

Abstract
Background:
Osteopontin (OPN) is a multifunctional proinflammatory matricellular protein overexpressed in multiple human cancers and associated with tumor progression and metastases. Thrombin cleavage of OPN reveals a cryptic binding site for α4β1 and α9β1 integrins.
Methods:
Thrombin cleavage-resistant OPNR153A knock-in (OPN-KI) mice were generated and compared to OPN deficient mice (OPN-KO) and wild type (WT) mice in their ability to support growth of melanoma cells. Flow cytometry was used to analyze tumor infiltrating leukocytes.
Results:
OPN-KI mice engineered with a thrombin cleavage-resistant OPN had reduced B16 melanoma growth and fewer pulmonary metastases than WT mice. The tumor suppression phenotype of the OPN-KI mouse was identical to that observed in OPN-KO mice and was replicated in WT mice by pharmacologic inhibition of thrombin with dabigatran. Tumors isolated from OPN-KI mice had increased tumor-associated macrophages with an altered activation phenotype. Immunodeficient OPN-KI mice (NOG-OPN-KI) or macrophage-depleted OPN-KI mice did not exhibit the tumor suppression phenotype. As B16 cells do not express OPN, thrombin-cleaved fragments of host OPN suppress host antitumor immune response by functionally modulating the tumor-associated macrophages. YUMM3.1 cells, which express OPN, showed less tumor suppression in the OPN-KI and OPN-KO mice than B16 cells, but its growth was suppressed by dabigatran similar to B16 cells.
Conclusions:
Thrombin cleavage of OPN, derived from the host and the tumor, initiates OPN’s tumor-promoting activity in vivo.
Keywords: B16 melanoma, Metastasis, Osteopontin, Thrombin inhibitor, Tumor-associated macrophages (TAMs)
Introduction
Osteopontin (OPN), a widely expressed non-collagenous matricellular protein, is one of the 5% most highly expressed genes in cancer microarray data sets [1]. OPN promotes invasive and metastatic progression of breast, lung, prostate, and ovarian cancers, glioblastoma (GBM) and malignant melanoma [2-4]. In metastatic breast cancer patients, elevated baseline plasma OPN levels are associated with poorer survival [5]. Increased OPN expression in primary melanoma is predictive of reduced relapse-free survival. High levels of circulating OPN and/or increased expression of OPN by tumor cells correlate with more advanced cancer and worse prognosis [3].
OPN (secreted phosphoprotein 1) is a bone matrix component and a member of the small integrin-binding ligand N-linked glycoproteins (SIBLINGs) [6]. It is encoded by a single-copy gene, spp1, with one full-length isoform (OPNa) and five OPN splice variants identified [7]. OPN contains multiple conserved functional domains, including an RGD sequence that binds to multiple integrins, a variant CD44 (vCD44)-binding domain and a heparan sulfate-binding domain. OPN is expressed by fibroblasts, hematopoietic and immune cells and its expression is markedly upregulated in inflammatory conditions such as cancer [8]. When released, OPN has pleiotropic cytokine- and chemokine-like functions [6, 8], e.g. it promotes leukocyte survival and differentiation, and regulates leukocyte adhesion, migration, and trafficking [9, 10]. This functional versatility of OPN is due to its multiple functional domains enabling it to interact with a diverse range of factors, including cell surface receptors (integrins, CD44), secreted proteases (matrix metalloproteases, urokinase plasminogen activator) and growth factor/receptor pathways (TGFα/EGFR, HGF/Met). Thus, OPN, either derived from the host and/or the cancer, impacts cell proliferation, survival, drug resistance, and stem-like behavior, and acts as a crucial autocrine and paracrine mediator of cellular crosstalk in the tumor microenvironment [3, 4, 11]. Recent data also suggest that it may play a role in tumor evasion [7]. However, the precise mechanism(s) that mediates OPN’s tumor-promoting activity in vivo remains unknown.
Just C-terminal to the RGD sequence in human OPN is a conserved thrombin cleavage site (Arg168 in human and Arg153 in mouse) that when cleaved exposes a previously cryptic integrin-binding site for α4β1 and α9β1 integrins at the new C-terminus, SVVYGLR [12] (SLAYGLR in mouse). Thrombin cleaves OPN at this site with moderate catalytic efficiency when compared to thrombin’s canonical substrates in the blood coagulation cascade, that will be effective in an extravascular compartment where the physiological intravascular thrombin inhibitor, antithrombin, is normally not present [13]. Thrombin-cleaved OPN-Arg (OPN-R) has enhanced α4β1-dependent cell-binding activity compared to full length OPN (OPN-FL) that is abolished when the C-terminal arginine in OPN-R is removed by either carboxypeptidase N (CPN), or carboxypeptidase B2 (CPB2, also known as thrombin-activatable fibrinolysis inhibitor, TAFI). This second cleavage converts OPN-R to OPN-Leu (OPN-L), suggesting that the successive proteolytic cleavages of OPN-FL by thrombin and CPN or CPB2 represent a sequential up- and down-regulation of OPN’s cell adhesion activity [14] (Figure 1A). In addition, the released C-terminal OPN fragment (OPN-CTF) acquires a new conformation-dependent chemotactic activity towards dendritic cells (DC) [15]. Thus, thrombin cleavage of OPN adds a further dimension to OPN’s functional capabilities.
Figure 1: Suppression of B16 tumor growth and metastasis in OPN-KO and OPN-KI mice.
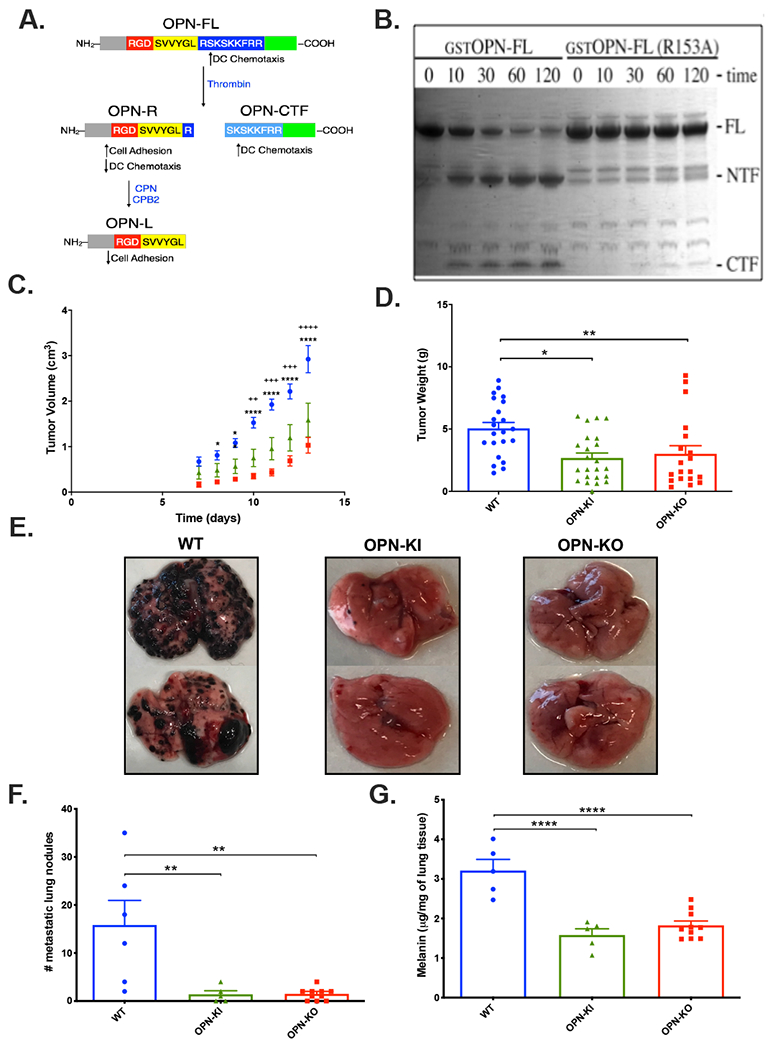
A. Model of OPN cleavages and its functional modulation by thrombin and carboxypeptidase N or B2. OPN-FL=OPN-full length; OPN-R=OPN-Arg; OPN-L=OPN-Leu. DC=dendritic cells. B. Time course (min) of thrombin treatment (10 nM) of E. coli produced GST-OPN-FL and GST-OPN-FLR153A analyzed by SDS-PAGE. FL=full length, NTF=N-terminal fragment, CTF=C-terminal fragment. C. Daily determination of B16 tumor volume grown on WT (blue), OPN-KI (green) and OPN-KO (red) mice. ++: p<0.01 WT vs. OPN-KI, +++: p<0.001 WT vs. OPN-KI, +++: p<0.0001 WT vs. OPN-KI, *: p<0.05 WT vs. OPN-KO, **: p<0.01 WT vs. OPN-KO, ****: p<0.0001 WT vs. OPN-KO. D. B16 tumor weight after sacrifice. *: p<0.05 WT vs. OPN-KI, **: p<0.01 WT vs. OPN-KO. E. Lungs from mice sacrificed 15 days after tumor injection i.v. F. Number of visible metastatic lung nodules in mice sacrificed 13 days after tumor injection i.v. G. Melanin content of lungs in mice sacrificed 13 days after tumor injection i.v. . All data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. **: p<0.01 WT vs. OPN-KI or OPN-KO, ****: p<0.0001 WT vs. OPN-KI or OPN-KO.
Using ELISAs specific for human OPN-R and OPN-L, we showed that levels of OPN-R and OPN-L are substantially elevated in the joint fluid of patients with inflammatory arthritis, demonstrating that these cleavages occur in vivo [10]. However, the role of thrombin cleavage of OPN in cancer biology has been largely overlooked because assays for thrombin-cleaved OPN fragments have only been developed recently. The previously reported association of OPN with cancer utilized assays for OPN that did not discriminate between intact and cleaved OPN. We showed that OPN-R and OPN-L are increased in the cerebral spinal fluid (CSF) of patients with GBM and non-GBM malignant gliomas [16]. OPN and the cleaved fragments promote motility and adhesion of U87 MG cells, a human GBM cell line, and confer resistance to apoptosis. Thus cleaved OPN fragments may promote GBM development.
To define the role of thrombin cleavage of OPN in vivo, we created a thrombin cleavage-resistant OPNR153A knock-in (OPN-KI) mouse. B16 melanoma is suppressed in mice deficient in OPN (OPN-KO, Spp1−/−) showing that tumor growth is dependent on the presence of OPN [17, 18]. We discovered that B16 melanoma growth and metastasis are grossly suppressed in the OPN-KI mouse, and the tumor suppression phenotype is identical to that observed in the OPN-KO mouse, demonstrating that thrombin cleavage of OPN is the critical step in mediating OPN’s tumor-promoting activity in vivo.
Materials and Methods
Details of mice, cell lines, cell assays and histology are described in the Supplemental Methods.
Generation of mice expressing thrombin-resistant osteopontin (Spp1R153A/R153A)
Homozygous C57BL/6J Spp1R153A/R153A mice were generated by Caliper Discovery Alliances and Services (Hanover, MD) and are designated as OPN-KI mice (Supplementary Table 1 and Supplementary Figures 1 and 2). Spp1−/− mice on a C57BL6J background, designated as OPN-KO mice, and wild type C57BL/6J mice (WT) were obtained from Jackson Laboratories (Sacramento, CA).
Immune-deficient mice carrying OPN-KI and OPN-KO genes
NOG-WT, NOG-OPN-KO and NOG-OPN-KI mice [19] were obtained from the Central Institute for Experimental Animals (CIEA; Kawasaki, Japan) and housed in a gnotobiotic facility at the VA Palo Alto Health Care System (VAPAHCS). These mice possess an intact OPN gene (NOG-WT), are deficient in OPN (NOG-OPN-KO) or carry the R153A mutation in the OPN gene rendering OPN thrombin-resistant (NOG-OPN-KI).
Mouse experiments
The mice were housed at VAPAHCS and experiments were performed under protocols approved by the VAPAHCS Institutional Animal Care and Use Committee in accordance with NIH guidelines. Animals were randomized to the different groups and analysis was blinded. All experiments were repeated at least twice independently and all mice that entered the experiments were accounted for and included in the analysis.
Tumor inoculation
B16 or YUMM3.1 cells (2 x 106 cells) were inoculated subcutaneously into WT, OPN-KI and OPN-KO mice. Mice were monitored daily for tumor growth by determining tumor size with calipers and the volume calculated using the equation (width x width x length)/2. For the metastasis model, B16 cells (0.5x106 cells) were injected via tail vein into mice. After sacrifice, lungs were isolated and the number of metastatic nodules on surface was counted on both sides, and the left lobe was used for melanin determination.
Dabigatran Etexilate (DE) treatment
DE (Boehringer Ingelheim, Ridgefield, CT) was administered orally [20] from 4 days before inoculation of B16 cells.
Depletion of macrophages by clodronate
Macrophages were depleted from mice by injecting clodronate (150 μl, 5mg/ml) or control liposomes retro-orbitally (Clodrosome, Brentwood, TN) every second day [21]. After 4 days, B16 cells were inoculated subcutaneously and given clodronate or control liposomes on alternate days until sacrifice.
Proteins and reagents
The different recombinant fragments of human OPN including mutated forms were produced as previously described [15, 16].
Flow cytometric analysis of leukocytes
Cells from tumors were stained with either a mixture of antibodies against CD3, CD11b, CD11c, CD19, CD38, CD45, CD161, CD206, EGR-2, F4/80, Gr-1, MHC II and Sytox green [22, 23], or a mixture of antibodies against CD3, CD19, CD45, and Sytox green and analyzed on a flow cytometer (BD LSR II). Data analysis was carried out with FlowJo (Becton Dickinson, San Jose, CA). The gating strategy is shown in Supplementary Figure 3 and antibody source in Supplementary Table 2.
Statistics
Data are expressed as mean ± SEM. Statistical analyses were performed using GraphPad Prism software. As indicated in the figure legends, unpaired 2-tailed Student t tests were used to make statistical comparisons between 2 groups. Comparisons between multiple groups were performed by 1-way ANOVA with either Tukey’s test for post hoc analysis or, when comparing all samples to control, Dunnett’s test. A P value of <0.05 was considered statistically significant and only those comparisons are marked.
Results
Suppression of B16 tumor growth in OPN-KO and OPN-KI mice
To confirm that the substitution of Arg-153 with Ala-153 in the thrombin cleavage site in OPN renders it resistant to thrombin cleavage, we expressed a mutant OPN (OPNR153A) in E. coli and demonstrated that it is resistant to thrombin cleavage (Figure 1B). OPN-KI mice carrying that substitution, similar to OPN-KO mice, were fertile and healthy before challenge, without any phenotypic changes observable in complete blood count (CBC), clinical chemistry, liver function and kidney function parameters (Supplementary Figures 4 and 5).
As growth of murine melanoma B16 tumors is suppressed in OPN-KO compared to WT mice [17, 18], we tested if the same phenotype would be observed in OPN-KI mice. B16 tumor growth was suppressed in OPN-KO or OPN-KI mice (Figure 1C). Tumor weight was lower in OPN-KI mice (mean 2.7±0.4g, n=23) and OPN-KO mice (mean 3.0±0.7g, n=19) than WT mice (mean 5.1±0.5g, n=22; OPN-KI vs. WT p=0.0032 and OPN-KO vs. WT p=0.0187) (Figure 1D). Results in male and female mice showed no difference in the reduction of tumor size in OPN-KI mice versus WT mice (Supplementary Figure 6). Similarly, varying the number of B16 cells inoculated did not remove the difference in tumor size in OPN-KI mice versus WT mice (Supplementary Figure 6). There was no difference in tumor growth or weight between OPN-KO and OPN-KI mice, showing that prevention of thrombin cleavage of OPN is equivalent to its complete deficiency.
Suppression of B16 metastasis in OPN-KI mice is similar to OPN-KO mice
In addition to primary tumors, OPN-KO mice also exhibit reduced tumor metastasis compared to WT mice in a hematogenous pulmonary metastasis model [24]. We tested whether OPN-KI mice display a similar phenotype. After sacrifice, clinical inspection revealed that the tumor burden was reduced in both OPN-KI and OPN-KO mice when compared to WT mice (Figure 1E). Quantitation by counting metastatic pulmonary nodules showed a comparable reduction in both OPN-KI mice (1.4±0.7/lung, n=5) and OPN-KO mice (1.5±0.4/lung, n=10) compared to WT mice (15.8±5.1/lung, n=6; OPN-KI vs. WT: p=0.0061; OPN-KO vs WT: p=0.0016; Figure 1F). There was less melanin in the lungs from OPN-KI mice (1.6±0.2μg/mg lung tissue, n=5) and OPN-KO mice (1.82±0.11μg/mg lung tissue, n=10) than WT mice (3.2±0.3μg/mg lung tissue, n=6; OPN-KI vs. WT: p=<0.0001; OPN-KO vs WT: p=<0.0001; Figure 1G). The number of metastatic pulmonary nodules and melanin content were reduced equivalently, suggesting that both the process by which the B16 cells exited the vasculature and established tumors in the extravascular compartment as well as the growth of those tumors were inhibited by the prevention of thrombin cleavage of OPN to an extent similar to complete absence of OPN. Thus, thrombin cleavage-resistant OPN-KI mice phenocopy OPN-KO mice in the B16 melanoma tumor models.
Inhibition of thrombin in WT mice phenocopies the OPN-KI tumor suppression phenotype
Since B16 tumor growth and metastasis were suppressed in thrombin cleavage-resistant OPN-KI mice similarly to OPN-KO mice, we investigated if blocking thrombin cleavage of OPN with an oral thrombin inhibitor also reduces tumor growth in WT mice. Mice were treated with the orally active thrombin inhibitor, dabigatran etexilate (DE), compounded into their chow or matched control chow [20] from 4 days before B16 inoculation until sacrifice. WT mice treated with DE showed tumor suppression (Figure 2A) similar to that observed in OPN-KI mice, irrespective of whether the OPN-KI mice had been treated with DE (Figure 2B). The tumor volume after sacrifice was lower in WT mice treated with DE (1.0±0.2 cm3, n=14 vs. 2.5±0.4 cm3, n=21, p=0.0017; Figure 2C). Tumor volume of OPN-KI mice were similar with and without DE treatment (0.73±0.3 cm3, n=10 vs. 0.77±0.5 cm3, n=7; p=0.9999; Figure 2C) and were also similar to DE-treated WT mice. The tumor weights observed in DE-treated WT mice (1.0±0.2g vs. 2.5±0.4g, p=0.0018) were similar to those found in OPN-KI mice (0.8±0.9g vs. 0.73±0.9g; p=0.9716; Figure 2D). We confirmed that this dosing regimen blocked thrombin activity by measuring activated partial thromboplastin time (aPTT) (Figure 2E) and noted a trend of increased anticoagulation to be inversely correlated with tumor weight (Figure 2F, r=−0.37). Thus, inhibition of thrombin in WT mice replicates the thrombin cleavage-resistant OPN-KI phenotype. Treatment of OPN-KI mice with DE did not further reduce B16 tumor burden, suggesting that there were no additional effects of thrombin inhibition beyond its prevention of OPN cleavage.
Figure 2: Inhibition of thrombin in WT phenocopies the OPN-KI phenotype.
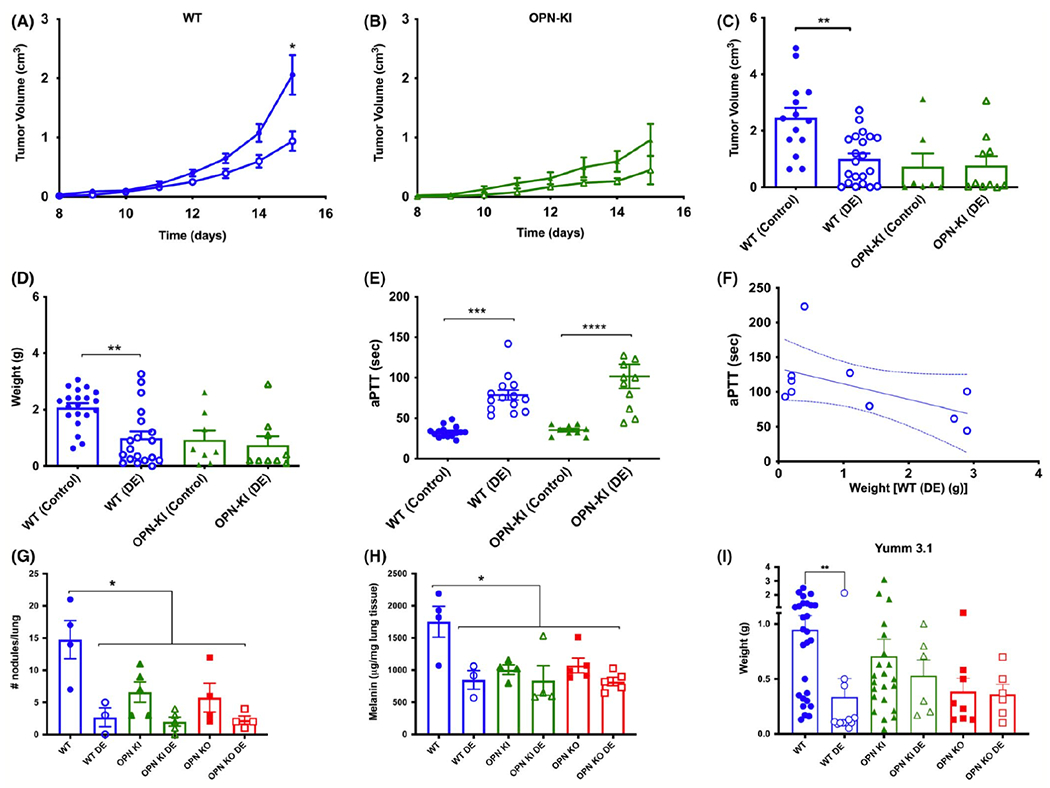
A. Daily determination of B16 tumor growth implanted on the flank of WT mice either fed on DE-containing chow (WT (DE) open circles) or control chow (WT (Control) closed circles). B. B16 tumor weights after sacrifice in OPN-KI mice either on DE-containing chow (OPN-KI (DE) open triangles) or control chow (OPN-KI (Control) closed triangles). C. B16 tumor volumes after sacrifice. D. B16 tumor weights after sacrifice. E. aPTT determined from blood collected at the time of sacrifice. In A – E, data are shown as mean ± SEM. Statistical significance was calculated by Student’s t-test comparing each genotype with and without DE in the chow. *: p<0.05 Control vs. DE, **: p<0.01 Control vs. DE, ****: p<0.0001 Control vs. DE F. Correlation of aPTT prolongation and tumor weights in WT mice treated with DE. G. Number of visible nodules counted in the lungs of mice sacrificed 13 days after inoculation in WT, OPN-KI and OPN-KO mice fed either DE or control chow in the metastasis model. H. Melanin content of lungs at 13 days after inoculation. In G and H, data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. *: p<0.05 WT vs. any other group. I. Weight of YUMM3.1 tumors sacrificed 23 days after inoculation in WT, OPN-KI and OPN-KO mice fed either DE or control chow in the flank model. Statistical significance was calculated by Student’s t-test comparing each genotype with and without DE in the chow. **: p<0.01 Control vs. DE.
In the pulmonary metastasis model, DE-treated mice exhibited fewer metastatic nodules/lung and reduced melanin content than control WT mice (Figure 2G, H). While DE treatment had no effect on local tumor growth in OPN-KI (Figure 2B) and OPN-KO mice, there was a trend for DE-treated OPN-KI or OPN-KO mice to exhibit fewer pulmonary metastases and decreased melanin content compared to untreated OPN-KI or OPN-KO mice respectively (Figure 2G, H).
B16 cells do not express OPN but express OPN-binding integrins
B16 cells have no detectable OPN mRNA expression [24] and we confirmed that OPN was not expressed by PCR (data not shown). Thus, B16 cells do not produce OPN and the suppression of B16 tumor growth in OPN-KO and OPN-KI mice compared to WT mice is due to thrombin cleavage of the host OPN in WT mice. Thrombin-cleaved OPN reveals a cryptic integrin-binding site for integrins α4β1 and α9β1 [12], and we demonstrated by FACS that B16 cells express those integrins on the cell surface (Supplementary Figure 7B). We hypothesized that thrombin-cleaved OPN fragments may directly modulate B16 tumor growth and metastasis by binding and signaling via α9β1 and α4β1 integrins.
To investigate the role of tumor cell-produced OPN, we used YUMM3.1 cells as full length OPN was present in YUMM3.1 cell medium (data not shown). Tumor growth for YUMM3.1 cells in OPN-KI (0.74±0.15g) and OPN-KO mice (0.37±0.18g) was lower than that in WT (0.94±0.15g) but did not reach statistical significance (Figure 2I), suggesting that tumor-derived OPN is able to partially compensate for the loss of thrombin-cleavable OPN in the OPN-KI mouse or absence of OPN in the OPN-KO mouse, but insufficient to elicit the full tumor suppression phenotype in this model.
Treatment with DE significantly suppressed YUMM3.1 tumor growth in WT mice (0.37±0.23g vs. 0.94±0.15g, p=0.0091: Figure 2I), to an extent similar to that found in DE-treated OPN-KI mice (0.53±0.14g) and OPN-KO mice (0.36±0.09g). Thus YUMM3.1 cells produce OPN and their growth is less suppressed than B16 cells in OPN-KI and OPN-KO mice but is suppressed in DE-treated WT mice.
Effects of thrombin-cleaved OPN fragments on B16 cells
We treated B16 tumor cells with different thrombin-cleaved OPN fragments, including OPN-R, OPN-L and OPN-CTF [15]. Compared to bovine serum albumin (BSA), all OPN fragments increased cell adhesion (Figure 3A). There was a ~3 fold increase in adhesion with OPN-R compared to OPN-FL, confirming the importance of thrombin cleavage of OPN [25, 26]. Both OPN-L and OPN-RRAA (in which the RGD sequence in OPN-R has been mutated to RAA) had reduced adhesion compared to OPN-R, indicating that both the RGD domain and thrombin cleavage-exposed integrin-binding site (SVVYGLR) at its C-terminus are important in cell adhesion, consistent with involvement of both α4 and β1 integrins [25, 26]. OPN fragments did not increase B16 cell proliferation compared to OPN-FL but all increased proliferation compared to BSA (Figure 3B).
Figure 3: Different OPN cleavage products produce different responses from B16 cells.
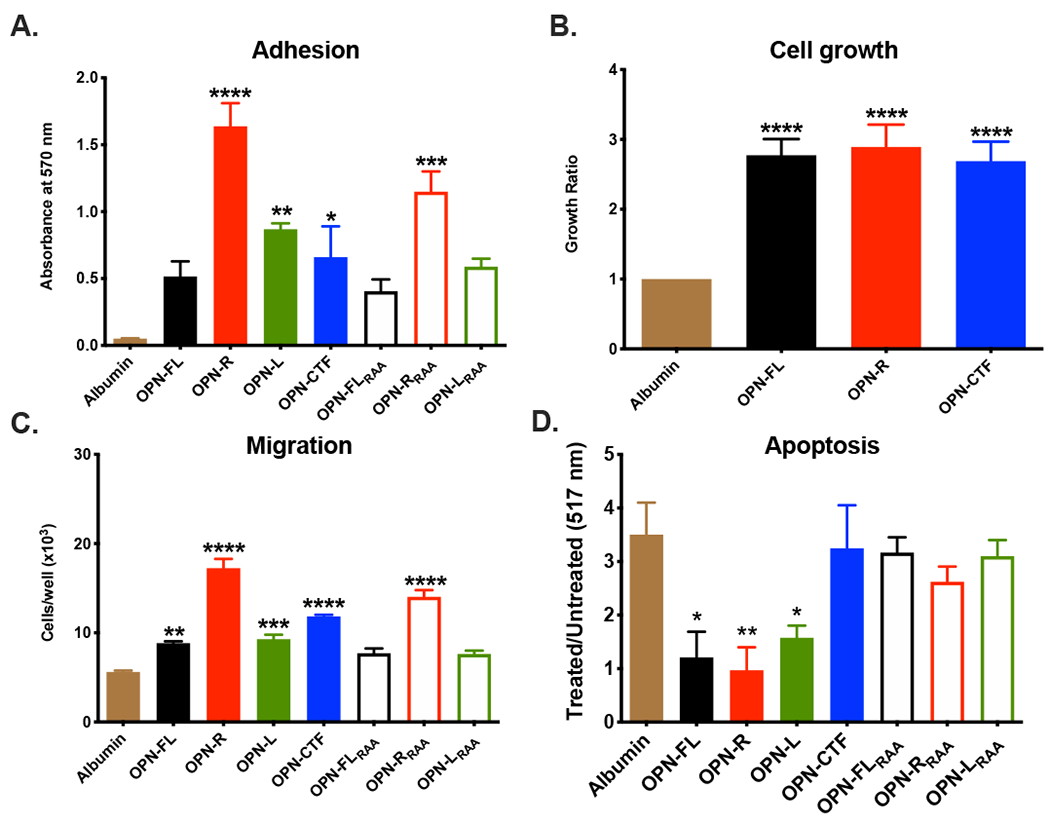
The effects of OPN fragments on B16 cell adhesion (A), growth (B), migration (C and apoptosis (D) were determined. Data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA comparing all OPN fragments to albumin (negative control) followed by Dunnett’s multiple comparison test. *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.001. OPN-FL=OPN full length, OPN-R=OPN-Arg, OPN-L=OPN-Leu, OPN-CTF=OPN-C-terminal fragment. Subscript RAA denotes substitution of the RGD sequence by RAA in that OPN fragment.
OPN enhances chemotaxis [27] so we tested the effects of the OPN fragments on B16 cell migration. Compared to BSA, OPN-FL, OPN-R, OPN-L, OPN-CTF and OPN-RRAA all increased migration (Figure 3C). When compared to OPN-FL, chemotaxis was increased by both OPN-R (~2-fold) and OPN-RRAA (~1.5-fold), while OPN-FLRAA OPN-LRAA and OPN-L did not enhance chemotaxis compared to OPN-FL, again indicating that both RGD and the cleavage-exposed SVVYGLR at the C-terminus in OPN-R play a role in increasing cell migration. Interestingly, OPN-CTF, which does not have the integrin-binding sites, also increased cell migration ~1.5-fold compared to OPN-FL suggesting that OPN-CTF is functionally active and may interact independently with a different receptor on B16 cells.
We also studied the effect of OPN and its fragments on B16 cell apoptosis. OPN-FL, OPN-R and OPN-L reduced apoptosis equally compared to BSA, while OPN-CTF, OPN-FLRAA, OPN-RRAA and OPN-LRAA had no anti-apoptotic effect, showing that OPN confers a protective anti-apoptotic effect in B16 cells mediated solely by its RGD domain that is not affected by thrombin cleavage (Figure 3D). Thus, B16 tumor suppression in the OPN-KI mice is not due to the loss of anti-apoptotic effect in OPNR153A resulting in increased tumor cell apoptosis.
While thrombin-cleaved OPN fragments modestly affected B16 tumor cell adhesion and chemotaxis, given the dramatic in vivo phenotype, we hypothesized that thrombin-cleaved OPN fragments may modulate the host antitumor immune response to promote tumor growth and metastasis.
B16 tumor suppression phenotype is lost in immune-deficient NOG-OPN-KO and NOG-OPN-KI mice
We generated OPN-KO and OPN-KI mice in the severely immunodeficient NOD/Shi-scid/IL-2Rγnull (NOG) mice to create NOG-OPN-KO and NOG-OPN-KI mice respectively [19]. These mice lack T cells, B cells, NK cells, and exhibit dysfunction of macrophages and dendritic cells. If thrombin-cleaved OPN fragments modulate host antitumor immune response, we would expect the tumor suppression phenotype in OPN-KO and OPN-KI mice to be lost in their NOG counterparts. We tested these mice in our B16 tumor and metastasis models.
There were no differences in B16 tumor size or weight between NOG control mice, NOG-OPN-KO and NOG-OPN-KI mice, even when we titrated the dose of the B16 inoculum (Figure 4A, B, Supplementary Figure 8). We also found no gender specific differences (Supplementary Figure 9). In the pulmonary metastasis model, there were no differences in metastatic pulmonary nodules or melanin content between NOG control mice, NOG-OPN-KO, and NOG-OPN-KI mice (Figure 4C, D). Thus, tumor suppression in OPN-KI mice requires an intact immune system.
Figure 4: B16 growth in immune-deficient mice is not affected by OPN status.
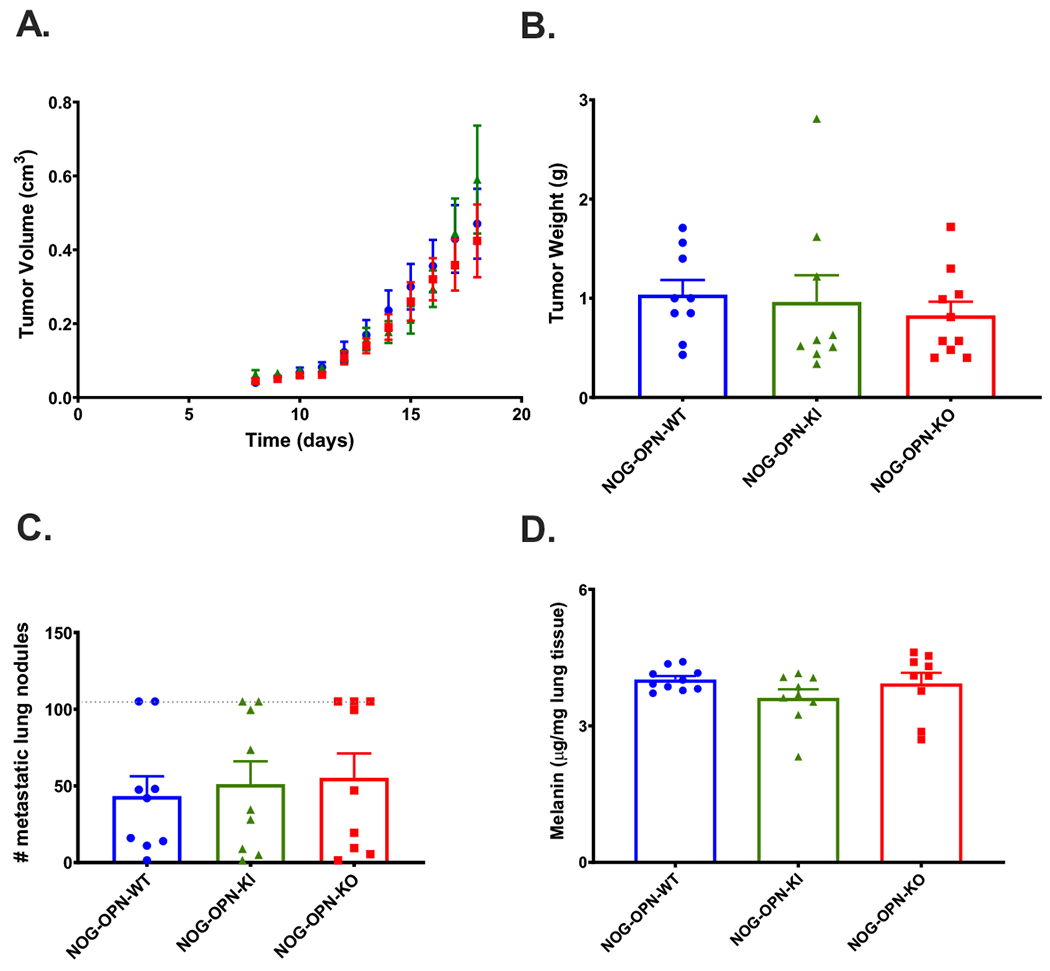
A. Daily determination of B16 tumor volume grown on NOG-WT (blue circles), NOG-OPN-KI (green triangles) and NOG-OPN-KO (red squares) mice in the flank model. B. B16 tumor weights after sacrifice in NOG-WT, NOG-OPN-KI and NOG-OPN-KO mice in the flank model. C. Number of visible metastatic lung nodules counted in the lungs of mice sacrificed 13 days after tumor injection i.v. Dotted line represents maximum number of nodules that could be counted. D. Melanin content of lungs. All data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test.
Changes in macrophage phenotypes in tumor-associated macrophages (TAMs) from OPN-KI and OPN-KO mice compared to WT mice
Histological examination did not show significant differences between the B16 tumors from the different mouse genotypes (Figure 5A). There was no difference in numbers of T cells determined by anti-CD3 antibody in immunohistological studies (Figure 5C). However, immunohistological studies and flow cytometric analysis showed significantly more F4/80+ cells in tumors from OPN-KO and OPN-KI than WT mice (Figure 5B, D) suggesting that tumor suppression might be mediated in part via modulation of the macrophage response.
Figure 5: B16 tumors from OPN-KI and OPN-KO mice contain more macrophages.
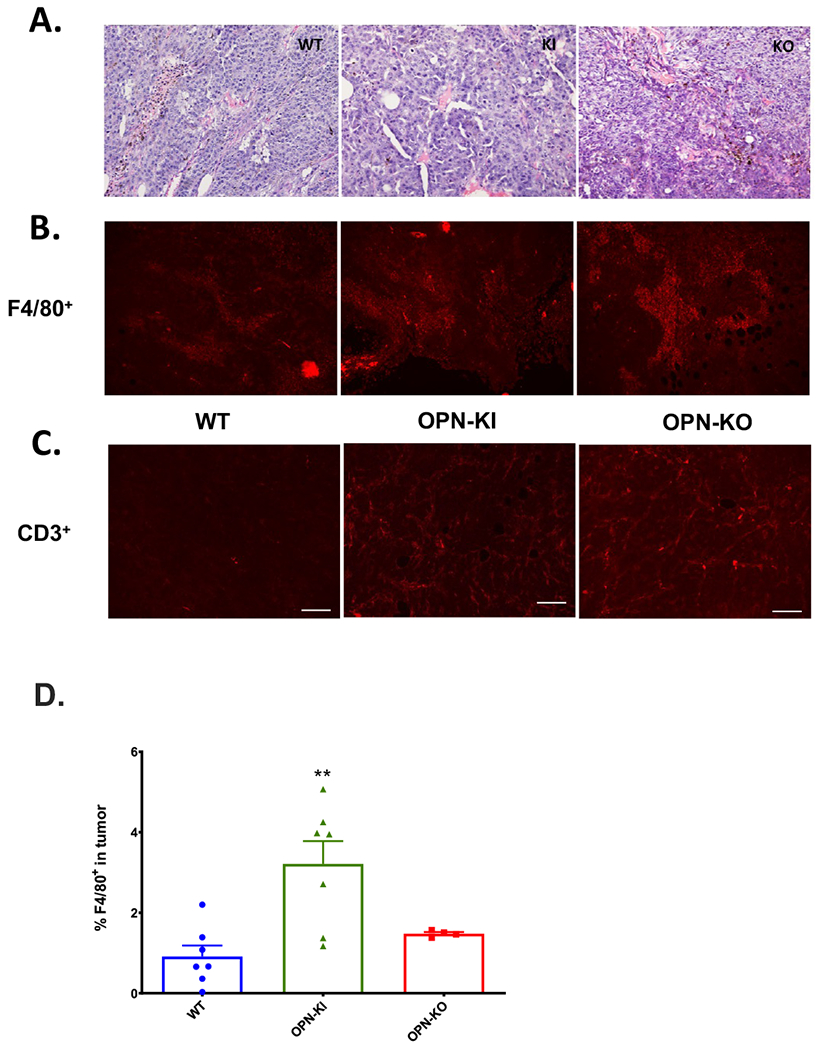
A.H&E staining of tumor sections from WT, OPN-KI and OPN-KO mice. B. Tumor sections from WT, OPN-KI and OPN-KO mice stained with anti-F4/80 antibody. C. Tumor sections from WT, OPN-KI and OPN-KO mice stained with anti-CD3 antibody. Scale bars = 100nm. D. The percentage of F4/80+ cells present in tumors from OPN-KI, OPN-KO and WT mice determined by flow cytometry. All data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. **: p<0.01 WT vs. OPN-KI.
We analyzed infiltrating macrophages and lymphocytes in the tumors from the different genotypes by flow cytometry. In tumors from OPN-KI and OPN-KO mice compared to WT mice, overall tumor-associated macrophages (TAMs; CD45+F4/80+) were significantly increased (WT: 50.7±1.6% of CD45+; OPN-KI: 66.5±5.9%, p=0.0168 and OPN-KO: 58.3±1.1%, p<0.05, n=5 per group, Figure 6A). There were no differences in numbers of M1 macrophages defined as EGR2−CD38+ macrophages (Figure 6B). The fraction of M2 macrophages in the TAMs defined as either EGR2+CD38− or CD206+CD11b+ macrophages was decreased (Figure 6C-D), while the fraction of TAMs defined as CD11b+CD11c− CD206+Ly-6G− macrophages was increased by ~2-fold (WT: 4.7±0.65%, OPN-KI: 9.2±1.5%, p=0.0399 and OPN-KO: 9.0±1.3%, p=0.0495, n=5 per group, Figure 6E). Therefore, in addition to an increase in the total number of TAMs in B16 tumors in OPN-KI and OPN-KO mice, there was a switch from M2 macrophages to TAMs with a different activation profile in these tumors. There were no differences in numbers of infiltrating neutrophils, B- and T-cells in tumors from OPN-KI and OPN-KO mice compared to WT (Figure 6F-H). A model of the changes in TAMs is proposed in Figure 6I.
Figure 6: Changes in macrophages in tumors from OPN-KI and OPN-KO mice compared to WT mice.
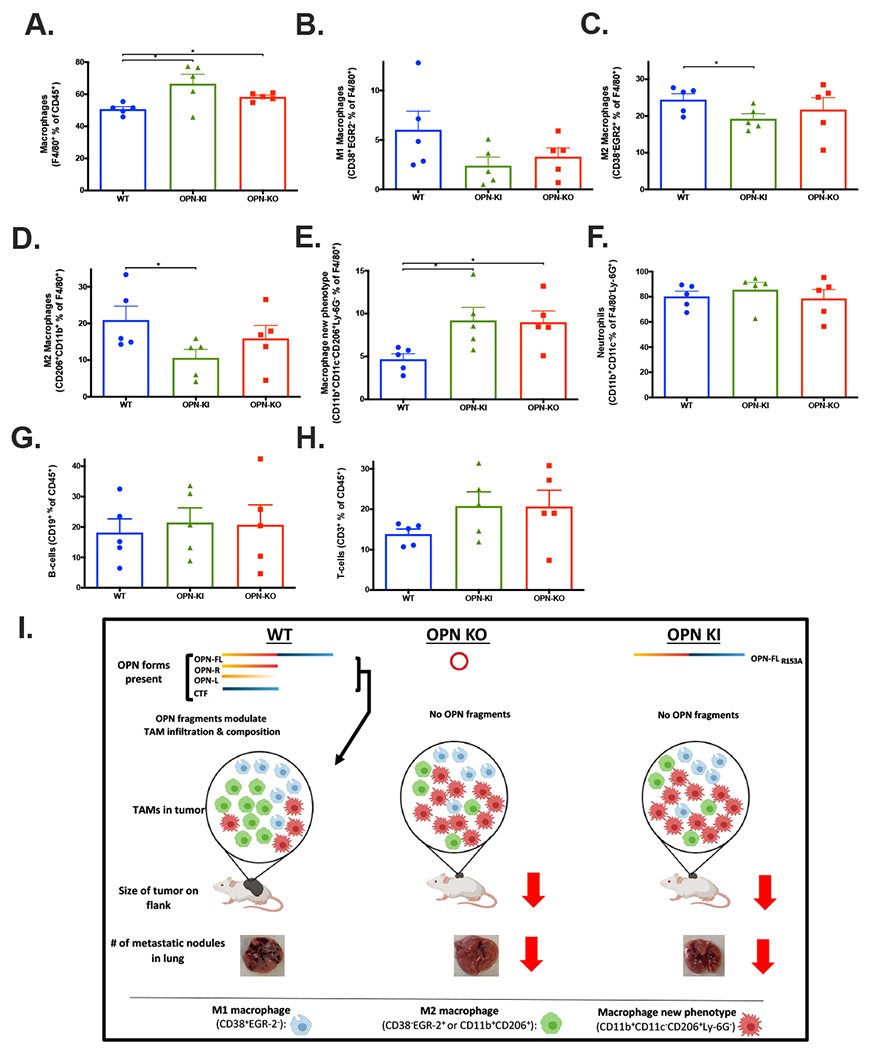
Infiltrating macrophages (A), M1 macrophages (B), M2 macrophages (C and D), Macrophages with new phenotype (E), neutrophils (F), B-cells (G) and T-cells (H) were determined by flow cytometry in tumor samples from the flank model in WT, OPN-KI and OPN-KO mice. All data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. The experiment (n=5) shown is representative of two independent experiments. Model of thrombin +/− CPN/CPB2 cleavage(s) of OPN and their effect on macrophages, tumor growth and metastasis. (I). Cleaved OPN fragments affect the infiltration and composition of macrophages in the tumor maintaining tumor-promoting M2 macrophages (green cells) in WT mice. These OPN fragments are absent in either OPN KO or thrombin-resistant OPN-KI mice, leading to decrease in M2 macrophages and replacement by macrophages with a different activation phenotype (red cells) leading to tumor suppression.
Depletion of macrophages by clodronate reverses the B16 tumor suppression phenotype in OPN-KI mice
Since WT mice tumors had fewer macrophages, we investigated whether tumor suppression in OPN-KI mice is mediated by macrophages by depleting them with clodronate [28]. Treatment with clodronate did not alter B16 tumor growth in WT mice whereas the tumor suppression phenotype in OPN-KI mice was reversed by macrophage depletion. (Figure 7A, B). Clodronate treatment increased B16 tumor volume (clodronate vs. control: 2.88±0.2 cm3, n=10 vs. 1.0±0.25 cm3, n=11, p=<0.0001) and tumor weight (clodronate vs. control: 2.25±0.23 g vs. 1.0±0.26 g, p=<0.003) in OPN-KI mice to levels similar to WT (clodronate vs. control: 3.1±0.15 cm3, n=13 vs. 3.15±0.2 cm3, n=14, p=0.9930 (Figure 7C) and clodronate vs. control: 2.8±0.09 g vs. 2.8±0.14 g, p=>0.9999 (Figure 7D). The F4/80+ macrophage cell population was reduced in both the bone marrow and tumor compartments of WT and OPN-KI mice treated with clodronate compared to controls, while the number of F4/80+ cells in blood was unaffected (Figures 7E-G). This suggests that tumor suppression in OPN-KI mice was due to increased macrophages in the tumors. While macrophages were depleted by clodronate in WT mice, there was no increase in tumor size.
Figure 7: Depletion of macrophages by clodronate reverses the B16 tumor suppression phenotype in OPN-KI mice.
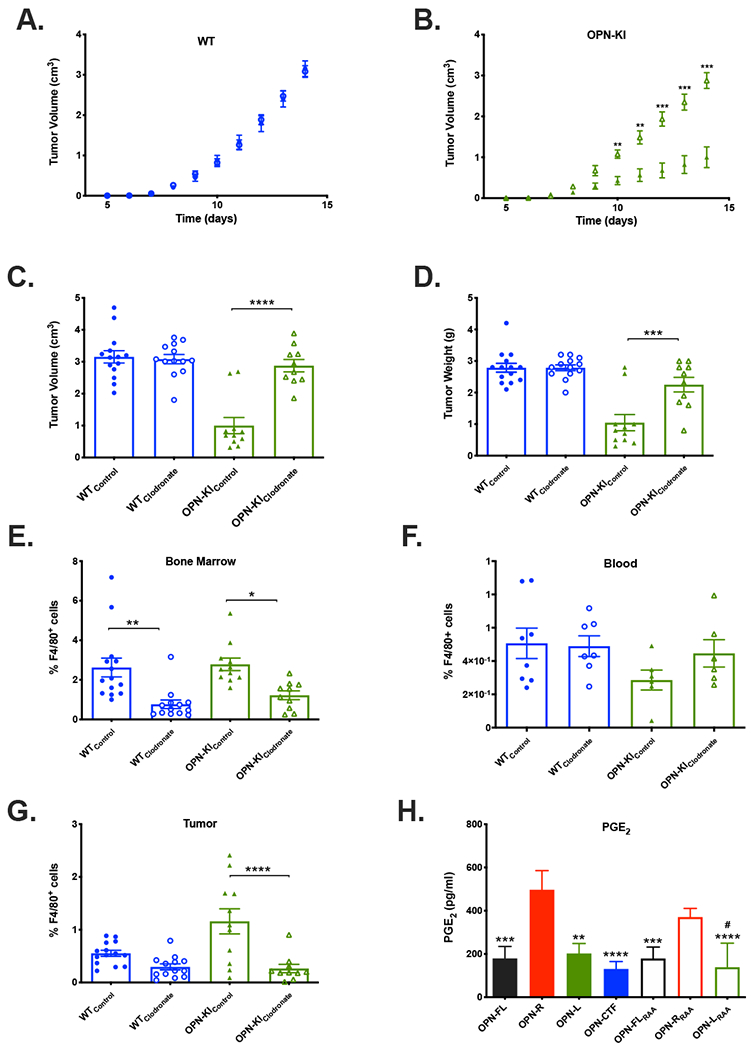
A. WT mice were treated with either clodronate (open circles) or control (closed circles) liposomes and the tumor volumes measured daily. B. OPN-KI mice were treated with either clodronate (open triangles) or control (closed triangles) liposomes and the tumor volumes measured daily. C. WT or OPN-KI mice were treated with either clodronate or control liposomes and the tumor volumes measured after sacrifice. D. WT or OPN-KI mice treated with either clodronate or control liposomes and the tumor weights measured after sacrifice. The percentage of F4/80+ cells was determined in the bone marrow (E), blood (F) and tumors (G) from WT or OPN-KI mice treated with either clodronate or control liposomes 14 days after inoculation with B16 cells. All data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. *: p<0.05 control vs. clodronate, **: p<0.01 p<0.05 control vs. clodronate, ***: p<0.001 p<0.05 control vs. clodronate, ****: p<0.001 p<0.05 control vs. clodronate. H. Production of PGE2 by RAW cells in response to OPN fragments (BSA control subtracted). Data are shown as mean ± SEM. Statistical significance was calculated by one-way ANOVA followed by Tukey’s post-hoc test. **: p<0.01 vs. OPN-R, ***: p<0.001 vs. OPN-R, ****: p<0.001 vs. OPN-R, #: p<0.05 vs. OPN-RRAA. Subscript RAA denotes substitution of the RGD sequence by RAA in that OPN fragment.
Thus, the data on clodronate depletion of macrophages plus that from the NOG immune deficient mice suggest that suppression of B16 tumor growth and metastasis in OPN-KI and OPN-KO mice is due to an enhanced host-antitumor immune response mediated by increased F4/80+ TAMs with an altered activation phenotype (Fig. 6I).
RAW cell responses to different OPN forms
RAW cells, a murine macrophage line, have been used as a model for TAMs [17] and we demonstrated that they expressed α4β1 and α9β1 integrins by flow cytometry (Supplementary Figure 7). All OPN fragments increased RAW cell adhesion compared to BSA with no differences between them (Supplementary Figure 10). We investigated prostaglandin E2 (PGE2) production as it is a mediator that has been reported to induce tumor angiogenesis and enhance B16 cell growth [17, 29]. OPN-R and OPN-RRAA both increased PGE2 secretion in RAW cells by >2 fold compared to OPN-FL (Figure 7H), indicating that the increase in PGE2 secretion was mediated by the α4β1 and α9β1 integrin-binding site revealed in OPN-R upon thrombin cleavage.
Discussion
Although OPN has been extensively investigated in vitro as a thrombin substrate [12-14], here we show for the first time that OPN is a pathophysiological substrate of thrombin in the B16 melanoma model and that thrombin cleavage of OPN is critical in initiating OPN’s tumor-promoting effects in vivo. Our findings that OPN-KI mice phenocopy OPN-KO mice in tumor suppression (Figure 1) and that the direct thrombin inhibitor dabigatran replicates the tumor suppression phenotype in WT mice (Figure 2) show that intact OPN-FL, despite the presence of its multiple functional domains, does not promote tumor progression by itself. The initiation of this activity requires its cleavage by thrombin and it is the resistance of OPNR153A to thrombin cleavage, and not the increased levels of total OPN, in the OPN-KI mouse that accounts for its tumor suppression phenotype. OPN can be derived from the tumor and/or the host. YUMM3.1 cells were derived from a mouse melanoma carrying the BRAF V660E and Cdkn2 deficient mutations relevant to human melanoma [30]. YUMM3.1 cells show some tumor suppression in the OPN-KI and OPN-KO mice but less than B16 cells. In contrast to B16 cells, YUMM3.1 cells express OPN, indicating that tumor-derived OPN is insufficient by itself to elicit the complete tumor suppression phenotype, and that host-derived OPN is required. The levels of the thrombin-cleaved OPN fragments may be the critical determinant in this process.
This tumor suppression phenotype in the OPN-KI mouse is lost in the immune deficient NOG-OPN-KI mice (Figure 4) and is reversed following macrophage depletion by clodronate (Figure 7), indicating that it is mediated by changes in TAMs. Macrophage activation has been broadly divided into a proinflammatory M1 state involved in responses of type 1 helper T cells to pathogens and an “alternatively activated” M2 state involved in type 2 helper T cell responses including humoral immunity and wound healing [31]. TAMs promote tumor initiation and progression [32] and are typically biased from M1 to M2 [33]. Indeed, higher numbers of M2 TAMs are associated with worse clinical prognosis [34]. The binary M1/M2 activation states, however, are insufficient to describe the much broader complexity of stimuli and responses by macrophages in vivo [35]. In response to cues in the tumor microenvironment, TAMs undergo dynamic functional reprogramming [36]. Our results showed that there was a significant change in the TAM’s activation phenotype, with a switch from the tumor-promoting M2 phenotype in WT mice to a phenotype defined by CD45+F4/80+CD11b+CD11c− CD206+Ly-6G− in the OPN-KI and OPN-KO mice (Figure 6). The functional characterization of this new macrophage activation state remains to be defined. Our results are consistent with studies in other cancer models with OPN-deficient mice. In a metastatic breast cancer model, OPN deficiency suppressed tumor growth and was associated with switching to a more immunosuppressive cellular subsets in myeloid-derived suppressor cells [37]. In a glioblastoma model, M2 macrophages were reduced in tumors from OPN-KO mice [38]. There was no investigation, however, of thrombin cleavage of OPN in these studies. Finally, we demonstrated that thrombin-cleaved OPN-R enhanced secretion of PGE2 by macrophages (Figure 7H). PGE2 induced tumor angiogenesis and enhanced B16 cell growth [17, 18, 29] and inhibition of PGE2 production by COX-1 inhibitors reduced B16 metastasis [39]. It should be noted that the initiation of the tumor-promoting activity of OPN by thrombin cleavage may not be mediated solely by OPN-R and PGE2. We have previously shown that thrombin cleavage of OPN disrupts a pro-chemotactic sequence in intact OPN, and this loss of its pro-chemotactic activity is compensated by the release of OPN-CTF, which assumes a new conformation and enhances chemokine-induced migration of dendritic cells [25]. Here, we also showed that OPN-CTF is more active than OPN-FL in its chemotactic activity towards B16 cells (Figure 3C). Thus, OPN-CTF, which does not have any known integrin-binding sites, is functionally active but its cellular receptor remains to identified. In contrast, overexpression of OPN in human melanoma cells results in integrin-dependent increases in migration and growth with integrin αV involved in the OPN response [40, 41]. Future studies are needed to define how thrombin-cleaved OPN fragments, be it OPN-R, OPN-CTF or OPN fragments that have undergone further proteolytic processing [42], modulate the activation states in TAMs and promote tumor progression and metastasis (Figure 6I).
While macrophage depletion reversed the immunosuppression phenotype in OPN-KI mice, it is notable that macrophage depletion had no effect on tumor growth in the WT mice (Fig. 7C and 7D). Even when tumor inocula of different sizes were compared, using 1x106 or 0.5x106 B16 cells instead of 2x106 cells, there was no significant increase in tumor growth with macrophage depletion (supplementary figure 6). The data suggest that in WT mice, thrombin cleavage of OPN leads to suppression of the host-anti-tumor immune response, that may involve both macrophage-dependent and macrophage-independent pathways, thus macrophage depletion does not lead to enhanced tumor growth in the WT mice. The macrophage-independent pathway(s) remains to be defined.
Our finding that thrombin cleavage of OPN modulates macrophage subsets in the B16 tumor model may be applicable to other pathological conditions. Metabolic-associated fatty liver disease (MAFLD), related to the increasing prevalence of obesity and associated insulin resistance, represents a spectrum of disease states ranging from steatosis to non-alcoholic steatohepatitis (NASH). Kupffer cells, the resident hepatic macrophages, are reduced in MAFLD and replaced by macrophages originating from the bone marrow [43]. These monocyte-derived macrophages exist in two subsets with distinct activation states - one resembles the homeostatic Kupffer cells, while the other resembles lipid-associated macrophages from obese adipose tissues and scar-associated macrophages in the fibrotic human liver [43]. These lipid-associated macrophages express OPN, which is a serum biomarker for NASH [44] and may be linked to the development of hepatic fibrosis. Blocking OPN has a protective effect in a mouse NASH model [45]. In addition, thrombin-cleaved OPN has been reported to induce collagen production in cardiac fibroblasts and may play a role in cardiac fibrosis [46]. Whether the OPN-KI mouse will show decreased hepatic or cardiac fibrosis in these mouse models remains to be determined.
Cancer cells express a variety of cancer procoagulants including tissue factor [47] that activate the blood coagulation cascade and lead to thrombin generation with cross-talk between coagulation activation, inflammation and cancer cell biology [48]. Hirudin, a parenteral direct thrombin inhibitor, prevented metastasis of B16 cells [49]. A Cochrane systematic review showed a survival benefit of parenteral heparin in cancer patients, particularly in patients with limited small cell lung cancers [50], although whether anticoagulation has a direct beneficial effect on cancer patient survival remains controversial [51]. Our data support the notion that thrombin cleavage of OPN, be it derived from the tumor and/or the host, leads to suppression of the host-antitumor immune response, and cancer cells exploit this mechanism to enhance their survival. Dabigatran, a direct orally active anticoagulant (DOAC), replicated the tumor suppression phenotype in both local tumor growth as well as metastasis in WT mice (Figure 2 and 3). Rivaroxaban, a DOAC targeting factor Xa in the clotting cascade, also promotes host-antitumor immunity by blocking factor Xa-protease-activated receptor 2 signaling in TAMs [52]. DOACs are now increasingly used clinically and have a superior therapeutic profile compared to warfarin, the conventional vitamin K antagonist [53]. As such, dabigatran and other DOACs might be considered as an adjunct to conventional chemotherapy in cancer treatment, if these observations are generalizable to other cancers. However, there is a higher incidence of bleeding complications in cancer patients upon anticoagulation, likely related to the abnormal tumor vasculature [54]. Thus, the development of a non-anticoagulant drug that effectively blocks thrombin cleavage of OPN without compromising the integrity of the blood clotting cascade would represent an attractive novel adjunctive therapy in cancer treatment.
In summary, this is the first direct demonstration of the role for thrombin cleavage of OPN in vivo. Thrombin cleavage of OPN, derived from either the tumor and/or the host, is required for the initiation of OPN’s tumor-promoting activity. Thrombin-cleaved OPN fragments have a major pathophysiological impact on cancer biology and may offer a unique opportunity to develop novel immunotherapeutic agents for cancer treatment by enhancing the host-antitumor immune response mediated by TAMs. Their clinical application may not be limited to cancer therapy since thrombin-cleaved OPN may play a pathophysiological role in other clinical conditions. Whether thrombin cleavage of OPN plays a physiological role, for example in buttressing the host immune response in sepsis, remains to be defined.
Supplementary Material
Essentials:
Thrombin cleavage of osteopontin (OPN) suppresses anti-tumor immunity promoting B16 tumor growth
Thrombin-cleaved OPN fragments modulate the tumor-associated macrophage phenotype
Dabigatran treatment replicates the tumor suppression phenotype in WT mice
A new link in the crosstalk between thrombosis, inflammation and immunity
Acknowledgements
The authors thank Marcus Gatto for tail vein injections, Jing Jin for help with aPTT assays, and Brandon Carter for assistance with the flow cytometry.
Financial support:
Funding for the creation of the OPN KI mouse was provided by the Palo Alto Institute for Research and Education (PAIRE) at VAPAHCS. This work was supported by NIH 1R01 HL57530, the Maureen Lyles D’Ambrogio Endowed Professorship at the Stanford University School of Medicine and VA grant BX005148 (all to LL); mentored and financially supported by Stanford’s SPARK Translational Research Program. These studies were also supported by the CCHD doctoral program of the FWF (W1205-B09), the Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development, Austria, via the Christian Doppler Society (to BW).
Footnotes
Conflict of interests: M.I. is an employee of CIEA. L.L., J.M. and T.M. are the inventors of a relevant patent. The other authors declare no competing interests.
References
- 1.Atai NA, Bansal M, Lo C, Bosman J, Tigchelaar W, Bosch KS, Jonker A, De Witt Hamer PC, Troost D, McCulloch CA, Everts V, Van Noorden CJ, Sodek J. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology. 2011; 132: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiodoni C, Colombo MP, Sangaletti S. Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010; 29: 295–307. [DOI] [PubMed] [Google Scholar]
- 3.Lamort AS, Giopanou I, Psallidas I, Stathopoulos GT. Osteopontin as a Link between Inflammation and Cancer: The Thorax in the Spotlight. Cells. 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shevde LA, Samant RS. Role of osteopontin in the pathophysiology of cancer. Matrix biology : journal of the International Society for Matrix Biology. 2014; 37: 131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singhal H, Bautista DS, Tonkin KS, O’Malley FP, Tuck AB, Chambers AF, Harris JF. Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 1997; 3: 605–11. [PubMed] [Google Scholar]
- 6.Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008; 19: 333–45. [DOI] [PubMed] [Google Scholar]
- 7.Moorman HR, Poschel D, Klement JD, Lu C, Redd PS, Liu K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers (Basel). 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osteopontin Uede T., intrinsic tissue regulator of intractable inflammatory diseases. Pathol Int. 2011; 61: 265–80. [DOI] [PubMed] [Google Scholar]
- 9.Grassinger J, Haylock DN, Storan MJ, Haines GO, Williams B, Whitty GA, Vinson AR, Be CL, Li S, Sorensen ES, Tam PP, Denhardt DT, Sheppard D, Choong PF, Nilsson SK. Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with alpha9beta1 and alpha4beta1 integrins. Blood. 2009; 114: 49–59. [DOI] [PubMed] [Google Scholar]
- 10.Sharif SA, Du X, Myles T, Song JJ, Price E, Lee DM, Goodman SB, Nagashima M, Morser J, Robinson WH, Leung LLK. Thrombin-activatable carboxypeptidase B-cleavage of osteopontin regulates neutrophil survival and synoviocyte binding in rheumatoid arthritis. Arthritis and rheumatism. 2009; 60: 2902–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, Reinhardt F, Harris LN, Hylander BL, Repasky EA, Weinberg RA. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008; 133: 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, Higashiyama S, Saitoh Y, Yamakido M, Taooka Y, Sheppard D. The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem. 1999; 274: 36328–34. [DOI] [PubMed] [Google Scholar]
- 13.Myles T, Leung LL. Thrombin hydrolysis of human osteopontin is dependent on thrombin anion-binding exosites. J Biol Chem. 2008; 283: 17789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, Pearl RG, Leung LL. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem. 2003; 278: 51059–67. [DOI] [PubMed] [Google Scholar]
- 15.Shao Z, Morser J, Leung LL. Thrombin cleavage of osteopontin disrupts a pro-chemotactic sequence for dendritic cells, which is compensated by the release of its pro-chemotactic C-terminal fragment. J Biol Chem. 2014; 289: 27146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi Y, Shao Z, Sharif S, Du XY, Myles T, Merchant M, Harsh G, Glantz M, Recht L, Morser J, Leung LL. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J Biol Chem. 2013; 288: 3097–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kale S, Raja R, Thorat D, Soundararajan G, Patil TV, Kundu GC. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene. 2014; 33: 2295–306. [DOI] [PubMed] [Google Scholar]
- 18.Kale S, Raja R, Thorat D, Soundararajan G, Patil TV, Kundu GC. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene. 2015; 34: 5408–10. [DOI] [PubMed] [Google Scholar]
- 19.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002; 100: 3175–82. [DOI] [PubMed] [Google Scholar]
- 20.Sparkenbaugh EM, Chantrathammachart P, Mickelson J, van Ryn J, Hebbel RP, Monroe DM, Mackman N, Key NS, Pawlinski R. Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood. 2014; 123: 1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Levesque JP. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010; 116: 4815–28. [DOI] [PubMed] [Google Scholar]
- 22.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One. 2015;10:e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, Frank AC, Scholich K, Pierre S, Syed SN, Olesch C, Ringleb J, Oren B, Doring C, Savai R, Jung M, von Knethen A, Levkau B, Fleming I, Weigert A, Brune B. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J Exp Med. 2017; 214: 2695–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nemoto H, Rittling SR, Yoshitake H, Furuya K, Amagasa T, Tsuji K, Nifuji A, Denhardt DT, Noda M. Osteopontin deficiency reduces experimental tumor cell metastasis to bone and soft tissues. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2001; 16: 652–9. [DOI] [PubMed] [Google Scholar]
- 25.Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer research. 2003; 63: 6751–7. [PubMed] [Google Scholar]
- 26.Katagiri YU, Murakami M, Mori K, Iizuka J, Hara T, Tanaka K, Jia WY, Chambers AF, Uede T. Non-RGD domains of osteopontin promote cell adhesion without involving alpha v integrins. Journal of cellular biochemistry. 1996; 62: 123–31. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi C, Rittling S, Hayata T, Amagasa T, Denhardt D, Ezura Y, Nakashima K, Noda M. Serum osteopontin, an enhancer of tumor metastasis to bone, promotes B16 melanoma cell migration. Journal of cellular biochemistry. 2007; 101: 979–86. [DOI] [PubMed] [Google Scholar]
- 28.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of immunological methods. 1994; 174: 83–93. [DOI] [PubMed] [Google Scholar]
- 29.Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim Biophys Acta. 2015; 1851: 414–21. [DOI] [PubMed] [Google Scholar]
- 30.Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment cell & melanoma research. 2016; 29: 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon S Alternative activation of macrophages. Nature reviews Immunology. 2003; 3: 23–35. [DOI] [PubMed] [Google Scholar]
- 32.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010; 141: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010; 22: 231–7. [DOI] [PubMed] [Google Scholar]
- 34.Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Advanced drug delivery reviews. 2016; 99: 180–5. [DOI] [PubMed] [Google Scholar]
- 35.Natoli G, Monticelli S. Macrophage activation: glancing into diversity. Immunity. 2014; 40: 175–7. [DOI] [PubMed] [Google Scholar]
- 36.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014; 40: 274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangaletti S, Tripodo C, Sandri S, Torselli I, Vitali C, Ratti C, Botti L, Burocchi A, Porcasi R, Tomirotti A, Colombo MP, Chiodoni C. Osteopontin shapes immunosuppression in the metastatic niche. Cancer research. 2014; 74: 4706–19. [DOI] [PubMed] [Google Scholar]
- 38.Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, Grami Z, Kong LY, Ling X, Caruso H, Zhou S, Wang YA, Fuller GN, Huse J, Gilboa E, Kang N, Huang X, Verhaak R, Li S, Heimberger AB. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. 2019; 129: 137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lucotti S, Cerutti C, Soyer M, Gil-Bernabe AM, Gomes AL, Allen PD, Smart S, Markelc B, Watson K, Armstrong PC, Mitchell JA, Warner TD, Ridley AJ, Muschel RJ. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest. 2019; 129:1845–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin M, Soikkeli J, Jahkola T, Virolainen S, Saksela O, Holtta E. Osteopontin promotes the invasive growth of melanoma cells by activating integrin alphavbeta3 and down-regulating tetraspanin CD9. Am J Pathol. 2014; 184: 842–58. [DOI] [PubMed] [Google Scholar]
- 41.Samanna V, Wei H, Ego-Osuala D, Chellaiah MA. Alpha-V-dependent outside-in signaling is required for the regulation of CD44 surface expression, MMP-2 secretion, and cell migration by osteopontin in human melanoma cells. Exp Cell Res. 2006; 312: 2214–30. [DOI] [PubMed] [Google Scholar]
- 42.Ellert-Miklaszewska A, Wisniewski P, Kijewska M, Gajdanowicz P, Pszczolkowska D, Przanowski P, Dabrowski M, Maleszewska M, Kaminska B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene. 2016; 35: 6366–77. [DOI] [PubMed] [Google Scholar]
- 43.Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, Pavie B, Roels J, Vanneste B, De Prijck S, Vanhockerhout M, Binte Abdul Latib M, Devisscher L, Hoorens A, Bonnardel J, Vandamme N, Kremer A, Borghgraef P, Van Vlierberghe H, Lippens S, Pearce E, Saeys Y, Scott CL. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glass O, Henao R, Patel K, Guy CD, Gruss HJ, Syn WK, Moylan CA, Streilein R, Hall R, Mae Diehl A, Abdelmalek MF. Serum Interleukin-8, Osteopontin, and Monocyte Chemoattractant Protein 1 Are Associated With Hepatic Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Hepatology communications. 2018; 2: 1344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honda M, Kimura C, Uede T, Kon S. Neutralizing antibody against osteopontin attenuates non-alcoholic steatohepatitis in mice. J Cell Commun Signal. 2020; 14: 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herum KM, Romaine A, Wang A, Melleby AO, Strand ME, Pacheco J, Braathen B, Duner P, Tonnessen T, Lunde IG, Sjaastad I, Brakebusch C, McCulloch AD, Gomez MF, Carlson CR, Christensen G. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. Journal of the American Heart Association. 2020; 9: e013518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hisada Y, Mackman N. Cancer cell-derived tissue factor-positive extracellular vesicles: biomarkers of thrombosis and survival. Current opinion in hematology. 2019; 26: 349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruf W, Rothmeier AS, Graf C. Targeting clotting proteins in cancer therapy - progress and challenges. Thromb Res. 2016; 140 Suppl 1: S1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niers TM, Bruggemann LW, VANS GL, Liu RD, Versteeg HH, Buller HR, VANN CJ, Reitsma PH, Spek CA, Richel DJ. Long-term thrombin inhibition promotes cancer cell extravasation in a mouse model of experimental metastasis. J Thromb Haemost. 2009; 7: 1595–7. [DOI] [PubMed] [Google Scholar]
- 50.Akl EA, Rohilla S, Barba M, Sperati F, Terrenato I, Muti P, Schunemann HJ. Anticoagulation for the initial treatment of venous thromboembolism in patients with cancer. The Cochrane database of systematic reviews. 2008: CD006649. [DOI] [PubMed] [Google Scholar]
- 51.O’Rorke MA, Murray LJ, Hughes CM, Cantwell MM, Cardwell CR. The effect of warfarin therapy on breast, colorectal, lung, and prostate cancer survival: a population-based cohort study using the Clinical Practice Research Datalink. Cancer causes & control: CCC. 2015; 26: 355–66. [DOI] [PubMed] [Google Scholar]
- 52.Graf C, Wilgenbus P, Pagel S, Pott J, Marini F, Reyda S, Kitano M, Macher-Goppinger S, Weiler H, Ruf W. Myeloid cell-synthesized coagulation factor X dampens antitumor immunity. Sci Immunol. 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chai-Adisaksopha C, Hillis C, Isayama T, Lim W, Iorio A, Crowther M. Mortality outcomes in patients receiving direct oral anticoagulants: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost. 2015; 13: 2012–20. [DOI] [PubMed] [Google Scholar]
- 54.Prandoni P, Lensing AW, Piccioli A, Bernardi E, Simioni P, Girolami B, Marchiori A, Sabbion P, Prins MH, Noventa F, Girolami A. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood. 2002; 100: 3484–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.