

Abstract
Background
Congenital ISG15 deficiency is a rare autoinflammatory disorder that is driven by chronically elevated systemic interferon levels and predominantly affects central nervous system and skin.
Methods and results
We have developed induced pluripotent stem cell‐derived macrophages and endothelial cells as a model to study the cellular phenotype of ISG15 deficiency and identify novel treatments. ISG15–/– macrophages exhibited the expected hyperinflammatory responses, but normal phagocytic function. In addition, they displayed a multifaceted pathological phenotype featuring increased apoptosis/pyroptosis, oxidative stress, glycolysis, and acylcarnitine levels, but decreased glutamine uptake, BCAT1 expression, branched chain amino acid catabolism, oxidative phosphorylation, β‐oxidation, and NAD(P)H‐dependent oxidoreductase activity. Furthermore, expression of genes involved in mitochondrial biogenesis and respiratory chain complexes II–V was diminished in ISG15–/– cells. Defective mitochondrial respiration was restored by transduction with wild‐type ISG15, but only partially by a conjugation‐deficient variant, suggesting that some ISG15 functions in mitochondrial respiration require ISGylation to cellular targets. Treatment with itaconate, dimethyl‐itaconate, 4‐octyl‐itaconate, and the JAK1/2 inhibitor ruxolitinib ameliorated increased inflammation, propensity for cell death, and oxidative stress. Furthermore, the treatments greatly improved mitochondria‐related gene expression, BCAT1 levels, redox balance, and intracellular and extracellular ATP levels. However, efficacy differed among the compounds according to read‐out and cell type, suggesting that their effects on cellular targets are not identical. Indeed, only itaconates increased expression of anti‐oxidant genes NFE2L2, HMOX1, and GPX7, and dimethyl‐itaconate improved redox balance the most. Even though itaconate treatments normalized the elevated expression of interferon‐stimulated genes, ISG15–/– macrophages maintained their reduced susceptibility to influenza virus infection.
Conclusions
These findings expand the cellular phenotype of human ISG15 deficiency and reveal the importance of ISG15 for regulating oxidative stress, branched chain amino acid metabolism, and mitochondrial function in humans. The results validate ruxolitinib as treatment for ISG15 deficiency and suggest itaconate‐based medications as additional therapeutics for this rare disorder.
Keywords: 4‐octyl itaconic acid, apoptosis, ATP, branched chain amino acid amino transferase 1, dimethyl itaconic acid, induced pluripotent stem cells, inflammation, ISG15, itaconate, itaconic acid, mitochondrial biogenesis, NRF2, oxidative stress, pyroptosis, reactive oxygen species, ruxolitinib
ISG15 deficiency triggers hyperinflammation, apoptosis/pyroptosis, and oxidative stress, while reducing mitochondrial respiration and biogenesis, branched chain amino acid catabolism, and viral infectivity.
Itaconates and the JAK1/2 inhibitor ruxolitinib ameliorate this dysregulated phenotype, identifying them as potential therapeutic options for this rare disorder.
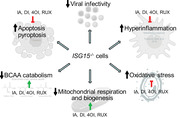
1. INTRODUCTION
Type I interferon (IFN‐I) signalling is activated by viral infection and results in tightly regulated antiviral innate immune responses. 1 Inborn disbalance of this system can lead to type I interferonopathy, which is characterized by persistent upregulation of interferon‐stimulated genes (ISGs) resulting in autoinflammation and autoimmunity. 1 , 2 , 3 Thus, monogenic type I interferonopathies can result from enhanced detection of intracellular nucleic acids leading to chronic induction of IFN‐I responses (familial chilblain lupus, Aicardi–Gutieres syndrome 4 ), increased sensitization of IFN‐I signalling (STING associated vasculopathy with onset in infancy [SAVI] 5 ), enhanced transcription of ISGs due to gain of function of STAT2 activation, 6 , 7 or due to loss of negative regulation of interferon α/β receptor (IFNAR) signalling by USP18. The latter may be caused by loss‐of‐function mutations in the USP18 gene itself 8 or in the gene encoding ISG15, which is required for stabilization of the USP18 protein in humans. 9
ISG15 deficiency originally came to clinical attention because affected individuals had an unusual susceptibility to mycobacterial infections, which was subsequently found to be due to inadequate IFN‐γ synthesis resulting from lack of free (unconjugated) ISG15. 10 Subsequently it was found that affected individuals may also, or predominantly, suffer from organ pathology resulting from chronic IFN‐I driven inflammation in CNS (seizures, intracranial calcifications 9 ) and/or skin ulcerations 11 , 12 . It remains to be clarified why one or the other clinical feature predominates in a given patient. Martin‐Fernandez et al. recently identified CD14–CD16+ monocytes as the peripheral blood cell type that drives chronic hyperinflammation and also showed that histiocytes in affected skin tissue are chronically activated. 11 In the same report, we showed activation of IFN‐I signalling and hyperinflammation in IFN‐α stimulated vascular endothelial cells (ECs) and HaCaT keratinocytes.
ISG15 exists either as free molecule, both intracellularly and extracellularly, or in conjugates with lysine residues of target proteins reminiscent of ubiquitination (“ISGylation”). ISGylation is executed by ISG15‐activating enzymes, ISG15‐conjugating enzymes, and ISG15 E3 ligases. 13 ISG15 expression and conjugation to targets is induced by a variety of processes, including infection, IFN‐α and ‐β signalling, ischemia, DNA damage, and aging. The current concept of human ISG15 deficiency is that IFN‐driven hyperinflammation and a reduced susceptibility to certain viral infections are the cardinal features of its cellular phenotype, 9 , 14 whereby the clinically intriguing lower viral infectivity may be explained by chronically elevated IFN‐I signalling and ISG expression. However, work in a variety of models has suggested that cellular functions of conjugated and free ISG15 go far beyond limiting IFN responses. 15 , 16 For instance, ISG15 has been implicated in regulating apoptosis, 17 cell cycle, 18 and autophagy, 13 and cells from ISG15–/– mice show depressed mitochondrial respiration and reduced levels of mitochondrial reactive oxygen species (ROS). 19 However, many of these studies were conducted in small animal models, which may not entirely reflect the human situation. This is exemplified by work using ISG15–/– mice, which do not manifest hyperinflammation or spontaneous skin lesions, likely because ISG15 is not required for USP18 stabilization in rodents. 15 In addition, it is not known to what extent the various functions of ISG15 that have been described in a broad array of models actually contribute to the phenotype of a representative single model. This uncertainty would also suggest that treatments could be designed in a more targeted manner once the full phenotype is known.
Janus kinase (JAK) inhibitors such as ruxolitinib (RUX) are emerging as disease‐modifying treatment for type I interferonopathies, particularly for patients with severe disease. 20 However, these medications are associated with high cost and may not be accessible to some patients or not be available in resource‐limited settings. In addition, they have not been evaluated for the treatment of ISG15 deficiency and it is not clear whether they would cover the whole spectrum of disease manifestations, since not all consequences of ISG15 loss may be due to lack of inhibition of IFNAR signalling but, rather, to absent ISGylation of protein targets other than USP18. Itaconic acid (IA) is a readily available, low‐cost dicarboxylic acid that has been used for decades in the synthesis of industrial polymers. It was recently identified as an immunomodulatory substance that occurs naturally in myeloid cells, notably activated macrophages. 21 , 22 Exogenously applied IA has well‐documented IFN‐reducing, immunomodulatory, 23 , 24 , 25 and ROS‐reducing properties, which have been demonstrated, for example, in animal models of ischemic reperfusion injury (e.g., 26 ) and influenza A virus infection of human macrophages. 25 Alkylated variants of IA such as dimethyl‐ and 4‐octyl‐IA (DI, 4OI) have more potent anti‐inflammatory and cytoprotective properties, likely due to greater electrophilicity. 24 Consequently, there are ongoing efforts to develop optimized IA derivatives into disease‐modifying treatments for acute and chronic inflammatory diseases.
Considering the uncertainty of the full extent of the cellular phenotype of ISG15 deficiency and the need for additional treatment options, we have established a cellular disease model featuring cell types relevant to systemic inflammation (induced pluripotent stem cell [iPSC]‐derived macrophages and vascular endothelial cells) and cutaneous pathology (immortalized dermal fibroblasts and HaCaT keratinocytes). While placing the main focus on macrophages, we have used these diverse cell types to identify key phenotypic features resulting from loss of ISG15. Moreover, considering their well‐documented ability to reduce IFN‐I expression, 24 we used this model to test whether itaconates and RUX could ameliorate aspects of the pathological phenotype.
2. MATERIALS AND METHODS
2.1. Cell culture
Wild‐type (WT) (MHHi001‐A) 27 and ISG15–/– (MHHi001‐A‐3) hiPSCs 28 were maintained on mouse embryonic fibroblasts (CF1 MEF), which were used as feeder cells as previously described 29 or under feeder‐free conditions on Geltrex‐coated tissue culture flasks (TPP, Trasadingen, Switzerland) in E8 medium and passaged as single cells using Accutase™ (PAA, Pasching, Austria) with a seeding density of 3.6 × 104 cells/cm2. For differentiation toward macrophages embryoid body (EB) formation was induced. hiPSCs were washed gently with phosphate‐buffered saline (PBS) and collagenase IV was then added to each well and incubated for 15–25 min in an incubator (37°C, 5% CO2) for detachment. Cells should be kept in fragments at this stage and resuspended in KO DMEM medium (without basic fibroblast growth facor). The cell suspension was transferred to a 6‐well suspension plate with 10 µM Rho‐associated kinase (ROCK) inhibitor (Y‐27632, Hölzel Diagnostica, Cologne, Germany) added. These plates were placed on a shaker at 85 rpm for 5 days. The medium was changed once or twice during this time and after 5 days, EBs > 200 µm diameter were picked using an upright low magnification microscope and transferred to a normal 6‐well plate containing X‐vivo differentiation medium I (Biozym (LONZA), Hessisch Oldendorf, Germany). 15–25 EBs were seeded per well and placed in an incubator for 8 days. After that, fresh X‐vivo differentiation medium I was added to the well, making sure that EBs were attached to the surface. At this stage EBs will start producing macrophages for up to 3–4 months, but the production rate will decrease with time. Differentiation toward ECs was performed as previously described. 11 , 30 Briefly, EC differentiation was induced by treatment with 25 ng/ml BMP4 (Bio‐Techne, Minneapolis, USA) and the WNT pathway activator CHIR 90221 (7.5 µM) in N2B27 medium (Thermo Fischer Scientific, Massachusetts, USA) for 2 days. From day 3 to 7, cultures were maintained in StemPro‐34 medium (Thermo Fischer Scientific, Massachusetts, USA) supplemented with 260 ng/ml rhVEGF‐A165 and 2 µM Forskolin (Sigma‐Aldrich, St. Louis, USA) with daily medium change. CD31+ cells were purified by MACS using CD31 MicroBead Kit (Miltenyi). hiPSC‐ECs were cultured on fibronectin (Corning, New York, USA) coated plates in ECGM‐2 medium (PromoCell, Darmstadt, Germany). hTert‐immortalized dermal fibroblasts from a healthy control and an ISG15‐deficient patient, as well as ISG15–/– fibroblasts transduced with WT or ΔGG ISG15 were kindly provided by Dusan Bogunovic. 14 In HaCaT cells the ISG15 locus was ablated with the CRISPR/Cas9 system using two guide RNAs (gRNA) cloned separately into vector PX458_pSpCas9‐2A‐GFP (Addgene, Watertown, MA); gRNA1: GCGCAGATCACCCAGAAGAT; gRNA2: GGTAAGGCAGATGTCACAGG. 1 × 106 cells were transfected with 1 µg of each targeting plasmid using Lipofectamine 3000 (Thermo Fischer Scientific, Massachusetts, USA) and green fluorescent protein (GFP) positive cells were isolated the next day by fluorescent activated cell sorting (FACS). The cell suspensions were then serially diluted (1:10) to obtain single cell clones, which were picked manually and expanded clonally. Homozygosity of the ISG15 knock‐out was confirmed by PCR of genomic DNA, resulting in a single band of 378 bp in homozygous KO clones (fwd_TTTCTTCCGCTCACTCTGGG and rev_GTTCGTCGCATTTGTCCACC) and the absence of the 5′ (fwd_TTTCTTCCGCTCACTCTGGG, rev_GAGGATCTCAGGGGTGACCT) and 3′ (fwd_AGAGGACAGACAGGAGGGAG, rev_GTTCGTCGCATTTGTCCACC) junction of the WT allele, and by absence of ISG15 protein expression by western blot (Figure S3H).
2.2. RT‐qPCR analysis
mRNA was extracted using the Nucleospin RNA purification kit (Macherey‐Nagel, Düren, Germany) and reverse transcribed with PrimeScript™ kit (Takara, Shiga, Japan) according to the manufacturers’ protocols. Gene expression was analysed using the SensiFast™ SYBR® No‐ROX Kit (Bioline, Taunton, MA, USA) and a LightCycler480 instrument (Roche, Mannheim, Germany). The 2–ΔΔCT method was used to measure relative mRNA expression of genes, 31 using HPRT mRNA as internal control. Primer sequences are listed in Table S1.
2.3. Western blotting
Cells were lysed using RIPA buffer supplemented with freshly added protease and phosphatase inhibitors. Protein concentrations were measured by bicinchoninic acid (BCA) assay, equal amounts of protein were separated by SDS‐PAGE, transferred to a nitrocellulose membrane using semi‐dry blotting method, incubated with the respective antibodies, and bands visualized with Amersham enhanced chemiluminescence western blot detection reagent (GE Healthcare Science, Pittsburgh, USA). iNTAS imaging device (iNTAS Science Imaging, Göttingen, Germany) was used for imaging and measurement of relative band intensities. The following antibodies were used to detect the respective proteins: Human ISG15 (sc‐166755, 1:500); pSTAT1 (58D6, 1:1000); IFIT1 (14769S, 1:1000); MX1 (37849S, 1:1000); pAKT (4060S, 1:2000) (Cell Signaling Technology, MA, USA); β‐Actin (ab49900, 1:20 000) (Abcam, Cambridge, UK).
2.4. Enzyme linked immunosorbent assay
Cells were cultured in 12‐well plates, supernatants or cells were collected and sandwich ELISA were performed according to the manufacturers’ protocols using the following kits: IL‐1β (Biolegend; cat# 437004), IL‐10 (Biolegend; cat# 430601), IP‐10 (PeproTech GmbH; cat# 900‐K39), and GSK3β (Abnova/Fisher scientific; cat# KA2908) according to the manufacturers’ protocols.
2.5. Mitochondrial ROS measurement
Cells were seeded at a density of 2 × 105 cells per well in a 12‐well plate, incubated for 5 min with medium containing 5 µM of MitoSOX ™ Red (mitochondrial superoxide indicator, Invitrogen, cat# M36008) and then washed with PBS. Cells were then resuspended in cold PBS and mitochondrial ROS was measured via the phycoerythrin (PE) channel using a BD™ LSR‐II flow cytometer.
2.6. Phagocytosis assay
Cells were cultured in a 96‐well plate at a density of 2 × 104 cells per well. PBMCs from healthy volunteers, 25 WT, and ISG15 –/– macrophages were kept in RPMI media supplemented with M‐CSF for 5 days, for terminal differentiation into the M2 activation state. After removal of nonadherent cells, phagocytosis assay was performed according to the manufacturers’ protocols (Phagocytosis Assay–Zymosan Substrate kit; Abcam, ab211156).
2.7. ATP measurement assay
Cells were seeded in a 12‐well plate at a density of 2 × 105 cells per well. After completion of the respective experiment, supernatants were collected for extracellular ATP measurement and RIPA buffer was added to obtain cell lysates for intracellular ATP measurements. ATP was measured according to the manufacturers’ protocol using ATP Determination Kit (ThermoFisher Scientific, A22066). Luminescence was measured using a luminometer (Centro XS3 LB960, Berthold Technologies, Bad Wildbad, Germany).
2.8. Single‐cell RNA sequencing
Macrophages were isolated from EB‐containing culture by FACS using CD11b, CD14, CD45 and CD163 as macrophage markers. 32 Cells were washed with 1x PBS and 1x binding buffer and incubated for 15 min with propidium iodide (PI) solution (Apoptosis Detection Kit, eBioscience, cat# 88‐8005‐74). Live and single cells were sorted by FACS based PI staining and single cell suspensions prepared in 1x PBS (10x Genomics). The single cell suspension was loaded onto a Chromium Single Cell Controller (10x Genomics). Barcoding and cDNA synthesis were performed according to the manufacturer's instructions. Quality was checked using the Agilent Bioanalyzer High Sensitivity Assay. Libraries were sequenced on Illumina NovaSeq 6000 2 × 50 paired‐end kits using the following read lengths: 26 bp Read1 for cell barcode and Unique Molecular Identifier (UMI), 8 bp I7 index for sample index and 89 bp Read2 for transcript. Cell Ranger 1.3 (http://10xgenomics.com) was used to process Chromium single cell 3′ RNA‐seq output. First, “cellranger mkfastq” demultiplexed the sequencing samples based on the 8 bp sample index read to generate fastq files for the Read1 and Read2, followed by extraction of 16 bp cell barcode and 10 bp UMI. Second, “cellranger count” aligned the Read2 to the mouse reference genome (mm10) using STAR. Then, aligned reads were used to generate data matrix only when they have valid barcodes and UMI, map to exons (Ensembl GTFs GRCm38.p4) without PCR duplicates. Valid cell barcodes were defined based on UMI distribution. Cells were removed when they had <500 unique feature counts and >25% reads mapped to mitochondrial expression genome (Figure S17). After filtering and doublet removal, the following cell numbers remained: WT con = 1894 cells, WT IFN‐α = 741, WT IFN‐α + IA = 2590, WT IFN‐α + 4OI = 3045, KO con = 2364, KO IFN‐α = 2107, KO IFN‐α + IA = 2378 and KO IFN‐α + 4OI = 2279.
Single‐cell gene expression data analysis including filtering, normalization, and clustering was processed using Seurat V3.1 (https://satijalab.org/seurat/) in the R environment for computing (www.bioconductor.org). Raw counts were normalized by the global‐scaling normalization method “LogNormalize”.
2.9. Bulk RNA sequencing
Total RNA was isolated using Nucleospin RNA purification kit (Macherey‐Nagel, Düren, Germany). RNA quality was checked on an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies; Waldbronn, Germany), and only samples with RNA Integrity Number ≥8 were used. The RNA sequencing library was generated from 100 ng total RNA using NEBNext® Single Cell/Low Input RNA Library according to the manufacturer´s protocols (New England BioLabs). The libraries were sequenced on an Illumina NovaSeq 6000, using the NovaSeq 6000 S1 Reagent Kit (100 cycles, paired‐end run 2 × 50 bp) with an average of 5 × 107 reads per sample. Sequencing reads were mapped against Homo sapiens hg38 reference genome via STAR 2.7.3a tool.
2.10. Small non‐coding RNA sequencing
Using aliquots from the same total RNA used for long RNAseq, quality and integrity of RNA was checked on an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies; Waldbronn, Germany). NEBNext® Multiplex Small RNA Library Prep Set for Illumina was used for library preparation. 200 ng of total RNA was used as input and the libraries were sequenced on an Illumina NovaSeq 6000 using the NovaSeq 6000 S1 Reagent Kit (100 cycles, paired end run 2 × 50 bp) with an average of 2 × 107 reads per RNA sample. FASTAQ files were then obtained to annotate the transcripts as described in the OASIS 2.0 web tool tutorial. 33 Briefly, FASTAQ files were compressed by OASIS compressor uploaded for annotation using the OASIS web tool and reference genome (Homo sapiens – hg38). Potential targets of the miRNAs were predicted using the TargetScan, miRDB and miRTarBase programs using the miRWalk 3.0 tool, keeping only those targets that were predicted by all three programs. Potential targets were uploaded to the WebGestalt platform for estimation of enriched pathways, using a threshold of false discovery rate (FDR) of <.05.
2.11. Seahorse Mito Stress assay
SeahorseXF Mito Stress assay (Seahorse XFe/XF analyser, Agilent) was used to measure mitochondrial function. It is a plate‐based live‐cell assay that measures oxygen consumption rate (OCR) in real time. Briefly, cells were seeded in a Seahorse XF cell culture microplate and subjected to IFN‐α stimulation and/or treatment as indicated. Cells were washed with Seahorse XF DMEM medium (supplemented with 8 mM glucose, 1 mM pyruvate and 2 mM glutamine, pH 7.4) and then kept in this medium in a non‐CO2 incubator at 37°C for 60 min. The Seahorse cartridge has four injection ports (A‐D). Injection solutions of 1 µM oligomycin (port A), 1.5 µM carbonyl cyanide‐4 (trifluoromethoxy) phenylhydrazone (FCCP) (port B), 1 µM rotenone (port C), and 1 µM antimycin (port D) were prepared in Seahorse medium and added to the respective ports once incubation of the cartridge was complete. The cartridge was then loaded into the Seahorse instrument and calibrated for 15–20 min. Following calibration, the cell culture microplate was loaded to run the assay. Once the assay was complete, WAVE software generated the report, which was then exported to an Excel file.
2.12. Crystal violet staining
Crystal violet staining was used to quantify the cells in each well after the completion of Seahorse assay. Briefly, cells were washed twice with PBS, 300 µl of crystal violet staining solution was added to each well, and the plate was incubated for 10 min. Afterward, it was washed three times with PBS, and 200 µl of 1% SDS was added to each well and the plate was kept on a rocking shaker for 30 min. Last, 100 µl of experimental lysate was transferred to a new 96‐well plate and absorbance at 570 nm was measured with a spectrophotometer. Seahorse data were normalized with OD of each well for final calculations of OCR.
2.13. Fluorometric assay
A caspase multiplex fluorometric assay kit (Abcam; cat# ab219915) was used to measure caspase activity to detect cells undergoing apoptosis. Briefly, cells were seeded in a 96‐well plate and after completion of the experiment, medium was removed and assay loading solution (caspase substrate and assay buffer) was added to each well. The plate was then incubated in the dark for 60 min and fluorescence was measured with a fluorescence microplate reader.
2.14. Glutathione measurements
GSH/GSSG Ratio Detection Assay Kit II (Abcam, ab205811) was used to detect reduced (GSH) and oxidized (GSSG) glutathione levels. Cells were seeded in a 12‐well plate and after completion of the experiment, medium was removed and cells were washed with cold PBS. Cells were lysed, homogenized for 15 min, and clear supernatant was collected and transferred to a new tube. For GSH detection, 50 µl of GSH assay mixture was added to standards and samples. For GSSG measurement, 50 µl of total glutathione assay mixture was added to standards and samples. After incubation of 15 min, fluorescence was measured at Ex/Em = 490/520 nm with fluorescence microplate reader.
2.15. FAM‐FLICA caspase‐1 assay
To detect pyroptosis, the FAM‐FLICA caspase‐1 assay kit (Biozol/Immunochemistry; cat# ICT‐98) was used according to the manufacturer's protocol. Briefly, cells were grown in 12‐well plates at a density of 2 × 105 cells per well. Once IFN‐α stimulation and treatment were complete, cells were collected in FACS tubes and washed with 10x apoptosis wash buffer. FLICA was added to each sample at 1:30 dilution and samples were incubated for 1 h. Afterward, samples were washed and resuspended in wash buffer and analyzed using a BD™ LSR‐II flow cytometer (BD Biosciences, San Jose, CA).
2.16. LDH release assay
Cytotoxicity Detection KitPLUS (Roche, Mannheim, Germany) was used to assess LDH release as a measure of cytotoxicity. Cells were seeded in a 96‐well plate and after the completion of treatment, 5 µl of lysis solution was added and incubated for 15 min. Meanwhile catalyst and dye solution were mixed to make a reaction mixture, and 100 µl of this reaction mixture was added to each well containing supernatants and incubated for 30 min in the dark at room temperature. Last, 50 µl of stop solution was added to each well and after shaking for 10 s, OD was measured at 490 nm using an ELISA plate reader.
2.17. MTT assay
Yellow tetrazolium salt (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide, or MTT) kit (Life Technologies, Carlsbad, California) was used to measure cell respiration as a measure of cell viability according to the manufacturer's protocol. Briefly, cells were seeded in a 96‐well plate and after completion of the treatment 100 µl of MTT labelling reagent (final concentration 0.5 mg/ml) was added to the cells and incubated at 37°C for 60 min. MTT was removed from the wells and 100 µl of solubilization solution (DMSO) was added to each well and, once purple formazan crystals were formed and completely solubilized, the plate was measured at 540 nm wavelength using a spectrophotometer.
2.18. Microarray analysis
RNA was extracted using the RNeasy kit (Qiagen), RNA quality was checked with a Bioanalyzer 2100 (Agilent Technologies) and samples with RNA integrity number >7 were used for microarray analysis. Total RNA/Poly‐A RNA Control Mixture was prepared by adding poly‐A RNA controls supplied by Affymetrix. This poly‐A RNA was then used to prepare double stranded cDNA. In the next step, complimentary RNA (cRNA) was synthesized and amplified by in vitro transcription of the ds‐cDNA template using T7 RNA polymerase. Enzymes, salts, inorganic phosphates, and unincorporated nucleotides were removed to prepare the cRNA for 2nd‐cycle ds‐cDNA synthesis. The quality and yield of cRNA was assessed. Later, sense‐strand cDNA was synthesized by reverse transcription of cRNA. The cRNA template was hydrolysed leaving ds‐cDNA, which was then purified. cDNA was fragmented and then labelled by terminal deoxynucleotidyl transferase (TdT) using the Affymetrix proprietary biotin‐linked DNA Labelling Reagent. Cartridge Array Hybridization was conducted on the GeneChip® Instrument (Affymetrix) using Thermo Fisher Scientific microarrays (Clariom™ S Assay, human) [252‐254]. Files were analyzed for differential gene expression using the Transcriptome Analysis Console (TAC4.0.2) Software (ThermoFisher Scientific). Metaboanalyst software (https://www.metaboanalyst.ca/) was used for differential expression analysis. TAC files were also processed for functional enrichment analysis using WEB‐based GEne SeT AnaLysis Toolkit (Webgestalt, http://www.webgestalt.org/), defining enriched pathways as those having a false discovery rate (FDR) <.05. KEGG gene sets were downloaded from GSEA online portal. R‐Bioconductor package “fgsea” was used to pre‐rank the differentially expressed genes and annotate them against different pathways.
2.19. Targeted metabolomics by mass spectrometry
The mass spectrometric measurements for the targeted metabolomics screen were performed on an AB SCIEX 5500 QTrap™ mass spectrometer (AB SCIEX, Darmstadt, Germany) using the MxP® Quant500 kit (Biocrates Life Sciences, Innsbruck, Austria), following the manufacturer's protocols (https://biocrates.com/mxp‐quant‐500‐kit). 5 × 106 WT or ISG15 –/– dermal fibroblasts were used per sample and processed according to Biocrates application note 2001‐1, using extraction buffer containing 10 mM phosphate buffer/85% ethanol and adding three freeze‐thaw‐sonication cycles to maximize cell lysis. 34 Output files were generated by MetIDQ™ software (Biocrates Life Sciences, Innsbruck, Austria) and analyzed further using Metaboanalyst software (https://www.metaboanalyst.ca/).
2.20. 13C stable isotope assisted metabolic flux assay
Cells were seeded in 6‐well plates and 25 mM glucose (12C or 13C6) and 4 mM of glutamine (12C or 13C5) containing medium was added to each well. After IFN‐α stimulation, the cells were washed with 0.9% NaCl, and 400 µl of −20°C methanol (Chromasolve) was added to the cells. Subsequently, 400 µl of Millipore water containing 1 µg/ml D6 glutaric acid (internal standard) was added. Cells were scraped and the suspension was transferred to an Eppendorf tube containing 400 µl of −20°C chloroform (Chromasolve). The tubes were vortexed for 20 min at 4°C and then centrifuged for 5 min at 17 000 g. 250 µl of the polar (top) phase was transferred to a mass spec (MS) vial. Samples were dried using a refrigerated vacuum centrifuge set to −4°C. Before removing the vials, they were warmed to room temperature to avoid condensation. Samples were then analyzed by gas chromatography‐mass spectrometry (GC‐MS).
2.21. GC‐MS measurement
Metabolites were derivatized using an Axel Semrau Chronect Robotic Pal TRC. Dried polar metabolites were dissolved in 15 µl of 2% methoxyamine hydrochloride in pyridine at 55°C under shaking. After 90 min, an equal volume of N‐(tert‐butyldimethylsilyl)‐N‐methyltrifluoroacetamide (MTBSTFA, Sigma‐Aldrich) was added and held for 30 min at 55°C under continuous shaking. One microliter sample was injected into an SSL injector at 270°C in splitless mode. GC‐MS analysis was performed using an Agilent 7890A GC equipped with a 30‐m ZB‐35 + 5‐m Duraguard capillary column (Phenomenex). Helium was used as carrier gas at a flow rate of 1 ml/min. The GC oven temperature was held at 100°C for 2 min, increased to 300°C at 10°C/min, and finally held for 4 min at 300°C. The GC was connected to an Agilent 5975C inert XL MSD, operating under electron ionization at 70 eV. The MS source was held at 230°C and the quadrupole at 150°C. The detector was operated in selected ion monitoring. The total run time per sample was 26.00 min.
2.22. Mass isotopomer distribution analysis
All GC‐MS chromatograms were processed using MetaboliteDetector. 35 Chemical formulas for mass isotopomer distribution (MID) determination were taken from Reference. 36 Weighted carbon contribution was calculated with the following formula: 1/n∗ΣMi*i, where n is the number of carbons of the molecule of interest and Mi the ith mass isotopomer.
2.23. Sample preparation for IA measurements by HPLC‐MS/MS
iPSC‐derived macrophages were cultivated in 6‐well plates as described above and stimulated with IFN‐α for 24 h at 37°C and 5% CO2. Subsequently, the medium was aspirated and cells were washed once with 1xPBS. After carefully removing the supernatant, cells were extracted with 1000 µl of extraction reagent (methanol/acetonitrile/water; 2/2/1; v/v; spiked with 0.1 µM 13C2‐citrate and 13C5‐IA, 0.2 µM 13C6‐cis‐aconitate and 1 µM 13C3‐lactate as internal standards). The suspensions were transferred to 2 ml safe‐lock reaction tubes (0030120094 Eppendorf), vortexed for 30 s and frozen at −20°C overnight to complete protein precipitation. The subsequent sample preparation and HPLC‐MS/MS assay using a Kinetex C18 column (00D‐4462‐Y0, Phenomenex) on a Nexera chromatography system (Shimadzu, Japan) coupled to a QTRAP5500 triple quadrupole/linear ion trap mass spectrometer (Sciex, Framingham, MA, USA) was performed essentially as described in reference. 37
2.24. Influenza A virus infection
The 2009 pandemic field isolate (A/Giessen/06/09 [H1N1]) was used for viral infection. Briefly, macrophages were seeded at a density of 2 × 105 cells/well in 12‐well plates and incubated overnight at 37°C. The following day, cells were infected with influenza A virus at a multiplicity of infection (MOI) of 1. Cells were kept in DMEM media containing virus (without FCS) for 2 h and then washed with PBS to remove unbound virus and fresh medium containing all the treatments was added to the respective wells. Once the experiment was complete, RA1 lysis buffer was used to collect the cells.
2.25. Statistical analysis
All experiments featured 3–4 biological replicates. Data are expressed as means ± standard error of mean (SEM) unless stated otherwise. GraphPad Prism 8.0.2 was used for statistical analyses. One‐way ANOVA with Tukey's test to adjust for multiple testing was performed to assess significant difference of means among three or more groups. Student's t‐test was performed to assess significance of differences in means between two groups. P values <.05 were considered significant, using the following abbreviations: *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001.
DESeq2 tool (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) was used for differential expression analysis of bulk RNAseq data. Microarray data were analyzed using Transcriptome Analysis Console (TAC4.0.2) Software (ThermoFisher Scientific).
3. RESULTS
3.1. Type I IFN signature and hyperinflammation in ISG15 deficiency
WT and ISG15–/– macrophages were validated phenotypically and functionally in that they expressed CD45, CD11b, and CD14. In addition, they demonstrated phagocytic activity similar to M2‐type macrophages derived from human PBMC, which could be inhibited by cytochalasin‐D, a classic inhibitor of phagocytosis (Figure 1A–D).
FIGURE 1.
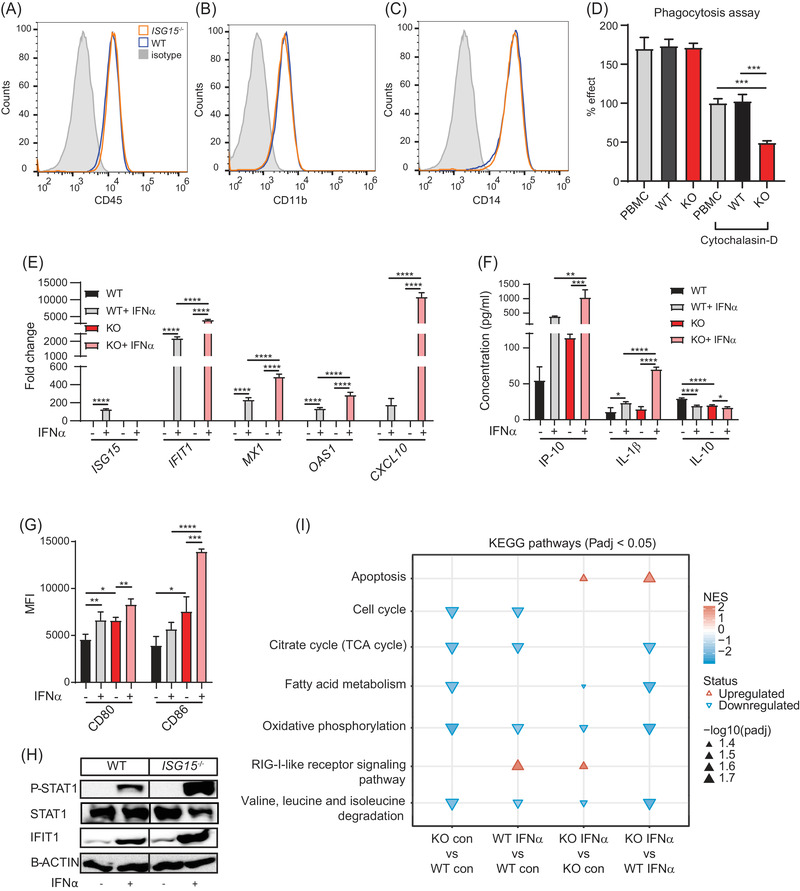
Phenotypic and functional characterization of iPSC‐derived macrophages. (A–C) Flow cytometry analysis demonstrating similar expression of CD45 (A) and the classic macrophage markers CD11b (B) and CD14 (C) on WT and ISG15–/– (KO) iPSC‐derived macrophages. (D) Phagocytosis assay demonstrating similar phagocytic activity of human PBMC and WT and ISG15–/– iPSC‐derived macrophages. The phagocytosis inhibitor cytochalasin‐D was added as positive control. 100% = cytochalasin‐D treated PBMC. (E‐H) Pro‐inflammatory phenotype of ISG15–/– iPSC‐derived macrophages. WT and ISG15–/– macrophages were stimulated with 1000 IU/ml of IFN‐α for 24 h as indicated on the x‐axes. (E) Increased expression of ISG15, IFIT1, MX1, OAS1, and CXCL10 mRNA in ISG15–/– cells after IFN‐α stimulation (RT‐qPCR). (F) Higher concentrations of IP‐10 and IL‐1β, but lower concentrations of IL‐10 in cell culture supernatants of ISG15–/– cells (ELISA). (G) Higher cell surface expression of CD80 and CD86 on ISG15–/– cells with and without IFN‐α stimulation (flow cytometry; MFI, mean fluorescent intensity). (H) Higher levels of phosphorylated STAT1 (P‐STAT1) and IFIT1 in ISG15–/– cells after IFN‐α stimulation (western blot of cell lysates). (I) Gene set enrichment analysis based on KEGG pathways (FDR < .05) for the pairwise comparisons indicated below the figure. Selection of the most relevant pathways in the enrichment analysis is shown in Figure S5E. The normalized enrichment score (NES) is calculated by ranking gene expression differences and normalizing for size of each gene set. Significance of enrichment/depletion is indicated by the size of the triangles. Abbreviations: con, unstimulated; IFNα, IFNα stimulated. *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
Increased IFN‐I expression is a cardinal sign of ISG15 deficiency. 9 To verify the presence of an IFN‐I signature and associated hyperinflammation in iPSC‐derived macrophages, we stimulated ISG15–/– and WT cells with IFN‐α2b. Indeed, expression of ISG mRNAs ISG15, IFIT1, MX1, OAS1, and CXCL10 was higher in ISG15–/– than WT macrophages (Figure 1E and Figure S1A). Also, in cell culture supernatants of ISG15–/– cells there were higher levels of the CXCL10 gene product, IP10 (a pro‐inflammatory chemokine), and the classic pro‐inflammatory cytokine IL‐1β, whereas the anti‐inflammatory cytokine IL‐10 was down‐regulated (Figure 1F). Moreover, expression of the pro‐inflammatory surface markers CD80 and CD86 was higher on unstimulated and IFN‐α stimulated ISG15–/– cells, and intracellular levels of phosphorylated STAT1 (P‐STAT1 = activated STAT1) and IFIT1 proteins (Figure 1 G,H) were higher in stimulated ISG15–/– cells. Thus, the ISG15–/– iPSC‐derived macrophages possessed the strong IFN‐I signature which is characteristic of patients affected by ISG15 deficiency. The ISG15–/– macrophages also demonstrated a more general hyperinflammatory phenotype in that the induction of IFIT1 and CXCL10 mRNA and IL‐1β protein by LPS was significantly stronger than in WT cells, even though the absolute degree of induction of IFIT1 and CXCL10 mRNA was considerably less in both WT and ISG15–/– than after IFN‐α stimulation (Figure S1C–E).
Additionally, we assessed inflammation in iPSC‐derived ECs (Figure S2), the transformed keratinocyte cell line HaCaT (Figure S3), and immortalized dermal fibroblasts (Figure S4) and found the expected hyperinflammatory phenotype of ISG15–/– cells in all cases.
3.2. Transcriptomic profiling and functional enrichment analysis reveals additional phenotypic features of ISG15–/– cells
We then used transcriptome profiling to search for additional phenotypic differences between ISG15–/– and WT macrophages, both at the bulk and single‐cell level. Principle component analysis (PCA) of transcriptomes obtained by bulk RNAseq revealed clear differences between ISG15–/– and WT cells both with and without IFN‐α stimulation (Figure S5B). Expression of the enzyme that catalyses IA synthesis during macrophage activation (ACOD1) is IFN‐inducible. 38 In agreement with this, the RNAseq analysis revealed that IFN‐α stimulation results in higher levels of ACOD1 mRNA in ISG15–/– macrophages, which was corroborated by higher intracellular IA concentration (Figure S5C,D).
Considering their key roles in regulating gene expression, we analyzed the miRNA data obtained by small RNA sequencing of the same samples. Indeed, a PCA showed distinct miRNA expression patterns in ISG15–/– and WT macrophages with and without IFN‐α stimulation (Figure S6A). PCA of the mRNA microarray data from iPSC‐derived ECs also showed distinct expression patterns in ISG15–/– and WT cells with and without IFN‐α stimulation (Figure S7A). Thus, loss of ISG15 led to distinct transcriptome patterns in both macrophages and ECs, and a distinct ISG15–/– transcriptomic phenotype was apparent even without IFN‐α stimulation.
We then used gene set enrichment analysis (GSEA) based on KEGG pathways to identify pathways whose dysregulation might contribute to the ISG15–/– phenotype. Compared to IFN‐α stimulated WT macrophages, IFN‐α stimulated ISG15–/– macrophages featured increased apoptosis, but depletion of cell cycle, fatty acid metabolism, oxidative phosphorylation, PPAR signalling, purine metabolism, pyrimidine metabolism, pyruvate metabolism, TCA cycle, and valine, leucine and isoleucine degradation (Figure S5E and Figure 1I). Increased IFN‐I signalling was not identified in this enrichment analysis, but it was clearly evident in a dedicated differential expression analysis (see Figure S1A for the IFN‐I related differentially expressed genes). The miRNA expression patterns of the iPSC‐derived ISG15–/– macrophages revealed enrichment of the PI3K‐AKT, FoxO, and MAPK signalling pathways along with various cancer‐related pathways, whereby PI3K‐AKT and FoxO signalling pathways were most closely associated with ISG15 deficiency, as they were enriched in ISG15–/– cells both with and without IFN‐α stimulation (Figure S6D–G).
GSEA of microarray data from the iPSC‐derived ECs revealed interferon‐driven and pro‐inflammatory pathways including toll‐like receptor signalling, TNF signalling, and RIG‐I‐like signalling among the most significantly enriched in IFN‐α stimulated ISG15–/– ECs compared to IFN‐α stimulated WT cells, and also some evidence of depletion of energy metabolism and amino acid biosynthesis (Figure S7B).
Taken together, this GSEA suggested that apart from the expected hyperinflammation and IFN‐I signature, increased apoptosis, but decreased PI3K‐AKT signalling, energy generation, nucleotide synthesis, and branched chain amino acid (BCAA) catabolism were cardinal cellular features of ISG15 deficiency and confirmed that a distinct phenotype was apparent without IFN‐α stimulation at least in the ISG15 –/– macrophages.
3.3. Increased ROS production and reduced AKT phosphorylation modulate GSK3β signalling and apoptosis‐related genes
Considering the increased inflammation and suggestions of mitochondrial dysfunction in ISG15–/– cells seen in the GSEA, we then measured mitochondrial ROS. Indeed, ROS levels were higher in ISG15–/– macrophages both before and after IFN‐α stimulation (Figure 2A,B). Since ROS modulate activity of the PI3K‐AKT pathway 39 and depletion of PI3K activity was seen in the GSEA, we measured AKT phosphorylation (P‐AKT = activated AKT). Indeed, P‐AKT levels were lower in IFN‐α stimulated and, less so, unstimulated ISG15–/– macrophages (Figure 2C). AKT inactivates glycogen synthase kinase (GSK3β) by phosphorylation. 40 It was therefore plausible that the decreased AKT phosphorylation activated GSK3β signalling. Indeed, expression of GSK3β mRNA was higher in IFN‐α stimulated ISG15–/– than WT cells, and GSK3β protein expression was higher in ISG15–/– cells also without IFN‐α stimulation (Figure 2D,E). GSK3β activity affects expression of pro‐ and anti‐apoptotic genes. 41 Consistent with the observed increased GSK3β levels, the ISG15–/– macrophages featured higher transcription of BAX (a pro‐apoptotic gene) but lower transcription of the anti‐apoptotic gene BCL2 (Figure 2E).
FIGURE 2.
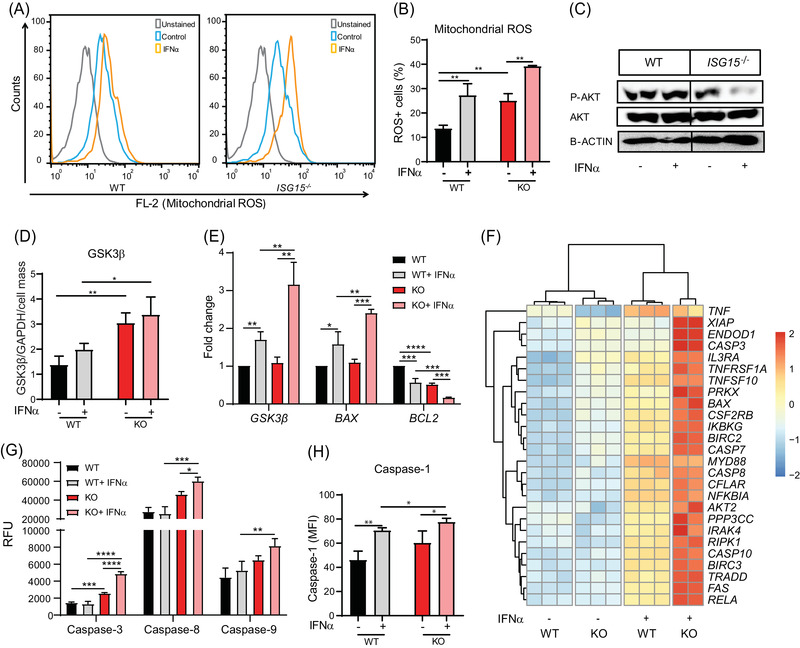
Increased mitochondrial ROS production, reduced AKT phosphorylation, and induction of GSK3β, apoptotic genes, apoptosis, and pyroptosis in ISG15–/– iPSC‐derived macrophages. WT and ISG15–/– macrophages were stimulated with 1000 IU/ml of IFN‐α for 24 h as indicated on the x‐axes. (A,B) Mitochondrial ROS levels are higher in ISG15–/– than in WT macrophages with and without IFN‐α stimulation (flow cytometry). (C) Lower levels of phosphorylated (P‐AKT), but not unphosphorylated, AKT in lysates of IFN‐α stimulated ISG15–/– cells (western blot). (D) Increased levels of GSK3β protein in unstimulated and IFN‐α stimulated ISG15–/– cells (in‐cell ELISA normalized against GAPDH and crystal violet staining). (E) Increased expression of GSK3β mRNA and BAX but lower expression of BCL2 mRNA in IFN‐α stimulated ISG15–/– cells (RT‐qPCR). (F) Hierarchical clustering analysis based on differentially expressed genes (FDR < .05) as defined by KEGG pathway apoptosis (RNA sequencing). (G,H) Increased activity of caspases 3, 8, 9, and 1 in IFN‐α stimulated ISG15–/– cells: (G) fluorometry (RFU, relative fluorescent units); (H) flow cytometry (MFI, mean fluorescent intensity). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.4. Transcriptomic signatures reveal increased oxidative stress and apoptosis in ISG15–/– macrophages
In order to understand the underlying mechanism(s) of the apparent pro‐apoptotic phenotype of the ISG15–/– macrophages, we analyzed transcriptomes of the IFN‐α stimulated ISG15–/– and WT macrophages further. Specifically, ISG15–/– macrophages exhibited higher expression of CASP3, CASP7, CASP8, CASP10, BAX, RELA, and FAS mRNA (all of which are functionally related to oxidative stress and apoptosis) as well as, for instance, higher expression of the TNF superfamily and NFκB‐related factors (Figure 2F). We then measured the activity of initiator caspases, CASP‐8 and CASP‐9, and the effector caspase, CASP‐3, via fluorometric assay. Indeed, activity of all three was higher in ISG15–/– cells, thus confirming their pro‐apoptotic phenotype (Figure 2G).
Expression of CASP1, which induces pyroptotic cell death, 42 , 43 was higher in IFN‐α stimulated ISG15–/– macrophages (Figure 2H). This also explained the higher levels of IL‐1β seen in Figure 1F, as this cytokine is released by cleaved CASP1 and CASP3. Activated CASP1 also cleaves gasdermin‐D (GSDMD), which forms membrane pores resulting in pyroptosis. 44 Increased expression of GSDMD mRNA was evident in ISG15–/– macrophages (Figure S1B). Consistently, ISG15–/– cells exhibited elevated CASP1 activity after LPS stimulation (Figure S1F). Together, these findings suggested that increased oxidative stress and propensity for apoptosis and pyroptosis are central features of the cellular phenotype of ISG15 deficiency.
3.5. ISG15 deficiency reduces mitochondrial respiration and expression of mitochondrial genes
The enrichment analysis had suggested reduced oxidative phosphorylation and citrate cycle (TCA cycle) activity in ISG15–/– cells (Figure 1I and Figure S5E). Moreover, mitochondrial ROS production may reflect dysfunction of the respiratory chain. We therefore used the Seahorse XF extracellular flux analyser to compare oxidative phosphorylation capacity between ISG15–/– and WT macrophages. Indeed, mitochondrial respiration was lower in ISG15–/– than WT macrophages (Figure 3A,B). In agreement with this, intracellular ATP levels were lower in ISG15–/– cells (Figure 3G). The elevated extracellular ATP levels in ISG15–/– cells (Figure 3G) were consistent with their propensity for increased apoptosis/pyroptosis shown above (Figure 2 G,H). In addition, fatty acid oxidation was lower in ISG15–/– macrophages (Figure 3C,D), which agreed well with depletion of fatty acid oxidation seen in the GSEA (Figure S5E). We therefore analyzed the acylcarnitine profiles which were available in a targeted metabolomic data set from the WT and ISG15–/– fibroblasts (Figure S8). Indeed, acylcarnitine profiles in WT and ISG15–/– cells were distinct. This was mostly due to higher concentrations in the ISG15–/– cells, indicating a general back‐up of the pathway. In order to assess alternative energy production pathways, we also measured glycolytic activity but found that IFN‐α stimulated ISG15–/– macrophages featured higher glycolysis (Figure 3E,F). To test whether ISG15 is required for optimal energy metabolism in a different cell type and whether this requires its ISGylation function, we assessed mitochondrial respiration in immortalized ISG15–/– fibroblasts and in ISG15–/– fibroblasts stably expressing WT or conjugation‐deficient (ΔGG) ISG15 (Figure S9A,B). As in the iPSC‐derived macrophages, OCR was markedly lower in the ISG15–/– cells. Of note, transduction of ISG15–/– cells with the WT allele led to complete rescue of OCR, whereas intermediate OCR was measured in the ISG15–/– cells expressing the ΔGG mutant. In contrast, expression of the IFN‐regulated mRNAs ACOD1, IFIT1, and CXCL10 normalized to the same extent in cells transduced with WT or ΔGG ISG15 (Figure S9C–E), which confirmed previous findings that up‐regulation of these IFN‐driven genes in ISG15 deficiency results from loss of USP18‐mediated inhibition of IFNAR signalling rather than defective ISG15 conjugation. 12
FIGURE 3.
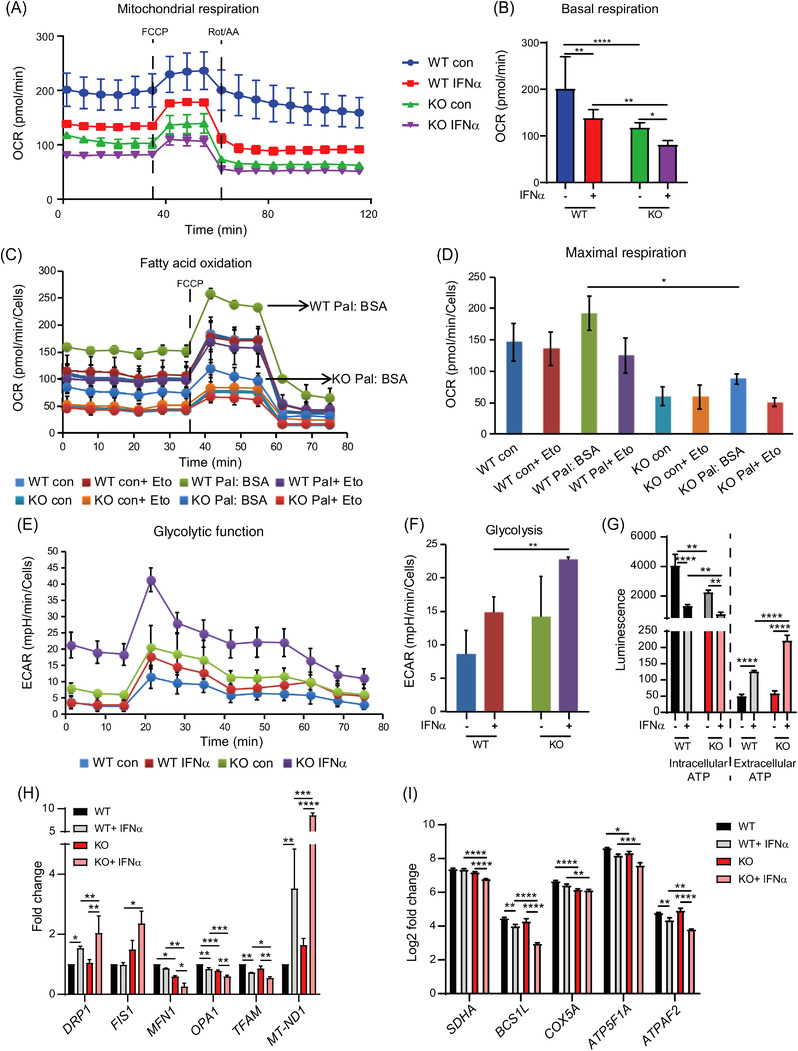
Reduced mitochondrial respiration, fatty acid oxidation (FAO), and increased glycolysis in ISG15–/– cells. WT and ISG15–/– macrophages were stimulated with IFN‐α (1000 IU/ml) and oxygen consumption rate (OCR), extracellular acidification rate (ECAR), ATP levels, and expression of selected genes were measured after 24 h. (A,B) OCR is lower in ISG15–/– than in WT macrophages and decreases in both cell types after IFN‐α stimulation (Seahorse XF Extracellular Flux Analyzer using XF Cell Mito Stress Test). (C,D) Differences in FAO between ISG15–/– and WT cells (Seahorse XF Extracellular Flux Analyzer, XF Cell Mito Stress Test). The FAO inhibitor etomoxir (Eto) was added to selected groups 15 min prior to starting the XF assay. Palmitate:BSA or Control:BSA was added to selected groups at start of the XF assay to quantify FAO by measuring the resulting change in OCR. OCR was higher in the WT Pal:BSA group than in the KO Pal:BSA group, indicating lower FAO in the ISG15–/– cells (marked by arrows). (E,F) Higher ECAR in ISG15–/– than in WT cells after IFN‐α stimulation, indicating higher glycolysis (Seahorse XF Extracellular Flux Analyser, Glycolysis Stress Test). (G) Lower intracellular but higher extracellular ATP levels in IFN‐α stimulated ISG15–/– cells (luciferase assay). (H) IFN‐α stimulated ISG15–/– macrophages feature higher expression of mitochondrial fission genes but lower expression of mitochondrial fusion genes (RT‐qPCR). The mitochondrial biogenesis gene TFAM is downregulated more in the ISG15–/– than the WT cell. Expression of the mitochondrial ROS‐responsive gene Mt‐ND1 (a component of respiratory chain complex I) is markedly higher in IFN‐α stimulated ISG15–/– than in WT cells. (I) Expression of respiratory chain complex II‐V genes is lower in unstimulated and stimulated ISG15–/– than WT cells (RNA sequencing). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
These results suggest that some, but not all, functions of ISG15 in mitochondrial respiration require its conjugation to cellular targets. In search for potential mechanisms relating to metabolic fluxes, we measured the uptake rate of glucose and glutamine in combination with stable‐isotope‐assisted metabolomics in dermal fibroblasts. Uptake of glutamine (but not glucose) was lower in ISG15–/– fibroblasts than in WT cells (Figure S10A,B). In line with these results, glutamine contributed less carbon to TCA cycle intermediates in ISG15–/– fibroblasts than in WT cells, while the contribution from glucose increased (Figure S10C,D). The latter could, however, only partially compensate for the decreased contribution by glutamine, as the absolute increase in carbon contribution from glucose was very small (compare relative carbon contributions in c and d). Reduced glutamine uptake, leading to lower glutamine oxidation, could therefore contribute to the mitochondrial dysfunction in ISG15–/– cells. In addition, it may contribute to the reduced purine and pyrimidine metabolism seen in the GSEA, as glutamine is a precursor of both.
High ROS levels can affect mitochondrial biogenesis, and genes associated with mitochondrial fusion and fission might therefore be affected. 45 Indeed, expression of the fission‐associated genes DRP1 and FIS1 was increased in IFN‐α stimulated ISG15–/– macrophages compared to the WT cells (Figure 3H). Additionally, the mitochondrial fusion genes MFN1 and OPA1 were downregulated in unstimulated and stimulated ISG15–/– macrophages and there was also reduced expression of TFAM in stimulated ISG15–/– macrophages, one of the most important regulators of mitochondrial biogenesis 46 (Figure 3H). In addition, expression of the mitochondrial gene Mt‐ND1, which encodes a subunit of respiratory chain complex I and is induced in response to elevated ROS, 47 was higher in ISG15–/– macrophages after IFN‐α stimulation, thus confirming the innately higher oxidative stress level in these cells. Moreover, expression of nuclear genes encoding components of mitochondrial respiratory chain complexes II‐V was also lower in IFN‐α stimulated ISG15–/– macrophages, suggesting that the observed defect in mitochondrial respiration may, at least in part, be due to reduced levels of these components (Figure 3I).
Analysis of the macrophage miRNA data further supported the above findings: miR‐98, which has anti‐apoptotic and anti‐oxidative stress functions, 48 was down‐regulated in IFN‐α stimulated ISG15–/– macrophages, whereas expression of miR‐302b, which is induced by oxidative stress, 49 was higher in ISG15–/– than in WT macrophages (Figure S6B,C).
Taken together, the above results suggest that a heterogeneous mitochondrial dysfunction is a cardinal feature of the cellular phenotype of ISG15 deficiency.
3.6. Reduced branched‐chain amino acid amino transferase 1 (BCAT1) expression and branched chain amino acid (BCAA) metabolism in ISG15 deficient cells
BCAT1 is responsible for catabolism of BCAA, which results in formation of branched chain α‐ketoacids (BCKA) and glutamate. 50 After transamination, BCKA are converted to acetyl‐CoA, which is later oxidized in the TCA cycle. It has been reported that higher BCAT1 expression correlates with reduced oxidative stress. 51 Indeed, the macrophage RNAseq data revealed reduced levels of BCAT1 mRNA (Figure 4A,B) and also of mRNA encoding branched‐chain α‐ketoacid dehydrogenase kinase (BCKDK), another key enzyme in BCAA catabolism 52 (Figure 4C). We verified mRNA and protein expression of BCAT1 in an independent experiment and, indeed, its expression was markedly reduced in ISG15–/– macrophages (Figure 4D,E). Analysis of amino acid concentrations in the targeted metabolomics data set from ISG15–/– and WT dermal fibroblasts indicated a tendency toward lower BCAA catabolism, as reflected by higher concentrations of valine, leucine, and isoleucine in ISG15–/– cells (Figure 4F). These findings suggest that a relative BCAT1 deficiency contributes to the observed reduced mitochondrial respiration in ISG15–/– cells after IFN‐α stimulation.
FIGURE 4.
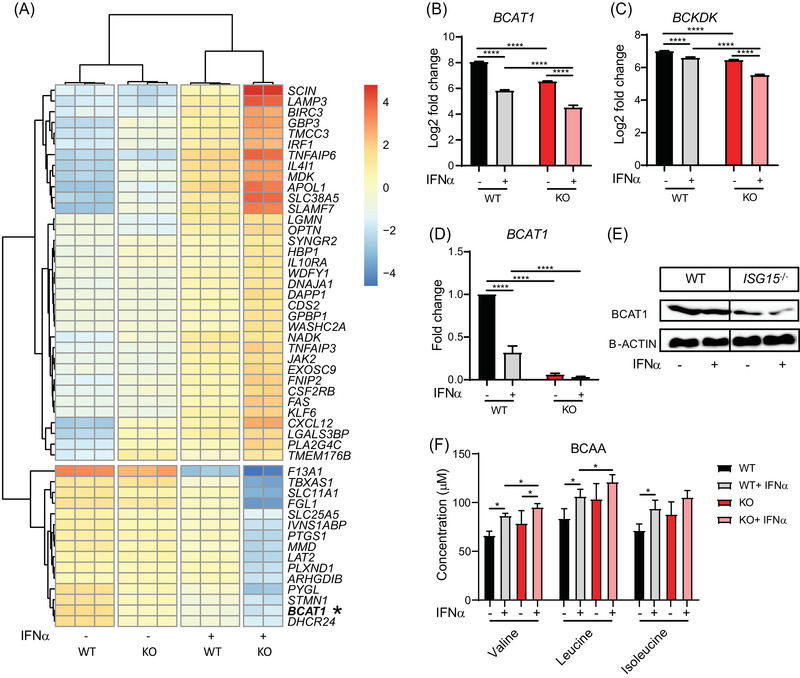
Reduced BCAT1 and BCKDK expression and branched chain amino acid (BCAA) catabolism in ISG15–/– cells. (A–E) ISG15–/– and WT macrophages were stimulated with IFN‐α (1000 IU/ml) for 24 h as indicated on the x‐axes. (A) Hierarchical biclustering analysis of the 50 most significantly differentially expressed genes across all samples (RNA sequencing). (B,C) Lower expression of BCAT1 and BCKDK mRNA in unstimulated and stimulated ISG15–/– than WT cells (RNA sequencing). (D,E) Lower expression of BCAT1 mRNA (RT‐qPCR) and protein (western blot) in stimulated and unstimulated ISG15–/– than WT cells. (F) BCAA analysis. WT and ISG15–/– dermal fibroblasts were grown in the presence or absence of IFN‐α (1000 IU/ml), and concentrations of the indicated BCAA were measured by mass spectrometry after 24 h. Valine and leucine concentrations are significantly higher in IFN‐α stimulated ISG15–/– than WT cells, and there is a tendency for increase in isoleucine. *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.7. Further validation of the ISG15–/– phenotype by analysis of gene expression in iPSC‐derived EC
The phenotypic features of iPSC‐derived macrophages were further verified by microarray analysis of iPSC‐derived EC. As in the macrophages, the greatest degree of transcriptional change (compared to unstimulated WT cells) was detected in IFN‐α stimulated ISG15–/– cells (Figure S11A–C). Specifically, there was upregulation of genes related to type I IFN signature (e.g., IFIT2, MX2, IFI27 and ISG20), hyperinflammation (e.g., TLR3, CXCL10 and CXCL11) (Figure S11D), and downregulation of genes related to PI3K‐AKT signalling (AKT1) and ROS scavenging (HMOX1, NQO1) (Figure S11E) in IFN‐α stimulated ISG15–/– cells as compared to WT cells. Additionally, there was higher expression of apoptosis associated genes (CASP9, CASP1, CASP3, CASP7, CASP10, CASP8, TNFSF10, and FAS) and the pyroptosis mediator GSDMD (Figure S11D,F), but reduced expression of mitochondrial biogenesis genes (MFN1, OPA1) and BCAT1 in IFN‐α stimulated ISG15–/– EC as compared to WT cells (Figure S11E). These results underscored the importance of ISG15 to regulate oxidative stress and apoptosis in different cell types and underscored the shared features of ISG15 deficiency between macrophages and EC.
3.8. IA and derivatives reduce type I IFN signature, hyperinflammation, and ROS production
In order to evaluate IA and derivatives as treatments for the dysfunctional phenotype of ISG15–/– cells, we determined nontoxic doses of IA (10 mM) and DI (.5 mM) (Figure S12A,B), while using concentrations of 4OI (25 µM) and RUX (1 µM) that are generally known to be nontoxic. RUX is a well‐characterized JAK1/2 inhibitor that reduces type I IFN responses 53 and we used it as positive control and comparator for the relative effectiveness of the IA compounds. Of note, when these concentrations were verified to be nontoxic in the MTT assay, less formazan precipitate formation (indicating lower NAD(P)H‐dependent oxidoreductase activity) was obtained with untreated ISG15–/– than with untreated WT macrophages, which was reduced further by IFN‐α (Figure S12C). This suggested that loss of ISG15 compromises not only mitochondrial respiration (see Figure 3A) but also extramitochondrial NAD(P)H availability. The latter may be in part due to depressed synthesis of its adenine component, as suggested by depletion of purine synthesis in the GSEA of WT and ISG15–/– macrophages shown in Figure S5A, and it may also relate to depletion of pyruvate metabolism (in which NADP+ is a cofactor) in the same analysis. Of note, the treatments brought the MTT signal of IFN‐α stimulated ISG15–/– cells up to the level of unstimulated ISG15–/– cells, but not up to the level of WT cells. In contrast, when unstimulated ISG15–/– macrophages were treated, the MTT signal increased to near WT levels, indicating that the compounds could rectify oxidoreductase activity in these cells when there was no additional IFN‐signalling (Figure S12D). These non‐toxic doses were then used to test effects of the compounds on hyperinflammation in IFN‐α stimulated ISG15–/– macrophages. Treatment with all four compounds led to significantly reduced expression of IFIT1, DDX58, IFIH1, and CXCL10 mRNA and IP‐10 and IL‐1β protein (Figure 5A–F), but to increased levels of the predominantly anti‐inflammatory IL‐10 (Figure 5G). Likewise, all four compounds reduced the expression of P‐STAT1 and IFIT1 (Figure 5H). A normalization of IFN‐I responses by IA and 4OI was also verified in single‐cell transcriptomes (Figure S13A–C). There was a comparable reduction of IFN‐I responses and hyperinflammation in ECs (Figure S13D–H), HaCaT cells (Figure S14A–E) and fibroblasts (Figure S14F–I). The treatment effect was also evident in LPS‐stimulated macrophages, but RUX was not as effective as the itaconates, which is consistent with the lesser induction of IFN‐I signalling by LPS than by IFN‐α (Figure S15). Treatment with all four compounds also reduced mitochondrial ROS production in ISG15–/– macrophages to a comparable extent (Figure 5I).
FIGURE 5.
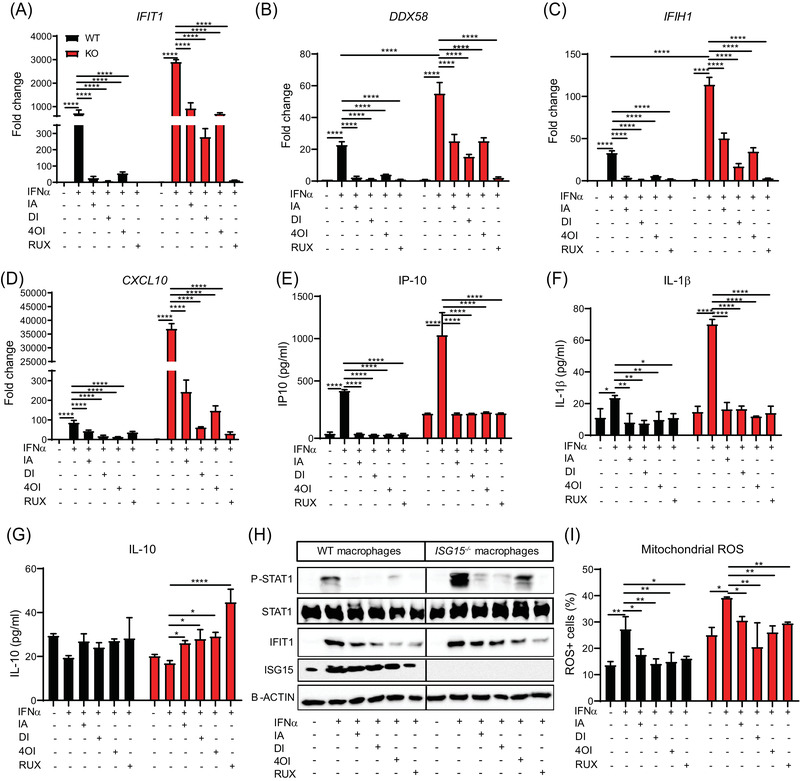
Treatment with itaconate and derivatives reduces type I IFN signature, hyperinflammation, and ROS production in ISG15–/– iPSC‐derived macrophages. WT and ISG15–/– macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (IA, 10 mM), DI (0.5 mM), 4OI (25 µM) and RUX (1 µM) for 16 h. (A–G) Treatment‐induced reduction of the indicated ISGs and proinflammatory cytokines, but enhancement of IL‐10 levels: (A–D) RT‐qPCR; (E–G) ELISA of cell culture supernatants. (H) Treatment‐induced reduction of phosphorylated STAT1 (P‐STAT1) and IFIT1 expression in IFN‐α stimulated ISG15–/– and WT macrophages (western blot of cell lysates). The blot also demonstrates the absence of ISG15 protein in the ISG15–/– cells. (I) Treatment‐induced reduction of mitochondrial ROS+ cells (flow cytometry). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.9. IA and derivatives induce AKT phosphorylation and reduce GSK3β expression
The results shown in Figure 2C had suggested reduced AKT phosphorylation in ISG15–/– macrophages, which correlated with increased ROS levels. Consistent with their anti‐ROS activity shown above, the treatments increased AKT phosphorylation in the ISG15–/– macrophages (Figure 6A). Likewise, they reduced levels of GSK3β mRNA and protein (Figure 6B,C) and BAX mRNA expression, but increased BCL2 expression (Figure 6D,E). Thus, IA and derivatives reprogram the AKT/GSK3 signalling pathway to reduce levels of pro‐apoptotic mediators.
FIGURE 6.
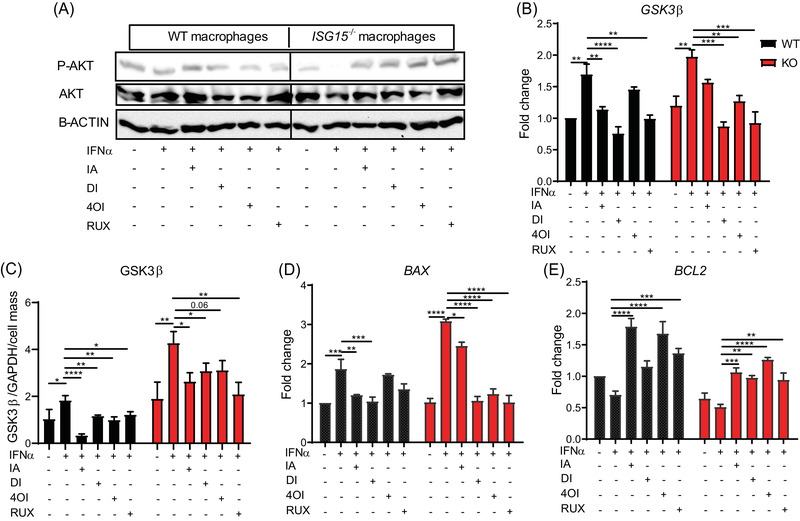
Treatment with itaconate and derivatives induces AKT phosphorylation and reduces GSK3β signalling and apoptotic genes in ISG15–/– macrophages. WT and ISG15–/– macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and then treated with itaconate (IA, 10 mM), DI (0.5 mM), 4OI (25 µM), and RUX (1 µM) for 16 h. (A–E) The treatments increase phosphorylated AKT levels in IFN‐α stimulated ISG15–/– cells, but decrease GSK3β mRNA and protein levels, as well as BAX mRNA, whereas BCL2 mRNA expression increases upon treatment: (A) western blot of cell lysates; (B,D,E) RT‐qPCR; (C) ELISA of cell lysates normalized to GAPDH and cell number as determined by crystal violet staining. *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.10. IA and derivatives reduce apoptosis and pyroptosis
Considering the downregulation of GSK3β and BAX expression in ISG15–/– macrophages treated with IA and derivatives, we analyzed the single cell‐RNA sequencing data of iPSC‐derived macrophages under treatment with the four compounds for expression of genes involved in apoptosis. In agreement with the bulk RNAseq data (Figure 2F) and microarray analysis of iPSC‐derived EC (Figure S11F), there was strong upregulation of, for example, CASP3, CASP7, CASP8, CASP10, FAS, BAX, RELA and TNFSF10 in ISG15–/– macrophages stimulated with IFN‐α, which was effectively reversed by the treatments (Figure 7A). Additionally, we found in the microarray data of ECs that the pro‐apoptotic differentially expressed genes (CASP9, CASP1, CASP3, CASP7, CASP10 and CASP8) were upregulated by IFN‐α in ISG15–/– cells, but were effectively downregulated by the treatments (Figure S11F). To confirm these findings, we also measured caspase 3, 8, and 9 activity. Indeed, caspase 3, 8, and 9 activity increased in ISG15–/– macrophages after stimulation with IFN‐α, which was reduced by the treatments (Figure 7B–D). Moreover, increased caspase 1 activity in ISG15–/– macrophages stimulated with IFN‐α was reduced by treatment with the itaconates (Figure 7E). Consistent with this, the microarray data of ECs revealed that the treatments could at least partially reverse the strong induction of GSDMD in ISG15–/– ECs (Figure S11D).
FIGURE 7.
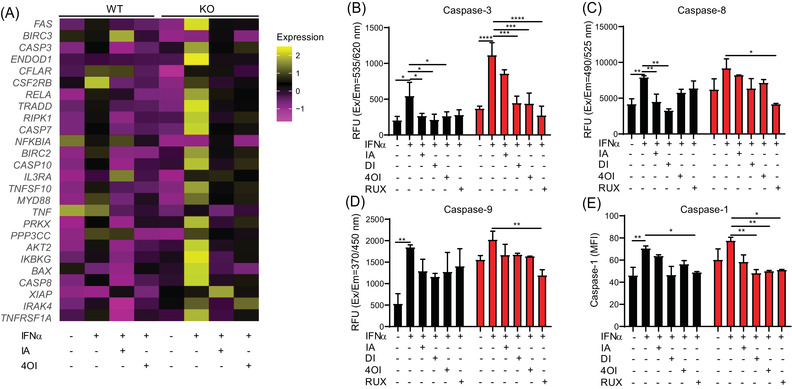
Reduction of apoptosis and pyroptosis in ISG15–/– iPSC‐derived macrophages treated with itaconate and derivatives. WT and ISG15–/– macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (IA, 10 mM), DI (0.5 mM), 4OI (25 µM) and RUX (1 µM) for 16 h. (A) The heatmap shows relative expression of differentially expressed (FDR < .05) genes in the KEGG pathway apoptosis (single cell‐RNA sequencing). The pro‐apoptotic signature of the ISG15–/– cells is effectively reduced by treatment with itaconate and 4OI. (B–E) Reduction of caspase 3, 8, 9, and 1 activity by the treatments: (B–D) fluorometry (RFU, relative fluorescent units); (E) flow cytometry (MFI, mean fluorescent intensity). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.11. Impact of itaconates on the dysfunctional mitochondrial phenotype
We had observed lower mitochondrial respiration in ISG15–/– than in WT macrophages (Figure 3A), but only a partial rescue of cellular oxidoreductase activity (MTT assay) by all 4 compounds (Figure S12C,D). Nonetheless, the compounds reduced expression of the mitochondrial fission genes DRP1 and FIS1 and increased expression of the fusion genes MFN1 and OPA1 (Figure 8A). Also, the treatments increased relative expression of TFAM but they decreased Mt‐ND1 expression (Figure 8A,B). In addition, expression of the mitochondrial biogenesis genes MFN1 and OPA1 was reduced in ISG15–/– ECs stimulated with IFN‐α, which was reversed by the treatments (Figure S11E). Interrogation of the bulk RNAseq data had shown that expression of 5 genes encoding respiratory electron chain components was reduced in ISG15–/– macrophages (Figure 3I). We measured their expression by RT‐qPCR in an independent treatment experiment (Figure 8B). This verified their downregulation in ISG15–/– macrophages and revealed that itaconate treatments increased their expression. The catalytic subunit of SDH (SDHA), of which itaconate is a competitive inhibitor, 54 formed a notable exception in that IA and DI did not affect its expression significantly. We also observed rescue of mitochondrial respiration by RUX and 4OI in WT cells but only partial rescue in ISG15–/– cells (Figure 8C–F). Of note, only 4OI and RUX corrected the defect in mitochondrial respiration, whereas addition of IA even led to a complete loss of reserve respiratory capacity in WT and ISG15–/– cells (Figure 8C–F; green lines in C and E), likely due to the additional inhibition of SDH activity. Nonetheless, all four compounds corrected the reduced intracellular and elevated extracellular ATP levels seen in ISG15–/– cells, demonstrating that their positive impact on global cell energy balance is substantial (Figure 8G,H). Together with the results of the MTT assay (Figure S12C,D), these results suggest that stimulation of mitochondrial biogenesis and improved mitochondrial and cytosolic energy homeostasis are possible mechanisms by which itaconates and RUX provide at least a partial rescue of cell respiration.
FIGURE 8.
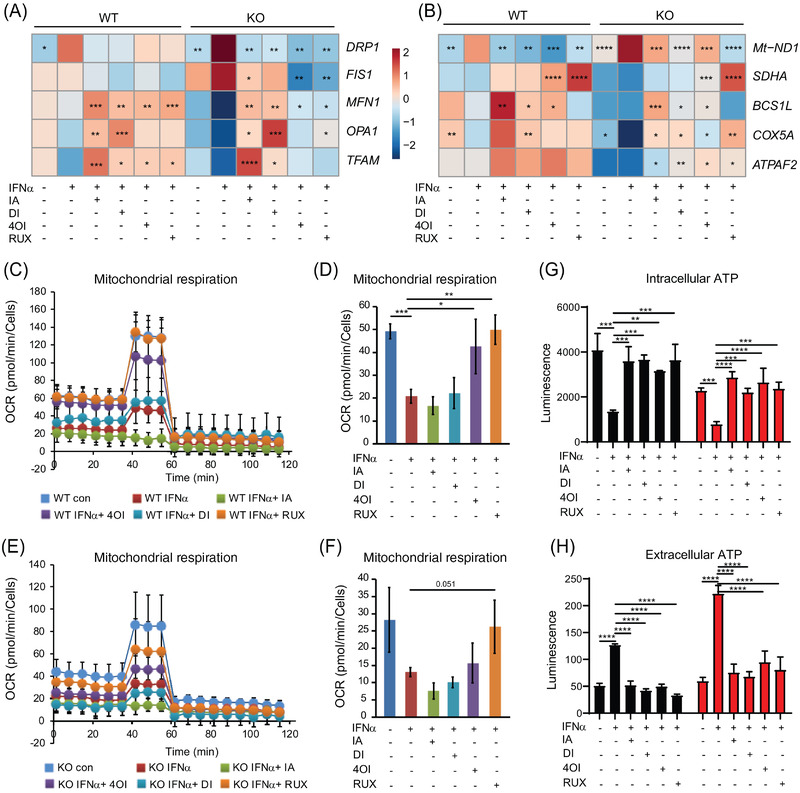
Rescue of the mitochondrial phenotype by treatment with itaconates and RUX. WT and ISG15–/– iPSC‐derived macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (IA, 10 mM), DI (0.5 mM), and 4OI (25 µM) for 16 h. (A) Expression changes of mitochondrial biogenesis genes upon treatment (RT‐qPCR). Treatments reduce expression of mitochondrial fission genes DRP1 and FIS1, increase expression of fusion genes MFN1 and OPA1 and mitochondrial biogenesis gene TFAM. (B) Expression changes of respiratory chain complex I–V genes upon treatment (RT‐qPCR). Treatments decrease expression of ROS‐responsive gene Mt‐ND1 (component of complex I) but increase expression of respiratory chain complex II–V genes. Statistical significance was determined with respect to the untreated, IFN‐α stimulated WT or ISG15–/– group. (C–F) OCR is restored by treatment with RUX and 4OI in IFN‐α stimulated WT cells (C,D), whereas partial rescue of OCR is observed by treatment with RUX and 4OI in IFN‐α stimulated ISG15–/– cells (E,F) (Seahorse XF Extracellular Flux Analyzer, XF Cell Mito Stress Test). Seahorse XF extracellular flux analyser using XF cell mito stress test) (G,H) Treatment with all four compounds rectifies abnormal intra‐ and extracellular ATP levels in IFN‐α stimulated ISG15–/– cells (luciferase assay). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
3.12. Treatment with itaconates enhances BCAT1 expression and BCAA catabolism and improves cellular redox balance in ISG15–/– cells
We had observed reduced BCAT1 expression and BCAA catabolism in ISG15–/– macrophages (Figure 4A,B and Figure S5F). The treatments increased BCAT1 expression, which was seen in the single‐cell RNAseq data of macrophages, by immunoblot, and in a dedicated RT‐qPCR analysis (Figure 9A–C). Analysis of the microarray data of WT and ISG15–/– ECs also confirmed that the treatments enhanced BCAT1 expression, albeit less than in macrophages (Figure S11E). To verify the functional relevance of IA treatment for BCAT1 activity, we analyzed BCAA concentrations in the targeted metabolomics data set from WT and ISG15–/– dermal fibroblasts. Indeed, IA treatment resulted in significantly reduced concentrations of the three BCAA in IFN‐α stimulated ISG15–/– cells, suggesting increased catabolism (Figure 9D).
FIGURE 9.
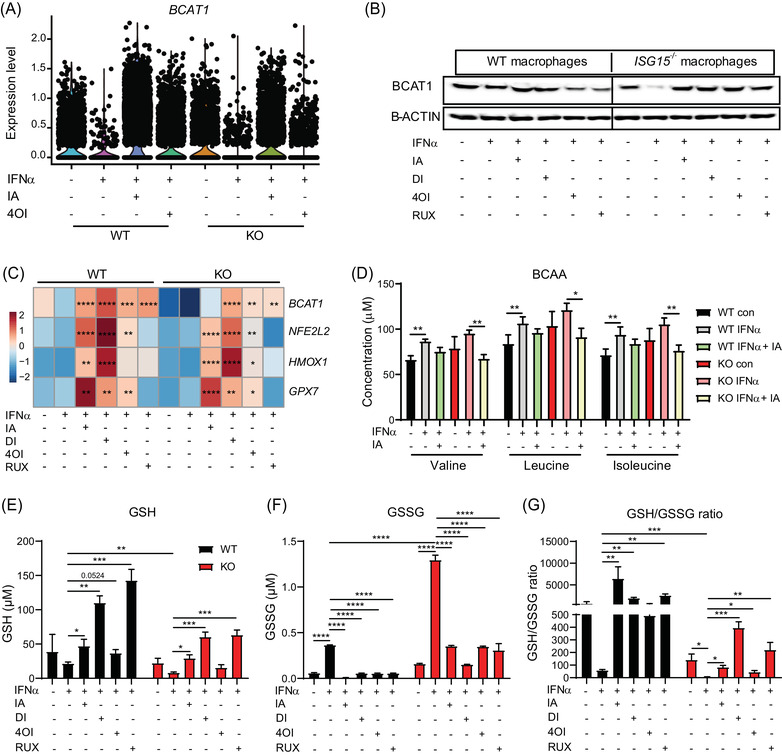
Itaconates and RUX rescue BCAT1 expression and redox balance in IFN‐α stimulated ISG15–/– cells, but only itaconates induce expression of anti‐oxidant genes NFE2L2, HMOX1, and GPX7. (A–E) WT and ISG15–/– iPSC‐derived macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (IA, 10 mM), DI (0.5 mM), 4OI (25 µM), and RUX (1 µM) for 16 h. (A,B) Treatments rescue IFN‐α induced downregulation of BCAT1 mRNA (scRNAseq, each dot represents one cell) and BCAT1 protein (western blot of cell lysates). (C) Itaconates and RUX upregulate BCAT1 mRNA in IFN‐α stimulated WT and ISG15–/– cells, but only itaconates increase expression of anti‐oxidant genes NFE2L2, HMOX1, and GPX7 (RT‐qPCR). Statistical significance was determined with respect to the untreated, IFN‐α stimulated WT or ISG15–/– group. (D) WT and ISG15–/– dermal fibroblasts were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (25 mM) for 16 h. Concentrations of BCAA valine, leucine, and isoleucine were measured by mass spectrometry. Itaconate treatment reduces levels of all three in IFN‐α treated ISG15–/– cells, indicating increased BCAT1 activity. (E–G) WT and ISG15–/– iPSC‐derived macrophages were stimulated with IFN‐α (1000 IU/ml) for 8 h and treated with itaconate (IA, 10 mM), DI (0.5 mM), 4OI (25 µM), and RUX (1 µM) for 16 h. GSH (reduced glutathione) and GSSG (oxidized glutathione) concentrations were measured with a fluorescence microplate reader (Ex/Em = 490/520 nm). Treatments increase GSH and decreased GSSG concentrations, resulting in improved redox balance (higher GSH/GSSG ratio). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001, one‐way ANOVA followed by Tukey's post hoc test
BCAT1 also induces the NRF2 pathway and expression of ROS scavengers. 51 In agreement with this, itaconate treatments increased expression of NFE2L2 (encoding NRF2), and the NRF2 target genes HMOX1 and GPX7 (Figure 9C). However, this effect was more likely due to direct activation of NRF2 signalling by the itaconates, as RUX (which increased BCAT1 mRNA and protein levels but is not a direct NRF2 agonist) did not affect expression of these mRNAs. To assess the impact of ISG15 deficiency and the treatments on net redox state of the cells, we measured reduced (GSH) and disulfide (GSSG) glutathione as bioindicators (Figure 9E–G). Indeed, GSH/GSSG ratio (indicating a favourable redox balance) was significantly lower in the ISG15–/– cells, but increased under treatment with all four compounds, with DI exerting the strongest effect, followed by RUX. These findings support the hypothesis that treatment of ISG15‐deficient cells with itaconates improves cellular redox balance by mechanisms involving NRF2 signalling and BCAT1 activity.
3.13. Reduction of IFN expression by treatment with IA does not lead to increased viral replication
Patients with ISG15 deficiency do not have an increased risk of viral infections, and ISG15–/– cells are less susceptible to certain viral infections, possibly due to the persistently elevated IFN levels. 14 We tested whether treatment with the compounds would raise cellular susceptibility to influenza A virus, one of the most important respiratory pathogens in all age groups. As expected, viral infectivity was lower in ISG15–/– than in WT macrophages. Remarkably, despite a striking reduction of the excess induction of ISGs in the infected ISG15–/– cells by treatment with both IA and DI, viral infectivity remained reduced (Figure S16).
4. DISCUSSION
Using a variety of human cells types and analytical techniques, we identified an increased propensity for cell death coupled with major defects in mitochondrial homeostasis, energy generation, and redox balance as cardinal features of the cellular phenotype of ISG15 deficiency. Moreover, we identified itaconates and the JAK1/2 inhibitor RUX as effective treatments for most aspects of this multifaceted phenotype.
The fact that the conjugation‐deficient mutant (ΔGG) only partially restored mitochondrial respiration in ISG15–/– cells suggests that ISG15 participates in a complex regulatory network governing cell respiration in which some of its functions require conjugation to target proteins. Indeed, ISG15‐conjugated proteins in murine and human cells include several enzymes involved in carbohydrate energy metabolism. 55 Our observation of increased glycolysis in ISG15–/– cells agrees well with recent findings by Yan et al. that ISG15 limits glycolysis by covalent binding to glycolytic enzymes. 56 A functional connection between ISG15 and branched chain amino acid catabolism has not been described. BCAT1 is the key enzyme responsible for conversion of branched chain amino acids toward branched chain keto acids and the formation of macromolecular precursors. 57 These authors also showed that BCAT1 has additional “moonlighting” functions in that it contributes to efficient function of the TCA cycle by maintaining levels of α‐ketoglutarate and 2‐OH‐glutarate. In addition, BCAT1 induces ROS scavengers and attenuates ROS accumulation, 51 and its expression in tumour cells is associated with enhanced proliferation and decreased apoptosis. Together with these studies, our findings suggest a central role for a relative BCAT1 deficiency in the full manifestation of the cellular phenotype of ISG15 deficiency and it may also play a role in the decreased apoptosis observed in ISG15–/– cells, as BCAT1 is generally thought to have anti‐ROS and anti‐apoptotic effects. 51
Work on bone‐marrow derived macrophages from ISG15–/– mice showed that ROS levels were actually lower in cells from KO mice during infection with vaccinia virus. 19 ROS levels, as a physiological by‐product of oxidative phosphorylation, usually drop along with mitochondrial respiration. Our results suggest the polar opposite, and it is conceivable that this may be yet another scenario where ISG15 has different effects in humans than in mice. The underlying mechanism of the increased ROS levels is not clear at this time. We favour a combination of decreased ROS scavengers (due to compromised BCAT1/NRF2 activity) and increased IFN levels, as it is well known that the latter raise intracellular ROS levels. In addition, they likely relate to an underlying mitochondrial dysfunction, as ISG15 interacts directly with mitochondrial proteins responsible for autophagy, mitochondrial function, and apoptosis. 13 , 19 , 58 Furthermore, expression of genes supporting mitochondrial biogenesis and encoding respiratory chain complexes II‐V was diminished in ISG15‐deficient cells. Also considering the propensity for apoptosis in ISG15–/– cells, it is plausible that release of DNA from dysfunctional mitochondria into the cytosol contributes to the hyperinflammation of ISG15 deficiency. Even though not investigated in detail, our data also suggested increased autophagy and mitophagy due to loss of ISG15. Taken together, our results therefore suggest mitochondrial dysfunction of diverse causes and consequences as a cardinal, previously unrecognized feature of human ISG15 deficiency. Its clinical significance is uncertain at this time and will require detailed prospective investigations of affected kindreds. In analogy to other associations between oxidative stress and mitochondrial dysfunction and epilepsy, 59 it is tempting to speculate that the mitochondrial pathology may be one factor contributing to the high risk of seizures in these children.
Overall, itaconates and RUX had similar beneficial effects on nearly all aspects of this multifaceted phenotype of ISG15 deficiency, suggesting that they either acted through their previously documented anti‐IFN effects 24 , 25 or affected the same processes by different mechanisms. The latter may be the case with pyroptosis, as IA has been shown to inhibit it by directly interacting with NLRP3 and thus interfering with inflammasome activation. 60 Interestingly, itaconates are well‐documented activators of the anti‐oxidative NRF2 signalling pathway 24 and were significantly more effective than RUX in inducing NRF2 target genes, the ROS scavengers HMOX1 and GPX7. 61 Irrespective of the observed differences between itaconates and RUX, or even among the itaconates themselves, it should be noted that all four compounds greatly improved overall cell energy state (intracellular ATP) and redox balance (GSH/GSSG ratio), and normalized all parameters of hyperinflammation. Taken together, our data therefore suggest that IA‐derived treatments may be at least equivalent to RUX, and that type I interferonopathies should be considered as one disease indication as therapeutics based on IA progress toward clinical applications. Notably, treatment with IA and DI reduced IFN‐I levels but did not alter the reduced infectivity of ISG15–/– macrophages by influenza A virus. It is conceivable that the increased IFN‐signalling alone accounts for the reduced susceptibility of ISG15‐deficient cells for certain viruses. However, our results suggest strongly that this is not the case, but that other ISG15‐regulated mechanisms are at work. IA has antiviral properties 62 and one might therefore argue that this masked an increased viral infectivity that would be expected under normalized IFN levels. However, we can exclude this possibility because neither IA nor DI affect HA mRNA levels in human macrophages. 25 From a clinical perspective it appears remarkable that, inferring from this reductionist model, there is no evidence to suspect that targeted treatment of ISG15 deficiency would increase the risk of viral infections of the patients.
Taken together, our results suggest (i) that ISG15 deficiency leads to a complex cellular phenotype that extends beyond the well‐described hyperinflammatory manifestations to encompass a mitochondrial dysfunction of potentially broad consequences, and (ii) that pharmacological agents based on IA merit further investigation as alternate treatment options.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Supporting information
Figure S1. Hyperinflammatory phenotype of ISG15‐/‐ hiPSC‐derived macrophages
Figure S2. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ hiPSC‐derived ECs
Figure S3. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ HaCaT keratinocytes
Figure S4. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ dermal fibroblasts
Figure S5. IFN‐α stimulation effects major changes in transcriptomes in WT and ISG15‐/‐ iPSC‐derived macrophages.
Figure S6. miRNA populations in WT and ISG15‐/‐ iPSC‐derived macrophages differ according to genotype and IFN‐α stimulation
Figure S7. Differences in transcriptomes due to IFN−α stimulation of iPSC‐derived WT and ISG15‐/‐ ECs
Figure S8. Differences in acylcarnitine populations in WT and ISG15‐/‐ fibroblasts support the existence of a global defect in energy metabolism in ISG15‐/‐ cells
Figure S9. Full restoration of mitochondrial respiration in ISG15‐/‐ fibroblasts requires the ISGylation ability of ISG15
Figure S10. Stable‐isotope assisted metabolic flux studies of WT and ISG15‐/‐ dermal fibroblasts reveal decreased glutamine uptake and glutamine‐derived carbon contribution to the TCA cycle
Figure S11. Microarray analysis reveals differential impact of itaconate treatments on gene expression in WT and ISG15‐/‐ iPSC‐derived ECs
Figure S12. A,B. Determination of nontoxic doses of IA and DI in IFN‐α stimulated immortalized dermal fibroblasts for treatment with itaconate and derivatives. C,D. Effects of itaconates and RUX on NAD(P)H‐dependent oxidoreductase activity in unstimulated and stimulated WT and ISG15‐/‐ macrophages
Figure S13. A–C. Single‐cell RNAseq analysis of iPSC‐derived macrophages. D‐H. Treatment with itaconate and derivatives reduces type I IFN signature and hyperinflammation in iPSC‐derived ISG15‐/‐ ECs.
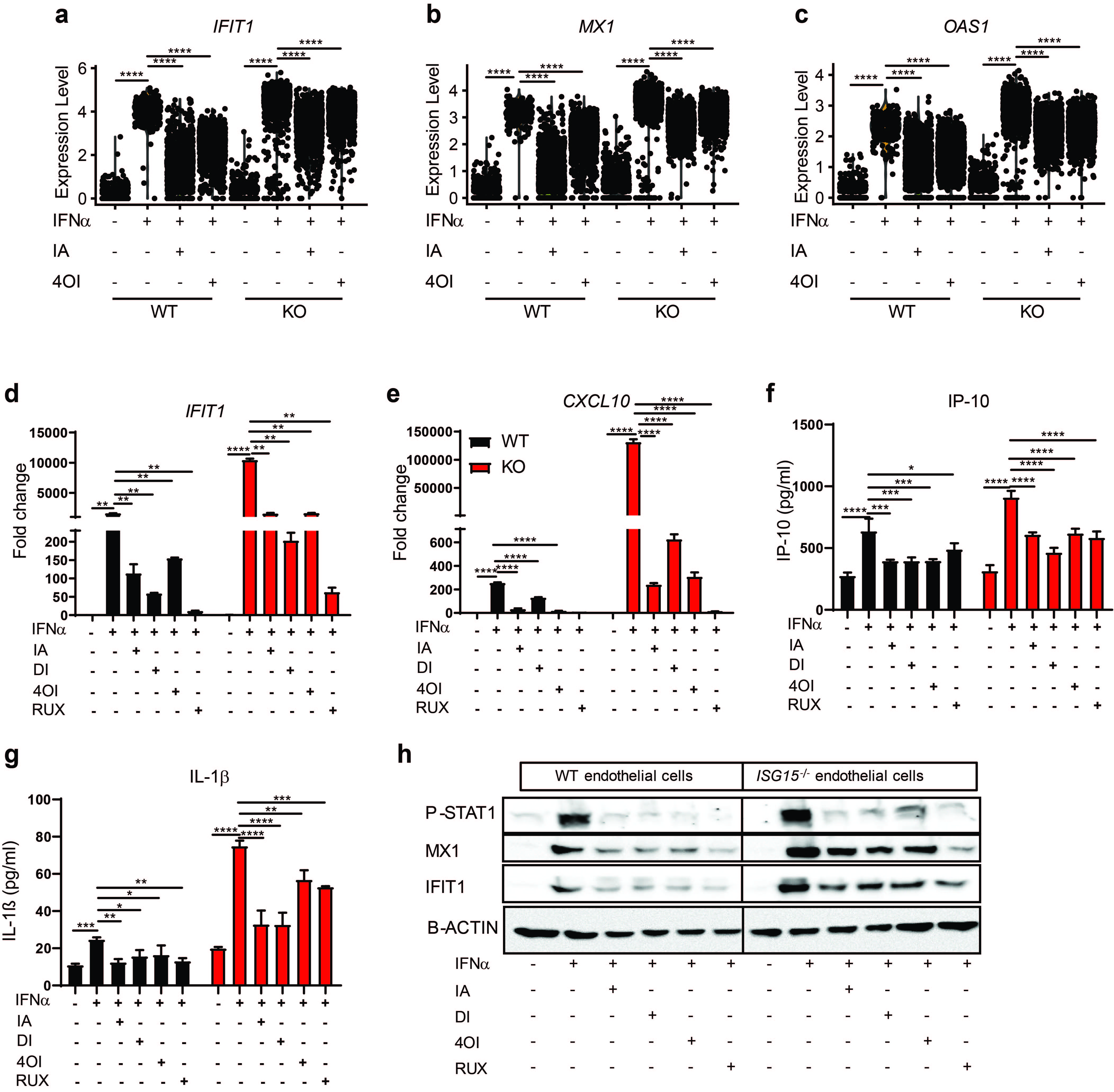{kind=link}
Figure S14. Itaconate and derivatives reduce type I IFN signature and hyperinflammation in ISG15‐/‐ HaCaT cells and dermal fibroblasts
Figure S15. Treatment with itaconate and derivatives reduces type I IFN signature and hyperinflammation in LPS‐stimulated iPSC‐derived ISG15‐/‐ macrophages
Figure S16. Treatment with itaconate and DI reverts hyperinflammation in influenza A virus infected ISG15‐/‐ iPSC‐derived macrophages but does not increase viral infectivity
Figure S17. Basic quality data of single‐cell RNAseq analysis of iPSC‐derived macrophages
{kind=link}
Table S1. List of RT‐qPCR primers
ACKNOWLEDGEMENTS
We thank Theresa Bucheggar for helping with iPSC and macrophage cell culture, Matthias Ballmaier for single cell sorting, Oliver Dittrich‐Breiholz and staff of the Research Core Unit Genomics (Hannover Medical School) for library preparation of sc‐RNA sequencing samples, Julia Dahlmann and Emilia von Riegen for helping with iPSC‐derived EC cell culture, Maren Scharfe and Petra Hagendorff for helping with RNA sequencing, and Mareile Schönfeld for helpful discussion and support. Funding is gratefully acknowledged from the Helmholtz Association's Cross‐Programm Initiative on Personalised Medicine (iMed, to F.P.). S.F.H.W. was supported by a predoctoral fellowship from the Higher Education Commission of Pakistan and the German Academic Exchange Service (DAAD).
Waqas SF‐H, Sohail A, Nguyen AHH, et al. ISG15 deficiency features a complex cellular phenotype that responds to treatment with itaconate and derivatives. Clin Transl Med. 2022;12:e931. 10.1002/ctm2.931
REFERENCES
- 1. Rodero MP, Crow YJ. Type I interferon‐mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. 2016;213(12):2527‐2538. 10.1084/jem.20161596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee‐Kirsch MA. The type I interferonopathies. Annu Rev Med. 2017;68:297‐315. 10.1146/annurev-med-050715-104506 [DOI] [PubMed] [Google Scholar]
- 3. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238(1):91‐98. 10.1111/j.1749-6632.2011.06220.x [DOI] [PubMed] [Google Scholar]
- 4. Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35(3):235‐243. 10.1007/s10875-015-0147-3 [DOI] [PubMed] [Google Scholar]
- 5. Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507‐518. 10.1056/NEJMoa1312625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duncan CJA, Thompson BJ, Chen R, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol. 2019;4(42). 10.1126/sciimmunol.aav7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gruber C, Martin‐Fernandez M, Ailal F, et al. Homozygous STAT2 gain‐of‐function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med. 2020;217(5):e20192319. 10.1084/jem.20192319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meuwissen ME, Schot R, Buta S, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo‐TORCH syndrome. J Exp Med. 2016;213(7):1163‐1174. 10.1084/jem.20151529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wen Y, Zhang X, Dong L, Zhao J, Zhang C, Zhu C. Acetylbritannilactone modulates MicroRNA‐155‐mediated inflammatory response in ischemic cerebral tissues. Mol Med. 2015;21(1):197‐209. 10.2119/molmed.2014.00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bogunovic D, Byun M, Durfee LA, et al. Mycobacterial disease and impaired IFN‐gamma immunity in humans with inherited ISG15 deficiency. Science. 2012;337(6102):1684‐1688. 10.1126/science.1224026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martin‐Fernandez M, Bravo García‐Morato M, Gruber C, et al. Systemic type I IFN inflammation in human ISG15 deficiency leads to necrotizing skin lesions. Cell Rep. 2020;31(6):107633. 10.1016/j.celrep.2020.107633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malik MNH, Waqas SF, Zeitvogel J, et al. Congenital deficiency reveals critical role of ISG15 in skin homeostasis. J Clin Invest. 2022;132(3):e141573. 10.1172/JCI141573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Thery F, Wu NC, et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat Commun. 2019;10(1):5383. 10.1038/s41467-019-13393-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Speer SD, Li Z, Buta S, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7:11496. 10.1038/ncomms11496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hermann M, Bogunovic D. ISG15: in sickness and in health. Trends Immunol. 2017;38(2):79‐93. 10.1016/j.it.2016.11.001 [DOI] [PubMed] [Google Scholar]
- 16. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018;16(7):423‐439. 10.1038/s41579-018-0020-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao H, Wang M, Cao B, Zhou H, Zhang Z, Mao X. Interferon‐stimulated gene 15 induces cancer cell death by suppressing the NF‐kappaB signaling pathway. Oncotarget. 2016;7(43):70143‐70151. 10.18632/oncotarget.12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vuillier F, Li Z, Commere PH, Dynesen LT, Pellegrini S. USP18 and ISG15 coordinately impact on SKP2 and cell cycle progression. Sci Rep. 2019;9(1):4066. 10.1038/s41598-019-39343-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baldanta S, Fernández‐Escobar M, Acín‐Perez R, et al. ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLoS Pathog. 2017;13(10):e1006651. 10.1371/journal.ppat.1006651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu ZX, Song HM. Toward a better understanding of type I interferonopathies: a brief summary, update and beyond. World J Pediatr. 2020;16(1):44‐51. 10.1007/s12519-019-00273-z [DOI] [PubMed] [Google Scholar]
- 21. Michelucci A, Cordes T, Ghelfi J, et al. Immune‐responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. 2013;110(19):7820‐7825. 10.1073/pnas.1218599110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cordes T, Michelucci A, Hiller K. Itaconic acid: the surprising role of an industrial compound as a mammalian antimicrobial metabolite. Annu Rev Nutr. 2015;35:451‐473. 10.1146/annurev-nutr-071714-034243 [DOI] [PubMed] [Google Scholar]
- 23. O'Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. 2019;19(5):273‐281. 10.1038/s41577-019-0128-5 [DOI] [PubMed] [Google Scholar]
- 24. Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti‐inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113‐117. 10.1038/nature25986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sohail A, Iqbal AA, Sahini N, Chen F, Pessler F. Itaconate and derivatives reduce interferon responses and inflammation in influenza A infection. PLoS Pathog. 2022;18(1):e1010219. 10.1371/journal.ppat.1010219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cordes T, Lucas A, Divakaruni AS, Murphy AN, Cabrales P, Metallo CM. Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Mol Metab. 2020;32:122‐135. 10.1016/j.molmet.2019.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haase A, Gohring G, Martin U. Generation of non‐transgenic iPS cells from human cord blood CD34(+) cells under animal component‐free conditions. Stem Cell Res. 2017;21:71‐73. 10.1016/j.scr.2017.03.022 [DOI] [PubMed] [Google Scholar]
- 28. Merkert S, Jaboreck MC, Engels L, et al. Generation of two human ISG15 knockout iPSC clones using CRISPR/Cas9 editing. Stem Cell Res. 2020;50:102135. 10.1016/j.scr.2020.102135 [DOI] [PubMed] [Google Scholar]
- 29. Lachmann N, Ackermann M, Frenzel E, et al. Large‐scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Reports. 2015;4(2):282‐296. 10.1016/j.stemcr.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olmer R, Engels L, Usman A, et al. Differentiation of human pluripotent stem cells into functional endothelial cells in scalable suspension culture. Stem Cell Reports. 2018;10(5):1657‐1672. 10.1016/j.stemcr.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2^(‐delta delta CT) method for quantitative real‐time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3(3):71‐85. [PMC free article] [PubMed] [Google Scholar]
- 32. Bertani FR, Mozetic P, Fioramonti M, et al. Classification of M1/M2‐polarized human macrophages by label‐free hyperspectral reflectance confocal microscopy and multivariate analysis. Sci Rep. 2017;7(1):8965. 10.1038/s41598-017-08121-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rahman RU, Gautam A, Bethune J, et al. Oasis 2: improved online analysis of small RNA‐seq data. BMC Bioinformatics. 2018;19(1):54. 10.1186/s12859-018-2047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arshad H, Alfonso JCL, Franke R, et al. Decreased plasma phospholipid concentrations and increased acid sphingomyelinase activity are accurate biomarkers for community‐acquired pneumonia. J Transl Med. 2019;17:365. 10.1186/s12967-019-2112-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hiller K, Hangebrauk J, Jäger C, Spura J, Schreiber K, Schomburg D. MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal Chem. 2009;81(9):3429‐3439. 10.1021/ac802689c [DOI] [PubMed] [Google Scholar]
- 36. Wegner A, Weindl D, Jäger C, et al. Fragment formula calculator (FFC): determination of chemical formulas for fragment ions in mass spectrometric data. Anal Chem. 2014;86(4):2221‐2228. 10.1021/ac403879d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winterhoff M, Chen F, Sahini N, et al. Establishment, validation, and initial application of a sensitive LC‐MS/MS assay for quantification of the naturally occurring isomers itaconate, mesaconate, and citraconate. Metabolites. 2021;11(5):270. 10.3390/metabo11050270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. 2019;19(5):273‐281. 10.1038/s41577-019-0128-5 [DOI] [PubMed] [Google Scholar]
- 39. Yin W, Park JI, Loeser RF. Oxidative stress inhibits insulin‐like growth factor‐I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3‐Kinase‐Akt and MEK‐ERK MAPK signaling pathways. J. Biol. Chem. 2009;284(46):31972‐31981. 10.1074/jbc.M109.056838. [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duda P, Akula SM, Abrams SL, et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells. 2020;9(5):1110. 10.3390/cells9051110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Z, Havasi A, Gall J, et al. GSK3beta promotes apoptosis after renal ischemic injury. J Am Soc Nephrol. 2010;21(2):284‐294. 10.1681/ASN.2009080828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miao EA, Rajan JV, Aderem A. Caspase‐1‐induced pyroptotic cell death. Immunol Rev. 2011;243(1):206‐214. 10.1111/j.1600-065X.2011.01044.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu X, Zhang Z, Ruan J, et al. Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153‐158. 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26(1):99‐114. 10.1038/s41418-018-0212-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: the yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel). 2018;7(1):13. 10.3390/antiox7010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo A, Li K, Xiao Q. Fibroblast growth factor 19 alleviates palmitic acid‐induced mitochondrial dysfunction and oxidative stress via the AMPK/PGC‐1alpha pathway in skeletal muscle. Biochem Biophys Res Commun. 2020;526(4):1069‐1076. 10.1016/j.bbrc.2020.04.002 [DOI] [PubMed] [Google Scholar]
- 47. Yusnita Y, Norsiah MD, Rahman AJ. Mutations in mitochondrial NADH dehydrogenase subunit 1 (mtND1) gene in colorectal carcinoma. Malays J Pathol. 2010;32(2):103‐110. [PubMed] [Google Scholar]
- 48. Yu S, Zhai J, Yu J, Yang Q, Yang J. miR‐98‐5p protects against cerebral ischemia/reperfusion injury through anti‐apoptosis and anti‐oxidative stress in mice. J Biochem. 2020;169(2):195‐206. 10.1093/jb/mvaa099 [DOI] [PubMed] [Google Scholar]
- 49. Fang YC, Yeh CH. Inhibition of miR‐302 suppresses hypoxia‐reoxygenation‐induced H9c2 cardiomyocyte death by regulating Mcl‐1 expression. Oxid Med Cell Longev. 2017;2017:7968905. 10.1155/2017/7968905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tonjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild‐type IDH1. Nat Med. 2013;19(7):901‐908. 10.1038/nm.3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y, Zhang J, Ren S, et al. Branched‐chain amino acid metabolic reprogramming orchestrates drug resistance to EGFR tyrosine kinase inhibitors. Cell Rep. 2019;28(2):512‐525.e516. 10.1016/j.celrep.2019.06.026 [DOI] [PubMed] [Google Scholar]
- 52. Tian Q, Yuan P, Quan C, et al. Phosphorylation of BCKDK of BCAA catabolism at Y246 by Src promotes metastasis of colorectal cancer. Oncogene. 2020;39(20):3980‐3996. 10.1038/s41388-020-1262-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fremond ML, Rodero MP, Jeremiah N, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173‐activating mutations in 3 children. J Allergy Clin Immunol. 2016;138(6):1752‐1755. 10.1016/j.jaci.2016.07.015 [DOI] [PubMed] [Google Scholar]
- 54. Chen F, et al. Citraconate inhibits ACOD1 (IRG1) catalysis, reduces interferon responses and oxidative stress, and modulates inflammation and cell metabolism. Nature Metabolism. Published online June 2, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giannakopoulos NV, Luo JK, Papov V, et al. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem Biophys Res Commun. 2005;336(2):496‐506. 10.1016/j.bbrc.2005.08.132 [DOI] [PubMed] [Google Scholar]
- 56. Yan S, Sui M, Tian H, et al. IRF3 reduces adipose thermogenesis via ISG15‐mediated reprogramming of glycolysis. J Clin Invest. 2021;131(7):e144888. 10.1172/JCI144888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Papathanassiu AE, Ko JH, Imprialou M, et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun. 2017;8:16040. 10.1038/ncomms16040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Albert M, Becares M, Falqui M, Fernandez‐Lozano C, Guerra S. ISG15, a small molecule with huge implications: regulation of mitochondrial homeostasis. Viruses. 2018;10(11):629. 10.3390/v10110629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang N, Guan QW, Chen FH, et al. Antioxidants targeting mitochondrial oxidative stress: promising neuroprotectants for epilepsy. Oxid Med Cell Longev. 2020;2020:6687185. 10.1155/2020/6687185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hooftman A, O'Neill LAJ. The immunomodulatory potential of the metabolite itaconate. Trends Immunol. 2019;40(8):687‐698. 10.1016/j.it.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 61. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401‐426. 10.1146/annurev-pharmtox-011112-140320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Daniels BP, Kofman SB, Smith JR, et al. The nucleotide sensor ZBP1 and Kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity. 2019;50(1):64‐76.e4. 10.1016/j.immuni.2018.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hyperinflammatory phenotype of ISG15‐/‐ hiPSC‐derived macrophages
Figure S2. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ hiPSC‐derived ECs
Figure S3. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ HaCaT keratinocytes
Figure S4. IFN‐α inducible hyperinflammatory phenotype of ISG15‐/‐ dermal fibroblasts
Figure S5. IFN‐α stimulation effects major changes in transcriptomes in WT and ISG15‐/‐ iPSC‐derived macrophages.
Figure S6. miRNA populations in WT and ISG15‐/‐ iPSC‐derived macrophages differ according to genotype and IFN‐α stimulation
Figure S7. Differences in transcriptomes due to IFN−α stimulation of iPSC‐derived WT and ISG15‐/‐ ECs
Figure S8. Differences in acylcarnitine populations in WT and ISG15‐/‐ fibroblasts support the existence of a global defect in energy metabolism in ISG15‐/‐ cells
Figure S9. Full restoration of mitochondrial respiration in ISG15‐/‐ fibroblasts requires the ISGylation ability of ISG15
Figure S10. Stable‐isotope assisted metabolic flux studies of WT and ISG15‐/‐ dermal fibroblasts reveal decreased glutamine uptake and glutamine‐derived carbon contribution to the TCA cycle
Figure S11. Microarray analysis reveals differential impact of itaconate treatments on gene expression in WT and ISG15‐/‐ iPSC‐derived ECs
Figure S12. A,B. Determination of nontoxic doses of IA and DI in IFN‐α stimulated immortalized dermal fibroblasts for treatment with itaconate and derivatives. C,D. Effects of itaconates and RUX on NAD(P)H‐dependent oxidoreductase activity in unstimulated and stimulated WT and ISG15‐/‐ macrophages
Figure S13. A–C. Single‐cell RNAseq analysis of iPSC‐derived macrophages. D‐H. Treatment with itaconate and derivatives reduces type I IFN signature and hyperinflammation in iPSC‐derived ISG15‐/‐ ECs.
Figure S14. Itaconate and derivatives reduce type I IFN signature and hyperinflammation in ISG15‐/‐ HaCaT cells and dermal fibroblasts
Figure S15. Treatment with itaconate and derivatives reduces type I IFN signature and hyperinflammation in LPS‐stimulated iPSC‐derived ISG15‐/‐ macrophages
Figure S16. Treatment with itaconate and DI reverts hyperinflammation in influenza A virus infected ISG15‐/‐ iPSC‐derived macrophages but does not increase viral infectivity
Figure S17. Basic quality data of single‐cell RNAseq analysis of iPSC‐derived macrophages
Table S1. List of RT‐qPCR primers