

Abstract
Fate decisions in the embryo are controlled by a plethora of microenvironmental interactions in a three‐dimensional niche. To investigate whether aspects of this microenvironmental complexity can be engineered to direct myogenic human‐induced pluripotent stem cell (hiPSC) differentiation, we here screened murine cell types present in the developmental or adult stem cell niche in heterotypic suspension embryoids. We identified embryonic endothelial cells and fibroblasts as highly permissive for myogenic specification of hiPSCs. After two weeks of sequential Wnt and FGF pathway induction, these three‐component embryoids are enriched in Pax7‐positive embryonic‐like myogenic progenitors that can be isolated by flow cytometry. Myogenic differentiation of hiPSCs in heterotypic embryoids relies on a specialized structural microenvironment and depends on MAPK, PI3K/AKT, and Notch signaling. After transplantation in a mouse model of Duchenne muscular dystrophy, embryonic‐like myogenic progenitors repopulate the stem cell niche, reactivate after repeated injury, and, compared to adult human myoblasts, display enhanced fusion and lead to increased muscle function. Altogether, we provide a two‐week protocol for efficient and scalable suspension‐based 3D derivation of Pax7‐positive myogenic progenitors from hiPSCs.
Keywords: embryoids, hiPSCs, MuSCs, myogenic progenitors, Pax7
Subject Categories: Methods & Resources, Musculoskeletal System, Stem Cells & Regenerative Medicine
A heterotypic suspension embryoid model enables scalable generation of Pax7+ skeletal muscle progenitors in a defined microenvironment.
Introduction
Pax7 positive skeletal muscle progenitors have an extensive self‐renewal capacity and hold great promise for cell therapy of muscular dystrophy (Pawlikowski et al, 2009). However, sourcing large quantities of freshly isolated human skeletal muscle progenitors (hskMPs) is challenging. Importantly, myogenic progenitors lose their regenerative potential after in vitro expansion in conventional 2D myoblast culture and hskMPs progressively downregulate the expression of Pax7 over early passages (Bentzinger et al, 2013; Romagnoli et al, 2020). Removal of myogenic progenitors from their adult niche promotes commitment to differentiation, while those resident in a biologically faithful microenvironment are able to self‐renew and maintain quiescence (Flamini et al, 2018). Thus, a protocol for the large‐scale generation of human myogenic progenitors expressing Pax7 would open new avenues for the development of stem cell‐based therapeutics.
Human‐induced pluripotent stem cells (hiPSCs) represent a nearly unlimited source for the derivation of tissue progenitors. Myogenic hiPSC differentiation can be induced by ectopic expression of transcription factors (Darabi et al, 2012; Rao et al, 2012; Albini et al, 2013; Abujarour et al, 2014; Shoji et al, 2015), or using small molecules and growth factors that modulate pathways involved in mesoderm specification and commitment to the skeletal muscle lineage (Barberi et al, 2007; Yu et al, 2007; Shelton et al, 2014; Chal et al, 2015, 2016; Caron et al, 2016). Similar to classic hskMP culture, transgene‐free protocols for myogenic hiPSC differentiation are commonly adapted to 2D culture environments and are not focused on the sole generation and maintenance of Pax7 positive progenitors (Shelton et al, 2014; Chal et al, 2016; Choi et al, 2016). Akin to the regulation of adult skeletal muscle progenitors, myogenic fate decisions in the embryo are controlled by complex microenvironmental interactions mediated by diverse cell types (Chal & Pourquie, 2017). Collectively these extrinsic signals modulate batteries of intertwined pathways specific to each developmental stage. We hypothesized that recreating aspects of this microenvironmental complexity could improve the efficiency of myogenic hiPSC specification and preserve Pax7 expression and embryonic stem cell characteristics that are favorable for downstream applications.
Results
Identification of heterotypic embryoids that are permissive for Pax7 induction
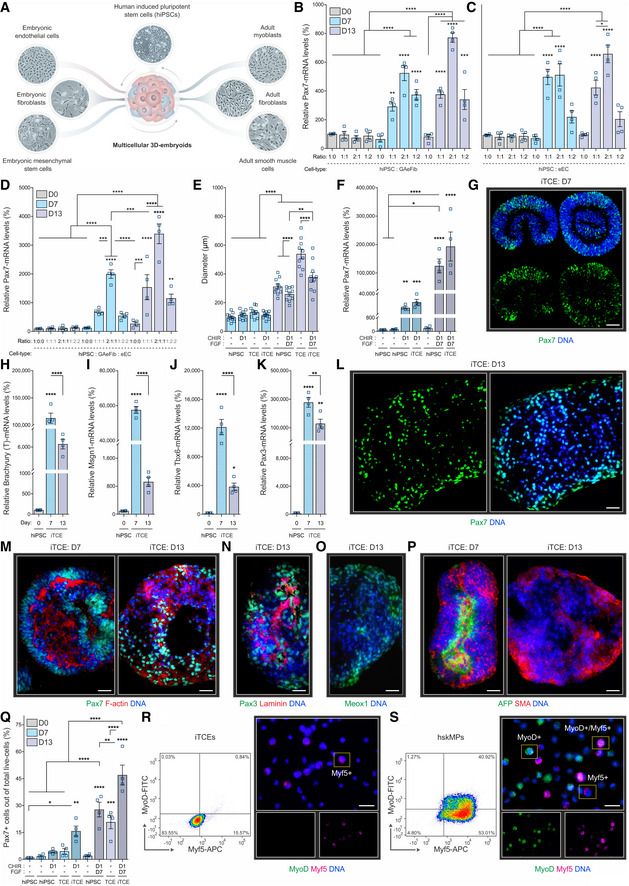
To identify cell types with permissive effects on mesoderm induction and commitment to the myogenic lineage, we screened murine embryonic fibroblasts, mesenchymal stem cells, endothelial cells, as well as adult smooth muscle cells, myoblasts, and fibroblasts in combination with human‐induced pluripotent stem cells (hiPSCs) in suspension embryoids generated using a horizontal shaker platform (Simunovic & Brivanlou, 2017) (Fig 1A and Appendix Fig S1A). As a readout for this experiment, we assessed the expression of the conserved myogenic marker Pax7 by quantitative PCR (Bryson‐Richardson & Currie, 2008; Bentzinger et al, 2012). Embryonic or adult fibroblasts and embryonic mesenchymal stem cells did not increase human Pax7 mRNA expression at 7, 13, or 21 days after embryoid formation with hiPSCs in any of the tested ratios, while adult smooth muscle cells had minor effects at day 7 (Appendix Fig S1B–E). Interestingly, hiPSCs combined with growth‐arrested embryonic fibroblasts (GAeFib), embryonic endothelial cells (eEC), and adult myoblasts (aMyo) led to a 4‐ to 8‐fold induction of Pax7 at day 7 and 13 (Fig 1B and C, and Appendix Fig S1F and G). To test for additive or synergistic effects, we generated four‐component embryoids of hiPSCs with GAeFib, eEC, and aMyo. However, we did not observe a significant increase in Pax7 expression compared to two‐component embryoids containing hiPSCs with GAeFib, eEC, or aMyo (Appendix Fig S1H). To reduce the probability of cross‐inhibition by one of the cell types, we subsequently tested three‐component embryoid conditions. No additive effects were observed for eEC and aMyo when combined with hiPSCs (Appendix Fig S1I). In contrast, the combination of hiPSCs with GAeFib and aMyo in a 2:1:1 ratio resulted in a more than 10‐fold induction of Pax7 expression at day 7 and 13 (Appendix Fig S1J). Furthermore, hiPSCs in combination with GAeFib and eEC at a 2:1:1 ratio induced Pax7 expression at day 13 more than 30‐fold (Fig 1D). Thus, three‐component embryoids (TCEs) of GAeFib, eEC, and hiPSCs are most permissive for Pax7 induction.
Figure 1. Three‐component embryoids (TCEs) are permissive for myogenic specification.
-
ASchematic outlining embryonic and adult cell lines that were screened for their effects on the induction of the myogenic marker Pax7 in heterotypic human‐induced pluripotent stem cell (hiPSC) suspension embryoids.
-
B, CExpression of the human Pax7 mRNA in embryoids containing different ratios of hiPSCs and growth‐arrested embryonic fibroblasts (GAeFib) or hiPSCs and embryonic endothelial cells (eEC). Mixtures of the respective cell types were analyzed before (D0), and 7 and 13 days (D7 and D13) after aggregation.
-
DHuman Pax7 mRNA expression in TCEs containing hiPSCs, GAeFib, and eEC.
-
E, FSize and human Pax7 mRNA expression in hiPSC mono‐aggregates or in TCEs when combined with biochemical induction of the Wnt and FGF pathways (iTCEs).
-
GRepresentative Pax7 immunostaining of sections of D7 iTCEs.
-
H–KmRNA expression of the mesoderm marker brachyury (T), the paraxial mesoderm markers mesogenin 1 (Msgn1) and T‐box 6 (Tbx6), and the dermomyotome marker Pax3 in hiPSCs and iTCEs at D7 and D13.
-
LRepresentative Pax7 immunostaining of a D13 iTCE.
-
M–PRepresentative stainings for actin, the extracellular matrix component laminin, the mesoderm marker homeobox 1 (Meox1), the endoderm marker alpha‐fetoprotein (AFP), and the mesoderm marker smooth muscle actin (SMA) in D7 or D13 iTCEs.
-
QFlow cytometry quantification of the number of Pax7 positive cells in hiPSC mono‐aggregates, TCEs, and iTCEs.
-
R, SRNA fluorescence in situ hybridization for the myogenic commitment markers Myf5 and MyoD in cells derived from D13 iTCEs (R) and primary adult human skeletal muscle myogenic progenitors (hskMPs) (S) by flow cytometry and in representative post‐sort cytospins.
Data information: G, L, M–P, R, S. Scale bars = 25 μm. B–F, H–K, Q. Data are represented as means ± SEM from n ≥ 4 independent experiments. B–S. hiPSC donor 1. R, S. Representative sorts of n = 3 repeats are provided in Appendix Table S2. P‐values are *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA followed by a Bonferroni post hoc test.
In order to independently reproduce our screen for cell types that are permissive for myogenic specification in 3D embryoids using an alternative readout, we used hiPSCs from a different donor and quantified the number of Pax7 positive (Pax7+) cells after enzymatic release by flow cytometry (Appendix Fig S2A). Due to its high efficacy in our pilot screen, we focused on a 2:1 ratio of hiPSCs and supportive cell types. As observed before, GAeFib, aMyo, and eECs were most permissive in inducing the generation of Pax7+ cells at the 13‐day timepoint (Appendix Fig S2B–H). Moreover, as previously observed, four‐component embryoids containing hiPSCs, GAeFib, eECs, and aMyo did not lead to synergistic or additive effects at any of the timepoints (Appendix Fig S2I). However, similar to our expression‐based screen, when different permutations of three‐component embryoids were analyzed, hiPSCs, GAeFib, and eECs allowed for the highest yield of Pax7+ cells at day 13 after aggregation (Appendix Fig S2J–L). Altogether, these experiments confirm that TCEs containing GAeFibs and eECs facilitate the differentiation of hiPSCs into Pax7+ cells in multiple hiPSC lines.
In light of translational considerations in the context of potential downstream applications such as cell therapy, we also tested whether xeno‐free TCEs containing hiPSCs with human growth‐arrested fibroblasts (hGAFib) and human endothelial cells (hECs) are permissive for Pax7 expression. Similar to TCEs containing murine support cells, fully humanized embryoids showed the strongest induction of Pax7 expression at a 2:1:1 ratio of hiPSCs, hGAFib, and hECs at the 13‐day timepoint after aggregation (Appendix Fig S2M–O). Therefore, the permissive effect of fibroblasts and endothelial cells on myogenic differentiation of hiPSCs in TCEs is not limited by species barriers.
WNT/FGF stimulation of TCEs supports hiPSC commitment to the mesodermal lineage
Established protocols for the derivation of myogenic cells from hiPSCs are performed in 2D culture and frequently rely on activation of the Wnt pathway through glycogen synthase kinase‐3 (GSK3) inhibitors in the early stages of differentiation and subsequently switch to basic fibroblast growth factor (bFGF) stimulation (Barberi et al, 2007; Yu et al, 2007; Shelton et al, 2014; Chal et al, 2015, 2016; Caron et al, 2016). Suggesting increased differentiation at the expense of proliferation, chemically induced TCEs (iTCEs) treated with the WNT pathway activator CHIR99021 from day 1 to 7 of aggregation and stimulated with bFGF from day 7 to 13, displayed a reduced growth rate (Fig 1E and Appendix Fig S2P). Notably, expression analysis revealed an almost 2000‐fold induction of Pax7 in iTCEs at the 13‐day timepoint (Fig 1F). At day 7, iTCEs contained abundant epithelial‐like structures with distinct domains of Pax7+ cells (Fig 1G). iTCEs also expressed high levels of the early mesoderm marker brachyury (T) (Showell et al, 2004), the paraxial mesoderm markers mesogenin 1 (Msgn1) (Chalamalasetty et al, 2014) and T‐box 6 (Tbx6) (Chapman et al, 1996), and the dermomyotome marker Pax3 (Goulding et al, 1994; Fig 1H–K). Between day 7 and 13, iTCEs underwent an epithelial–mesenchymal transition‐like process and invaginated (Fig 1G and L). Pax7+ cells in iTCEs at day 13 were more dispersed than at day 7 and preferentially resided in actin‐poor regions (Fig 1M). Expression of the myogenic commitment marker Myf5 increased in iTCEs at day 13, while MyoD was not detectable (Appendix Fig S3A and B). Immunostainings revealed that iTCEs contain high amounts of the muscle extracellular matrix component laminin, while Pax3 and homeobox 1 (Meox1) (Candia et al, 1992) positive cells confirmed mesodermal induction (Fig 1N and O). In contrast to day 13, iTCEs at day 7 stained positive for the primitive endoderm marker alpha‐fetoprotein (Kwon et al, 2006) (AFP) in their center while peripheral regions were positive for the mesoderm marker smooth muscle actin (SMA) (Bharathan et al, 2017; Fig 1P). A transient endoderm induction in day 7 iTCEs was confirmed by Gata4 (Narita et al, 1997) expression (Appendix Fig S3C). The ectoderm marker Fgf5 (Hebert et al, 1991) and the neural commitment marker Neurog1 (Ma et al, 1998) were expressed at low levels at all timepoints in iTCEs (Appendix Fig S3D and E). Immunostaining revealed that the center of day 7 iTCEs contained small amounts of cells positive for the ectoderm marker Tuj1 (Easter et al, 1993), while no staining was observed for the neural progenitor markers nestin (Frederiksen & McKay, 1988) or Sox2 (Ellis et al, 2004; Appendix Fig S3F). In summary, two‐week differentiation of hiPSCs in iTCEs induces mesodermal commitment and leads to the generation of Pax7+ cells.
The 3D microenvironment in iTCEs determines the efficacy of Pax7 induction
Enzymatic release of cells from day 13 embryoids and quantification by flow cytometry revealed 127% and 70% more Pax7+ cells in iTCEs when compared to TCEs or chemically stimulated hiPSC mono‐aggregates respectively (Figs 1Q and Appendix Fig S4A). Using hiPSC lines from two donors different from our initial expression screen, we confirmed a yield of ~40–50% of Pax7+ cells from iTCEs (Appendix Fig S4B and C). Notably, when hiPSCs with GAeFib and eEC were not aggregated but plated for 13 days in 2D culture and treated with the same Wnt/FGF induction scheme as iTCEs, only 3.6% of the cells were observed to be Pax7+ (Appendix Fig S4D and Appendix Table S1). When the two established transgene‐free 2D myogenic hiPSC differentiation protocols (Shelton et al, 2014; Choi et al, 2016) were stopped after two weeks, between 0.1% and 0.6% Pax7+ cells could be detected in the cultures (Appendix Fig S4E and F, and Appendix Table S1). Moreover, when the established protocols (Shelton et al, 2014; Choi et al, 2016) were completed, they yielded between 7.8% and 10.8% Pax7+ cells at day 30 and 50, respectively (Appendix Fig S4G and H, and Appendix Table S1). Thus, the 3D context is essential for the speed and efficiency of Pax7 induction in iTCEs.
iTCE‐derived Pax7 positive cells display embryonic characteristics
To interrogate the number of cells expressing the myogenic commitment markers Myf5 and MyoD, we quantified enzymatically liberated cells from iTCEs stained by RNA fluorescence in situ hybridization (FISH) by flow cytometry. This revealed expression of Myf5 in around 16% of cells in iTCEs, while MyoD was below 1% (Fig 1R, Appendix Fig S4I, and Appendix Table S2 and S3). In contrast, 2D cultured myogenic progenitors isolated from adult human skeletal muscle (hskMPs) contained between 40 and 50% of Myf5/MyoD+ cells (Fig 1S and Appendix Table S2 and S3). These results demonstrate that iTCE‐derived Pax7+ cells express low levels of myogenic commitment factors.
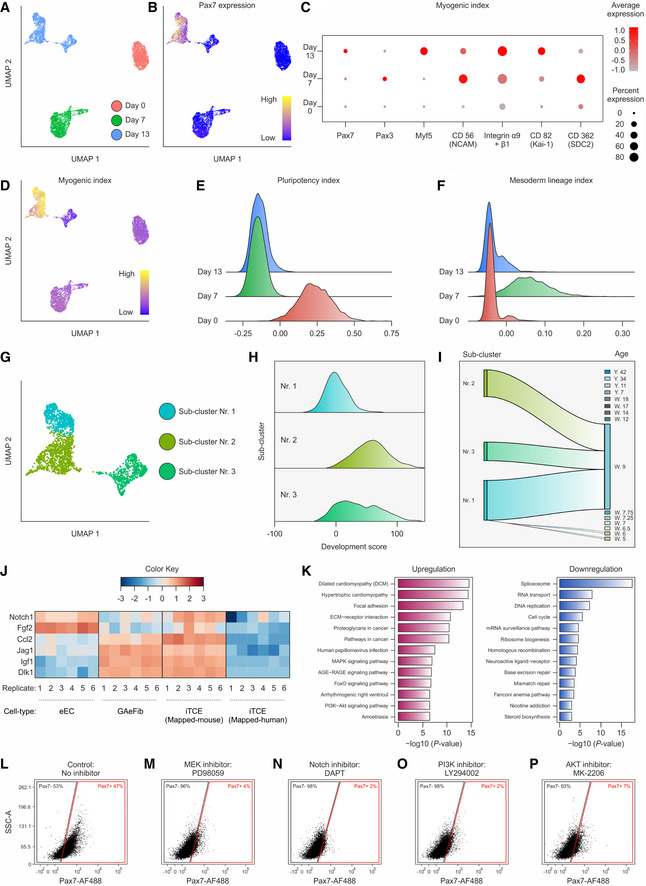
In order to understand how heterologous GAeFibs and eECs promote myogenic specification of hiPSCs, we performed single‐cell RNA sequencing of 2D cultured hiPSCs (day 0) and of 7‐ and 13‐day iTCEs. This experiment revealed an increasing complexity of human cells in the embryoids over the differentiation time course (Fig 2A). Mapping of Pax7 expression showed a distinct localization to a portion of the 13‐day cluster (Fig 2B). To better characterize the identity of cells in iTCEs, we defined a myogenic expression index based on published markers that is composed of Pax7, Pax3, Myf5, CD56 (NCAM), integrin α9β1, CD82 (Kai‐1), and CD362 (SDC2) (Fig 2C). Expression of the myogenic index mapped to the same region in the day 13 cluster as Pax7 (Fig 2D). An index composed of pluripotency markers (Wu et al, 2018) showed highest expression at day 0, while the mesodermal lineage index was most expressed by cells in day 7 iTCEs (Figs 2E and F, and Appendix Fig S5A and B; Wu et al, 2018). Assessment of the neuro‐ectoderm lineage index in iTCEs revealed an induction of genes on day 7 that was decreased by day 13 (Appendix Fig S5C), while the hematopoietic‐endothelial lineage index showed a lower overall induction of genes (Appendix Fig S5D). Further analysis of the day 13 iTCE cluster revealed that it contains 3 sub‐clusters (Fig 2G). Importantly, sub‐clusters 1 and 2 in day 13 iTCEs contained the myogenic cell population (Fig 2D and G). Collectively genes of the pluripotency index mapped to the day 0 cluster (Appendix Fig S5E), while genes of the mesodermal and neuro‐ectoderm lineage index mapped to the day 7 iTCE cluster (Appendix Fig S5F and G). Lastly, the hematopoietic‐endothelial lineage index mapped to sub‐cluster 3 of day 13 iTCEs (Appendix Fig S5H). Thus, mesodermal and neuro‐ectodermal markers are expressed by day 7 iTCEs but downregulated by day 13. In contrast, myogenic markers map strongest to sub‐cluster 1 and 2 and hematopoietic‐endothelial genes to sub‐cluster 3 of day 13 iTCEs.
Figure 2. Transcriptome analysis and mechanism of myogenic specification in iTCEs.
-
A2D UMAP projection visualizing human single‐cell transcriptomes of hiPSCs before aggregation (D0) and from day 7 and 13 iTCEs. Cells are colored by timepoint.
-
BExpression of the human Pax7 mRNA overlaid on the 2D UMAP projection.
-
CExpression of the myogenic index composed of selected human myogenic markers at D0 and in D7 and D13 iTCEs.
-
DEnrichment of the myogenic index overlaid on the 2D UMAP projection.
- E, F
-
GSub‐clusters in D13 iTCEs.
-
HHistograms of the enrichment of human myogenic development score (Xi et al, 2020) among the three sub‐clusters of day 13 iTCEs.
-
IMapping of the three sub‐clusters of day 13 iTCEs to myogenic cells across the human myogenic development score (Xi et al, 2020).
-
JExpression heatmap of extracellular ligands expressed by eECs, GAeFib, and iTCEs. Mapping to the human genome is shown as a control.
-
KTop KEGG pathways based on differentially expressed genes in iTCEs compared to Wnt/FGF pathway induced hiPSC mono‐aggregates.
-
L–PFlow cytometry quantification for Pax7 in D13 iTCEs incubated with vehicle (Control, no inhibitor) or inhibitors of MEK, Notch, PI3K, and AKT.
Data information: A–P. hiPSC donor 1. L–P. Representative sorts of n = 3 repeats are provided in Appendix Table S4.
Assignment of the human myogenic developmental score based on the database compiled by Xi et al (2020) revealed that sub‐cluster nr. 2 at day 13 contains the most mature population of cells expressing myogenic markers in iTCEs (Fig 2H). Moreover, iTCE‐derived cells from all day 13 sub‐clusters mapped to fetal week 9 of human development (Fig 2I). Thus, similar to previously developed myogenic hiPSC differentiation protocols, iTCEs contain cells expressing myogenic markers that display transcriptional characteristics resembling skeletal muscle progenitors at the embryonic‐to‐fetal transition period between 7 and 12 weeks post‐fertilization (Xi et al, 2020).
Extracellular matrix and paracrine signals in iTCEs
To study the cellular cross‐talk in iTCEs, we performed bulk RNA sequencing of GAeFibs and eECs in 2D mono‐culture. This experiment revealed ligands activating Notch, FGF, and IGF1/PI3K/AKT signaling (Figs 2J and Appendix Fig S6A). Moreover, when gene set enrichment analysis using the hallmark database was performed by comparing bulk‐sequenced iTCEs to WNT/FGF‐stimulated hiPSC mono‐aggregates, we observed that the supportive cell types induce genes involved in focal adhesion, ECM‐receptor interaction, and the MAPK and PI3K/AKT pathways (Figs 2K and Appendix Fig S6B). In agreement with increased differentiation and lineage commitment, genes involved in DNA replication and cell cycle were downregulated in iTCEs compared to WNT/FGF‐stimulated hiPSC mono‐aggregates. Importantly, MAPK and PI3K/AKT function downstream of FGF and IGF1 receptors (Tao et al, 2007; Ornitz & Itoh, 2015). To validate these observations, we subsequently exposed iTCEs to small molecular inhibitors of the FGF and IGF1 receptors, the MAPK, Notch, and PI3K/AKT pathways during the two‐week differentiation time course. The yield of Pax7+ cells from iTCEs was reduced from 47% in the control to 20% for an FGF receptor inhibitor, 5% for an IGF1 receptor inhibitor, 4% for a MEK inhibitor, 2% for a Notch inhibitor, 2% for a PI3K inhibitor, and 7% for an AKT inhibitor (Fig 2L–P, Appendix Fig S6C and D, and Appendix Table S4 and S5). Therefore, GAeFibs and eECs generate a distinct structural extracellular environment in iTCEs and stimulate the FGF‐MAPK, IGF1/PI3K/AKT, and Notch pathways to direct mesodermal fate decisions and myogenic specification in hiPSCs.
Flow cytometry isolation of eMPs from iTCEs
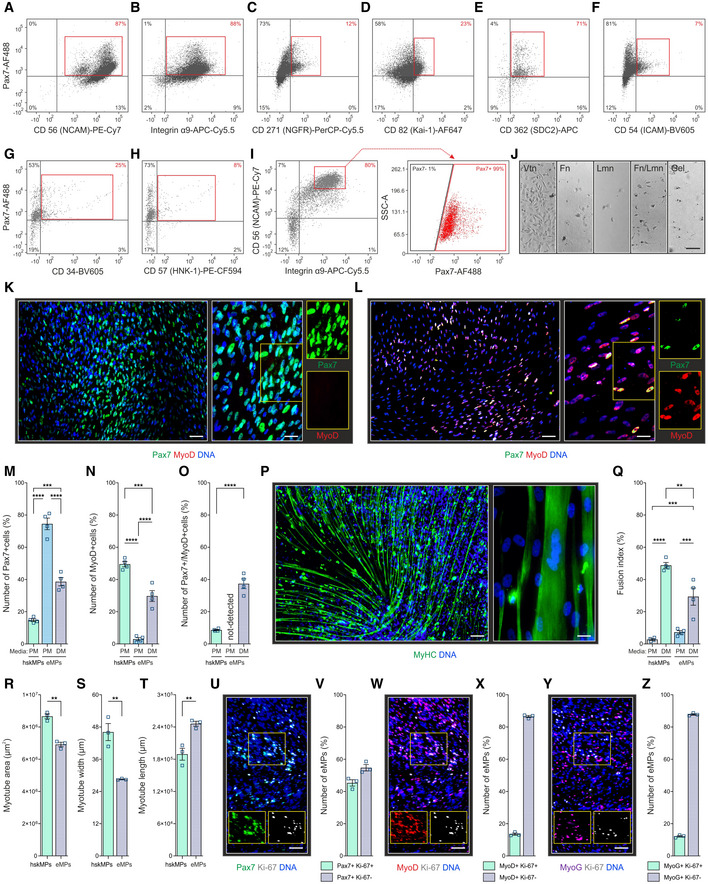
To isolate live Pax7+ cells and examine their myogenic potential, we tested for co‐expression of Pax7 with a panel of cell surface markers previously reported to be expressed by adult or embryonic myogenic progenitors by flow cytometry (Fig 3A–F and Appendix Table S6). After enzymatic liberation from day 13 iTCEs, CD56 (Schubert et al, 1989) and integrin α9 (Magli et al, 2017) detected 87% and 88% of Pax7+ cells, respectively (Fig 3A and B, Appendix Table S6). The enrichment was smaller when CD271 (Hicks et al, 2018), CD82 (Uezumi et al, 2016), CD362 (Magli et al, 2017), and CD54 (Magli et al, 2017) were used (Fig 3C–F and Appendix Table S6). A lower percentage of Pax7+ cells was positive for the marker CD34, which has been associated with adipogenic differentiation of human muscle progenitors (Pisani et al, 2010), and the neural crest marker CD57 (Lipinski et al, 1983; Fig 3G and H, and Appendix Table S6). Notably, combination of CD56 and integrin α9 antibodies allowed for enrichment of a population of > 99% Pax7 positive eMPs from day 13 iTCEs (Fig 3I and Appendix Table S6).
Figure 3. Purification and characterization of iTCE‐derived embryonic‐like myogenic progenitors (eMPs).
-
A–FFlow cytometry quantification for Pax7 and myogenic progenitor surface markers in cells derived from D13 iTCEs.
-
G, HFlow cytometry quantification of Pax7, CD34 marking human muscle progenitors with adipogenic potential, and the neural crest marker CD57 in cells derived from D13 iTCEs.
-
IFlow cytometry quantification of Pax7, integrin α9, and CD56 in cells isolated from D13 iTCEs.
-
JRepresentative brightfield images of the adhesion of integrin α9 and CD56 positive flow cytometrically sorted eMPs isolated from D13 iTCEs on vitronectin (Vtn), fibronectin (Fn), laminin (Lmn), and gelatine (Gel) in the presence of ROCK inhibitor.
-
KRepresentative immunostainings for Pax7 and MyoD in eMPs plated on Vtn and maintained in proliferation media (PM) after isolation.
-
LRepresentative immunostainings for Pax7 and MyoD in eMPs plated on Vtn that were first maintained in PM and then switched to differentiation media (DM).
-
M–OQuantification of the number of Pax7 (M), MyoD (N) and Pax7/MyoD double‐positive (O) cells in hskMPs and eMPs in PM and after switching to DM.
-
PRepresentative immunostaining for the muscle fiber marker myosin heavy chain (MyHC) of eMPs maintained in DM.
-
QQuantification of the fusion index of hskMPs and eMPs in PM and DM.
-
R–TQuantification of the total myotube area (R), myotube width (S), and myotube length (T) of hskMPs and eMPs in DM.
-
U–ZRepresentative immunostainings and quantification of eMPs in DM expressing Pax7 (V), MyoD (X), and MyoG (Z), as well as the proliferation marker Ki67.
Data information: K, L, P. Left‐hand scale bar = 75 μm, right‐hand scale bar = 50 μm. J. Scale bar = 40 μm. U, W, Y. Scale bar = 75 μm. M–O, Q–T, V, X, Z, Data are represented as means ± SEM from n = 4 (M–O, Q) and n = 3 (R–T, V, X, Z) independent experiments. A–I. Representative sorts of n = 3 repeats are provided in Appendix Table S6. A–Z. hiPSC donor 1. P‐values are *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA followed by a Bonferroni post hoc test.
iTCE‐derived eMPs partially resist differentiation
Following sorting of live CD56 and integrin α9 positive cells from iTCEs, the cells adhered most efficiently on vitronectin‐coated dishes compared to fibronectin, laminin, or gelatine (Fig 3J). After isolation and 2D culture on vitronectin, 80% of CD56 and integrin α9 positive embryonic‐like myogenic progenitor cells (eMPs) sorted from iTCEs stained positive for Pax7 but remained negative for the commitment marker MyoD (Fig 3K). In growth factor‐depleted differentiation medium, the number of eMPs expressing Pax7 dropped but was still higher than in proliferating hskMPs (Fig 3L and M). While remaining lower than in hskMPs, MyoD positive cells became more abundant in eMPs cultures in differentiation media (Fig 3N and O).
To determine whether eMPs are candidates for sustained engraftment in a cell therapy setting, we assessed their ability to resist differentiation. After prolonged exposure to differentiation media, eMPs upregulated myosin heavy chain (MyHC) and formed multinucleated myotubes (Fig 3P). With ~30%, the fusion index of eMPs was lower than hskMPs (Fig 3Q). Moreover, the total area covered by myotubes was 20% lower when differentiated eMPs were compared to hskMPs (Fig 3R). While the width of myotubes generated by differentiated eMPs was 38% smaller than hskMPs, their length was increased by 30% (Fig 3S and T). Under differentiation conditions, 45% of eMP‐derived cells that resist fusion and still express Pax7 stained positive for the proliferation marker Ki‐67, while 55% were negative (Fig 3U and V). However, the majority of eMP‐derived cells expressing MyoD or MyoG under differentiation conditions were Ki67 negative (Fig 3W–Z). Thus, while a fraction of the cells remains resistant to differentiation, eMPs are bona fide muscle progenitors capable of fusion.
The number of Pax7+ eMPs in 2D culture was higher under both proliferation and differentiation conditions when benchmarked to established transgene‐free hiPSC differentiation protocols (Shelton et al, 2014; Choi et al, 2016) after two weeks and at their 30‐ and 50‐day endpoints respectively (Appendix Fig S7A). The spontaneous fusion index of eMPs in proliferation media was not different from the established protocols at any timepoint (Appendix Fig S7B). Moreover, eMPs in differentiation media fused to the same degree as cells at day 30 of the Choi et al protocol, while they produced more differentiated myotubes than Shelton et al at day 50. Therefore, compared to the mixed cultures obtained from established transgene‐free hiPSC differentiation protocols, eMPs allow for efficient segregation of proliferation and differentiation in vitro.
eMPs fuse to muscle fibers and engraft as stem cells that can be reactivated
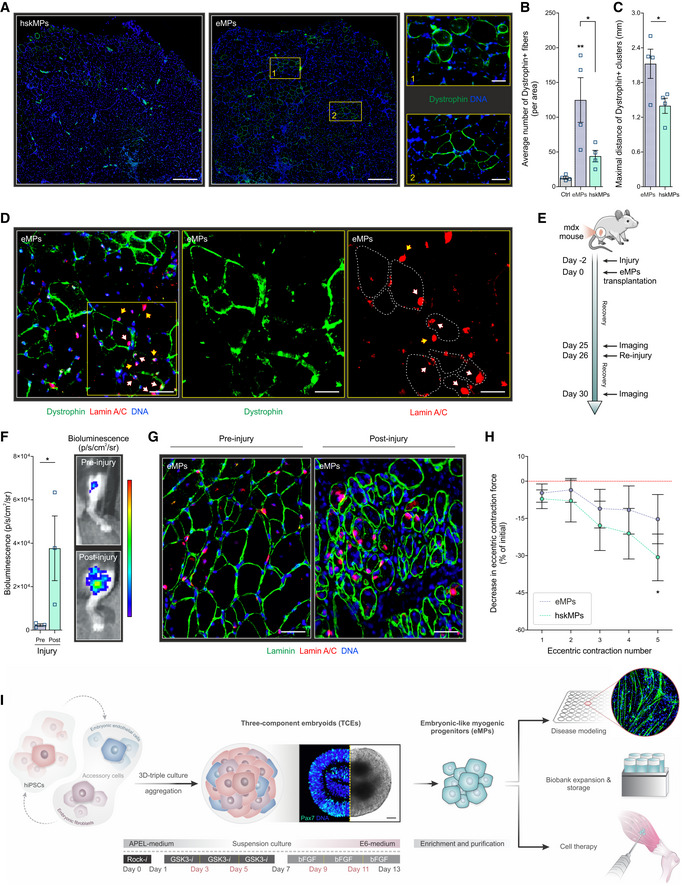
To test their ability to engraft in a cell therapy setting, we injected freshly sorted eMPs into muscles of the mdx mouse model of Duchenne muscular dystrophy (Sicinski et al, 1989). Following maximization of regeneration by cardiotoxin, transplanted eMPs generated 183% more dystrophin positive fibers than hskMPs 10 days after engraftment (Fig 4A and B). The number of spontaneously reverting fibers in sham transplanted mdx mice was 13% and contributed only marginally to these results. Indicating an extensive migratory capacity in the host tissue, dystrophin positive fibers were spaced out 52% further apart in the eMP‐injected muscles when compared to hskMPs (Fig 4A and C). Co‐immunostaining for human nuclear Lamin A/C and CD56 showed that a fraction of eMPs maintained stem cell characteristics and remained unfused after engraftment into muscles of mdx mice (Appendix Fig S8A). eMPs frequently engrafted into the satellite cell position under the basal‐lamina in the periphery of muscle fibers with restored dystrophin expression (Fig 4D and Appendix Fig S8B and C). To assess their ability to reactivate after transplantation, we derived eMPs from an hiPSC line expressing luciferase. Following a second cardiotoxin injury 25 days after engraftment of luciferase eMPs, a 1,532% increase in the amount of bioluminescence was observed compared to the pre‐injury condition (Fig 4E and F). Immunostaining for human nuclear Lamin A/C and laminin revealed extensive eMP fusion to muscle fibers in the post‐injury condition, as well as cells that remained unfused in the periphery of muscle fibers (Fig 4G). These results support the notion that eMPs have a higher fusion and migratory capacity than hskMPs, engraft into the stem cell niche, and can sustain multiple rounds of regeneration in a dystrophic skeletal muscle environment.
Figure 4. Engraftment of eMPs in mdx mice.
-
ARepresentative immunostainings for dystrophin in cross‐sections of muscles of mdx mice after transplantation of hskMPs or eMPs. Scale bars in the two left‐hand side images = 300 μm. Scale bars in inserts 1 and 2 = 50 μm.
-
BNumber of dystrophin positive fibers in mdx muscles injected with vehicle (Ctrl), eMPs and hskMPs.
-
CMaximal distance of clusters of dystrophin positive fibers in cross‐sections of mdx muscles transplanted with eMPs and hskMPs.
-
DRepresentative immunostainings for the human nuclear marker lamin A/C and dystrophin in muscles of mdx mice after transplantation of eMPs. Dashed lines outline dystrophin positive fibers. Yellow arrows depict cells in the satellite cell position in the periphery of muscle fibers and white arrows show human myonuclei. Scale bars = 50 μm.
-
ESchematic outlining the re‐injury experiment of mdx muscles transplanted with eMPs derived from a luciferase hiPSC line.
-
FQuantification and representative images of luciferase bioluminescence before (pre‐injury) and after re‐injury (post‐injury) of eMP transplanted muscles.
-
GRepresentative immunostainings for laminin and human lamin A/C in cross‐sections of muscles of mdx mice four days after re‐injury. Scale bars = 50 μm.
-
HEvolution of eccentric force production of mdx muscles transplanted with eMPs or hskMPs.
-
IScheme summarizing the procedure for the generation of eMPs and potential downstream applications.
Data information: A–C. 10 days post‐transplantation. D. 25 days post‐transplantation. F–G. Pre‐injury 25 days and post‐injury 30 days after transplantation. H. 17 days post‐transplantation. B, C. n = 4 mice per condition. F. n = 3 mice per condition. H. n = 5 mice per condition. Data are represented as means ± SEM. P‐values are *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA followed by a Bonferroni post hoc test for multiple comparisons or students t‐test for binary comparisons. A–C, H. hiPSC donor 1. D, F–G. hiPSC donor 4.
Eccentric contraction is the main mechanical trigger leading to membrane damage in mdx mice (Blaauw et al, 2010). Therefore, we compared the eccentric force generation of mdx muscles 17 days after transplantation with hskMPs or eMPs. With increasing numbers of eccentric contractions, eMP transplantation supported the maintenance of a higher force generation when compared to contralateral muscles treated with hskMPs (Fig 4H). Altogether, these observations show that eMPs are suitable for cell therapy of recessive genetic muscle diseases and, through their ability to engraft into the host stem cell compartment, are likely to have sustained therapeutic effects.
iTCE production can be scaled to the bioreactor level
Due to the requirement of large tissue culture surfaces and the absence of essential niche signals, 2D scale‐up culture of primary skeletal muscle progenitors for transplantation therapy in human patients is presently challenging. To demonstrate that iTCE suspension cultures can be scaled to larger volumes, we quantitatively repeated our protocol using roller bottles containing 250 ml of media (Appendix Fig S9A). Similar to the horizontal shaker method, we observed a significant induction of Pax7 expression over the 13‐day differentiation time course in roller bottle cultures (Appendix Fig S9B). With a starting number of 1x108 total cells in the form of iTCEs, this scale‐up method yielded on average 5.4 × 108 and 3.8 × 108 cells after 7 and 13 days of culture, respectively (Appendix Fig S9C). The overall viability of iTCE‐derived cells after enzymatic liberation at day 13 was ≥ 91% (Appendix Fig S9D). Moreover, the yield of CD56 and integrin α9 double‐positive eMPs from scale‐up cultures was ≥ 62% (Appendix Fig S9E). Thus, iTCE‐derived eMPs can efficiently be produced in scale‐up culture.
In summary, by mimicking features of the embryonic environment in heterotypic 3D embryoids, we have significantly improved the efficiency and duration of established biochemical protocols for myogenic differentiation of hiPSCs. Our streamlined method allows for the two‐week derivation of pure Pax7+ MPs from hiPSCs that map to the embryonic‐to‐fetal transition period. We demonstrate that the supportive cell types in iTCEs generate a highly specialized structural 3D niche environment that induces mesoderm specification and myogenic commitment in hiPSCs by activating the FGF‐MAPK, Notch, and IGF1/AKT/PI3K pathways.
Discussion
Our results demonstrate that the induction of Pax7 expression in iTCEs depends on the FGF‐MAPK, Notch, and IGF1/PI3K/AKT pathways in a distinct structural extracellular matrix environment promoting the formation of focal adhesions. Importantly, all three pathways identified in our study have previously been shown to promote mesoderm specification in hiPSCs (Engels et al, 2014; Shelton et al, 2014; Choi et al, 2016; Kumar et al, 2019). However, we demonstrate that 2D adaptation of our protocol is highly inefficient. These findings demonstrate that soluble signals alone are not able to mimic the embryonic microenvironment and that direct 3D cell‐cell contacts and adhesion to the structural extracellular matrix are key elements for the generation of Pax7+ cells in iTCEs.
One of the primary reasons for the limited efficacy of hskMP transplantation in human trials is the use of adult donor hskMPs after prolonged expansion in 2D culture, which downregulates Pax7 expression and reduces the engraftment potential of the cells (Bentzinger et al, 2013; Skuk & Tremblay, 2015; Romagnoli et al, 2020). Pax7+ eMPs isolated from iTCEs fuse to muscle fibers, but also generate cells in the satellite cell position that maintain stemness markers and can reactivate following repeated rounds of regeneration. While published transgene‐free protocols for the derivation of myogenic cells from hiPSCs typically result in mixed cultures of progenitors and more differentiated cells, our study provides a means to enrich for embryonic‐like Pax7+ cells that can partially resist differentiation and have an engraftment potential that leads to sustained genetic correction.
To establish our protocol and facilitate the distinction of human cells by transcriptomics, we used widely available immortalized murine support cell lines to generate heterotypic embryoids with hiPSCs. However, for downstream applications in patients, xeno‐free approaches would be favorable. Importantly, we show that similar to murine GAeFib and eECs, human growth‐arrested fibroblasts and endothelial cells are also highly permissive for the specification of hiPSCs toward Pax7+ myogenic progenitors. Moreover, recent protocols allow for the generation of fibroblast‐like cells and endothelial cells from hiPSCs (Du et al, 2015; Patsch et al, 2015). Future studies addressing heterotypic suspension embryoids containing support cells derived from autologous or non‐immunogenic hiPSCs will further facilitate translational applications.
Altogether, our method provides a novel means for the generation of large quantities of Pax7+ human myogenic progenitors in a biologically faithful 3D niche environment. Moreover, the scalability of embryoid suspension cultures allows for production at the bioreactor level and, next to much‐needed cell therapy applications for genetic skeletal muscle diseases, will allow for extensive in vitro modeling and screening applications (Fig 4I).
Materials and Methods
Cell lines and 2D cell culture conditions
The human‐induced pluripotent stem cell (hiPSC) lines 603 (donor 1) and 482 (donor 2) were purchased from Fujifilm Cellular Dynamics, Inc (FCDI), HYS0103 (ACS‐1020) hiPSCs (donor 3) were obtained from ATCC, and luciferase‐expressing hiPSCs from RxCell Inc (donor 4). hiPSCs from FCDI were cultivated in essential‐8 medium (A1517001, Thermo Fisher Scientific) on vitronectin (VTN‐N, A14700, Thermo Fisher Scientific) coated dishes. HYS0103 hiPSCs were initially cultured in pluripotent stem cell SFM XF/FF (ATCC ACS‐3002) media on CellMatrix basement membrane gel (ATCC ACS‐3035). Subsequently, the medium was switched to essential‐8 medium (A1517001, Thermo Fisher Scientific) on vitronectin (VTN‐N, A14700, Thermo Fisher Scientific) coated dishes. Luciferase‐expressing hiPSCs were initially cultured in mTeSR Plus medium (100‐0276, STEMCELL Technologies). Subsequently, the cells were switched to essential‐8 medium (A1517001, Thermo Fisher Scientific) on vitronectin (VTN‐N, A14700, Thermo Fisher Scientific) coated dishes. Human skeletal muscle cells (hskMPs, LZ‐CC‐2580, Lonza) between passages 4 and 6 were plated on fibronectin‐coated (F2006, Sigma‐Aldrich) dishes and expanded in skeletal muscle cell growth medium (SKM‐M, AmsBio). Murine embryonic endothelial cells (eEC, C166, ATCC CRL‐2581), murine adult myoblasts (aMyo, C2C12, ATCC CRL‐1772), murine adult smooth muscle cells (aSMC, MOVAS, ATCC CRL‐2797), murine embryonic fibroblasts (eFib, NIH‐3T3, ATCC CRL‐1658), murine adult fibroblasts (aFib, NOR‐10, ATCC CCL‐197), murine embryonic mesenchymal stem cells (eMSC, OP‐9, ATCC CRL‐2749), human endothelial cells (hEC, HUVEC, ATCC CRL‐1730), human fibroblasts (HDFa, ATCC PCS‐201‐012), and human neuronal progenitor cells (hNPCs, BXS0117, ATCC ACS‐5003) were obtained from ATCC, and mitomycin C‐treated murine embryonic fibroblasts (GAeFib, PMEF‐CF‐1) were purchased from Merck Millipore. eEC, aMyo, and eFib were cultured in DMEM‐High Glucose 4.5 mg/l (11995065, ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS, 10270098, ThermoFisher Scientific) and 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific). aSMC were cultured in DMEM‐High Glucose 4.5 mg/l (11995065, ThermoFisher Scientific) supplemented with 0.2 mg/ml geneticin (G‐418, 10131027, ThermoFisher Scientific), 10% FBS (10270098, ThermoFisher Scientific), and 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific). aFib were cultured in DMEM containing 4.5 mg/l glucose (11995065, ThermoFisher Scientific) supplemented with 20% FBS and 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific). eMSC were cultured in alpha‐minimum essential medium (α‐MEM, without ribonucleosides and deoxyribonucleosides and with 2.2 g/l sodium bicarbonate, 12561056, ThermoFisher Scientific), supplemented with 20% FBS (10270098, ThermoFisher Scientific), and 1% antibiotic/antimycotic solution (15140122 ThermoFisher Scientific). GAeFib were cultured in DMEM with 2 mM l‐glutamine (11965084, ThermoFisher Scientific) and 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific) on gelatine (G1393, Sigma‐Aldrich) coated dishes. hECs were cultured in F‐12K medium (ATCC 30‐2004), 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific), 10% FBS (10270098, ThermoFisher Scientific), 100 μg/ml heparin sodium (H3393, Sigma‐Aldrich), and 30 μg/ml endothelial cell growth supplement (ECGS, 211‐GS, Merck Millipore). Human fibroblasts were cultivated in fibroblast basal medium (ATCC PCS‐201‐030) supplemented with fibroblast growth kit‐low serum medium (ATCC PCS‐201‐041), 1% antibiotic/antimycotic solution (15140122, ThermoFisher Scientific), and treated with Mitomycin C (Merck Millipore) to generate human growth‐arrested fibroblasts (hGAFib). hNPCs were cultured in DMEM:F‐12 (ATCC 30‐2006) on CellMatrix basement membrane gel (ATCC ACS‐3035) supplemented with a growth kit for neural progenitor cell expansion (ATCC ACS‐3003). hNPCs differentiation was induced using a dopaminergic neuron differentiation kit (ATCC ACS‐3004). hiPSCs and hNPCs were passaged using StemPro accutase (A1110501, Thermo Fisher Scientific). hskMPs, hECs, hGAFib, and all murine cell lines were passaged using the TrypLE express enzyme (12605010, Thermo Fisher Scientific). All cell lines were regularly tested for mycoplasma contamination.
Embryoid formation and 3D cell culture conditions
hiPSCs alone or in combination with support cell types were prepared for 3D‐aggregation by adding a total of 5.5 × 106 cells per well in Corning Costar ultra‐low attachment 6‐well plates (CLS3471, Sigma‐Aldrich) in STEMdiff APEL2 medium (05270, STEMCELL Technologies) supplemented with 10 μM ROCK inhibitor (Y‐27632, 1254, Tocris Bioscience) with constant shaking (Orbi‐Shaker CO2, Benchmark Scientific) at 95 rpm at 37°C and 8% CO2 overnight. For Wnt/FGF pathway stimulation, the medium was then replaced with STEMdiff APEL2 medium (05270, STEMCELL Technologies), supplemented with 2.5 μM of GSK3‐inhibitor CHIR99021 (Tocris Bioscience) for 6 days. On day 7, the embryoids were switched to essential 6 medium (A1516401, Thermo Fisher Scientific), supplemented with 20 ng/ml of bFGF (FGF‐2, PHG0023, Thermo Fisher Scientific) until day 13. Culture medium was changed every other day. In conditions without Wnt/FGF pathway stimulation, DMSO was used as a vehicle control. For iTCE scale‐up culture, a total of 1 × 108 cells were added to Corning 850 cm² polystyrene roller bottles (CLS430851, Sigma‐Aldrich) in 250 ml of STEMdiff APEL2 medium (05270, STEMCELL Technologies) supplemented with 10 μM ROCK inhibitor (Y‐27632, 1254, Tocris Bioscience) under constant rolling conditions (FlexiRoll, BioTool AG) at 32 rpm at 37°C and 8% CO2 overnight. Subsequently, media composition was similar to the horizontal shaker protocol.
Small molecular inhibitors
iTCEs at day 3 to 5 after 3D‐aggregation were exposed to 20 μM MEK inhibitor (PD98059, 72172, STEMCELL Technologies), 10 μM Notch inhibitor (DAPT, 72082, STEMCELL Technologies), 10 μM PI3K inhibitor (LY294002, 72152, STEMCELL Technologies), 3 μM AKT inhibitor (MK‐2206, HY‐10358), 100 nM FGF‐R inhibitor (PD173074, 72164, STEMCELL Technologies), and 3 μM IGF‐1R inhibitor (Tyrphostin, AG‐1024, 143283, Abcam) in the culture medium during the entire differentiation time course.
Myogenic 2D differentiation
Integrin α9/CD56 positive flow cytometrically isolated eMPs were plated on vitronectin (VTN‐N, A14700, Thermo Fisher Scientific) coated plates in the presence of 10 μM ROCK inhibitor (Y‐27632, 1254, Tocris Bioscience). eMPs were maintained in proliferation medium (PM, SkGM2 medium, CC‐3160, Lonza) supplemented with 20 ng/ml of bFGF (FGF‐2, PHG0023, Thermo Fisher Scientific). For differentiation, following 11 days of maintenance in PM, eMPs were switched to differentiation medium (DM, N2‐media containing insulin‐transferrin‐selenium, ITS‐G, Thermo Fisher Scientific) and N‐2 Supplement (17502048, Thermo Fisher Scientific) for 10 days. Extracellular matrix proteins other than vitronectin were fibronectin (F2006, Sigma‐Aldrich), gelatine (G1393, Sigma‐Aldrich), and laminin (LN‐211, BioLamina). hskMP were obtained from adult human skeletal muscle (Lonza, LZ‐CC‐2580) and plated on fibronectin. hskMP were either maintained in PM (Skeletal Muscle Cell Growth Medium, AmsBio, SKM‐M medium) or switched to DM (Dulbecco's Modified Eagle Medium/F‐12, DMEM/F‐12, 11320033, Thermo Fisher Scientific) supplemented with 2% horse serum (26050088, Thermo Fisher Scientific) for 4 days. Quantification of myotubes was performed using the automated MetaXpress software version 6.6 (Molecular Devices) for segmentation of myosin heavy chain and Hoechst. The fusion factor was quantified as the percentage of nuclei within myotubes. Myogenic 2D differentiation of hiPSCs according to Shelton et al (2014) was performed as described. Briefly, hiPSCs were seeded on Matrigel‐coated tissue culture plates in essential 8 medium (A1517001, Thermo Fisher Scientific) containing 10 μM ROCK inhibitor (Y‐27632, 1254, Tocris Bioscience). The medium was subsequently replaced with essential 6 medium (A1516401, Thermo Fisher Scientific), supplemented with 10 μM GSK3‐inhibitor CHIR99021 (Tocris Bioscience) for 2 days. CHIR99021 was washed out at day 2, and cells were cultivated in un‐supplemented essential 6 medium until day 12. The medium was replaced with StemPro‐34 SFM media (10639011, Thermo Fisher Scientific) supplemented with 10 ng/ml of bFGF (FGF‐2, PHG0023, Thermo Fisher Scientific) from days 12 to 20. From days 20 to 35, cells were returned to essential 6 medium. Finally, the medium was replaced with N2‐media containing insulin‐transferrin‐selenium (ITS‐G, Thermo Fisher Scientific) and N‐2 Supplement (17502048, Thermo Fisher Scientific) until the endpoint of the experiment. Cultured cells were fed with fresh medium every day. Myogenic 2D differentiation of hiPSCs according to Choi et al was performed as described (Choi et al, 2016). Briefly, hiPSCs were cultivated with N2‐media supplemented with 3 μM CHIR99021. At day 4, 10 μM DAPT (72082, STEMCELL Technologies) was supplemented to the N2‐media and maintained until day 12. From days 12 to 30, cells were maintained in the N2‐media and culture media were changed every other day.
Immunofluorescence
Cells were washed in PBS (10010023, Thermo Fisher Scientific) for 5 min before fixation in 4% paraformaldehyde (PFA, 28908, Thermo Fisher Scientific) for 15 min. Fixed cells were washed three times with PBS (10010023, Thermo Fisher Scientific) and permeabilized in 0.1% Triton X‐100 (T8787, Sigma‐Aldrich) for 15 min at room temperature. The cells were then blocked with 4% bovine serum albumin (BSA, 001000162, Jackson ImmunoResearch, IgG‐free) in PBS (10010023, Thermo Fisher Scientific) and 4% goat‐serum (16210064, Thermo Fisher Scientific) for 1 h at room temperature. Samples were incubated with primary antibody at 4°C overnight or at room temperature for 2 h. After three PBS (10010023, Thermo Fisher Scientific) washes, cells were incubated with corresponding secondary antibody and 40, 6‐diamidino‐2‐phenylindole (DAPI, D1306, Thermo Fisher Scientific) for 45 min at room temperature. After PBS washing (10010023, Thermo Fisher Scientific), the sample was dried and mounted with mounting buffer (ProLong Diamond Antifade Mountant, P36965, Thermo Fisher Scientific). Tibialis anterior (TA) muscles were and frozen in isopentane cooled with liquid nitrogen. Cryosectioning was performed at 10 μm. Sections were allowed to air‐dry for 10 min, fixed for 10 min with 4% paraformaldehyde (PFA, 28908, Thermo Fisher Scientific) and subsequently permeabilized in 0.1% Triton X‐100 (T8787, Sigma‐Aldrich) for 15 min. After blocking in PBS (10010023, Thermo Fisher Scientific) with 4% goat‐serum (16210064, Thermo Fisher Scientific) for 3 h at room temperature, cryo‐sections were incubated with primary antibodies for 3 h at room temperature in blocking solution. Slides were then incubated for 1 h at room temperature with secondary antibodies and counterstained with DAPI (D1306, Thermo Fisher Scientific). For Pax7 staining, antigen retrieval was performed with two successive incubations of hot citric acid 0.01 M pH 6 for 10 min. The signal obtained from the Pax7 antibody was further amplified using a goat‐anti mouse IgG1‐biotin (115065205, Jackson ImmunoResearch) followed by a Streptavidin Alexa‐555 (S32355, Life Tech) treatment, together with other secondary antibodies and DAPI (D1306, Thermo Fisher Scientific). Embryoids were washed with PBS (10010023, Thermo Fisher Scientific) and fixed with 4% paraformaldehyde (PFA, 28908, Thermo Fisher Scientific) for 30 min. Fixed samples were soaked in 30% sucrose for at least 24 h at 4°C. For freezing, embryoids were embedded in gelatine (G1393, Sigma‐Aldrich) solution (15% sucrose, 7.5% gelatine in PBS). Samples were frozen in dry ice‐cooled isopentane and stored at −80°C until sectioning. Cryo‐sections of 10 μm were stained using the same procedure as TA muscles. Imaging was carried out using DMI6000 inverted microscope (Leica), a VS120 slide scanner (Olympus), or a DMI 4000B microscope (Leica) and analyzed either using the VS‐ASW FL software measurement tools or the LAS AF software.
Antibodies and staining probes
The following antibodies and staining probes were used: Pax7 (34360, Abcam), Pax7 (528428, DSHB), Pax3 (381801, ThermoFisher Scientific), Meox1/Mox‐1 (75895, Abcam), Tuj1/βIII‐Tubulin (ectodermal), a‐SMA (mesodermal), and AFP (endodermal) markers from the 3‐germ Layer immunocytochemistry kit (A25538, ThermoFisher Scientific), Tuj1/βIII‐Tubulin (18207, Abcam), CytoPainter‐Phalloidin‐iFluor 488 reagent (176753, Abcam) labeling F‐actin, MyoD (SC304, Santa Cruz), MyoD (PA523078, ThermoFisher Scientific), MyHC (MF20, MAB4470, R&D Systems), Dystrophin (15277, Abcam), Dystrophin (MANDYS106‐clone 2C6) (MABT827, Millipore), Lamin A/C (clone JOL2, 40567, Abcam), Nestin (Rat‐401) (6142, Abcam), Sox2 (97959, Abcam), MyoG (clone EPR4789, ab124800, Abcam), Ki‐67 (ab833, Abcam), CD56 (75813, Abcam), and Laminin (LC‐C96142‐100, Lifespan Bioscience). The following antibodies were used for flow cytometry: CD56 (clone B159, 557747, BD Biosciences), CD57 (Clone HNK‐1, 562488, BD Biosciences), CD54 (ICAM, Clone HA58, 740404, BD Biosciences), CD271 (NGFR, Clone C40‐1457, 560834, BD Biosciences), CD82 (Kai‐1, 564341, BD Biosciences), CD34 (Clone 8G12‐HPCA2, 745247, BD Biosciences), CD362 (SDC2, FAB2965A, R&D Systems), and Integrin α9 (clone Y9A2, 351602, BioLegend). Hoechst (B2261, Sigma‐Aldrich) was used to stain DNA. RNA visualization was performed using the PrimeFlow RNA Assay Kit (88‐18009‐204) from Thermo Fisher Scientific according to the manufacturer’s instructions and was analyzed using an LSRFortessa SORP (H647800N0001, BD Biosciences). Briefly, dissociated cells from embryoids and hskMPs were fixed and permeabilized in Fixation Buffer 1 on ice for 30 min, followed by incubation in permeabilization‐buffer on ice for 30 min. Then, samples were fixed in Fixation Buffer 2 at room temperature for 1 h. To detect cellular RNA targets, sequential hybridizations were performed in a dry incubator pre‐calibrated to 40°C. Target probe sets were hybridized for 2 h, and the signal was amplified using hybridizations with PreAmplifier and then Amplifier for 1.5 h each. Eventually, cells were hybridized with fluorochrome‐conjugated Label Probes for 1 h. Probes were: MyoD‐AF488 (VA4‐3082435), Myf5‐AF647 (VA1‐11144), and GADPH‐AF750 (VA6‐10337). For visualization, stained cells were centrifuged onto a slide using a cytospin 4 (Thermo Fisher Scientific).
Isolation of cells from embryoids
Embryoids were washed with PBS (10010023, Thermo Fisher Scientific) and dissociated using StemPro accutase (A1110501, Thermo Fisher Scientific) for 5 min and TrypLE express enzyme (12605010, Thermo Fisher Scientific) for 3 min. For flow cytometry, the suspension was filtered, centrifuged, and reconstituted in FACS‐buffer (1% BSA/PBS). Cells were incubated with primary antibodies at room temperature for 1 h, washed, and labeled for 45 min with secondary antibodies for analysis. For quantification of intracellular antigens, the cells were fixed and permeabilized with the FIX&PERM Cell Fixation & Cell Permeabilization kit (GAS003, Life Technology).
Fluorescence‐activated cell sorting
Flow cytometry analysis and sorting was performed using an LSRFortessa SORP (H647800N0001, BD Biosciences) and a MoFlo Astrios EQ (106694, Beckman Coulter) respectively. The data on the LSRFortessa SORP were acquired and recorded with the BD FACSDiva software version 8.0.2. The data of the MoFlo Astrios EQ were acquired and recorded with the software Summit version 6.3.1.16945. All data were subsequently analyzed with FCSExpress Flow Cytometry version 6.06.0014 (4193, De Novo Software). Cell viability was assessed for all the experiments using the LIVE/DEAD fixable aqua dead cell stain kit (L34957, Thermo Fisher Scientific). The purity of sorted samples was verified by flow cytometry re‐analysis.
RNA extraction
RNA was extracted using the Agencourt‐RNAdvance tissue kit (A32645, Beckman Coulter) following the manufacturer's instructions. Two rounds of cRNA synthesis starting with 5 ng of total RNA were performed using the MessageAmp II aRNA amplification kit (AM1751, Life Technologies) and MessageAmp II‐biotin enhanced aRNA amplification kit (AM1791, Life Technologies) according to the manufacturer's instructions. RNA was quantified using the Quant‐iT RiboGreen RNA Assay Kit (R11490, Invitrogen) using a Spectramax M2 (Molecular Devices). Total RNA quality was assessed using Fragment Analyzer‐96 with DNF‐471‐0500 Standard Sensitivity RNA Analysis Kit (Agilent Technologies).
Quantitative RT‐PCR
RNA samples were subjected to reverse transcription using Prime Script reverse transcriptase (2680B, Takara) with random 6‐mer primers according to the manufacturer’s instructions using peqSTAR 96 universal gradient thermocycler (Peqlab). Quantitative PCR was performed by using the LightCycler 480 (Roche Diagnostics), 384‐well PCR plates, and the LightCycler DNA green master mix (05573092001, Roche Molecular Systems). All reactions were conducted in quadruplicate with each PCR consisting of 2 μl of the diluted cDNA, 0.5 μl primers, and 2 μl master mix. The reaction volume was adjusted to 10 μl with nuclease‐free water. The non‐template control (NTC) reactions contained water instead of cDNA as template. Taqman probes (ThermoFisher Scientific) were: Pax7 (Hs00242962_m1), Brachyury (T) (Hs00610080_m1), Msgn1 (Hs03405514_s1), Tbx6 (Hs00365539_m1), Pax3 (Hs00240950_m1), Myf5 (Hs00929416_g1), MyoD (Hs02330075_g1), Fgf5 (Hs03676587_s1), Gata4 (Hs00171403_m1), and Neurog1 (Hs01029249_s1).
Transcriptomics
Embryoids were washed with PBS and dissociated using StemPro accutase (A1110501, Thermo Fisher Scientific) and TrypLE express enzyme (12605010, Thermo Fisher Scientific). The dissociated cells were resuspended in 0.05% BSA/PBS, and cell viability was assessed by trypan blue (15250061, Thermo Fisher Scientific). 50 ng of total RNA (RNA quality number (RQN) = 10) was used to generate QuantSeq libraries using the 3’ mRNA‐Seq library prep kit FWD for Illumina (Lexogen, GmbH, Vienna, Austria) with 20 cycles of PCR amplification. Libraries were quantified using Quant‐iT Picogreen (Invitrogen, Carlsbad, California, USA) on a Spectramax M2 (Molecular Devices, Sunnyvale, California, USA). The size pattern was controlled using a Fragment Analyzer‐96 with the DNF‐474‐0500 High Sensitivity NGS Fragment Analysis Kit (Agilent Technologies, Santa Clara, California, USA). Libraries (average size: 295 bp) were pooled at an equimolar ratio and clustered at a concentration of 9 pmol on a single read sequencing flow cell. 65 cycles of sequencing were performed on an Illumina HiSeq 2500 in rapid mode using the 50 cycles SBS kit (Illumina Inc., San Diego, California, USA). Sequencing raw data (bcl) were demultiplexed and transformed into fastq files using CASAVA v1.8.2 (Illumina Inc., San Diego, California, USA). The fastq files were then aligned against the reference genomes hs_GRCH38.p2 and GRCm38 using RNAstar v2.5.3a (https://github.com/alexdobin/STAR). To eliminate reads that mapped to both genomes, “human” and “mouse” bam files were processed using Picard Tools (FilterSamReads). The counts per gene from the new bam files containing only reads uniquely mapping to a single genome were generated using HTSeq‐count v2.16.2 (https://htseq.readthedocs.io). For differential expression and pathway enrichment analyses, genes with more than 3 counts per million in at least 6 samples were selected; RNA sequencing data were normalized by the trimmed mean of M‐values (TMM) method as implemented in function calcNormFactors in edgeR (Robinson et al, 2010). Differentially expressed genes were defined by fitting a quasi‐likelihood negative binomial generalized log‐linear model to count data using glmQLFTest function in edgeR. The over‐representation of gene ontology (GO) terms and KEGG pathways for the differentially expressed genes (< 5% FDR) was tested using the topGO and topKEGG functions as implemented in LIMMA (Ritchie et al, 2015). All genome‐wide statistical analyses were performed using R version 3.6.3 (R CoreTeam, https://cran.r‐project.org) and Bioconductor version 3.10 (https://bioconductor.org). For network analysis, extracellular ligands were selected for high expression in iTCEs and, after mapping to the mouse genome and to eECs or GAeFib, were used to define the connected component of the protein‐protein interaction network (http://string‐db.org/). Each node represents a protein and each edge represents a high confidence protein‐protein interaction (interaction score > 0.7). A maximum number of 50 interactions in the first shell was shown. Nodes were colored according to pathways with an inflation parameter of 3. Single‐cell transcriptome profiling was performed using the Chromium Single Cell 3’ kit from 10X Genomics (PN‐120237; 10X Genomics; Leiden, Netherlands). In brief, 8,000 single cells were loaded into each channel of a Chromium Single Cell B Chip (PN‐1000009; 10X Genomics; Leiden, the Netherlands). Reverse transcription and library preparation were performed following the manufacturer’s recommendations (10X Genomics; Leiden, the Netherlands). Libraries were sequenced paired‐end (26 cycles for Read 1; and 101 cycles for Read 2) on a HiSeq 2500 sequencer (Illumina, San Diego, USA) using the High Output SBS kit V4 (FC‐401‐4003; Illumina, San Diego, USA). Raw FASTQ files were processed using Cell Ranger 2.0.2 and the pre‐built human reference (GRCh38) provided by 10x Genomics. The Seurat R package 3.1.1 was used for data analysis (Stuart et al, 2019). Outlier cells with extreme (top and bottom 1%) numbers of genes detected were excluded, and low‐quality cells with more than 10% of their unique molecular identifiers (UMIs) associated with mitochondrial genes were filtered out. Raw UMI counts were divided by total UMI counts per cell, multiplied by 10,000, and natural‐log transformed to obtain normalized data. Potential doublets were identified per sample using the scDblFinder 1.1.8 package from Bioconductor and excluded from downstream analyses. Furthermore, cells from day 0 and day 7 iTCEs were down‐sampled as previously described (Hie et al, 2019), to match N = 2,255 cells retained from day 13 iTCEs following quality control. Next, highly variable genes were identified per sample using the FindVariableFeatures functionality from Seurat, with the selection method set to mean.var.plot (mvp). Given that batch and timepoint (Day 0, 7, and 13) were fully confounded, no batch correction was applied. Instead, we used the concatenated data based on the union of highly variable genes in downstream analyses. The dimensionality of data was reduced by keeping the top 20 principal components (PC) of highly variable genes, which were subsequently used as input for 2D visualization by uniform manifold approximation and projection (UMAP) using the default settings in Seurat. Using the top 20 PCs, a shared nearest neighbor (SNN) graph was constructed between pairs of cells by calculating the neighborhood overlap as quantified by Jaccard similarity, with the k parameter indicating the number of neighbors set to the square root of the total number of cells. The SNN graph was subsequently partitioned into sub‐clusters using the Louvain algorithm with the resolution parameter set to 0.8. Enrichment was computed of selected gene sets per cell using the AddModuleScore functionality of Seurat. Default functionalities of Seurat were used for plotting, and occasionally adapted the output using ggplot2 v3.3.5 (https://ggplot2.tidyverse.org). The ENHANCE denoising algorithm (https://github.com/yanailab/enhance) was used to remove the high levels of technical noise in the data and was applied for plotting the expression of individual genes or enrichment of selected gene modules. Developmental scores of the day 13 iTCE cell population were computed using data provided by Xi et al (2020). Postnatal genes were defined as upregulated in stage 5 satellite cells (SCs) compared to stage 1 and 2 embryonic skeletal muscle progenitor cells (SMPCs), and embryonic genes as genes upregulated in stage 1 and 2 embryonic SMPCs compared to stage 5 SCs. Normalized expression data were converted to z‐scores having mean zero and unit variance, and the developmental score per cell as the sum of weighted z‐scores for genes with log‐fold‐change> 0 (upregulated in stage 5 SCs vs. stage 1 and 2 SMPCs), minus the sum of weighted z‐scores for genes with log‐fold‐change < 0 (upregulated in stage 1 and 2 SMPCs vs. stage 5 SCs) was computed. Each gene was weighted by abs (AUC‐0.5)*2 as provided by Xi et al (2020), depicting its predictive power in classifying the two groups (i.e., stage 1 and 2 embryonic SMPCs compared to stage 5 SCs). Intuitively, a higher value of developmental score indicates a cell with a developmental “age” closer to postnatal SCs relative to embryonic SMPCs. To compare the day 13 iTCE cell population to skeletal muscle progenitor and stem cells at distinct stages of human development, we used the data reported by Xi et al (2020) from the Gene Expression Omnibus under accession number GSE147457 and used the metadata provided by the authors to reproduce reference transcriptome data at various stages of human development as described in their original publication. We next used the scmap 1.12.0 package from Bioconductor, which allows projecting cells from one dataset (Day 13 iTCE cell population from our study) onto cell types from the dataset by Xi et al (2020). In brief, each developmental stage was represented by its centroid, i.e., the median value of gene expression for each gene across all cells in that stage. The similarities between each cell from the day 13 iTCE cell population in our study and all centroids in the Xi et al (2020) data were calculated using Pearson, Spearman, and Cosine correlation. Each cell was then assigned to the developmental stage corresponding to the highest similarity value. By default, scmap requires that at least two similarity measures agree with each other, otherwise cells were marked as unassigned. Additionally, if the maximum similarity value across all three similarities was below 0.7, the cells were also marked as unassigned.
Mice
Animal experiments were performed using male C57BL/10ScSn‐Dmdmdx/J (Mdx mice) according to the Swiss regulation on animal experimentation for the care and use of laboratory animals and approved by the ethical committee of the Canton de Vaud under licenses VD2620, VD2764, VD3002, VD3199, and VD3282, and by the animal committee of the Centre hospitalier universitaire Sainte‐Justine of the Université de Montréal under the license 2020‐2668 in accordance with the Canadian Council on Animal Care guidelines. Mice had access to water and food ad libitum. All the mice were studied between 4 and 8 weeks. Tacrolimus/FK‐506 (S5003‐500MG, Selleck Chemicals) diluted in 70% EtOH was delivered continuously through a pre‐equilibrated Alzet osmotic pump (model 1002, Charles River) delivering 2.5 mg/kg/day implanted subcutaneously 5 days before cell transplantation. Two days before transplantation, the TA muscle of one leg was injected through the skin with 50 μl of 20 μM cardiotoxin (L8102, Latoxan). On the day of the transplantation, 25,000 integrin α9/CD56 positive flow cytometrically purified eMPs and 25,000 hskMPs were prepared under sterile conditions and were injected into the injured TA muscle in 50 μl of 0.9% NaCl. Bioluminescence images were detected using the Xenogen in vivo Imaging System 200 (Caliper Life Sciences, Inc.) according to the manufacturer's recommendation. Briefly, 10 min after intraperitoneal administration of Furimazine (Nano‐Glo Luciferase Assay System, N1110, Promega) in 100 μl PBS, images were acquired and analyzed using the Living Image software (Caliper Life Sciences, Inc.).
Skeletal muscle force measurements
For the assessment of eccentric force, the legs were skinned from the feet to the upper knee and the TA fascia was removed. Then, the mice were placed on the warmed platform of the 1300A: 3‐in‐1 Whole Animal System (Aurora Scientific), and the knee was firmly fixed at a 90‐degree angle using a limb clamp. The TA distal tendon was sectioned, attached to a 3‐0 silk suture, and connected to the force lever. Electrodes were placed in proximity to the deep fibular nerve on the lateral side of the knee. Several 10 Hz stimulations were performed to determine the optimal length of the muscle. Muscles were stimulated at 50 Hz to determine the initial maximal force. Finally, the eccentric contraction (ECC) protocol was performed by stimulation at 50 Hz for 350 ms while stretching the muscle at 15% of its maximal length. Stimulations were performed 5 times with two minutes of rest between contractions, and maximal force was recorded at each contraction.
Statistics
All mice were randomized according to body weight before interventions. Sample size determination was based on the expected effect size and variability that was previously observed for similar readouts in the investigator’s labs. Imaging readouts but not in vivo treatments were blinded. All other statistical analyses were performed using GraphPad Prism (Version 7, GraphPad Software). A two‐sample unpaired Student’s t‐test was used for two‐group comparisons. For comparison of more than two groups, one‐way or two‐way ANOVA was used, according to the experimental design, and followed by Bonferroni multiple‐comparison testing. All data are expressed as mean ± SEM.
Author contributions
Omid Mashinchian: Conceptualization; Data curation; Formal analysis; Supervision; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Filippo De Franceschi: Data curation; Formal analysis; Investigation; Methodology. Sina Nassiri: Formal analysis; Methodology. Joris Michaud: Formal analysis; Methodology. Eugenia Migliavacca: Data curation; Formal analysis; Methodology. Patrick Aouad: Data curation; Formal analysis; Methodology. Sylviane Metairon: Data curation; Formal analysis; Methodology. Solenn Pruvost: Data curation; Formal analysis; Methodology. Sonia Karaz: Data curation; Methodology. Paul Fabre: Data curation; Methodology. Thomas Molina: Data curation; Methodology. Pascal Stuelsatz: Data curation; Methodology. Nagabhooshan Hegde: Data curation; Investigation; Methodology. Emmeran Le Moal: Resources; Writing—original draft. Gabriele Dammone: Data curation; Methodology. Nicolas A Dumont: Resources; Formal analysis; Supervision; Writing—original draft. Matthias P Lutolf: Supervision; Methodology; Writing—original draft; Writing—review and editing. Jerome N Feige: Conceptualization; Supervision; Validation; Investigation; Methodology; Writing—original draft; Project administration; Writing—review and editing. C Florian Bentzinger: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
OM, CFB, and JNF initiated and managed the project. OM, SN, SK, PS, GD, and NH designed and conducted experiments, and analyzed data. FDF and ELM provided support with flow cytometry. PA and JM provided support with imaging. SN and EM performed bioinformatic analysis. SM and SP provided support with transcriptomics. NAD, PF, and TM performed cell transplantation and muscle force measurements. OM, MPL, CFB, and JNF interpreted the results and wrote the manuscript.
Disclosure and competing interests statement
All authors except S.N., P.A., P.F., T.M., E.L.M., N.A.D., and M.P.L. are or were employees of the Société des Produits Nestlé S.A., Switzerland.
Supporting information
Appendix
Acknowledgments
We are grateful to the Nestle Research community for fruitful discussion and support, in particular the Musculo‐Skeletal Health department, Eric Rolland and Ed Baetge. We thank Alessio Palini and Federico Sizzano for expert advice on flow cytometry, José Sanchez for help with mouse husbandry, Frederic Raymond and Patrick Descombes for technical support with genomics, and the laboratory of April D. Pyle at UCLA for support with cross‐referencing of our data single‐cell sequencing data to human myogenic progenitors. O.M., J.N.F. and C.F.B are supported by the Fondation Suisse de Recherche sur les Maladies Musculaires (FSRMM). T.M., E.L.M, and P.F. are supported by scholarships from the Fonds de Recherche du Québec ‐ Santé (FRQS). N.A.D. is supported by the Canadian Institutes of Health Research (CIHR, PJT‐156408 and MY4‐155369), the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN‐2018‐05979), the FRQS (Dossiers 35015 and 296512), the ThéCell network (supported by the FRQS), and the Canada Foundation for Innovation (37622). C.F.B. is supported by CIHR (PJT‐162442), NSERC (RGPIN‐2017‐05490), FRQS (Dossiers 36789 and 296357), the ThéCell Network (supported by the FRQS), the Canadian Stem Cell Network, and a research chair of the Centre de Recherche Médicale de l’Université de Sherbrooke (CRMUS).
The EMBO Journal (2022) 41: e110655.
Contributor Information
Jerome N Feige, Email: jerome.feige@rd.nestle.com.
C Florian Bentzinger, Email: cf.bentzinger@usherbrooke.ca.
Data availability
Sequencing data discussed in this publication has been deposited in NCBI's Gene Expression Omnibus (GEO) under the accession numbers GSE201424 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201424) and GSE202308 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE202308).
References
- Abujarour R, Bennett M, Valamehr B, Lee TT, Robinson M, Robbins D, Le T, Lai K, Flynn P (2014) Myogenic differentiation of muscular dystrophy‐specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl Med 3: 149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV, Puri PL (2013) Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep 3: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberi T, Bradbury M, Dincer Z, Panagiotakos G, Socci ND, Studer L (2007) Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat Med 13: 642–648 [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, Rudnicki MA (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4: a008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, von Maltzahn J, Rudnicki MA (2013) The emerging biology of muscle stem cells: implications for cell‐based therapies. BioEssays 35: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharathan SP, Manian KV, Aalam SM, Palani D, Deshpande PA, Pratheesh MD, Srivastava A, Velayudhan SR (2017) Systematic evaluation of markers used for the identification of human induced pluripotent stem cells. Biol Open 6: 100–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauw B, Agatea L, Toniolo L, Canato M, Quarta M, Dyar KA, Danieli‐Betto D, Betto R, Schiaffino S, Reggiani C (2010) Eccentric contractions lead to myofibrillar dysfunction in muscular dystrophy. J Appl Physiol 108: 105–111 [DOI] [PubMed] [Google Scholar]
- Bryson‐Richardson RJ, Currie PD (2008) The genetics of vertebrate myogenesis. Nat Rev Genet 9: 632–646 [DOI] [PubMed] [Google Scholar]
- Candia AF, Hu J, Crosby J, Lalley PA, Noden D, Nadeau JH, Wright CV (1992) Mox‐1 and Mox‐2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development 116: 1123–1136 [DOI] [PubMed] [Google Scholar]
- Caron L, Kher D, Lee KL, McKernan R, Dumevska B, Hidalgo A, Li J, Yang H, Main H, Ferri G et al (2016) A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy‐affected skeletal muscles. Stem Cells Transl Med 5: 1145–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chal J, Al Tanoury Z, Hestin M, Gobert B, Aivio S, Hick A, Cherrier T, Nesmith AP, Parker KK, Pourquie O (2016) Generation of human muscle fibers and satellite‐like cells from human pluripotent stem cells in vitro . Nat Protoc 11: 1833–1850 [DOI] [PubMed] [Google Scholar]
- Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A, Bousson F, Zidouni Y, Mursch C, Moncuquet P et al (2015) Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol 33: 962–969 [DOI] [PubMed] [Google Scholar]
- Chal J, Pourquie O (2017) Making muscle: skeletal myogenesis in vivo and in vitro . Development 144: 2104–2122 [DOI] [PubMed] [Google Scholar]
- Chalamalasetty RB, Garriock RJ, Dunty WC Jr, Kennedy MW, Jailwala P, Si H, Yamaguchi TP (2014) Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 141: 4285–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman DL, Agulnik I, Hancock S, Silver LM, Papaioannou VE (1996) Tbx6, a mouse T‐Box gene implicated in paraxial mesoderm formation at gastrulation. Dev Biol 180: 534–542 [DOI] [PubMed] [Google Scholar]
- Choi I, Lim HoTae, Estrellas K, Mula J, Cohen T, Zhang Y, Donnelly C, Richard J‐P, Kim Y, Kim H et al (2016) Concordant but varied phenotypes among Duchenne muscular dystrophy patient‐specific myoblasts derived using a human iPSC‐based model. Cell Rep 15: 2301–2312 [DOI] [PubMed] [Google Scholar]
- Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, Perlingeiro RC (2012) Human ES‐ and iPS‐derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10: 610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du SH, Tay JC, Chen C, Tay FC, Tan WK, Li ZD, Wang S (2015) Human iPS cell‐derived fibroblast‐like cells as feeder layers for iPS cell derivation and expansion. J Biosci Bioeng 120: 210–217 [DOI] [PubMed] [Google Scholar]
- Easter SS Jr, Ross LS, Frankfurter A (1993) Initial tract formation in the mouse brain. J Neurosci 13: 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, Pevny L (2004) SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci 26: 148–165 [DOI] [PubMed] [Google Scholar]
- Engels MC, Rajarajan K, Feistritzer R, Sharma A, Nielsen UB, Schalij MJ, de Vries AA, Pijnappels DA, Wu SM (2014) Insulin‐like growth factor promotes cardiac lineage induction in vitro by selective expansion of early mesoderm. Stem Cells 32: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamini V, Ghadiali RS, Antczak P, Rothwell A, Turnbull JE, Pisconti A (2018) The satellite cell niche regulates the balance between myoblast differentiation and self‐renewal via p53. Stem Cell Rep 10: 970–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen K, McKay RD (1988) Proliferation and differentiation of rat neuroepithelial precursor cells in vivo . J Neurosci 8: 1144–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M, Lumsden A, Paquette AJ (1994) Regulation of Pax‐3 expression in the dermomyotome and its role in muscle development. Development 120: 957–971 [DOI] [PubMed] [Google Scholar]
- Hebert JM, Boyle M, Martin GR (1991) mRNA localization studies suggest that murine FGF‐5 plays a role in gastrulation. Development 112: 407–415 [DOI] [PubMed] [Google Scholar]
- Hicks MR, Hiserodt J, Paras K, Fujiwara W, Eskin A, Jan M, Xi H, Young CS, Evseenko D, Nelson SF et al (2018) ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat Cell Biol 20: 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B, Cho H, DeMeo B, Bryson B, Berger B (2019) Geometric sketching compactly summarizes the single‐cell transcriptomic landscape. Cell Syst 8: 483–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Lee JH, Suknuntha K, D'Souza SS, Thakur AS, Slukvin II (2019) NOTCH activation at the hematovascular mesoderm stage facilitates efficient generation of T cells with high proliferation potential from human pluripotent stem cells. J Immunol 202: 770–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Fraser ST, Eakin GS, Mangano M, Isern J, Sahr KE, Hadjantonakis AK, Baron MH (2006) Tg(Afp‐GFP) expression marks primitive and definitive endoderm lineages during mouse development. Dev Dyn 235: 2549–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski M, Braham K, Caillaud JM, Carlu C, Tursz T (1983) HNK‐1 antibody detects an antigen expressed on neuroectodermal cells. J Exp Med 158: 1775–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen Z, del Barco BI, de la Pompa JL, Anderson DJ (1998) neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20: 469–482 [DOI] [PubMed] [Google Scholar]
- Magli A, Incitti T, Kiley J, Swanson SA, Darabi R, Rinaldi F, Selvaraj S, Yamamoto A, Tolar J, Yuan C et al (2017) PAX7 targets, CD54, integrin alpha9beta1, and SDC2, allow isolation of human ESC/iPSC‐derived myogenic progenitors. Cell Rep 19: 2867–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita N, Bielinska M, Wilson DB (1997) Wild‐type endoderm abrogates the ventral developmental defects associated with GATA‐4 deficiency in the mouse. Dev Biol 189: 270–274 [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsch C, Challet‐Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K et al (2015) Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol 17: 994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski B, Lee L, Zuo J, Kramer RH (2009) Analysis of human muscle stem cells reveals a differentiation‐resistant progenitor cell population expressing Pax7 capable of self‐renewal. Dev Dyn 238: 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani DF, Dechesne CA, Sacconi S, Delplace S, Belmonte N, Cochet O, Clement N, Wdziekonski B, Villageois AP, Butori C et al (2010) Isolation of a highly myogenic CD34‐negative subset of human skeletal muscle cells free of adipogenic potential. Stem Cells 28: 753–764 [DOI] [PubMed] [Google Scholar]
- Rao L, Tang W, Wei Y, Bao L, Chen J, Chen H, He L, Lu P, Ren J, Wu LU et al (2012) Highly efficient derivation of skeletal myotubes from human embryonic stem cells. Stem Cell Rev 8: 1109–1119 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli C, Zonefrati R, Sharma P, Innocenti M, Cianferotti L, Brandi ML (2020) Characterization of skeletal muscle endocrine control in an in vitro model of Myogenesis. Calcif Tissue Int 107: 18–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert W, Zimmermann K, Cramer M, Starzinski‐Powitz A (1989) Lymphocyte antigen Leu‐19 as a molecular marker of regeneration in human skeletal muscle. Proc Natl Acad Sci USA 86: 307–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton M, Metz J, Liu J, Carpenedo RL, Demers SP, Stanford WL, Skerjanc IS (2014) Derivation and expansion of PAX7‐positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Rep 3: 516–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji E, Sakurai H, Nishino T, Nakahata T, Heike T, Awaya T, Fujii N, Manabe Y, Matsuo M, Sehara‐Fujisawa A (2015) Early pathogenesis of Duchenne muscular dystrophy modelled in patient‐derived human induced pluripotent stem cells. Sci Rep 5: 12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showell C, Binder O, Conlon FL (2004) T‐box genes in early embryogenesis. Dev Dyn 229: 201–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder‐Cook AS, Barnard EA, Darlison MG, Barnard PJ (1989) The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244: 1578–1580 [DOI] [PubMed] [Google Scholar]
- Simunovic M, Brivanlou AH (2017) Embryoids, organoids and gastruloids: new approaches to understanding embryogenesis. Development 144: 976–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuk D, Tremblay JP (2015) Cell therapy in muscular dystrophies: many promises in mice and dogs, few facts in patients. Expert Opin Biol Ther 15: 1307–1319 [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Pinzi V, Bourhis J, Deutsch E (2007) Mechanisms of disease: signaling of the insulin‐like growth factor 1 receptor pathway–therapeutic perspectives in cancer. Nat Clin Pract Oncol 4: 591–602 [DOI] [PubMed] [Google Scholar]
- Uezumi A, Nakatani M, Ikemoto‐Uezumi M, Yamamoto N, Morita M, Yamaguchi A, Yamada H, Kasai T, Masuda S, Narita A et al (2016) Cell‐surface protein profiling identifies distinctive markers of progenitor cells in human skeletal muscle. Stem Cell Rep 7: 263–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Matthias N, Lo J, Ortiz‐Vitali JL, Shieh AW, Wang SH, Darabi R (2018) A myogenic double‐reporter human pluripotent stem cell line allows prospective isolation of skeletal muscle progenitors. Cell Rep 25: 1966–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Langerman J, Sabri S, Chien P, Young CS, Younesi S, Hicks M, Gonzalez K, Fujiwara W, Marzi J et al (2020) A human skeletal muscle atlas identifies the trajectories of stem and progenitor cells across development and from human pluripotent stem cells. Cell Stem Cell 27: 158–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga‐Otto K, Antosiewicz‐Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920 [DOI] [PubMed] [Google Scholar]