

Abstract
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is considered the etiological agent of the disease that caused the COVID-19 pandemic, and for which there is currently no effective treatment. This pandemic has shown that the rapid identification of therapeutic compounds is critical (when a new virus with high transmissibility occurs) to prevent or reduce as much as possible the loss of human lives. To meet the urgent need for drugs, many strategies were applied for the discovery, respectively the identification of potential therapies / drugs for SARS-CoV-2. Molecular docking and virtual screening are two of the in silico tools/techniques that provided the identification of few SARS-CoV-2 inhibitors, removing ineffective or less effective drugs and thus preventing the loss of resources such as time and additional costs. The main target of this review is to provide a comprehensive overview of how in-silico tools have been used in the crisis management of anti-SARS-CoV-2 drugs, especially in virtual screening of substances used in the treatment of SARS-CoV-2 infection and analysis of compounds with known action on structurally similar proteins from other viruses; also, completions were added to the way in which these methods came to meet the requirements of biomedical research in the field. Moreover, the importance and impact of the topic approached for researchers was highlighted by conducting an extensive bibliometric analysis.
Keywords: Virtual screening, Bibliometric analysis, Treatment of SARS-CoV-2 infection, Proteins, Viruses, Molecular docking
Graphical Abstract

1. Introduction
Since the development of the modern digital era, numerous scientific fields have made a sudden transition to data-dependent research methods [1]. Bioinformatics is a field of science that was developed as a result of this progress [2], [3]. Molecular docking, virtual screening (VS), and molecular dynamics represent important tools that can be used in bioinformatics. Experiments conducted in vivo or in vitro are expensive and time-consuming, but they provide accurate results. By comparison, in silico ones do not yield information that is as accurate. However, they are faster and less expensive, making them widely used in research in the pharmaceutical industry and other scientific fields [2], [4].
Molecular docking studies, as a part of in silico analysis, are carried out using a biomedical informatics program that attempts to predict as accurately as possible how a ligand (a small molecule or a protein) interacts with a receptor represented by proteins or genetic materials [5]. These studies are used to identify compounds with therapeutic potential. Furthermore, they accurately determine the interaction between the ligand and the protein, and help establish a link between the chemical structure and the activity of the studied compound [6]. The Protein Data Bank (PDB) is a repository of three-dimensional structures of proteins that are used as receptors for molecular docking studies [7]. Selection of the receptor is a crucial step in the docking process, and if there are multiple sources for a protein, the higher resolution receptor will be selected [8].
The docking of numerous molecules on a target protein and the assessment of these compounds based on the score supplied using an algorithm is the basis of VS. Molecular dynamics allows the introduction of variables such as protein flexibility, behavior in the presence of water and ions and the evolution of the system over time. By associating VS / molecular docking with molecular dynamics, the obtained results are significantly closer to the in-vivo ligand-protein interaction mode [9]. Although investment in drug research and development is on an upward trend, the number of new drugs introduced into therapy declined significantly in recent years [10]. The regulatory agencies authorize only a small percentage of the drugs tested. Therefore, pharmaceutical companies tend to use drugs that are already used in therapy by finding new indications for them. A recent example is the repositioning of drugs to treat SARS-CoV-2 infection since the onset of the pandemic [11], [12], [13].
The present research aims to provide an overview of how in silico tools have been used in the crisis management of anti-SARS-CoV-2 drugs, especially in virtual screening of substances used in the treatment of SARS-CoV-2 infection, also considering the analysis of compounds with known action on structurally similar proteins from other viruses, and the way these methods were usable in the biomedical research. Moreover, the importance of the topic for researchers and the impact of the information in the field were underlined by conducting an extensive bibliometric analysis. Using scientific mapping technology to visually assess knowledge structure and research trends facilitates the access of future authors to the most prolific journals while also revealing the most relevant and productive researchers in the field. Identifying the terms with a high occurrence and the citation average for the articles in which they are contained highlights the latest developments in this field. The keywords representing antivirals and other chemical substances are useful tools in understanding the current trends in molecules used for docking or included in databases subjected to virtual screening.
2. Bibliometric analysis
In order to assess the importance of the topic, an extensive bibliometric analysis was performed, highlighting the most important parameters describing literature such as data source, journal, impact factor, number of citations, most prolific authors, etc.
2.1. Data source
A scientific literature search was performed on the Scopus database, the largest abstract and citation database to date. The following search parameters were used: articles that were written in English with the following keywords in the title, abstract, and keywords: SARS-CoV-2, molecular docking, virtual screening. 575 results were identified, of which 539 (93.74 %) were original articles, 16 (2.78 %) reviews, and the rest of them were included in the following categories: book chapter, conference paper, and letter. Most of the articles were classified in the following subject areas: “Biochemistry, Genetics and Molecular Biology- 304 articles”, “Chemistry-175″, “Pharmacology, Toxicology, and Pharmaceuticals-171″, “Computer Science-117″, and “Medicine-104″; other subject areas had < 100 articles.
2.2. Leading countries in publishing SARS-CoV-2 and molecular docking papers
India, the United States, China, and Saudi Arabia published most of the articles (75.88 %) in this field. Despite ranking third in terms of the number of published articles, China has a far higher average citation/document than India and the United States. Although Italy has only 25 published articles, the average citation/document is 20. Pakistan also stands out with an average citation/document of 26.41 for 22 articles. India had a high total link strength (TLS) score because it collaborated with a significant number of countries (48) in the publication process. Table 1 shows all the countries that had more than 20 published papers in the topic.
Table 1.
Countries with >20 published papers.
| Country | Papers No/ % |
Citations | Average citation / document |
TLS |
|---|---|---|---|---|
| India | 200/37.11 | 2885 | 14.43 | 104 |
| United States | 76/14.10 | 1321 | 17.38 | 70 |
| China | 69/12.80 | 1918 | 27.80 | 48 |
| Saudi Arabia | 64/11.87 | 552 | 8.63 | 91 |
| Egypt | 31/5.75 | 288 | 9.29 | 40 |
| Brazil | 26/4.82 | 345 | 13.27 | 10 |
| Iran | 25/4.64 | 133 | 5.32 | 7 |
| Italy | 25/4.64 | 500 | 20.00 | 20 |
| United Kingdom | 25/4.64 | 282 | 11.28 | 44 |
| Germany | 22/4.08 | 380 | 17.27 | 26 |
| Pakistan | 22/4.08 | 581 | 26.41 | 15 |
Legend: TLS, total link strength: a measure of the degree of collaboration with other countries.
VOSviewer was used to create the network map of country co-authorship for articles matching the presented search terms ( Fig. 1). Countries that frequently co-author articles are included in different clusters:
-
•
The red cluster includes Bangladesh, China, Germany, Iran, Italy, Pakistan, and Spain.
-
•
The green cluster includes Egypt, Nigeria, South Africa, Turkey, and the United Kingdom.
-
•
The blue cluster includes India, Saudi Arabia, and South Korea.
-
•
The yellow cluster includes the United States and Brazil.
Fig. 1.

Bubble map of co-authorship by the country for the selected search terms. Only countries with > 10 published articles were presented (17 countries in 4 clusters).
The bubble size reflects the TLS, which means that more collaborative countries have a larger bubble. The strength of the link (LS) between two countries is shown by the line's thickness. The line connecting two countries will be thicker if they frequently publish together because the LS score for those publications is higher. Researchers from India (TLS 104), Saudi Arabia (91), and the United States (70) closely collaborated with a significant proportion of foreign academics. Indian researchers had the highest level of collaboration with researchers from Saudi Arabia, followed up by experts from the United States and South Korea.
2.3. Journals
Over 200 journals have published at least 1 article matching the search terms. The journals that showed the most interest regarding in-silico studies are presented in Table 2. All the information regarding the impact factor (IF) was obtained using the Journal Citations Reports website. When analyzed in terms of the IF, the highest-ranking journal was the International Journal of Molecular Sciences (5.924) and the lowest ranking was the Journal of Molecular Graphics and Modeling (2.518).
Table 2.
Top 10 journals according to the number of articles published that fit the search parameters.
| Journal | Papers | Total citations | Average citation / document | Publisher | Impact factora |
TLS |
|---|---|---|---|---|---|---|
| Journal of Biomolecular Structure and Dynamics | 104 | 1966 | 18.90 | Taylor and Francis Ltd. | – | 46 |
| Computers in Biology and Medicine | 21 | 191 | 9.10 | Elsevier | 4.589 | 24 |
| Molecules | 18 | 274 | 15.22 | Multidisciplinary Digital Publishing Institute (MDPI) | 4.412 | 21 |
| Journal of Chemical Information and Modeling | 15 | 373 | 24.87 | ACS Publications | 4.956 | 39 |
| International Journal of Molecular Sciences | 12 | 188 | 15.67 | MDPI | 5.924 | 13 |
| Molecular Diversity | 11 | 89 | 8.09 | Springer | 2.943 | 10 |
| Journal of Molecular Graphics and Modeling | 10 | 89 | 8.90 | Elsevier | 2.518 | 7 |
| Frontiers in Chemistry | 9 | 42 | 4.67 | Frontiers Media | 5.221 | 12 |
| Pharmaceuticals | 9 | 60 | 6.67 | MDPI | 5.863 | 5 |
| Scientific Reports | 8 | 112 | 14.00 | Nature | 4.380 | 6 |
Impact factors are given according to year 2020.
Furthermore, to assess whether molecular docking and virtual screening are within the scope of interest of these journals outside of the context of SARS-CoV-2, the term SARS-CoV-2 has been removed from the search criteria, and the number of papers from 2012 to the present is analyzed ( Fig. 2). Several variables are considered while selecting a journal to publish a scientific paper, including the topic area in which the journal shows interest, in order to increase the chances of successful publication.
Fig. 2.

The number of articles fitting the following search terms “molecular docking, virtual screening” from 2012 to present in the journals presented in Table 2.
2.4. Prolific authors
Of the 539 articles analysed, 2868 authors were identified. Table 3 presents the top ten writers ordered by the number of published papers. Furthermore, they have 82 (15.2 %) publications with 1393 citations.
Table 3.
Prolific authors based on the number of articles published.
| Author | Papers | Total citations | Average citation / document |
|---|---|---|---|
| Kumar S. | 12 | 285 | 23.75 |
| Sharma P. | 9 | 150 | 16.67 |
| Sharma S. | 9 | 109 | 12.11 |
| Chandra S. | 8 | 134 | 16.75 |
| Joshi T. | 8 | 134 | 16.75 |
| Prakash A. | 8 | 285 | 35.63 |
| Ahmad S. | 7 | 46 | 6.57 |
| Islam M.A. | 7 | 57 | 8.14 |
| Kumar V. | 7 | 65 | 9.29 |
| Wei D.-Q. | 7 | 128 | 18.29 |
Fig. 3 shows the network map of co-authorship authors, created by VOSViewer; only authors with a minimum of 5 articles were represented. The bubble map shows the relationship between the authors, highlighting the groups of authors who published together. There were generated six clusters (e.g., the red cluster including 11 authors, the yellow cluster 4, the green cluster 9, the blue cluster 5, the purple cluster 3, and the cyan cluster 1). The most extensive set of interrelated data featured 29 interconnected writers.
Fig. 3.

Bubble map of co-authorship. Only authors with > 5 published articles were presented (33 authors).
2.5. Most cited articles
Table 4 shows the most cited papers, the authors' names, the journal in which they published, and the publication's IF. 7 out of the 10 articles were published in 2020, in journals with IF ranging from 3.39 (Journal of Biomolecular Structure and Dynamics) to 11.614 (Acta Pharmaceutica Sinica B). Considering that nearly all authors require access to high-quality information when writing a paper, the number of citations is a good measure for quantifying an article's quality. The IF of the journal in which an article is published is also a reliable predictor of the quality of the material contained.
Table 4.
Top 10 most cited articles.
| Authors (year) |
Title | Journal | IFa | Citations | Ref. |
|---|---|---|---|---|---|
| Wu C. (2020) | Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods | Acta Pharmaceutica Sinica B | 11.413 | 1093 | [14] |
| Wang J. (2020) | Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study | Journal of Chemical Information and Modeling | 4.956 | 254 | [15] |
| Ton A.-T. (2020) | Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds | Molecular informatics | 3.353 | 251 | [16] |
| Elmezayen A.D. (2020) | Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes | Journal of Biomolecular Structure and Dynamics | – | 210 | [17] |
| Khan S.A. (2021) | Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach | Journal of Biomolecular Structure and Dynamics | – | 191 | [18] |
| Khan R.J. (2021) | Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3 C-like proteinase and 2′-O-ribose methyltransferase | Journal of Biomolecular Structure and Dynamics | – | 179 | [19] |
| Gentile D. (2020) | Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study | Marine drugs | 4.762 | 164 | [20] |
| Pant S. (2020) | Peptide-like and small-molecule inhibitors against Covid-19 | Journal of Biomolecular Structure and Dynamics | – | 164 | [21] |
| Sarma P. (2021) | In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain) | Journal of Biomolecular Structure and Dynamics | – | 143 | [22] |
| Kumar Y. (2020) | In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation-based drug-repurposing | Journal of Infection and Public Health | 3.718 | 126 | [23] |
| Sinha S.K. (2021) | An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets | Journal of Biomolecular Structure and Dynamics | – | 103 | [24] |
Impact factors are given according to year 2020.
2.6. Co-occurrence of the keywords
Topics with high citations included drug screening (178 publications, 29.35 average citation/publication), drug repositioning (115, 25.57), and molecular dynamics simulation (171, 13.32). Antiviral drugs were of high-interest terms like remdesivir (52, 36.9), favipiravir (19, 74.84), darunavir (21, 82.90), lopinavir (37, 55,57), having a high average citation rate/document > 35, but also a high occurrence. Chloroquine (29, 55.86) and hydroxychloroquine (32, 14.81) had a high occurrence among the terms related to drugs). Other drug terms presented nelfinavir (24, 23.92), cobicistat (14, 25.93), simeprevir (15, 32.67), paritaprevir (12, 26.00), saquinavir (17, 30.53), amprenavir (10, 21.40), atazanavir (12,17.25) indinavir (20, 21,10), nilotinib (11, 25.91), raltegravir (12, 46.42), grazoprevir (12, 19.67), tipranavir (14, 27.93). However, they had lower presence in publications, but still a high average citation score/ publication. Umifenovir (11, 6.82), ribavirin (16, 11.69), and sofosbuvir (10, 9.40) had low occurrences and average citation scores/ publication compared to the other drug terms. The assessment of the words related to phytochemicals (30, 12.57) concluded that flavonoids (14, 8.57), luteolin (10, 7), quercetin (16, 7.31), rutoside (15, 9.13), Chinese medicine (11, 21.91) had on the average, lower occurrence and lower average citation score/ document compared to synthetic drugs.
3. The origin of SARS-CoV-2
Coronaviridae, Arteriviridae, Mesoniviridae, and Roniviridae comprise the order Nidovirales, of which coronaviruses are the largest group. The Coronaviridae family is further classified into two subfamilies, Torovirinae and Coronavirinae. The Coronavirinae subfamily is subdivided into 4 genera [25], as it is presented in Fig. 5. The form of presentation has been chosen as to highlight the taxonomic hierarchy in biological classification of SARS-CoV-2.
Fig. 5.

A brief taxonomy of coronaviruses.
HCoV-229E and HCoV-NL63 of the genus Alphacoronavirus usually cause common colds, but they can produce complications in immunocompromised patients or the elderly. Betacoronaviruses like HCoV-OC43 and HCoV-HKU1 are non-virulent, while SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) may cause severe symptoms. Alphacoronavirus and Betacoronavirus also contain animal viruses. Furthermore, only the viruses found in animals have been identified as Gammacoronavirus and Deltacoronavirus [26], [27], [28].
SARS-CoV-2 was identified by genetic sequencing [29]. Formerly known as the novel coronavirus 2019 (2019-nCoV), SARS-CoV-2 is a new betaCoV belonging to the same subgenus as SARS-CoV and MERS-CoV viruses that caused epidemics with fatality rates of 10 % and 35 %, respectively [30].
The origin of SARS-CoV-2 remains unknown, and the generally accepted hypothesis is that the disease was transmitted from animal to human (zoonotic transmission). In silico-related studies describing the interactions between human ACE-2 receptor homologues, TLRs and SARS-CoV-2 spike glycoprotein [31] showed that the glycoprotein can be considered phylogenetically as close to the coronavirus found in bats, binding mighty to the ACE2 receptor protein (both animal − bat, and human). In the same study, it was revealed that cell surface TLRs (i.e., TLR4) are probably the most involved in recognizing the patterns of molecules induced by SARS-CoV-2, which produce the characteristic inflammatory responses. Thus, the data come to support the zoonotic origin (from a bat) hypothesis of SARS-CoV-2 [32].
The genome of the new coronavirus is 96.2 % similar to RaTG13, a β-CoV found in bats, and 79.5 % to SARS-CoV [33], [34]. The majority of the SARS-CoV-2 genome is represented by open reading frames (ORF1a and ORF2b), which will be translated into two pp1a and pp1ab polyproteins. The two polyproteins will be cleaved by papain-like protease (PLpro) and main-protease viral proteases (3CL pro, M pro) to create 16 non-essential proteins (nsp). The rest of the genome will be translated into four structural proteins spike (S), Membrane (M), Envelope (E), and Nucleocapsid (N) [35].
4. Drug repositioning
The process of developing new drugs is costly and time-consuming. The pharmaceutical industry is experiencing issues due to the low number of novel pharmaceuticals launched onto the market in recent years. To address this issue, pharmaceutical companies have resorted to the following methods: improved formulation (modified release), improved bioavailability by co-administration of two pharmaceuticals (e.g. pharmacokinetic enhanced protease inhibitors with ritonavir or cobicistat [36]), and drug repositioning (Fig. 1) (finding new indications for drugs already approved for human use) [10], [37].
The latter method was used at the onset of the COVID-19 pandemic when in the absence of a viable treatment, the only therapeutic option was to use drugs already on the market. Many of the compounds used against SARS-CoV-2 have proven ineffective (e.g. ivermectin), and the efficacy of others has decreased due to the evolution of the virus (sfotrovimab has low activity against the subvariant Omicron BA.2.) [12], [38]. Remdesivir was the only repositioned drug that remained as a first-line treatment among the drugs included in the WHO therapeutic guidelines for COVID-19 therapy. Ritonavir-boosted Nirmatrelvir, Bebtelovimab, Molnupiravir, and Sotrovimab were developed de novo for the treatment of SARS-CoV-2 infection [38], [39].
5. Molecular docking
Molecular docking is the process of simulating how a receptor will interact with a ligand. The ligand is usually a small molecule that is thought to have a pharmacological effect, while the receptor is a protein or nucleic acid molecule [5]. Because molecular docking predicts the three-dimensional structure of the ligand-protein and the orientation of the ligand, this method can be used in the discovery of new drugs or in drug repositioning. The binding potential of the ligand to the receptor is calculated using different algorithms that consider possible interactions (e.g., columbic, van der Waals) [40], [41].
The foundations of molecular docking have been laid since 1980, and considerable progress has been made to date. The software is far more accurate and accessible. Thus, academic researchers conduct molecular docking studies more frequently ( Fig. 7) [42], [43], [44]. If the target protein's structure has not been identified, the homology modeling approach can be utilized, which entails building a protein with an unknown structure from a homologous one whose structure is known. [41].
Fig. 6.

The advantages of drug repositioning compared to de novo drug design.
Fig. 7.

Number of articles/years regarding molecular docking (data from PubMed).
The way a ligand binds to a certain protein (position prediction) gives information about the interactions between them. Binding energy is often used to compare ligands with similar chemical structures (the hypothesis being that an analog or a substance with a higher affinity than the native ligand may cause competitive inhibition). Furthermore, the goal of VS is to discover new ligands with therapeutic potential [32], [45].
Following the docking studies, a checkup is necessary to see if the approach used or the software functioned appropriately. The verification process involves re-docking the native ligand, and calculating root mean square deviation (RMSD) [45]. Calculating the RMSD determines the distance between a pair of superimposed atoms, the value of which is calculated for heavy backbone atoms. The docking protocol is considered better in molecular docking when the RMSD value is low. [46].
The pandemic has brought to light a significant issue affecting humanity, namely the absence of therapies for new and quickly spreading illnesses. In such cases, one alternative is to find new therapeutic indications for authorized drugs, and molecular docking is a helpful tool to achieve this. [47].
6. Virtual screening (VS)
Pharmaceutical companies employ high-throughput screening (HTS) to identify novel compounds. HTS is an automated process that uses robots paired with high-sensitivity sensors to examine a large number of molecules in a short time [48]. However, HTS is an expensive method that requires special equipment, which has led to the need to find a more economical alternative. Thus, VS has been developed, through which numerous compounds are docked on a protein, being previously downloaded from a database ( Table 5) that can be open access. A protein whose crystallographic structure is known is commonly used, but there is also the possibility of generating a protein by homology modeling [49], [50], [51].
Table 5.
Compound databases.
| Database | Availability | No. of compounds |
|---|---|---|
| PubChem | Open access | 11,000,000 |
| ChEMBL | 2200,000 | |
| ZINC | 2300,000 | |
| DrugBank | 14,624 | |
| SWEETLEAD | 4442 | |
| TCMSP | 3311 | |
| Asinex | 575,302 | |
| CAS Antiviral COVID19 | 50,000 | |
| ChemDiv | Commercial | 1522,032 |
| Enamine | 2157,315 | |
| Princeton Biomolecular Research | 1533,024 |
The most significant benefit of VS is the reduction in the number of substances that must be subjected to expensive experiments. The most significant disadvantage is that a compound with pharmacologic potential can be omitted by the algorithm [52]. VS methods are usually classified into ligand-based VS (LBVS) and structure-based VS (SBVS). In the case of LBVS, a ligand is used as a reference, and the database is searched for molecules with similar characteristics. SBVS approaches are substantially more complex since they consider a protein macromolecule. Ligand classification is based on the degree of the ligand fit in the protein active site (ligand-protein match) [53], [54].
7. Molecular dynamics
Molecular dynamics simulations are in-silico methods that predict the behavior of a system over time. The rapid evolution of computer hardware has also benefited the field of molecular dynamics. Simulations, which once needed the most advanced computers of the time, can be done today on a computer with mid-range specifications [55], [56]. The increased availability of these simulations is mirrored in the number of published articles ( Fig. 8).
Fig. 8.

Number of articles/years related to molecular dynamics (data from PubMed).
Docking or VS experiments usually have a mobile ligand-rigid protein, although proteins are mobile in the physiological environment. The association of these studies with molecular dynamics is used to improve the accuracy of the results [57], [58].
8. Targeted proteins using in silico methods
8.1. 3-chymotrypsin like protease (3CLpro, Mpro)
The SARS-CoV-2 genome contains at least six ORFs, of which ORF1ab encodes two polyproteins, pp1a and pp1ab. These will be cleaved by the 3CLpro and PLpro ( Fig. 9) and as a result, 16 non-structural proteins (nsp) which are essential to the virus's life cycle are formed [59], [60]. 3CLpro is a dimer composed of two symmetrical protomers further subdivided into three domains. Domain I is represented by residue 8–101, domain II by residue 102–184, and domain III by residue 201–303. A catalytic dyad represented by histidine41 (belongs to domain I) and cysteine145 (belongs to domain II) forms the active site of 3CLpro [61].
Fig. 9.

Mechanism of action of 3-chymotrypsin like protease- 3CLpro and papain-like protease PLpro, pp1a and pp1ab- polyprotein 1a and 1ab, nsps- non-structural proteins.
3CLpro has no known structural analog in the human body, so discovering a therapeutic agent that targets the mechanism of action of 3CLpro will not show negative effects in the host cell [62], [63], [64]. Table 5 summarizes some of the SBVS studies performed searching for compounds with inhibitory potential on 3Clpro. The PDB ID of the target protein and the software used for molecular docking were coupled with molecular dynamics.
The crystal structure of 3CLpro SARS-CoV-2 with the PDB ID: 6LU7 was published on 2020–02–05 by Jin et al., 6LU7 has a resolution of 2.16 Å and is co-crystallized with a molecule that functions as a protease inhibitor. This molecule was obtained by computer-aided drug design, following the analysis of a large number of compounds (over 10,000) [85]. Many protease inhibitors used in the treatment of HIV/SIDA and hepatitis C ( Table 6) have been identified as possible inhibitors of 3CLpro protease by docking studies and docking studies combined with VS [86].
Table 6.
A summary of in-silico studies targeting 3CLpro.
| Target | Docking software | Molecular dynamics | Drugs tested | Promising anti-viral ligands | Ref. |
|---|---|---|---|---|---|
| 6LU7 | AutoDock Vina | – | ZINC database. 10 FDA-approved drugs | ZINC32960814, ZINC12006217, ZINC03231196 | [65] |
| 6LU7 | Autodock Vina | – | TCMSP database | Puerarin, bicuculline, luteolin | [66] |
| 6LU7 | Glide | DESMOND | Seaweed Metabolite Database | Nigricanoside A, Nigricanoside B, Callophysin A | [67] |
| 6M2N | AutoDock Vina | YASARA | 32 phytochemicals | Amentoflavone, Gallocatechin |
[68] |
| 6LU7 | AutoDock Vina | GROMACS | 92 phytochemicals | Hyperin, lupinifolin, rutin | [69] |
| 6LU7 | LeDock software | GROMACS | ZINC database | Viomycin, capastat | [70] |
| 6LU7 | AutoDock Tool | AMBER 16 | Phytochemicals | Demethoxycurcumin, Bisdemethoxycurcumin, Scutellarin, Quercetin, Myricetin | [71] |
| 6Y2F | AutoDock Vina | – | 263 phytochemicals, 75 FDA-approved drugs | Alloyohimbine Gummadiol, Asparagamine A Vincapusine, Simeprevir Ledipasvir, Paritaprevir, Glecaprevir, Daclatasvir | [72] |
| 6LU7 | MOE, Autodock Vina | – | FDA-approved Drug Library from Selleckchem | Oxytetracycline, Naringin, Kanamycin, Cefpiramide, Salvianolic Acid B, Teniposide, Etoposide, Doxorubicin | [73] |
| 6LU7 | AutoDock Vina | – | 154 phytochemicals | Benzoylgedunin, Glycyrrhizic Acid, Limonin, Obacunone | [74] |
| 6M03 | AutoDock Vina | GROMACS | PubChem database. | 6-Deaminosinefungin, UNII-O9H5KY11SV | [75] |
| 6LU7 | DOCK 6 | AMBER 16 | SWEETLEAD database of drug molecules | Indinavir, Ivermectin, Cephalosporin-Derivatives, Neomycin and Amprenavir | [76] |
| 6LU7 | AutoDock Vina | GROMACS | Zinc databse, drugbank database | Tideglusib, Nilotinib, Amentoflavone | [77] |
| 6LU7 | Schrödinger’s Covalent Docking | AMBER 18 | Asinex Focused Covalent library 116- FDA-approved drugs | Simeprevir, Paritaprevir | [78] |
| 6LU7 | The smina software | AMBER 18 | CAS Antiviral COVID19 database | 4-(morpholin-4-yl)− 1,3,5-triazin-2-amine derivatives: 2001083–68–5 and 2001083–69–6 | [79] |
| 6LU7 | Maestro | AMBER 18 | 31 FDA approved drugs TCM | Saquinavir TCM5280805, TCM5280445, TCM5280343, TCM5280863, TCM5458190, |
[80] |
| 6LU7 | Glide | Schrödinger software | Over 1000 analogs were prepared by the Maestro tool. | 25 unnamed Hydroxyethylamine Analogs | [81] |
| 6LU7 | UCSF Chimera platform | AMBER 18 | 97 compounds from marine and terrestrial fungi | Quinadoline, Scedapin C, Polyketide isochaetochromin D1 | [82] |
| 6LU7 | AutoDock vina | – | Bioactive alkaloids (62) and terpenoids (100) | 0-Hydroxyusambarensine, Cryptoquindoline, 6-oxoisoiguesterin, 22-hydroxyhopan3-one |
[83] |
| 6LU7 | Autodock4 | GROMACS | 93 ligands Generative Adversarial Network | 6 unnamed compounds | [84] |
Legend: FDA, Food and Drug Administration; MOE, Molecular Operating Environment; TCMSP, Traditional Chinese Medicine System Pharmacology Database; YASARA, Yet another scientific artificial reality application
Protease inhibitors that are conventionally used in the treatment of HIV / AIDS have a relatively high affinity for 3CLpro, making them suitable candidates for inhibiting the SARS-CoV-2 protease. Darunavir and Saquinavir have affinities of − 7.7 / − 9.5 kcal/mol, respectively. Grazopavir and Paritaprevir, two NH3 / 4 A protease inhibitors used to treat HCV, have affinities to 3Clpro of − 8.1 / − 9.5 kcal/mol, respectively. According to docking studies on 3CLpro and the fact that they act on the protease of another RNA virus (HCV/ HIV/SIDA), these compounds may also interfere with SARS-CoV-2 3CLpro.
8.1.1. A quick look at nirmatrelvir
The mechanism of action is specific to this class and prevents 3CLpro from processing the pp1a and pp1ab polyproteins [106]. It is coupled with ritonavir, a human immunodeficiency virus type 1 (HIV-1) protease inhibitor that plays a vital role in lowering the metabolization rate of Nirmatrelvir, by inhibition of the CYP3A isoenzymes [107].
According to WHO, it is the most effective therapy for high-risk patients to date (e.g., unvaccinated or immune-suppressed patients). The drug is administered orally in the early stages of the disease. The pharmaceutic concept of the drug is not new, and it is based on a compound developed for the treatment of SARS-CoV. The Emergency use authorization was granted within a year after Phase 1 clinical studies got started. [108].
In terms of in-silico docking, Nirmatrelvir was docked to the 6Y2F protein, and the authors observed that it binds to the following residues through hydrogen bonds: Cys145, Glu166, and Gln189 [109]. Another docking study on the protein with the PDB ID: 7SI9 reported that Nirmatrelvir interacts with the protein via hydrogen bonds with the following amino acids: Gly143, Phe140, and Glu 166 [110].
8.2. Papain-like protease (PLpro)
Another SARS-CoV-2 protease that anti-virals might target is PLpro , because it is involved in processing the viral polyproteins (Fig. 4) of SARS-CoV-2, allowing viral spread [111]. It has been demonstrated that it also interfere with host cell proteins by modulating the host viral response to facilitate the spread of the virus [112].
Fig. 4.

Bubble map of 111 terms with at least 20 occurrences. Bubble size is directly proportional to word occurrence. The bubble color indicates the average citation count received by publications containing the word (title/abstract). If the bubbles are in closer proximity, the two words have more frequent co-occurrences.
Proteases are one of the primary targets that researchers are examining in the battle against viral infections. In silico studies on PLpro have been conducted to discover inhibiting drugs for this dual-effect protease. Table 7 presents a series of in-silico studies aimed at discovering PLpro inhibitors.
Table 7.
A summary of protease inhibitors docked on 6LU7.
| Docking software |
Ligand | Affinity | Mechanism | Used in | References |
|---|---|---|---|---|---|
| Target | |||||
| AutoDock Vina 6LU7 |
Ciluprevir | -9,1 | NH3/4 A protease inhibitor |
Hepatitis C | [87], [88], [89] |
| Grazoprevir | -8,1 | [87], [90] | |||
| Simeprevir | -9,0, − 9.0 | [87], [91] | |||
| Paritaprevir | -9,5 | [87], [92] | |||
| Faldaprevir | -8,4 | Of interest in treating hepatitis. |
[87], [93] | ||
| Indinavir | -8,5 -8.3 |
Protease inhibitor | HIV/AIDS | [87], [94], [95] | |
| Saquinavir | -8,5, -8,9 -9.5 |
[87], [94], [96] | |||
| Nelfinavir | − 9.1 | [97], [98] | |||
| -7.9 | |||||
| Atazanavir | -8.1 | [94], [99] | |||
| Darunavir | -7.7 | [94], [100] | |||
| Ritonavir | -7.3 -7.6 |
[101], [102] | |||
| Brecanavir | -8,1 | [87], [103] | |||
| Lopinavir | -8,1, − 9.3, − 8.4 -7.9 − 8.2 |
[87], [94], [97], [101], [104], [105] |
Legend: HIV, human immunodeficiency virus; AIDS, acquired immunodeficiency syndrome.
8.3. RNA-dependent RNA polymerase (RdRp)
RdRp is a non-structural protein with a key role in genomic transcription and replication. The analysis of the crystallographic structure of RdRp shows the distinct shape of the right hand consisting of a palm, fingers, and thumb. [129], [130]. Involved in genomic transcription and replication, RdRp is considered a valuable therapeutic target ( Fig. 11). RdRp has no structural homologs in the human cell, so drugs that act specifically on this protein should, in theory, not cause side effects induced by the drug's mechanism of action [131], [132].
Fig. 10.
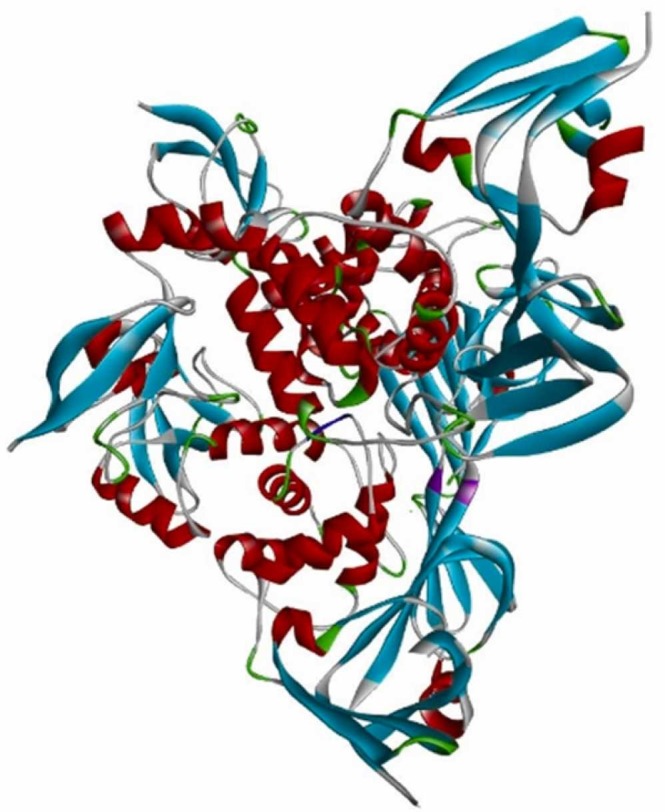
The 3D structure of PLpro (PDB ID: 6W9C).
Fig. 11.

Nucleoside analogs mechanism of action. RNA, ribonucleic acid; RdRp, RNA-dependent RNA polymerase.
In the research process of RdRp inhibitors, the most promising drug class is that of nucleoside analogs [133]. VS studies sought to find RdRp inhibitors, and the most promising compounds were the already approved anti-virals and used as nucleoside analogs in RNA viral infections. The fact that the active sites of these enzymes retain their fundamental properties in most RNA viruses increases the interest in RdRp-targeted anti-viral for the treatment of SARS-CoV-2 infection [134], [135]. Until the crystallographic structure of RdRp was identified, molecular docking studies were based on homology models to build the protein needed to perform these studies [136], [137], [138]. After the publication of the crystallographic structure of RdRp (e.g. PDB ID: 7BW4 [139] 6M71 [140], homology models have diminished in significance. Table 8 reviews SBVS studies focused on finding RdRp inhibitors.
Table 8.
A summary of in-silico studies targeting 3CLpro.
| Target | Docking software | Molecular dynamics | Drugs tested | Promising anti-viral ligands | Ref. |
|---|---|---|---|---|---|
| 6W9C | AutoDock Tools | GROMACS | Multiple drug databases | MFCD00832476, MFCD02180753, Bemcentinib, Pacritinib Ergotamine | [113] |
| 7JN2 | Maestro | Desmond | Database generated by AI integration | A3659, A3777, A3777 | [114] |
| 7CMD | MOE | GROMACS | In-house database contains 35,000 compounds | Unnamed | [115] |
| 6WX4 | Maestro | AMBER | 149 polyphenols | Leucopelargonidin, Taxifolin, Morin, Eriodictyol, Myricetin, Enterodiol | [116] |
| 6WX4 | YASARA | YASARA | 14,000 phytochemicals | Baicalin, Hesperidin, Naringen, Flemiflavanone D, Euchrestaflavanone A | [117] |
| 7CJM | AutoDock Vina | AMBER | FoodComEx database | Triamterene, Estrone, Ibuprofen, Chlorpheniramine | [118] |
| 6W9C) | AutoDock Tools | GROMACS | ChEBML database | Azadirachtin-H Azadirachtin-I Azadirachtin-Q Azadirachtin | [119] |
| 7JN2 | Glide | Desmond | SuperNatural Database | SN00334175, SN00162745 | [120] |
| 6W9C) | AutoDock Vina AutoDock Tools iGEMDOCK |
LARMD | Phytochemicals of Nigella sativa | Campesterol, Cycloeucalenol, Alphaspinasterol, Taraxerol, | [121] |
| 6W9C | AutoDock Vina too | GROMACS | 40 related to S. Sonchilofolius and L. Meyenii | Rutin | [122] |
| 6XAA | AutoDock Vina | AMBER | MPD3 database + reported natural anti-viral compounds | Gycyrrhizin, Azadirachtani, Mycophenolic acid, Kushenol-w, 6-azauridine | [123] |
| 6WX4 | Maestro | – | ChEMBl 2390 phase III and IV clinical trial drugs | Curcumin, Afatinib | [124] |
| 6W9C | AutoDock Vina Swiss Dock |
GROMACS | ZINC15 drug database – 291 FDA approved drugs | Thiamine, Levodopa Naloxone | [125] |
| 7JIW | AutoDock Vina | AMBER | FooDB database |
Diosbulbinoside D, Estrone-2,3-quinone, Cyanidin 5-O-β-D-glucoside |
[118] |
| 7JRN | CDOCKER | – | 9032 drugs from TCM database | Hinokiflavone, Morelloflavone, Methylochnaflavon, Amentoflavone, Ginkgetin, Isoginkget, Sciadopitysin, Podocarpusflavone A, Cryptomerin | [126] |
| 6W9C | UCSF Chimera platform | AMBER | 97 terrestrial fungi metabolites | Norquinadoline A, Scedapin C, Quinadoline B, Cytochalasin Z8 | [127] |
| 6W9C | Chimera-AutoDock Vina plugin | Desmond | 46 bioactive flavonoids | Naringin, Hesperidin | [128] |
FDA, Food and Drug Administration; MOE, Molecular Operating Environment; TCMSP, Traditional Chinese Medicine System Pharmacology Database; YASARA, Yet another scientific artificial reality application.
8.3.1. Remdesivir
Remdesivir is a nucleotide analog designed to target RBA viruses (e.g., hepatitis C). It is a prodrug that is bioactivated in the body and functions as an adenosine triphosphate analog (ATP). Fig. 5 summarizes the mechanism of action. The suggested administration model is one dose on the first day, followed by two more on subsequent days. According to research, it must be administered in the early stages of the infection [154], [155].
Since the pandemic's beginning, in-silico studies that recommended remdesivir for the treatment of COVID-19 have begun to emerge, using the keywords, ”molecular docking" and, remdesivir". Table 9 summarizes several studies involving remdesivir. Most of these studies used remdesivir as a ligand control.
Table 10.
Docking studies that include Remdesivir.
| Target | Molecular docking software | Binding affinity Kcal/mol |
Hydrogen bond with | Ref |
|---|---|---|---|---|
| 6M71 | Autodock Vina | -9 | ASN781, HIS133, SER709, TYR129 LYS A47, ASP711 | [156] |
| 6M71 | Autodock Vina | − 8 | LYS47, TYR129, SER709, ASP711, ASN781 | [146] |
| 6M71 | Autodock Vina | − 7.1 | Lys621, Cys622, Asp761, Lys798, Glu811. | [157] |
| 7VH8 | AutoDock 4.2 | -8.56 | GLY143 CYS145 LEU167 TYR54 CYS145 GLU166 | [158] |
| 6M71 | AutoDock Vina | -7.8 | Gly143, Ser144 (2), Cys145, Glu166, Asn142 | [159] |
| 7BV2 | AutoDockVina | -7.2 | ILE23, LEU126, GLY48 | [160] |
| 7BTF | DOCK 6 | -8.8 | ALA 550, LYS 55, ARG 55, CYS 813, SER814, GLN 815 | [161] |
Legend: ASN, asparagine; HIS, histidine; SER, serine; TYR, tyrosine; LYS, lysine; ASP, aspartic acid; ALA, alanine; ARG, arginine; CYS, cysteine; GLN, glutamine; GLY, glycine; LEU, leucine; GLU, glutamic acid; ILE, isoleucine.
Table 9.
A summary of in-silico studies targeting RdRp.
| Target | Docking software | Molecular dynamics | Drugs tested | Promising anti-viral ligands | Ref. |
|---|---|---|---|---|---|
| 7BV2 | AutoDock Vina, Glide, rDock | GROMACS | 1615 FDA-approved drugs, ZINC database |
Leucal, Natamycin, Folic Acid | [101] |
| 7BW4 | DockThor, Autodock Vina, PatchDock | GROMACS | 48 antivirals | Ribavirin, Zanamivir, Penciclovir | [141] |
| 6M71 | AutoDock Vina | GROMACS | 92 phytochemicals | Hesperidin, Rutin, Quercetin | [69] |
| 7BTF | AutoDock Vina | NAMD VMD |
Novel adenosine derivatives | 4 unamed adenosine derivates | [142] |
| 6M71 | Autodock 4.0.1. | – | 65 FDA approved small molecule | Raltegravir, Indinavir, Tipranavir, Dolutegravir, Etravirine |
[143] |
| 6M71 | Moe-Dock | GROMACS | 63 anti-viral drugs approved by the FDA | Ledipasvir, Remdesivir, Paritaprevir | [144] |
| 6M71 | AutoDock Vina | – | Flavonoids, phenolic acids, and terpenes | p-Coumaric Acid, Ellagic Acid, Kaempferol, Quercetin | [145] |
| 6M71 | Discovery studio | Desmond | Zinc15 DataBank | Paritaprevir, Glecaprevir, Velpatasvir, Remdesivir, Ribavirin | [146] |
| 7BTF | Autodock Vina | – | FDA Drug library | Ketazolam, Methylnaltrexone, Ethynodiol Diacetate | [147] |
| 7BV2 | Maestro | GROMACS | 480 tested polyphenos were retrieved from Phenol-Explorer 3.6 | Cyanidin 3-O-rutinoside, Petunidin 3,5-O-diglucoside, Delphinidin 3-O-rutinoside | [148] |
| 6M71 | Autodock vina | YASARA | ZINC database | Grazoprevir, Ledipasvir, Galidesivir | [149] |
| 7BV2 | AutoDock Vina | GROMACS | Asinex EliteSynergy and BioDesign libraries | Las 51620435, Las 51620429 | [150] |
| 7BV2 | Schrodinger Glide | Desmond | FDA drugs drugbank | Rosoxacin, Levomefolic, Etodolac | [151] |
| 6M71 | Schrodinger Glide | GROMACS | 75 FDA approved antiviral drugs | Ritonavir, Dolutegravir, Tenofovir, Tinofoviralafenamide, Boceprevir, Catechin and Zanamivir | [152] |
| 6M71 | Schrodinger Glide | – | FDA approved drug database | Ornipressin, Otosiban, Lanreotide, Argiprestocin, Demoxytocin, Carbetocin, Lypressin, Examorelin, Colistin, Polymyxin B1 | [153] |
Legend: FDA, Food and Drug Administration; MOE, Molecular Operating Environment; YASARA, Yet another scientific artificial reality application.
9. Conclusions and future perspectives
In-silico docking is continually changing, and previously unavailable approaches due to hardware limitations or a lack of target proteins have become widely available. Researchers' interest in this topic has grown due to the large number of macromolecules published on the Protein Data Bank (190404) and the fast advancement of hardware components. In order to discover new, innovative drug substances, pharmaceutical companies are increasingly using HTS, but the high costs make this method require a more economical alternative. The in-silico counterpart of HTS is VS, which enables the test of a large number of compounds in a short amount of time.
VS, more specifically, SBVS in combination with molecular dynamics, has the potential to play a vital role in the reuse or even development of medicinal compounds. Several experiments were conducted to find pharmacological compounds that may act on this RNA virus during the pandemic. Drugs that act on the target structures of other RNA viruses were among the drugs considered. Drugs such as protease inhibitors used in treating HIV / AIDS or hepatitis C have been subjected to docking studies. Compounds of natural origin have been rigorously tested against all SARS-CoV-2 targets in the hope of finding a potential inhibitor.
Currently, in-silico studies are being utilized to limit the number of more costly in-vitro / in-vivo candidates. The introduction of a standard approach is a first step that must be made before the usefulness of in-silico procedures rises, especially when it comes to molecular docking. Because most programs employ different algorithms and scoring techniques, reproducibility of findings from one program to another is nearly impossible. Artificial intelligence (AI) and deep learning (DL) have become more prevalent in recent years, and public interest in these technologies has increased. One of the most significant advancements in in-silico research is the incorporation of AI into the drug development process. These approaches are becoming more widely available and are highly accurate. However, it has been observed that in silico methods are extremely useful when it comes to researching the therapeutic potential of different molecules, analyzing the interactions between viral targets and their potential/possible inhibitors, as well as designing new agents usable in prevention or therapy. The applicability of these methods of drug screening, is of real use both in the field of designing innovative drugs for treating various aspects of COVID-19 and regarding their applicability in the event of a future pandemic. In summary, the development of all these biomedical tools mentioned above opens new opportunities in testing and discovering innovative therapies, contributing to obtaining fundamental results with immediate usability in adopting precise intervention strategies / protocols in cases of need or emergency.
Funding
The APC was funded by the University of Oradea. The research was funded by Romanian Ministry of Research, Innovation and Digitisation through Programme 1—Development of the National Research and Development System, Subprogramme 1.2—Institutional Performance—Projects for funding the excellence in RDI, Contract No. 29 PFE/30.12.2021 with University of Oradea, Oradea, Romania.
CRediT authorship contribution statement
Paul Andrei Negru: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. Denisa Claudia Miculas: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft. Tapan Behl: Supervision, Methodology. Alexa Florina Bungau: Visualization, Data curation, Investigation. Simona Gabriela Bungau: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Visualization, Writing – review & editing.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors also thank the University of Oradea, for supporting the publication of this manuscript.
Data Availability
All data used in this research are supported by the References inserted in the References section.
References
- 1.Vellido A. The importance of interpretability and visualization in machine learning for applications in medicine and health care. Neural Comput. Appl. 2020;32:18069–18083. doi: 10.1007/s00521-019-04051-w. [DOI] [Google Scholar]
- 2.Mehmood M.A., Sehar U., Ahmad N. Use of bioinformatics tools in different spheres of life sciences. J. Data Min. Genom. Proteom. 2014;5:1. [Google Scholar]
- 3.Xia X. Bioinformatics and drug discovery. Curr. Top. Med Chem. 2017;17:1709–1726. doi: 10.2174/1568026617666161116143440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behl T., Kaur I., Sehgal A., Singh S., Bhatia S., Al-Harrasi A., Zengin G., Babes E.E., Brisc C., Stoicescu M., et al. Bioinformatics accelerates the major tetrad: a real boost for the pharmaceutical industry. Int. J. Mol. Sci. 2021:22. doi: 10.3390/ijms22126184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dias R., de Azevedo J., Walter F. Molecular docking algorithms. Curr. Drug Targets. 2008;9:1040–1047. doi: 10.2174/138945008786949432. [DOI] [PubMed] [Google Scholar]
- 6.Pinzi L., Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019;20:4331. doi: 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sussman J.L., Lin D., Jiang J., Manning N.O., Prilusky J., Ritter O., Abola E.E. Protein data bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr D. Biol. Crystallogr. 1998;54:1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- 8.Grosdidier A., Zoete V., Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menchon G., Maveyraud L., Czaplicki G. In Computational Drug Discovery and Design. Springer; 2018. Molecular dynamics as a tool for virtual ligand screening; pp. 145–178. [DOI] [PubMed] [Google Scholar]
- 10.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 11.Pizzorno A., Padey B., Terrier O., Rosa-Calatrava M. Drug repurposing approaches for the treatment of influenza viral infection: reviving old drugs to fight against a long-lived enemy. Front. Immunol. 2019:10. doi: 10.3389/fimmu.2019.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parvathaneni V., Gupta V. Utilizing drug repurposing against COVID-19 – efficacy, limitations, and challenges. Life Sci. 2020;259 doi: 10.1016/j.lfs.2020.118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassanipour S., Arab-Zozani M., Amani B., Heidarzad F., Fathalipour M., Martinez-de-Hoyo R. The efficacy and safety of Favipiravir in treatment of COVID-19: a systematic review and meta-analysis of clinical trials. Sci. Rep. 2021;11:11022. doi: 10.1038/s41598-021-90551-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ton A.-T., Gentile F., Hsing M., Ban F., Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020;39:2000028. doi: 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmezayen A.D., Al-Obaidi A., Şahin A.T., Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2021;39:2980–2992. doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan S.A., Zia K., Ashraf S., Uddin R., Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2021;39:2607–2616. doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- 19.Khan R.J., Jha R.K., Amera G.M., Jain M., Singh E., Pathak A., Singh R.P., Muthukumaran J., Singh A.K. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021;39:2679–2692. doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gentile D., Patamia V., Scala A., Sciortino M.T., Piperno A., Rescifina A. Putative inhibitors of SARS-CoV-2 main protease from A library of marine natural products: a virtual screening and molecular modeling study. Mar. Drugs. 2020;18:225. doi: 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pant S., Singh M., Ravichandiran V., Murty U.S.N., Srivastava H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2021;39:2904–2913. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarma P., Shekhar N., Prajapat M., Avti P., Kaur H., Kumar S., Singh S., Kumar H., Prakash A., Dhibar D.P., et al. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain) J. Biomol. Struct. Dyn. 2021;39:2724–2732. doi: 10.1080/07391102.2020.1753580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar Y., Singh H., Patel C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health. 2020;13:1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha S.K., Shakya A., Prasad S.K., Singh S., Gurav N.S., Prasad R.S., Gurav S.S. An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J. Biomol. Struct. Dyn. 2021;39:3244–3255. doi: 10.1080/07391102.2020.1762741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses. 2015:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burrell C.J., Howard C.R., Murphy F.A. Coronaviruses. Fenner White’S. Med. Virol. 2017:437–446. doi: 10.1016/B978-0-12-375156-0.00031-X. [DOI] [Google Scholar]
- 27.Van Der Hoek L. Human coronaviruses: what do they cause? Antivir. Ther. 2007;12:651–658. doi: 10.1177/135965350701200s01.1. [DOI] [PubMed] [Google Scholar]
- 28.Loeffelholz M.J., Tang Y.-W. Laboratory diagnosis of emerging human coronavirus infections – the state of the art. Emerg. Microbes Infect. 2020;9:747–756. doi: 10.1080/22221751.2020.1745095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keretsu S., Bhujbal S.P., Cho S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020;10:17716. doi: 10.1038/s41598-020-74468-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behl T., Kaur I., Aleya L., Sehgal A., Singh S., Sharma N., Bhatia S., Al-Harrasi A., Bungau S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total Environ. 2022:808. doi: 10.1016/j.scitotenv.2021.152072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhury S., Moulick D., Saikia P., Mazumder M.K. Evaluating the potential of different inhibitors on RNA-dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2: a molecular modeling approach. Med. J. Armed Forces India. 2021;77:S373–S378. doi: 10.1016/j.mjafi.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludwig S., Zarbock A. Coronaviruses and SARS-CoV-2: a brief overview. Anesth. Analg. 2020:131. doi: 10.1213/ANE.0000000000004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, evaluation, and treatment of coronavirus (COVID-19). Statpearls [internet] 2022. [PubMed]
- 35.Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H., Kumar S., Bhattacharyya A., Kumar H., Bansal S., et al. Drug targets for corona virus: a systematic review. Indian J. Pharm. 2020;52:56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marin R.-C., Streinu-Cercel A., Moleriu L.C., Bungau S.G. Analysis of virological response to therapy and resistance profile in treatment-experienced and naive HIV-1 infected Romanian patients receiving regimens containing darunavir boosted with ritonavir or cobicistat. Biomed. Pharmacother. 2022;150 doi: 10.1016/j.biopha.2022.113077. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S., Kumar S. In: In In Silico Drug Design. Roy K., editor. Academic Press; 2019. Chapter 6 - Molecular Docking: A Structure-Based Approach for Drug Repurposing; pp. 161–189. [Google Scholar]
- 38.What's New in the Guidelines. Available online: 〈https://www.covid19treatmentguidelines.nih.gov/about-the-guidelines/whats-new/〉 (accessed on May 10, 2022).
- 39.Babadaei M.M.N., Hasan A., Vahdani Y., Bloukh S.H., Sharifi M., Kachooei E., Haghighat S., Falahati M. Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. J. Biomol. Struct. Dyn. 2021;39:3771–3779. doi: 10.1080/07391102.2020.1767210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng X.-Y., Zhang H.-X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pagadala N.S., Syed K., Tuszynski J. Software for molecular docking: a review. Biophys. Rev. 2017;9:91–102. doi: 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuntz I.D., Blaney J.M., Oatley S.J., Langridge R., Ferrin T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-X. [DOI] [PubMed] [Google Scholar]
- 43.Sousa S.F., Fernandes P.A., Ramos M.J. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 44.Elokely K.M., Doerksen R.J. Docking challenge: protein sampling and molecular docking performance. J. Chem. Inf. Model. 2013;53:1934–1945. doi: 10.1021/ci400040d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guedes I.A., de Magalhães C.S., Dardenne L.E. Receptor–ligand molecular docking. Biophys. Rev. 2014;6:75–87. doi: 10.1007/s12551-013-0130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kufareva I., Abagyan R. In Homology Modeling. Springer; 2011. Methods of protein structure comparison; pp. 231–257. [Google Scholar]
- 47.Gol S., Pena R.N., Rothschild M.F., Tor M., Estany J. A polymorphism in the fatty acid desaturase-2 gene is associated with the arachidonic acid metabolism in pigs. Sci. Rep. 2018;8:1–9. doi: 10.1038/s41598-018-32710-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szymański P., Markowicz M., Mikiciuk-Olasik E. Adaptation of high-throughput screening in drug discovery—toxicological screening tests. Int. J. Mol. Sci. 2012;13:427–452. doi: 10.3390/ijms13010427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ripphausen P., Nisius B., Peltason L., Bajorath J., Vadis Q. Virtual screening? a comprehensive survey of prospective applications. J. Med. Chem. 2010;53:8461–8467. doi: 10.1021/jm101020z. [DOI] [PubMed] [Google Scholar]
- 50.Muegge I., Oloff S. Advances in virtual screening. Drug Discov. Today.: Technol. 2006;3:405–411. doi: 10.1016/j.ddtec.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bajorath J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002;1:882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 52.McInnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 53.Gimeno A., Ojeda-Montes M.J., Tomás-Hernández S., Cereto-Massagué A., Beltrán-Debón R., Mulero M., Pujadas G., Garcia-Vallvé S. The light and dark sides of virtual screening: what is there to know? Int. J. Mol. Sci. 2019;20:1375. doi: 10.3390/ijms20061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng T., Li Q., Zhou Z., Wang Y., Bryant S.H. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14:133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansson T., Oostenbrink C., van Gunsteren W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002;12:190–196. doi: 10.1016/S0959-440X(02)00308-1. [DOI] [PubMed] [Google Scholar]
- 56.Hollingsworth S.A., Dror R.O. Molecular dynamics simulation for all. Neuron. 2018;99:1129–1143. doi: 10.1016/j.neuron.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nichols S.E., Baron R., Ivetac A., McCammon J.A. Predictive power of molecular dynamics receptor structures in virtual screening. J. Chem. Inf. Model. 2011;51:1439–1446. doi: 10.1021/ci200117n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu M., Yuan M., Luo M., Bu X., Luo H.-B., Hu X. Binding of curcumin with glyoxalase I: Molecular docking, molecular dynamics simulations, and kinetics analysis. Biophys. Chem. 2010;147:28–34. doi: 10.1016/j.bpc.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., Lu B.G., Kuchel N.W., Grohmann C., Shibata Y., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. Embo J. 2020;39 doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.ul Qamar M.T., Alqahtani S.M., Alamri M.A., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020;10:313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreira J.C., Fadl S., Villanueva A.J., Rabeh W.M. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 Protease 3-chymotrypsin-like protease. Front Chem. 2021;9:491. doi: 10.3389/fchem.2021.692168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiao J., Li Y.-S., Zeng R., Liu F.-L., Luo R.-H., Huang C., Wang Y.-F., Zhang J., Quan B., Shen C. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shankar U., Kumar A. Discovery of new hydroxyethylamine analogs against 3CLproProtein target of SARS-CoV-2: molecular docking. Mol. Dyn. Simul., Struct. -Act. Relatsh. Stud. 2020 [Google Scholar]
- 64.Alfhad H., Saftarina F., Kurniawan B. Dampak infeksi SARS-Cov-2 terhadap penderita hipertensi. Majority. 2020:9. [Google Scholar]
- 65.Abdusalam A.A.A., Murugaiyah V. Identification of potential inhibitors of 3CL protease of SARS-CoV-2 from ZINC database by molecular docking-based virtual screening. Front Mol. Biosci. 2020:7. doi: 10.3389/fmolb.2020.603037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu J., Gao L., Liang H., Chen S.-D. In silico screening of potential anti–COVID-19 bioactive natural constituents from food sources by molecular docking. Nutrition. 2021;82 doi: 10.1016/j.nut.2020.111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muteeb G., Alshoaibi A., Aatif M., Rehman M.T., Qayyum M.Z. Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Appl. Biol. Chem. 2020;63:79. doi: 10.1186/s13765-020-00564-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swargiary A., Mahmud S., Saleh M.A. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: an in silico approach to combat COVID-19. J. Biomol. Struct. Dyn. 2022;40:2067–2081. doi: 10.1080/07391102.2020.1835729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mosquera-Yuqui F., Lopez-Guerra N., Moncayo-Palacio E.A. Targeting the 3CLpro and RdRp of SARS-CoV-2 with phytochemicals from medicinal plants of the Andean Region: molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022;40:2010–2023. doi: 10.1080/07391102.2020.1835716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q., Zhao Y., Chen X., Hong A. Virtual screening of approved clinic drugs with main protease (3CLpro) reveals potential inhibitory effects on SARS-CoV-2. J. Biomol. Struct. Dyn. 2022;40:685–695. doi: 10.1080/07391102.2020.1817786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma A., Goyal S., Yadav A.K., Kumar P., Gupta L. In-silico screening of plant-derived antivirals against main protease, 3CLpro and endoribonuclease, NSP15 proteins of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022;40:86–100. doi: 10.1080/07391102.2020.1808077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bahadur Gurung A., Ajmal Ali M., Lee J., Abul Farah M., Mashay Al-Anazi K. Structure-based virtual screening of phytochemicals and repurposing of FDA approved antiviral drugs unravels lead molecules as potential inhibitors of coronavirus 3C-like protease enzyme. J. King Saud. Univ. - Sci. 2020;32:2845–2853. doi: 10.1016/j.jksus.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meyer-Almes F.-J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and structure based drug design. Comput. Biol. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vardhan S., Sahoo S.K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput. Biol. Med. 2020;124 doi: 10.1016/j.compbiomed.2020.103936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohammad T., Shamsi A., Anwar S., Umair M., Hussain A., Rehman M.T., AlAjmi M.F., Islam A., Hassan M.I. Identification of high-affinity inhibitors of SARS-CoV-2 main protease: Towards the development of effective COVID-19 therapy. Virus Res. 2020;288 doi: 10.1016/j.virusres.2020.198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koulgi S., Jani V., Uppuladinne M., Sonavane U., Nath A.K., Darbari H., Joshi R. Drug repurposing studies targeting SARS-CoV-2: an ensemble docking approach on drug target 3C-like protease (3CLpro. J. Biomol. Struct. Dyn. 2021;39:5735–5755. doi: 10.1080/07391102.2020.1792344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olubiyi O.O., Olagunju M., Keutmann M., Loschwitz J., Strodel B. High throughput virtual screening to discover inhibitors of the main protease of the coronavirus SARS-CoV-2. Molecules. 2020;25:3193. doi: 10.3390/molecules25143193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alamri M.A., Tahir ul Qamar M., Mirza M.U., Bhadane R., Alqahtani S.M., Muneer I., Froeyen M., Salo-Ahen O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021;39:4936–4948. doi: 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aouidate A., Ghaleb A., Chtita S., Aarjane M., Ousaa A., Maghat H., Sbai A., Choukrad Mb, Bouachrine M., Lakhlifi T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2021;39:4522–4535. doi: 10.1080/07391102.2020.1779130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khan A., Ali S.S., Khan M.T., Saleem S., Ali A., Suleman M., Babar Z., Shafiq A., Khan M., Wei D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro. J. Biomol. Struct. Dyn. 2021;39:4659–4670. doi: 10.1080/07391102.2020.1779128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar S., Sharma P.P., Shankar U., Kumar D., Joshi S.K., Pena L., Durvasula R., Kumar A., Kempaiah P., Poonam, et al. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: molecular docking, molecular dynamics simulation, and structure–activity relationship studies. J. Chem. Inf. Model. 2020;60:5754–5770. doi: 10.1021/acs.jcim.0c00326. [DOI] [PubMed] [Google Scholar]
- 82.Quimque M.T.J., Notarte K.I.R., Fernandez R.A.T., Mendoza M.A.O., Liman R.A.D., Lim J.A.K., Pilapil L.A.E., Ong J.K.H., Pastrana A.M., Khan A., et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2021;39:4316–4333. doi: 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gyebi G.A., Ogunro O.B., Adegunloye A.P., Ogunyemi O.M., Afolabi S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2021;39:3396–3408. doi: 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Macchiagodena M., Pagliai M., Procacci P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020;750 doi: 10.1016/j.cplett.2020.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 86.Narkhede R.R., Cheke R.S., Ambhore J.P., Shinde S.D. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. Eurasia J. Med. Oncol. 2020;4:185–195. [Google Scholar]
- 87.Hakmi M., Bouricha E.M., Kandoussi I., Harti J.E., Ibrahimi A. Repurposing of known anti-virals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. Bioinformation. 2020;16:301–306. doi: 10.6026/97320630016301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ciluprevir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/9853710〉 (Accessed on.
- 89.Ciluprevir. Available online: 〈https://go.drugbank.com/drugs/DB05868〉 (Accessed on 12 May 2022).
- 90.Grazoprevir. Available online: 〈https://go.drugbank.com/drugs/DB11575〉 (Accessed on 11 May 2022).
- 91.Simeprevir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/24873435〉 (Accessed on 10 May 2022).
- 92.Paritaprevir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/45110509〉 (Accessed on 12 May 2022).
- 93.Kamal S.M. In: Hepatitis C in Developing Countries. Kamal S.M., editor. Academic Press; 2018. Chapter 6 - Hepatitis C treatment in the era of direct-acting antiviral agents: challenges in developing countries; pp. 209–246. [Google Scholar]
- 94.Qiao Z., Zhang H., Ji H.-F., Chen Q. Computational view toward the inhibition of SARS-CoV-2 spike glycoprotein and the 3CL protease. Computation. 2020;8:53. doi: 10.3390/computation8020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Indinavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/5362440〉 (Accessed on 10 May 2022).
- 96.Saquinavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/441243〉 (Accessed on 11 May 2022).
- 97.Sisakht M., Mahmoodzadeh A., Darabian M. Plant-derived chemicals as potential inhibitors of SARS-CoV-2 main protease (6LU7), a virtual screening study. Phytother. Res.: PTR. 2021;35:3262–3274. doi: 10.1002/ptr.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nelfinavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/#query=Nelfinavir〉 (Accessed on 10 May 2022).
- 99.Atazanavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/148192〉 (Accessed on 11 May 2022).
- 100.Darunavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/213039〉 (Accessed on 11 May 2022).
- 101.Hosseini, M., Chen, W., Wang, C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. 2020. [DOI] [PMC free article] [PubMed]
- 102.Hsu A., Granneman G.R., Bertz R.J.Ritonavir. Clin. Pharmacokinet. 1998;35:275–291. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 103.Brecanavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/5743186〉 Accessed on 12 May 2022).
- 104.Reiner Ž., Hatamipour M., Banach M., Pirro M., Al-Rasadi K., Jamialahmadi T., Radenkovic D., Montecucco F., Sahebkar A. Statins and the COVID-19 main protease: in silico evidence on direct interaction. Arch. Med. Sci. 2020;16:490–496. doi: 10.5114/aoms.2020.94655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lopinavir. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/92727〉 (Accessed on 10 May 2022).
- 106.Lamb Y.N. Nirmatrelvir plus ritonavir: first approval. Drugs. 2022;82:585–591. doi: 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hung Y.-P., Lee J.-C., Chiu C.-W., Lee C.-C., Tsai P.-J., Hsu I.-L., Ko W.-C. Oral nirmatrelvir/ritonavir therapy for COVID-19: the dawn in the dark? Antibiotics. 2022;11:220. doi: 10.3390/antibiotics11020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.WHO recommends highly successful COVID-19 therapy and calls for wide geographical distribution and transparency from originator. Available online: 〈https://www.who.int/news/item/22–04-2022-who-recommends-highly-successful-covid-19-therapy-and-calls-for-wide-geographical-distribution-and-transparency-from-originator〉 (accessed on May 25, 2022).
- 109.Ahmad B., Batool M., Ain Qu, Kim M.S., Choi S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021;22:9124. doi: 10.3390/ijms22179124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chalmers, G. Computational Study of Paxlovid in Ligand GA. 2022.
- 111.Yan S., Wu G. Spatial and temporal roles of SARS-CoV PLpro—a snapshot. FASEB J. 2021;35 doi: 10.1096/fj.202002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jade D., Ayyamperumal S., Tallapaneni V., Joghee Nanjan C.M., Barge S., Mohan S., Nanjan M.J. Virtual high throughput screening: potential inhibitors for SARS-CoV-2 PL(PRO) and 3CL(PRO) proteases. Eur. J. Pharm. 2021;901 doi: 10.1016/j.ejphar.2021.174082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang L.C., Zhao H.L., Liu J., He L., Yu R.L., Kang C.M. Design of SARS-CoV-2 Mpro, PLpro dual-target inhibitors based on deep reinforcement learning and virtual screening. Future Med. Chem. 2022;14:393–405. doi: 10.4155/fmc-2021-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tian X., Zhao Q., Chen X., Peng Z., Tan X., Wang Q., Chen L., Yang Y. Discovery of novel and highly potent inhibitors of SARS CoV-2 papain-like protease through structure-based pharmacophore modeling, virtual screening, molecular docking, molecular dynamics simulations, and biological evaluation. Front Pharm. 2022;13 doi: 10.3389/fphar.2022.817715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rudrapal M., Issahaku A.R., Agoni C., Bendale A.R., Nagar A., Soliman M.E.S., Lokwani D. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021:1–17. doi: 10.1080/07391102.2021.1944909. [DOI] [PubMed] [Google Scholar]
- 117.Rehman M.F.U., Akhter S., Batool A.I., Selamoglu Z., Sevindik M., Eman R., Mustaqeem M., Akram M.S., Kanwal F., Lu C., et al. Effectiveness of natural antioxidants against SARS-CoV-2? insights from the in-silico world. Antibiotics. 2021:10. doi: 10.3390/antibiotics10081011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhowmick S., AlFaris N.A., Zaidan Altamimi J., Alothman Z.A., Patil P.C., Aldayel T.S., Wabaidur S.M., Saha A. Identification of bio-active food compounds as potential SARS-CoV-2 PLpro inhibitors-modulators via negative image-based screening and computational simulations. Comput. Biol. Med. 2022;145 doi: 10.1016/j.compbiomed.2022.105474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kumar K.A., Sharma M., Dalal V., Singh V., Tomar S., Kumar P. Multifunctional inhibitors of SARS-CoV-2 by MM/PBSA, essential dynamics, and molecular dynamic investigations. J. Mol. Graph. Model. 2021;107 doi: 10.1016/j.jmgm.2021.107969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jupudi S., Rajagopal K., Murugesan S., Kumar B.K., Raman K., Byran G., Chennaiah J., Muthiah Vp, Dasan P B., Sankaran S. Identification of Papain-Like Protease inhibitors of SARS CoV-2 through HTVS, molecular docking, MMGBSA and molecular dynamics approach. South Afr. J. Bot. 2021 doi: 10.1016/j.sajb.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Siddiqui S., Upadhyay S., Ahmad R., Gupta A., Srivastava A., Trivedi A., Husain I., Ahmad B., Ahamed M., Khan M.A. Virtual screening of phytoconstituents from miracle herb Nigella sativa targeting nucleocapsid protein and papain-like protease of SARS-CoV-2 for COVID-19 treatment. J. Biomol. Struct. Dyn. 2020:1–21. doi: 10.1080/07391102.2020.1852117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goyzueta-Mamani L.D., Barazorda-Ccahuana H.L., Mena-Ulecia K., Chávez-Fumagalli M.A. Antiviral activity of metabolites from peruvian plants against SARS-CoV-2: an In Silico approach. Molecules. 2021:26. doi: 10.3390/molecules26133882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Muhseen Z.T., Hameed A.R., Al-Hasani H.M.H., Ahmad S., Li G. Computational determination of potential multiprotein targeting natural compounds for rational drug design against SARS-COV-2. Molecules. 2021;26:674. doi: 10.3390/molecules26030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Delre P., Caporuscio F., Saviano M., Mangiatordi G.F. Repurposing known drugs as covalent and non-covalent inhibitors of the SARS-CoV-2 papain-like protease. Front Chem. 2020;8 doi: 10.3389/fchem.2020.594009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rajpoot S., Alagumuthu M., Baig M.S. Dual targeting of 3CLpro and PLpro of SARS-CoV-2: a novel structure-based design approach to treat COVID-19. Curr. Res. Struct. Biol. 2021;3:9–18. doi: 10.1016/j.crstbi.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li L., Ma L., Hu Y., Li X., Yu M., Shang H., Zou Z. Natural biflavones are potent inhibitors against SARS-CoV-2 papain-like protease. Phytochemistry. 2022;193 doi: 10.1016/j.phytochem.2021.112984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Quimque M.T.J., Notarte K.I.R., Fernandez R.A.T., Mendoza M.A.O., Liman R.A.D., Lim J.A.K., Pilapil L.A.E., Ong J.K.H., Pastrana A.M., Khan A. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2021;39:4316–4333. doi: 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kumar S., Paul P., Yadav P., Kaul R., Maitra S.S., Jha S.K., Chaari A. A multi-targeted approach to identify potential flavonoids against three targets in the SARS-CoV-2 life cycle. Comput. Biol. Med. 2022;142 doi: 10.1016/j.compbiomed.2022.105231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schaurich D., Munhoz O.L., Ramos Junior A., Dalmolin A., Oliveira G., Cremonese L. Clinical progression of COVID-19 coinfection in people living with the human immunodeficiency virus: scoping review. Rev. Bras. De. Enferm. 2021:75. doi: 10.1590/0034-7167-2020-1380. [DOI] [PubMed] [Google Scholar]
- 130.Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science. 2002;296:1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 131.Machitani M., Yasukawa M., Nakashima J., Furuichi Y., Masutomi K. RNA-dependent RNA polymerase, RdRP, a promising therapeutic target for cancer and potentially COVID-19. Cancer Sci. 2020;111:3976–3984. doi: 10.1111/cas.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Campagnola G., McDonald S., Beaucourt S., Vignuzzi M., Peersen O.B. Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J. Virol. 2015;89:275–286. doi: 10.1128/JVI.01574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J., Li J., Wan M., YuWen X., Li H., et al. RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Venkataraman S., Prasad B.V.L.S., Selvarajan R. RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses. 2018;10:76. doi: 10.3390/v10020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Won J.-H., Lee H. The current status of drug repositioning and vaccine developments for the COVID-19 pandemic. Int. J. Mol. Sci. 2020;21:9775. doi: 10.3390/ijms21249775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arba M., Wahyudi S.T., Brunt D.J., Paradis N., Wu C. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-cov-2. Comput. Biol. Med. 2021;129 doi: 10.1016/j.compbiomed.2020.104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Iftikhar H., Ali H.N., Farooq S., Naveed H., Shahzad-ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput. Biol. Med. 2020;122 doi: 10.1016/j.compbiomed.2020.103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Molavi Z., Razi S., Mirmotalebisohi S.A., Adibi A., Sameni M., Karami F., Niazi V., Niknam Z., Aliashrafi M., Taheri M., et al. Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed. Pharmacother. 2021;138 doi: 10.1016/j.biopha.2021.111544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X., Wang Q., Sun Y., Fan Z., Qi J., et al. Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 2020:31. doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grahl M.V.C., Alcará A.M., Perin A.P.A., Moro C.F., Pinto É.S.M., Feltes B.C., Ghilardi I.M., Rodrigues F.V.F., Dorn M., da Costa J.C., et al. Evaluation of drug repositioning by molecular docking of pharmaceutical resources available in the Brazilian healthcare system against SARS-CoV-2. Inform. Med. Unlocked. 2021;23 doi: 10.1016/j.imu.2021.100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sonousi A., Mahran H.A., Ibrahim I.M., Ibrahim M.N., Elfiky A.A., Elshemey W.M. Novel adenosine derivatives against SARS-CoV-2 RNA-dependent RNA polymerase: an in silico perspective. Pharmacol. Rep. 2021;73:1754–1764. doi: 10.1007/s43440-021-00300-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Indu P., Rameshkumar M.R., Arunagirinathan N., Al-Dhabi N.A., Valan Arasu M., Ignacimuthu S. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: a molecular docking and drug repurposing approach. J. Infect. Public Health. 2020;13:1856–1861. doi: 10.1016/j.jiph.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pirzada R.H., Haseeb M., Batool M., Kim M., Choi S. Remdesivir and ledipasvir among the FDA-approved antiviral drugs have potential to inhibit SARS-CoV-2 replication. Cells. 2021;10:1052. doi: 10.3390/cells10051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shaldam M.A., Yahya G., Mohamed N.H., Abdel-Daim M.M., Al Naggar Y. In silico screening of potent bioactive compounds from honeybee products against COVID-19 target enzymes. Environ. Sci. Pollut. Res. 2021;28:40507–40514. doi: 10.1007/s11356-021-14195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Singh S.K., Upadhyay A.K., Reddy M.S. Screening of potent drug inhibitors against SARS-CoV-2 RNA polymerase: an in silico approach. 3 Biotech. 2021;11:93. doi: 10.1007/s13205-020-02610-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Akinlalu A.O., Chamundi A., Yakumbur D.T., Afolayan F.I.D., Duru I.A., Arowosegbe M.A., Enejoh O.A. Repurposing FDA-approved drugs against multiple proteins of SARS-CoV-2: An in silico study. Sci. Afr. 2021;13 doi: 10.1016/j.sciaf.2021.e00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wu Y., Crich D., Pegan S.D., Lou L., Hansen M.C., Booth C., Desrochers E., Mullininx L.N., Starling E.B., Chang K.Y., et al. Polyphenols as Potential Inhibitors of SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Molecules. 2021;26:7438. doi: 10.3390/molecules26247438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Begum F., Srivastava A.K., Ray U. Repurposing nonnucleoside antivirals against SARS-CoV2 NSP12 (RNA dependent RNA polymerase): In silico-molecular insight. Biochem. Biophys. Res. Commun. 2021;571:26–31. doi: 10.1016/j.bbrc.2021.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nagar P.R., Gajjar N.D., Dhameliya T.M. In search of SARS CoV-2 replication inhibitors: virtual screening, molecular dynamics simulations and ADMET analysis. J. Mol. Struct. 2021;1246 doi: 10.1016/j.molstruc.2021.131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baby K., Maity S., Mehta C.H., Suresh A., Nayak U.Y., Nayak Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: an in silico drug repurposing for COVID-19. F1000Research. 2020:9. doi: 10.12688/f1000research.26359.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mishra C.B., Pandey P., Sharma R.D., Malik M.Z., Mongre R.K., Lynn A.M., Prasad R., Jeon R., Prakash A. Identifying the natural polyphenol catechin as a multi-targeted agent against SARS-CoV-2 for the plausible therapy of COVID-19: an integrated computational approach. Brief. Bioinforma. 2020;22:1346–1360. doi: 10.1093/bib/bbaa378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ahmad J., Ikram S., Ahmad F., Rehman I.U., Mushtaq M. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) &x2013; a drug repurposing study. Heliyon. 2020:6. doi: 10.1016/j.heliyon.2020.e04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Therapeutics and COVID-19. Available online: 〈https://www.who.int/teams/health-care-readiness/covid-19/therapeutics〉 (Accessed on 25 May 2022).
- 155.Pubchem, Remdesivir, Compound summary. Available online: 〈https://pubchem.ncbi.nlm.nih.gov/compound/Remdesivir〉 (Accessed on 02 November 2021).
- 156.Alizadehmohajer N., Behmardi A., Najafgholian S., Moradi S., Mohammadi F., Nedaeinia R., Haghjooy Javanmard S., Sohrabi E., Salehi R., Ferns G.A., et al. Screening of potential inhibitors of COVID-19 with repurposing approach via molecular docking. Netw. Model. Anal. Health Inform. Bioinforma. 2022;11:11. doi: 10.1007/s13721-021-00341-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Khan A., Khan M., Saleem S., Babar Z., Ali A., Khan A.A., Sardar Z., Hamayun F., Ali S.S., Wei D.-Q. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdiscip. Sci.: Comput. Life Sci. 2020;12:335–348. doi: 10.1007/s12539-020-00381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Patnin S., Makarasen A., Vijitphan P., Baicharoen A., Chaivisuthangkura A., Kuno M., Techasakul S. Computational screening of phenylamino-phenoxy-quinoline derivatives against the main protease of SARS-CoV-2 using molecular docking and the ONIOM method. Molecules. 2022;27:1793. doi: 10.3390/molecules27061793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Veerasamy R., Karunakaran R. Molecular docking unveils the potential of andrographolide derivatives against COVID-19: an in silico approach. J. Genet. Eng. Biotechnol. 2022;20:58. doi: 10.1186/s43141-022-00339-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rao S.J.A., Shetty N.P. Structure-based screening of natural product libraries in search of potential antiviral drug-leads as first-line treatment to COVID-19 infection. Microb. Pathog. 2022;165 doi: 10.1016/j.micpath.2022.105497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koulgi S., Jani V., Uppuladinne M.V.N., Sonavane U., Joshi R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020;10:26792–26803. doi: 10.1039/D0RA04743K. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this research are supported by the References inserted in the References section.