

Abstract
Background & Aims:
PARP1 is a key mediator of cellular stress responses and critical in multiple physiological and pathophysiological processes of cells. However, whether it is involved in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) remains elusive.
Methods:
We analysed PARP1 activity in the liver of mice on a high fat diet (HFD), and samples from NAFLD patients. Gain- or loss-of-function approaches were used to investigate the roles and mechanisms of hepatic PARP1 in the pathogenesis of NAFLD.
Results:
PARP1 is activated in fatty liver of HFD-fed mice. Pharmacological or genetic manipulations of PARP1 are sufficient to alter the HFD-induced hepatic steatosis and inflammation. Mechanistically we identified peroxisome proliferator-activated receptor α (PPARα) as a substrate of PARP1-mediated poly (ADP-ribosyl)ation. This poly(ADP-ribosyl)ation of PPARα inhibits its recruitment to target gene promoters and its interaction with SIRT1, a key regulator of PPARα signaling, resulting in suppression of fatty acid oxidation upregulation induced by fatty acids. Moreover, we show that PARP1 is a transcriptional repressor of PPARα gene in human hepatocytes, and its activation suppresses the ligand (fenofibrate)-induced PPARα transactivation and target gene expression. Importantly we demonstrate that liver biopsies of NAFLD patients display robust increases in PARP activity and PPARα poly(ADP-ribosyl)ation levels.
Conclusions:
Our data indicate that PARP1 is activated in fatty liver, which prevents maximal activation of fatty acid oxidation by suppressing PPARα signaling. Pharmacological inhibition of PARP1 may alleviate PPARα suppression and therefore have therapeutic potential for NAFLD.
Lay summary:
PARP1 is activated in the non-alcoholic fatty liver of mice and patients. Inhibition of PARP1 activation alleviates lipid accumulation and inflammation in fatty liver of mice.
Keywords: Non-alcoholic fatty liver disease, Poly(ADP-ribose) polymerase 1, PPARα, Transcriptional regulation, High fat diet
Introduction
Non-alcoholic fatty liver disease (NAFLD) covers a broad spectrum of liver abnormalities ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), fibrosis and cirrhosis [1]. The prevalence of NAFLD is increasing globally, and becoming the predominant cause of chronic liver disease in many parts of the world [2]. The hallmark of NAFLD is an over-accumulation of triglycerides-rich lipid droplets in the hepatocytes of liver due to deregulation of hepatic lipid metabolism, including increased fatty acid (FA) uptake and synthesis as well as insufficient disposal (i.e., oxidation and secretion). In addition to triglycerides, lipotoxic lipids often accumulate in the fatty liver, leading to serious liver injuries through activation of oxidative stress, cell death and inflammatory responses [3], which may in turn worsen lipid metabolism deregulation, and create a vicious cycle to amplify liver damage and drive NAFLD progression [4–9]. Therefore, maintenance of hepatic lipid homeostasis, for example upregulating fatty acid oxidation (FAO) when there is increased FA availability, is critical for breaking this vicious cycle and alleviation of liver damage. Fibrates, the synthetic agonists of peroxisome proliferator-activated receptor (PPAR)α, are used in some clinical trials for NAFLD therapy as they can increase FAO in hepatocytes [10–13]. However, due to unrecognized regulatory mechanisms of PPARα in human fatty liver, their effects on steatosis, inflammation and fibrosis of NAFLD patients are limited [11,12,14,15].
Poly(ADP-ribose) polymerase (PARP) 1, the most abundant member of the PARP family, is a eukaryotic nuclear enzyme accounting for about 90% of cellular PARP activity. Upon activation, it catalyzes polymerization of ADP-ribose units from donor NAD+ molecules on target proteins to form linear or branched polymers. PARP1 is a cellular stress sensor that can be activated by multiple stresses, such as oxidative, metabolic and genotoxic stresses, and in response, directs cells to specific fates according to the type and strength of stress stimuli [16]. Our previous work has shown that PARP1 mediates oxidative stress-induced liver damage and cell death [17]. Recently, Mukhopadhyay et al. also showed that PARP1 is a key mediator of carbon tetrachloride (CCl4)-and bile duct ligation-induced hepatic inflammation and fibrosis [18]. These findings suggest that activation of PARP1 is detrimental to the liver. PARP1 is a major cellular NAD+ consumer, its inhibition increases cellular NAD+/sirtuin activity, and thus promotes mitochondrial biosynthesis and oxidation in skeleton muscle and brown adipose tissues [19]. Moreover, PARP1 deficiency seems to alter metabolic responses to high fat diet (HFD) of mice, despite some contradictory results [20,21]. Nevertheless, whether PARP1 is involved in the pathogenesis of lipid metabolic disorder and NAFLD remains unclear. In this report, we provided the first evidence supporting that liver PARP1 is activated by chronic HFD feeding, which in turn, promotes lipid accumulation and inflammation in the liver by inhibiting HFD-induced FAO upregulation through poly(ADP-ribosyl)ation of PPARα. Additionally we found that NAFLD patients relative to normal subjects display a robust increase in PARP activity and poly(ADP-ribosyl)ation of PPARα. In human hepatocytes and HepaRG cells, PARP1 not only suppresses PPARα expression, but also inhibits the fenofibrate-induced PPARα transactivation and expression of FAO-related genes.
Materials and methods
All human studies and animal procedures conformed to the National Institutes of Health (NIH) guidelines, and were approved by the Ethics Committee of Union Hospital, Huazhong University of Science and Technology, China, and conducted in accordance with the Declaration of Helsinki.
Animal studies
Male 8–10 week old C57BL/6 mice were purchased from Beijing University (Beijing, China). Male 8–10 week old PARP1 knockout (PARP1−/−, 129S-Parp1tm1Zqw/J, PKO) mice and their wild-type (WT) 129Sv littermates were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). PPARα knockout (PPARα−/−, C57BL/6 background) mice were kindly provided by Dr. Hongliang Li. All mice were kept in a temperature controlled room on a 12 h light/dark cycle, 60% humidity, with access to food and water ad libitum. Mice were acclimatized to laboratory conditions for 1 week before the study.
C57BL/6 mice were randomly divided into a chow- or a HFD-fed group. In the chow group, mice were fed with the standard chow diet (D12450B, Research Diets, New Brunswick, NJ). In the HFD group, mice were fed a diet containing 60% calories as fat (D12492, Research Diets, New Brunswick, NJ) for 24 h, 4 weeks, 8 weeks and 12 weeks, respectively. In the PARP1 inhibitor-treated group, mice were fed with HFD for 8–10 weeks. Two weeks after HFD initiation, mice randomly received intraperitoneal injection of N-(6-oxo-5,6-dihydrophenanthoursi din-2-yl)-2-(N,N-dimethylamino) acetamide (PJ34, 10 mg/kg/day, ALX-270–289, Alexis Biochemicals, Lausanne, Switzerland), or vehicle (normal saline) once a day, and fed with HFD for another 6–8 weeks.
PARP1−/− mice and their WT 129Sv littermates were fed with chow or HFD for 8 weeks. PPARα−/− mice and their WT C57BL/6 littermates were fed with chow or HFD for 4 weeks. 2 weeks after HFD initiation, mice on HFD were treated with PJ34 or saline for another 2 weeks.
C57BL/6 mice were randomly divided into a chow- or methionine-choline deficient diet (MCD)-fed group for 6 weeks. Two weeks after MCD initiation, mice randomly received intraperitoneal injection of PJ34 (10 mg/kg/day) or vehicle (normal saline) once a day.
After the experiments, all mice were anesthetized with pentobarbital (50 mg/kg, Sigma, St. Louis, MO). Livers were excised and weighed immediately. The liver and serum samples were used for the following experiments.
Human liver tissue preparation
Paraffin-embedded human liver tissues of patients with NAFLD (n = 12) and healthy subjects (n = 12) were collected from liver biopsy samples. All liver biopsies were read by a single hepatopathologist who was blinded to clinical data. NAFLD activity (NAS) was scored using the NASH Clinical Research Network Histologic Scoring System. Immunohistochemistry for poly(ADP-ribose) polymer (PAR) were performed. The PAR-positive area was quantified with the use of NIH imaging software.
A total of 72 histologically normal liver tissue specimens (all were surgically resected) were obtained from the tissue bank, Clinic Center of Human Genomic Research, HUST. People with a history of alcoholic liver disease, chronic viral hepatitis, autoimmune hepatitis, drug-induced liver disease, primary biliary cirrhosis, biliary obstruction, hemochromatosis, Wilson’s disease, and α-1 antitrypsin deficiency-associated liver disease were excluded. The main clinical and laboratory data of 72 human subjects were summarized in Supplementary Table 1. Samples were stored at −80 °C until use.
Histological analyzes
Liver tissues were either fixed in 10% neutral buffered formalin (HT5011, Sigma, St. Louis, MO) or embedded in OCT compound (Tissue-Tek® OCT™ Compound, Sakura Finetek USA) and then frozen in dry ice. Liver tissues were cut with a microtome (Leica, Heidelberg, Germany) into sequential slices of 5 μm and subjected to hematoxylin and eosin (H&E) staining. For Oil Red O staining, frozen sections (10 μm) were stained with Oil Red O (O0625, Sigma, St. Louis, MO) and counterstained with Mayer’s hematoxylin to visualize intracellular lipid droplets. All digital images were obtained with a light microscope (Olympus, Tokyo, Japan).
Glucose tolerance tests and insulin tolerance tests
Glucose tolerance tests were performed 4 weeks after HFD feeding by glucose gavage (1 g of glucose/kg body weight) after an overnight fast. For insulin tolerance tests, mice were anesthetized with pentobarbital 5 h after food removal and insulin (0.75 unit insulin/kg body weight) was injected intraperitoneally. Blood samples were taken from the tail vein at the indicated time points. Blood glucose was determined using the One-Touch Accu-Chek Glucometer (Roche).
Cell lines and culture
Primary hepatocytes were isolated from mice, purified and cultured as previously described [1]. Briefly, mice were anesthetized and perfused with freshly prepared perfusion medium (Invitrogen, Carlsbad, CA, USA) via the portal vein and then followed by digestion buffer (collagenase IV, 0.1 mg/ml, Worthington, NJ, USA). The perfused livers were removed from the mice and dispersed with hepatocyte wash medium (Invitrogen, Carlsbad, CA, USA). The viable cells were counted by trypan blue exclusion assay and then seeded onto fibronectin-coated plates (Corning, Rochester, NY).
Human hepatocytes (#5200) were purchased from ScienCell Research Laboratories (San Diego, CA) and cultured according to the manufacturer’s instructions. Human undifferentiated HepaRG cells (HPRGC10) were purchased from Gibco (Thermo Fisher Scientific) and cultured as previously described [2]. Human hepatoma cell line HepG2 cells were purchased from the American Type Culture Collection (USA). Cells were cultured at 37 °C with 5% CO2 in humidified atmosphere. After 24 h serum starvation, cells were exposed to oleic acid (O1008, Sigma, St. Louis, MO), linoleic acid (L1376, Sigma, St. Louis, MO), palmitic acid (P5585, Sigma, St. Louis, MO), stearic acid (85679, Sigma, St. Louis, MO), fenofibrate (F6020, Sigma, St. Louis, MO), 3AB (A0788, Sigma, St. Louis, MO), PJ34 (ALX-270-289, Alexis Biochemicals, San Diego, CA, USA.), hydrogen peroxide (Sigma, St. Louis, MO), or the relevant vehicle. For Oil Red O staining, HepG2 cells were cultured in a 24-well plate and incubated in the oleic acid-containing medium (600 μmol/L) for 48 h. This medium was used because it induces lipid droplet formation in cultured cells. All the cells were proved to be mycoplasma-free prior to the experiments, as determined by repetitive fluorescent staining for cytoplasmic DNA.
LC-MS/MS analysis of PARP1-associating proteins
For LC-MS/MS analysis of PARP1-associating proteins, HepG2 cells were transfected with the FLAG-PARP1-encoding vector or an empty vector for 48 h. Nuclear fractions were extracted, mixed with EZview red anti-FLAG M2 affinity gels (Sigma-Aldrich, USA), and rotated overnight at 4 °C. The beads were washed with Tris-buffered saline, and immunoprecipitated proteins were eluted by incubation in Tris-buffered saline containing 1 mg/ml 3× FLAG peptide (Sigma-Aldrich, USA) for 30 min on ice. The eluted proteins were subjected to LC-MS/MS analysis. Data were analyzed with ProteinPilot software (AB SCIEX, Framingham, USA).
Chromatin immunoprecipitation (ChIP) and re-ChIP assay
ChIP assays were performed as previously described [18]. Hepatocytes were fixed with 1% formaldehyde for 15 min and stopped by adding glycine to a final concentration of 125 mM. Then cells were washed, harvested, lysed, and sonicated to shear chromatin to DNA fragments of 0.5–1 kb. Lysates were centrifuged, and an aliquot (20 μl) of supernatant was saved as input DNA. Supernatants were then immunoprecipitated with indicated antibodies or an IgG as a negative control. Finally, DNA was purified and concentrated using the QIAquick PCR purification kit (Qiagen, CA, USA). Purified DNA was analyzed by conventional and realtime PCR with specific primers (Supplementary Table 6) for the ApoA1 gene promoter. An equal volume of non-precipitated (input) genomic DNA was amplified as positive control. In re-ChIP assays, chromatin was firstly immunoprecipitated with an anti-PPARα antibody, then eluted with 100 μl of elution buffer with 10 mM DTT at 37 °C for 30 min, diluted (25-fold) with dilution buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100), and finally reimmunoprecipitated with IgG or an antibody against PARP1, PAR, Sirt1, PGC1α, or RXR (Santa Cruz). Real-time PCR was performed using 1 μg of template DNA with primers specific for Cpt1α, Cyp4a11, or Acox1 (Supplementary Table 6). The input chromosomal DNA and ChIP DNA with non-specific IgG were subjected to the same PCR amplification. PCR products were separated on an ethidium bromide-stained 2% agarose gel.
Statistical analysis
Values are shown as mean ± SEM of at least three independent experiments. The significance of differences was estimated by one-way ANOVA, followed by Student-Newmann-Keuls multiple comparison tests. p <0.05 was considered significant. All statistical analyzes were performed with SPSS software (version 11.0, SPSS Inc). Additional experimental procedures are provided in the Supplementary materials and methods.
Results
PARP1 is activated in the liver of mice fed a high fat diet
Given that in skeletal muscle PARP1 activity is regulated by nutritional fluctuations [19], we first determined if similar regulation occurs in liver. Overall, liver vs. skeletal muscle expressed lower levels of PARP1 mRNA (Fig. 1A) [19]. Western blotting using a PAR antibody showed that hepatic PARP1 auto-poly(ADP-ribosyl)ation levels (indicative of global PARP1 activity) did not differ among the three groups of mice (fasted for 24 h, fed ad libitum, and re-fed for 24 h after fasting) (Fig. 1B). On the other hand and consistent with a previous report [19], the gastrocnemius from the fasted mice manifested a significant decrease in PARP1 auto-poly(ADP-ribosyl)ation levels compared to that from the free fed controls or the re-fed mice (Fig. 1B). These results indicate that liver and muscle PARP1 activities are differentially regulated by nutritional states.
Fig. 1. PARP1 mediates high fat diet (HFD)-induced hepatic steatosis in mice.
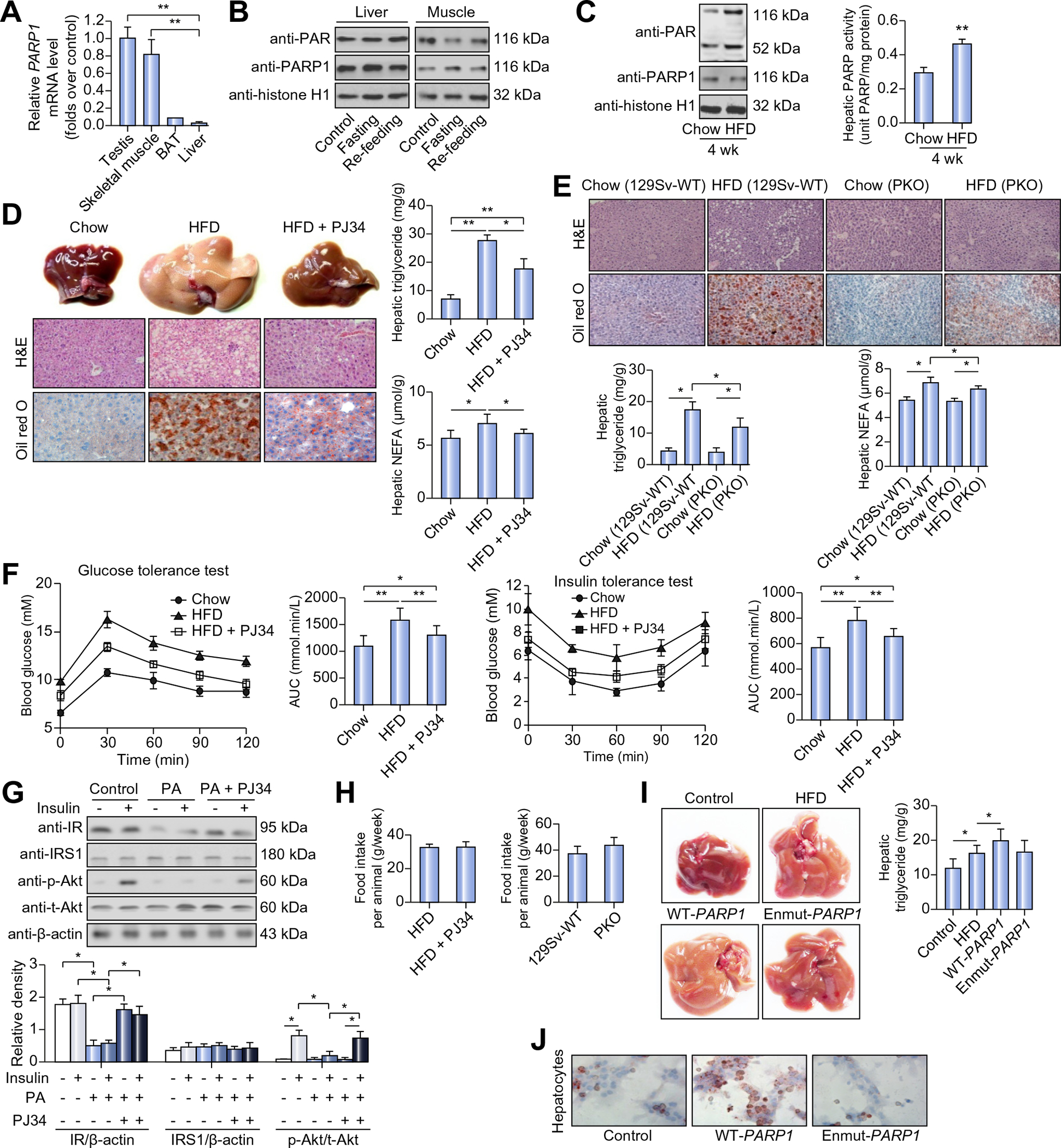
(A) PARP1 mRNA expression in C57BL/6 mice. **p <0.05. (B) Poly(ADP-ribosyl)ation levels of PARP1 analyzed by Western blotting, in nuclear extracts from livers and gastrocnemius muscles of mice fed ad libitum, fasted (24 h) or re-fed (24 h) after 24 h fasting. (C) Poly(ADP-ribosyl)ation levels (left) and PARP activity (right) in nuclear extracts from livers of mice fed with chow diet or HFD for 4 weeks. **p <0.01 represents HFD group vs. chow group. (D) C57BL/6 mice were fed with chow diet or HFD for 12 weeks, or fed with HFD for 12 weeks in combination with 10 week-PJ34 injection (10 mg/kg/day, n = 8 each). Representative photographs of liver samples were shown. Hematoxylin and eosin (H&E) and Oil Red O staining of liver sections were analyzed (original magnification: 40×). Triglyceride and NEFA concentrations in livers were analyzed. *p <0.05, **p <0.01. (E) 129Sv-WT or PARP1−/− (PKO) mice were fed with chow diet or HFD for 8 weeks. H&E and Oil Red O staining of liver sections were analyzed (original magnification: 40×. n = 8 each). Triglyceride and NEFA concentrations in livers were analyzed. *p <0.05. (F) C57BL/6 mice were fed with chow diet or HFD for 12 weeks, or fed with HFD for 12 weeks in combination with 10 week-PJ34 injection (10 mg/kg/day). A glucose tolerance test (1 g/kg body weight, glucose intraperitoneal injection) and an insulin tolerance test (0.75 units/kg body weight, insulin intraperitoneal injection) were performed (n = 8 each). The corresponding area under the curve (AUC) of blood glucose levels in each group was calculated. *p <0.05, **p <0.01. (G) Western blotting analysis of insulin receptor, IRS1, Akt and phosphorylated Akt expression in hepatocytes treated with vehicle or PJ34 (10 μmol/L, 24 h) in the absence or presence of palmitic acid (PA, 30 μmol/L, 24 h) or/and insulin (100 nM, 30 min). *p <0.05. (H) Food intake was monitored during HFD feeding. (I) C57BL/6 mice were infected with an adenovirus vector containing the wild-type human PARP1 gene (WT-PARP1) or mutant PARP1 gene lacking enzymatic activity (enmut-PARP1), and fed on HFD or chow diet for 7 weeks (n = 7 each). Representative photographs of liver samples were shown. Triglyceride concentrations in livers were analyzed. *p <0.05. (J) Oil Red O staining of hepatocytes transfected with WT-PARP1 or enmut-PARP1 construct (original magnification: 200×). Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
Since chronic overnutrition is a major cause of metabolic syndrome and associated NAFLD, we next examined the effect of HFD (60 kCal% of fat) on hepatic PARP1 activation in C57BL/6 mice. Compared to chow-fed controls, the mice on HFD for 4 weeks displayed significant increases in PARP activity and PARP1 auto-poly (ADP-ribosyl)ation levels (Fig. 1C), indicating that HFD activates PARP1 in the liver. Similar results were obtained from the mice on HFD for 8 and 12 weeks (data not shown). Furthermore, HFD feeding for 8 weeks failed to activate hepatic PARP in PARP1 knockout (PARP1−/−, PKO) mice (Supplementary Fig. 1A), also indicating a critical role of PARP1 in mediating HFD-induced PARP activation. In this study, we measured mRNA expression of all PARP family members and protein expression of all nuclear members in mouse liver. As shown in Supplementary Fig. 1B, HFD feeding did not alter the expression of these members.
PARP1 mediates high fat diet-induced hepatic steatosis in mice
To determine whether PARP1 is involved in the pathogenesis of hepatic lipid metabolic disorder and steatosis induced by HFD, we first treated C57BL/6 mice with a PARP1 inhibitor N-(6-Ox o-5,6-dihydrophenan-thridin-2-yl)-N,N-dimethylacetamide (PJ34) for 10 weeks during HFD feeding. Oil Red O staining of liver sections and enzymatic colorimetric assays of extracted lipids showed that PJ34-treated mice relative to untreated animals displayed a significant decrease in hepatic lipid accumulation (Fig. 1D). Consistently, PKO mice compared to WT littermates also had a significant decrease in hepatic lipid accumulation after 8 weeks of HFD feeding (Fig. 1E). In the PARP1 pharmacological inhibition study, we also observed that PJ34 treatment decreased serum levels of non-esterified fatty acids (NEFAs), the major substrate source of hepatic steatosis, in HFD-fed mice (Table 1). Serum levels of triglyceride, total cholesterol and glucose were also decreased in the PJ34-treated mice (Table 1). PJ34 treatment attenuated HFD-induced intolerance of glucose and insulin in HFD mice (Fig. 1F and G). Moreover, in primary hepatocytes, palmitic acid-induced suppression of insulin receptor and insulin-stimulated phosphorylation of Akt were restored after PJ34 treatment. In this study, we determined the effects of PJ34 on the food intake of mice on HFD, and did not find any difference between PJ34-treated mice and saline controls (Fig. 1H). However, PKO mice on HFD displayed a marginally increased food intake (p = 0.059, n = 6) as compared to their WT counterparts (Fig. 1H). Inhibition or deletion of PARP1 did not affect the ratio of liver/body weight in mice on HFD (Supplementary Fig. 1C).
Table 1.
Serum lipids (mmol) in mice (mean ± SEM).
| Parameters | Groups |
||
|---|---|---|---|
| Chow | HFD | HFD + PJ34 | |
|
| |||
| Glucose | 5.12 ± 0.45 | 7.37 ± 0.59 | 5.91 ± 0.98* |
| TG | 1.02 ± 0.11 | 1.36 ± 0.14 | 1.16 ± 0.12* |
| TC | 2.22 ± 0.18 | 5.58 ± 0.76 | 4.21 ± 0.82* |
| LDL | 1.90 ± 0.13 | 2.90 ± 0.20 | 2.53 ± 0.28* |
| HDL | 0.72 ± 0.06 | 1.37 ± 0.24 | 1.40 ± 0.17 |
| NEFA | 0.83 ± 0.06 | 1.31 ± 0.08 | 1.12 ± 0.04* |
TG, triglycerides; TC, total cholesterol; LDL, low-density lipoprotein; HDL, high-density lipoprotein; NEFA, non-esterified fatty acids.
p <0.05 represents HFD + PJ34 group vs. HFD group.
To further explore the effects of PARP1 on HFD-induced hepatic steatosis, we used adenoviruses to overexpress human wild-type PARP1 gene (WT-PARP1) and mutant PARP1 that lacks enzymatic activity (enmut-PARP1) in the liver of C57BL/6 mice fed a HFD for 7 weeks. As expected, hepatic overexpression of WT-PARP1 but not enmut-PARP1 significantly increased hepatic triglyceride content in the HFD-fed mice (Fig. 1I). Similar results were obtained when WT-PARP1 or enmut-PARP1 was overexpressed in isolated mouse primary hepatocytes that were analyzed after a 3 h treatment with palmitic acid (PA, 16:0) to mimic overnutrition (Fig. 1J). Collectively these results indicate that hepatocyte overexpression of PARP1 exacerbates overnutrition-associated hepatic steatosis.
In addition to HFD-induced hepatic steatosis model, we also investigated the effect of PARP1 inhibition on MCD-induced NASH in C57BL/6 mice. Oil Red O staining of liver sections and enzymatic colorimetric assays of extracted lipids showed that PJ34-treated MCD mice displayed a significant decrease in hepatic content of triglycerides and NEFA as compared to saline-MCD mice (Supplementary Fig. 1D).
PARP1 inhibits hepatic FAO pathway upregulation in mice challenged with high fat diet
Hepatic steatosis develops when hepatic FAO and/or lipid export are insufficient to counteract the FA uptake and de novo lipogenesis-induced increase. PARP1 is involved in the transcriptional regulation of multiple genes [16]. To determine if it is implicated in the regulation of these lipid metabolic pathways, we measured hepatic mRNA and protein levels of relevant genes in HFD-fed mice treated with or without PARP1 inhibitor PJ34. As shown in Fig. 2A and Supplementary Fig. 1E, PJ34-treated HFD-fed mice displayed significant increases in hepatic mRNA and protein levels of genes involved in FAO (Acox1, Cyp4a10, CPT1a, MCAD and LCAD) and FA transport (CD36 and FATP2) as compared to saline-HFD controls. Interestingly, HFD feeding alone also slightly increased expression of several genes involved in FAO and fatty acid transport (Fig. 2A, Supplementary Fig. 1E). Consistent with the augmented expression of hepatic FAO genes, PJ34-treated HFD mice had increased fasting serum levels of ketone bodies, acetoacetate and β-hydroxybutyrate, which are markers of hepatic FAO (Fig. 2B). Similarly, PKO vs. littermate control mice on HFD also displayed increases in hepatic expression levels of FAO-related genes and fasting serum levels of ketone bodies (Fig. 2C and D; Supplementary Fig. 1F). These pharmacological and genetic data suggest that PARP1 inhibition increases hepatic FAO in mice on HFD. In line with these results, treatment with PJ34 increased the expression of Acox1, Cyp4a10, CPT1a and FATP2 genes in MCD fed mice, as compared to saline-MCD controls. However, the expression of FA synthesis genes were not changed (Supplementary Fig. 1G).
Fig. 2. PARP1 inhibition increases hepatic FAO in mice on HFD.
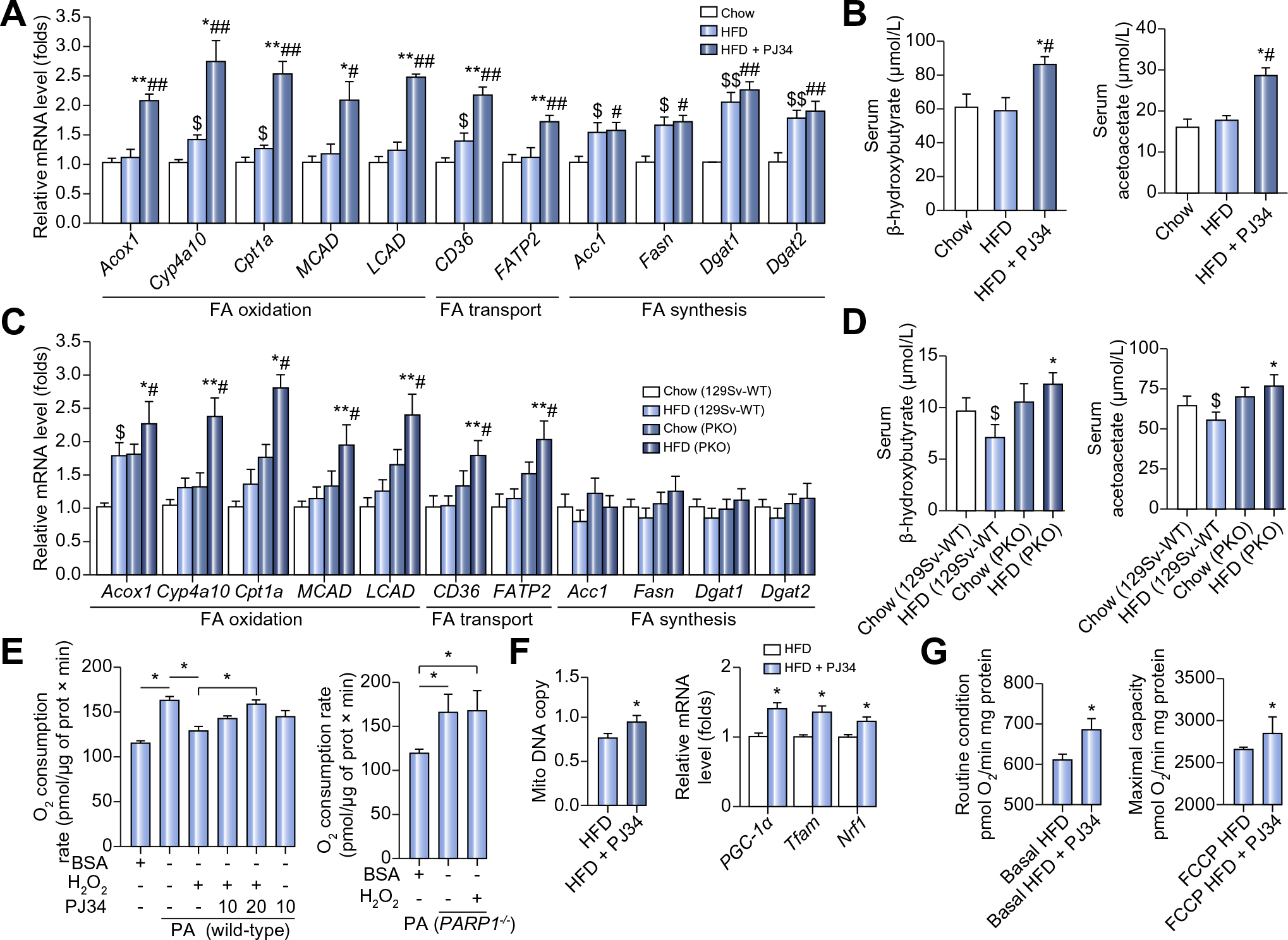
(A and B) Hepatic mRNA levels of genes (A), serum levels of β-hydroxybutyrate and acetoacetate (B) in C57BL/6 mice fed with chow diet or HFD for 8 weeks, or fed with HFD for 8 weeks in combination with 6 week-PJ34 injection (10 mg/kg/day, n = 8 each). *p <0.05, **p <0.01 represent HFD group vs. HFD + PJ34 group, #p <0.05, ##p <0.01 represent chow group vs. HFD + PJ34 group, $p <0.05, $$p <0.01 represent chow group vs. HFD group. (C and D) Hepatic mRNA levels of genes (C), serum levels of β-hydroxybutyrate and acetoacetate (D) in 129Sv-WT or PARP1−/− (PKO) mice fed with chow diet or HFD for 8 weeks (n = 8 each). *p <0.05, **p <0.01 represent HFD (PKO) vs. HFD (WT) group. #p <0.05 represent chow (PKO) group vs. HFD (PKO) group, $p <0.05 represent chow (129Sv-WT) group vs. HFD (129Sv-WT) group. (E) Cellular oxygen consumption analyzed in wild-type or PARP1−/− hepatocytes treated with H2O2 in the absence or presence of PJ34, (n = 4). *p <0.05. (F) Relative mitochondrial DNA content, and mRNA levels of the indicated genes analyzed in livers from HFD-fed C57BL/6 mice with or without PJ34 treatment (n = 8 each). *p <0.05 represents HFD + PJ34 group vs. HFD group. (G) Oxygen flux in mitochondrial isolated from livers of mice fed with HFD for 8 weeks, or fed with HFD for 8 weeks in combination with 6 week-PJ34 injection (10 mg/kg/day, n = 8 each). Oxygen consumption was shown for routine respiration and maximally uncoupled state respiration after addition of FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone. *p <0.05 represents HFD + PJ34 group vs. HFD group. Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
We did not observe any effects of PJ34 or PARP1 deletion on hepatic expression levels of genes related to de novo lipogenesis (ACC, FAS, DGAT1, DGAT2) (Fig. 2A and C; Supplementary Fig. 1E and F). Treatment with PJ34 or PARP1 deletion did not alter hepatic content of malonyl-CoA, an intermediate of FA synthesis, in HFD mice either (Supplementary Fig. 1H). These findings together indicate that it is unlikely that PARP1 promotes hepatic steatosis by increasing hepatic de novo lipogenesis. In addition, PARP1 does not seem to induce hepatic steatosis via suppressing hepatic VLDL-mediated lipid export because PARP1 inhibition by PJ34 decreased instead of increased hepatic VLDL-TG production during the triton block experiment (1.878 ± 0.064 mM in PJ34 group vs. 2.064 ± 0.043 mM in saline group, p = 0.013).
To provide direct evidence in support of an inhibitory role for PARP1 on FAO, oxygen consumption rates were measured in isolated primary hepatocytes that were treated with H2O2, a well known PARP1 activator [19], alone or together with 10 μM or 20 μM PJ34 for 6 h prior to the addition of 200 μM palmitate to stimulate FAO. As expected, in primary hepatocytes from WT mice, H2O2 treatment suppressed palmitate-stimulated oxygen consumption, and this inhibitory effect was reversed by PJ34 treatment (Fig. 2E). In PARP1-deficient hepatocytes, H2O2 failed to suppress palmitate-induced oxygen consumption (Fig. 2E). These results directly demonstrate that activation of PARP1 suppresses hepatocyte FAO upregulation induced by fatty acids.
In line with this in vitro finding, in vivo studies showed that PJ34 treatment significantly increased hepatic mitochondrial DNA content and expression levels of PGC-1α, Tfam and Nrf1 in HFD-fed mice (Fig. 2F). More importantly the mitochondrial oxygen consumption was also significantly increased after PJ34 treatment (Fig. 2G).
PARP1 interacts with PPARα to suppress hepatic FAO upregulation and promotes hepatic steatosis in mice on high fat diet
To investigate the factors mediating PARP1 effects on hepatic FAO, we conducted an unbiased "interactome" screen to identify PARP1-interacting proteins. Human hepatoma HepG2 cells were transfected with a FLAG-PARP1-expressing plasmid or an empty vector for 48 h. Nuclear extracts were immunoprecipitated with an anti-FLAG antibody, and the eluted proteins were subjected to LC-MS/MS analysis. To identify and eliminate false positives, nuclear proteins from cells transfected with the empty vector were served as negative controls. In addition to confirming several of the previously identified targets [17,22], we found that PPARα was a candidate of PARP1-interacting proteins (Fig. 3A). To further validate this finding, nuclear extracts from primary hepatocytes were immunoprecipitated with an anti-PPARα or anti-PARP1 antibody. We observed that endogenous PARP1 was specifically co-immunoprecipitated with the anti-PPARα antibody, or vice visa (Fig. 3B). In contrast to PPARα, two lipogenesis transcriptional factors SREBP1c and ChREBP were not precipitated with the anti-PARP1 antibody (Fig. 3B). Additionally, by forced expression of His-tagged PPARα fragments or Flag-tagged PARP1 fragments in HepG2 cells, we mapped the PARP1 binding site to the C-terminal ligand-binding domain (LBD) of PPARα and the PPARα-binding site to the central BRCA1 C-terminus/automodification domain of PARP1 using immunoprecipitation assays (Fig. 3C).
Fig. 3. PARP1 interacts with PPARα to suppress hepatic FAO upregulation and promotes hepatic steatosis in mice on HFD.
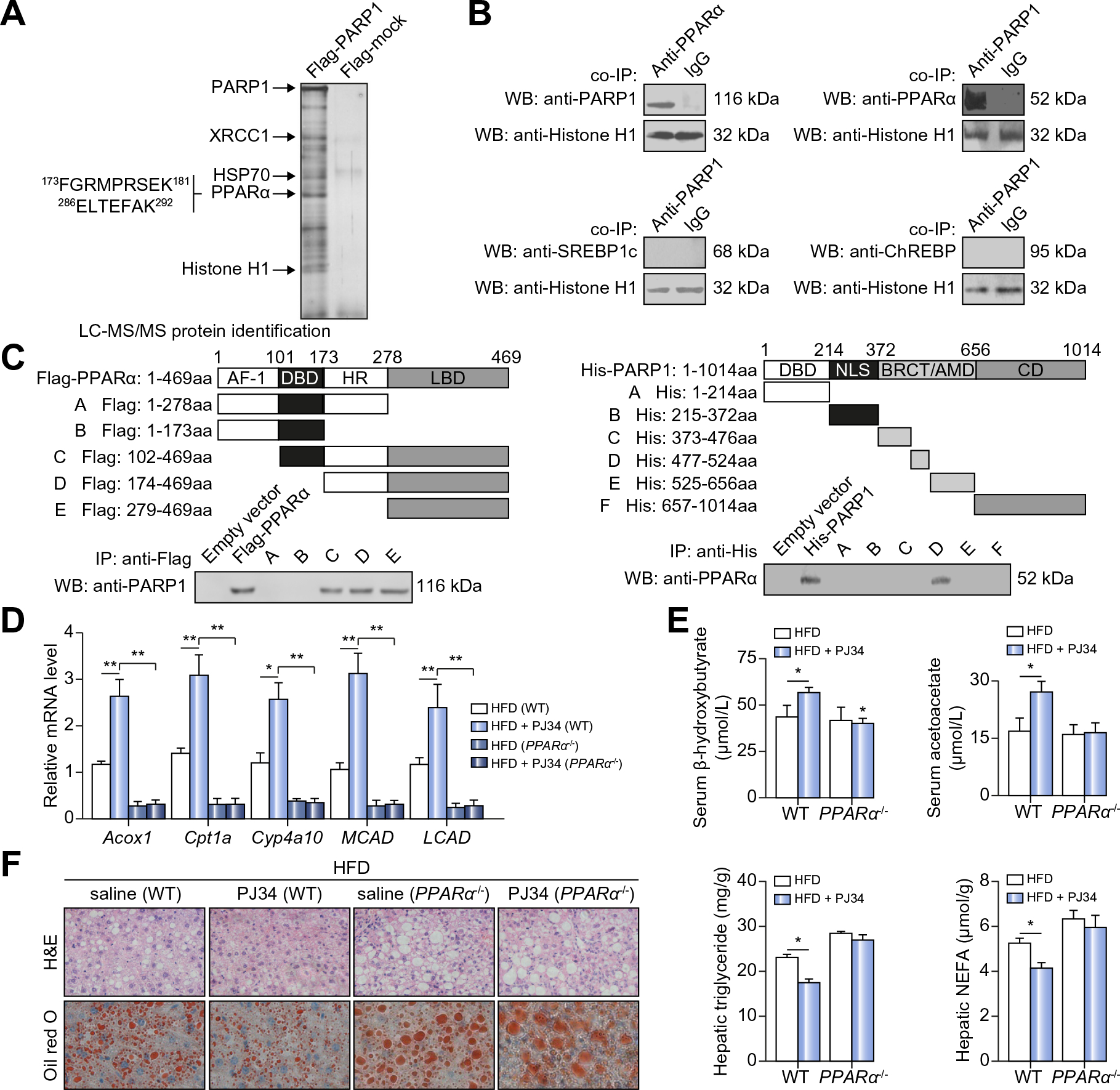
(A) Protein extracts from HepG2 cells transfected with the Flag-PARP1 encoding vector or an empty vector were subjected to immunoprecipitation (IP) using the anti-Flag antibody. The IP-protein complex was then respectively subjected to SDS–PAGE and analyzed by LC-MS/MS analysis. Two identified peptide sequences of PPARα were shown. (B) Co-IP of PPARα-bound proteins in nuclear extracts from hepatocytes, followed by Western blotting with anti-PARP1 antibody. Co-IP of PARP1-bound proteins in nuclear extracts from hepatocytes, followed by Western blotting with anti-PPARα antibody, anti-SREBP1c antibody or anti-ChREBP antibody respectively. Non-specific IgG served as a negative control. (C) Flag-tagged human PPARα deletion mutants were transfected into HepG2 cells respectively. Co-IP assay demonstrated the specific binding of PARP1 to C-terminal LBD of PPARα (left). His-tagged human PARP1 deletion mutants were transfected into HepG2 cells respectively. Co-IP assay demonstrated the specific binding of PPARα to BRCT/AMD domain of PARP1 (right). (D–F) C57BL/6-WT or PPARα−/− mice fed on HFD for 4 weeks were randomly received vehicles or PJ34 (10 mg/kg/day, 2 weeks after HFD feeding, n = 6 each) treatment. (D) Hepatic mRNA levels of genes and (E) serum levels of β-hydroxybutyrate and acetoacetate were analyzed. *p <0.05, **p <0.01. (F) H&E and Oil Red O staining of liver sections were analyzed (original magnification: 40×). Triglyceride and NEFA concentrations in livers were analyzed. *p <0.05 represents HFD + PJ34 group vs. HFD group. Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
Considering that PPARα is the master regulator of hepatic FAO [10], we explored whether PPARα is implicated in mediating the effects of PARP1 on hepatic FAO and lipid accumulation. After treating with PJ34 for 2 weeks, HFD-fed WT C57BL/6 mice displayed significant increases in hepatic expression levels of FAO-related genes and serum levels of β-hydroxybutyrate and acetoacetate (Fig. 3D and E). These effects were not observed in PPARα knockout (PPARα−/−) mice (Fig. 3D and E). Moreover, PJ34 treatment suppressed the HFD-induced hepatic lipid (triglyceride and NEFA) accumulation in WT but not PPARα−/− mice (Fig. 3F). Consistently, knockdown of PPARα by siRNA suppressed the upregulation of FAO genes, Acox1 and Cyp4a10, in PKO hepatocytes (Supplementary Fig. 1I). Thus, PPARα is required for PARP1 to suppress FAO upregulation and to induce lipid accumulation in the liver of mice on HFD.
PARP1-mediated poly(ADP-ribosyl)ation of PPARα suppresses its transactivation
PARP1 enzyme modifies nuclear proteins by catalyzing protein poly(ADP-ribosyl)ation [15]. We speculated that PPARα may be poly(ADP-ribosyl)ated by PARP1 in hepatocytes, resulting in its functional inhibition. To test this possibility, nuclear extracts from WT or PARP1−/− hepatocytes were immunoprecipitated with an anti-PPARα antibody, followed by immunoblotting with an anti-PAR antibody. Indeed, the endogenous PPARα was poly (ADP-ribosyl)ated in WT but not in PARP1−/− hepatocytes (Fig. 4A). Importantly, incubation of recombinant PPARα with PARP1, NAD+ and nicked DNA (PARP1 activator) led to poly (ADP-ribosyl)ation of PPARα in a cell free system (Fig. 4A). These results together demonstrate that PPARα is poly(ADP-ribosyl) ated by PARP1.
Fig. 4. Poly(ADP-ribosyl)ation of PPARα suppresses its transactivation.
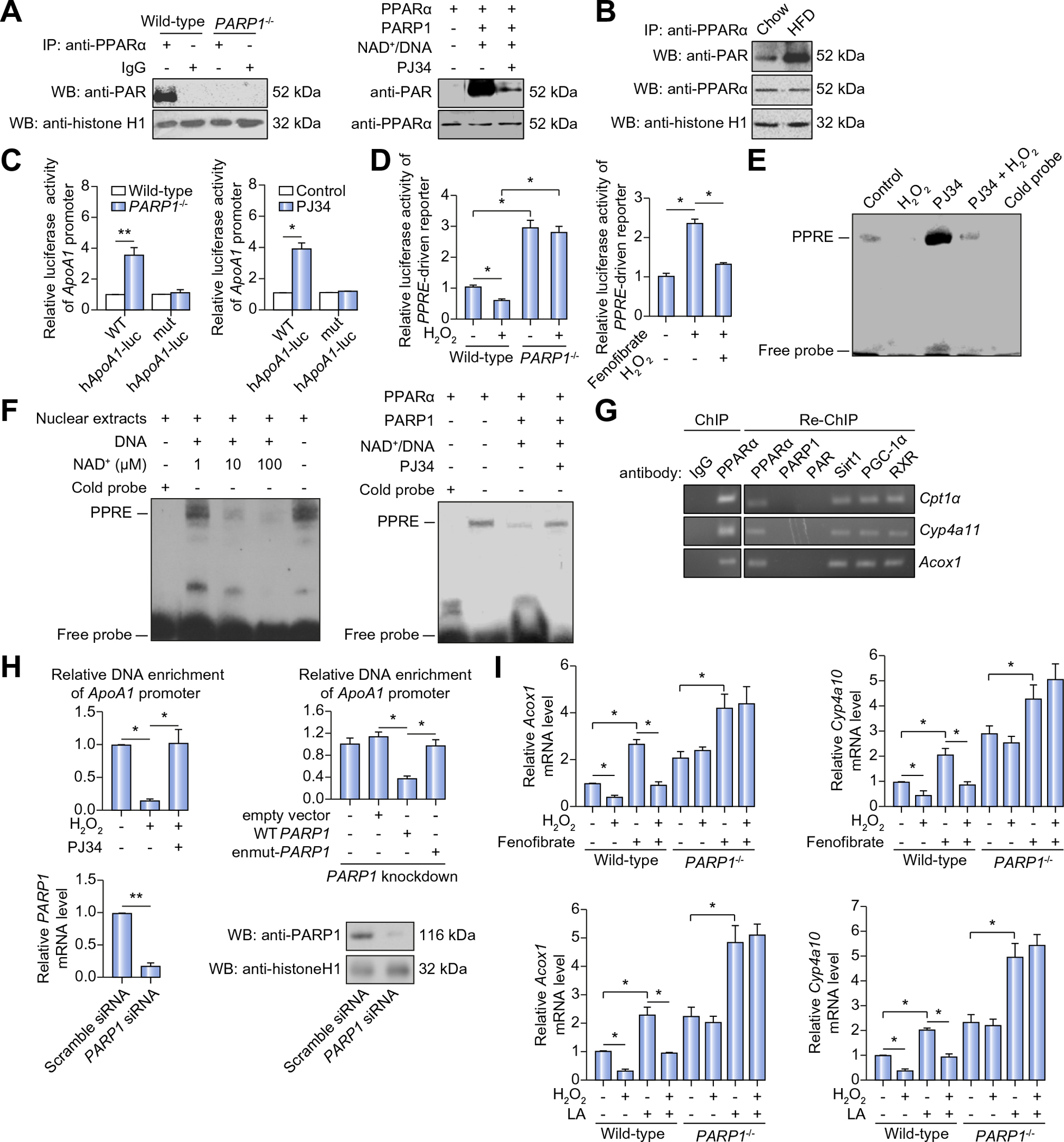
(A) Poly(ADP-ribosyl)ation levels of PPARα in nuclear extracts from wild-type (WT) or PARP1−/− hepatocytes were determined by immunoprecipitation (IP) with anti-PPARα antibody, followed by Western blotting using anti-PAR antibody (left). Recombinant PPARα proteins were incubated as indicated. Poly(ADP-ribosyl)ation levels of PPARα was analyzed (right). (B) C57BL/6-WT mice were fed with chow diet or HFD for 8 weeks. Poly (ADP-ribosyl)ation levels of PPARα in liver on HFD were determined by IP with anti-PPARα antibody, followed by Western blot assay using anti-PAR antibody. (C) The WT hApoA1 (WT-hApoA1) or the mutant hApoA1 (mut-hApoA1) luciferase promoter was transfected into WT or PARP1−/− hepatocytes, followed by treatment with vehicles or PJ34 (10 μmol/L, 24 h). Luciferase activities were analyzed (n = 4). *p <0.05, **p <0.01 represent PARP1−/− group vs. WT group. (D) PPRE-driven luciferase reporter was transfected into WT or PARP1−/− hepatocytes, followed by treatment with vehicles, H2O2 (0.3 mmol/L, 2 h) or fenofibrate (100 μmol/L, 24 h). Luciferase activities were analyzed (n = 3). *p <0.05. (E) EMSA assay of PPARα-PPRE complex formation in nuclear extracts from WT hepatocytes treated with vehicle or PJ34 (10 μmol/L, 24 h) in the absence or presence of H2O2 (0.3 mmol/L, 2 h). (F) Nuclear extracts from hepatocytes were incubated with active DNA and NAD+ respectively, then subjected to EMSA. Recombinant PPARα proteins were incubated as indicated, and subjected to EMSA. (G) Chromatin was first IP with anti-IgG antibody, or anti-PPARα antibody and then re-IP with anti-PPARα antibody, anti-PARP1 antibody, anti-PAR antibody, anti-Sirt1 antibody, anti-PGC-1α or anti-RXR antibody respectively. (H) ChIP assays using anti-PPARα antibody for amplification of human ApoA1 promoters in HepG2 cells treated with vehicle, PJ34 (10 mol/L, 24 h) in the presence or absence of H2O2 (0.3 mmol/L, 2 h) (left), and in PARP1-knockdown (using PARP1 siRNA) HepG2 cells transfected with full-length (WT) PARP1, or enzymatic-mutant (en-mutant) PARP1 constructs (right), (n = 4). *p <0.05, **p <0.01. (I) mRNA levels of Acox1 and Cyp4a10 in WT or PARP1−/− hepatocytes treated with fenofibrate (100 μmol/L, 24 h) or linoleic acid (LA, 30 μmol/L, 24 h) in the presence or absence of H2O2 (0.3 mmol/L, 2 h), (n = 3). *p <0.05. Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
Given that HFD activates PARP1, we hypothesized that poly (ADP-riboyl)ation of PPARα might be increased in the liver of mice on HFD. Indeed, WT mice on HFD vs. chow diet displayed a significant increase in poly(ADP-riboyl)ation of PPARα (Fig. 4B). Unlike PPARα, two lipogenic transcriptional factors SREBP1c and ChREBP were not poly(ADP-ribosyl)ated by PARP1 (Supplementary Fig. 1J). Next, we examined the effects of PPARα poly(ADP-ribosyl)ation on its transactivation using a luciferase reporter assay. Primary hepatocytes from WT or PKO mice were transfected with a luciferase reporter driven by the promoter of human ApoA1 gene [23] with either WT or mutated (mut) PPRE motif. Transfection with the WT- but not mut-construct induced higher luciferase activity in PARP1−/− than WT hepatocytes (Fig. 4C). In line with this, treatment with PJ34 increased luciferase activity of WT- but not mut-construct in WT hepatocytes (Fig. 4C). These results indicate that PPRE mediates the effects of PARP1 on human ApoA1 promoter activation. To solidify this finding, WT or PARP1−/− hepatocytes were transfected with three tandem repeats of PPRE (3 × PPRE)-driven luciferase reporter construct, followed by H2O2 treatment and luciferase activity assays. Exposure to H2O2 promoted PPARα poly(ADP-ribosyl)ation (Supplementary Fig. 1K), which was associated with a significant suppression of the PPRE-driven luciferase activity in WT but not PARP1−/− hepatocytes (Fig. 4D). Moreover, fenofibrate (PPARα agonist)-dependent induction of luciferase activity was also blunted when WT hepatocytes were exposed to H2O2 (Fig. 4D). Collectively, our data support that poly(ADP-ribosyl)ation of PPARα suppresses its transactivation.
PPARα regulates gene transcription via binding to its consensus response element in the target promoter [24]. To further define how PPARα poly(ADP-ribosyl)ation inhibits its function, we studied the effects of PPARα poly(ADP-ribosyl)ation on its sequence-specific DNA binding capacity using EMSA assays with a biotin-labeled PPRE probe. Exposure to H2O2 resulted in a significant decrease in PPARα-PPRE complex formation in nuclear extracts from WT hepatocytes, and this decrease was partially reversed by simultaneous treatment with PJ34 (Fig. 4E). Additionally, incubation of nuclear extracts from WT hepatocytes with NAD+ and nick DNA for 30 min, a condition that promoted endogenous PPARα poly(ADP-ribosyl)ation, caused a NAD+ concentration-dependent decrease in PPARα-PPRE complex formation (Fig. 4F). Moreover, incubation of recombinant PPARα with PARP1, NAD+ and nick DNA also resulted in a significant decrease in PPARα-PPRE complex formation in a cell free system (Fig. 4F). Altogether, these results suggest that poly(ADP-ribosyl) ation decreases DNA binding capacity of PPARα.
In addition to reduced binding capacity, PPARα poly(ADP-ribosyl)ation may also prevents its recruitment to target promoters. To test this, recruitment of PPARα to the promoters of Cpt1a, Cyp4a11 and Acox1 in HepG2 cells was examined using re-ChIP (ChIP on ChIP) assays. After the first round of ChIP using an anti-PPARα antibody, no qPCR products were detected while the second round ChIP using an anti-PAR antibody showed that poly(ADP-ribosyl)ated PPARα did not bind to the promoters selectively (Fig. 4G). In agreement, ChIP assays with an anti-PPARα antibody revealed that exposure to H2O2 led to a significant decrease in recruitment of PPARα to ApoA1 promoter in HepG2 cells, which was reversed by PJ34 treatment (Fig. 4H). Furthermore, forced expression of WT-PARP1 gene suppressed PPARα recruitment to ApoA1 promoter in PARP1-knockdown HepG2 cells, while transfection with the enmut-PARP1 construct did not (Fig. 4H). Taken together, the data suggest that activation of PARP1 suppresses PPARα recruitment to target promoters. To determine the functional significance of PARP1-associated reduction in PPARα recruitment to its target promoters, we examined the effects of PARP1 on PPARα target gene expression. Exposure to H2O2 suppressed not only the basal, but also the PPARα ligand, fenofibrate or linoleic acid (LA), -induced expression of Acox1 and Cyp4a10 genes in WT hepatocytes, but not in PARP1−/− hepatocytes (Fig. 4I). Thus, PARP1-induced PPARα poly(ADP-riboyl) ation suppresses PPARα function.
Poly(ADP-ribosyl)ation suppresses the association of PPARα with SIRT1
SIRT1, an NAD+-dependent deacetylase, is an important PPARα signaling regulator. In the nucleus, SIRT1 deacetylates PGC-1α, and associates with PPARα in its target promoter to facilitate gene transcription [25]. In this study, administration of PJ34 reversed the HFD-induced reduction of NAD+/NADH ratio (Fig. 5A), suggesting that the stimulatory effects of PARP1 inhibitor (PJ34) on PPARα signaling might be mediated through activation of NAD+/SIRT1. Consistent with this speculation, SIRT1 silencing completed blocked PJ34-induced expression of PPARα target genes in HepG2 cells (Fig. 5B). These results prompted us to link PARP1 to SIRT1 on PPARα signaling. HepG2 cells were thus pretreated with H2O2 for 3 h alone or together with PJ34, followed by ChIP assays. Exposure to H2O2 resulted in decreased recruitment of SIRT1 and PGC-1α to the promoters of Acox1 and MCAD, which was reversed by PJ34 treatment (Fig. 5C). These results indicate that activation of PARP1 prevents recruitment of SIRT1 and PGC-1α to PPARα target promoters. To further explore the underlying mechanisms, we tested whether SIRT1 and PGC-1α can be poly(ADP-ribosyl)ated by PARP1 in hepatocytes. Co-immunoprecipitation assays showed that neither SIRT1 nor PGC-1α was pulled down by an anti-PARP1 antibody (Supplementary Fig. 2A). Additionally, immunoprecipitation assays using an anti-PAR antibody demonstrated that SIRT1 and PGC-1α were not poly(ADP-ribosyl)ated (Supplementary Fig. 2A). These results suggest that SIRT1 and PGC-1α are not substrates of PARP1.
Fig. 5. Poly(ADP-ribosyl)ation suppresses the association of PPARα with SIRT1.
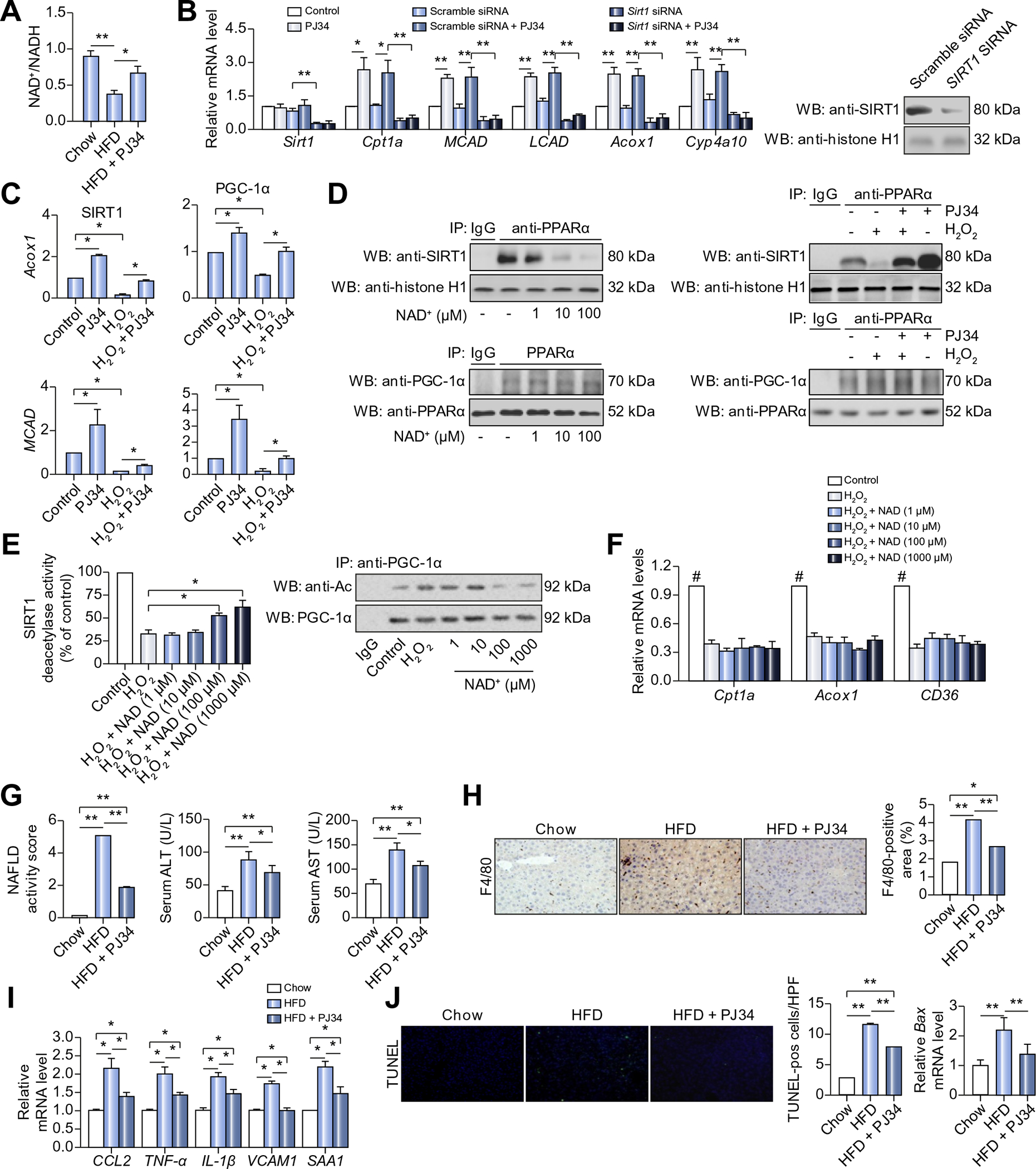
(A) C57BL/6 mice were fed with chow diet or HFD for 8 weeks, or fed with HFD for 8 weeks in combination with 6 week-PJ34 injection (10 mg/kg/day, n = 8 each). NAD+/NADH ratio in liver of mice was measured. *p <0.05. (B) mRNA levels of genes in hepatocytes transfected with Sirt1 siRNA (50 nmol/L, 48 h) or scramble siRNA, followed by treatment with vehicles or PJ34 (10 mol/L, 24 h), (n = 3). *p <0.05, **p <0.01. (C) ChIP assays using anti-Sirt1 antibody or anti-PCG-1α for amplification of Acox1 or MCAD promoters in HepG2 cells treated with vehicles or H2O2 (0.3 mmol/L, 2 h) after pretreatment with PJ34 (10 mol/L, 24 h) (n = 3). *p <0.05. (D) Nuclear extracts from HepG2 cells were subjected to IP assay with anti-PPARα antibody, followed by Western blotting using anti-Sirt1 antibody or anti-PGC-1α antibody. HepG2 cells was treated with different concentration of NAD+, or treated with vehicles or H2O2 (0.3 mmol/L, 2 h) after pretreatment with PJ34 (10 mol/L, 24 h). Non-specific IgG served as a negative control. (E) SIRT1 deacetylase activity was analyzed in HepG2 cells treated with in different concentration of NAD+, in the presence or absence of H2O2 (0.3 mmol/L, 2 h) (left), (n = 4). *p <0.05. Nuclear extracts from HepG2 cells were subjected to IP assay with anti-PGC1α antibody, followed by Western blotting using anti-acetylation (Ac) antibody (right). (F) mRNA levels of genes in HepG2 cells treated with different concentration of NAD+, (n = 4). #p <0.05 represent control group vs. H2O2 group. (G-J) C57BL/6 mice were fed with chow diet or HFD for 12 weeks, or fed with HFD for 12 weeks in combination with 10 week-PJ34 injection (10 mg/kg/day, n = 8 each). (G) NAFLD activity score (NAS), and serum levels of alanine transaminase (ALT) and aspartate transaminase (AST) in mice. *p <0.05, **p <0.01. (H) Liver sections from mice were immunohistochemically stained with anti-F4/80 antibody. *p <0.05, **p <0.01. (I) mRNA levels of the indicated genes in livers of mice were analyzed. *p <0.05, **p <0.01. (J) TUNEL staining of liver sections, and mRNA expression of Bax in the liver were analyzed. *p <0.05, **p <0.01. Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
Next, we examined the effects of PPARα poly(ADP-ribosyl) ation on the association between PPARα and SIRT1 or PGC-1α. Nuclear extracts from HepG2 cells were incubated with NADP+ and nick DNA to promote endogenous PPARα poly(ADP-ribosyl) ation, and then subjected to co-immunoprecipitation assays using an anti-PPARα antibody. Incubation with NAD+ and nick DNA reduced the amount of SIRT1 pull-down by the anti-PPARα antibody (Fig. 5D), suggesting that PPARα was dissociated with SIRT1 after poly(ADP-ribosyl)ation. In line with this, exposure to H2O2 also reduced the amount of SIRT1 pull-down by the anti-PPARα antibody in the nuclear extracts from HepG2 cells, and this reduction was reversed by PJ34 treatment (Fig. 5D). Unlike SIRT1, incubation of nuclear extracts with NAD+ and nick DNA or exposure of cells to H2O2 did not affect the association between PPARα and PGC-1α (Fig. 5D). In this study, we also examined the acetylation of PGC-1α in liver of both WT and PPARα−/− mice on HFD, and found that HFD-induced increases in PGC-1α acetylation were attenuated by PJ34 treatment in both strains of mice, suggesting that deficiency in PPARα did not affect deacetylation of PGC-1α by SIRT1 (Supplementary Fig. 2B).
As poly(ADP-ribosyl)ated PPARα was dissociated with PPARα/SIRT1 complex, preventing the complex recruitment to PPARα target promoters, we reasoned that restoration of SIRT1 activity could not reverse the PARP1 activation-induced suppression of PPARα signaling. To test this hypothesis, HepG2 cells were firstly exposed to H2O2 to consume intracellular NAD+ [19], and then incubated with different concentrations of NAD+ to restore SIRT1 activity, followed by measurements of PPARα target gene expression. Although incubation with NAD+ restored H2O2 induced decrease in SIRT1 activity (more than 60%, Fig. 5E) and acetylation of PGC-1α (Fig. 5E), it did not alter PPARα target gene expression (Fig. 5F). These results indicate that poly(ADP-ribosyl) ation of PPARα, rather than inhibition of SIRT1 activity, mediates the PARP1 activation-induced suppression of PPARα signaling.
Activation of PARP plays a crucial role in the pathogenesis of hepatic inflammation in mice on high fat diet
Persistent inflammation is a critical driver of NAFLD progression to NASH, fibrosis, cirrhosis, and hepatocellular carcinoma [26]. PARP1 was shown to be a key mediator of CCl4- and bile duct ligation-induced hepatic inflammation [18], but its role in mediating NASH was not known. We found that C57BL/6 mice on HFD for 12 weeks displayed increased hepatic steatosis, ballooning and lobular inflammation as revealed by H&E staining of liver sections (Fig. 1D). These pathological alterations were significantly reduced when the HFD-fed mice were treated with PJ34 for 10 week (2 weeks post HFD initiation) (Fig. 1D). The NAFLD activity score (NAS) [27] of liver sections also decreased significantly in PJ34-treated mice (1.88 ± 0.07 vs. 5.08 ± 1.04, p <0.01) (Fig. 5G). In addition, PI34 treatment attenuated the HFD-induced elevation in serum aspartate and alanine transaminase (Fig. 5G), hepatic macrophage infiltration as revealed by immunohistochemical stain of liver section using an antibody against F4/80 (a marker of macrophages in mice) (Fig. 5H), and hepatic expression of chemokine (C-C motif) ligand (CCL2) (Fig. 5I). PJ34 also suppressed the HFD-induced increases in hepatic mRNA levels of TNF-α and IL-1β, two major pro-inflammatory cytokines implicated in the pathogenesis of NASH, as well as VCAM-1 and SAA-1 (Fig. 5I). Consistently, treatment with PJ34 effectively suppressed the MCD-induced increases in serum alanine transaminase levels as compared to saline controls (Supplementary Fig. 2C), indicating a protective effect of PJ34 on the MCD-induced liver damage. In line with this finding, the hepatic F4/80 positive cells and mRNA levels of CCL2, TNF-α and IL-1β were also decreased after PJ34 treatment (Supplementary Fig. 2D and E). Intriguingly, although PJ34 also reduced the hepatic F4/80 positive cells in HFD PPARα−/− mice when compared to saline controls, this effect was obviously weaker than it did on WT mice on HFD (Supplementary Fig. 2F).
Hepatocytes apoptosis is another important trigger of liver inflammation in the progression of NAFLD [28]. PARP1 is a pivotal regulator of apoptosis [16]. Consistently, TUNEL staining showed that PJ34 treatment significantly decreased cell apoptosis in HFD-fed mice (Fig. 5J). Moreover, hepatic expression of apoptotic gene Bax (Bcl-2-associated X protein) (was also suppressed after PJ34 treatment (Fig. 5J). Taken together, these results demonstrate that PARP1 is critically involved in the pathogenesis of hepatic inflammation induced by chronic HFD feeding.
PARP1 is robustly activated in the liver of NAFLD patients, and activation of PARP1 suppresses fenofibrate-induced PPARα activation in HepG2 cells
To explore clinical relevance of our findings in mice, we collected liver biopsy samples from twelve NAFLD patients. PARP1 activation status in these biopsies was assessed on paraffin sections using immunohistochemical staining with the anti-PAR antibody. Very impressively, liver sections from NAFLD patients vs. normal subjects displayed a 20-fold increase in PAR-positive cells (Fig. 6A). Notably, the frequency of PAR-positive cells increased more dramatically in NAFLD biopsies with high NAS compared to low NAS (Fig. 6B). These results indicate that PARP1 is activated in the liver of NAFLD patients and its activation is associated with NAFLD progression. In line with this finding, we found that NAFLD patients displayed a decreased hepatic NAD+/NADH ratio as compared to normal subjects (Fig. 6C).
Fig. 6. PARP1 is robustly activated in liver of NAFLD patients.
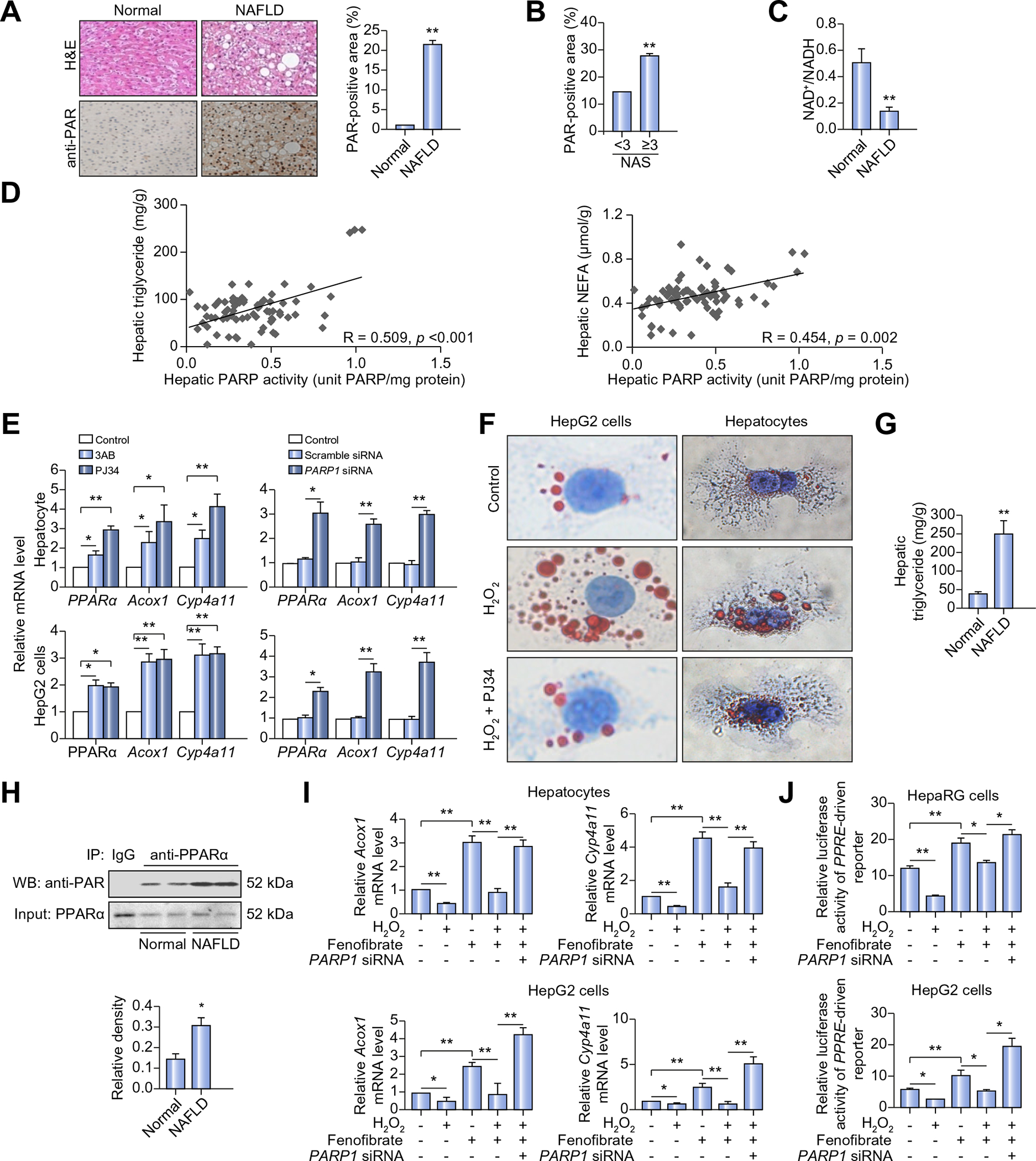
(A) H&E staining and immunohistochemical staining of poly(ADP-ribosyl)ation (PAR) in liver sections from NAFLD patients (n = 12) or normal controls (n = 12). Original magnification: 40×, **p <0.01 represents NAFLD group vs. normal group. (B) PAR-positive area was calculated based on NAFLD activity score (NAS) (NAS >3, n = 6; NAS <3, n = 6). **p <0.01. (C) NAD+/NADH ratio in liver sections from NAFLD patients (n = 12) or normal controls (n = 12) was analyzed. **p <0.01 represents NAFLD group vs. normal group. (D) Correlations of hepatic PARP activity with hepatic triglyceride or non-esterified fatty acid (NEFA) contents in 72 human subjects. Symbols represent individual person. (E) mRNA levels of genes in human primary hepatocytes and HepG2 cells treated with 3AB (7 mmol/L, 24 h) or PJ34 (10 μmol/L, 24 h), or transfected with PARP1 siRNA (50 nmol/L, 48 h) or scramble siRNA, (n = 4). *p <0.05, **p <0.01. (F) Oil Red O staining of human primary hepatocytes and HepG2 cells treated with vehicles or H2O2 (0.3 mmol/L, 2 h) after pretreatment with PJ34 (10 μmol/L, 24 h) original magnification: 200×. (G and H) 6 NAFLD patients and 6 normal controls were employed. (G) Triglyceride concentration in liver sections. **p <0.01 represents NAFLD group vs. normal group. (H) Nuclear extracts from the liver sections were subjected to IP assay with anti-PPARα antibody, followed by Western blotting using anti-PAR antibody. *p <0.05 represents NAFLD group vs. normal group. (I and J) Cells were treated as indicated, H2O2 (0.3 mmol/L, 2 h), fenofibrate (100 μmol/L, 24 h), PARP1 siRNA (50 nmol/L, 48 h), (n = 4). (I) mRNA levels of Acox1 and Cyp4a11 in human primary hepatocytes and HepG2 cells. *p <0.05, **p <0.01. (J) PPRE-driven luciferase reporter was transfected into undifferentiated HepaRG cells and HepG2 cells, and the luciferase activities were analyzed. *p <0.05, **p <0.01. Data are expressed as the mean ± SEM. One-way ANOVA followed by Student-Newmann-Keuls multiple comparison tests was used for all statistical analyzes.
To determine if PARP1 activity is associated with degrees of lipid deposition in human liver, we collected 72 human liver samples from the tissue bank of Clinic Center of Human Genomic Research, Huazhong University of Science and Technology, China. The main clinical and laboratory data of 72 human subjects are summarized in Supplementary Table 1. As expected, PARP activity in these liver samples was positively correlated with hepatic steatosis, i.e., with the content of triglyceride (r = 0.509, p <0.001) and NEFAs (r = 0.454, p = 0.002) (Fig. 6D). Not surprisingly, hepatic PARP activity was not associated with plasma levels of triglyceride (Supplementary Fig. 2G), because blood concentrations of triglyceride are balanced by its production from liver and intestine and its tissue clearance rates.
In human hepatocytes, PPARα is capable of activating its own transcription via functional PPREs in its promoter [24]. As a lower expression of PPARα is reported in human compared to rodent livers [23], we examined whether PARP1 was involved in the suppression of PPARα expression in primary human hepatocytes. Real-time RT-PCR assays showed that inhibition of PARP1 by PJ34 or 3AB caused a significant increase in PPARα transcription (Fig. 6E). Additionally, PARP1 knockdown in these cells resulted in around 3-fold elevation in the expression levels of PPARα and its targets, including Acox1 and CYP4a11 (Fig. 6E). To further confirm this finding, these experiments were repeated in HpeG2 cells, and the same results were obtained (Fig. 6E). These results indicate that PARP1 is a suppressor of PPARα expression in human hepatocytes. Given that PPARα promotes liver FAO in both rodents and human [29], we examined the effects of PARP1 activation on lipid accumulation in primary human hepatocytes and HepG2 cells. As shown in Fig. 6F, H2O2 treatment significantly increased oleic acid (OA)-induced lipid deposition in both types of cells, and this increase was reversed by PARP1 inhibitor PJ34. Since OA per se did not affect PARP1 activity (Supplementary Fig. 2H), this result strengthened a critical role for PARP1 activation in mediating the oxidative stress-induced steatosis of human hepatocytes.
Since PPARα is poly(ADP-ribosyl)ated by PARP1, and NAFLD patients manifest a robust increase in hepatic PARP activity, we hypothesized that poly(ADP-ribosy)ation of PPARα might be increased in the liver of NAFLD patients. To test this hypothesis, we compared the poly(ADP-ribosy)ation levels of PPARα between liver samples from normal subjects (hepatic triglyceride content: 41.33 ± 7.21 mg/g, n = 6) and those from NAFLD patients (hepatic triglyceride content: 202.75 ± 35.28 mg/g, n = 6) (Fig. 6G). Western blotting using the anti-PAR antibody showed that NAFLD patients had much higher hepatic poly(ADP-ribosyl) ation levels of PPARα than healthy controls (Fig. 6H).
Clinical trials demonstrate limited effects of PPARα ligands, fibrates, on fatty liver therapy [10,11]. Since activation of PARP1 suppresses the PPARα ligands (fenofibrate and LA)-induced PPARα target gene expression in mouse hepatocytes (Fig. 4I), we tested if this is also the case in human hepatocytes. Realtime RT-PCR assays showed that H2O2 also suppressed the fenofibrate-induced expression of Acox1 and Cyp4a11 genes in human hepatocytes and HepG2 cells, and this suppression was effectively abolished when PARP1 was knocked down (Fig. 6I). Moreover, we also found that exposure of undifferentiated HepaRG cells and HepG2 cells to H2O2 suppressed the fenofibrate-induced increases in luciferase activities of a reporter driven by 3 × PPRE, and this suppression was abrogated when PARP1 was knocked down (Fig. 6J). Taken together, these results suggest that activation of PARP1 suppresses fenofibrate-induced activation of PPARα signaling in human hepatic cells.
Discussion
One major finding of this study is that hepatic PARP1 activity is activated in mouse and human steatotic liver. In mice, we show that activation of PARP1 mediates the HFD-induced hepatic steatosis and inflammation. Mechanistically we demonstrate that PPARα is a substrate of PARP1. Poly(ADP-ribosyl)ation of PPARα by PARP1 prevents PPARα to activate its target gene expression, thereby suppressing FAO pathway upregulation in response to HFD challenge, worsening overnutrition-induced lipid deposition in hepatocytes. Our results suggest that highly activated PARP1 in the steatotic liver may attenuate clinical efficacy of PPARα agonists, like fibrates, and co-administration of PARP1 inhibitors may improve efficacy of fibrates in NAFLD treatment.
In NAFLD, oxidative stress causes mitochondrial damage and suppresses FAO in liver [30,31]. In the present study, we established PARP1 as a key mediator of oxidative stress-induced liver damage, and identified PPARα signaling as the central pathway inhibited by PARP1. PPARα is a master regulator of genes involved in mitochondrial and peroxisomal FA β-oxidation [10,32]. PPARα signaling can be regulated by intracellular NAD+ levels, since SIRT1, an important regulator of PPARα activity, is an NAD+ dependent deacetylase [33]. Under physiological conditions, SIRT1 deacetylates PGC-1α and associates with PPARα on target promoters to facilitate gene transcription [33]. As deacetylation of PGC-1α is critical for PPARα activation, deficiency in SIRT1 dramatically suppresses PPARα signaling and FAO in liver [24]. Inhibition of PARP1 increases intracellular NAD+ levels and SIRT1 activity, and thus leading to an upregulation of PPARα signaling and mitochondrial oxidation [19]. In support of this, knockdown of SIRT1 suppresses the PARP1 inhibition-induced upregulation of PPARα targets in muscle cells [19] and HepG2 cells (Fig. 5B). Therefore, it is speculated that activation of PARP1 suppresses PPARα signaling via downregulation of SIRT1 activity. However, our work may not support this notion since restoration of NAD+/SIRT1 activity and deacetylation of PGC-1α does not increase PPARα target expression when PARP1 is activated (Fig. 5F). Instead, we have shown that poly(ADP-ribosyl)ation of PPARα by PARP1 inhibits PPARα association with SIRT1 and recruitment of PPARα/SIRT1/PGC-1α complex to its target promoter, thus suppressing the transcription (Fig. 7). Our results demonstrate that poly(ADP-ribosyl)ation of PPARα per se, rather than suppression of NAD+/SIRT1 activity, impairs PPARα signaling.
Fig. 7. Proposed mechanism of PARP1 activation on fatty acid oxidation.
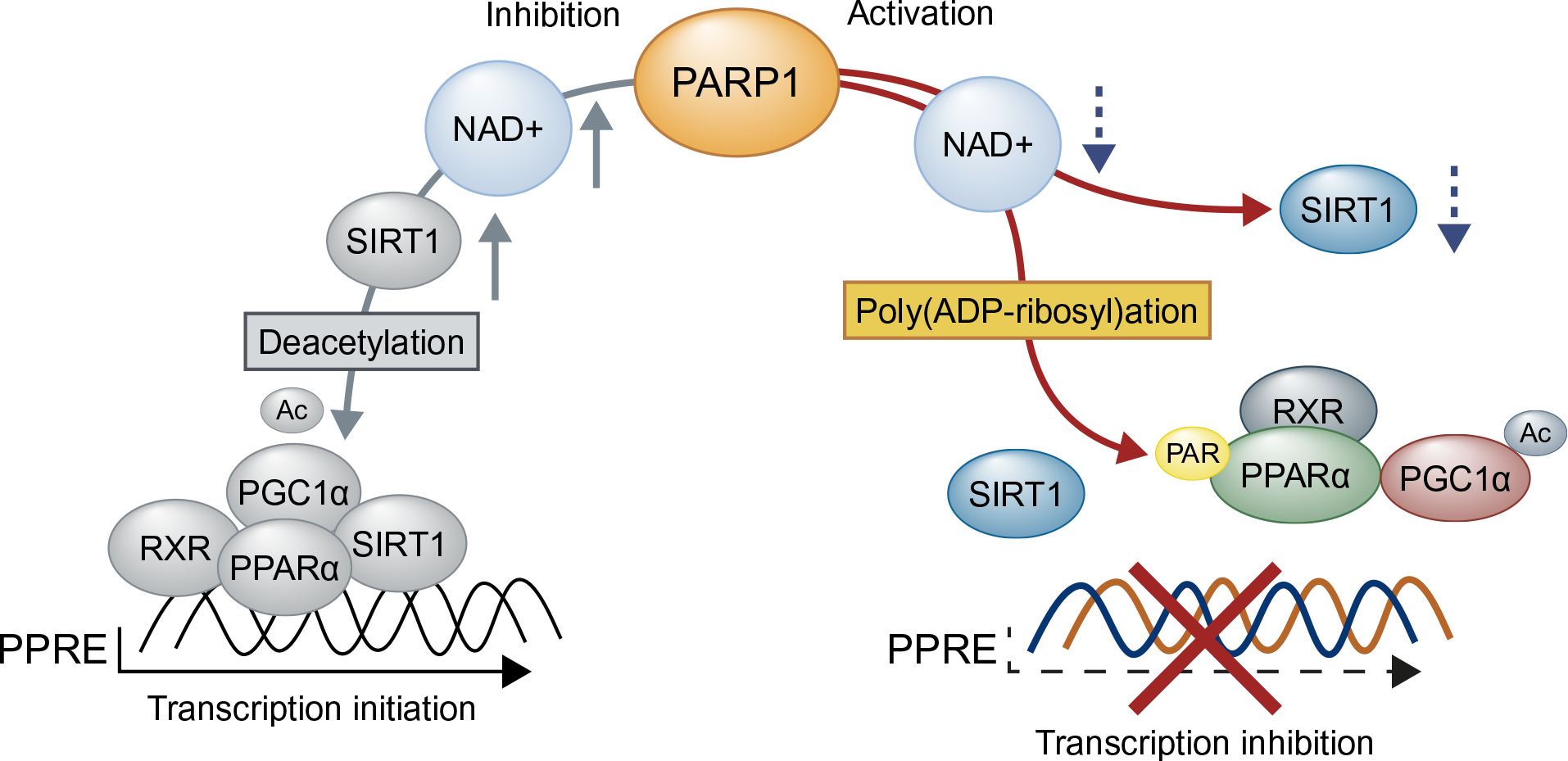
Activation of PARP1 promotes PPARα poly(ADP-ribosyl)ation in the nucleus. Poly(ADP-ribosyl) ation suppresses PPARα recruitment to the target promoter and dissociates PPARα/PGC1α/SIRT1 complex, leading to downregulation of fatty acid oxidation and related gene expression in hepatocytes.
It was reported that normal PPARα signaling protects against HFD-induced hepatic steatosis in mice [10,32,34]. Despite these animal findings, some clinical pilot studies showed that PPARα agonists (fibrates) have limited protective effects on human NAFLD [11]. This phenomenon is usually attributed to the lower hepatic expression of PPARα in humans than rodents, though contradictory results have been recently reported [29,35]. As a self-regulated gene, activation of PPARα is capable of increasing its own expression in human hepatocytes [24]. Here we demonstrate that PARP1-mediated poly(ADP-ribosyl)ation also suppresses PPARα gene transcription in human hepatocytes (Fig. 6E), implying that aberrant activation of PARP1 may contribute importantly to the downregulation of hepatic PPARα expression in NAFLD patients, especially in those with severe NAFLD because their hepatic PARP activities are much higher than subjects with simple steatosis. Consistent with this, Staels et al. demonstrate that liver PPARα expression decreases with progressive stages of liver fibrosis in patients with NASH. Besides suppression of PPARα expression, activation of PARP1 significantly suppresses the ligand (fenofibrate)-induced FAO gene expression and PPARα transactivation in human hepatocytes and HepaRG cells (Fig. 6I and J). Altogether, these observations suggest that activation of PARP1 may account for, at least partially, the blunted response of hepatic steatosis to fibrates in NAFLD patients. Thus administration of PARP1 inhibitors should enhance the protective effects of PPARα ligands, such as fibrates, on NAFLD.
It has been demonstrated that activation of PPARα can promote oncogenesis in rodents [36]. This finding has raised a safety concern regarding the clinical use of PPARα agonists. Recently, Morimura et al. showed that compared to WT controls, PPARα humanized mice displays a dramatically decreased hepatocellular carcinoma incidence (5% vs. 71%) after long-term Wy14,643 treatment [37], indicating a different response of human PPARα to the agonist stimulation. Importantly, until now, no clinical trials or meta-analyzes have shown that long-term use of fibrates can increase the incidence of cancer in patients with hyperlipidemia [10]. Nevertheless, whether these findings can be extended to the diseases other than hyperlipidemia is still unclear. Therefore, the effects of PPARα agonist on the oncogenesis of patients with different kinds of diseases should be studies in the future, especially when in combination with potential PARP1 inhibitors.
NASH is a major cause of liver damage in NAFLD. Oxidative stress is an important mechanism contributing to the pathogenesis and propagation of inflammation in NAFLD [38]. As a downstream effecter of oxidative stress, PARP1 promotes inflammation in various tissues through interactions with, or poly(ADP-ribosyl) ation of, some redox-sensitive transcription factors, such as NF-κB and AP-1 [39], both of which are implicated in the pathogenesis of NASH [40]. PARP1 was also shown to promote CCl4- and bile duct ligation-induced liver inflammation [18], and mediate oxidative stress-induced liver damage [17]. Consistently we found that PARP activity was dramatically increased in NAFLD patients, especially in those with high NAS (Fig. 6A). In rodents, we showed that inhibition of PARP1 suppressed liver inflammation induced by chronic HFD feeding (Fig. 5H and I). Thus, we for the first time demonstrate that PARP1 is also critically involved in the pathogenesis of NASH. On the other hand, PPARα not only controls FAO, but also transrepresses pro-inflammatory signaling pathways through physical interaction with p65, c-Jun and C/EBP in the liver [41]. Recently, Loyer et al. report that suppression of miR-21 in non-parenchymal cells attenuates hepatic inflammation through induction of PPARα expression [42]. Indeed, it has been reported that PPARα-dependent regulation of inflammatory genes requires the presence of non-parenchymal cells [43]. These findings, along with the fact that non-parenchymal cells contribute critically to hepatic inflammation [40], illustrate that PPARα in non-parenchymal cells plays an important role in the protection against NASH. In line with this, we showed that deletion of PPARα significantly, although not completely, attenuated PJ34-induced suppression of F4/80 positive cells in liver on HFD (Supplementary Fig. 2F).
Increased oxidative stress is an important mechanism contributing to the development and progression of NAFLD. In this study, we observed that administration of anti-oxidants, SOD and CAT, effectively suppressed the palmitic acid-induced PARP1 activation in primary hepatocytes (Supplementary Fig. 2I), strengthening the role for oxidative stress in the SFA-induced PARP1 activation. However, as PARP1 is a cellular stress sensor which can be activated by various stimuli [16], this finding cannot rule out the potential contribution of other pathogenic factors, such as inflammatory mediators, gut-derived endotoxin, lipotoxicity and etc., on the persistent activation of PARP1 in the process of NAFLD. The connection between PARP1 and diverse pathogenic factors in the progression of NAFLD should be explored in the future.
In conclusion, PARP1 is activated in rodent and human fatty liver. Hepatic PARP1 activation inhibits FAO pathway upregulation through poly(ADP-ribosyl)ation of PPARα, worsening hepatic steatosis and inflammatory responses associated with overnutrition. Biochemically, poly(ADP-ribosyl)ation of PPARα prevents its association with SIRT1 and recruitment to target promoters. In human hepatic cells, PARP1 inhibits ligand-induced PPARα activation and FAO gene expression. Our data for the first time link PARP1 to NAFLD development and progression, and suggest that PARP1 is a potential therapeutic target for NAFLD.
Supplementary Material
Financial support
This work was supported by the grants from National Natural Science Foundation of China (No: 81570405, No: 81670241, No: 81370263, No: 81170239 and No: 81500348).
Footnotes
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2016.11.020.
References
- [1].Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol 2013;10:645–655. [DOI] [PubMed] [Google Scholar]
- [2].Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686–690. [DOI] [PubMed] [Google Scholar]
- [3].Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774–788. [DOI] [PubMed] [Google Scholar]
- [4].Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836–1846. [DOI] [PubMed] [Google Scholar]
- [5].Boursier J, Diehl AM. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog 2015;11 e1004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fukunishi S, Sujishi T, Takeshita A, Ohama H, Tsuchimoto Y, Asai A, et al. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J Clin Biochem Nutr 2014;54:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2015;124:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kohli R, Pan X, Malladi P, Wainwright MS, Whitington PF. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J Biol Chem 2007;282:21327–21336. [DOI] [PubMed] [Google Scholar]
- [9].Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and independent fatty liver. J Biol Chem 2012;287:40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015;62:720–733. [DOI] [PubMed] [Google Scholar]
- [11].Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis 2008;40:200–205. [DOI] [PubMed] [Google Scholar]
- [12].Keating GM. Fenofibrate: a review of its lipid-modifying effects in dyslipidemia and its vascular effects in type 2 diabetes mellitus. Am J Cardiovasc Drugs 2011;11:227–247. [DOI] [PubMed] [Google Scholar]
- [13].Zambon A, Cusi K. The role of fenofibrate in clinical practice. Diab Vasc Dis Res 2007;4:S15–S20. [DOI] [PubMed] [Google Scholar]
- [14].Bajaj M, Suraamornkul S, Hardies LJ, Glass L, Musi N, DeFronzo RA. Effects of peroxisome proliferator-activated receptor (PPAR)-alpha and PPAR-gamma agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia 2007;50:1723–1731. [DOI] [PubMed] [Google Scholar]
- [15].Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology 1996;23:1464–1467. [DOI] [PubMed] [Google Scholar]
- [16].Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly (ADP-ribose) and PARP-1. Genes Dev 2012;26:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang C, Zhang F, Wang L, Zhang Y, Li X, Huang K, et al. Poly(ADP-ribose) polymerase 1 promotes oxidative-stress-induced liver cell death via suppressing farnesoid X receptor alpha. Mol Cell Biol 2013;33: 4492–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mukhopadhyay P, Rajesh M, Cao Z, Horvath B, Park O, Wang H, et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology 2014;59:1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 2011;13:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Devalaraja-Narashimha K, Padanilam BJ. PARP1 deficiency exacerbates diet-induced obesity in mice. J Endocrinol 2010;205:243–252. [DOI] [PubMed] [Google Scholar]
- [21].Erener S, Mirsaidi A, Hesse M, Tiaden AN, Ellingsgaard H, Kostadinova R, et al. ARTD1 deletion causes increased hepatic lipid accumulation in mice fed a high-fat diet and impairs adipocyte function and differentiation. FASEB J 2012;26:2631–2638. [DOI] [PubMed] [Google Scholar]
- [22].Huang D, Yang C, Wang Y, Liao Y, Huang K. PARP-1 suppresses adiponectin expression through poly(ADP-ribosyl)ation of PPAR gamma in cardiac fibroblasts. Cardiovasc Res 2009;81:98–107. [DOI] [PubMed] [Google Scholar]
- [23].Kersten S Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res 2008;2008 132960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol 2002;16:1013–1028. [DOI] [PubMed] [Google Scholar]
- [25].Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest 2015;125:501–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–1321. [DOI] [PubMed] [Google Scholar]
- [28].Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rakhshandehroo M, Hooiveld G, Muller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One 2009;4 e6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nguyen D, Samson SL, Reddy VT, Gonzalez EV, Sekhar RV. Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: novel protective role of glutathione. Aging Cell 2013;12:415–425. [DOI] [PubMed] [Google Scholar]
- [31].Wei Y, Clark SE, Thyfault JP, Uptergrove GM, Li W, Whaley-Connell AT, et al. Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am J Pathol 2009;174:1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr 2011;141:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev 2006;20:2913–2921. [DOI] [PubMed] [Google Scholar]
- [34].Carmona MC, Louche K, Nibbelink M, Prunet B, Bross A, Desbazeille M, et al. Fenofibrate prevents Rosiglitazone-induced body weight gain in ob/ob mice. Int J Obes (Lond) 2005;29:864–871. [DOI] [PubMed] [Google Scholar]
- [35].Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res 2010;2010:612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 1980;283:397–398. [DOI] [PubMed] [Google Scholar]
- [37].Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 2006;27:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ba X, Garg NJ. Signaling mechanism of poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am J Pathol 2011;178:946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015;61:1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pawlak M, Bauge E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, et al. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 2014;60:1593–1606. [DOI] [PubMed] [Google Scholar]
- [42].Loyer X, Paradis V, Henique C, Vion AC, Colnot N, Guerin CL, et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut 2015;65:1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Szalowska E, Tesfay HA, van Hijum SA, Kersten S. Transcriptomic signatures of peroxisome proliferator-activated receptor alpha (PPARalpha) in different mouse liver models identify novel aspects of its biology. BMC Genomics 2014;15:1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.