

Abstract
Necroptotic cell death is mediated by a super-molecular complex called necrosome which consists of receptor-interacting protein kinase 1 and 3 (RIPK1, RIPK3) and mixed-lineage kinase domain-like protein (MLKL). The role of these kinases has been extensively investigated in the regulation of necroptosis. However, whether the protein phosphatase is involved in necroptosis is still largely unknown. Here, we identified protein phosphatase 6 catalytic subunit (PPP6C) promotes TNF-induced necroptosis by genome-wide CRISPR/Cas9 library screening. We found that PPP6C deficiency protects cells from TNF-induced necroptosis in a phosphatase-activity-dependent manner. Mechanistically, PPP6C acts as a TGF-β activated kinase 1 (TAK1) phosphatase to inactivate its kinase activity. Deletion of PPP6C leads to hyperactivation of TAK1 and reduced RIPK1 kinase activity upon TNF stimulation. We further showed that heterozygous deletion of Ppp6c in mouse gastrointestinal tract alleviates necroptosis-related tissue injury and inflammation. Thus, our study identifies PPP6C as an important regulator of necroptosis and highlights a central role of phosphatase in the regulation of necroptosis-related diseases.
Subject terms: Kinases, Cell signalling
Introduction
Necroptosis, a lytic form of programmed cell death, is characterized by organelle swelling, plasma membrane rupture, and release of intracellular components that could elicit an innate immune response [1, 2]. In general, necroptosis is mediated by the receptor-interacting protein kinase 1 (RIPK1)-RIPK3-mixed-lineage kinase domain-like protein (MLKL) signaling cascade, and triggered by various stimuli, such as tumor necrosis factor (TNF), Fas, as well as TRAIL ligands [3, 4]. RIPK3 and MLKL are the crucial effectors of necroptosis, since RIPK1 is sometimes dispensable, for example, in Toll-like Receptor 3 or 4 (TLR3 or TLR4)-induced necroptosis [5, 6]. TNF is a widely-used cytokine that induces canonical necroptosis. Under TNF stimulation, tumor necrosis factor receptor 1 (TNFR1) triggers the recruitment of TNFR-associated factor 2/5 (TRAF2/5), cellular inhibitors of apoptosis (cIAPs), TNFR1-associated death domain protein (TRADD), and RIPK1 to form complex I [7, 8]. Within the complex I, RIPK1 is rapidly conjugated with ubiquitin chains which create a scaffold to recruit and activate TGF-β activated kinase 1 (TAK1)/TAK1-binding protein 2/3 (TAB2/3) complex and the inhibitor of nuclear factor-κB (NF-κB) kinase (IKK) complex (NEMO-IKKα/β), which then activates the mitogen-activated protein kinase (MAPK) and canonical NF-κB signaling pathways to prevent cell death through promoting the transcription of pro-survival genes [9]. Accordingly, when cIAPs, NEMO or TAK1 is inhibited or depleted, RIPK1 dissociates from the membrane and moves into cytoplasm to form complex II with RIPK3, Fas-associated via death domain (FADD), and caspase-8, inducing RIPK1 kinase activity-dependent apoptosis [10]. If caspase-8 is genetically depleted or inhibited by the use of Z-VAD, RIPK1 autophosphorylates and recruits RIPK3 via its homotypic RIP homology interaction motif (RHIM), causing oligomerization and autophosphorylation of RIPK3 [11–14]. Subsequently, RIPK3 phosphorylates MLKL within its C-terminal pseudokinase domain, which provokes the killing activity of MLKL by forming active oligomers that translocate to and rupture the plasma membrane [12, 15–19].
TAK1 is considered as a central regulator of cell death and has been demonstrated to play a pro-survival role via the NF-κB and MAPK activation [20, 21]. Recent studies unveiled that TAK1 also regulates necroptosis independent of its role in NF-κB activation [10, 22, 23]. Previous studies showed that TAK1 directly or indirectly via P38 and MAPK activated protein kinase 2 (MAPKAPK2, also known as MK2) axis phosphorylates RIPK1 at S320/321 to inhibit RIPK1 activation [24–26]. IKKα/β were also reported to inhibit necroptosis by regulating RIPK1 phosphorylation at S25 [27]. In addition, cells are more sensitive to RIPK1-dependent apoptosis upon TNF stimulation in the absence of functional TAK1 [23]. Thus, TAK1 plays an anti-necroptosis role from diverse perspectives. However, one research found that deletion of Tab2 in mouse embryonic fibroblast (MEF) cells results in sustained activation of TAK1, but unexpectedly aggravates necroptosis following TNF stimulation [28]. These studies suggest that TAK1 plays multiple roles in the determination of cell fate upon TNF treatment.
Protein phosphatase 6 (PP6), a member of the phosphoprotein phosphatase (PPP) family of Ser/Thr protein phosphatases, is a trimetric holoenzyme, consisting of catalytic, structural, and regulatory subunits with the catalytic subunit designated PPP6C [29]. PPP6C is highly conserved throughout eukaryotes including human, and exhibits multiple roles in regulating several cellular processes. PPP6C was previously identified as a negative regulator of NF-κB signaling by stabilizing NF-κB inhibitor ε (IκBε) and dephosphorylating TAK1 [30, 31]. In this study, we identified PPP6C as a positive regulator of necroptosis through loss-of-function screening by utilizing a pooled genome-scale CRISPR/Cas9 library in L929 cells. We found that depletion of Ppp6c enhances TAK1-IKKα/β axis activity which promotes phosphorylation of RIPK1 at S25, and results in the inhibition of RIPK1 kinase activity and preventing TNF-mediated RIPK1-dependent necroptosis.
Materials and methods
CRISPR/Cas9 knockout library screening
We used the mouse GeCKOv2 CRISPR knockout pooled library to identify genes that regulate TNF-induced necroptisis in L929 cells. The library contains 130,209 unique sgRNA sequences targeting 20,611 mouse genes with six guides per gene (Addgene #1000000053). L929 cells were transduced with the lentivirus library at <0.3 multiplicity of infection to barcode individual cells. Puromycin was added at a final concentration of 6 μg/ml in the cell culture medium 24 h post infection for 5 days to generate a mutant cell pool. The cells were then treated with TZ (10 ng/ml TNF + 20 µM Z-VAD-FMK) for 8 h to deplete non-resistant cells. After 6-round TZ selection, at least 3 × 107 cells were used for genomic DNA extraction. sgRNA sequences were amplified with forward primer GTAGAAAGTA ATAATTTCTTGGGTAGTTTGCAG and reverse primer CACTCCTTTCAAGACCTAGCTAGCGAATTCA. Then, the amplified DNA fragments were sequenced on an Illumina Hiseq.
Cell culture and cell lines
HEK293T and MEF cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin. L929 and HT29 cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% FBS, 100 U/ml penicillin/streptomycin. All cells were cultured under 5% CO2 at 37 °C. To generate Ppp6c knockout cells, L929 cells were infected with lentiCRISPRv2-sgPpp6c lentivirus and single clone was picked. To generate TAK1 knockout cells, HEK293T were infected with lentiCRISPRv2-sgTAK1 lentivirus and single clone was picked. To generate HA-PPP6C or HA-PPP6C-PD expressing cells, L929 or Ppp6c-KO L929 cells were infected with pCDH-HA-PPP6C or pCDH-HA-PPP6C-PD lentivirus. PPP6C-knockdown HT29 cells and Tab2-knockdown L929 cells were generated by shRNA. Cells were infected with pLKO.1-shPPP6C or pLKO.1-shTab2 lentivirus and selected for puromycin-resistant cells. Guide RNA and shRNA sequences are listed in Table S1. Ppp6cflox/flox MEFs were transduced with SV40 large T antigen-expressing lentivirus for immortalization and then with Cre-expressing lentivirus to generate Ppp6c KO MEFs. For transfection, plasmid DNA was transfected into HEK293T cells by Polyethylenimine (PEI). For lentivirus infection, viruses generated in HEK293T with indicated lentivirus plasmids and packaging plasmids psPAX2 (Addgene, #12260), pMD2.G (Addgene, #12259) were added to cell culture medium with 10 μg/ml Polybrene (Beyotime, C0351).
Luciferase reporter assay
Luciferase reporter assay was performed using a dual-luciferase reporter assay system. Plasmids encoding NF-κB-driven firefly luciferase reporter and CMV promoter-driven renilla luciferase were co-transfected with FLAG-PPP6C into HEK293T cells for 24 h. Cells were treated with TNF (10 ng/ml) for 6 h, and then cell lysis was subjected to luciferase activity measurement as described in dual-luciferase reporter assay kit (Promega, USA).
Immunoblotting and immunoprecipitation
Cells were collected in ice-cold lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% TritonX-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4 and protease inhibitor cocktail), and then centrifuged at 150,00 × g for 20 min at 4 °C. The supernatants were collected for immunoblotting or immunoprecipitation. For immunoprecipitation, 10 μl anti-FLAG or anti-HA magnetic beads were added to the supernatants and incubated at 4 °C for 6 h under gentle agitation. Then the beads were washed by washing buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% TritonX-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4 and protease inhibitor cocktail) for three times. Finally, the beads were mixed with SDS loading buffer and the eluted protein was used for immunoblotting. Antibody information is listed in Table S2.
RNA extraction and quantitative RT-PCR
Total RNA was isolated from indicated cells using the TRIzol reagent (Invitrogen, USA), according to the manufacture’s protocol. One microgram total RNA was used for cDNA synthesis using The PrimeScript™ RT reagent Kit with genomic DNA Eraser (Takara). Quantitative PCR was then performed using Hieff® qPCR SYBR Green Master Mix (Yeasen). Sequences of the primers are listed in Table S1.
Cell death and survival assay
For L929 cells, necroptosis was induced by the treatment with TZ (10 ng/ml mouse TNF + 20 μM Z-VAD-FMK). For HT29 cells, necroptosis was induced by TSZ treatment, which is a combination of 20 ng/ml TNF, 0.2 μM SM-164, and 20 μM Z-VAD-FMK. To measure the cell death after treatment, PI (propidium iodide) was directly added in the medium at a concentration of 5 μg/ml and incubated for 15 min. The visual cell death was photographed by an inverted fluorescence microscope and the percentage of PI incorporation was quantified by flow cytometer (Beckman CytoFlex S). Cell viability was measured by Cell Counting Kit 8 (CCK8).
Animal work
Ppp6c conditional knockout mice were a gift from Dr. Xingzhi Xu. Ppp6cflox/flox mice were bred to ShhCre/+ mice to generate ShhCre/+;Ppp6cf/+ mice. All mice were bred on a C57BL/6J genetic background. ShhCre/+;Ppp6cf/+ mice and their littermate controls Ppp6cflox/+ mice were bred by crossing ShhCre/+ with Ppp6cf/f. For systemic inflammatory response syndrome (SIRS) mouse model, male mice aged at 6–8 weeks were injected intravenously through tail vein with mouse TNF diluted in endotoxin-free PBS (0.25 μg/g per mouse). Control mice were injected with PBS. For DSS-induced colitis, male mice aged at 6–8 weeks were supplied with dextran sulfate sodium (DSS) in drinking water 3% (w/v) for 7 days followed by regular water for 1 day. DSS water was replaced on day 2 and day 4. Body weight was measured daily for up to 8 days, and the mice were then sacrificed to enable colon length measurement, histology analysis, and colonic proteins extraction for immunoblotting analysis. All mice were housed in specific pathogen-free condition with 12-h light/dark cycle and freely access to food and water at the Zhejiang University Laboratory Animal Center. All mouse experiments were approved by the Institutional Animal Care and Use Committee of Zhejiang University.
Histology
Mouse colons were fixed in 4% paraformaldehyde for 6 h. The fixed tissues were dehydrated in gradient ethanol, cleared in xylene, and embedded in paraffin. Five-micrometer sections were cut and mounted on adhesion microscope slides. Sections were then used for hematoxylin and eosin (H&E) or terminal dexynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNLE) staining using TUNEL Apoptosis Detection Kit (FITC) (Yeasen, Shanghai, China) according to the manufacturer’s instruction.
Immunofluorescence
L929 cells were plated on coverslips and treated with TNF (10 ng/ml) for the indicated time, and then fixed with pre-cooled methanol on ice for 10 min, followed by blocking with 3% BSA at room temperature for 30 min. Cells were incubated with primary antibodies diluted in 3% BSA at 1:500 for 2 h at room temperature. After washed for three times with PBST, cells were incubated with secondary antibodies in 3% BSA for 2 h. Nuclei were stained with 0.5 μg/ml of DAPI at room temperature for 10 min. Images were acquired on a confocal microscope.
Production of recombinant HA-mTNF proteins
N-terminal HA-tagged mouse TNF (mTNF) construct was obtained by cloning truncated mTNF cDNA (encoding Asp89-Leu235) into the pcDNA3 vector with an Igκ leader sequence and a HA tag at the N-terminus of mTNF. Conditional medium with recombinant HA-mTNF proteins was obtained by transfecting this construct into HEK293T cells for 60 h. Concentration of the recombinant HA-mTNF protein in the conditional medium was determined using commercial mTNF protein.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 8.0.1; GraphPad Software). Statistical analysis was performed using an unpaired Student’s t test (two groups) or one-way ANOVA analysis (multiple groups). A difference was considered significant if P < 0.05 and statistical significance was defined as P < 0.05 (*), P < 0.01(**), and P < 0.001(***).
Results
Genome-wide CRISPR/Cas9 library screening identified PPP6C as a positive regulator of necroptosis
To identify uncharacterized regulators of necroptosis, we conducted a positive-selection screening to enrich the cells acquired TNF + Z-VAD (TZ) resistance in L929 cells, which exhibit high sensitivity to TNF-induced necroptosis and are widely used in TNF-induced necroptosis study [32], by utilizing a pooled genome-scale CRISPR/Cas9 knockout (GeCKO) library. After 6 rounds of TZ selection, the sgRNA abundance in the enriched cells was determined by next-generation sequencing (NGS). The enrichment of sgRNAs was calculated and plotted (Fig. 1A). We identified a subset of sgRNAs targeting 79 genes significantly enriched (P < 0.05) in the TZ-treated cells when compared to vehicle control (Table S3). Among these sgRNAs, we observed the highest level of sgRNA enrichment targeting genes such as Tnfrsf1a, Ripk1, Ripk3, Mlkl, Tradd, Cyld, Spata2 (Fig. 1A), which have been previously reported as the regulators of necroptosis [7, 33–37]. The enrichment of these genes indicates that our screening was effective.
Fig. 1. Deletion of Ppp6c prevents TNF-induced necroptosis.

A Scatter diagram revealed that sgRNAs targeting Ppp6c were selected during TNF + Z-VAD-FMK (TZ) treatment in L929 cells. B, C Control and Ppp6c-KO L929 cells were treated with TZ for 6 h and analyzed by propidium iodide (PI) staining under a microscope (B) or quantified by flow cytometer (C). Scale bar, 50 μm. D Control and Ppp6c-KO L929 cells were treated with TZ for the indicated time and the cell viability was measured by CCK8. E Control and Ppp6c-KO L929 cells were treated with TZ for the indicated time and the activation of RIPK1, RIPK3 and MLKL proteins was monitored by immunoblot. F Control and Ppp6c-KO L929 cells were treated with TZ for the indicated time and cell lysates were resolved on non-reducing PAGE for immunoblot. G Control and PPP6C-knockdown HT29 cells were treated with TSZ for the indicated time and cell lysates were probed with indicated antibodies. H–J Immortalized Ppp6cf/f MEF cells were infected with control lentivirus or Cre-Lentivirus for 72 h and followed by the treatment with TSZ for 6 h. Cells were stained with PI and analyzed under a microscope (H) or lysed for immunoblot with indicated antibodies (I), or quantified by flow cytometer after PI staining (J). Scale bar, 50 μm. Data shown are representative of three independent experiments and presented as means ± SDs of triplicates (C, D, J). **p < 0.01, ***p < 0.001, with an unpaired Student’s t-test (C, D, J).
In this screening, Ppp6c, the catalytic subunit of protein phosphatase 6 [38], was identified as a positively selected gene upon TZ treatment since 5 sgRNAs targeting Ppp6c were enriched in TZ-treated cells (Fig. S1A), implying that loss of Ppp6c protected L929 cells from TZ-induced necroptosis. To investigate the function of Ppp6c in necroptosis, we generated Ppp6c knockout (KO) L929 cell subclones using CRISPR/Cas9. Deletion of Ppp6c in L929 cells did no significantly affect the cell proliferation (Fig. S1B). Then, we treated Ppp6c-KO L929 cells and control cells with TZ for 6 hours and then stained with PI which is usually used to evaluate plasma membrane integrity. Consistent with the result of CRISPR/Cas9 screening, we found that Ppp6c-KO L929 cells were resistant to TZ-induced necroptosis (Fig. 1B, C) and displayed a higher cell viability under TZ treatment (Fig. 1D). It is well established that necroptosis signaling pathway is tightly regulated by a kinase cascade RIPK1-RIPK3 to phosphorylate MLKL [12–14, 19, 39]. To determine whether PPP6C signals through this cascade, we treated Ppp6c-KO and control L929 cells with TZ for 2, 4, and 6 h, and then lysed cells for western blot analysis. We found that the phosphorylation level of RIPK1 (S166) [14], RIPK3 (S232) [13], and MLKL (S345) [19] in Ppp6c-KO L929 cells was remarkably reduced compared with the control cells (Fig. 1E). Consistently, MLKL oligomerization was greatly inhibited upon the removal of Ppp6c in L929 cells (Fig. 1F). To evaluate whether these findings were conserved in human cells, we generated PPP6C-knockdown human HT29 cells using two independent shRNAs. Consistently, shRNA-mediated knockdown of PPP6C conferred HT29 cells to the resistance to TSZ-induced necroptosis by analyzing plasma membrane integrity (Fig. S1C, D). In addition, PPP6C-knockdown HT29 cells exhibited a higher cell viability (Fig. S1E), reduced phosphorylation level of RIPK1 (S166), and MLKL (S358, corresponding to mouse S345) [12] (Fig. 1G), and less MLKL oligomerization upon TSZ treatment (Fig. S1F). Furthermore, we isolated MEF cells from Ppp6cflox/flox mouse embryos and infected the immortalized Ppp6cflox/flox MEF cells with control or Cre-lentivirus to generate Ppp6c-KO MEF cells. Consistent with previous report [40], Ppp6c deletion reduced cell proliferation in MEF cells (Fig. S1G). Notably, Ppp6c deficiency protected MEF cells from TSZ-induced necroptosis during the early stage of the treatment (Fig. 1H, I). However, longtime exposure to TSZ eventually resulted in the cell death of Ppp6c-KO MEF cells (Fig. 1J), while Ppp6c deletion provided a long-term protection in L929 cells treated with TZ (Fig. S1H). Taken together, these data suggested that Ppp6c deficiency inhibits RIPK1-RIPK3-MLKL cascade-mediated necroptotic cell death.
Phosphatase activity of PPP6C is required for its role in regulating necroptosis
PPP6C is the catalytic subunit of PP6, which exhibits multiple roles in regulating several cellular processes [29]. We were wondering whether its catalytic activity is required for its role in TNF-induced necroptosis. We first generated PPP6C-overexpressing L929 cells and found that overexpression of PPP6C sensitized L929 to TNF-induced necroptosis (Fig. 2A–C), which is consistent with its positive role in TNF-induced necroptosis. Then, we generated a PPP6C reintroducing (add-back) stable L929 cells (Re-PPP6C) by transducing Ppp6c-KO L929 cells with lentivirus HA-PPP6C. Of note, the protein level of add-back PPP6C was similar with the endogenous PPP6C protein (Fig. S2A). As expected, PPP6C re-expression restored the sensitivity of Ppp6c-KO L929 cells to TZ-induced necroptotic cell death (Fig. 2D, E), in conjunction with the expression of phosphorylated RIPK1 (S166), RIPK3 (S232) and MLKL (S345) (Fig. 2F), and the oligomeric MLKL (Fig. 2G). The catalysis of PPP6C requires several active residues including Asp84, associated with salt bridge formation and Arg85, related to phosphoryl group binding [41, 42]. We substituted Asp84 and Arg85 to Asn and Ala, respectively, to produce a D84N-R85A phosphatase-dead mutant (PPP6C-PD) (Fig. S2B). We found that only wild-type (WT) PPP6C, but not PPP6C-PD, restored the sensitivity of Ppp6c-KO L929 cells to TZ-induced necroptotic cell death indicated by PI staining (Fig. 2D, E). Consistently, the phosphorylation levels of RIPK1 (S166), RIPK3 (S232), and MLKL (S345), and MLKL oligomerization were increased in WT PPP6C but not PPP6C-PD re-expression L929 cells treated with TZ (Fig. 2H, I). Together, these results indicate that PPP6C regulates necroptosis depending on its phosphatase activity.
Fig. 2. Phosphatase activity of PPP6C is required for its role in regulating necroptosis.

A, C Control and PPP6C-overexpression L929 cells were treated with TZ for 4 h, then stained with PI and analyzed under a microscope (A), or quantified by flow cytometer (C). Scale bar, 50 μm. B Control and PPP6C-overexpression L929 cells were treated with TZ for 4 h and cell lysates were probed with indicated antibodies. D, E Ppp6c-KO cells were transduced with PPP6C or its phosphatase dead (PD) lentivirus and followed by the treatment with TZ for the indicated time. Cells were then stained with PI and analyzed under a microscope (D) or quantified by flow cytometer (E). Scale bar, 50 μm. F, G Ppp6c-KO L929 cells were transduced with PPP6C lentivirus and followed by the treatment with TZ for the indicated time. Cell lysates were resolved on SDS-PAGE (F) or non-reducing PAGE (G) for immunoblot with indicated antibodies. H, I Ppp6c-KO L929 cells were transduced with PPP6C or PPP6C-PD lentivirus and followed by the treatment with TZ for the indicated time. Cell lysates were resolved on non-reducing PAGE (H) or SDS-PAGE (I) for immunoblot with indicated antibodies. Data shown are representative of three independent experiments and presented as means ± SDs of triplicates (C, E). **p < 0.01, ***p < 0.001, with an unpaired Student’s t-test (C) or one-way ANOVA analysis (E).
RIPK3 and MLKL are the key components in TNF-induced necroptotic signaling. We found that both the protein and mRNA levels of RIPK3 and MLKL were reduced in Ppp6c-KO L929 and PPP6C knockdown HT29 cells (Fig. S2A, C–E). As expected, re-expression of WT PPP6C but not PPP6C-PD in Ppp6c-KO L929 cells restored the expression of RIPK3 and MLKL (Fig. S2A, E). Our results suggest that Ppp6c depletion reduces the expression of RIPK3 and MLKL, which might partially contribute to the resistance to TNF-induced necroptosis.
The resistance to TNF-induced necroptosis in Ppp6c deficient L929 cells is dependent on the TAK1-IKK signaling
PPP6C was previously revealed to oppose the activation of NF-κB at multiple steps [43, 44]. To elucidate the molecular mechanism by which PPP6C regulates necroptosis, we first investigated whether PPP6C regulates TNF-mediated NF-kB activity. By examining the nuclear translocation of P65, the phosphorylation levels of both IKKα/β and IκBα, and the responsive genes of NF-κB pathway, such as A20, Tnf, IκB, and cIAP1, we found that Ppp6c-KO L929 cells displayed a slightly faster and higher NF-κB activation compared with the control cells after TNF stimulation (Fig. S3A–D). In addition, we found that the expression of PPP6C suppressed NF-κB-driven luciferase activity induced by TNF, TAK1, and IKKα/β, but not by P65 (Fig. S3E). Thus, these data suggest that PPP6C represses TNF-induced NF-κB activity.
To evaluate whether NF-κB activity is required for PPP6C-regulated necroptosis, we treated L929 cells with several chemical inhibitors targeting the components in NF-κB pathway together with TZ. We found that blocking NF-κB pathway by two different inhibitors, maslinic acid (targeting P65) [45] and BAY 11-7082 (targeting IκBα) [46], significantly increased cell death in control cells but had little effect on Ppp6c-KO L929 cells (Fig. 3A, B, Fig. S3F). Intriguingly, TAK1 inhibitor 5Z-7-Oxozeaenol [47] or IKKα/β inhibitor IKK16 [48] completely abolished the resistance of Ppp6c-KO cells to TZ-induced necroptosis (Fig. 3C, D, Fig. S3G). Co-treatment with the RIPK1 inhibitor Necrostatin-1 (Nec1) fully reversed this sensitization to necroptosis induced by TAK1 and IKK inhibitors in Ppp6c-KO L929 cells (Fig. 3E–G). TAK1 has been reported to phosphorylate RIPK1 at S320/321 directly, or indirectly via its downstream pathway such as P38-MK2, which inhibits RIPK1 kinase activity and prevents necroptosis [24–26]. To test whether P38-MK2 signaling cascade is involved in the resistance to TZ-induced necroptosis in Ppp6c-KO L929 cells, we treated the cells with TZ in the presence of adezmapimod or MK2-IN-1, which inhibits P38 and MK2 respectively to prevent TNF-induced P38 and MK2 activation. We found that pharmacologic inhibition of P38 or MK2 did not interfere with the resistance to TZ-induced necroptosis in Ppp6c-KO L929 cells (Fig. 3H–J, Fig. S3H). Together, our results suggest that the resistance to TZ-induced necroptosis in Ppp6c-KO L929 cells is dependent on the TAK1-IKK signaling activity, but not P38-MK2, or IκBα-P65 signaling cascade.
Fig. 3. Inhibition of TAK1 or IKK activity restores TNF-induced necroptosis in Ppp6c deficient L929 cells.

A, B Control and Ppp6c-KO L929 cells were pretreated with P65 inhibitor (Maslinic acid, 20 μM) or IκBα inhibitor (BAY117085, 10 μM) for 30 min and then treated with TZ for 4 h. Cells were stained with PI, and analyzed under a microscope (A) or quantified by flow cytometer (B). Scale bar, 50 μm. (C, D) Control and Ppp6c-KO L929 cells were pretreated with TAK1 inhibitor (5Z-7-Oxozeaenol, 1 μM) or IKKα/β inhibitor (IKK16, 1 μM) for 30 min and then treated with TZ for 4 h. Cells were stained with PI, and analyzed under a microscope (C) or quantified by flow cytometer (D). Scale bar, 50 μm. E–G Ppp6c-KO L929 cells were pretreated with TAK1 inhibitor (5Z-7-Oxozeaenol, 1 μM) or IKKα/β inhibitor (IKK16, 1 μM) in the presence or absence of Necrostatin-1 (10 μM) for 30 min, and then treated with TZ for 4 h. Cells were stained with PI and analyzed under a microscope (E) or quantified by flow cytometer (F). Cell lysates were probed with indicated antibodies (G). Scale bar, 50 μm. H–J Control and Ppp6c-KO L929 cells were pretreated with P38 inhibitor (Adezmapimod, 10 μM) or MK2 inhibitor (MK2-IN-1, 10 μM) for 30 min, and then treated with TZ for 4 h. And cells were stained with PI and analyzed under a microscope (H) or quantified by flow cytometer (I), and cell lysates were probed with indicated antibodies (J). Scale bar, 50 μm. Data shown are representative of three independent experiments and presented as means ± SDs of triplicates (B, D, F, I). **p < 0.01, ***p < 0.001, with an unpaired Student’s t-test (B, D, I) or one-way ANOVA analysis (F).
PPP6C regulates necroptosis by dephosphorylating TAK1
It has been reported that PPP6C negatively regulates IL-1 signaling via dephosphorylating TAK1 at T187 [31], a key phosphorylation site that determines whether TAK1 is activated [49]. To dissect the molecular mechanism that causes the resistance to TZ-induced necroptosis in Ppp6c-KO L929 cells, we examined the phosphorylation status of TAK1. The result revealed that the phosphorylation level of TAK1 at T187 was enhanced and sustained longer in Ppp6c-KO L929 cells in response to TNF stimulation (Fig. 4A). In addition, we found a prolonged IKKα/β activation, and a lower activity of RIPK1 indicated by the reduced S166 phosphorylation in Ppp6c-KO L929 cells following TNF stimulation (Fig. 4B). Thus, these results imply that Ppp6c depletion impairs the RIPK1 kinase activity in TNF-treated L929 cells. To further explore whether the inhibition of RIPK1 kinase activity is dependent on TAK1-IKKα/β axis, we made use of pharmacologic inhibitors. We found that the inhibition of TAK1 or IKKα/β significantly increased the phosphorylation of RIPK1 at S166 in TNF-treated Ppp6c-KO L929 cells (Fig. 4C, D), while the inhibition of IκBα or P65 had no such effect (Fig. S4A, B). These results indicate that RIPK1 kinase activity is regulated by TAK1-IKKα/β cascade in Ppp6c-KO L929 cells.
Fig. 4. PPP6C interacts with and dephosphorylates TAK1.

A, B Control and Ppp6c-KO L929 cells were treated with TNF (10 ng/ml) for the indicated time, and then lysed for immunoblot with indicated antibodies. C, D Control and Ppp6c-KO L929 cells were pretreated with TAK1 inhibitor (5Z-7-Oxozeaenol, 1 μM) (C) or IKKα/β inhibitor (IKK16, 1 μM) (D) for 30 min and then treated with TNF (10 ng/ml) for the indicated time. Cell lysates were probed with indicated antibodies. E HEK293T cells were transfected with TAK1 and TAB1 plasmids together with different dose of PPP6C plasmid. Cell lysates were probed with indicated antibodies 24 h after transfection. F HEK293T cells were transfected with TAK1 and TAB1 plasmids together with PPP6C or PPP6C-PD mutant plasmids. Cell lysates were probed with indicated antibodies 24 h after transfection. G HEK293T cells were transfected with the indicated plasmids and then lysed for co-immunoprecipitation assay as indicated. H Ppp6c-KO L929 cells were transduced with HA-PPP6C lentivirus and then treated with TNF (10 ng/ml) for the indicated time, and lysed for co-immunoprecipitation assay as indicated. I Control and Ppp6c-KO L929 cells were stimulated with 1 μg/ml HA-mTNF for 5 min. TNFR1 complex I was immunoprecipitated by anti-HA antibody and analyzed by immunoblotting. J Control and Ppp6c-KO L929 cells were transduced with FLAG-RIPK1 lentivirus and then treated with TNF (10 ng/ml) for the indicated time. FLAG-RIPK1 proteins were immunoprecipitated with anti-FLAG antibody and probed with anti-p-RIPK1 (S25) antibody.
TAK1 was reported as a substrate of PPP6C [31]. Consistent with previous finding, PPP6C overexpression reduced the phosphorylation of TAK1 at T187 in a dose-dependent manner (Fig. 4E), and the phosphatase dead mutant of PPP6C had no such activity (Fig. 4F). In addition, co-immunoprecipitation assay showed that PPP6C was associated with TAK1 but not IKKα/β or RIPK1 (Fig. 4G), implying that PPP6C may regulate RIPK1 kinase activity through TAK1, rather than by dephosphorylating IKKα/β or RIPK1. To investigate whether the interaction between PPP6C and TAK1 was regulated by TNF stimulation, we re-expressed HA-PPP6C in Ppp6c-KO L929 cells and performed a co-immunoprecipitation assay. The result showed that the endogenous TAK1 was associated with HA-PPP6C and the interaction was enhanced by TNF treatment in L929 cells (Fig. 4H). Importantly, we found that endogenous PPP6C was recruited to TNFR1 complex under TNF stimulation in L929 cells, and TAK1 was marked reduced in TNFR1 complex I when Ppp6c was depleted in L929 cells (Fig. 4I). Previous research found that RIPK1 is a direct substrate of IKKα/β [22], and IKKα/β phosphorylate RIPK1 at S25 to prevent RIPK1 kinase activation [27]. In accordance with a higher IKKα/β activity, enhanced phosphorylation of RIPK1 at S25 was detected in Ppp6c-KO L929 cells upon TNF treatment (Fig. 4J). To examine whether PPP6C regulates the activity of IKKα/β or RIPK1, we used TAK1-KO HEK293T cells to rule out the interference of TAK1 on the activity of IKKα/β or RIPK1. We transfected TAK1-KO HEK293T cells with IKKα/β, RIPK1, and PPP6C plasmids and found that PPP6C has little effect on the phosphorylation levels of IKKα/β or RIPK1 when TAK1 is deficient (Fig. S4C). As a core regulator of TNF-induced necroptosis, RIPK1 can be phosphorylated at multiple sites, whether PPP6C could dephosphorylate RIPK1 at other IKKα/β-independent sites needs further investigation. Taken together, these data suggest that Ppp6c deficiency inhibits RIPK1 kinase activity, at least in part, through acting on TAK1-IKKα/β signaling axis to increase phosphorylation of RIPK1 at S25, which ultimately prevents TNF-induced necroptosis.
TAB2 is essential for the resistance to TNF-induced necroptotic cell death in Ppp6c deficient cells
A previous study has shown that TAK1-binding protein 2 (TAB2), is necessary for TAK1 inactivation by recruiting PPP6C to the TAK1 complex [50] and deficiency in TAB2 leads to sustained TAK1 activation but unexpectedly sensitized cells to necroptosis [28]. To investigate whether TAB2 involves in the resistance to necroptosis in Ppp6c-KO L929 cells, we used shRNA to deplete Tab2 in WT and Ppp6c-KO L929 cells. The knockdown efficiency was determined by quantitative RT-PCR (Fig. S5A). Consistent with the previous report, we found that Tab2-depleted L929 cells had elevated TAK1 phosphorylation (Fig. S5B) and were more sensitive to TNF-induced necroptosis (Fig. 5A–C, Fig. S5C). However, to our surprise, knockdown of Tab2 also remarkably increased TNF-induced necroptosis in Ppp6c-KO L929 cells (Fig. 5D–F). These results suggest that Tab2 depletion abolished the resistance to necroptosis in Ppp6c-KO L929 cells. Since both Ppp6c and Tab2 depletion resulted in the increased TAK1 activity, but led to opposite effect on TZ-induced necroptosis. To further investigate this, we stimulated cells with TNF, and found that TAK1 and IKKα/β were sustainably activated in Tab2-depletion cells just like that in Ppp6c-KO L929 cells. If the increased necroptotic cell death in Tab2-depleted cells was caused by hyperactivation of TAK1, one would expect that inhibition of TAK1 would reduce TNF-induced necroptotic cell death in Tab2-depleted cells. However, inhibition of TAK1 by 5Z-7-Oxozeaenol did not prevent TNF-induced necroptotic cell death in Tab2-depleted cells (Fig. 5G, H). In addition, the increased RIPK1 kinase activity indicated by the phosphorylation level at S166 of RIPK1 in Tab2-depleted cells was not affected by TAK1 inhibition during TNF stimulation (Fig. 5I). Together, these results suggest that TAB2 may play a more complicated regulatory role in the necroptosis signaling and depletion of Tab2 sensitizes Ppp6c-KO cells to TNF-induced necroptosis.
Fig. 5. TAB2 is required for the resistance to TNF-induced necroptotic cell death in PPP6C deficient cells.

A, B Control and Tab2-knockdown L929 cells were treated with TZ for the indicated time, and analyzed under a microscope (A) or quantified by flow cytometer (B) after stained with PI. Scale bar, 50 μm. C Control and Tab2-knockdown L929 cells were treated with TZ for the indicated time and then lysed for immunoblot with indicated antibodies. D, E Control and Tab2-knockdown Ppp6c-KO L929 cells were treated with TZ for the indicated time, and analyzed under a microscope (D) or quantified by flow cytometer (E) after stained with PI. Scale bar, 50 μm. F Control and Tab2-knockdown Ppp6c-KO L929 cells were treated with TZ for the indicated time and then lysed for immunoblot with indicated antibodies. G, H Control and Tab2-knockdown L929 cells were pretreated with TAK1 inhibitor (5Z-7-Oxozeaenol, 1 μM) in the presence or absence of Necrostatin-1 (10 μM) for 30 min, and then treated with TZ for 4 h. Cells were stained with PI and analyzed under a microscope (G) or quantified by flow cytometer (H). Scale bar, 50 μm. I Control and Tab2-knockdown L929 cells were pretreated with TAK1 inhibitor (5Z-7-Oxozeaenol, 1 μM) for 30 min, and then treated with TNF (10 ng/ml) for the indicated time. Cell lysates were probed with indicated antibodies. Data shown are representative of three independent experiments and presented as means ± SDs of triplicates (B, E, H). **p < 0.01, ***p < 0.001, with one-way ANOVA analysis (B, E, H).
Ppp6c loss alleviates necroptosis-related tissue injury and inflammation
Necroptosis is an inflammatory form of programmed cell death and dysregulated necroptosis leads to intestinal inflammation [51]. It has been demonstrated that necroptosis is essential for TNF-induced SIRS, as both Ripk3-/- and Mlkl-/- mice are protected from TNF-induced SIRS [52, 53]. We utilized a TNF-SIRS mouse model to determine whether PPP6C regulates the onset of systemic inflammatory response syndrome. It is important to note that the cecum was described as particularly sensitive to TNF-induced injury in the TNF-SIRS model [54], so we used ShhCre driver to delete Ppp6c in gastrointestinal tract. Lineage tracing study from ShhCre/+;Rosa26tdTomato mice confirmed the Cre activity in mouse gastrointestinal tract (Fig. S6A). We crossed the ShhCre/+ mice with Ppp6cflox/flox mice to obtain Ppp6c intestine knockout mice. However, it is likely that ShhCre/+-mediated Ppp6c knockout mice are embryonic lethal, we did not obtain ShhCre/+;Ppp6cflox/flox offspring. Therefore, ShhCre/+;Ppp6flox/+ mice were used for further investigation. Their littermates, Ppp6cflox/+ mice, were used as the control. We found that ShhCre/+;Ppp6flox/+ mice showed partial protection from TNF-induced cecum damage compared with the littermate control mice after intravenous injection of mouse TNF (Fig. 6A). In addition, analysis of the cecum lysate showed that TNF-induced phosphorylation of MLKL in control mice was higher than that in ShhCre/+;Ppp6flox/+ mice (Fig. 6B). These data further support that loss of PPP6C prevent TNF-induced cecum injury through inhibition of necroptosis. In addition, we employed a DSS-induced colitis mouse model to further investigate the role of PPP6C in regulating the inflammatory diseases. We found that upon DSS treatment, ShhCre/+;Ppp6cflox/+ mice showed less weight loss compared to the control (Fig. S6D), and markedly longer colons than that in DSS-treated control mice (Fig. S6B, C, E, F). Consistently, H&E staining also revealed less inflammation and mucosal epithelium damage in ShhCre/+;Ppp6cflox/+ mice (Fig. S6G). Consistent with TNF-SIRS model, MLKL was less phosphorylated in ShhCre/+;Ppp6cflox/+ mice (Fig. S6H), indicating that the necroptotic pathway was compromised in ShhCre/+;Ppp6cflox/+ mice. Moreover, reduced apoptotic cell death of intestinal cells was found in DSS-treated ShhCre/+;Ppp6cflox/+ mice by TUNEL staining (Fig. S6I). It is worth noting that whether necroptosis or the kinase activity of RIPK1 is necessary for DSS-induced colitis is still debatable [53, 55], so we speculate that PPP6C might play a more complex role in regulating tissue injury and inflammation beyond its ability to promote necroptosis. Collectively, these results indicate that Ppp6c depletion alleviates necroptosis-related tissue injury and inflammation.
Fig. 6. PPP6C loss alleviates TNF-induced necroptosis in SIRS model.
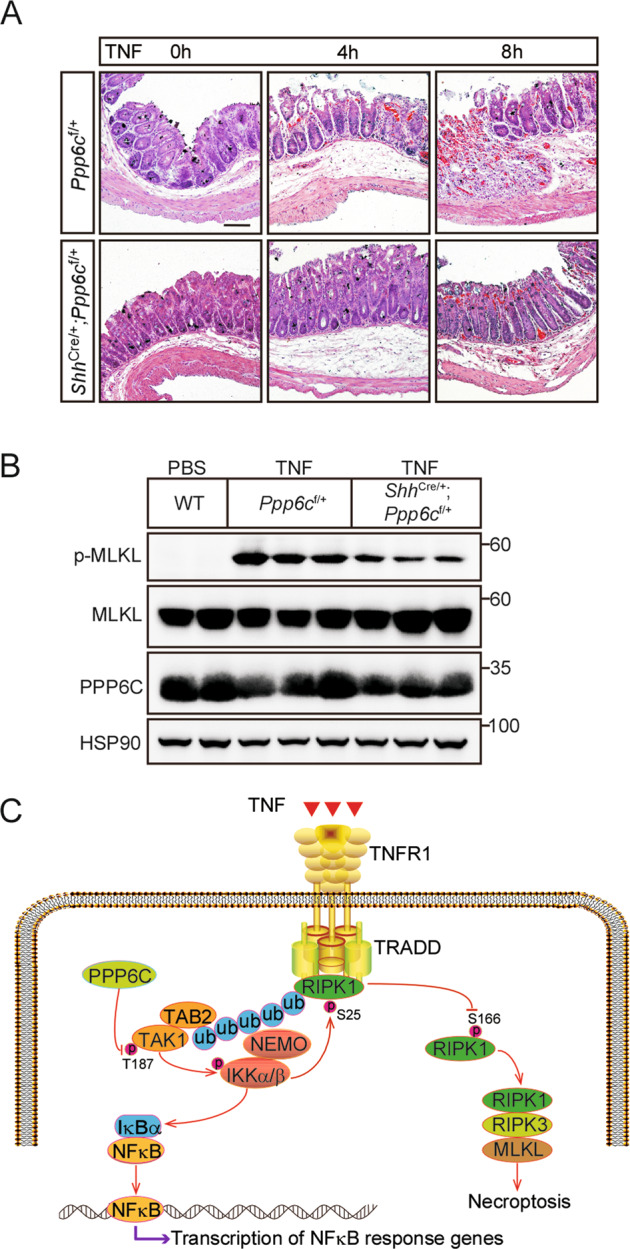
A Representative H&E staining of cecum from Ppp6cf/+ and ShhCre/+;Ppp6cf/+ mice (n = 3 each group at a time point) injected with TNF for the indicated time. Scale bar, 100 μm. B Cecal proteins from Ppp6cf/+ and ShhCre/+;Ppp6cf/+ mice injected with TNF for 8 h were analyzed by immunoblot with indicated antibodies. C A schematic model shows that PPP6C positively regulates TNF-induced necroptosis via TAK1-IKKα/β axis.
Discussion
In this study, we performed a positive-selection screen to identify necroptosis regulators in L929 cells by using CRISPR/Cas9 gene knockout library against 20,611 genes of the mouse genome and identified 79 genes by positive sgRNA enrichment. As expected, we found a strong enrichment of sgRNA targeting TNF receptor Tnfrsf1a and known necrosome components Ripk1, Ripk3, Mlkl and some moderate enrichments of sgRNAs targeting genes related to RIPK1 posttranslational modification, such as Cyld and Spata2. In addition, we identified Ppp6c as a novel mediator of necroptosis. Intriguingly, we noticed some similar screens for necroptosis resistance were previously performed in several publications [35, 56–62], but there is a remarkable lack of overlap between the regulators identified in those genetic screens except a few key factors such as Ripk1, Ripk3, and Mlkl. Similarly, little overlap of screen hits exists between our study and other reported screens. We suspect that those differences in screening strategies among different groups might partly contribute to the discrepancy of screen hits. So different screening strategies may complement each other and help us get a more complete understanding of necroptosis. More importantly, the nonoverlapping hits among many of these screens also suggest that some of the regulators influence necroptosis in a context-dependent manner except the core RIPK1-RIPK3-MLKL axis.
Necroptosis is a process tightly regulated by a variety of precise phosphorylation, and its key regulators RIPK1, RIPK3, and MLKL are all phosphorylated at multiple positions. Many kinases such as TAK1, MK2, IKKα/β, CK1, and TAM [24, 25, 27, 63, 64] were previously reported to play a crucial role in the necroptosis signaling pathway. However, how the necroptosis is regulated by the phosphatase which acts on the opposition to the kinase is still rarely reported. In this study, we revealed that Ppp6c is a novel positive regulator of necroptosis by genome-wide CRISPR/Cas9 library screening. We showed that the deficiency of Ppp6c protects cells from TNF-induced necroptotic cell death. Accordingly, deletion of Ppp6c increases TNF-induced IKKα/β activation which inhibits RIPK1 kinase activity. TNF-induced necroptotic cell death could be restored by TAK1 or IKKα/β inhibitor instead of NF-κB inhibitor in Ppp6c-KO L929 cells. Although we showed that PPP6C interacts with and dephosphorylates TAK1, we could not rule out the possibility that PPP6C acts directly on RIPK1 which is phosphorylated at multiple sites. Thus, our study reveals the molecular mechanism how PPP6C controls the activity of RIPK1 kinase and regulates TNF-induced necroptosis, at least in part, via the TAK1-IKKα/β cascade (Fig. 6C), suggesting that the phosphatase also plays important roles in the process of necroptosis.
Global deletion of Ppp6c leads to embryonic lethality in mice [40], suggesting that Ppp6c plays essential functions during the normal development. PPP6C has been implicated in DNA damage repair, oocyte, and lymphocyte development, virus infection, innate immune responses, carcinogenesis, etc [29, 65, 66]. Here, we showed that the colon epithelium-specific knockdown of Ppp6c alleviates necroptosis-related tissue injury and inflammation. Interestingly, we found Ppp6c deletion reduced cell proliferation in MEFs while deletion of Ppp6c in L929 cells had no such effect. Similarly, one study reported that shRNA-mediated PPP6C knockdown in HepG2 cells has little influence on proliferation rate [67], but another research showed that in vivo infection of lentivirus shRNA targeting Ppp6c in epidermis leads to an enhanced proliferation of keratinocyte [68]. As a multifunctional phosphatase, PPP6C has diverse important functions and controls various physiological processes. A functional PP6 holoenzymes usually consists of its catalytic subunit (PPP6C) and other regulatory subunits. Its diverse functions may be regulated by these regulatory subunits.
We found that the depletion of Ppp6c in MEF cells only delays necroptotic cell death and Ppp6c-KO MEF cells eventually died after longtime exposure to TNF. We postulate that there might be other phosphatases that play a compensatory role in the absence of Ppp6c in MEF cells. As an evolutionarily conserved and ubiquitously expressed protein phosphatase, PP6 is highly homologous with protein phosphatase 2A (PP2A) and protein phosphatase 4 (PP4) [29], however, PP6 has received less attention than its close relative PP2A. Since PP6 is closely related to PP2A and PP4, whether PP2A or PP4 has a similar regulatory function on necroptosis is worthy of further investigation. Noteworthy, PPM1A and PPM1B were identified as IKKβ phosphatases to synergistically terminate TNF-induced IKKβ and NF-κB activation [69]. Only PPM1B was showed to negatively regulate necroptosis through dephosphorylating RIPK3 while PPM1A did not regulate necroptosis [70]. These studies indicate that two close phosphatases with similar structures may still not have an overlap function. Further studies are needed to determine the mechanism of phosphatases in the regulation of cell death.
TAK1 is considered as a pro-survival regulator via promoting the activation of NF-κB pathway [23]. Accumulating evidence suggests that TAK1 also regulates apoptosis and necroptosis pathway independent on its role in NF-κB activation. PPP6C was reported to be recruited to the TAK1 complex in a TAB2-dependent manner [50]. Similar to PPP6C deletion, deficiency of TAB2 leads to sustained TAK1 activation. However, TAB2 deficiency sensitized cells to necroptosis [28], which is opposite to PPP6C inactivation. Our data showed that TAB2 is essential for Ppp6c-KO cells to survive under TZ treatment, since Tab2 knockdown abolished the Ppp6c deletion-mediated resistance to TNF-induced necroptosis. Our data suggest that Tab2 knockdown promotes the activation of both TAK1 and RIPK1, and leads to RIPK1 kinase-dependent necroptosis. Considering the fact that Ppp6c removal leads to prolonged TAK1 activation but still protects cells from necroptosis, we speculate that TAK1 hyperactivation in Tab2 knockdown cells does not necessarily lead to hypersensitivity to TNF-induced necroptosis. It is possible that the deficiency of Tab2 promotes RIPK1 kinase activity and leads to necroptosis in a TAK1-independent manner. Intriguingly, in contrast to the previous finding and ours, a recent work showed that ablation of Tab2 blocks, instead of promotes, TAK1 activation in cardiomyocytes [71]. It is likely that the discrepancy is caused by different cell types and warrants further investigation.
In summary, the previous studies have revealed that a variety of kinases play essential roles in regulating necroptosis, while the functions of phosphatases were rarely studied in the pathway. Herein, we identified PPP6C as a positive regulator of necroptosis, and revealed a previously unknown function of PPP6C in regulating programed cell death.
Supplementary information
supplementary figure legend and tables CDDIS-22-0857R2
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31970727), the Natural Science Foundation of Zhejiang Provincial (No. LZ22C050002), and the National Key Research and Development Program of China (No. 2019YFA0802003).
Author contributions
YZ, MW, and HS conceived conception and experimental design; YZ carried out most of the experiments; QZ performed animal study; BJ performed flow cytometry analysis; ZH and YL contributed to intellectual inputs; LM provided reagents; HS and YZ wrote the manuscript.
Materials availability
Materials are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Ethics approval
All experiments were performed under the approval of the Ethics Committee at the Zhejiang University.
Footnotes
Edited by Professor Jianke Zhang
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yonggang Zou, Qi Zheng.
Contributor Information
Ming Wu, Email: iwuming22@zju.edu.cn.
Hai Song, Email: haisong@zju.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-022-05076-1.
References
- 1.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 2.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 4.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaiser WJ, Sridharan H, Huang CZ, Mandal P, Upton JW, Gough PJ, et al. Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33. doi: 10.1038/nature20559. [DOI] [PubMed] [Google Scholar]
- 7.Hsu HL, Xiong J, Goeddel DV. The Tnf receptor 1-associated protein tradd signals cell-death and Nf-kappa-B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 8.Hsu HL, Huang JN, Shu HB, Baichwal V, Goeddel DV. TNF-Dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–96. doi: 10.1016/S1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 9.Bertrand MJM, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 10.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–92. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21:1709–20. doi: 10.1038/cdd.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun LM, Wang HY, Wang ZG, He SD, Chen S, Liao DH, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 13.Chen WZ, Zhou ZR, Li LS, Zhong CQ, Zheng XR, Wu XR, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288:16247–61. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HY, Sun LM, Su LJ, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Su LJ, Quade B, Wang HY, Sun LM, Wang XD, Rizo J. A plug release mechanism for membrane permeation by MLKL. Structure. 2014;22:1489–500. doi: 10.1016/j.str.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Li WJ, Ren JM, Huang DL, He WT, Song YL, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NF-kappa B antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 21.Chang LF, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNF alpha-induced cell death by inducing c-FLIPL turnover. Cell. 2006;124:601–13. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 22.Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappa B-independent role of IKK alpha/IKK beta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell. 2015;60:63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 23.Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell Death Differ. 2014;21:1667–76. doi: 10.1038/cdd.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geng JF, Ito Y, Shi LY, Amin P, Chu JC, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;8:359. doi: 10.1038/s41467-017-00406-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell. 2017;66:698-+. doi: 10.1016/j.molcel.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol. 2017;19:1237–47. doi: 10.1038/ncb3608. [DOI] [PubMed] [Google Scholar]
- 27.Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun. 2019;10:1729.. doi: 10.1038/s41467-019-09690-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, et al. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol. 2014;204:607–23. doi: 10.1083/jcb.201305070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohama T. The multiple functions of protein phosphatase 6. Bba-Mol Cell Res. 2019;1866:74–82. doi: 10.1016/j.bbamcr.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Stefansson B, Ohama T, Daugherty AE, Brautigan DL. Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochem-Us. 2008;47:1442–51. doi: 10.1021/bi7022877. [DOI] [PubMed] [Google Scholar]
- 31.Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, et al. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem. 2006;281:39891–6. doi: 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang ZH, Wu XN, He P, Wang XK, Wu JF, Ai TT, et al. A non-canonical PDK1-RSK signal diminishes pro-caspase-8-mediated necroptosis blockade. Mol Cell. 2020;80:296-+. doi: 10.1016/j.molcel.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Yuan JY, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–53. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J, Jitkaew S, Cai ZY, Choksi S, Li QN, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad Sci USA. 2012;109:5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–U132. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei R, Xu LW, Liu JP, Li YX, Zhang P, Shan B, et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev. 2017;31:1162–76. doi: 10.1101/gad.299776.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bastians H, Ponstingl H. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J Cell Sci. 1996;109:2865–74. doi: 10.1242/jcs.109.12.2865. [DOI] [PubMed] [Google Scholar]
- 39.Xie T, Peng W, Yan CY, Wu JP, Gong XQ, Shi YG. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013;5:70–8. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 40.Ogoh H, Tanuma N, Matsui Y, Hayakawa N, Inagaki A, Sumiyoshi M, et al. The protein phosphatase 6 catalytic subunit (Ppp6c) is indispensable for proper post-implantation embryogenesis. Mechanisms Dev. 2016;139:1–9. doi: 10.1016/j.mod.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Hosing AS, Valerie NCK, Dziegielewski J, Brautigan DL, Larner JM. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J Biol Chem. 2012;287:9230–9. doi: 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammond D, Zeng K, Espert A, Bastos RN, Baron RD, Gruneberg U, et al. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J Cell Sci. 2013;126:3429–40. doi: 10.1242/jcs.128397. [DOI] [PubMed] [Google Scholar]
- 43.Bouwmeester T. A physical and functional map of the human TNF-alpha, NF-kappa, B signal transduction pathway. Nat Cell Biol. 2004;6:97. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 44.Stefansson B, Brautigan DL. Protein phosphatase 6 subunit with conserved sit4-associated protein domain targets I kappa B epsilon. J Biol Chem. 2006;281:22624–34. doi: 10.1074/jbc.M601772200. [DOI] [PubMed] [Google Scholar]
- 45.Fukumitsu S, Villareal MO, Fujitsuka T, Aida K, Isoda H. Anti-inflammatory and anti-arthritic effects of pentacyclic triterpenoids maslinic acid through NF-B inactivation. Mol Nutr Food Res. 2016;60:399–409. doi: 10.1002/mnfr.201500465. [DOI] [PubMed] [Google Scholar]
- 46.Mori N, Yamada Y, Ikeda S, Yamasaki Y, Tsukasaki K, Tanaka Y, et al. Bay 11-7082 inhibits transcription factor NF-kappa B and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100:1828–34. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]
- 47.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–90. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 48.Waelchli R, Bollbuck B, Bruns C, Buhl T, Eder J, Felfel R, et al. Design and preparation of 2-benzamido-pyrimidines as inhibitors of IKK. Bioorg Med Chem Lett. 2006;16:108–12. doi: 10.1016/j.bmcl.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 49.Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–68. doi: 10.1074/jbc.M407537200. [DOI] [PubMed] [Google Scholar]
- 50.Broglie P, Matsumoto K, Akira S, Brautigan DL, Ninomiya-Tsuji J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem. 2010;285:2333–9. doi: 10.1074/jbc.M109.090522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–71. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 52.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 53.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tracey KJS. Shock and tissue injury induced recombinant human cachectin. Science. 1986;234:470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 55.Alvarez-Diaz S, Preaudet A, Samson AL, Nguyen PM, Fung KY, Garnham AL, et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ. 2021;28:1466–76. doi: 10.1038/s41418-020-00673-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hitomi JI, Christofferson DE, Ng A, Yao JH, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He SD, Wang L, Miao L, Wang T, Du FH, Zhao LP, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 58.Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Callow MG, Watanabe C, Wickliffe KE, Bainer R, Kummerfield S, Weng JL, et al. CRISPR whole-genome screening identifies new necroptosis regulators and RIPK1 alternative splicing. Cell Death Dis. 2018;9:261. doi: 10.1038/s41419-018-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fauster A, Rebsamen M, Willmann KL, Cesar-Razquin A, Girardi E, Bigenzahn JW, et al. Systematic genetic mapping of necroptosis identifies SLC39A7 as modulator of death receptor trafficking. Cell Death Differ. 2019;26:1138–55. doi: 10.1038/s41418-018-0192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong Y, Li LL, Zhang LT, Cui YY, Wu CY, Li H, et al. The bromodomain protein BRD4 positively regulates necroptosis via modulating MLKL expression. Cell Death Differ. 2019;26:1929–41. doi: 10.1038/s41418-018-0262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo HY, et al. MLKL requires the inositol phosphate code to execute necroptosis. Mol Cell. 2018;70:936. doi: 10.1016/j.molcel.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanna-Addams S, Liu SZ, Liu H, Chen S, Wang ZG. CK1 alpha, CK1 delta, and CK1 epsilon are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc. Natl Acad Sci USA. 2020;117:1962–70. doi: 10.1073/pnas.1917112117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan HL, Amin PP, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell. 2019;75:457-+. doi: 10.1016/j.molcel.2019.05.022. [DOI] [PubMed] [Google Scholar]
- 65.Li M, Shu HB. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell. 2020;11:584–99. doi: 10.1007/s13238-020-00729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ni GX, Ma Z, Wong JP, Zhang ZG, Cousins E, Ben Major M, et al. PPP6C negatively regulates STING-dependent innate immune responses. Mbio. 2020;11:e01728–20. doi: 10.1128/mBio.01728-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boylan JM, Salomon AR, Tantravahi U, Gruppuso PA. Adaptation of HepG2 cells to a steady-state reduction in the content of protein phosphatase 6 (PP6) catalytic subunit. Exp Cell Res. 2015;335:224–37. doi: 10.1016/j.yexcr.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan S, Xu ZY, Lou FZ, Zhang LY, Ke F, Bai J, et al. NF-kappa B-induced microRNA-31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat Commun. 2015;6:7652. doi: 10.1038/ncomms8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun WJ, Yu Y, Dotti G, Shen T, Tan XJ, Savoldo B, et al. PPM1A and PPM1B act as IKK beta phosphatases to terminate TNF alpha-induced IKK beta-NF-kappa B activation. Cell Signal. 2009;21:95–102. doi: 10.1016/j.cellsig.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen WZ, Wu JF, Li LS, Zhang ZM, Ren JM, Liang YJ, et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol. 2015;17:4. doi: 10.1038/ncb3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yin HF, Guo XY, Chen Y, Zeng YC, Mo XL, Hong SQ, et al. TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis. J Clin Invest. 2022;132:e152297. doi: 10.1172/JCI152297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary figure legend and tables CDDIS-22-0857R2
Data Availability Statement
Materials are available from the corresponding author upon reasonable request.