

Abstract
Epithelial–mesenchymal transition (EMT) is involved in the pathophysiology of lung cancer (LC) and COPD, and the latter is an important risk factor for LC. We hypothesised that the EMT gene expression profile and signalling cascade may differ in LC patients with COPD from those with no respiratory diseases. In lung tumour specimens obtained through video-assisted thoracoscopic surgery from LC (n=20, control group) and LC-COPD patients (n=30), gene expression (quantitative real-time PCR amplification) of EMT markers SMAD3, SMAD4, ZEB2, TWIST1, SNAI1, ICAM1, VIM, CDH2, MMP1 and MMP9 was detected. In lung tumours of LC-COPD compared to LC patients, gene expression of SMAD3, SMAD4, ZEB2 and CDH2 significantly declined, while no significant differences were detected for the other analysed markers. A significant correlation was found between pack-years (smoking burden) and SMAD3 gene expression among LC-COPD patients. LC-COPD patients exhibited mild-to-moderate airway obstruction and a significant reduction in diffusion capacity compared to LC patients. In lung tumour samples of patients with COPD, several markers of EMT expression, namely SMAD3, SMAD4, ZEB2 and CDH2, were differentially expressed suggesting that these markers are likely to play a role in the regulation of EMT in patients with this respiratory disease. Cigarette smoke did not seem to influence the expression of EMT markers in this study. These results have potential clinical implications in the management of patients with LC, particularly in those with underlying respiratory diseases.
Short abstract
The downregulation of the epithelial–mesenchymal transition repressor SMAD pathway may favour a pro-tumoural micro-environment in patients with chronic airway diseases, namely COPD, which could be targeted therapeutically https://bit.ly/39oXnoG
Introduction
Nowadays, lung cancer (LC) continues to be a major cause of mortality worldwide [1–3]. Despite the progress in the diagnosis and treatment of LC that has been achieved in the last decades [2], the pathophysiology and underlying biology remain to be fully understood. Importantly, patients with underlying obstructive lung diseases, namely COPD, are placed at a greater risk of developing tumours in their lungs and airways [3, 4].
Several biological mechanisms have been shown to participate in the greater risk for LC development in COPD patients [5]. For instance, increased oxidative stress [6], inflammation including a differential inflammatory profile [7], epigenetics abnormalities [8] and the pattern of expression of stroma markers [9] have all been demonstrated to be differentially expressed in tumours of patients with mild-to-moderate COPD. In addition, overall survival was also shown to be significantly altered on the basis of the inflammatory profile in lung tumours of patients with underlying COPD compared to LC patients without COPD [10].
Epithelial–mesenchymal transition (EMT) is a biological process characterised by the loss of polarity and cell–cell adhesion of epithelial cells to gain invasive characteristics. The transformed epithelial cells become mesenchymal stem cells, which can differentiate into a great variety of cell types [11, 12]. EMT is a physiological process involved in organ development in the embryo and in wounds [11]. Recently, a role of EMT in the pathogenesis of COPD has also been demonstrated in the literature [13]. Furthermore, tumour development and progression also rely on the expression of EMT within the lungs of LC patients [13–16]. Whether a differential expression profile of EMT markers can be observed in LC patients with COPD, even in those at early stages of their disease, remains to be answered.
We hypothesised that EMT gene expression profile and signalling cascade may differ in lung tumours of patients with COPD from those without this respiratory disease. Thus, our objectives were to explore the following in a clinical observational study of lung tumours in patients with and without COPD: 1) gene expression of EMT signalling markers; 2) EMT gene expression profile of specific markers such as intracellular adhesion molecule 1 (ICAM1), cadherin 2 (CDH2) and vimentin (VIM); and 3) correlations between clinical and biological variables. A group of LC patients with no COPD was included as a control group for the purpose of comparisons.
Methods
See specific details in the online supplementary material.
Study population
All patients were prospectively and consecutively recruited from the Lung Cancer Clinic at Hospital del Mar (Barcelona, Spain). All the patients were part of the Lung Cancer Mar Cohort. For this observational investigation, 50 patients with LC were consecutively recruited during the years 2018–2020. Patients were further subdivided according to the presence or absence of COPD: n=30 patients with COPD (LC-COPD group) and n=20 patients with no COPD (LC control group). This was a prospective controlled clinical investigation, in which the World Medical Association guidelines for research on human beings (Seventh revision of Declaration of Helsinki, Fortaleza, Brazil, 2013) were followed. The institutional Ethics Committee on Human investigations (protocol #2008/3390/I, 4 February 2008, at Hospital del Mar-IMIM, Barcelona, Spain) approved all the procedures and study protocol. All patients invited to participate in the study signed their written informed consent.
In all cases, preoperative staging was performed using chest and upper abdominal computed tomography scan and fluoro-deoxy-glucose positron emission tomography/computed tomography body-scan. When there was suspected mediastinal lymph node involvement, a fiberoptic bronchoscopy with endobronchial ultrasound and trans-tracheal biopsy of the suspected nodes were performed. In case of negative results, a surgical exploration of the mediastinum: cervical video-assisted mediastinal lymphadenectomy and/or anterior mediastinotomy were performed, the latter depending on the location of the suspected nodes. Notwithstanding, in all surgical cases, intra-operative systematic hilar and mediastinal lymphadenectomy (at least, ipsilateral paratracheal, subcarinal and ipsilateral pulmonary ligament) was performed as previously recommended [17, 18].
Sample collection and preservation
Lung samples were obtained from tumours following standard technical procedures during video-assisted thoracoscopic surgery in the surgery room. The fresh samples were carefully transported to the Pathology Department, located at a very short distance (<5 min). In all patients, the expert pulmonary pathologists selected tumour lung specimens of ∼10×10 mm2 area from the fresh samples. For all the recruited patients, fragments of tumour specimens were immediately snap-frozen to be subsequently stored at −80°C until further use in the laboratory experiments.
Biological experiments
Figure 1 illustrates the flow of the signalling markers analysed in the investigation. The sequence of experiments was as follows. 1) RNA isolation: RNA was isolated from 30–50 µg frozen tumour samples using 500 µL TRIzol reagent (Cat. 15596026; Thermo Fisher Scientific, Waltham, MA, USA). 2) RNA reverse transcription: Invitrogen® cDNA Synthesis Kit (Cat.18018044; Thermo Fisher Scientific, Carlsbad, CA, USA) was used to prepare cDNA templates following the manufacturer's instructions. 3) Quantitative real-time PCR amplification, performed using commercial gene expression assays for human studies.
FIGURE 1.

Epithelial–mesenchymal transition (EMT) pathway schematic diagram. SM....AD: mothers against decapentaplegic homolog; TF: transcription factors; P: phosphate group; ZEB: zinc finger E-box binding homeobox; CDH: cadherin; ICAM: intercellular adhesion molecule; MMP: matrix metalloproteinase.
Statistical analysis
Sample size was calculated on the basis of four target markers (CDH2, ZEB2, SMAD4 and SMAD3). Accepting an alpha risk of 0.05 and a beta risk of 0.2 in a two-sided test, 16, 16, 13 and 16 subjects were required in each group to identify a statistically significant difference ≥ 0.5, 2, 1.7 and 3 units in the mean value and a standard deviation of 0.5, 2, 1.5 and 3 in the expression of the genes CDH2, ZEB2, SMAD4 and SMAD3, respectively. As 30 and 20 patients were included in the LC-COPD and LC control group of patients, respectively, the total number of patients was sufficient to attain an 80% power.
The normality of the study variables was examined using the Shapiro–Wilk test. For an initial descriptive analysis of clinical parameters, qualitative variables were described as frequencies (number and percentage) and quantitative variables as mean±sd. Potential differences between LC and LC-COPD and between smokers and never-smokers as a whole (with no distinction between COPD and non-COPD patients) were assessed using t-test. The Chi-squared test was used to assess differences between the two groups for the categorical variables including the driver mutations and expression or no gene expression for the markers SMAD3, SMAD4, ZEB2, TWIST1, SNAI1, ICAM1, VIM, CDH2, MMP1 and MMP9.
In each group of patients, potential correlations between clinical and biological variables were explored using the Pearson's correlation coefficient. All the statistical analyses were performed using the Statistical Package for the Social Sciences (Portable SPSS, PASW statistics 22.0 version for Windows, SPSS Inc., Chicago, IL, USA). Correlations are displayed in graphical correlation matrices, obtained from R package corrplot (https://cran.r-project.org/web/packages/corrplot/index.html), in two different colours: blue for positive correlations and red for negative ones. Statistical significance was established at p≤0.05 for all the comparisons.
Results
Clinical characteristics of the study patients
Clinical and functional characteristics of all the study patients are expressed in table 1. Age and sex did not significantly differ between the two study groups, while body mass index was greater in the LC-COPD patients than in LC patients (table 1). The number of ex-smokers and pack-year variables were significantly higher in the LC-COPD patients than in the LC patients (table 1). LC-COPD patients exhibited mild-to-moderate airway obstruction along with a significant reduction in diffusion capacity compared to LC patients (table 1). As expected, the majority of the COPD patients were in Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages I and II (96.6%). Tumour, nodes, metastasis (TNM) staging or histological subtypes did not significantly differ between the study groups. Adenocarcinoma was the predominant histopathological subtype in both groups of patients (table 1). The levels of blood parameters such as total leukocytes, neutrophils, lymphocytes, albumin, total proteins and body weight loss did not significantly differ between the two study groups of patients.
TABLE 1.
Clinical characteristics of the study groups of patients
| Anthropometric variables | LC control | LC-COPD |
| Subjects n | 20 | 30 |
| Age years | 65±7 | 69±8 |
| Male/female n | 13/7 | 26/4 |
| BMI kg·m−2 | 25.6±4 | 27.9±4* |
| Smoking history n (%) | ||
| Current | 6 (30) | 6 (20) |
| Ex-smoker | 6 (30) | 24 (80)* |
| Never-smoker | 8 (40) | 0 (0)* |
| Smoking pack-years | 37±37 | 61±18* |
| Lung function parameters | ||
| FEV1 | 83±17 | 68±14*** |
| FEV1/FVC % | 75±6 | 59±9*** |
| DLCO % | 82±16 | 60±12*** |
| KCO % | 83±14 | 63±15*** |
| GOLD stage n (%) | ||
| GOLD stage I | NA | 4 (13) |
| GOLD stage II | NA | 25 (83) |
| GOLD stage III | NA | 1 (3) |
| GOLD stage IV | NA | 0 (0) |
| TNM staging n (%) | ||
| Stage I | 8 (40) | 10 (33) |
| Stage II | 5 (25) | 14 (47) |
| Stage III | 7 (35) | 5 (17) |
| Stage IV | 0 (0) | 1 (3) |
| Histological diagnosis n (%) | ||
| Squamous cell carcinoma | 4 (20) | 8 (27) |
| Adenocarcinoma | 11 (55) | 21 (70) |
| Others | 5 (25) | 1 (3) |
| Blood parameters | ||
| Total leukocytes per μL | 8.53±2.8×103 | 8.85±2.4×103 |
| Total neutrophils per μL | 5.48±2.6×103 | 5.51±2×103 |
| Total lymphocytes per μL | 2.24±0.6×103 | 2.15±0.8×103 |
| Albumin g·dL−1 | 4.39±0.4 | 4.28±0.4 |
| Total proteins g·dL−1 | 7.22±0.5 | 7.05±0.4 |
| Body weight loss kg, n (%) | ||
| 0 | 18 (90) | 29 (97) |
| 1–5 | 1 (5) | 0 (0) |
| 6–10 | 1 (5) | 1 (3) |
Continuous variables are reported as mean±sd, categorical variables are expressed as the number of patients per group and the respective percentage in each group with respect to the total population. LC: lung cancer; BMI: body mass index; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide; KCO: Krogh transfer factor; GOLD: Global Initiative for Chronic Obstructive Lung Disease; NA: not applicable; TNM: tumour, nodes, metastasis. Statistical significance: *: p<0.05; ***: p<0.001 between LC-COPD and LC patients.
The proportions of tumour driver mutations of the genes Kirsten rat sarcoma virus (KRAS), epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) did not significantly differ between the two groups of patients (table 2).
TABLE 2.
Driver mutation status of the study groups of patients
| Mutation status | LC control | LC-COPD |
| Subjects n | 20 | 30 |
| KRAS, n (%) | ||
| No mutation | 10 (76.9) | 12 (75.0) |
| Mutation | 3 (23.1) | 4 (25.0) |
| EGFR, n (%) | ||
| No mutation | 11 (84.6) | 14 (87.5) |
| Mutation | 2 (15.4) | 2 (12.5) |
| ALK, n (%) | ||
| No mutation | 13 (100.0) | 16 (100.0) |
| Mutation | 0 (0) | 0 (0) |
Categorical variables are expressed as the number of patients per group and the respective percentage in each group with respect to the total population. LC: lung cancer; KRAS: Kirsten rat sarcoma virus; EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma kinase. Statistical significance: no statistically significant differences were found between LC-COPD and LC patients.
EMT signalling markers
The gene expression profile of all the analysed genes is depicted in figure 2. LC-COPD patients are represented in green colour, while LC patients are shown in red colour (figure 2). In tumours of LC-COPD patients, SMAD3 gene expression was significantly reduced compared to tumours in LC patients (figure 3a). The number of patients who did not show any expression of SMAD3 in their tumours did not significantly differ between the two study groups (figure 3a). SMAD4 gene expression was also significantly lower in tumours of LC-COPD than in LC patients (figure 3b). The number of patients who did not show any expression of SMAD4 in their tumours did not significantly differ between the two study groups (figure 3b). ZEB2 gene expression levels significantly declined in tumours of LC-COPD patients compared to the LC patients (figure 3c). The number of patients who did not show any expression of ZEB2 in their tumours did not significantly differ between LC-COPD and LC patients (figure 3c). No significant differences were seen in TWIST1 gene expression between LC and LC-COPD patients (figure 3d). SNAI1 gene expression did not show any significant differences between LC and LC-COPD patients (figure 3e). When all the patients (n=50) were analysed according to smoking history, no significant differences were detected in the expression of these genes, except for ZEB2 between smokers (current and ex-smokers) and never-smokers (supplementary figure S1).
FIGURE 2.
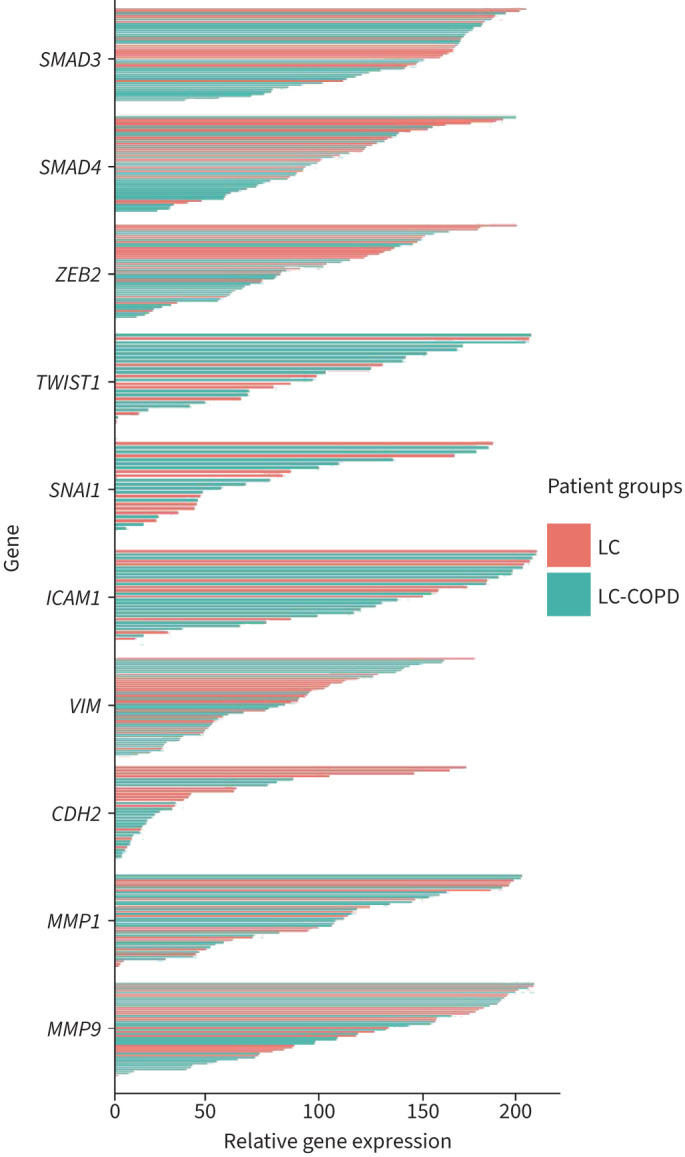
Gene expression profiles of the analysed genes in the two groups of patients. Lung cancer (LC) patients are depicted as red lines, while LC-COPD patients are represented as green lines. The gene expression levels obtained from the quantitative PCR results were normalised to GAPDH expression levels for each sample. SMAD: mothers against decapentaplegic homolog; ZEB: zinc finger E-box binding homeobox; CDH: cadherin; ICAM: intercellular adhesion molecule; MMP: matrix metalloproteinase; VIM: vimentin.
FIGURE 3.

Quantitative real-time PCR amplification analysis of gene expression of epithelial–mesenchymal transition (EMT) between lung cancer (LC) and LC-COPD patients (graphs), and comparisons of the number of patients expressing the target genes of EMT (tables). a) SMAD3 (mothers against decapentaplegic homolog 3); b) SMAD4; c) ZEB2 (zinc finger E-box binding homeobox 2); d) TWIST1; e) SNAI1; f) ICAM1 (intercellular adhesion molecule 1); g) VIM (vimentin); h) CDH2 (cadherin 2); i) MMP1 (matrix metalloproteinase 1); j) MMP9. Graph data are presented as mean±sd. Two-tailed t-tests were used to assess significance of differences: *: p<0.05; **: p<0.01.
Expression of markers of EMT
Gene expression levels of ICAM1 in tumours did not significantly differ between the two study groups (figure 3f). No significant differences in tumour VIM gene expression were observed between LC and LC-COPD patients (figure 3g). VIM gene expression was detected in all the tumour samples from both groups (figure 3g). In tumours of LC-COPD patients, CDH2 gene expression was significantly reduced compared to tumours of LC patients (figure 3h). The number of patients who did not show any expression of CDH2 in their tumours did not significantly differ between the two patient groups (figure 3h). MMP1 and MMP9 gene expression in tumours did not significantly differ between the two study patient groups (figure 3i and j, respectively). The number of patients who did not show any expression of MMP1 or MMP9 in their tumours did not significantly differ between LC and LC-COPD patients (figure 3i and j, respectively). When all patients (n=50) were analysed according to smoking history, no significant differences were detected in the expression of these genes between smokers (current and ex-smokers) and never-smokers (supplementary figure S2). Among LC patients, significant positive correlations were found between VIM and lung function parameters (forced expiratory volume in 1 s/forced vital capacity, diffusing capacity of the lung for carbon monoxide (DLCO) and Krogh transfer factor (KCO), respectively, figure 4). In LC-COPD patients, the number of pack-years positively correlated with SMAD3 gene expression (figure 4 and supplementary figure S2). Moreover, among the same group of patients, CDH2 gene expression positively correlated with blood neutrophil and leukocyte counts (figure 4).
FIGURE 4.

Clinical correlations in each group of patients. a) Correlation matrix of the clinical and analytical variables in lung cancer (LC) patients. b) Correlation matrix of the clinical and analytical variables in LC-COPD patients. In the matrices, positive correlations are represented as blue dots, while negative correlations are represented as red dots. Black dots within the circles represent p-values >0.05. Colour intensity and the size of the circle are proportional to the correlation coefficients, as indicated on the y-axis on the right-hand side of the graphs. FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide; KCO: Krogh transfer factor; SMAD: mothers against decapentaplegic homolog; VIM: vimentin; CDH: cadherin.
Discussion
In the current investigation, the main findings were that gene expression of the EMT signalling markers SMAD3, SMAD4 and ZEB2 was significantly lower in the tumours of patients with LC-COPD than in LC control patients. In addition, a significant decline in the EMT marker CDH2 was also observed in the tumours of patients with underlying COPD compared to non-COPD patients. Driver mutations were similarly distributed across the two study groups. A differential profile of gene expression of EMT signalling markers was observed among the patients with underlying COPD in this study.
During EMT, polarised epithelial cells transform into fibroblast-like mesenchymal cells in a process in which the cells can move and invade tissues [19]. Several growth factors initiate and maintain EMT within tissues, particularly transforming growth factor (TGF)-β [20, 21]. TGF-β has been consistently shown to increase in the systemic compartment and lungs of patients with LC, particularly in those with COPD [9, 22]. In a previous investigation [23], we clearly demonstrated that TGF-β expression levels were significantly greater in lung tumours compared to non-tumour control tissues, and in patients with COPD those levels were, indeed, significantly higher than in tumour specimens of non-COPD patients.
TGF-β signalling involves several ligands and SMAD2/3 protein expression, which subsequently activate SMAD4 to translocate to the nucleus to activate or repress gene expression [24]. Disruptions in the expression of the SMAD signalling pathway may impact on the functions of TGF-β cytokine as was shown to occur in bronchial epithelial cells of COPD patients that were also exposed to cigarette smoke [25] and in myofibroblasts of the airway remodelling process in healthy smokers and COPD patients [26]. Similarly, in the current investigation, gene expression of SMAD3 and SMAD4 was significantly reduced in the tumours of the patients with underlying COPD. This signalling pathway may play an important role in the initiation of a profibrogenic effect through TGF-β signalling in patients with COPD, thus favouring lung carcinogenesis [12, 27]. As such the downregulation of the EMT repressor SMAD pathway may favour a pro-tumoural micro-environment in patients with chronic airways diseases, namely COPD, which could be targeted therapeutically. On the other hand, EMT-related protein expression increased in the peripheral leading edge of nonsmall cell lung cancer (NSCLC) and was related to the tumour characteristics that were associated with a poor prognosis in the patients [28]. Furthermore, cigarette smoke also downregulates SMAD gene expression [25], which may also enhance the profibrogenic status reported in patients with COPD. Importantly, the expression of SMAD3 was higher in LC-COPD patients with a greater burden of cigarette smoke exposure (positive correlation). These are relevant findings that are consistent with previous knowledge on the persistent increase in SMAD3 expression induced by cigarette smoke [29]. Nonetheless, the analysis between smokers and never-smokers of the entire population of patients did not yield any relevant results in the current study.
CDH2 is a transmembrane protein involved in cell adhesion with multiple biological roles including its contribution to cancer development and metastasis [30]. In the present study, CDH2 gene expression levels were significantly lower in tumours of patients with underlying COPD than in those without this respiratory disease. In a previous investigation that was based exclusively on patients with COPD, gene expression of CDH2 was increased in the bronchial epithelial cells compared to cells obtained from healthy subjects [31]. Differences in the methodologies employed (laboratory techniques and sample types, cells versus tumour biopsies) to identify levels of CDH2 gene expression as well as the number of patients (greater in the current study) analysed in each investigation are likely to account for the discrepancies encountered between the two studies [31].
ZEB2 transcription factor is involved in the process of human carcinogenesis and correlates well with invasiveness and metastasis [32]. In mammary epithelial cells, ZEB2 favoured EMT expression, while repressing E-cadherin expression [33]. It has also been demonstrated that the targeting of ZEB2 expression avoided the migration and invasion of LC cells [34]. Moreover, ZEB2 has also been involved in the response to therapy in LC patients [35]. Importantly, gene expression of ZEB2 also decreased in bronchial epithelial cells from patients with COPD compared to control cells [31]. The authors demonstrated that bronchial epithelial cells from COPD patients underwent EMT during normal baseline conditions, and this may have favoured other biological processes in those patients [31]. In the present investigation, a significant decline in ZEB2 gene expression was also detected in the tumours of patients with underlying COPD. The results obtained in the present study are very consistent with those previously reported by our group and others [34, 36], and should be the focus of future research to better identify LC development in patients with chronic respiratory diseases, with a special emphasis on COPD due to its high prevalence worldwide.
SNAI1 is a transcription factor that represses the transcription of membrane adhesion molecules and interacts with SMADs to induce EMT [37]. TWIST1 is another transcription factor that along with SNAI1 downregulates epithelial gene expression to activate EMT [38]. In patients with moderate-to-severe COPD, the expression of SNAI1 and TWIST1 was higher in the bronchial epithelium specimens than in those of the healthy subjects [39]. VIM, an intermediate filament, is expressed in mesenchymal cells. In the airways of patients with COPD, expression levels of VIM were increased compared to samples obtained from the healthy controls [40]. Collectively, these findings imply that differences exist in the expression of EMT markers such as TWIST1, SNAI1 and VIM in the airways of patients with COPD compared to a group of healthy subjects. In the current investigation, however, no significant differences were detected in the gene expression levels of these markers between the two groups of LC patients. These results imply that COPD per se may not have influenced EMT expression in this cohort of patients. The specific localisation of the samples analysed across the different investigations [24] and the degree of the COPD itself may explain the discrepancies encountered across studies in the gene expression profile of the target EMT markers.
Study limitations
A limitation is related to the lack of expression of some of the analysed markers in the tumours of the two study groups of patients. In spite of the fact that samples were always run in duplicate and two identical gene expression experiments were performed, no expression could be detected in the tumour samples of a few patients in both groups. Assessment of protein expression levels of EMT signalling or of additional markers (e.g. inhibitor SMAD) would have been of interest and could be the subject of research in future investigations. The fact that non-tumour lung specimens from patients with no COPD were not available in the study might be another limitation. However, for ethical reasons those samples could not be obtained. Whether different results might have been obtained in patients with more advanced COPD should be assessed in future investigations using other approaches (e.g. lung volume reduction surgery). Another limitation may be related to the assessment of the potential influence of cigarette smoking on the expression of EMT, which should be investigated in a larger cohort of patients, particularly in follow-up investigations. As not all smokers develop COPD, monitoring of smokers through time would be of interest.
Conclusions
In lung tumour samples of patients with COPD, several markers of EMT expression, namely SMAD3, SMAD4, ZEB2 and CDH2, were differentially expressed suggesting that these markers are likely to play a role in the regulation of EMT in patients with this respiratory disease. Cigarette smoke did not seem to influence the expression of EMT markers in this study. These results have potential clinical implications in the management of patients with LC, particularly in those with underlying respiratory diseases.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00105-2022.SUPPLEMENT (521KB, pdf)
Acknowledgements
The authors are thankful to Xavier Duran for his advice on the statistical analyses performed in the investigation.
Provenance: Submitted article, peer reviewed.
Author contributions: Conception and design: E. Barreiro, V. Curull, A. Sánchez-Font, A. Rodríguez-Fuster and R. Aguiló; patient assessment and recruitment, and sample collection: V. Curull, A. Sánchez-Font, A. Rodríguez-Fuster and R. Aguiló; molecular biology analyses: Y. Xia, J. Zha and M. Guitart; statistical analyses and data interpretation: Y. Xia, J. Zha and E. Barreiro; manuscript drafting and intellectual input: E. Barreiro, Y. Xia and J. Zha; manuscript writing final version: E. Barreiro.
Conflict of interest: E. Barreiro is the Chief Editor of ERJ Open Research. The remaining authors have nothing to disclose.
Support statement: This study has been supported by CIBERES and Project FIS 18/00075 of the Instituto de Salud Carlos III (ISCIII) and cofunded by the European Union, Spanish Ministry of Science and Innovation and Sociedad Española de Neumología y Cirugía Torácica 2020. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Cayuela L, López-Campos JL, Otero R, et al. . The beginning of the trend change in lung cancer mortality trends in Spain, 1980–2018. Arch Bronconeumol (Engl Ed) 2021; 57: 115–121. doi: 10.1016/j.arbr.2020.04.011 [DOI] [PubMed] [Google Scholar]
- 2.Hirsch FR, Scagliotti GV, Mulshine JL, et al. . Lung cancer: current therapies and new targeted treatments. The Lancet 2017; 389: 299–311. doi: 10.1016/S0140-6736(16)30958-8 [DOI] [PubMed] [Google Scholar]
- 3.de-Torres JP, Wisnivesky JP, Bastarrika G, et al. . Exploring the impact of lung cancer screening on lung cancer mortality of smokers with obstructive lung disease: analysis of the NLST-ACRIN cohort. Arch Bronconeumol (Engl Ed) 2021; 57: 36–41. doi: 10.1016/j.arbr.2020.03.022 [DOI] [PubMed] [Google Scholar]
- 4.Skillrud DM, Offord KP, Miller DW. Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled study. Ann Intern Med 1986; 105: 503–507. doi: 10.7326/0003-4819-105-4-503 [DOI] [PubMed] [Google Scholar]
- 5.Durham AL, Adcock IM. The relationship between COPD and lung cancer. Lung Cancer 2015; 90: 121–127. doi: 10.1016/j.lungcan.2015.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang J, Curull V, Wang X, et al. . Increased PARP activity and DNA damage in NSCLC patients: the influence of COPD. Cancers 2020; 12: 3333. doi: 10.3390/cancers12113333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang J, Ramis-Cabrer D, Curull V, VCet al. . Immune cell subtypes and cytokines in lung tumor microenvironment: influence of COPD. Cancers 2020; 12: 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundar IK, Mullapudi N, Yao H, et al. Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Curr Opin Pulm Med 2011; 17: 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang J, Ramis-Cabrer D, Curull V, et al. . Markers of stroma in lung cancer: influence of COPD. Arch Bronconeumol (Engl Ed) 2021; 57: 130–137. doi: 10.1016/j.arbr.2020.09.005 [DOI] [PubMed] [Google Scholar]
- 10.Tang J, Ramis-Cabrer D, Curull V, et al. . B cells and tertiary lymphoid structures influence survival in lung cancer patients with resectable tumors. Cancers 2020; 12: 2644. doi: 10.3390/cancers12092644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 2005; 233: 706–720. doi: 10.1002/dvdy.20345 [DOI] [PubMed] [Google Scholar]
- 12.Dong B, Wu Y. Epigenetic regulation and post-translational modifications of SNAI1 in cancer metastasis. Int J Mol Sci 2021; 22: 11062. doi: 10.3390/ijms222011062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahmood MQ, Shukla SD, Ward C, et al. . The underappreciated role of epithelial mesenchymal transition in chronic obstructive pulmonary disease and its strong link to lung cancer. Biomolecules 2021; 11: 1394. doi: 10.3390/biom11091394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walters EH, Shukla SD, Mahmood MQ, et al. . Fully integrating pathophysiological insights in COPD: an updated working disease model to broaden therapeutic vision. Eur Respir Rev 2021; 30: 200364. doi: 10.1183/16000617.0364-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibue T, Robert AW. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017; 14: 611–629. doi: 10.1038/nrclinonc.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Justyna W. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Physiol Behav 2017; 176: 139–148.28363838 [Google Scholar]
- 17.Armstrong P, Congleton J, Fountain SW, et al. . BTS guidelines: guidelines on the selection of patients with lung cancer for surgery. Thorax 2001; 56: 89–108. doi: 10.1136/thorax.56.2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Leyn P, Dooms C, Kuzdzal J, et al. . Revised ESTS guidelines for preoperative mediastinal lymph node staging for non-small-cell lung cancer. Eur J Cardiothorac Surg 2014; 45: 787–798. doi: 10.1093/ejcts/ezu028 [DOI] [PubMed] [Google Scholar]
- 19.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cicenas J, Kvederaviciute K, Meskinyte I, et al. . KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers 2017; 9: 42. doi: 10.3390/cancers9050042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Tilló E, Siles L, de Barrios O, et al. . Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res 2011; 1: 897–912. [PMC free article] [PubMed] [Google Scholar]
- 22.Vandamme N, Denecker G, Bruneel K, et al. . The EMT transcription factor ZEB2 promotes proliferation of primary and metastatic melanoma while suppressing an invasive, mesenchymal-like phenotype. Cancer Res 2020; 80: 2983–2995. doi: 10.1158/0008-5472.CAN-19-2373 [DOI] [PubMed] [Google Scholar]
- 23.Mateu-Jimenez M, Curull V, Pijuan L, et al. . Systemic and tumor Th1 and Th2 inflammatory profile and macrophages in lung cancer: influence of underlying chronic respiratory disease. J Thorac Oncol 2017; 12: 235–248. doi: 10.1016/j.jtho.2016.09.137 [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–172. doi: 10.1038/cr.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Springer J, Scholz FR, Peiser C, et al. . SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smoke. Biol Chem 2004; 385: 649–653. doi: 10.1515/BC.2004.080 [DOI] [PubMed] [Google Scholar]
- 26.Eapen MS, Lu W, Hackett TL, et al. . Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: potential role of epithelial–mesenchymal transition. ERJ Open Res 2021; 7: 00876-2020. doi: 10.1183/23120541.00876-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Na YR, Lee JS, Lee SJ, et al. . Interleukin-6-induced Twist and N-cadherin enhance melanoma cell metastasis. Melanoma Res 2013; 23: 434–443. doi: 10.1097/CMR.0000000000000021 [DOI] [PubMed] [Google Scholar]
- 28.Mahmood MQ, Ward C, Muller HK, et al. . Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): a mutual association with airway disease. Med Oncol 2017; 34: 45. doi: 10.1007/s12032-017-0900-y [DOI] [PubMed] [Google Scholar]
- 29.Sailland J, Grosche A, Baumlin N, et al. . Role of Smad3 and p38 signalling in cigarette smoke-induced CFTR and BK dysfunction in primary human bronchial airway epithelial cells. Sci Rep 2017; 7: 10506. doi: 10.1038/s41598-017-11038-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gheldof A, Hulpiau P, Van Roy F, et al. . Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci 2012; 69: 2527–2541. doi: 10.1007/s00018-012-0935-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chua HL, Bhat-Nakshatri P, Clare SE, et al. . NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 2007; 26: 711–724. doi: 10.1038/sj.onc.1209808 [DOI] [PubMed] [Google Scholar]
- 32.You J, Li Y, Fang N, et al. . MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One 2014; 9: e91827. doi: 10.1371/journal.pone.0091827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui J, Pan G, He Q, et al. . MicroRNA-545 targets ZEB2 to inhibit the development of non-small cell lung cancer by inactivating Wnt/β-catenin pathway. Oncol Lett 2019; 18: 2931–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishioka M, Venkatesan N, Dessalle K, et al. . Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir Res 2015; 16: 72. doi: 10.1186/s12931-015-0232-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Ann Rev Biochem 1990; 59: 237–252. [DOI] [PubMed] [Google Scholar]
- 36.Mateu-Jimenez M, Curull V, Rodríguez-Fuster A, et al. . Profile of epigenetic mechanisms in lung tumors of patients with underlying chronic respiratory conditions. Clin Epigenetics 2018; 10: 7. doi: 10.1186/s13148-017-0437-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vincent T, Neve EPA, Johnson JR, et al. . A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat Cell Biol 2009; 11: 943–950. doi: 10.1038/ncb1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ansieau S, Bastid J, Doreau A, et al. . Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008; 14: 79–89. doi: 10.1016/j.ccr.2008.06.005 [DOI] [PubMed] [Google Scholar]
- 39.Mahmood MQ, Walters EH, Shukla SD, et al. . β-catenin, twist and snail: transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci Rep 2017; 7: 10832. doi: 10.1038/s41598-017-11375-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gohy ST, Hupin C, Fregimilicka C, et al. . Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J 2015; 45: 1258–1272. doi: 10.1183/09031936.00135814 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00105-2022.SUPPLEMENT (521KB, pdf)