose monohydrate (Millipore Sigma, # M5895)
Erlenmeyer Flasks, 125 ml, sterile (VWR, #89095-258)
Conical tubes, 15 and 50 ml, polypropylene (VWR, #21008)
Microcentrifuge tubes, 1.7 ml (VWR, #87003-294)
Micro Bio-Spin columns, 0.8 ml (Bio-Rad, #7326204)
Amicon Ultra-0.5 Centrifugal Filter Units, MWCO 100 kDa (Millipore Sigma, #UFC510096)
Disposable Serological Pipets 5 ml, 10 ml, 25 ml, VWR #89130
Liquid nitrogen and appropriate vessel for freezing tubes of 1.5 mL to 50 mL volume
Uranyl formate (Electron Microscopy Sciences, #22450)
Formvar/carbon 400 mesh copper grids (Ted Pella, #01754-F)
Graphene-oxide (GO)-coated Quantifoil R1.2/1.3 300 mesh Au holey-carbon grids (in house, provided by Feng Wang (Palovcak et al., 2018).
Liquid ethane, (Matheson, #UN1035).
Whatman qualitative filter paper, grade 1 for preparation of negative-stain EM grids (Millipore Sigma, #WHA1001090)
Whatman qualitative filter paper, grade 597 for cryoEM grid freezing using a Vitrobot Mark IV (FEI) (Millipore Sigma, #WHA10311807)
Buffers
1M TRIS pH 7.4
TBS (50 mM TRIS pH 7.4 and 150 mM NaCl
Lysis Buffer (50 mM TRIS pH 7.4, 150 mM NaCl, 1% DDM (Inalco), 1 mM EDTA, 1mM sodium orthovanadate, 1 mM NaF, one tablet of cOmplete EDTA-free Protease Inhibitor Cocktail Mini Tablets per 50 ml lysis buffer, and 1 mg/ml DNase
TBS-DDM (50 mM TRIS pH 7.4, 150 mM NaCl and 0.5 mM DDM (Anatrace))
HBS-DDM (50 mM HEPES pH 7.4, 150 mM NaCl and 0.5 mM DDM (Anatrace))
FLAG elution buffer (TBS-DDM+ 0.25 mg/ml FLAG peptide)
Software
MotionCor2 (Zheng et al., 2017)
CryoSPARC2 (Punjani, Rubinstein, Fleet, & Brubaker, 2017)
RELION3 (Scheres, 2012)
UCSF PyEM v0.5 (“Daniel Asarnow, Eugene Palovcak, & Yifan Cheng. (2019). asarnow/pyem: UCSF pyem v0.5 (v0.5). Zenodo. https://doi.org/10.5281/zenodo.3576630,”)
UCSF Chimera (Pettersen et al., 2004)
Equipment
Incubator at 37 °C and 8% CO2 with shaker (Heracell VIOS 160i, Thermo Scientific)
Tissue culture hood
Table-top microcentrifuge (5424 R, Eppendorf)
Table-top centrifuge (for 15/50 ml conical tubes, Allegra X-15R, Beckman Coulter)
Nutating mixer (Fisher scientific)
Superose 6 10/300 Increase column (GE Healthcare, now Cytiva, #29-0915-96)
Akta purification system (Akta Pure, Cytiva)
FEI-Tecnai T12
4k CCD camera (Gatan)
Glow discharger (PELCO easyGlow, Ted Pella)
GPU-equipped workstation (at least 11GB of GPU memory, at least 128GB of system memory, at least 4TB of fast SSD storage and 20TB of spinning disc storage)
Vitrobot Mark IV (FEI)
300-keV Titan Krios (Thermo Fisher Scientific) with K3 direct electron detector (Gatan) and BioQuantum energy filter (Gatan)
Tweezers, Style N5AC, Electron Microscopy Sciences (#0203-N5AC-PO)
−80 °C freezer
3.2. Protocol
3.2.1. Transfection of Expi293 cultures
Expi293F cells were cultured in Expi293 Expression Medium and were transfected with the expression constructs using the ExpiFectamine 293 Transfection Kit. Maintain 30 ml suspension cell cultures in 125 ml flasks in Expi Expression Medium at 37 °C and 8% CO2 shaking at 120 rpm at a cell density between 0.5 and 4 × 106 cells/ml. Cells double approximately once every 24 hours. Separate cultures for HER2 and HER3 expression were prepared. Following optimization, we found that two 30 ml expression flasks per receptor yielded enough samples for heterodimer reconstitution to allow preparation of at least 12 cryo-EM grids. The purification protocol below was optimized for a total of four 30 ml cell pellets.
-
1
One day prior to transfection, seed 30 ml of Expi293F suspension cultures into 125 ml flasks at a density of 2 × 106 cells/ml to reach a density of ~4 × 106 cells/ml at the time of transfection. Depending on the number of flasks needed, plan to have sufficient cultures at ~4 × 106 cells/ml on this day to seed transfection cultures. For example, we typically seed multiple 30 ml cultures at 0.5 × 106 cells/ml on Fridays and seed transfection cultures on Mondays by diluting approximately 1:1 with fresh medium.
-
2
In the afternoon, prepare transfection mix according to the manufacturer’s protocol and add to cells. We transfect the suggested 30 ug of plasmid DNA for each 30 ml culture.
-
3
The next morning (16–18 hours after transfection), add 150 μl Transfection Enhancer 1 and 1.5 ml Transfection Enhancer 2 to each 30 ml flask of transfected cells according to the manufacturer’s protocol. At this stage, any other desired supplement can be added to the expression cultures. As described below, for expression of the HER2 constructs, we obtained best results by growing cells in 10 μM canertinib. Canertinib is a covalent inhibitor of HER2 and was only added at this stage of the purification.
-
4
Grow cells for another 24 hours.
-
5
Harvest cell pellets via centrifugation of the cultures at 1500 × g for 5 min.
-
6
Flash freeze cell pellets in liquid nitrogen and store at −80 °C.
3.2.2. Purification and sample preparation for negative stain and cryo-EM
-
7
Thaw cells on ice and resuspend each 30 ml cell pellet in 10 ml of the Lysis Buffer and combine lysates of HER2 and HER3 in a 50 ml conical tube.
-
8
Allow cells to lyse for 2–3 hours by gently rocking the lysate at 4 °C on a nutating mixer.
-
9
In the meantime, prepare NRG1β-coated anti-FLAG resin. Transfer 0.5 ml anti-FLAG resin for four 30 ml pellets to a 15 ml conical tube. Equilibrate the resin in TBS and add 1 mg NRG1β (EGF-like domain) for 0.5 ml anti-FLAG resin. Incubate by gently rocking at 4 °C for 1 hour and then wash the beads with 3 × 10 ml TBS using cycles of centrifugation (500 × g for 2 min at 4 °C) and resuspension in TBS. Remove excess TBS. Beads can be kept on ice until receptor lysates are ready.
-
10
Remove cell debris via centrifugation at 4,000 × g for 20 min at 4 °C and transfer the clarified lysate (supernatant) into a fresh 50 ml conical tube.
-
11
Add the NRG1β-bound anti-FLAG resin and incubate by gently rocking at 4 °C overnight.
-
12
The next morning, wash resin by centrifugation at 500 × g for 2 min at 4 °C and resuspension in 10 ml TBS-DDM in 3 serial cycles. Remove as much of the buffer from the beads as possible.
-
13
Add 4 bed volumes (2 ml for 500 μl resin) of FLAG elution buffer to the resin and incubate by gently rocking for 1 h at 4 °C.
-
14
Tape a Bio-Spin column at the top of a fresh 15 ml conical tube and elute anti-FLAG resin suspension stepwise through the column by gravity flow. Run additional two column volumes of FLAG elution buffer over the column and collect the flow-through in the same tube.
-
15
Equilibrate 500 ul of Amylose resin with TBS-DDM and load the eluate obtained in the previous step on this resin.
-
16
Incubate for 2 hours gently rocking at 4 °C.
-
17
Wash resin by centrifugation at 500 × g for 2 min at 4 °C and resuspension in TBS-DDM in 2 serial cycles. Add 4 column volumes of TBS-DDM + 20 mM maltose and incubate overnight gently rocking at 4 °C.
-
18
Elute through a Bio-Spin column, as described in step 14, into a fresh 15 ml conical tube. Run additional two column volumes of TBS-DDM + 20 mM maltose over the column and collect the flow-through into the same tube.
-
19
Concentrate the eluate using Amicon Ultra-0.5 centrifugal concentration units with 100 kDa cutoff filters to 400 μl via repeated spins at 4000 × g for 4 min and 4 °C.
-
20
Load the concentrated sample on the Superose 6 10/300 Increase column equilibrated in TBS-DDM using an Akta Pure chromatography instrument.
-
21
Collect peak fractions corresponding to the elution volume of ~ 14 ml. In our experience, the sample may be flash frozen in liquid nitrogen and stored at −80 °C at this point.
-
22
To prepare the grids for negative stain EM (NS-EM), we added 3 μl of sample equivalent in concentration to a 4 mAU SEC peak, which we empirically determined to be an ideal protein concentration for NS-EM imaging. For example, for the SEC profile shown in Figure 2d, we took the 2 ml peak fraction of peak 2 (peak height of ~0.4 mAU) and concentrated the sample to 200 μl to prepare negative stain grids. Samples were applied to negatively glow-discharged carbon coated copper grids, stained with 0.75% uranyl-formate, and imaged on an FEI-Tecnai T12 with an 4k CCD camera (Gatan). Note that it is important to empirically determine the optimal protein concentration for NS-EM imaging based on an individual protein and chromatography system. This empirical concentration can then be used to estimate the concentration for cryo-EM imaging on Graphene Oxide (GO)- coated holey carbon grids.
-
23
The same sample was then further concentrated to 20–40 μl to prepare GO grids for cryo-EM. The resulting sample concentration for cryo-EM is 5–10 fold higher than that prepared for the NS-EM, and is equivalent to a peak of 20–40 mAU range. See section 4 for further rationale and details. Sample was applied to graphene-oxide coated Quantifoil R1.2/1.3 300 mesh Au holey-carbon grids prepared as previously described (Palovcak et al., 2018), blotted using a Vitrobot Mark IV (FEI) and plunge frozen in liquid ethane (no glow discharge, 30 second wait time, room temperature, 100% humidity, 4–8 seconds blot time and 0 blot force using Whatman qualitative filter paper, grade 597).
3.2.3. Optional receptor crosslinking
We were able to increase the homogeneity and quality of NS-EM class averages further by mildly crosslinking the HER2/HER3/NRG1β sample with a non-specific, amine-reactive crosslinker glutaraldehyde (Figure 2e). Further rationale to apply crosslinking, was to increase the chance of observing density for intracellular domains. As discussed below, crosslinking resulted in a slight overall improvement of the extracellular domain resolution, but only marginally improved the density of the kinase domains after initial processing.
Since glutaraldehyde is an amine-reactive crosslinker, it is important not to perform the crosslinking reaction in buffers that contain primary amines such as TBS. Therefore, prior to crosslinking, we buffer-exchanged the sample over the second affinity step of the purification into the HBS-DDM buffer.
Follow steps 1–16 of the HER2/HER3/NRG1β purification protocol above (sections 3.21+3.2.2)
Wash resin by centrifugation at 500 × g for 2 min at 4 °C and resuspension in HBS-DDM in 3 serial cycles.
Add 4 column volumes of HBS-DDM + 20 mM maltose and incubate overnight at 4 °C on a rotator.
Transfer into a Bio-Spin column and elute into a fresh 15 ml conical tube by gravity flow. Run additional two column volumes of HBS-DDM + 20 mM maltose over the column and collect the flow-through in the same tube.
Concentrate the eluate using Amicon Ultra-0.5 centrifugal concentration units with 100 kDa cutoff filters to 400 μl. Spin at 3000 × g at 4 °C and repeated spins of 4 min.
Crosslink the sample on ice by adding 40 μl of aqueous 2% glutaraldehyde stock to the 400 μl of receptor from the previous step. Quench the reaction after 40 min by adding 40 μl of 1 M TRIS pH 7.4 buffer.
Load the sample on the Superose 6 10/300 Increase column equilibrated in TBS-DDM.
Collect peak fractions. In our experience, the HER2/HER3/NRG1β preparations may be flash frozen in liquid nitrogen and stored at −80°C.
For preparation of EM samples, see steps 23 and 24 of the sample transfection and purification protocol (sections 3.21+3.2.2)
3.4. Analysis and sample optimization
We screened various constructs of HER2 and HER3 to maximize the yields of NRG1β-induced heterodimers. Using the wild type sequence of both receptors, we were able to purify sufficient amounts of the heterodimer after tandem affinity purification for characterization by NS-EM (Figure 2c). 2D NS-EM class averages revealed heart-shaped particles, resembling those previously seen for the EGF-bound extracellular EGFR dimer, providing first structural characterization of the HER2/HER3 heterodimers, and showing, for the first time, that HER3 adopts an extended conformation upon ligand binding (discussed in detail in (Diwanji et al., 2021)). However, these initial samples appeared insufficiently homogeneous for further imaging by cryo-EM. We could never obtain high enough sample yields, even from a 120 ml Expi293 cell expression culture. We reasoned that the limiting factor is the amount of HER2 that was pulled down with HER3 in the first step of the purification, suggesting that the WT heterodimer complex might be inherently weak. This observation is consistent with previous findings that the extracellular domains of HER2 and HER3 do not heterodimerize in solution (Ferguson et al., 2000) and suggests that heterodimerization may be reliant on considerable contributions from the transmembrane and/or intracellular receptor domains.
To further increase the stability of the heterocomplex, yields of the purification and the chance of visualizing the kinase domains, we rationally introduced stabilizing mutations into the HER2 and HER3 kinase domains. The best yields were observed with HER2 constructs featuring the G778D (GD) mutation which confers independence from the cytosolic chaperone, Hsp90 (Xu et al., 2005). We introduced two mutations in the HER3 kinase domain: E928G (EG) and Q809R (QR). These cancer-associated mutations reside in the interface of the asymmetric kinase dimer and increase kinase/kinase and kinase/JM interactions between the activator (HER3) and the receiver (HER2) kinase, respectively (Figure 2a) (Hanker et al., 2021; Littlefield et al., 2014). In particular, there is good evidence that the oncogenic HER3 E928G mutation drives the formation of active asymmetric kinase dimers by increasing the affinity of the two kinase domains and stabilizing the active conformation of HER2 (Hanker et al., 2021; Littlefield et al., 2014). The introduction of stabilizing kinase mutations yielded sufficient amounts of heterocomplex from 4× 30 ml expression cultures to be visualized by SEC. Figure 2d shows a representative SEC profile of the crosslinked HER2-GD/HER3-EG-QR/NRG1β sample on a Superose 6 10/300 Increase column with the NRG1β-bound heterodimer eluting as the main species around 14 ml (Figure 2d, marked with (*)). NS-EM 2D class averages of this peak showed the expected heart-shaped structure of the HER2/HER3 extracellular domain dimer with improved overall homogeneity and structural detail (Figure 2e). In the presence of these mutations, we observed that addition of the HER2 inhibitor, canertinib, further increases the yield and quality of the HER2/HER3/NRG1β sample with improved resolution of transmembrane domains and intracellular domains. As described above, to obtain canertinib-bound HER2, a final concentration of 10 μM canertinib was added to the HER2 expression culture contemporaneously with the enhancers. Thus, introduction of stabilizing mutations in the receptor intracellular domains, crosslinking, and the use of the HER2 kinase inhibitor improved overall homogeneity of the samples as judged by the NS-EM 2D class average analysis (Figure 2e).
4. Sample preparation for freezing and imaging by cryo-EM.
4.1. Grid selection for cryo-EM analysis of the HER2/HER3/NRG1β complex
In addition to the heart-shaped extracellular domain density observed in the 2D class averages obtained by NS-EM analysis of the purified HER2/HER3/NRG1β complex, we observed a density that we interpreted as a micelle and the kinase domains (Figure 2c+e, ICD). However, we were unable to resolve this density at high resolution by cryo-EM. Other recent studies that have reported on cryo-EM analysis of full-length RTKs similarly failed to resolve the intracellular regions of the receptors. Likewise, in our accompanying paper describing the structural analysis of the unliganded HER3 by cryo-EM (Diwanji…Verba Chapter 20 of this volume), we only resolved the extracellular domain by cryo-EM, despite observing the extra density in the NS-EM 2D average analysis.
There are likely several reasons for the lack of high resolution in the intracellular region, which is currently a universal problem in the cryo-EM analysis of RTKs (Huang et al., 2021; Li et al., 2019; Uchikawa et al., 2019). One possibility is that the transmembrane domain junction between the extracellular and intracellular domains of RTKs is too flexible to stabilize these domains relative to each other, with the computational analysis biasing resolution of the extracellular domains due to their typically larger molecular weight and distinct structural features. This scenario, however, does not explain why the density corresponding to the intracellular domains can be observed by NS-EM, for HER receptors for example (Figure 2e). We hypothesized that the intracellular domains might be more prone to unfolding at the air-water interface when samples are frozen on uncoated holey carbon grids, and that we could improve their resolution by using different grids for freezing. We turned to graphene oxide (GO)-coated grids because the GO coating creates a hydrophilic support layer on which proteins settle during freezing, similar to the conditions present during negative staining (Palovcak et al., 2018; Pantelic, Meyer, Kaiser, Baumeister, & Plitzko, 2010). The GO layer also eliminates one of two air-water interfaces that are present when proteins are frozen in thin ice on uncoated holey carbon grids in the absence of a support layer. We hypothesized that the similarities between NS and GO grids may recover additional density and enable reconstructions of full-length receptor complexes by cryo-EM.
Another major enabling property of GO grids is sample imaging at low protein concentration. This opens up the studies on protein complexes that have traditionally been difficult, or even impossible, to obtain at high enough amounts for structure determination. We found that a working concentration of HER receptor samples for the NS-EM is an equivalent to a 4 mAU elution peak on SEC and that the optimal density of particles on GO grids can be obtained at only ~ 5–10 fold higher than that concentration (20–40 mAU, Figure 3a+b). This corresponds to a concentration of the HER2/HER3/NRG1β complex equivalent to ~ 0.06–0.12 μM (0.017–0.034 mg/ml). Our typical SEC profile from the HER2/HER3/NRG1β complex purification from 120 ml of Expi293 cells yields a dimer peak with ~ 0.5–1 mAU at 280 nm over a 2 ml elution volume, which is equivalent to 20–40 mAU when concentrated to 50 μl (Figure 2d). Typically, such amounts would be insufficient for imaging on uncoated holey carbon grids, which is typically done at concentration ranges at least one order of magnitude higher (Huang et al., 2021; Passmore & Russo, 2016; Uchikawa et al., 2019). Of note is that in order to obtain ideal and reproducible concentrations of HER receptor samples for imaging by cryo-EM on GO grids, we follow concentration estimates based on SEC profiles and subsequent analysis of the NS-EM micrographs, rather than absorbance readings on a spectrophotometer, such as nanodrop, which we found quite unreliable at such low protein concentrations.
Figure 3: Cryo-EM imaging of the HER2/HER3/NRG1β complex on graphene oxide (GO) grids.

a, Summary of ideal grid preparation procedure for low-concentration, detergent-solubilized HER2/HER3 heterocomplex. b, Representative NS-EM and cryo-EM GO micrographs of the heterocomplex. The NS-EM micrograph was recorded at a sample concentration of 4 mAU of the SEC elution peak after concentrating the 0.4 mAU elution peak 10 fold. For GO grid preparation, we found that the optimal sample concentration corresponds to a 5–10 fold concentration of a sample that yielded good NS-EM grids. In the presented example, this corresponds to 30 mAU of the SEC elution peak. c. Medium magnification montage (360 × magnification) of GO grid square showing clear areas of GO and areas without GO.
4.2. Freezing samples on GO grids
GO grids do not need to be glow-discharged prior to sample application. DDM-solubilized receptors are plunge-frozen into liquid ethane using a Vitrobot Mark IV (FEI) with a wait time of 30 seconds. The waiting period is required to reduce the presence of round, micelle-like particles in recorded micrographs. This has been empirically observed but the exact process is unclear. For data collection, we found that if GO is not easily visible on medium magnification montages (210–360x magnification, Figure 3c), we often cannot find particles in these areas and obtain suboptimal datasets with slightly above 50% particle coverage even when grids with close to 100% GO coverage are used (Palovcak et al., 2018). We only make this observation for samples that contain detergent (DDM), suggesting that DDM loosens and removes GO flakes during grid preparation for cryo-EM. We therefore advise imaging the detergent-solubilized membrane proteins in areas with clearly visible GO to increase the yield of micrographs with good particle coverage (Figure 2b). If GO grids can be made in house, we suggest exploring the use of GO grids with thicker GO coating (grid preparation and how to vary GO thickness is described in detail in (Palovcak et al., 2018), which has increased our particle yields for various detergent-containing samples.
4.3. Structure determination of the HER2/HER3/NRG1β extracellular domain dimer and the full-length dimer complex.
Application of GO grids allowed us to collect several datasets for the HER2/HER3/NRG1β complex sample on a 300-keV Titan Krios (Thermo Fisher) with a K3 direct electron detector (Gatan) and a BioQuantum energy filter (Gatan). Image stacks were motion-corrected using MotionCor2 and imported into CryoSPARC2 for most processing steps (Diwanji et al., 2021; Punjani et al., 2017). Micrographs were automatically curated to select for Contrast Transfer Function (CTF)-fit resolution better than 4–5 Å and to remove micrographs with thick ice, and again manually curated to remove all micrographs without particles due to a lack of GO in the respective area (in our experience this reflected 40% of data; the efficiency can be improved by being more selective during data acquisition). For particle picking by both Gaussian blob and 2D class average-derived template picking, we split the whole micrograph stack into a low-defocus and a high-defocus stack to ensure that during particle inspections the very high and very low contrast particles are not excluded (Diwanji et al., 2021). For the non-crosslinked sample bound to the inhibitor canertinib, we obtained a resolution of 3.7 Å for the extracellular domain dimer module (Figure 4b left). Since NS-EM 2D class averages suggested improved sample homogeneity after glutaraldehyde crosslinking, we collected a dataset with 2035 micrographs using the crosslinked sample (Diwanji et al., 2021). Data were processed with CryoSPARC2 to reach a final resolution within the extracellular domain dimer of 2.9 Å, which is a significant improvement over the non-crosslinked sample (Figure 3b). Our final reconstruction of the crosslinked mutant HER2-S310F/HER3/NRG1β complex bound to canertinib reached a final resolution of 3.1 Å (Diwanji et al., 2021). Features of the structures obtained from these datasets and the in-depth details of cryo-EM processing are discussed in detail in (Diwanji et al., 2021).
Figure 4: Processing scheme for obtaining improved maps of full-length complexes.
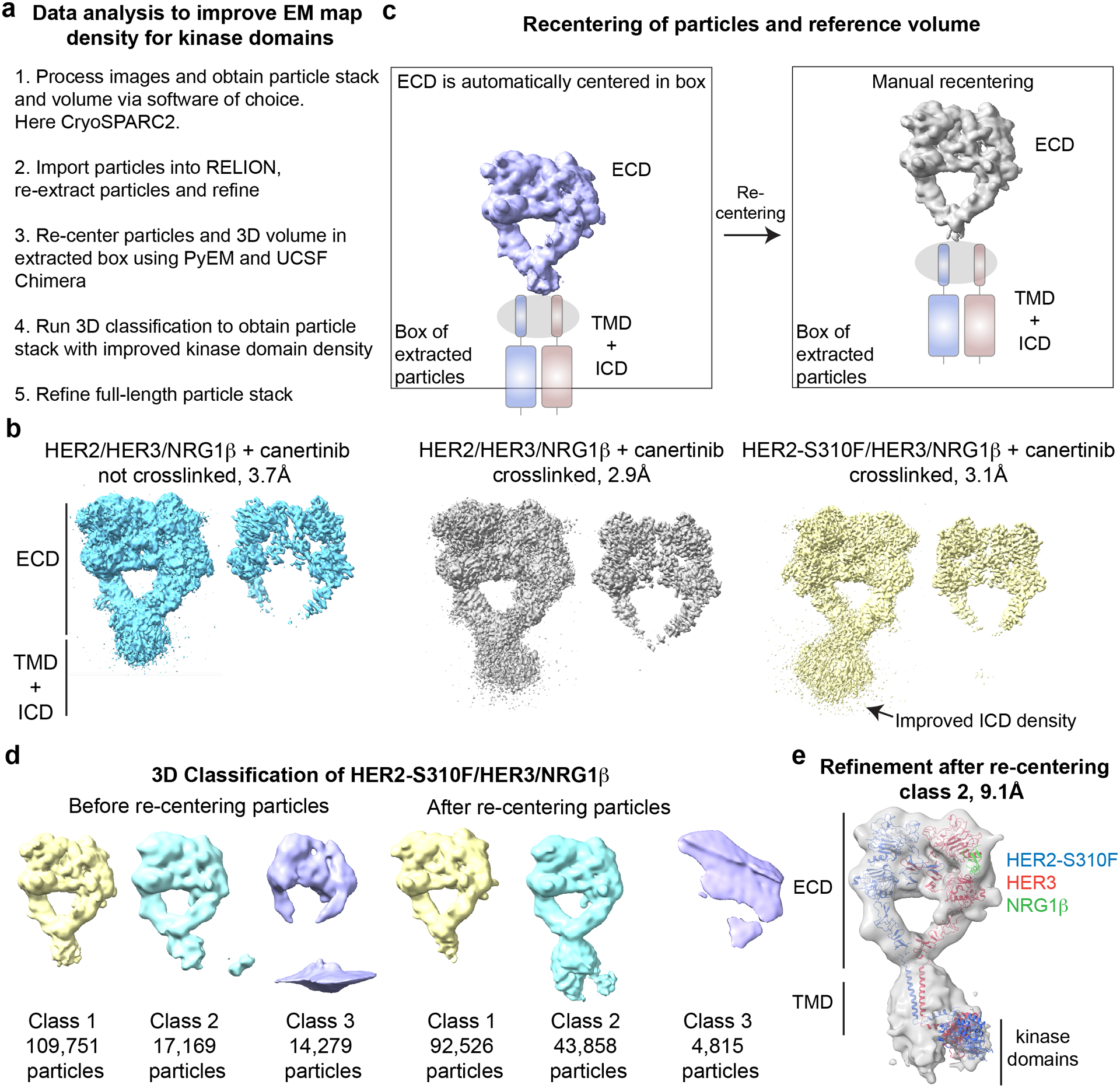
a, Overview of the data and the processing pipeline for the HER2/HER3/NRG1β and HER2-S310F/HER3/NRG1β complexes. b, Low- and high-contour EM maps of the complexes show best transmembrane and intracellular domain density for the crosslinked HER2-S310F/HER3/NRG1β complex. c, Illustration of the re-centering process using UCSF chimera and PyEM. d, 3D classification in RELION3 of the HER2-S310F/HER3/NRG1β complex before and after re-centering coordinates. e, 9.1 Å resolution full-length reconstruction of the HER2-S310F/HER3/NRG1β complex. Models used for fitting into cryo-EM map included: PDB ID: 7MN6 for the HER2-S310F/HER3/NRG1β extracellular domain dimer (Diwanji et al., 2021); PDB ID: 2N2A for the transmembrane domains and juxtamembrane-A (JM-A) segments (Bragin et al., 2016); and the model of the asymmetric HER2/HER3 kinase dimer generated based on PDB ID: 3PP0 (Aertgeerts et al., 2011) and PDB ID: 4RIW (Littlefield et al., 2014).
All high-resolution maps that we obtained of the extracellular domain module of the wild type and mutant HER2/HER3/NRG1β complexes (Diwanji et al., 2021), in addition to the density corresponding to the extracellular domains, featured density towards the expected transmembrane and intracellular domain regions (Figure 4b). These regions by and large were not seen in our reconstructions of unliganded HER3 obtained from a dataset collected on uncoated holey carbon grids (published in this edition). Notably, the density for the transmembrane and intracellular domain regions were more pronounced in the mutant HER2-S310F/HER3/NRG1β than in the wild type HER2/HER3/NRG1β complex, indicating that the disease mutation that stabilized the interactions between the extracellular domains has also stabilizing effects on the intracellular domain interactions (Figure 4b).
We hypothesized that the drop and eventual loss in resolution towards the kinase domains may be due to a loose connection between the extracellular, transmembrane and intracellular domains. In this case, during image alignment CryoSPARC2 focuses on aligning bulky, featureful extracellular domains thereby losing signal for adjacent transmembrane domains and kinase domains. To uncover a subpopulation of particles with matched orientation of the three subdomains, we exported a large particle stack of the most stable HER2-S310F/HER3/NRG1β complex from CryoSPARC2 into RELION3 using PyEM (“Daniel Asarnow, Eugene Palovcak, & Yifan Cheng. (2019). asarnow/pyem: UCSF pyem v0.5 (v0.5). Zenodo. https://doi.org/10.5281/zenodo.3576630,” ; Punjani et al., 2017; Scheres, 2012), re-extracted particles and performed 3D classification into 3 classes (Figure 4d left panel). Remarkably, this 3D classification revealed one class with density for only the extracellular domains at relatively high-resolution and another class with overall lower resolution but stronger density for putative transmembrane and kinase domains (Figure 4d left panel classes 1 and 2 with RELION estimated resolution of unrefined 3D classes of 7.3 Å and 12.0 Å, respectively). This observation might suggest that the extracellular and intracellular domains are loosely connected and the more one focuses on the extracellular domains in particle alignment the more definition is lost on the intracellular domains. It is, however, also possible that partial unfolding of transmembrane and intracellular domains at the air-water interface during freezing causes this reduction in cryo-EM density and that this phenomenon is slightly reduced on GO grids.
We consistently observe that CryoSPARC2 focuses on the bulky and featureful extracellular domains during particle picking and image alignment, centering the extracellular domains in the extraction box while placing putative intracellular domains towards the edge of the box (Figure 4c). This can result in gradual loss of cryo-EM density for the intracellular domains. Using UCSF Chimera (Pettersen et al., 2004) and PyEM (“Daniel Asarnow, Eugene Palovcak, & Yifan Cheng. (2019). asarnow/pyem: UCSF pyem v0.5 (v0.5). Zenodo. https://doi.org/10.5281/zenodo.3576630,”), we shifted coordinates of particles within the extracted box to center the micelle region instead (Figure 4c). Particles with adjusted coordinates were then re-extracted in RELION3 and again subjected to 3D classification. Remarkably, this re-centering process increased the density for the transmembrane and intracellular domain regions of the HER2-S310F/HER3/NRG1β complex. This particle stack and volume were refined and we were able to obtain a 9.1 Å resolution reconstruction of the full-length complex (Figure 4e).
Overall, while high-resolution maps of full-length RTKs remain challenging to obtain, we find that the combination of GO grids, and careful 3D classification with re-centering can significantly recover previously unresolved kinase domains at very low protein concentration.
Acknowledgements
This work was supported by the UCSF Program for Breakthrough Biomedical Research to K.V. and N.J., DFG German Research Foundation GZ: TR 1668/1-1 to R.T, and by the National Institutes of Health: NIH/NIGMS R35-GM139636 to N.J., pilot project grant within the U54 CA209891 to N.J, and NIH/NCI 1F30CA247147 to D.D.
References
- Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, et al. (2011). Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem, 286(21), 18756–18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alroy I, & Yarden Y (1997). The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand-receptor interactions. FEBS Lett, 410(1), 83–86. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, & Engelman JA (2014). ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell, 25(3), 282–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin PE, Mineev KS, Bocharova OV, Volynsky PE, Bocharov EV, & Arseniev AS (2016). HER2 Transmembrane Domain Dimerization Coupled with Self-Association of Membrane-Embedded Cytoplasmic Juxtamembrane Regions. J Mol Biol, 428(1), 52–61. [DOI] [PubMed] [Google Scholar]
- Chen PH, Unger V, & He X (2015). Structure of Full-Length Human PDGFRbeta Bound to Its Activating Ligand PDGF-B as Determined by Negative-Stain Electron Microscopy. J Mol Biol, 427(24), 3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asarnow Daniel, Palovcak Eugene, & Cheng Yifan. (2019). asarnow/pyem: UCSF pyem v0.5 (v0.5) Zenodo. 10.5281/zenodo.3576630. [DOI] [Google Scholar]
- Diwanji D, Thaker T, & Jura N (2019). More than the sum of the parts: Toward full-length receptor tyrosine kinase structures. IUBMB Life, 71(6), 706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwanji D, Trenker R, Thaker TM, Wang F, Agard DA, Verba KA, et al. (2021). Structures of the HER2-HER3-NRG1beta complex reveal a dynamic dimer interface. Nature, 600(7888), 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, & Lovly CM (2018). Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer, 17(1), 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, & Lemmon MA (2000). Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J, 19(17), 4632–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, et al. (2017). EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell, 171(3), 683–695 e618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. (2002). Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell, 110(6), 763–773. [DOI] [PubMed] [Google Scholar]
- Gutmann T, Kim KH, Grzybek M, Walz T, & Coskun U (2018). Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol, 217(5), 1643–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy PM, Platko JV, Cantley LC, Cerione RA, & Carraway KL 3rd. (1994). Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A, 91(17), 8132–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Brown BP, Meiler J, Marin A, Jayanthan HS, Ye D, et al. (2021). Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell, 39(8), 1099–1114 e1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, & Nikolov DB (2003). Eph signaling: a structural view. Trends Neurosci, 26(1), 46–51. [DOI] [PubMed] [Google Scholar]
- Huang Y, Ognjenovic J, Karandur D, Miller K, Merk A, Subramaniam S, et al. (2021). A molecular mechanism for the generation of ligand-dependent differential outputs by the epidermal growth factor receptor. Elife, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N, Shan Y, Cao X, Shaw DE, & Kuriyan J (2009). Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A, 106(51), 21608–21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E, Zorn JA, Huang Y, Barros T, & Kuriyan J (2015). A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem, 84, 739–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, & McCoy JM (1993). A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y), 11(2), 187–193. [DOI] [PubMed] [Google Scholar]
- Lee H, Lolicato M, Arrigoni C, & Minor DL Jr. (2021). Production of K2P2.1 (TREK-1) for structural studies. Methods Enzymol, 653, 151–188. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, & Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell, 141(7), 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Choi E, Yu H, & Bai XC (2019). Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat Commun, 10(1), 4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, & Jura N (2014). Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal, 7(354), ra114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Liu H, Chen X, Focia PJ, & He X (2007). Structural basis for stem cell factor-KIT signaling and activation of class III receptor tyrosine kinases. EMBO J, 26(3), 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cleveland T. E. t., Bouyain S, Byrne PO, Longo PA, & Leahy DJ (2012). A single ligand is sufficient to activate EGFR dimers. Proc Natl Acad Sci U S A, 109(27), 10861–10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi LZ, Grey MJ, Nishida N, Walz T, Lu C, & Springer TA (2008). Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry, 47(39), 10314–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi LZ, Lu C, Li Z, Nishida N, Walz T, & Springer TA (2011). Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol, 18(9), 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moasser MM (2007). The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene, 26(45), 6469–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky Y, Lax I, Tome F, Bleichert F, Unger VM, & Schlessinger J (2014). Structure, domain organization, and different conformational states of stem cell factor-induced intact KIT dimers. Proc Natl Acad Sci U S A, 111(5), 1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palovcak E, Wang F, Zheng SQ, Yu Z, Li S, Betegon M, et al. (2018). A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J Struct Biol, 204(1), 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelic RS, Meyer JC, Kaiser U, Baumeister W, & Plitzko JM (2010). Graphene oxide: a substrate for optimizing preparations of frozen-hydrated samples. J Struct Biol, 170(1), 152–156. [DOI] [PubMed] [Google Scholar]
- Passmore LA, & Russo CJ (2016). Specimen Preparation for High-Resolution Cryo-EM. Methods Enzymol, 579, 51–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem, 25(13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, & Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods, 14(3), 290–296. [DOI] [PubMed] [Google Scholar]
- Rush JS, Peterson JL, & Ceresa BP (2018). Betacellulin (BTC) Biases the EGFR To Dimerize with ErbB3. Molecular Pharmacology, 94(6), 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol, 180(3), 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierke SL, Cheng K, Kim HH, & Koland JG (1997). Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB3 (HER3) receptor protein. Biochem J, 322 (Pt 3), 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, et al. (1994). Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem, 269(20), 14661–14665. [PubMed] [Google Scholar]
- Sung LY, Chen CL, Lin SY, Li KC, Yeh CL, Chen GY, et al. (2014). Efficient gene delivery into cell lines and stem cells using baculovirus. Nat Protoc, 9(8), 1882–1899. [DOI] [PubMed] [Google Scholar]
- Sweeney C, & Carraway KL 3rd. (2000). Ligand discrimination by ErbB receptors: differential signaling through differential phosphorylation site usage. Oncogene, 19(49), 5568–5573. [DOI] [PubMed] [Google Scholar]
- Trenker R, & Jura N (2020). Receptor tyrosine kinase activation: From the ligand perspective. Curr Opin Cell Biol, 63, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikawa E, Choi E, Shang G, Yu H, & Bai XC (2019). Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, & Schlessinger J (1990). Signal transduction by receptors with tyrosine kinase activity. Cell, 61(2), 203–212. [DOI] [PubMed] [Google Scholar]
- van Lengerich B, Agnew C, Puchner EM, Huang B, & Jura N (2017). EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A, 114(14), E2836–E2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, & Ullrich A (1995). Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J, 14(17), 4267–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, & de Vos AM (1997). Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell, 91(5), 695–704. [DOI] [PubMed] [Google Scholar]
- Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, & Neckers L (2005). Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol, 12(2), 120–126. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, & Kuriyan J (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell, 125(6), 1137–1149. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, & Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods, 14(4), 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]