

Abstract
On September 1, 2020, the FDA granted approval for oral azacitidine (Onureg, CC-486) for continued treatment of adult patients with acute myeloid leukemia (AML) who achieved complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and who are not able to complete intensive curative therapy. Approval was based on improvement in overall survival using CC-486 300 mg daily in a two weeks on/two weeks off schedule in comparison to placebo (HR 0.69 [95% CI: 0.55, 0.86], p = 0.0009) in the randomized trial CC-486-AML-001 (QUAZAR) in adults ≥ 55 years old with AML in CR/CRi who did not complete standard intensive induction and post-remission therapy. Of note, the study was not designed to test CC-486 as maintenance after standard post-remission therapy nor as an alternative to standard post-remission therapy. Gastrointestinal toxicities, fatigue, and pneumonia were more common in patients treated with CC-486 compared to placebo. Additional studies are needed to establish safe dosing for patients with hepatic impairment. The pharmacokinetic parameters, recommended dose, and recommended schedule of CC-486 differ substantially from those of other azacitidine formulations; therefore, inappropriate substitutions between formulations pose a considerable risk for harm.
Introduction
For patients with newly diagnosed acute myeloid leukemia (AML) who are able to tolerate intensive therapy, standard frontline treatment typically involves induction with 7 days of cytarabine and 3 days of an anthracycline, or what is commonly referred to as the “7+3” regimen (1). Complete remission (CR) rates range from 40% to 60% in older adults and 60% to 80% in younger adults (2). However, it has long been recognized that after achieving first CR, nearly all patients relapse in the absence of further treatment (3), and additional post-remission therapy is required to prevent AML relapse. Prior randomized studies of maintenance versus consolidation showed that proceeding directly to maintenance therapy after induction resulted in worse outcomes, establishing intensive consolidation as an integral part of frontline AML therapy in patients in all age groups (4, 5). Additionally, use of allogeneic hematopoietic stem cell transplantation (HSCT) is associated with the lowest rate of AML relapses and better overall survival than consolidation with chemotherapy alone or autologous HSCT for patients with intermediate- and poor-risk AML (6, 7). Therefore, intensive post-remission therapy for AML consists of consolidation and/or, for patients with intermediate or high-risk disease, allogeneic HSCT.
In the United States, standard intensive consolidation consists of 3 to 4 cycles of high dose cytarabine (HiDAC) (8) though there has been debate regarding the number of cycles of consolidation required, especially in the older population. Nevertheless, data suggest that patients receiving more postremission therapy appear to have improved outcomes compared to those receiving less therapy. Even in older patients, the recent ASH guidelines note that treatment with antileukemic therapy demonstrates a survival benefit over best supportive care, and for those patients suitable for intensive therapy, the panel suggests intensive over less-intensive induction and recommends additional postremission therapy for those who are not suitable for allogeneic HSCT (9).
Given the risks of intensive chemotherapy for AML, it is not unexpected that some patients who responded to induction chemotherapy may have experienced adverse effects that precluded completing such intensive treatment in the post-remission phase. One potential option for these patients might be continued treatment for AML. Continued treatment is defined as an extended course of treatment after induction with the objective of controlling the AML disease burden while on therapy (10). Herein, we provide a summary of the FDA review (11) of the marketing application for oral azacitidine (CC-486) for continued treatment of adult patients with AML who achieved first CR or CRi following intensive induction chemotherapy and who are not able to complete intensive curative therapy.
Clinical Pharmacology
Azacitidine is a pyrimidine nucleoside analog of cytidine that inhibits DNA and RNA methyltransferases. CC-486 drug product is provided as 200 mg and 300 mg tablets. The mean oral bioavailability of CC-486 (300 mg) is approximately 11% relative to subcutaneous administration (75 mg/m2). The concentration-time profile for the oral formulation differs substantially from that of the subcutaneous formulation; even at the oral azacitidine maximal tolerated dose (MTD; 480 mg), the Cmax is only 32% and the AUC is only 35% of that for a 75 mg/m2 dose of the subcutaneous formulation (12, 13).
There were no formal dose-finding studies to optimize the CC-486 treatment regimen prior to use in the pivotal trial. The justification for the treatment regimen used, 300 mg daily x 14 days of a 28-day cycle, was the tolerability observed in studies of patients with active AML and MDS (12, 14).
Age (46 years to 93 years), sex, body weight (39.3 kg to 129 kg), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin 1 to 1.5 × ULN and any AST), and mild to severe renal impairment (CLcr 15 to 89 mL/min) had no clinically meaningful effect on the pharmacokinetics of CC-486, so dose adjustment is not needed for any of the characteristics. More frequent safety monitoring is recommended in patients with severe renal impairment. The effect of moderate or severe hepatic impairment on exposure was not studied.
Assessment of Efficacy
Pivotal Clinical Trial
Study CC-486-AML-001 (QUAZAR; NCT01757535) was a multicenter, randomized, double-blind, placebo-controlled study in patients ≥ 55 years old with AML in first CR or CRi after intensive induction chemotherapy with or without consolidation. Patients were excluded if they were candidates for HSCT at time of screening or if they had moderate or severe hepatic impairment. Patients were randomized 1:1 to receive 300 mg CC-486 or blinded placebo once daily (QD) for the first 14 days of each 28-day treatment cycle; all patients also received best supportive care. No crossover between treatment groups was allowed. Randomization was stratified by age at time of induction therapy (55 to 64 years versus ≥ 65 years), prior history of MDS or CMML (yes versus no), cytogenetic risk category at time of induction therapy (intermediate-risk versus poor-risk), and whether patients received consolidation therapy following induction (yes versus no).
There were 472 patients randomized to treatment with CC-486 tablets (n=238) or placebo (n=234). The study arms were balanced for key demographic and disease characteristics (Table 1). Of note, only 14% of the study population had adverse cytogenetic risk characteristics at diagnosis, and 9% had secondary AML. Regarding prior post-remission therapy, 20% had received no consolidation, 76% had received 1 or 2 cycles of consolidation, and 4% had received 3 cycles of consolidation. At study baseline, only 78% of the patients were in CR. Just 13% of the study population was treated in the United States.
Table 1:
QUAZAR – Demographics and Baseline Disease Characteristics
| CC-486 (N = 238) |
Placebo (N=234) |
|
|---|---|---|
| Age (years) | ||
| Median (range) | 68 (55, 86) | 68 (55, 82) |
| Age Category, n (%) | ||
| <65 years | 66 (28) | 68 (29) |
| ≥65 years to <75 years | 144 (61) | 142 (61) |
| ≥75 years | 28 (12) | 24 (10) |
| Sex, n (%) | ||
| Male | 118 (50) | 127 (54) |
| Female | 120 (50) | 107 (46) |
| Race, n (%) | ||
| White | 216 (91) | 197 (84) |
| Black or African American | 2 (1) | 6 (3) |
| Asian | 6 (3) | 20 (9) |
| Other | 12 (5) | 11 (5) |
| Not Collected or Reported | 2 (1) | 0 (0) |
| ECOG Performance Status, n (%) | ||
| 0 | 116 (49) | 111 (47) |
| 1 | 101 (42) | 106 (45) |
| 2 or 3 | 21 (9) | 17 (7) |
| NCCN Cytogenetic Risk Status at Diagnosis, n (%) | ||
| Intermediate Risk | 203 (85) | 203 (87) |
| Poor Risk | 35 (15) | 31 (13) |
| Type of AML, n (%) | ||
| Primary (de novo) | 213 (89) | 216 (92) |
| Secondary | 25 (11) | 18 (8) |
| Induction Response | ||
| CR | 187 (79) | 197 (84) |
| CRi | 51 (21) | 37 (16) |
| Consolidation following induction therapy | ||
| None | 52 (22) | 42 (18) |
| 1 cycle | 110 (46) | 102 (44) |
| 2 cycles | 70 (29) | 77 (33) |
| 3 cycles | 6 (3) | 13 (6) |
| Disease status at study baseline | ||
| CR | 185 (78) | 181 (77) |
| CRi | 44 (18) | 38 (16) |
| Not in CR or CRi | 9 (4) | 13 (6) |
| Not Reported | 0 | 2 (1) |
| Reported reason for ineligibility for transplantation1 | ||
| Age | 154 (65) | 152 (65) |
| Comorbidities | 52 (22) | 50 (21) |
| Donor not available | 37 (16) | 35 (15) |
| Subject decision | 19 (8) | 32 (14) |
| Performance status | 14 (6) | 9 (4) |
| Unfavorable cytogenetics | 6 (3) | 10 (4) |
| Other | 28 (12) | 21 (9) |
Source: Adapted from FDA Review (11)
Abbreviations: CR, complete remission; CRi, complete remission with incomplete blood count recovery; ECOG, Eastern Cooperative Oncology Group; NCCN, National Comprehensive Cancer Network
A subject may have had multiple reasons to be ineligible for transplantation
The primary endpoint was overall survival (OS) as measured from randomization to death. Assuming a median OS of 16 months in the placebo arm, a median OS of 22.9 months in the CC-486 arm, and a study duration of 60 months, 330 events among 460 subjects would be needed to detect an HR of 0.70 with 90% power at a one-sided alpha level of 0.025. As of the July 15, 2019, data cut-off date, there were 329 events among the 472 randomized patients. The median follow-up on study was 11.9 (range, 1.1, 62.5) months. There was a significant improvement in OS for the CC-486 arm in comparison to placebo (HR 0.69; 95% CI 0.55, 0.86; p=0.0009) (Figure 1). The median OS was 24.7 months in the CC-486 arm and 14.8 months in the placebo arm.
Figure 1. CC-486-AML-001 – Overall Survival: CC-486 (solid) vs Placebo (dashed).
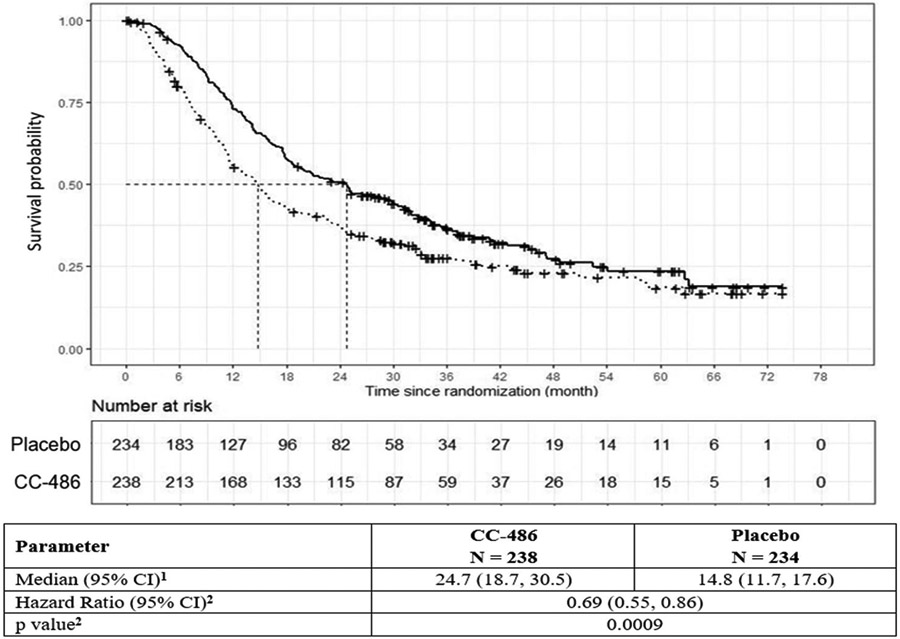
1Confidence intervals (CI) were calculated via the survival function itself without any logarithm transformation.
2Estimated with Cox proportional hazard model and log-rank test stratified by age at time of induction therapy (55 to 64 vs. ≥ 65 years), cytogenetic risk category at time of induction therapy (intermediate risk vs. poor risk) and received consolidation therapy following induction therapy (yes vs. no).
Source: Adapted from FDA Review (11).
The OS results were largely consistent across demographic and baseline disease status subgroups where the sizes of the subgroups were adequate for analysis (Table 2). There was a trend for a lack of treatment effect in the patients who had received 3 cycles of consolidation, but the number of patients in this subgroup was small (n = 19), so the confidence interval was quite wide (HR 1.37; 95% CI 0.37, 5.02). Additionally, the subgroup analysis by geographic region suggested substantial geographic variation in the treatment effect with lack of a treatment effect for patients in North America (Table 2). A by-country analysis, a Bayesian shrinkage analysis, and multivariate analyses failed to demonstrate significant differences in the treatment effect for the United States, specifically in comparison to Europe (11).
Table 2:
QUAZAR – Subgroup Analysis of OS
| Number of Patients | HR1 (95% CI) | ||
|---|---|---|---|
| CC-486 | Placebo | ||
| Overall | 238 | 234 | 0. 72 (0.58, 0.89) |
| Age Category, n (%) | |||
| < 65 years | 66 | 68 | 0.72 (0.46, 1.13) |
| ≥ 65 years | 172 | 166 | 0.71 (0.56, 0.92) |
| Sex, n (%) | |||
| Male | 118 | 127 | 0.74 (0.55, 1.00) |
| Female | 120 | 107 | 0.68 (0.50, 0.93) |
| Race, n (%) | |||
| White | 216 | 197 | 0.66 (0.53, 0.83) |
| Black or Other | 14 | 17 | 1.35 (0.53, 3.40) |
| Asian | 6 | 20 | 1.54 (0.43, 5.47) |
| Disease status at study baseline | |||
| CR | 185 | 181 | 0.72 (0.56, 0.92) |
| CRi | 44 | 38 | 0.87 (0.51, 1.50) |
| Consolidation following induction therapy | |||
| None | 52 | 42 | 0.55 (0.34, 0.89) |
| 1 cycle | 110 | 102 | 0.75 (0.55, 1.02) |
| 2 cycles | 70 | 77 | 0.69 (0.45, 1.04) |
| 3 cycles | 6 | 13 | 1.37 (0.37, 5.02) |
| Geographic Region | |||
| Europe | 167 | 147 | 0.60 (0.46, 0.77) |
| North America | 37 | 42 | 1.09 (0.65, 1.82) |
| Other | 34 | 45 | 0.90 (0.50, 1.64) |
Source: Adapted from FDA Review (11)
Abbreviations: CR, complete remission; CRi, complete remission with incomplete blood count recovery
Estimated with nonstratified Cox proportional hazard model
Additional Efficacy Considerations
The key secondary endpoint of QUAZAR was relapse-free survival (RFS), defined as the time from randomization to relapse or death. FDA noted that RFS would be an acceptable efficacy endpoint for AML when patients are in CR (10), but since only 78% of patients in QUAZAR were in CR at study baseline, RFS is not applicable to the randomized population. This endpoint was not considered further.
For patients on QUAZAR who developed blasts of 5-15% in the peripheral blood or marrow while on study, the dosage of blinded study drug was increased to daily for 21 days of the 28-day cycle. Forty-three patients in the CC-486 arm and thirty-five in the placebo arm had their dosage increased to 21 days after meeting these criteria. Of these patients, 4 (9%) in the CC-486 arm and 3 (9%) in the placebo arm were considered to have achieved subsequent CR.
Because QUAZAR included patients without overt AML after intensive therapy, alternative data were needed to confirm that CC-486 had an antileukemic effect. There were 36 patients with active AML treated with CC-486 at various doses and schedules on early phase single-arm trials (AZA-MDS-004, AZA-PH-US-2007-CL-005, and AZA-PH-US-2008-CL-008) (9). A CR was reported for one (3%) of the 36 patients treated.
Assessment of Safety
Safety in the Pivotal Clinical Trial
In QUAZAR, the median duration of exposure was 11.6 months (range 0.5 to 74.3) on the CC-486 arm and 5.8 months (range 0.7 to 71.4) on the placebo arm. Due to more rapid dropout in the placebo arm, the difference between study arms in the proportion of patients still on study was > 15% starting with cycle 6. Common (≥ 10%) adverse reactions occurring during Cycles 1-5 and through the full study period are shown in Table 3. For the full study period, postbaseline shifts to maximal grade 3 or 4 neutropenia occurred in 49% vs 23% and to maximal grade 3 or 4 thrombocytopenia for 21% vs 10% on the CC-486 and placebo arms, respectively.
Table 3.
CC-486-AML-001 – Common Adverse Reactions
| CC-486 N = 236 |
Placebo N = 233 |
|||
|---|---|---|---|---|
| All Grade | Grade ≥ 3 | All Grade | Grade ≥ 3 | |
| Cycles 1-5 | ||||
| Nausea | 58% | 2% | 19% | < 1% |
| Vomiting | 54% | 3% | 7% | 0% |
| Diarrhea | 38% | 3% | 14% | 1% |
| Constipation | 31% | < 1% | 21% | 0% |
| Fatigue a | 30% | 2% | 19% | 1% |
| Abdominal pain a | 14% | 1% | 10% | < 1% |
| Any Cycle | ||||
| Nausea | 65% | 3% | 24% | <1% |
| Vomiting | 60% | 3% | 10% | 0% |
| Diarrhea | 50% | 5% | 21% | 1% |
| Fatigue a | 44% | 4% | 25% | 1% |
| Constipation | 39% | 1% | 24% | 0% |
| Pneumonia a | 27% | 9% | 17% | 5% |
| Abdominal pain a | 22% | 2% | 13% | <1% |
| Arthralgia | 14% | 1% | 10% | <1% |
| Decreased appetite | 13% | 1% | 6% | 1% |
| Back pain | 12% | 1% | 10% | 1% |
| Febrile neutropenia | 12% | 11% | 8% | 8% |
| Dizziness | 11% | 0% | 9% | 0% |
| Pain in extremity | 11% | <1% | 5% | 0% |
Source: Adapted from FDA Review (11)
Grouped term based on MedDRA 22.0 terms: Abdominal pain – abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper, gastrointestinal pain; Fatigue – asthenia, fatigue; Pneumonia – Broad SMQ infective pneumonia
On the CC-486 arm, there was one (0.4%) on-treatment death (sepsis) that was considered at least possibly related to study drug. Overall, 8% of patients on the CC-486 arm discontinued treatment due to an adverse event, predominantly due to GI toxicity (nausea, vomiting, abdominal pain, or diarrhea). Dose reductions were required for 14%, mostly for neutropenia (6%), diarrhea (3.4%), thrombocytopenia (1.7%), and nausea (1.7%). Treatment interruptions were reported for 35% of patients on CC-486, largely due to neutropenia (20%), thrombocytopenia (8%), and nausea (6%).
Additional Safety Considerations
In AZA-MDS-003 (NCT01566695) (15), 216 patients with transfusion-dependent lower-risk myelodysplastic syndromes were randomized to CC-486 or placebo in a blinded fashion. One-hundred and seven patients received a median of 5 cycles of CC-486 300 mg daily x 21 days of a 28-day cycle, a duration longer than the approved regimen of 14 days of a 28-day cycle. Enrollment was discontinued early due to safety issues. Fatal adverse reactions occurred in 22% of patients in the CC-486 arm compared with 7% in the placebo arm. There was an imbalance in fatal infections occurring during Cycles 1-3 (12% vs 2% for CC-486 and placebo, respectively).
Regulatory Insights
Patients unable to complete intensive curative therapy are at high risk for early relapse. CC-486 is the first drug approved by FDA as continued therapy for AML in patients who are unable to complete curative post-remission therapy. In QUAZAR, use of CC-486 was associated with a significant improvement in survival over placebo (HR 0.69 (95% CI: 0.55, 0.86) for the intended population. Although enrollment in QUAZAR was limited to patients 55 years or older, based on the biology of AML, this efficacy outcome can be extrapolated to the full adult age range for the indication.
Although QUAZAR had been proposed as testing a maintenance strategy, the design was not consistent with this objective. The FDA defines maintenance therapy for AML as an extended but time-limited course of treatment given after achievement of CR with the objective of reducing the risk of relapse beyond the period of treatment (10). A trial designed to demonstrate the efficacy of maintenance therapy would need to include a specified induction and consolidation treatment followed by randomization to a predefined duration of treatment with placebo versus investigational maintenance therapy. QUAZAR enrolled patients with varying disease status at randomization (CR, CRi, or with elevated blasts), induction and consolidation were not prespecified, and treatment consisted of indefinite therapy until relapse or death. Hence, this study was not a test of post-consolidation maintenance.
QUAZAR also did not test CC-486 as an alternative to the standard 3-4 cycles of consolidation. Although there was considerable variability in the consolidation therapy before randomization, there were too few patients in QUAZAR who received 3 or 4 cycles to draw any conclusions regarding the efficacy of CC-486 as an alternative to consolidation. Nonetheless, the study demonstrated a survival benefit as continued therapy compared with placebo and addressed a substantial unmet need for additional treatments for patients who are unable to complete intensive curative therapy. It should be noted, however, that the current data do not support the use of CC-486 for remission induction in patients with overt AML.
The pharmacokinetic parameters of CC-486 following oral administration differ substantially from those of parenteral azacitidine formulations, as do the recommended dose and schedule for safe use. Using CC-486 at the 75 mg/m2 parenteral dose for oral administration would result in substantial underdosing and reduced efficacy for treatment of MDS, and administration of the parenteral formulation at the 300 mg oral dose could result in a potentially fatal overdose. Thus, substitutions between oral and parenteral azacitidine formulations pose a substantial risk for harm, and this risk is described in a warning in the label.
Despite the lower exposure with CC-486, toxicity is still substantial. In QUAZAR, 8% had permanent drug withdrawal, 14% had dose reductions, and 35% had treatment interruptions due to an adverse reaction, and there was one (0.4%) fatal event. The main adverse reactions of CC-486 stem from gastrointestinal intolerance and myelosuppression, consistent with its nature as a cytotoxic drug. Labeling includes instructions for frequent monitoring and dose modifications to mitigate these toxicities.
Although parenteral azacitidine is approved for the treatment of patients with MDS, CC-486 is not. In the randomized trial AZA-MDS-003, testing CC-486 at 300 mg daily x 21 days of a 28-day cycle compared to placebo for lower-risk MDS, the extended dosing regimen of CC-486 was associated with an unacceptable level of serious and fatal infections. Although it is unclear whether this toxicity was due to extended dosing regimen or to other factors inherent in the MDS population, this public health risk merited a warning in the label. At the present time, CC-486 is not approved for the treatment of MDS.
Conclusions
For patients with AML who achieve CR or CRi with intensive induction chemotherapy but who are unable to complete intensive curative therapy, the standard of care has been unclear. In QUAZAR, treatment with CC-486 resulted in an improvement in OS over placebo (HR 0.69 [95% CI 0.55, 0.86]). In general, the safety profile was consistent with oral cytotoxic therapies. Overall, with careful monitoring and appropriate dose modifications for toxicity, the clinical benefit of treatment with CC-486 outweighs the expected risks in the intended population.
Acknowledgements
The authors thank Dr. Rachel McMullen for expert project management.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors report no financial interests or relationships with the commercial sponsors of any products discussed in this report.
This is a U.S. Government work. There are no restrictions on its use.
References
- 1.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Acute Myeloid Leukemia, Version 3.2021 – March 2, 2021. Accessed October 13, 2021. Available from: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf. [Google Scholar]
- 2.Dohner H, Estey E, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2017;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 3.Cassileth PA, Harrington DP, Hines JD, Oken MM, Mazza JJ, McGlave P, et al. Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia. J Clin Oncol 1998;6:583–587. [DOI] [PubMed] [Google Scholar]
- 4.Cassileth PA, Lynch E, Hines JD, Oken MM, Mazza JJ, Bennett JM, et al. Varying intensity of postremission therapy in acute myeloid leukemia. Blood 1992;79(8):1924–1930. [PubMed] [Google Scholar]
- 5.Schlenk RF, Frohling S, Hartmann F, Fischer JTh, Glasmacher A, delValle F, et al. Intensive consolidation versus oral maintenance therapy in patients 61 years or older with acute myeloid leukemia in first remission: results of second randomization of the AML HD98-B treatment trial. Leukemia 2006;20:748–750. [DOI] [PubMed] [Google Scholar]
- 6.Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301(22):2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanada M, Matsuo K, Emi N, Naoe T. Efficacy of allogeneic hematopoietic stem cell transplantation depends on cytogenetic risk for acute myeloid leukemia in first disease remission: a metaanalysis. Cancer 2005;103(8):1652–1658. [DOI] [PubMed] [Google Scholar]
- 8.Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P, et al. , Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 1994;331(14):896–903. [DOI] [PubMed] [Google Scholar]
- 9.Sekeres MA, Guyatt G, Abel G, Alibhai S, Altman JK, Buckstein R, et al. , American Society of Hematology 2020 guidelines for treating newly-diagnosed acute myeloid leukemia in older adults. Blood Advances, 2020;4(15):3528–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.U.S. Food and Drug Administration, 2020, Guidance for Industry: Acute Myeloid Leukemia: Developing Drugs and Biological Products for Treatment. Accessed October 4, 2021. Available at: https://www.fda.gov/media/140821/download. [Google Scholar]
- 11.U.S. Food and Drug Administration, Oral Azacitidine Approval Multi-Discipline Review. Drugs@FDA [database on the internet]. Silver Spring (MD): U.S. Food and Drug Administration. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214120Orig1s000MultidisciplineR.pdf. [Google Scholar]
- 12.Garcia-Manero G, Gore SD, Cogle C, Ward R, Shi T, MacBeth KJ, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol 2011;29(18):2521–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laille E, Savona MR, Scott BL, Boyd TE, Dong Q, Skikne B. Pharmacokinetics of different formulations of oral azacitidine (CC-486) and the effect of food and modified gastric pH on pharmacokinetics in subjects with hematologic malignancies. J Clin Pharmacol 2014;54(6):630–639. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Manero G, Gore SD, Kambhampati S, Scott B, Tefferi A, Cogle CR, et al. Efficacy and safety of extended dosing schedules of CC-486 (oral azacitidine) in patients with lower-risk myelodysplastic syndromes. Leukemia 2016;30:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Manero G, Santini V, Almeida A, Platzbecker U, Jonasova A, Silverman LR, et al. Phase III, randomized, placebo-controlled trial of CC-486 (oral azacitidine) in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2021;39(13):1426–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]